 Natalia V. Segatto1
Natalia V. Segatto1 Mariana H. Remião1
Mariana H. Remião1 Kyle M. Schachtschneider2
Kyle M. Schachtschneider2 Fabiana K. Seixas1
Fabiana K. Seixas1 Lawrence B. Schook2,3
Lawrence B. Schook2,3 Tiago Collares1*
Tiago Collares1*- 1Biotechnology Graduate Program, Molecular and Cellular Oncology Research Group, Laboratory of Cancer Biotechnology, Technology Development Center, Federal University of Pelotas, Pelotas, Brazil
- 2Department of Radiology, University of Illinois at Chicago, Chicago, IL, United States
- 3Department of Animal Sciences, University of Illinois at Urbana–Champaign, Champaign, IL, United States
The screening of potential therapeutic compounds using phenotypic drug discovery (PDD) is being embraced once again by researchers and pharmaceutical companies as an approach to enhance the development of new effective therapeutics. Before the genomics and molecular biology era and the consecutive emergence of targeted-drug discovery approaches, PDD was the most common platform used for drug discovery. PDD, also known as phenotypic screening, consists of screening potential compounds in either in vitro cellular or in vivo animal models to identify compounds resulting in a desirable phenotypic change. Using this approach, the biological targets of the compounds are not taken into consideration. Suitable animal models are crucial for the continued validation and discovery of new drugs, as compounds displaying promising results in phenotypic in vitro cell-based and in vivo small animal model screenings often fail in clinical trials. Indeed, this is mainly a result of differential anatomy, physiology, metabolism, immunology, and genetics between humans and currently used pre-clinical small animal models. In contrast, pigs are more predictive of therapeutic treatment outcomes in humans than rodents. In addition, pigs provide an ideal platform to study cancer due to their similarities with humans at the anatomical, physiological, metabolic, and genetic levels. Here we provide a mini-review on the reemergence of PDD in drug development, highlighting the potential of porcine cancer models for improving pre-clinical drug discovery and testing. We also present precision medicine based genetically defined swine cancer models developed to date and their potential as biomedical models.
The Return of Phenotypic Drug Discovery
The pharmaceutical industry has increasingly invested in the research and development (R&D) of new potential drugs and has doubled these investments over the past 15 years, from $26.0 billion in 2000 to an estimated $58.8 billion in 2015 (PhRMA, 2016). Surprisingly, companies have produced on average less than one new drug per year since 1950, indicating that no strategy employed by pharmaceutical companies over this time period has increased their ability to discover and bring new drugs to market (Munos, 2009). Analysis of the drugs approved by the FDA between 1999 and 2008 indicates more first-in-class drugs have been approved using phenotypic screening assays than target-based approaches (Swinney and Anthony, 2011).
Phenotypic drug discovery (PDD) consists of screening potential compounds in either in vitro cellular (Mosmann, 1983) or in vivo animal models (Giacomotto and Segalat, 2010) to identify compounds resulting in a desirable phenotypic change. On the other hand, target-based drug discovery (TDD) relies on the identification of a target of interest believed to be “disease-modifying” and therefore related to a particular disease. The genomic era revolutionized the screening of new compounds in vitro and in vivo through the rise of comparative genomics, genome editing, and the consecutive emergence of TDD approaches. Even though TDD has several advantages, such as confirmation of the relevance of a target for a given disease, this approach has not effectively translated into the approval of new drugs as expected. Some researchers believe that one of the reasons for the decrease in drug discovery and innumerous failed clinical trials in the last 25 years may be due to the extensive use of TDD in the past decades (Sams-Dodd, 2005, 2013; Hellerstein, 2008). Therefore, researchers and pharmaceutical companies are once again embracing PDD as a way to improve development and screening of new effective therapeutics (Lee and Berg, 2013; Zheng et al., 2013; Warchal et al., 2015).
Pdd In Cancer Drug Discovery
Oncology is a huge market for pharmaceutical industries, and has grown to be the largest therapeutic area in terms of number of projects, investments in research and development (R&D), and number of clinical trials (Arrowsmith, 2012). However, discovering and approving new efficient cancer therapies is a challenge (Hoelder et al., 2012) mainly because cancer is a highly heterogeneous disease with multiple mechanisms of action. The hallmarks of cancer indicate that the transformation of normal cells into malignant cancer cells is a multistep process reflecting genetic alterations affecting six physiological processes. These processes include self-sufficient growth signaling, insensitivity to growth-inhibitory (antigrowth) signals, evasion of programmed cell death (apoptosis), limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis (Hanahan and Weinberg, 2000).
The majority of the cancer drugs developed in the past decades have been discovered using target-based approaches (Swinney and Anthony, 2011). This is expected, as our knowledge of the molecular basis of cancer is growing (Garraway and Lander, 2013) and as a consequence new cancer therapeutics are focused on targets known to be related to the above-mentioned multistep processes. For example, as kinases play key roles in signal transduction and regulation of a range of cellular activities, kinase inhibitors represent 21 out of the 29 approved drugs developed using TDD between 1999 and 2008 (Zhang et al., 2009; Swinney and Anthony, 2011).
