 Alejandro N. Rondón-Ortiz1
Alejandro N. Rondón-Ortiz1 Christian L. Lino Cardenas2,3
Christian L. Lino Cardenas2,3 Jimena Martínez-Málaga1,4Ana L. Gonzales-Urday1,4
Jimena Martínez-Málaga1,4Ana L. Gonzales-Urday1,4 Kuljeet S. Gugnani1
Kuljeet S. Gugnani1 Mark Böhlke1Timothy J. Maher1Alejandro J. Pino-Figueroa1*
Mark Böhlke1Timothy J. Maher1Alejandro J. Pino-Figueroa1*- 1Department of Pharmaceutical Sciences, MCPHS University, Boston, MA, United States
- 2Cardiovascular Research Center, Massachusetts General Hospital, Boston, MA, United States
- 3Scientific Consulting Group, BioMolecular-LC E.I.R.L, Arequipa, Peru
- 4Department of Pharmaceutical, Biochemical and Biotechnological Sciences, Catholic University of Santa Maria, Arequipa, Peru
Low-density lipoprotein receptor-related protein 1 (LRP1) is an endocytic receptor involved in the uptake of a variety of molecules, such as apoE, α2-macroglobulin, and the amyloid β peptide (Aβ), for either transcellular transport, protein trafficking or lysosomal degradation. The LRP1 gene can be transcribed upon activation of peroxisome proliferator receptor activated-γ (PPARγ) by the potent PPARγ agonist, rosiglitazone (RGZ). In previous studies, RGZ was shown to upregulate LRP1 levels in concentrations between 0.1 and 5 μM in HepG2 cells. In this study, we sought to replicate previous studies and to investigate the molecular mechanism by which high concentrations of RGZ reduce LRP1 levels in HepG2 cells. Our data confirmed that transcriptional activation of LRP1 occurred in response to RGZ at 3 and 10 μM, in agreement with the study reported by Moon et al. (2012a). On the other hand, we found that high concentrations of RGZ decreased both mRNA and protein levels of LRP1. Mechanistically, transcriptional dysregulation of LRP1 was affected by the downregulation of PPARγ in a time- and concentration-dependent manner. However, downregulation of PPARγ was responsible for only 40% of the LRP1 reduction and thereby the remaining loss of LRP1 (60%) was found to be through degradation in the lysosomal system. In conclusion, our findings demonstrate the mechanisms by which high concentrations of RGZ caused LRP1 levels to be reduced in HepG2 cells. Taken together, this data will be helpful to better explain the pharmacological modulation of this pivotal membrane receptor by PPARγ agonists.
Introduction
Low-density lipoprotein (LDL) receptor-related protein 1 (LRP1) is a transmembrane receptor that belongs to the LDL receptor family. LRP1 is ubiquitously expressed and has an important role in the transport and uptake of molecules (Lillis et al., 2008; Sagare et al., 2012). Indeed, LRP1 has been involved in the endocytosis of more than 40 structurally different ligands, including apoE, α2-macroglobulin, receptor associated protein (RAP) and the amyloid β peptide (Lillis et al., 2008; Ramanathan et al., 2015).
LRP1 consists of two chains that are non-covalently associated. The extracellular α-chain (515 KDa), composed of four ligand-binding clusters, is involved in the recognition and uptake of a diversity of LRP1 ligands. The transmembrane and cytoplasmic β-chain (85 KDa) binds adaptor proteins to facilitate mostly clathrin-mediated endocytosis (Lillis et al., 2008; Kanekiyo et al., 2013) and the more recently demonstrated caveolin-dependent endocytosis (Kanai et al., 2014).
At the transcriptional level, the LRP1 gene is regulated by PPARγ, due to the presence of the peroxisome proliferator response element (PPRE) on the LRP1 promoter region (Gauthier et al., 2003). PPARγ is a nuclear receptor involved in important biological processes, including adipogenesis and metabolism. This nuclear receptor is an interesting pharmacological target, since its activation regulates expression of proteins and enzymes involved in glucose metabolism, such as GLUT-2, GLUT-4, IRS-1, IRS-2, PI3K and others (Kim and Ahn, 2004; Ahmadian et al., 2013). Rosiglitazone (RGZ) is a member of the thiazolidinedione family and is a potent PPARγ agonist, known for its antidiabetic properties. However, RGZ has been associated with cardiovascular risks, resulting in a reconsideration of its therapeutic use (Nissen and Wolski, 2007; Hiatt et al., 2013). Indeed, results of some studies have indicated that RGZ modulates LRP1 expression by targeting PPARγ expression in cell culture models, including the HepG2 cell line (Gauthier et al., 2003; Moon et al., 2012a,b).
In this study, we proposed to replicate previous observations and to further explore the effects of RGZ on LRP1, by utilizing HepG2 cells. Our findings confirmed that transcriptional activation of LRP1 is induced by 3 and 10 μM RGZ, whereas higher concentrations (30–100 μM) reduce LRP1 mRNA levels. On the other hand, LRP1 protein levels remained steady in response to 0–10 μM RGZ, but concentrations of RGZ higher than 30 μM dramatically reduced LRP1 protein levels. We subsequently focused our investigation to identify the possible mechanisms by which high concentrations of RGZ decrease LRP1 levels. Indeed, we found that both mRNA and protein levels were negatively affected by two different mechanisms. LRP1 mRNA was reduced by the downregulation of PPARγ in a time- and concentration-dependent manner. Of interest, this mechanism was only responsible for 40% of the reduction of LRP1. The remaining LRP1 protein was found to undergo lysosomal degradation. These results might help to determine whether the side effects caused by RGZ, including those related to cardiovascular risk, are associated with LRP1 reduction.
Materials and Methods
Materials
MEM-alpha cell culture medium, OptiMEM reduced-serum medium, fetal bovine serum (FBS), penicillin and streptomycin were obtained from Gibco. Rosiglitazone, T0070907, MG132, bafilomycin A1, chloroquine diphosphate, pepstatin A and E64d were from Tocris. Anti-LRP1 (ab92544); anti-LDLR (ab52818) anti-β-actin (ab49900); secondary antibodies: HRP-conjugated goat anti-rabbit IgG (ab97051), HRP-conjugated goat anti-mouse IgG (ab97023), Alexa Fluor 488 conjugated goat anti-rabbit IgG (ab150077) and Alexa Fluor 594 conjugated goat anti-mouse IgG (ab150116), and concanamycin A were from Abcam. The anti-LC3α/β antibody (SC-398822) was purchased from Santa Cruz Biotechnologies. The anti-LAMP1 (D4O1S) and anti-caveolin-1(D46G3) were purchased from Cell Signaling. 6-Carboxyfluorescein-labeled (FAM) Aβ1-42 was obtained from Anaspec. TRIzol reagent and the transcriptor first strand cDNA synthesis kit were from Thermo Fisher and Roche, respectively. DMSO and other reagents were from Sigma–Aldrich.
Cell Culture
Hepatocellular carcinoma HepG2 cells were obtained from the American Type Culture Collection (HB-8065, ATCC). Cells were seeded in MEM-alpha medium, supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were then grown in a humidified environment with 5% CO2 until they were confluent. For experiments, HepG2 cells were seeded into 12-well (2.5 × 105 cells/well) or 6-well (5 × 105 cells/well) plates and were cultured for 48 h, for uptake and protein analysis experiments, respectively. Cells were then exposed to different concentrations of RGZ (3 nM–30 μM) in reduced-serum medium (OptiMEM). The final concentration of DMSO in each experiment was less than 0.5%. Other experiments were carried out in MEM containing 10% FBS, as explained in the Supplementary Information Section.
RNA Isolation and cDNA Synthesis
Following treatments, RNA was extracted from cells with TRIzol reagent. Isolated RNA was quantified utilizing a NanoDrop 2000 spectrophotometer (Thermo Fisher). cDNA was then prepared with the transcriptor first strand cDNA synthesis kit according to the manufacturer’s protocol. The final cDNA solution was stored at -20°C prior to analysis.
Quantitative Real-Time PCR (RT-qPCR)
cDNA from samples was analyzed and quantified by RT-qPCR using TaqMan assay probes for LRP1 (Hs00233856_m1), PPARG (Hs00234592_m1) and ACTB (Hs01060665_g1) in a StepOne Plus station (Applied Biosystems). This reaction was carried out in triplicate in microamp optical 96-well plates in a total volume of 10 μL/well. The relative LRP1 expression levels were calculated as the fold-change determined by the ΔΔCt method with ACTB representing a housekeeping gene.
