 Xuemei Zhao1†
Xuemei Zhao1† Yulong Liang
Yulong Liang- 1College of Pharmacy, Taishan Medical University, Tai’an, China
- 2State Key Laboratory of Genetic Engineering, Institute of Genetics, School of Life Sciences, Fudan University, Shanghai, China
- 3Michael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX, United States
Cyclophilin J (CyPJ), also called peptidylprolyl isomerase like 3, has been identified as a novel member of the cyclophilin family. Our previous research has resolved the three-dimensional structure of CyPJ and demonstrated the peptidylprolyl cis–trans isomerase (PPIase) activity of CyPJ, which can be inhibited by the common immunosuppressive drug cyclosporine A (CsA). Importantly, CyPJ is upregulated in hepatocellular carcinoma (HCC) and promotes tumor growth; CyPJ inhibition by CsA- or siRNA-based knockdown results in a remarkable suppression of HCC. These findings suggest that CyPJ may be a potential therapeutic target for HCC, and discovery of relevant inhibitors may facilitate development of a novel CyPJ-based targeting therapy. However, apart from the common inhibitor CsA, CyPJ has yet to be investigated as a target for cancer therapy. Here, we report structure-based identification of novel small molecule non-peptidic CyPJ inhibitors and their potential as antitumor lead compounds. Based on computer-aided virtual screening, in silico, and subsequently surface plasmon resonance analysis, 19 potential inhibitors of CyPJ were identified and selected for further evaluation of PPIase CyPJ inhibition in vitro. Thirteen out of 19 compounds exhibited notable inhibition against PPIase activity. Among them, the compound ZX-J-19, with a quinoxaline nucleus, showed potential for tumor inhibition; thus, we selected it for further structure–activity optimization. A total of 22 chemical derivatives with 2,3-substituted quinoxaline-6-amine modifications were designed and successfully synthesized. At least 2 out of the 22 derivatives, such as ZX-J-19j and ZX-J-19l, demonstrated remarkable inhibition of tumor cell growth, comparable to CsA but much stronger than 5-fluorouracil. These results indicate that these two small molecules represent novel potential lead compounds for CyPJ-based antitumor drug development.
Introduction
Cyclophilins constitute a superfamily of peptidylprolyl isomerases (PPIase), which catalyze the cis–trans isomerization of peptide bonds on the N-terminal side of proline residue (Harikishore and Yoon, 2015; Dunyak and Gestwicki, 2016). The cyclophilin family, which is comprised of more than 15 members, was named for its ability to bind the common immunosuppressive drug cyclosporine A (CsA) (Walsh et al., 1992). Cyclophilins have been shown to act as chaperones accelerating protein folding and maturation, as well as playing a critical role in signal transduction and the immune response (Gothel and Marahiel, 1999; Wang and Heitman, 2005; Lucke and Weiwad, 2011; Harikishore and Yoon, 2015). They have also been implicated in many pathological processes, including viral infection (Towers et al., 2003; Hopkins and Gallay, 2015; Dawar et al., 2017), rheumatoid arthritis (Kim et al., 2005; Pap, 2005), cardiovascular diseases (Satoh et al., 2009; Nigro et al., 2011; Xue et al., 2017), and cancers (Lee and Kim, 2010; Nigro et al., 2013; Wang et al., 2017).
Cyclophilin J (CyPJ), also known as peptidylprolyl isomerase like 3 (PPIL3), is a new member of the cyclophilin family, with human CyPJ being first reported in 2001 (Zhou et al., 2001). CyPJ is encoded by the CyPJ gene initially identified in the fetal human brain. Its protein sequence exhibits 50% identity to human cyclophilin A and 72% identity to cyclophilin isoform 10 of Caenorhabditis elegans (Zhou et al., 2001).