It is important to highlight that when researchers focus only on known targets for drug discovery, they lose the opportunity to identify potential drugs that may have different—and new—targeted mechanisms of action. Between 1999 and 2008, 19 of the approved cancer therapeutics were discovered using some level of phenotypic screening (Swinney and Anthony, 2011). Among them are the thalidomide analog Lenalidomide, for which the selection of second-generation analogs with anticancer activity was conducted using only phenotypic assays (Shortt et al., 2013). In addition, the observation that dimethyl sulfoxide (DMSO) caused growth arrest (Friend et al., 1971; Takase et al., 1992; Teraoka et al., 1996) and terminal differentiation of transformed cells (Friend et al., 1971) led researchers to test other polar, small-molecule solvent species for antitumor activity, resulting in the discovery of the histone deacetylase inhibitor Vorinostat (Marks and Breslow, 2007). Furthermore, Romidepsin, an anticancer agent approved for the treatment of cutaneous T-cell lymphoma was identified through phenotypic screening of microbial metabolites in tumor cell lines (Ueda et al., 1994).
In general, PDD remains a crucial approach in selecting, validating, and developing potential cancer drugs, even though it represents a minority of the investigational and recently approved oncology treatments (Moffat et al., 2014). PDD screening is only the first step in the long and expensive journey of drug discovery. The next stage, represented by animal experimentation in pre-clinical trials, must validate the promising results obtained in the initial screening. Therefore, both steps are complementary and equally important.
Importance of Animal Models in Drug Discovery
Animal models are essential tools in the drug development process. Due to ethical and regulatory issues, it is necessary to test new biomedical products in animal models during the pre-clinical phase before initiating human experimentation to evaluate efficacy, toxicity, and safety. However, many drugs that display promising results in pre-clinical animal studies do not produce the same response in humans (Mak et al., 2014). This is especially true for oncology due to difficulties in mirroring the heterogeneity and complex tumor characteristics in animal models (Huszthy et al., 2012; van Marion et al., 2016). Disregarding the approach used for drug discovery (TDD or PDD), there are several examples of candidate therapeutics failing in clinical trials even though they showed promising results in previous phases of drug development (Williams, 2013, 2015). In fact, about 85% of therapies tested in clinical trials fail (Ledford, 2011), with cancer therapeutics representing the largest proportion of these failures (Arrowsmith, 2011). Only 5% of agents that demonstrate anticancer activity in preclinical phases are approved after demonstrating sufficient efficacy in phase III testing (Hutchinson and Kirk, 2011).
One of the key takeaways from these failures is the need for improvements in the early steps of drug discovery, specifically in the use of adequate animal models in pre-clinical trials. Suitable animal models are crucial for the continued validation and discovery of new drugs, as compounds displaying promising results in phenotypic in vitro cell-based and in vivo small animal model screenings often fail in clinical trials.
Rodents are the most widely used platform for tumor pre-clinical screening and biological models in general. Some of their characteristics, such as small size, inexpensiveness, well-known genetics, and their ability to be easily genetically modified make them a standard tool for evaluating novel therapeutics. However, they are actually poor models for many human diseases (Seok et al., 2013; Burns et al., 2015), including cancer (de Jong and Maina, 2010). Consequently, difficulties translating the results obtained in pre-clinical studies to the clinical realm are quite common when rodents are chosen as the biological model. One example is the phase II clinical trial of IPI-926 (Saridegib) in patients with advanced chondrosarcoma. The trial was stopped early because no relevant effects were observed in humans treated with Saridegib compared to the placebo group (Wagner et al., 2013), even though medulloblastoma mice models treated with IPI-926 demonstrated a fivefold increase in survival (Lee et al., 2012). This work demonstrates the need for more relevant animal models as complementary tools for cancer drug discovery.
Even though genetically engineered mouse models are important tools for studying mechanisms associated with cancer biology (Jeong, 2016; Jiang and Yu, 2017; Perez-Guijarro et al., 2017) and represent improved models compared to wild mice models, they still have several limitations. We believe that the use of a suitable large animal model of cancer would improve the results of PDD for cancer therapeutics.
Pigs As Biological Models
Pigs have proven to be more predictive of therapeutic treatments in humans than rodents (Meurens et al., 2012; Schook et al., 2015a). Pigs provide an ideal platform to study cancer due to their similarities with humans at the anatomical, physiological, metabolic, and genetic levels (Table 1). The pig genome sequence was published in 2012, providing insights into their genetic similarities to humans (Groenen et al., 2012) and furthering their acceptance as a large animal biomedical model for human diseases (Prather, 2013; Schook et al., 2015a). In addition to the high homology between the pig and human genome, the swine genome also exhibits highly conserved epigenetic regulation demonstrated by the similar genome wide DNA methylation patterns observed between pigs and humans (Schachtschneider et al., 2015). Regarding their cancer genetics, a previous study using genetically engineered porcine cells showed that swine cells could be transformed by mutated oncogenes and tumor suppressor genes commonly found in human cancers (Adam et al., 2007). Furthermore, these transformed cells were able to form tumors following autologous injection (Adam et al., 2007). Also, normal murine cells can be transformed with fewer mutations (Rangarajan et al., 2004) compared to swine and human cells (Rangarajan et al., 2004; Adam et al., 2007). Together, this work demonstrates that porcine tumorigenic pathways are more similar to human pathways than rodents (Table 1).