Cell Viability
HepG2 cells were seeded into 96-well plates and were exposed to different concentrations of RGZ. Treatments were carried out with the control group consisting of DMSO-treated cells. Cell viability was determined by the colorimetric method utilizing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS, CellTiter 96®AQueous One Solution, Promega). Cell viability was determined at 490 nm and was calculated as percent of control.
Protein Extraction and Western Blot
HepG2 cells were rinsed twice with ice-cold PBS and proteins were extracted with M-PER and MEM-PER, for whole cell lysis and membrane isolation, respectively (both from Thermo Fisher). These lysis buffers contained Halt protease, phosphatase inhibitors and EDTA (Thermo Fisher). The protein concentration was determined by the colorimetric bicinchoninic acid assay (BCA assay, Thermo Fisher). Equal amounts of total protein from cell lysates were separated by SDS-PAGE (20 and 40 μg for LRP1 and LC3, respectively). Proteins from the gel were then electro-transferred onto 0.45 μm nitrocellulose and 0.2 μm PVDF membranes for LRP1 and LC3, respectively.
The membranes were then blocked for 1 h at room temperature (rt), with either 5% non-fat powdered milk dissolved in TBS-T or 5% bovine serum albumin in TBS-T, for the nitrocellulose and PVDF membranes, respectively. Following blocking, membranes were incubated overnight at 4°C with the primary antibodies anti-LRP1 (1:10,000), anti-LDLR (1:2,000), anti-caveolin1 (1:1,000) and anti-LC3 (1:500). The membranes were subsequently washed three times with TBS-T and were then exposed to HRP-conjugated goat anti-rabbit IgG (1:2,000) or HRP-conjugated goat anti-mouse IgG (1:2,000) for 1 h at rt. β-Actin was used as the loading control; the membranes were incubated with HRP-conjugated mouse polyclonal anti-β-actin (1:10,000) for 1 h at rt. SuperSignal West Pico chemiluminescent substrate (Thermo Fisher) was added to the membranes and they were incubated for 5 min. Bands were visualized with the C-DiGit blot scanner (Licor Technologies).
Cell Transfection
HepG2 cells were grown in MEM alpha medium. The LRP1 construct (Mini LRP-IV-EGFP) was kindly provided by Dr. Michel Khrestchatisky and Dr. Marion David (Université d’Aix-Marseille and Vect-Horus, Marseille, France, respectively; Perrot et al., 2012). Cells were transiently transfected with 1.5 μg of pDNA using Lipofectamine 2000 (Thermo Fisher), following manufacturer’s protocol.
Confocal Microscopy
For immunocytochemistry, HepG2 cells were cultured into 8-well Lab-TekTM II Chamber Slides (NuncTM) and were then treated with either RGZ or inhibitors. Cells were rinsed twice with ice-cold PBS, fixed with 4% paraformaldehyde in PBS (PFA, Boston Bioproducts) for 15 min at rt, and were permeabilized with 0.1% Triton-X (Sigma–Aldrich) for 10 min. The slides were blocked with 10% goat-serum, and 0.3 M glycine in PBS-Tween 20 (0.1%) for 1 h at rt. Subsequently, the antibodies anti-LRP1 (1:100), anti-LAMP1 (1:50) and anti-LC3 (1:50) were added and slides were incubated overnight at 4°C. The slides were then washed 3 times for 5 min each with PBS-T and were incubated with Alexa Fluor 488 conjugated goat anti-rabbit IgG and Alexa Fluor 594 conjugated goat anti-mouse IgG (each at 1:400 dilution) for 1 h at rt. Following immunostaining, slides were mounted with diamond mounting medium containing DAPI (Thermo Fisher). Slides were then visualized with the Leica TCS SP8 confocal microscopy station and the pictures were digitized with the Leica Application Suite X software. Recordings of the 3D-reconstruction can be found in the Supplementary Information Section. For time-lapse imaging microscopy, HepG2 cells were cultured in 35 mm μ-Dishes (Ibidi) overnight. Following transfection with the mini-LRP-IV-EGFP plasmid, cells were treated with either vehicle or RGZ (30 μM) for 22 h. Deep red lysotracker (Thermo Fisher) was added to visualize the lysosomal compartment. Single z-plane images were recorded every 3 min for the next 2 h from both control and RGZ-treated cells. Cells were visualized with the Leica TCS SP8 confocal microscopy station and pictures were digitized with the Leica Application Suite X software. Recordings of time-lapse imaging can be found in the Supplementary Information Section.
Aβ1-42 Preparation and Uptake
Synthetic FAM-Aβ1-42 peptide was resuspended as previously described (Fuentealba et al., 2010; Kanekiyo et al., 2013). Briefly, FAM-Aβ1-42 was resuspended in 1,1,1,3,3,3-hexafluoroisopropanol and aliquoted into amber tubes. The solvent was evaporated with a stream of nitrogen gas and tubes containing the dried film were stored at -20°C until they were utilized for uptake studies. For experiments, the films were resuspended in DMSO to obtain a 100 μM stock solution and were then diluted to 500 nM FAM-Aβ1-42 in OptiMEM. HepG2 cells were treated with RGZ (30 μM) for 24 or 48 h. Following this treatment, HepG2 cells were incubated with fresh FAM-Aβ1-42 (500 nM) for 30 min, 1 and 2 h. Cells were then lysed with M-PER, as above, and fluorescence intensity was monitored with the Synergy HT plate reader (Biotek), with excitation at 485/20 nm and emission at 528/20 nm. FAM-Aβ1-42 uptake is expressed as relative fluorescence units per mg protein (RFU/mg).
Statistical Analysis
All statistical analyses were conducted using GraphPad Prism 6. Data are reported as mean ± SEM for at least three independent experiments. Comparisons between two or multiple groups were performed by a two-tailed Student’s t-test and one-way analysis of variance (ANOVA), respectively. The ANOVA was followed by a post hoc Dunnet’s test, in which experimental groups were compared to controls (cells treated with vehicle). A value of p < 0.05 was considered to demonstrate a significant difference between treatments.
Results
Effect of RGZ on LRP1 Levels
Previous studies demonstrated the modulation of LRP1 by the transcription factor PPARγ in cells treated with RGZ (Gauthier et al., 2003; Moon et al., 2012a). In order to confirm the RGZ effect on LRP1 expression, we replicated the experiments previously reported, which utilized HepG2 cells (Moon et al., 2012a). In agreement with the literature, we observed the transcriptional activation of LRP1 mRNA in cells treated with RGZ at 3 and 10 μM for 24 h (Figure 1A). However, we did not observe increases in LRP1 protein levels following either 24 or 48 h exposures to these concentrations of RGZ (Figures 1C,D). Other variables were also tested, such as incubation time, RGZ concentration and presence of serum in the treatments, but we did not observe the LRP1 protein upregulation previously described (Moon et al., 2012a, Figure 1 and Supplementary Figure 1). In contrast, higher concentrations than 10 μM RGZ decreased both mRNA and protein LRP1 levels significantly after 24 and 48 h treatments. In addition, the effect of RGZ seemed to be specific to reduce LRP1, since LDLR (another member of the LDL receptor family that undergoes clathrin-dependent endocytosis) and caveolin-1 (a marker for caveolae-dependent endocytosis) were not altered in the presence of RGZ (Figures 1C,D). We established that these effects were not due to cytotoxicity, as cell viability was maintained at these RGZ concentrations (Figure 1B and Supplementary Figure 1).