Previously, we reported, for the first time, the three-dimensional structure of human CyPJ alone and in complex with CsA (Huang et al., 2005). We also identified its PPIase activity, which could be inhibited by CsA (Chen et al., 2015). The CyPJ protein contains four helices and one β-barrel composed of eight antiparallel β-strands. It also harbors a disulfide bridge and four segments with conformations strikingly different from those of CyPA (Huang et al., 2005). Specifically, His43, Arg44, and Gln52 are conserved active site residues located in the shallow pocket of CyPJ (Huang et al., 2005). Furthermore, a conserved water molecule is observed close to His43 and Gln52, while Arg44 is involved in hydrogen bonding interactions with CsA, which accounts for the observed inhibitory qualities of CsA on the PPIase activity within CyPJ (Chen et al., 2015). Importantly, we demonstrated that CyPJ is frequently upregulated in a small cohort of hepatocellular carcinoma (HCC), and CyPJ-based targeting leads to tumor suppression of HCC in a xenograft mouse tumor model (Chen et al., 2015). Several studies also report that CyPJ is upregulated in human glioma (Qi et al., 2005), estrogen receptor-negative breast cancer (Couch et al., 2016), and primary gastric adenocarcinoma (Gong et al., 2017), suggesting that the CyPJ enzyme may be an important and potential therapeutic target. However, apart from the common inhibitor of cyclophilins, CsA, CyPJ is yet to be investigated as a molecular target for cancer therapy.
A quinoxaline is an important class of nitrogen-containing heterocycle and this class of compounds plays an important role in drug development due to their potent pharmacological activity (Supplementary Table S1). For instance, marketed drugs, such as Echinomycin, Levomycin, Actinoleutin, Varenicline, Brimonidine, and Quinacillin, all contain a quinoxaline ring. In addition, quinoxaline derivatives have been reported as core motifs in medicinally active ingredients, acting as anti-protozoal, anti-proliferative, anti-inflammatory, anti-bacterial, anti-viral agents, and inhibitors of Jnk stimulatory phosphatase-1 (JSP-1), a special member of the dual-specificity protein phosphatase family 1 (Supplementary Table S1) (Hui et al., 2006; Zhang et al., 2007; Chen et al., 2011). More recently, evidence has emerged that quinoxaline-derived compounds might be capable of inhibiting tumor growth (Supplementary Table S1) (El Aissi et al., 2014; Gu et al., 2017; Miller et al., 2017). However, quinoxaline-based lead compounds have not been evaluated as inhibitors specific for target protein CyPJ to be applied in cancer therapy.
In this study, we aimed to identify novel potential inhibitors of CyPJ and characterize their possible antitumor activity, especially for HCC. To this end, we used structure-based in silico virtual screening (VS) followed by surface plasmon resonance and a PPIase inhibition assay to identify CyPJ inhibitors. Based on those results, we optimized antitumor activity of CyPJ inhibitors using quinoxaline nucleus-associated rational design. The evidence derived from the current study provides novel potential CyPJ inhibitors and highlights the potent inhibitory effects of these compounds on tumor cell growth.
Materials and Methods
General Information
Expression and purification of recombinant human CyPJ protein was carried out as described previously (Chen et al., 2015). CyPJ protein concentration was determined using the Bradford method. The substrates N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Suc-AAPF-pNA), CsA, 2,2,2-trifluoroethanol, and α-chymotrypsin were purchased from Sigma (St. Louis, MO, United States). The libraries of small molecular compounds used for VS were obtained from SPECS (200,000 compounds)1 and China Natural Products Database (CNPD, 50,000 compounds)2. The small organic compounds selected after VS were purchased from SPECS (Zoetermeer, Netherlands). Chemicals and solvents were either purchased from Sigma or purified by standard techniques. Analytical thin-layer chromatography (TLC) was performed on a Merck pre-coated TLC plate (silica gel 60 F254). Melting points were recorded on an X4-Data microscopic melting point apparatus and were uncorrected. Electrospray ionization-mass spectrometry (ESI-MS) was performed on a Bruker Esquire 3000 plus spectrometer. 1H NMR spectra were recorded on a Bruker Avance 400 spectrometer in CDCl3 using tetramethylsilane (TMS) as the internal standard. Elemental analysis was performed on Carlo-Erba 1106 (Carlo Erba, Italy).