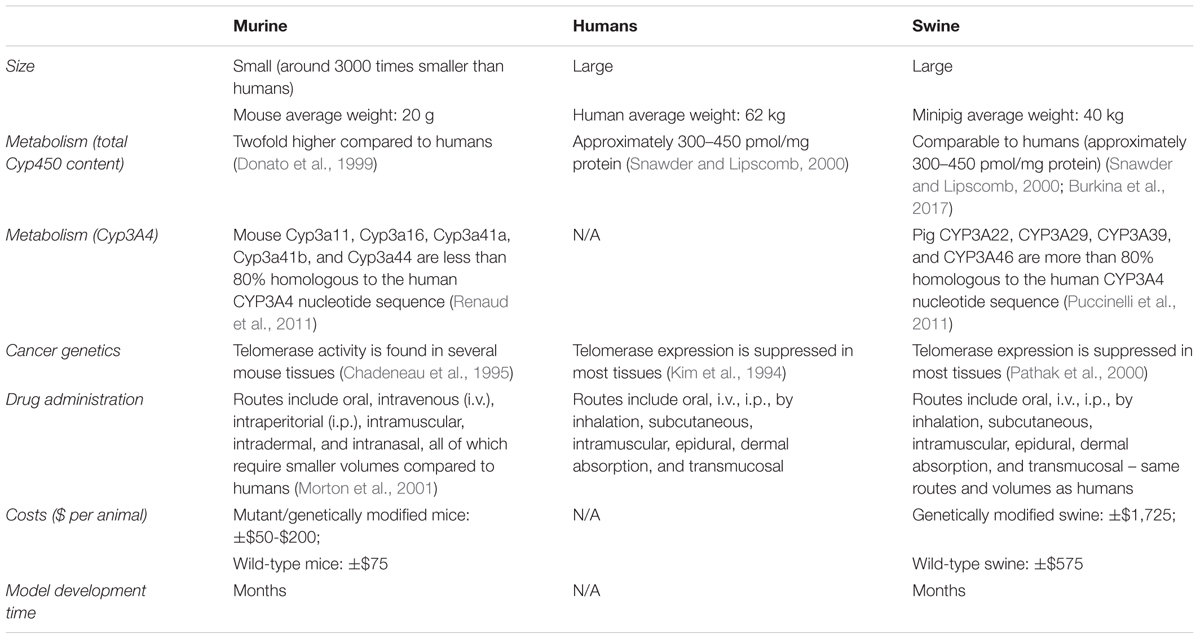
TABLE 1. Characteristics of murine and swine pre-clinical trial cancer models compared to humans.
Pigs provide an ideal large animal model for preclinical drug screening due to their metabolic similarities with humans. For instance, pigs have proven to be a suitable model for CYP3A-related drug metabolism. Cytochromes P450 enzymes (CYP) are known for their role in compound metabolism, and the CYP3A subfamily is responsible for metabolizing more than half of all drugs available on the market (Zuber et al., 2002). The swine xenosensor pregnane C receptors also displays high homology to the ones found in humans (Pollock et al., 2007; Gray et al., 2010). In addition, the CYP family receptors and enzymes display similar expression levels in pigs and humans (Nielsen et al., 2017). On the other hand, some rodents (e.g., rats) are not considered good models for CYP3A4 related metabolism due to the dissimilarities with humans in metabolism related to this enzyme, resulting in differential therapeutic metabolization (Martignoni et al., 2006). In contrast, swine models can and have been very useful for pharmacology and toxicology studies (Thorn et al., 2009, 2011).
Another beneficial feature of pigs that makes them a relevant biological model is the fact that they can live up to 10 years. For purposes of cancer drug development, this is especially relevant because it allows for therapeutic testing and posterior monitoring in a pre-clinical platform able to mimic multiple stages of tumor development, progression, invasion, and metastasis, thereby enabling the evaluation of the long-term effects of a variety of compounds. In addition, the large size of the pig is ideal not only for administration of therapeutics in the same manner as in human patients (Table 1), but also for the collection of larger volumes of bodily fluids (Helke and Swindle, 2013), allowing blood collection procedures to mirror those performed in humans. We thus hypothesize that the pig’s implementation as an ideal biomedical model for drug discovery could help fill the existing gap between early phenotypic screening of potential new therapeutics, identification of prognostic biomarkers, and testing of beneficial products and devices in human clinical patients.
Genetic engineering allows for the development of transgenic porcine models that recapitulate particular genetic alterations found in human diseases for translational biomedical research purposes. These models can be obtained using several techniques such as microinjection of DNA into the pronuclei of fertilized oocytes, lentiviral transgenesis (LVGT), sperm mediated gene transfer (SMGT), and somatic cell nuclear transfer (SCNT) using genetically modified nuclear donor cells (Aigner et al., 2010). Currently, transgenic pigs are increasingly being accepted as large animal models for several human diseases (Aigner et al., 2010), including neurodegenerative diseases (Kragh et al., 2009), cystic fibrosis (Rogers et al., 2008), cardiovascular diseases (Hao et al., 2006), diabetes mellitus (Renner et al., 2010), and cancer (Flisikowska et al., 2012; Schook et al., 2015b).
Genetically Defined Swine Models of Cancer
The advancement of genetic engineering techniques combined with knowledge of the pig genome sequence and its similarity to humans (Groenen et al., 2012) make genetic engineering a powerful tool for developing suitable transgenic porcine biological models for cancer drug discovery (Schook et al., 2016).
With the aim of producing a genetically defined porcine cancer model, Schook and collaborators developed the Oncopig Cancer Model (OCM), which contains Cre recombinase inducible mutated tumor suppressor and oncogene transgenes (TP53R167H and KRASG12D, respectively). Exposure to adenoviral vectors encoding Cre recombinase (AdCre) results in cellular transformation in a temporal and spatial manner that closely mimics the spontaneous tumor formation that occurs in humans (Schook et al., 2015b). The intramuscular injection of AdCre into the OCM results in the development of soft-tissue sarcomas (STSs) that display pathological characteristics of human leiomyosarcomas (Schook et al., 2015b). Furthermore, transcriptional profiling of Oncopig STS cell lines and tumors indicates Oncopig STS exhibits altered TP53 signaling, Wnt signaling activation, and signs of epigenetic reprogramming, all of which represent transcriptional hallmarks of human STS (Schachtschneider et al., 2017a). In addition, the transcriptional regulator FOSL1, which was previously identified as a potential human STS therapeutic target, was identified as a master regulator of Oncopig STS (Schachtschneider et al., 2017a).