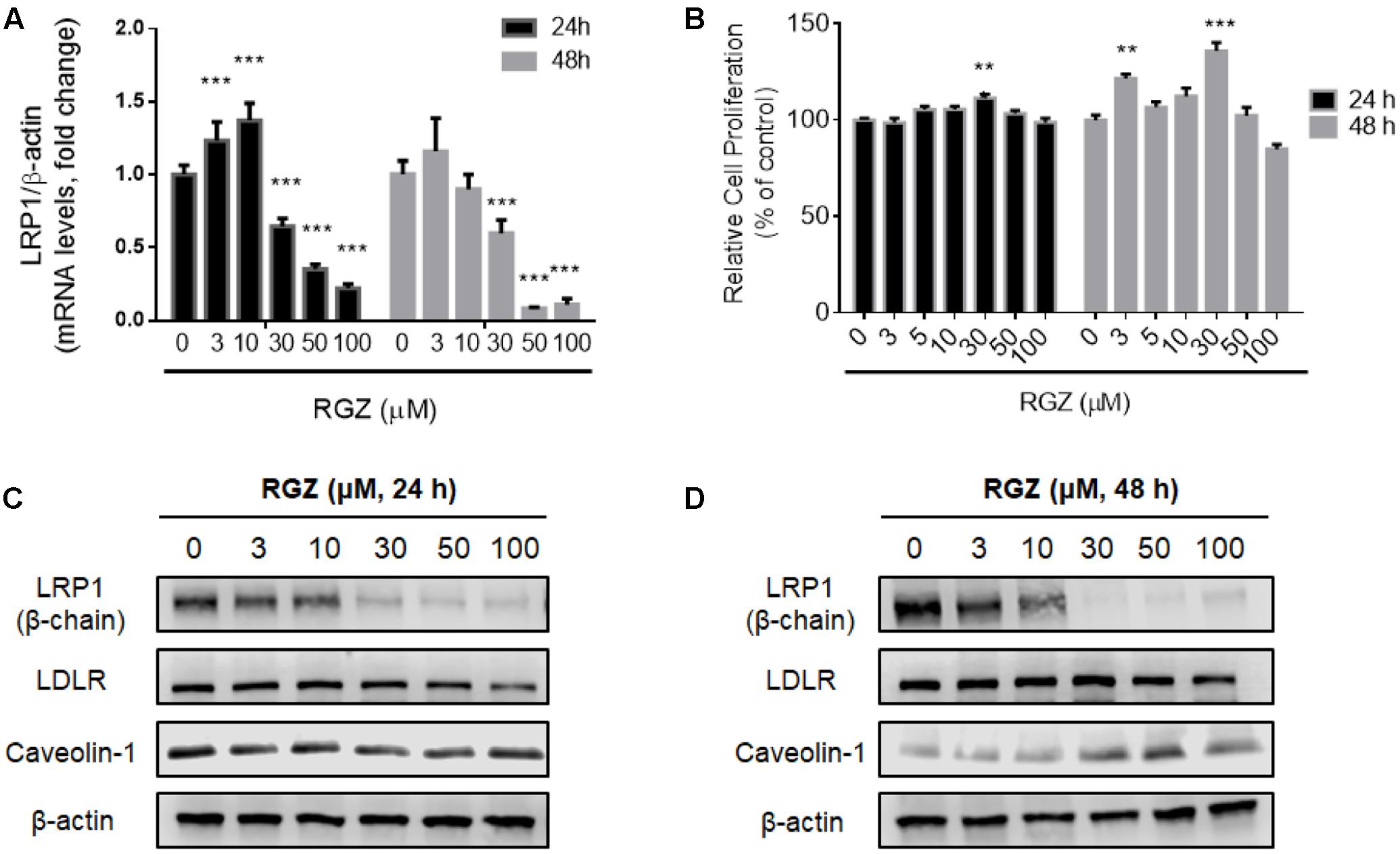
FIGURE 1. Effects of RGZ on LRP1 levels in HepG2 cells. RGZ (3 and 10 μM) increased LRP1 mRNA levels at 24 h. However, the highest concentration of RGZ (30 μM) reduced mRNA levels significantly and this effect was also observed at 48 h (A, n = 6). Cell proliferation assay demonstrated that RGZ did not cause cytotoxicity at these concentrations (B, n = 6). LRP1 protein levels remained steady (β-chain 85 Kda) in response to 3 and 10 μM RGZ compared to control for 24 and 48 h. Nonetheless, concentrations higher than 10 μM RGZ reduced LRP1 protein levels (C,D, representative blots). Data are presented as mean ± SEM. In the one-way ANOVA, followed by Dunnet’s test and compared to control group, p < 0.05 was considered significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
High Concentration Rosiglitazone Reduces both LRP1 mRNA and Protein Levels
The LRP1 gene contains the PPRE that is recognized by activated PPARγ to begin the transcription of LRP1 (Gauthier et al., 2003; Moon et al., 2012a,b; Kim et al., 2014). However, we observed that higher concentrations than 10 μM RGZ reduced LRP1 mRNA levels in a time- and concentration-dependent manner (Figure 1A). Previous studies demonstrated that high concentrations of PPARγ agonists, including RGZ and pioglitazone, reduce PPARγ levels (Hauser et al., 2000; Kilroy et al., 2009). We observed that LRP1 mRNA reduction is a consequence of the PPARγ downregulation resulting from exposures of cells to RGZ at concentrations higher than 10 μM in a time- and concentration-dependent manner, as shown in Figure 2A. In addition, we tested whether PPARγ antagonism by T0070907 (T007) affects LRP1 protein levels. Our observation was that PPARγ antagonism reduced LRP1 protein levels in a concentration-dependent manner (Figure 2B).
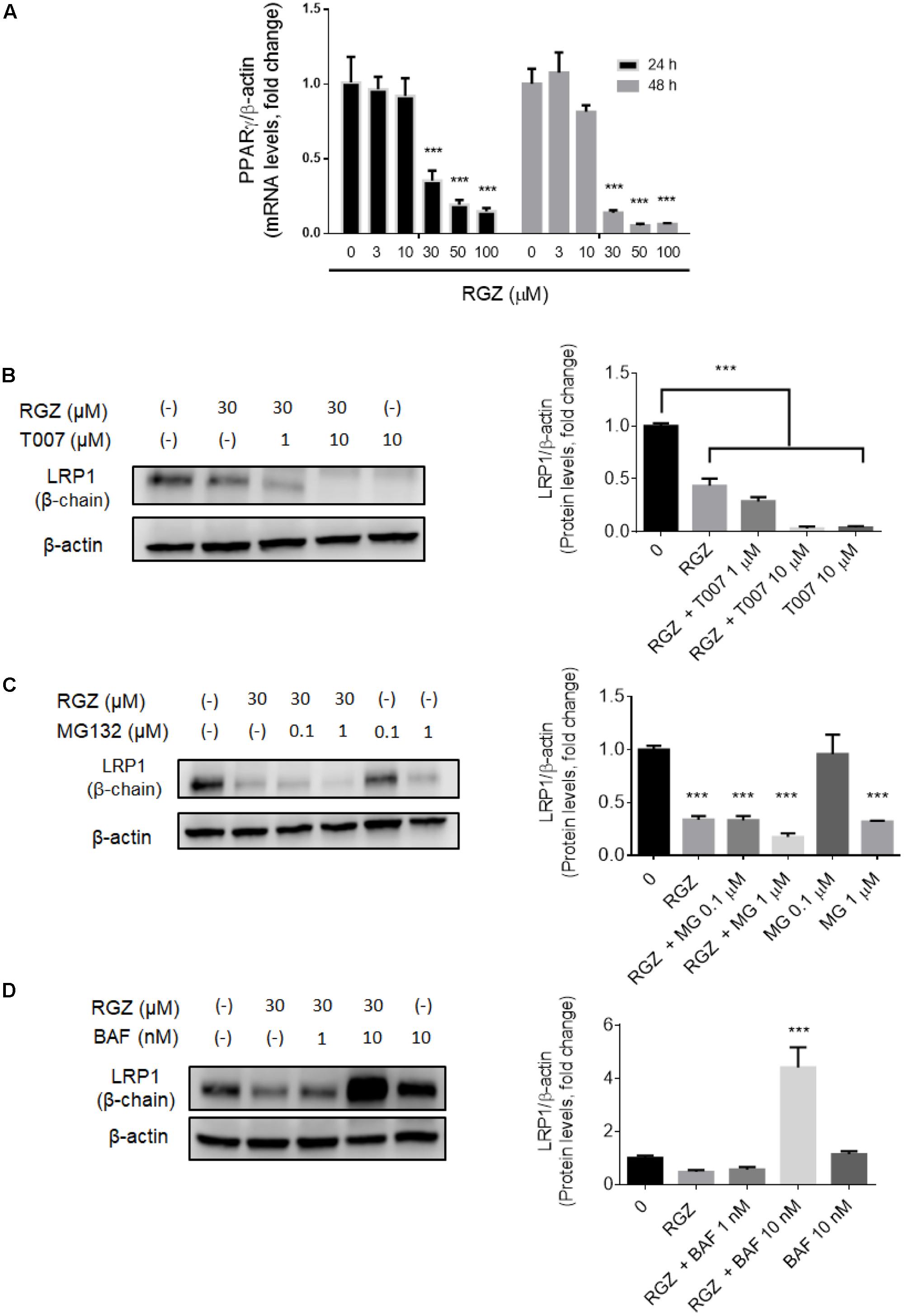
FIGURE 2. High concentrations of RGZ reduced both LRP1 mRNA and protein by two independent mechanisms in HepG2 cells. LRP1 mRNA reduction might be a consequence of PPARγ downregulation induced by RGZ in a time-and concentration-dependent manner (A, n = 6). This process is confirmed when PPARγ is antagonized by T007. T007 reduced LRP1 protein levels in a concentration-dependent manner (B, n = 4). UPS inhibition by MG132 did not prevent LRP1 degradation (C, n = 4). However, lysosomal degradation inhibitor (Bafilomycin A1, BAF) increased LRP1 by up to a four-fold change in RGZ-treated HepG2 cells compared to control group after 24 h incubation (D, n = 4). Data are presented as mean ± SEM. In the one-way ANOVA, followed by Dunnet’s test and compared to the control group, p < 0.05 was considered significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
While reduced LRP1 mRNA levels can be explained by PPARγ downregulation, we observed that high concentrations of RGZ reduced LRP1 protein levels to a greater extent than LRP1 transcription (Figure 1). This suggests that in addition to the transcriptional effect on LRP1 levels, a possible proteolytic mechanism might be affecting LRP1 protein levels in RGZ-treated HepG2 cells. We then investigated two proteolytic degradation mechanisms as possible explanations for these observations: the ubiquitin proteasome system (UPS) and lysosomal degradation.