Molecular Docking and Active Site Analysis
Previously, we obtained two crystal structures of CyPJ: the CyPJ/CsA complex at 2.4 Å (PDB: 2OJU) and the CyPJ crystal alone at 2.0 Å (PDB: 2OK3) (Chen et al., 2015). In order to better mimic the affinity between CyPJ and the relevant compounds, and accurately calculate their docking scores, we selected the crystal structure of the CyPJ/CsA complex (2OJU) for molecular docking analysis. Arg44, Gln52, Asn92, His110, and Tyr115 were conserved active site residues located in the shallow pocket of the protein structure, where Arg44 displayed a flexible conformation. Our compounds of interest were then each flexibly docked on the protein structure. In our docking calculation, potential energy maps of the receptor were calculated using default parameters.
In Silico Virtual Screening
Virtual screening in silico was carried out as described elsewhere with minor modifications (Li et al., 2006b). The structure of CyPJ/CsA complex (PDB: 2OJU) was used as a target. The DOCK 4.0 program suite was employed for primary screening of the small molecule databases SPECS and CNPD via 64CPU-SGI ORIGIN3800 (State Key Laboratory of computer-aided drug design, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China) and 392CPU-Shenwei I supercomputer (Shanghai Supercomputing Center) (Kuntz, 1992; Butler et al., 2016; Choi et al., 2017). Based on the X-ray structure of the CyPJ/CsA complex (Chen et al., 2015), Arg44, Gln52, Asn92, His110, and Tyr115 residues surrounding CsA were selected to generate a cavity. Our small molecules were then docked into this cavity, and ligand-binding quality was evaluated using a force-field scoring function. During the docking calculation, Kollman united-atom charges were assigned to the CyPJ protein, and Gasteiger–Marsili partial charges were assigned to the small molecules in the databases. Conformational flexibility of the compounds from the databases was considered during the docking search. Three thousand top-scoring compounds obtained by DOCK search were rescored by using the Consensus Score (CScore) method encoded in Sybyl6.8 (Sybyl molecular modeling package, version 6.8) (Houston and Walkinshaw, 2013; Campagna-Slater et al., 2014; Weill et al., 2014). Molecules with a CScore of ≥4 were visually analyzed. Finally, 74 compounds with the lowest energy and most favorable ligand orientation were selected.
Surface Plasmon Resonance (SPR) Assay
The binding affinity of the selected compounds to CyPJ was measured by SPR with a Biacore 3000 instrument (Biacore AB Corporation, Uppsala, Sweden) as previously described (Guo et al., 2005; Chen et al., 2007). Briefly, recombinant human CyPJ protein was immobilized on an activated sensor chip via amine coupling. CyPJ protein (10 μM) was coupled to a CM5 sensor chip surface (a carboxymethylated dextran surface; CM5 chip from Biacore, Inc., Piscataway, NJ, United States) in a buffer containing 10 mM sodium acetate (pH 4.0) using standard amine coupling chemistry according to the manufacturer’s instructions. Flow cell 1 (FC-1) was used as the control surface and flow cell 2 (FC-2) contained 9000 resonance units (RU) of CyPJ (1 RU corresponds to 1 pg of protein per mm2). The buffer stream was passed through FC-1 and FC-2 of the bi-channel at a flow rate of 40 μl/min. Compounds to be tested were purchased from SPECS (Netherlands). All chemicals were of the highest purity available, as certified by the vendor. All compounds were stocked in 100% dimethylsulfoxide at 10 mM and diluted at graded concentrations (0.3–10 μM) with HBS-EP buffer (0.01 M HEPES buffer, pH 7.4, containing 0.15 M NaCl, 3.4 mM EDTA, and 0.005% surfactant P20) containing 1% dimethylsulfoxide. The temperature of the instrument was set to 20°C, and the flow rate was set to 40 μl/min. Each compound (50 μl) was injected sequentially. NaOH (50 mM) was used to regenerate the surface of the CM5 chip. The 1:1 Langmuir binding model was used to determine the equilibrium dissociation constant (KD). For fast interactions, the steady-state model was used to determine KD values. All experiments were carried out in triplicate.