Two other cancer types have been developed to date in the OCM: hepatocellular carcinoma (HCC) and pancreatic cancer. By isolating and transforming Oncopig hepatocytes via exposure to AdCre in vitro, researchers were able to develop Oncopig HCC cell lines expressing TP53R167H and KRASG12D (Schachtschneider et al., 2017b). These cells display histopathological characteristics similar to human HCC and form tumors upon autologous injection into Oncopigs. Furthermore, human HCC transcriptional hallmarks were also observed in Oncopig HCC cells along with conserved gene expression profiles compared to human HCC cell lines (Schachtschneider et al., 2017b). The Oncopig pancreatic ductal adenocarcinoma (PDAC) model is still in development. However, the two most predominant pancreatic cancer histotypes (exocrine and neuroendocrine) have already been developed in this PDAC model via delivery of AdCre into the main pancreatic duct (Diaz et al., 2016).
In a recently published article, Schachtschneider et al. (2017c) presented multiple applications of the OCM as an innovative large animal translational oncology platform, ranging from the above-mentioned potential for therapeutic screening and development to its use for developing diagnostic imaging modalities. It also highlights the numerous advantages that swine models have over other commonly used biological models (Schachtschneider et al., 2017c), indicating large animal platforms can serve as predictable models of human therapeutic responses using PDD approaches (Figure 1).
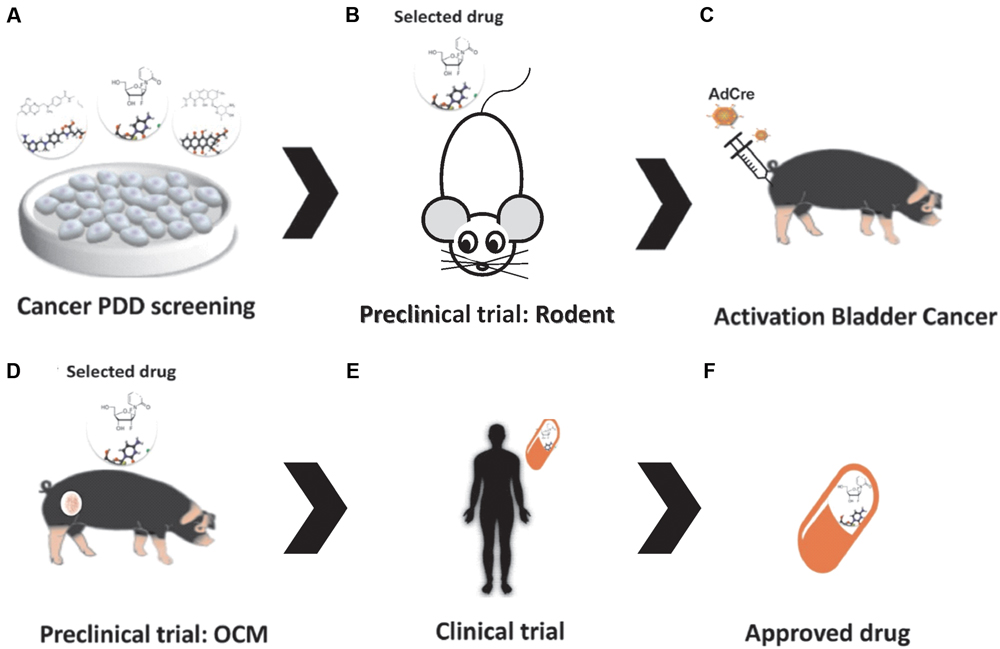
FIGURE 1. Drug discovery in the Oncopig Bladder Cancer Model. (A) Bladder Cancer PDD. (B) Pre-clinical trials: in vivo drug testing in rodent model. (C) Oncopig Bladder Cancer Model: intravesical AdCre injection activates expression of the mutated oncogenes (TP53R167H and KRASG12D), resulting in tumor formation. (D) Pre-clinical trial II: in vivo drug testing in the Oncopig Bladder Cancer Model. (E) Clinical trials. (F) Drug selection.
Regarding the ethical responsibilities of conducting animal experiments (Perry, 2007) and the three R’s paradigm (Reduce, Replace, and Refine) (Russell and Burch, 1959), the OCM provides an ideal platform capable of discretely inducting localized tumors that can be closely monitored to meet scientific goals while still minimizing the animal’s comorbidities and mortality.
In addition to the OCM, other transgenic swine cancer models have been developed. They include the human familial adenomatous polyposis model in which the APC gene was inactivated by introduction of a premature termination codon through electroporation of linearized vector DNA into mesenchymal stem cells followed by SCNT. The animals carrying the APC1311 mutation develop polyps in the colon and rectum after 1 year (Flisikowska et al., 2012). In addition, two models utilizing TP53 mutations have been developed: a genetically modified pig expressing mutant TP53R167H, which develops lymphomas and osteogenic tumors in homozygous individuals (Sieren et al., 2014), and TP53 knockout pigs that develop spontaneous osteosarcomas in older heterozygous animals and multiple large osteosarcomas in 7 to 8-month-old homozygous pigs (Saalfrank et al., 2016). More recently, a genetic model of intestinal cancer was developed using a Flp-recombinase inducible oncogene cassette containing KRASG12D, cMYC, and SV40LT in addition to a 4-hydroxytamoxifen (4-OHT) activator cassette controlled by an intestinal epithelium tissue-specific promoter. Activation of the oncogene cassette in vivo resulted in a duodenal neuroendocrine carcinoma with a lymph node metastasis in the minipig (Callesen et al., 2017). Finally, an attempt to develop a breast cancer model was performed using a recombinant adeno-associated virus (rAAV) mediated BRCA1 knockout. Unfortunately, these animals died before they were able to demonstrate any relevant phenotypic changes (Luo et al., 2011).