The ubiquitin proteasome system (UPS) is the primary mechanism for protein degradation in eukaryotic cells (Vilchez et al., 2014). Previous studies have demonstrated that the pharmacological activation of PPARγ promotes its own proteolytic degradation via UPS (Hauser et al., 2000; Kilroy et al., 2009). In addition, it has been proposed that UPS is the key regulator for LRP1 degradation in a number of cell lines, including HepG2 (Melman et al., 2002; Cal et al., 2013). The use of MG132, a potent UPS inhibitor, in HepG2 cells treated with RGZ (30 μM) for 24 h did not prevent LRP1 protein degradation, as shown in Figure 2C. We also observed that MG132 reduced LRP1 protein levels in a concentration-dependent manner (Figure 2C and Supplementary Figure 2). These results excluded the possibility that LRP1 degradation occurred by the UPS pathway in RGZ-treated HepG2 cells. This suggested that RGZ reduced the amount of LRP1 present by a mechanism other than UPS.
Since LRP1 has been demonstrated to be involved in the transport of its ligands to the lysosomal compartment for degradation (Laatsch et al., 2012; Gan et al., 2014; Zhao et al., 2015), we decided to study the status of lysosome proteolytic degradation in RGZ-treated HepG2 cells. Cells were incubated with bafilomycin A1 (BAF), a vacuolar H+-ATPase inhibitor which blocks lysosomal activity, in the presence or absence of RGZ (30 μM) for 24 h. Western blot analysis demonstrated increased LRP1 protein levels, up to a 4-fold change, in cells co-treated with BAF (10 nM) and RGZ (30 μM) compared to the control, as depicted in Figure 2D. These results suggest that during RGZ treatment, LRP1 undergoes proteolytic degradation through the lysosomal system.
High Concentration RGZ Promotes LRP1 Lysosomal Degradation in HepG2 Cells
A long exposure of RGZ-treated HepG2 cells to BAF might affect other non-specific cellular events, such as UPS activation and protein aggregation, as previously reported (Korolchuk et al., 2009; Rubinsztein et al., 2009). We reduced BAF incubation time to the last 4 h of the experiment (total time of 24 h). In addition, cells were exposed to a mixture of the cathepsin inhibitors pepstatin A (PepA) and E64d, each at 30 μM. The purpose of the PepA/E64d mixture was to reduce lysosomal activity. As expected, RGZ reduced LRP1 levels, determined both by immunocytochemistry and Western blot analysis. On one hand, BAF treatment significantly increased LRP1 signal both in the presence or absence of RGZ, but PepA/E64d did not prevent LRP1 degradation, probably because PepA and E64d act downstream from BAF (Figures 3A, 4A). This suggests that BAF is capable to prevent the lysosomal degradation of LRP1 caused by RGZ.
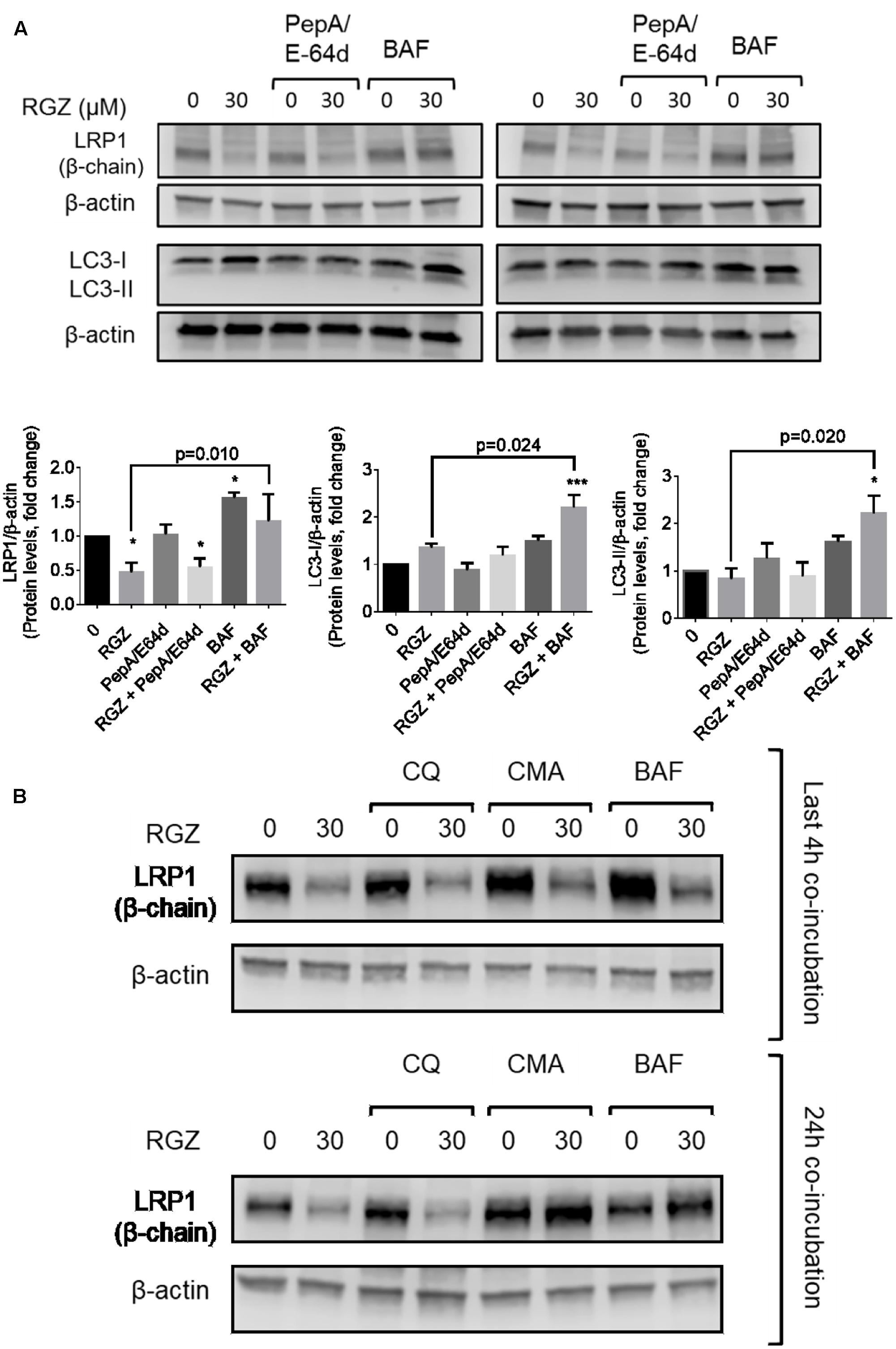
FIGURE 3. LRP1 protein levels are restored by lysosomal activity inhibitors in RGZ-treated HepG2 cells. BAF prevented LRP1 degradation in the last 4 h of exposure, whereas cathepsin inhibitors (PepA/E64d) failed to prevent LRP1 degradation. RGZ is associated with induced autophagy in cancer cells, as observed in the increase of LC3-II when lysosomal activity is blocked by BAF (A, n = 4). The lysosomotopic agent chloroquine (CQ) failed to prevent LRP1 degradation, while the potent vacuolar-type H+ ATPase inhibitor concanamycin A (CMA) prevented RGZ-induced LRP1 degradation (B, representative blot). Data are presented as mean ± SEM. In the one-way ANOVA, followed by Dunnet’s test and compared to the control group, p < 0.05 was considered significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). Then, a two-tailed Student’s t-test was performed to compare RGZ vs. RGZ and BAF and a p < 0.05 was considered significant.