PPIase Assay
The PPIase assay was carried out with minor modifications as described previously (Kofron et al., 1991). We used an α-chymotrypsin-coupled PPIase assay, where the substrate succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Suc-AAPF-pNA) is first converted to the trans conformation by PPIase, and can then be digested by α-chymotrypsin to release chromogenic p-nitroanilide; the latter compound is then monitored with a spectrometer. Briefly, the assay was performed under 8°C in a 100 μl system. The Suc-AAPF-pNA was dissolved in the tetrafluoroethylene containing 480 mM of LiCl (working concentration: 3.0 mM). The α-chymotrypsin was dissolved in 1 mM HCl (working concentration: 1.7 mM). When assayed, each test compound was diluted in 94 μl of assay buffer (50 mM HEPES, 100 mM NaCl; pH 8.0 at 0°C), and then mixed with 2 μl of CyPJ solution (5 μM). After equilibrating on ice for 3 h, 2 μl of α-chymotrypsin solution and 2 μl of Suc-AAPF-pNA were added to the assay mixture, and the absorbance at the wavelength 390 nm was recorded for 20 s on a Jasco V-550 Spectrophotometer (Jasco, Inc., Easton, MD, United States). Three independent experiments were performed for each test compound and the respective half maximal inhibitory concentration (IC50) was calculated with OriginPro 7.5 software (OriginLab, Northampton, MA, United States).
Synthesis of 2,3-Substituted Quinoxaline-6-Amine (Compound 3)
Briefly, benzaldehyde or furaldehyde (0.1 mol) was catalyzed by thiamine (vitamin B1, 0.02 mol) to generate 2-hydroxy-1,2-diphenylethanone or 1,2-di(furan-2-yl)-2-hydroxyethanone, respectively. The resulting alcohol hydroxyls were oxidized by Cu(NO3)2 to generate diones 1a and 1b in HOAc/H2O (1:1, V/V) at 70°C in 6 h. The selective acylation of pyrrole was catalyzed with oxalyl chloride in carbon disulfide to generate the dione 1c at -70°C. These 1,2-ethanedione derivatives (1a, 1b, and 1c, 0.3 mol each) were mixed with 4-nitro-o-phenylenediamine (0.5 mol) in AcOH (100 ml), and stirred at 110°C under normal atmosphere for 8 h. The mixture was then cooled to room temperature and poured into water (300 ml) to obtain 6-nitro-2, 3-disubstituted quinoxaline (compound 2). A reaction bottle was charged with compound 2 (0.1 mol) and Na2Sx (0.15 equiv) in ethanol (30 ml) under normal atmosphere. The mixture was vigorously stirred by refluxing for 12 h, and cooled to room temperature and poured into water (100 ml) after competition. The mixture was next extracted with 20 ml of EtOAc three times. The combined organic layer was washed with brine (20 ml), dried with Na2SO4, and the solvent was removed under reduced pressure. Finally, compound 3 was purified as an eluent by flash column chromatography using PE/EtOAc.
Synthesis of 2,3-Substituted Quinoxaline-6-Amine Derivatives (ZX-J-19 Derivatives)
The synthesis of 2,3-substituted quinoxaline-6-amine derivatives (i.e., ZX-J-19 derivatives) was performed as described elsewhere (Chen et al., 2011; Sridevi et al., 2011). Briefly, compound 3 (20 mmol) was treated with acyl chloride (30 mmol) in the presence of pyridine at room temperature in acetone. After compound 3 disappeared on TLC, the reaction mixture was poured into the water (50 ml), immediately cooled to room temperature, filtered, and purified to collect ZX-J-19 derivatives by flash column chromatography.