Future Approaches
We believe the use of porcine models in pre-clinical trials will increase the benefits of PDD. More punctually, the OCM could fulfill the needs of the pharmaceutical industry and academic researchers by providing a model more predictable of therapeutic responses in humans. These higher genetically defined swine cancer models have huge potential to serve as biomedical models in preclinical trials. Since the International Committee on Harmonization requires toxicity testing in two relevant animal species (Food and Drug Administration [FDA], 2010), these models can serve as translational models by testing the efficacy of new therapies that show promising results in small animal PDD screenings before moving to human clinical trials, ultimately decreasing the failure rates of clinical trials (Figure 1). In fact, although swine models are more expensive than rodents (Table 1), the use of pigs as a second animal model in pre-clinical trials is cheaper than using non-human primates. It can also provide a cost reduction in drug discovery by confirming the trial results before initiating a highly costly human clinical trial.
Furthermore, we hypothesize that intravesical injection of AdCre in the OCM platform could result in tumor formation in the bladder (Figure 1); resulting in a highly valuable bladder cancer model in which new compounds and immunotherapies such as recombinant BCG (a promising immunotherapeutic approach for bladder cancer (Begnini et al., 2015) could be tested. Our research group has tested the antitumoral activity of several compounds in bladder cancer cells lines using PDD approaches, including Brazilian red propolis (Begnini et al., 2014) and pyrazoline derivatives (Tessmann et al., 2017). A suitable animal model platform such as the OCM would further advance in vivo testing of the most promising compounds selected in these previous studies. To this end, we are currently evaluating the ability of OCM cells to mimic human bladder cancer cell line responses to commercially available therapeutics in vitro. We believe that this work will confirm the important role that the OCM can play in PDD of cancer drugs.
Author Contributions
NS, MR, KS, LS, FS, and TC had an equal participation in writing and approving the present manuscript.
Funding
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (Award ID: 311567/2016-9).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adam, S. J., Rund, L. A., Kuzmuk, K. N., Zachary, J. F., Schook, L. B., and Counter, C. M. (2007). Genetic induction of tumorigenesis in swine. Oncogene 26, 1038–1045. doi: 10.1038/sj.onc.1209892
Aigner, B., Renner, S., Kessler, B., Klymiuk, N., Kurome, M., Wunsch, A., et al. (2010). Transgenic pigs as models for translational biomedical research. J. Mol. Med. 88, 653–664. doi: 10.1007/s00109-010-0610-9
Arrowsmith, J. (2011). Trial watch: phase III and submission failures: 2007–2010. Nat. Rev. Drug Discov. 10, 87. doi: 10.1038/nrd3375
Begnini, K. R., Buss, J. H., Collares, T., and Seixas, F. K. (2015). Recombinant Mycobacterium bovis BCG for immunotherapy in nonmuscle invasive bladder cancer. Appl. Microbiol. Biotechnol. 99, 3741–3754. doi: 10.1007/s00253-015-6495-3
Begnini, K. R., Moura de Leon, P. M., Thurow, H., Schultze, E., Campos, V. F., Martins Rodrigues, F., et al. (2014). Brazilian red propolis induces apoptosis-like cell death and decreases migration potential in bladder cancer cells. Evid. Based Complement. Altern. Med. 2014:639856. doi: 10.1155/2014/639856
Burkina, V., Rasmussen, M. K., Pilipenko, N., and Zamaratskaia, G. (2017). Comparison of xenobiotic-metabolising human, porcine, rodent, and piscine cytochrome P450. Toxicology 375, 10–27. doi: 10.1016/j.tox.2016.11.014
Burns, T. C., Li, M. D., Mehta, S., Awad, A. J., and Morgan, A. A. (2015). Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: a systematic bioinformatics-based critique of preclinical models. Eur. J. Pharmacol. 759, 101–117. doi: 10.1016/j.ejphar.2015.03.021
Callesen, M. M., Árnadóttir, S. S., Lyskjaer, I., Ørntoft, M.-B. W., Høyer, S., Dagnaes-Hansen, F., et al. (2017). A genetically inducible porcine model of intestinal cancer. Mol. Oncol. 11, 1616–1629. doi: 10.1002/1878-0261.12136
Chadeneau, C., Siegel, P., Harley, C. B., Muller, W. J., and Bacchetti, S. (1995). Telomerase activity in normal and malignant murine tissues. Oncogene 11, 893–898.
de Jong, M., and Maina, T. (2010). Of mice and humans: Are they the same?–Implications in cancer translational research. J. Nucl. Med. 51, 501–504. doi: 10.2967/jnumed.109.065706
Diaz, A., Principe, D., DeCant, B., Grippo, P. J., Rund, L., and Schook, L. (2016). “Pigs as a new weapon against cancer: modeling solid tumors in porcine,” in Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, 2016 April 16–20, New Orleans, LA.