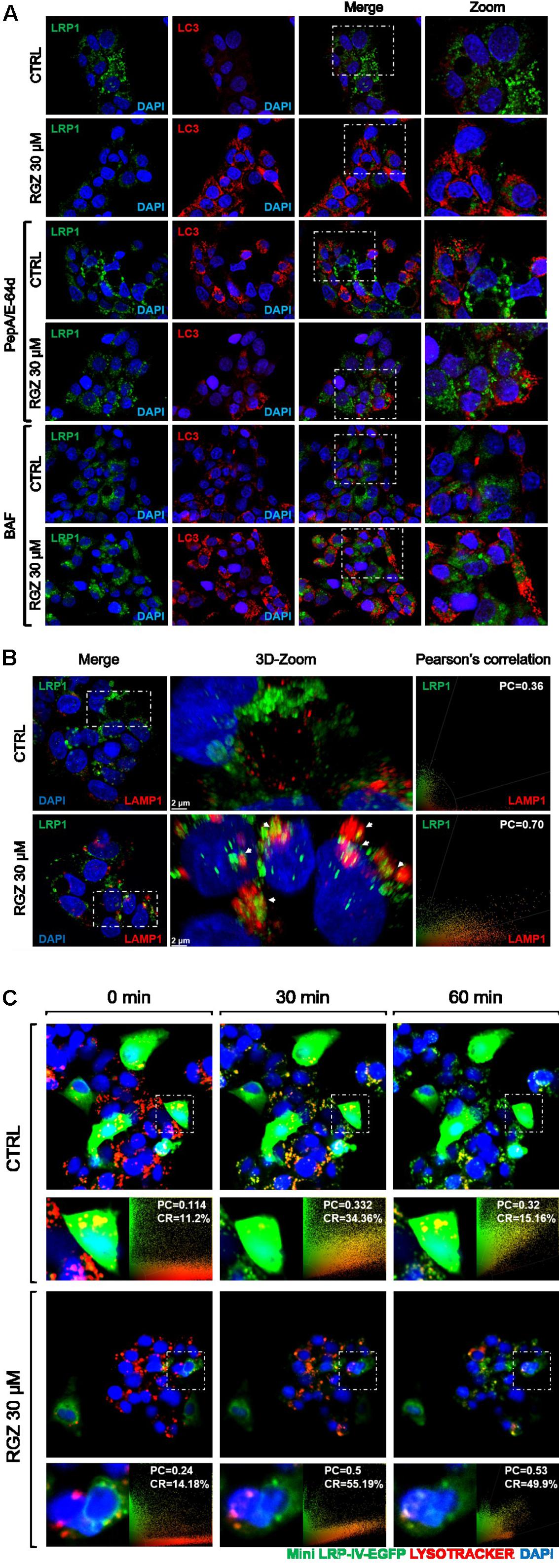
FIGURE 4. RGZ promotes LRP1 lysosomal degradation independently of autophagy. RGZ increased LC3 puncta in HepG2 cells, whereas LC3 did not co-localize with LRP1. This suggests that LRP1 is degraded by a process other than autophagy (A, representative slide). RGZ promoted LRP1/LAMP1 co-localization demonstrated by 3D-reconstruction confocal microscopy to a higher extent (PC = 0.70), compared with control (PC = 0.36); as observed in (B) (a representative z-projection one of 13 to 16 cuts). Deep red lysotracker was incubated with ectopically over-expressed fluorescent LRP1 construct (Mini-LRP-IV-EGFP) HepG2 cells after 22 h of RGZ treatment. Time-lapse imaging was used to monitor both lysotracker and GFP signal. Mini-LRP-IV-EGFP co-localized with positive lysosome compartment sensor and GFP signal was reduced in RGZ-treated cells compared with control, suggesting that RGZ promoted lysosomal degradation of ectopically over-expressed LRP1 (C). PC: Pearson’s co-localization, CR: Co-localization rate.
BAF inhibits the last stage of autophagy, consisting of lysosomal degradation (Klionsky et al., 2016); and RGZ has been associated with autophagy in cancer cell lines (Zhou et al., 2009; Vara et al., 2013). To assess whether autophagy induced by RGZ is responsible for LRP1 degradation, the autophagosome marker LC3 was analyzed by immunocytochemistry and Western blot. Our observations demonstrated that RGZ (30 μM) induced autophagy by the presence of LC3 puncta, monitored by immunocytochemistry and LC3-II generation by Western blot. Moreover, the presence of BAF induced LC3 accumulation (Figures 3A, 4A). In addition, reduced levels of LRP1 were also observed in EBSS-starved HepG2 cells, but BAF prevented LRP1 degradation (Supplementary Figure 3). LRP1 did not co-localize with LC3, suggesting that LRP1 degradation induced by RGZ is not an autophagy-dependent process, but RGZ might promote LRP1 lysosomal degradation (Figure 4A).
We found that BAF prevented LRP1 proteolytic degradation induced by RGZ (Figures 2D, 3A). We then tested whether other lysosomal activity inhibitors would prevent LRP1 degradation in 30 μM RGZ-treated cells. Our observations were that 10 nM concanamycin A (CMA), another vacuolar H+-ATPase inhibitor, had an effect similar to that of BAF in RGZ-treated cells. On the other hand, the lysosomotropic agent chloroquine (CQ, known to increase the lysosomal pH and thus reduce activity of some lysosome proteolytic enzymes) at 30 μM did not prevent RGZ-induced LRP1 degradation (Figure 3B). These observations suggest that lysosomal enzymes which are affected by CQ and PepA/E64d, are not responsible for LRP1 degradation. Taken together these results suggest that BAF and CMA, which act more upstream than CQ and PepA/E64d, prevented LRP1 lysosomal degradation caused by 30 μM RGZ.
High Concentration RGZ Promotes Intracellular Localization of LRP1 with Lysosomes
Since previous observations suggested that 30 μM RGZ might promote LRP1 lysosomal protein degradation (Figures 2D, 3B), we immunostained LRP1 and the lysosomal marker LAMP1 to determine LRP1 lysosomal localization. Our findings confirmed that LRP1 co-localizes with LAMP1 in the 3D-reconstruction we obtained using confocal microscopy (Figure 4B and Supplementary Figure 4). In addition, HepG2 cells were transfected with a functional LRP1 construct (Mini LRP-IV-EGFP; Perrot et al., 2012). Transfected HepG2 cells were then treated with 30 μM of RGZ for 22 h and we performed time-lapse imaging to monitor this fluorescent construct in the presence of lysotracker over a 1 h period. Lysotracker is a fluorescent dye used to label and to monitor acidic cellular compartments, including lysosomes. Our findings demonstrated that the mini-LRP-IV-EGFP construct co-localized with lysotracker to a greater extent in RGZ-treated cells than in the control. Moreover, the fluorescence intensity was constant in the control, while RGZ-treated cells displayed a reduced fluorescence intensity (Figure 4C). This suggested that RGZ induces lysosomal degradation of both endogenous LRP1 protein and ectopically overexpressed fluorescent Mini-LRP-IV-EGFP.
High Concentration RGZ Reduces LRP1 Activity
RGZ (30 μM) was demonstrated to reduce both LRP1 mRNA and protein levels, by the mechanisms of downregulation of PPARγ and protein lysosomal degradation, respectively (Figures 1–3). Given that the primary role of LRP1 is to endocytose its ligands at the cell surface (Perrot et al., 2012; Kanekiyo et al., 2013; Ramanathan et al., 2015) we evaluated LRP1 plasma membrane (PM) levels in cells treated with different concentrations of RGZ. Our results demonstrate that 30 μM RGZ reduced LRP1 PM levels, whereas 3 and 10 μM did not affect LRP1 PM levels. In addition, LDLR PM levels were not affected by RGZ, suggesting the specificity of RGZ to regulate LRP1 levels (Figure 5A).