Cell Lines and MTT-Based Cell Proliferation Assay
Human HCC cells SK-HEP1 and QGY; breast cancer cells HCC1954, BT474, and MDA-MB468; ovarian cancer cells SKOV3; and prostate cancer cells PC3 and LNCaP were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and cultured in T75 flasks with complete DMEM or RPMI 1640 medium (GIBCO, Invitrogen, Gaithersburg, CA, United States) supplemented with 10% FBS (Hyclone, New Zealand), and 1% penicillin/streptomycin (Solarbio, Beijing, China) at 37°C in a humidified atmosphere containing 5% CO2.
Cell proliferation was determined using methylthiazolyl diphenyl-tetrazolium bromide assay (MTT; Sigma–Aldrich). The cells were seeded onto 96-well plates in a total volume of 150 μl at a density of 3.5 × 104 cells/well. After incubation for 24 h, cells were treated with the selected compounds or positive control drugs 5-fluorouracil (5-FU) and CsA at the indicated dosages (0, 1.0, 5.0, 10, 50, and 250 μM). To avoid the effect of the solvent, the concentration of DMSO was less than 0.1% (v/v) in all experiments. After incubation at 37°C for 48 h, 10 μl of MTT solution (5 mg/ml) was added and continued to incubate for 4 h. Following removal of medium containing MTT, 200 μl of DMSO was added to dissolve the formazan crystals formed by live cells. Solution absorbance was measured at 490 nm with an absorbance microplate reader (BioTek, Winooski, VT, United States). The assay was repeated six times. The IC50 value was determined from plot of % viability against the dose of compounds added.
Statistical Analysis