Donato, M. T., Castell, J. V., and Gomez-Lechon, M. J. (1999). Characterization of drug metabolizing activities in pig hepatocytes for use in bioartificial liver devices: comparison with other hepatic cellular models. J. Hepatol. 31, 542–549. doi: 10.1016/S0168-8278(99)80049-X
Flisikowska, T., Merkl, C., Landmann, M., Eser, S., Rezaei, N., Cui, X., et al. (2012). A porcine model of familial adenomatous polyposis. Gastroenterology 143, 1173–1177. doi: 10.1053/j.gastro.2012.07.110
Food and Drug Administration [FDA] (2010). Guidance on M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. Available at: https://www.fda.gov/downloads/drugs/guidances/ucm073246.pdf
Friend, C., Scher, W., Holland, J. G., and Sato, T. (1971). Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc. Natl. Acad. Sci. U.S.A. 68, 378–382. doi: 10.1073/pnas.68.2.378
Garraway, L. A., and Lander, E. S. (2013). Lessons from the cancer genome. Cell 153, 17–37. doi: 10.1016/j.cell.2013.03.002
Giacomotto, J., and Segalat, L. (2010). High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 160, 204–216. doi: 10.1111/j.1476-5381.2010.00725.x
Gray, M. A., Pollock, C. B., Schook, L. B., and Squires, E. J. (2010). Characterization of porcine pregnane X receptor, farnesoid X receptor and their splice variants. Exp. Biol. Med. 235, 718–736. doi: 10.1258/ebm.2010.009339
Groenen, M. A., Archibald, A. L., Uenishi, H., Tuggle, C. K., Takeuchi, Y., Rothschild, M. F., et al. (2012). Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398. doi: 10.1038/nature11622
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/S0092-8674(00)81683-9
Hao, Y. H., Yong, H. Y., Murphy, C. N., Wax, D., Samuel, M., Rieke, A., et al. (2006). Production of endothelial nitric oxide synthase (eNOS) over-expressing piglets. Transgenic Res. 15, 739–750. doi: 10.1007/s11248-006-9020-8
Helke, K. L., and Swindle, M. M. (2013). Animal models of toxicology testing: the role of pigs. Expert Opin. Drug Metab. Toxicol. 9, 127–139. doi: 10.1517/17425255.2013.739607
Hellerstein, M. K. (2008). A critique of the molecular target-based drug discovery paradigm based on principles of metabolic control: advantages of pathway-based discovery. Metab. Eng. 10, 1–9. doi: 10.1016/j.ymben.2007.09.003
Hoelder, S., Clarke, P. A., and Workman, P. (2012). Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 6, 155–176. doi: 10.1016/j.molonc.2012.02.004
Huszthy, P. C., Daphu, I., Niclou, S. P., Stieber, D., Nigro, J. M., Sakariassen, P. O., et al. (2012). In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol. 14, 979–993. doi: 10.1093/neuonc/nos135
Hutchinson, L., and Kirk, R. (2011). High drug attrition rates–where are we going wrong? Nat. Rev. Clin. Oncol. 8, 189–190. doi: 10.1038/nrclinonc.2011.34
Jeong, J. H. (2016). Inducible mouse models for cancer drug target validation. J. Cancer Prev. 21, 243–248. doi: 10.15430/JCP.2016.21.4.243
Jiang, Y., and Yu, Y. (2017). Transgenic and gene knockout mice in gastric cancer research. Oncotarget 8, 3696–3710. doi: 10.18632/oncotarget.12467
Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, D., Ho, P. L. C., et al. (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. doi: 10.1126/science.7605428
Kragh, P. M., Nielsen, A. L., Li, J., Du, Y., Lin, L., Schmidt, M., et al. (2009). Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res. 18, 545–558. doi: 10.1007/s11248-009-9245-4
Ledford, H. (2011). Translational research: 4 ways to fix the clinical trial. Nature 477, 526–528. doi: 10.1038/477526a
Lee, J. A., and Berg, E. L. (2013). Neoclassic drug discovery: the case for lead generation using phenotypic and functional approaches. J. Biomol. Screen. 18, 1143–1155. doi: 10.1177/1087057113506118
Lee, M. J., Hatton, B. A., Villavicencio, E. H., Khanna, P. C., Friedman, S. D., Ditzler, S., et al. (2012). Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. U.S.A. 109, 7859–7864. doi: 10.1073/pnas.1114718109
Luo, Y., Li, J., Liu, Y., Lin, L., Du, Y., Li, S., et al. (2011). High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: developing a pig model for breast cancer. Transgenic Res. 20, 975–988. doi: 10.1007/s11248-010-9472-8
Mak, I. W., Evaniew, N., and Ghert, M. (2014). Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 6, 114–118.