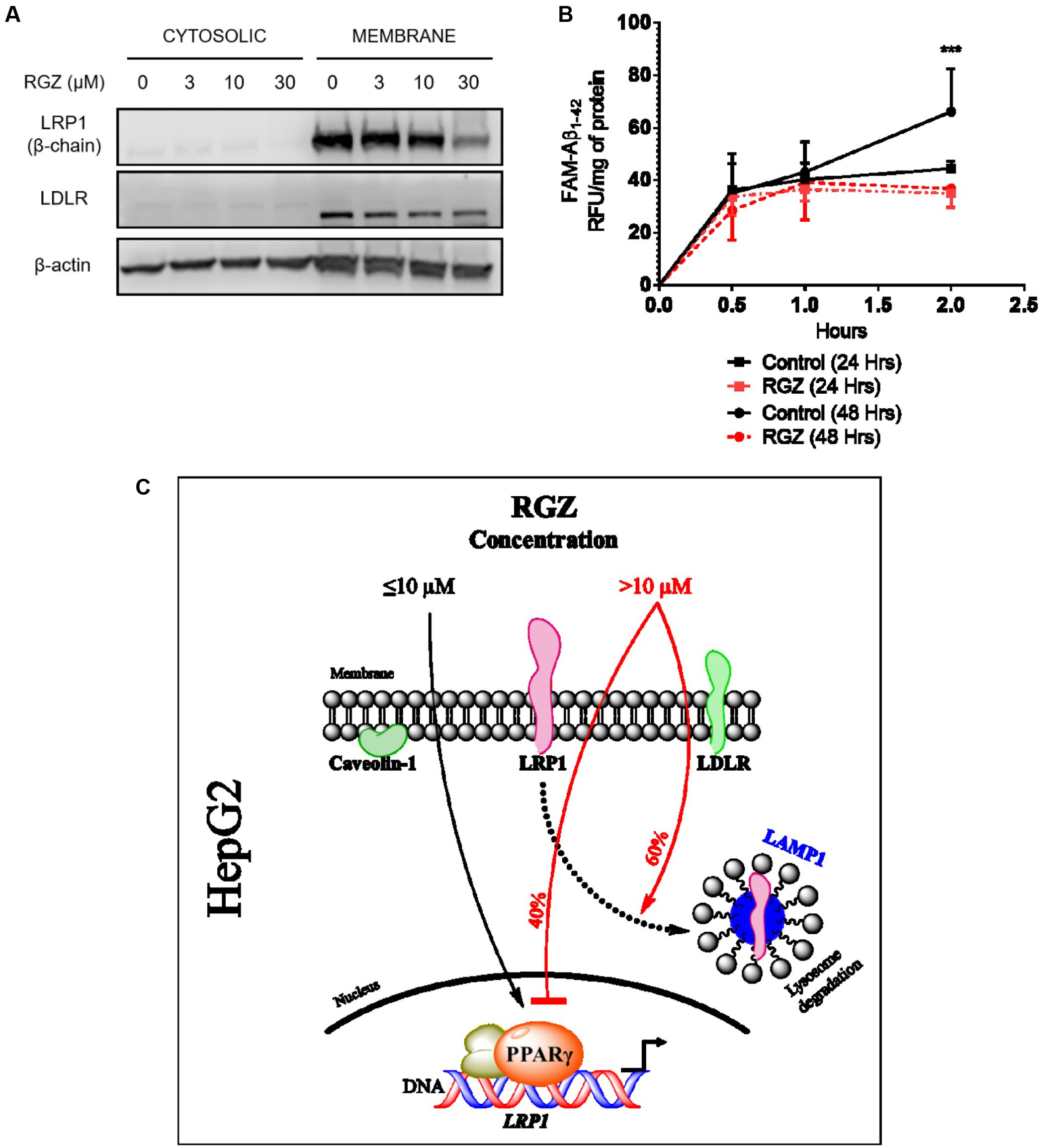
FIGURE 5. RGZ reduces LRP1 PM levels and impairs LRP1 activity. LRP1 at the cell surface area was significantly reduced by 30 μM RGZ, whereas LDLR PM levels remained steady in response to different concentrations of RGZ (A, representative blot). RGZ (30 μM), after 48 h of treatment, impaired FAM-Aβ1-42 peptide uptake during 2 h of incubation. This suggests that RGZ reduced LRP1 activity (B, n = 6). Summary representation of our study (C). Data are presented as mean ± SEM. A two-tailed Student’s t-test was performed to compare RGZ vs. CTRL at 2 h post-incubation with FAM-Aβ1-42 and a p < 0.05 was considered significant. (∗∗∗p < 0.001).
The fluorescent peptide Aβ1-42 (FAM-Aβ1-42) is one of the hallmarks of Alzheimer’s disease patients and this peptide was previously demonstrated to have a high affinity for LRP1 (Deane et al., 2004; Kanekiyo et al., 2013) Moreover, hepatic cells have an important role in the clearance and uptake of systemic Aβ via LRP1 endocytosis (Tamaki et al., 2006, 2007; Sagare et al., 2011). Thus, we tested whether the LRP1 reduction caused by 30 μM RGZ affected FAM-Aβ1-42 uptake in HepG2 cells. Our findings demonstrated that following 24 and 48 h incubations with RGZ, only the 2 h exposure of FAM-Aβ1-42, following the 48 h incubation, resulted in a significant difference in FAM-Aβ1-42 uptake compared to control (p < 0.001; Figure 5B). This impaired uptake correlated with lower LRP1 levels in the whole cell lysates and PM, presented in Figures 1, 2, and 5A.
Discussion
LRP1 is a ubiquitous transmembrane receptor, with a role in the internalization of more than 40 structurally different ligands, which are either transported to lysosomes for degradation or are transported transcellularly via endosomal trafficking (Sagare et al., 2012; Ramanathan et al., 2015). Previous reports have identified a conserved PPRE consensus sequence on the promoter region of the LRP1 gene, which can be transcriptionally activated by PPARγ agonists such as rosiglitazone (RGZ) in HepG2, HBMEC and SW872 cells (Gauthier et al., 2003; Moon et al., 2012a,b). A novel molecular interplay between LRP1 and PPARγ has recently been documented, suggesting that LRP1 might act as a co-activator of PPARγ to regulate glucose and lipid homeostasis in endothelial cells (Mao et al., 2017). In addition, LRP1 has been suggested to be involved in the metabolism and uptake of glucose, by interacting with insulin receptor-β (Jedrychowski et al., 2010; Liu et al., 2015). LRP1 PM residency is influenced by insulin signaling, as it has been observed that insulin-stimulated cells have increased LRP1 endocytic activity (Tamaki et al., 2007; Laatsch et al., 2009). Based on this evidence and on the role of LRP1 in cargo transport by endocytosis, there is no doubt that LRP1 dysregulation will affect important cell processes, and could be associated with disease states including diabetes, considering its effects on insulin and glucose metabolism. Moreover, low levels of LRP1 have been observed in Alzheimer’s disease patients (Zhao et al., 2015), in certain cancers (Perrot et al., 2012; Van Gool et al., 2015), in cases of reduced glucose uptake and metabolism (Tamaki et al., 2007; Jedrychowski et al., 2010; Liu et al., 2015) and in certain cardiovascular pathologies (Bown et al., 2011; Wild et al., 2012; Strickland et al., 2014).
RGZ is a synthetic PPARγ agonist, which has been used as an insulin sensitizer in Type 2 diabetes. Other beneficial properties have been attributed to RGZ; it is a renal protective agent, it is a cytoprotective agent and it attenuates the cognitive defects associated with Alzheimer’s disease (Pedersen et al., 2006; Risner et al., 2006; Morrison et al., 2017). However, cardiovascular risks have been associated with RGZ, resulting in a reconsideration of its therapeutic use (Nissen and Wolski, 2007; Hiatt et al., 2013). Of interest, RGZ seems to affect LRP1 levels in a concentration-dependent manner. Low concentrations of RGZ (0–5 μM) have been demonstrated to upregulate LRP1 expression at both the mRNA and protein levels, likely via PPARγ activation (Moon et al., 2012a). However, higher concentrations of RGZ (30–100 μM) seem to have the opposite effect on LRP1 expression. The mechanism by which these concentrations of RGZ reduce LRP1 levels is unclear. In this study, our goal was first to replicate previous observations; and second, to uncover the molecular mechanism by which RGZ at high concentrations reduces both LRP1 mRNA and protein levels.