The compound activities and MTT-based cell assays were repeated at least three times and the significance was assessed with unpaired Student’s t-test. The survival curve was created by the Kaplan–Meier method and analyzed by the log-rank test. A P < 0.05 was considered statistically significant.
Results
Identification of Small Molecule Inhibitors of CyPJ by Virtual Screening and SPR Analysis
To identify the inhibitory small molecules of CyPJ, first VS was conducted, in silico, using commercially available compound libraries. Crystal structures of CyPJ complexed with CsA were analyzed for potential inhibitory ligands (Huang et al., 2005; Chen et al., 2015). As illustrated in Figure 1, we screened SPECS and CNPD databases with a total of 250,000 small molecule compounds to determine their ability to dock at the catalytic site of CyPJ. We selected 74 compounds with the best docking scores. Among them, 63 compounds were commercially available and purchased from SPECS and their CyPJ binding was subsequently analyzed by SPR.
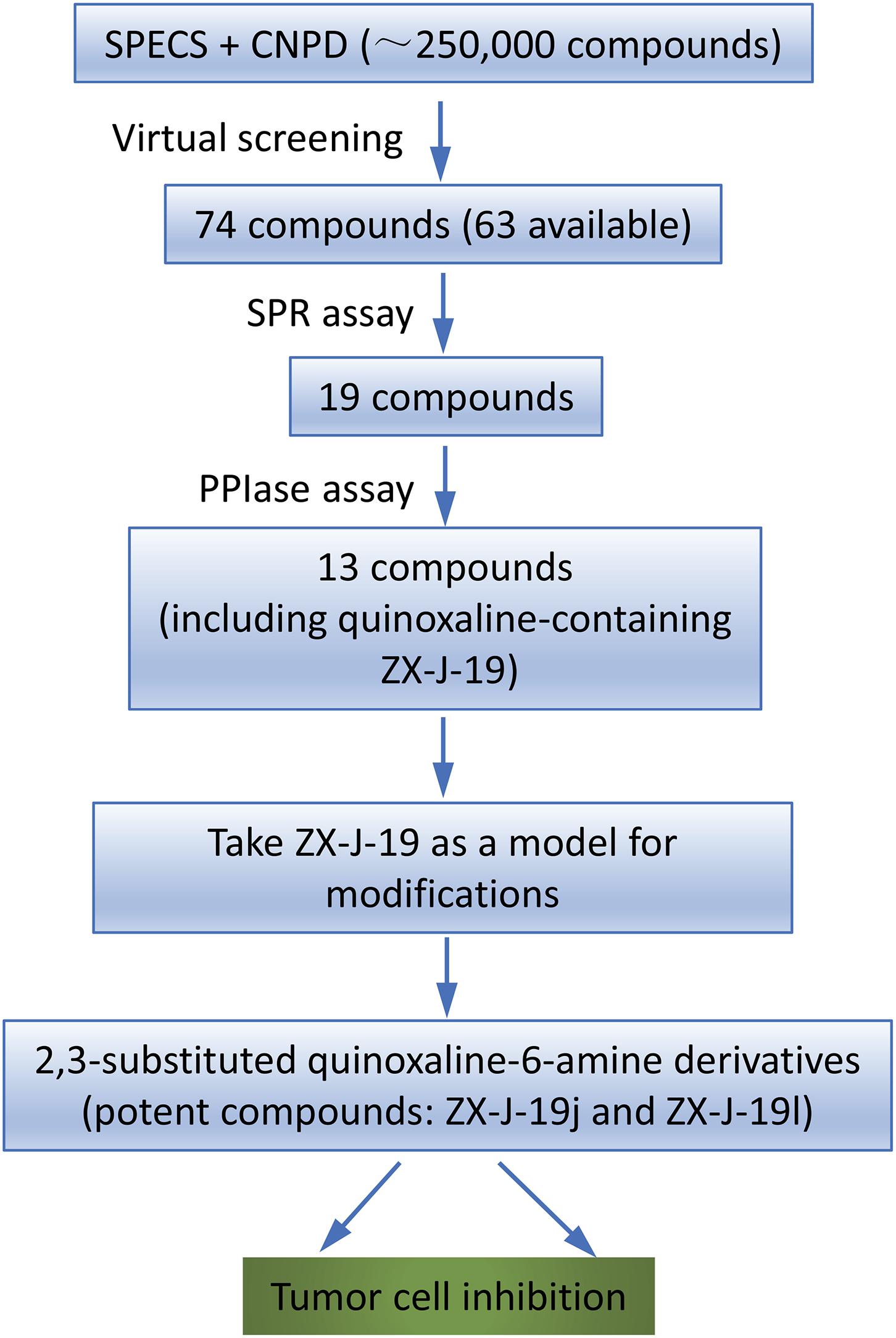
FIGURE 1. Flow chart of virtual screening and identification of CyPJ inhibitors. SPECS and CNPD are chemical libraries containing 200,000 and 50,000 compounds, respectively. SPR, surface plasmon resonance. PPIase, peptidylprolyl cis–trans isomerase.
Based on SPR analysis, 19 out of 63 compounds, i.e., ZX-J-1 to ZX-J-19 (Figure 2), were found to be capable of binding with CyPJ (Table 1). The binding of the above 19 compounds behaved in a concentration-dependent manner with Ki values ranging from 1 × 10-4 M to 9 × 10-9 M by VS, and with KD values ranging from 1 × 10-4 M to 9 × 10-8 M by SPR analysis (Table 1). These results suggested that these 19 compounds strongly interact with CyPJ.