Marks, P. A., and Breslow, R. (2007). Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90. doi: 10.1038/nbt1272
Martignoni, M., Groothuis, G. M., and de Kanter, R. (2006). Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2, 875–894. doi: 10.1517/17425255.2.6.875
Meurens, F., Summerfield, A., Nauwynck, H., Saif, L., and Gerdts, V. (2012). The pig: a model for human infectious diseases. Trends Microbiol. 20, 50–57. doi: 10.1016/j.tim.2011.11.002
Moffat, J. G., Rudolph, J., and Bailey, D. (2014). Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discov. 13, 588–602. doi: 10.1038/nrd4366
Morton, D. B., Jennings, M., Buckwell, A., Ewbank, R., Godfrey, C., Holgate, B., et al. (2001). Refining procedures for the administration of substances. Report of the BVAAWF/FRAME/RSPCA/UFAW Joint Working Group on Refinement. British Veterinary Association Animal Welfare Foundation/Fund for the Replacement of Animals in Medical Experiments/Royal Society for the Prevention of Cruelty to Animals/Universities Federation for Animal Welfare. Lab. Anim. 35, 1–41. doi: 10.1258/0023677011911345
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. doi: 10.1016/0022-1759(83)90303-4
Munos, B. (2009). Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 8, 959–968. doi: 10.1038/nrd2961
Nielsen, S. D., Bauhaus, Y., Zamaratskaia, G., Junqueira, M. A., Blaabjerg, K., Petrat-Melin, B., et al. (2017). Constitutive expression and activity of cytochrome P450 in conventional pigs. Res. Vet. Sci. 111, 75–80. doi: 10.1016/j.rvsc.2016.12.003
Pathak, S., Multani, A. S., McConkey, D. J., Imam, A. S., and Amoss, M. S. Jr. (2000). Spontaneous regression of cutaneous melanoma in sinclair swine is associated with defective telomerase activity and extensive telomere erosion. Int. J. Oncol. 17, 1219–1224. doi: 10.3892/ijo.17.6.1219
Perez-Guijarro, E., Day, C. P., Merlino, G., and Zaidi, M. R. (2017). Genetically engineered mouse models of melanoma. Cancer 123, 2089–2103. doi: 10.1002/cncr.30684
Perry, P. (2007). The ethics of animal research: a UK perspective. ILAR J. 48, 42–46. doi: 10.1093/ilar.48.1.42
PhRMA (2016). PhRMA Pharmaceutical Industry Profile 2016. Available at: http://phrma-docs.phrma.org/sites/default/files/pdf/biopharmaceutical-industry-profile.pdf
Pollock, C. B., Rogatcheva, M. B., and Schook, L. B. (2007). Comparative genomics of xenobiotic metabolism: a porcine-human PXR gene comparison. Mamm. Genome 18, 210–219. doi: 10.1007/s00335-007-9007-7
Prather, R. S. (2013). Pig genomics for biomedicine. Nat. Biotechnol. 31, 122–124. doi: 10.1038/nbt.2490
Puccinelli, E., Gervasi, P. G., and Longo, V. (2011). Xenobiotic metabolizing cytochrome P450 in pig, a promising animal model. Curr. Drug Metab. 12, 507–525. doi: 10.2174/138920011795713698
Rangarajan, A., Hong, S. J., Gifford, A., and Weinberg, R. A. (2004). Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6, 171–183. doi: 10.1016/j.ccr.2004.07.009
Renaud, H. J., Cui, J. Y., Khan, M., and Klaassen, C. D. (2011). Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicol. Sci. 124, 261–277. doi: 10.1093/toxsci/kfr240
Renner, S., Fehlings, C., Herbach, N., Hofmann, A., von Waldthausen, D. C., Kessler, B., et al. (2010). Glucose intolerance and reduced proliferation of pancreatic beta-cells in transgenic pigs with impaired glucose-dependent insulinotropic polypeptide function. Diabetes Metab. Res. Rev. 59, 1228–1238. doi: 10.2337/db09-0519
Rogers, C. S., Stoltz, D. A., Meyerholz, D. K., Ostedgaard, L. S., Rokhlina, T., Taft, P. J., et al. (2008). Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321, 1837–1841. doi: 10.1126/science.1163600
Russell, W. M. S., and Burch, R. L. (eds). (1959). The Principles of Humane Experimental Technique. London: Methuen & Co. Ltd.
Saalfrank, A., Janssen, K. P., Ravon, M., Flisikowski, K., Eser, S., Steiger, K., et al. (2016). A porcine model of osteosarcoma. Oncogenesis 5:e210. doi: 10.1038/oncsis.2016.19
Sams-Dodd, F. (2005). Target-based drug discovery: is something wrong? Drug Discov. Today 10, 139–147. doi: 10.1016/S1359-6446(04)03316-1
Sams-Dodd, F. (2013). Is poor research the cause of the declining productivity of the pharmaceutical industry? An industry in need of a paradigm shift. Drug Discov. Today 18, 211–217. doi: 10.1016/j.drudis.2012.10.010
Schachtschneider, K. M., Liu, Y., Makelainen, S., Madsen, O., Rund, L. A., Groenen, M. A. M., et al. (2017a). Oncopig soft-tissue sarcomas recapitulate key transcriptional features of human sarcomas. Sci. Rep. 7:2624. doi: 10.1038/s41598-017-02912-9
Schachtschneider, K. M., Madsen, O., Park, C., Rund, L. A., Groenen, M. A., and Schook, L. B. (2015). Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model. BMC Genomics 16:743. doi: 10.1186/s12864-015-1938-x
Schachtschneider, K. M., Schwind, R. M., Darfour-Oduro, K. A., De, A. K., Rund, L. A., Singh, K., et al. (2017b). A validated, transitional and translational porcine model of hepatocellular carcinoma. Oncotarget 8, 63620–63634. doi: 10.18632/oncotarget.18872
Schachtschneider, K. M., Schwind, R. M., Newson, J., Kinachtchouk, N., Rizko, M., Mendoza-Elias, N., et al. (2017c). The oncopig cancer model: an innovative large animal translational oncology platform. Front. Oncol. 7:190. doi: 10.3389/fonc.2017.00190
Schook, L. B., Collares, T. V., Darfour-Oduro, K. A., De, A. K., Rund, L. A., Schachtschneider, K. M., et al. (2015a). Unraveling the swine genome: implications for human health. Annu. Rev. Anim. Biosci. 3, 219–244. doi: 10.1146/annurev-animal-022114-110815
Schook, L. B., Collares, T. V., Hu, W., Liang, Y., Rodrigues, F. M., Rund, L. A., et al. (2015b). A genetic porcine model of cancer. PLOS ONE 10:e0128864. doi: 10.1371/journal.pone.0128864
Schook, L. B., Rund, L., Begnini, K. R., Remiao, M. H., Seixas, F. K., and Collares, T. (2016). Emerging technologies to create inducible and genetically defined porcine cancer models. Front. Genet. 7:28. doi: 10.3389/fgene.2016.00028
Seok, J., Warren, H. S., Cuenca, A. G., Mindrinos, M. N., Baker, H. V., Xu, W., et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 3507–3512. doi: 10.1073/pnas.1222878110
Shortt, J., Hsu, A. K., and Johnstone, R. W. (2013). Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene 32, 4191–4202. doi: 10.1038/onc.2012.599
Sieren, J. C., Meyerholz, D. K., Wang, X. J., Davis, B. T., Newell, J. D. Jr., Hammond, E., et al. (2014). Development and translational imaging of a TP53 porcine tumorigenesis model. J. Clin. Invest. 124, 4052–4066. doi: 10.1172/JCI75447
Snawder, J. E., and Lipscomb, J. C. (2000). Interindividual variance of cytochrome P450 forms in human hepatic microsomes: correlation of individual forms with xenobiotic metabolism and implications in risk assessment. Regul. Toxicol. Pharmacol. 32, 200–209. doi: 10.1006/rtph.2000.1424
Swinney, D. C., and Anthony, J. (2011). How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519. doi: 10.1038/nrd3480
Takase, K., Sawai, M., Yamamoto, K., Yata, J., Takasaki, Y., Teraoka, H., et al. (1992). Reversible G1 arrest induced by dimethyl sulfoxide in human lymphoid cell lines: kinetics of the arrest and expression of the cell cycle marker proliferating cell nuclear antigen in Raji cells. Cell Growth Differ. 3, 515–521.