We have confirmed that at 3 and 10 μM, RGZ activates LRP1 mRNA expression, as previously reported (Gauthier et al., 2003; Moon et al., 2012a). Higher concentrations than 10 μM reduce LRP1 mRNA levels, as a result of PPARγ downregulation, as shown in Figures 1 and 2. Previous studies reported that proteolytic degradation of PPARγ occurs via UPS in the presence of high concentrations of RGZ, and that degradation is prevented by the UPS inhibitors: MG132 and lactacystin (Hauser et al., 2000; Kilroy et al., 2009). Our observations suggest that in addition to PPARγ protein degradation via UPS, a transcriptional PPARγ downregulation takes place in cells treated with a high concentration of RGZ; as a consequence, PPARγ target genes are compromised.
High concentrations of PPARγ agonists, including RGZ, have been implicated to induce lysosomal degradation processes in cancer cells, for example autophagy (Zhou et al., 2009; Cerquetti et al., 2011). In addition, this statement corroborates the finding that PPARγ downregulation induced autophagy in hepatocellular carcinoma (Vara et al., 2013). Our results are consistent with others in the literature, since high concentrations of RGZ downregulated PPARγ, induced autophagy and promoted LRP1 lysosomal degradation (Figures 2–4, 5C).
This study demonstrates that RGZ reduced LRP1 levels at higher concentrations (>10 μM). These concentrations are higher than the maximum plasma concentration (Cmax) observed in healthy patients (652 ng/mL or 1.8 μM) following a dose of 8 mg/kg (Kirchheiner et al., 2006). However, the pharmacokinetic profile of this drug can be altered by affecting its metabolism. RGZ is metabolized primarily by CYP2C8 and to a lesser extent by CYP2C9 (Kirchheiner et al., 2006; Akhlaghi et al., 2017). CYP2C8 inhibition by gemfibrozil increased both RGZ Cmax by 22% and half-life from 3.6 h to 7.6 h (Niemi et al., 2003; Tornio et al., 2012). The CYP2C8 enzyme is genetically polymorphic and the variant CYP2C8∗3 encodes a protein with 2 linked amino acid substitutions, Arg139Lys (416G > A) and Lys399Arg (1196A > G). This variant has been reported to be present in 13–15% of the Caucasian population and it has considerably reduced oxidative capacity (Dai et al., 2001). However, the CYP2C8∗3 variant conferred a higher activity than wild-type CYP2C8 on RGZ metabolism (Kirchheiner et al., 2006). On the other hand, another genetic variant of CYP2C8 has affected metabolism and exacerbated cerivastatin side effects (Ishikawa et al., 2004). Thus, this study might be helpful to interpret the genetic status of RGZ’s metabolic enzymes of patients who have been medicated with RGZ. In this context, future studies might be required to determine the association of LRP1 reduction and the side-effects observed in RGZ-prescribed patients, including the cardiovascular risk-related effects.
Conclusion
The results of this study provide new insights into how LRP1 levels are affected by higher concentrations of RGZ. Our findings demonstrate that LRP1 reduction occurs by the mechanisms of both PPARγ downregulation and lysosomal degradation, responses to exposures to high concentrations of RGZ. LRP1 reduction might be associated with some adverse side effects observed in RGZ-prescribed patients.
Author Contributions
Participated in the experimental design and data analysis: AR-O, CLC, and AP-F. Conducted the experiments: AR-O and CLC, with help of JM-M, AG-U, and KG. Contributed with new reagents and analytical tools: MB, TM, and AP-F. Wrote the first draft of the manuscript: AR-O. Contributed to the writing of the manuscript: AR-O, CLC, MB, TM, and AP-F. All authors read and approved the final version of the manuscript.
Funding
This research project was completely funded by the MCPHS University School of Pharmacy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Dr. Michel Khrestchatisky and Dr. Marion David (Université d’Aix-Marseille and Vect-Horus, Marseille, France) for providing the LRP1 construct (Mini LRP-IV-EGFP).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2017.00772/full#supplementary-material
References
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi: 10.1038/nm.3159
Akhlaghi, F., Matson, K. L., Mohammadpour, A. H., Kelly, M., and Karimani, A. (2017). Clinical pharmacokinetics and pharmacodynamics of antihyperglycemic medications in children and adolescents with type 2 diabetes mellitus. Clin. Pharmacokinet. 56, 561–571. doi: 10.1007/s40262-016-0472-6
Bown, M. J., Jones, G. T., Harrison, S. C., Wright, B. J., Bumpstead, S., Baas, A. F., et al. (2011). Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am. J. Hum. Genet. 89, 619–627. doi: 10.1016/j.ajhg.2011.10.002
Cal, R., García-Arguinzonis, M., Revuelta-López, E., Castellano, J., Padró, T., Badimon, L., et al. (2013). Aggregated low-density lipoprotein induces LRP1 stabilization through E3 ubiquitin ligase CHFR downregulation in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 33, 369–377. doi: 10.1161/ATVBAHA.112.300748
Cerquetti, L., Sampaoli, C., Amendola, D., Bucci, B., Masuelli, L., Marchese, R., et al. (2011). Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp. Cell Res. 317, 1397–1410. doi: 10.1016/j.yexcr.2011.02.014
Dai, D., Zeldin, D. C., Blaisdell, J. A., Chanas, B., Coulter, S. J., Ghanayem, B. I., et al. (2001). Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11, 597–607. doi: 10.1097/00008571-200110000-00006
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/Amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
Fuentealba, R. A., Liu, Q., Zhang, J., Kanekiyo, T., Hu, X., Lee, J.-M., et al. (2010). Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Aβ42 uptake and lysosomal trafficking. PLOS ONE 5:e11884. doi: 10.1371/journal.pone.0011884
Gan, M., Jiang, P., McLean, P., Kanekiyo, T., and Bu, G. (2014). Low-density lipoprotein receptor-related protein 1 (LRP1) regulates the stability and function of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor in neurons. PLOS ONE 9:e113237. doi: 10.1371/journal.pone.0113237
Gauthier, A., Vassiliou, G., Benoist, F., and McPherson, R. (2003). Adipocyte low density lipoprotein receptor-related protein gene expression and function is regulated by peroxisome proliferator-activated receptor γ. J. Biol. Chem. 278, 11945–11953. doi: 10.1074/jbc.M212989200
Hauser, S., Adelmant, G., Sarraf, P., Wright, H. M., Mueller, E., and Spiegelman, B. M. (2000). Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J. Biol. Chem. 275, 18527–18533. doi: 10.1074/jbc.M001297200
Hiatt, W. R., Kaul, S., and Smith, R. J. (2013). The cardiovascular safety of diabetes drugs–insights from the rosiglitazone experience. N. Engl. J. Med. 369, 1285–1287. doi: 10.1056/NEJMp1309610
Ishikawa, C., Ozaki, H., Nakajima, T., Ishii, T., Kanai, S., Anjo, S., et al. (2004). A frameshift variant of CYP2C8 was identified in a patient who suffered from rhabdomyolysis after administration of cerivastatin. J. Hum. Genet. 49, 582–585. doi: 10.1007/s10038-004-0188-6
Jedrychowski, M. P., Gartner, C. A., Gygi, S. P., Zhou, L., Herz, J., Kandror, K. V., et al. (2010). Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J. Biol. Chem. 285, 104–114. doi: 10.1074/jbc.M109.040428
Kanai, Y., Wang, D., and Hirokawa, N. (2014). KIF13B enhances the endocytosis of LRP1 by recruiting LRP1 to caveolae. J. Cell Biol. 204, 395–408. doi: 10.1083/jcb.201309066
Kanekiyo, T., Cirrito, J. R., Liu, C. C., Shinohara, M., Li, J., Schuler, D. R., et al. (2013). Neuronal clearance of amyloid-B by endocytic receptor LRP1. J. Neurosci. 33, 19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013