Teraoka, H., Mikoshiba, M., Takase, K., Yamamoto, K., and Tsukada, K. (1996). Reversible G1 arrest induced by dimethyl sulfoxide in human lymphoid cell lines: dimethyl sulfoxide inhibits IL-6-induced differentiation of SKW6-CL4 into IgM-secreting plasma cells. Exp. Cell Res. 222, 218–224. doi: 10.1006/excr.1996.0027
Tessmann, J. W., Buss, J., Begnini, K. R., Berneira, L. M., Paula, F. R., de Pereira, C. M. P., et al. (2017). Antitumor potential of 1-thiocarbamoyl-3,5-diaryl-4,5-dihydro-1H-pyrazoles in human bladder cancer cells. Biomed. Pharmacother. 94, 37–46. doi: 10.1016/j.biopha.2017.07.060
Thorn, H. A., Hedeland, M., Bondesson, U., Knutson, L., Yasin, M., Dickinson, P., et al. (2009). Different effects of ketoconazole on the stereoselective first-pass metabolism of R/S-verapamil in the intestine and the liver: important for the mechanistic understanding of first-pass drug-drug interactions. Drug Metab. Dispos. 37, 2186–2196. doi: 10.1124/dmd.109.028027
Thorn, H. A., Lundahl, A., Schrickx, J. A., Dickinson, P. A., and Lennernas, H. (2011). Drug metabolism of CYP3A4, CYP2C9 and CYP2D6 substrates in pigs and humans. Eur. J. Pharm. Sci. 43, 89–98. doi: 10.1016/j.ejps.2011.03.008
Ueda, H., Manda, T., Matsumoto, S., Mukumoto, S., Nishigaki, F., Kawamura, I., et al. (1994). FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J. Antibiot. 47, 315–323. doi: 10.7164/antibiotics.47.315
van Marion, D. M., Domanska, U. M., Timmer-Bosscha, H., and Walenkamp, A. M. (2016). Studying cancer metastasis: existing models, challenges and future perspectives. Crit. Rev. Oncol. Hematol. 97, 107–117. doi: 10.1016/j.critrevonc.2015.08.009
Wagner, A. H. P., Okuno, S., Eriksson, M., Shreyaskumar, P., Ferrari, S., Casali, P., et al. (2013). “Results from a phase 2 randomized, placebo-controlled, double blind study of the hedgehog (HH) pathway antagonist IPI-926 in patients (PTS) with advanced chondrosarcoma (CS),” in Proceedings of the Connective Tissue Oncology Society 18th Annual Meeting, New York, NY.
Warchal, S. J., Unciti-Broceta, A., and Carragher, N. O. (2015). Next-generation phenotypic screening. Future Med. Chem. 7, 2131–2141. doi: 10.1017/CBO9781107415324.004
Williams, R. (2013). Discontinued drugs in 2012: oncology drugs. Expert Opin. Investig. Drugs 22, 1627–1644. doi: 10.1517/13543784.2013.847088
Williams, R. (2015). Discontinued in 2013: oncology drugs. Expert Opin. Investig. Drugs 24, 95–110. doi: 10.1517/13543784.2015.971154
Zhang, J., Yang, P. L., and Gray, N. S. (2009). Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. doi: 10.1038/nrc2559
Zheng, W., Thorne, N., and McKew, J. C. (2013). Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 18, 1067–1073. doi: 10.1016/j.drudis.2013.07.001
Keywords: PDD, animal model, swine, Oncopig cancer model, cancer
Citation: Segatto NV, Remião MH, Schachtschneider KM, Seixas FK, Schook LB and Collares T (2017) The Oncopig Cancer Model as a Complementary Tool for Phenotypic Drug Discovery. Front. Pharmacol. 8:894. doi: 10.3389/fphar.2017.00894
Received: 28 September 2017; Accepted: 22 November 2017;
Published: 05 December 2017.
Edited by:
Satish Kitambi, Karolinska Institute (KI), SwedenReviewed by:
Cristiana Tanase, “Victor Babes" National Institute of Pathology, RomaniaRahul K. Keswani, Exelead, Inc., United States
Copyright © 2017 Segatto, Remião, Schachtschneider, Seixas, Schook and Collares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiago Collares, dGlhZ28uY29sbGFyZXNAdWZwZWwuZWR1LmJy