Kilroy, G. E., Zhang, X., and Floyd, Z. E. (2009). PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity 17, 665–673. doi: 10.1038/oby.2008.616
Kim, H. I., and Ahn, Y. H. (2004). Role of peroxisome proliferator-activated receptor-gamma in the glucose-sensing apparatus of liver and beta-cells. Diabetes Metab. Res. Rev. 53(Suppl. 1), S60–S65. doi: 10.2337/diabetes.53.2007.S60
Kim, H. J., Moon, J. H., Kim, H. M., Yun, M. R., Jeon, B. H., Lee, B., et al. (2014). The hypolipidemic effect of cilostazol can be mediated by regulation of hepatic low-density lipoprotein receptor-related protein 1 (LRP1) expression. Metabolism 63, 112–119. doi: 10.1016/j.metabol.2013.09.006
Kirchheiner, J., Thomas, S., Bauer, S., Tomalik-Scharte, D., Hering, U., Doroshyenko, O., et al. (2006). Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin. Pharmacol. Ther. 80, 657–667. doi: 10.1016/j.clpt.2006.09.008
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Korolchuk, V. I., Mansilla, A., Menzies, F. M., and Rubinsztein, D. C. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527. doi: 10.1016/j.molcel.2009.01.021
Laatsch, A., Merkel, M., Talmud, P. J., Grewal, T., Beisiegel, U., and Heeren, J. (2009). Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 204, 105–111. doi: 10.1016/j.atherosclerosis.2008.07.046
Laatsch, A., Panteli, M., Sornsakrin, M., Hoffzimmer, B., Grewal, T., and Heeren, J. (2012). Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLOS ONE 7:e29385. doi: 10.1371/journal.pone.0029385
Lillis, A. P., Van Duyn, L. B., Murphy-Ullrich, J. E., and Strickland, D. K. (2008). LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918. doi: 10.1152/physrev.00033.2007
Liu, C. C., Hu, J., Tsai, C.-W., Yue, M., Melrose, H. L., Kanekiyo, T., et al. (2015). Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 35, 5851–5859. doi: 10.1523/JNEUROSCI.5180-14.2015
Mao, H., Lockyer, P., Li, L., Ballantyne, C. M., Patterson, C., Xie, L., et al. (2017). Endothelial LRP1 regulates metabolic responses by acting as a co-activator of PPARγ. Nat. Commun. 8:14960. doi: 10.1038/ncomms14960
Melman, L., Geuze, H. J., Li, Y., McCormick, L. M., Van Kerkhof, P., Strous, G. J., et al. (2002). Proteasome regulates the delivery of LDL receptor-related protein into the degradation pathway. Mol. Biol. Cell 13, 3325–3335. doi: 10.1091/mbc.E02
Moon, J. H., Kim, H. J., Kim, H. M., Yang, A. H., Lee, B.-W., Kang, E. S., et al. (2012a). Upregulation of hepatic LRP1 by rosiglitazone: a possible novel mechanism of the beneficial effect of thiazolidinediones on atherogenic dyslipidemia. J. Mol. Endocrinol. 49, 165–174. doi: 10.1530/JME-12-0119
Moon, J. H., Kim, H. J., Yang, A. H., Kim, H. M., Lee, B.-W., Kang, E. S., et al. (2012b). The effect of rosiglitazone on LRP1 expression and amyloid β uptake in human brain microvascular endothelial cells: a possible role of a low-dose thiazolidinedione for dementia treatment. Int. J. Neuropsychopharmacol. 15, 135–142. doi: 10.1017/S1461145711001611
Morrison, M. C., Yakala, G. K., Liang, W., Wielinga, P. Y., Salic, K., van Koppen, A., et al. (2017). Protective effect of rosiglitazone on kidney function in high-fat challenged human-CRP transgenic mice: a possible role for adiponectin and miR-21? Sci. Rep. 7:2915. doi: 10.1038/s41598-017-02444-2
Niemi, M., Backman, J. T., Granfors, M., Laitila, J., Neuvonen, M., and Neuvonen, P. J. (2003). Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia 46, 1319–1323. doi: 10.1007/s00125-003-1181-x
Nissen, S. E., and Wolski, K. (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 356, 2457–2471. doi: 10.1056/NEJMoa072761
Pedersen, W. A., McMillan, P. J., Kulstad, J. J., Leverenz, J. B., Craft, S., and Haynatzki, G. R. (2006). Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 199, 265–273. doi: 10.1016/j.expneurol.2006.01.018
Perrot, G., Langlois, B., Devy, J., Jeanne, A., Verzeaux, L., Almagro, S., et al. (2012). LRP-1-CD44, a new cell surface complex regulating tumor cell adhesion. Mol. Cell. Biol. 32, 3293–3307. doi: 10.1128/MCB.00228-12
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Risner, M. E., Saunders, A. M., Altman, J. F. B., Ormandy, G. C., Craft, S., Foley, I. M., et al. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. doi: 10.1038/sj.tpj.6500369
Rubinsztein, D. C., Cuervo, A. M., Ravikumar, B., Sarkar, S., Korolchuk, V. I., Kaushik, S., et al. (2009). In search of an “autophagomometer”. Autophagy 5, 585–589. doi: 10.4161/auto.5.5.8823
Sagare, A. P., Deane, R., and Zlokovic, B. V. (2012). Low-density lipoprotein receptor-related protein 1: a physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 136, 94–105. doi: 10.1016/j.pharmthera.2012.07.008
Sagare, A. P., Winkler, E. A., Bell, R. D., Deane, R., and Zlokovic, B. V. (2011). From the liver to the blood-brain barrier: an interconnected system regulating brain amyloid-β levels. J. Neurosci. Res. 89, 967–968. doi: 10.1002/jnr.22670
Strickland, D. K., Au, D. T., Cunfer, P., and Muratoglu, S. C. (2014). Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler. Thromb. Vasc. Biol. 34, 487–498. doi: 10.1161/ATVBAHA.113.301924
Tamaki, C., Ohtsuki, S., Iwatsubo, T., Hashimoto, T., Yamada, K., Yabuki, C., et al. (2006). Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid beta-peptide by the liver. Pharm. Res. 23, 1407–1416. doi: 10.1007/s11095-006-0208-7
Tamaki, C., Ohtsuki, S., and Terasaki, T. (2007). Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1-40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855. doi: 10.1124/mol.107.036913
Tornio, A., Niemi, M., Neuvonen, P. J., and Backman, J. T. (2012). Drug interactions with oral antidiabetic agents: pharmacokinetic mechanisms and clinical implications. Trends Pharmacol. Sci. 33, 312–322. doi: 10.1016/j.tips.2012.03.001
Van Gool, B., Dedieu, S., Emonard, H., and Roebroek, A. J. M. (2015). The matricellular receptor LRP1 forms an interface for signaling and endocytosis in modulation of the extracellular tumor environment. Front. Pharmacol. 6:271. doi: 10.3389/fphar.2015.00271
Vara, D., Morell, C., Rodríguez-Henche, N., and Diaz-Laviada, I. (2013). Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis. 4:e618. doi: 10.1038/cddis.2013.141
Vilchez, D., Saez, I., and Dillin, A. (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5:5659. doi: 10.1038/ncomms6659
Wild, J. B., Stather, P. W., Sylvius, N., Choke, E., Sayers, R. D., and Bown, M. J. (2012). Low density lipoprotein receptor related protein 1 and abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 44, 127–132. doi: 10.1016/j.ejvs.2012.05.009
Zhao, Z., Sagare, A. P., Ma, Q., Halliday, M. R., Kong, P., Kisler, K., et al. (2015). Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987. doi: 10.1038/nn.4025
Keywords: LRP1, rosiglitazone, PPARγ, protein degradation, lysosomal degradation, bafilomycin A1
Citation: Rondón-Ortiz AN, Lino Cardenas CL, Martínez-Málaga J, Gonzales-Urday AL, Gugnani KS, Böhlke M, Maher TJ and Pino-Figueroa AJ (2017) High Concentrations of Rosiglitazone Reduce mRNA and Protein Levels of LRP1 in HepG2 Cells. Front. Pharmacol. 8:772. doi: 10.3389/fphar.2017.00772
Received: 25 July 2017; Accepted: 12 October 2017;
Published: 14 November 2017.
Edited by:
Marc Poirot, Institut National de la Santé et de la Recherche Médicale, FranceReviewed by:
Nicolas Etique, University of Reims Champagne-Ardenne, FranceWeicheng Liang, The Chinese University of Hong Kong, Hong Kong
Copyright © 2017 Rondón-Ortiz, Lino Cardenas, Martínez-Málaga, Gonzales-Urday, Gugnani, Böhlke, Maher and Pino-Figueroa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alejandro J. Pino-Figueroa, YWxlamFuZHJvLnBpbm8tZmlndWVyb2FAbWNwaHMuZWR1