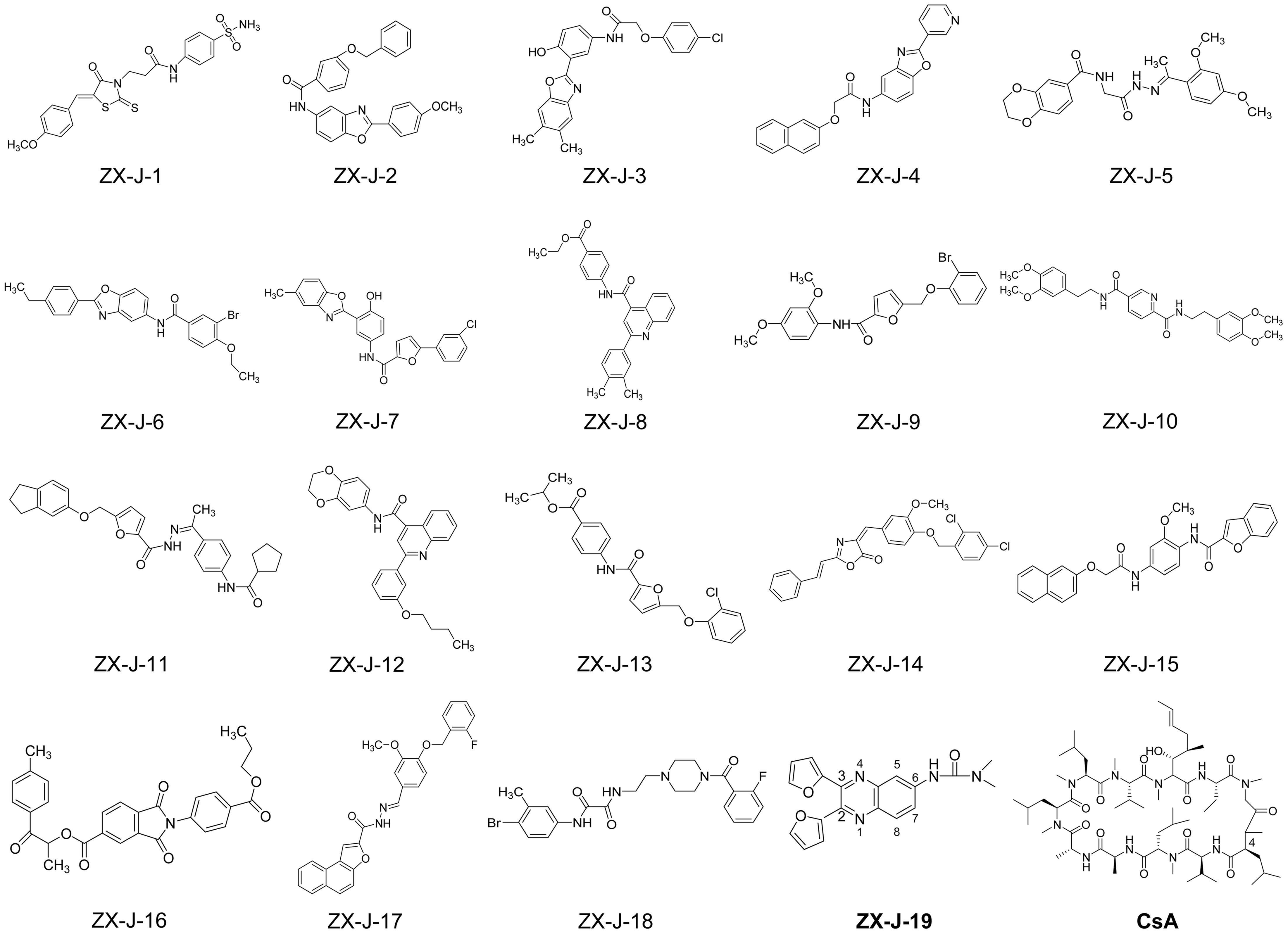
FIGURE 2. Chemical structures of 19 potential CyPJ inhibitors identified with in silico screening and SPR analysis. Following in silico screening and SPR analysis, 19 ZX-J compounds numbered 1–19 were identified and selected for further investigation. The numbers in the compound ZX-J-19 represent the positions of the carbons in the quinoxaline ring. CsA, cyclosporine A, is a well-established cyclophilin protein inhibitor.
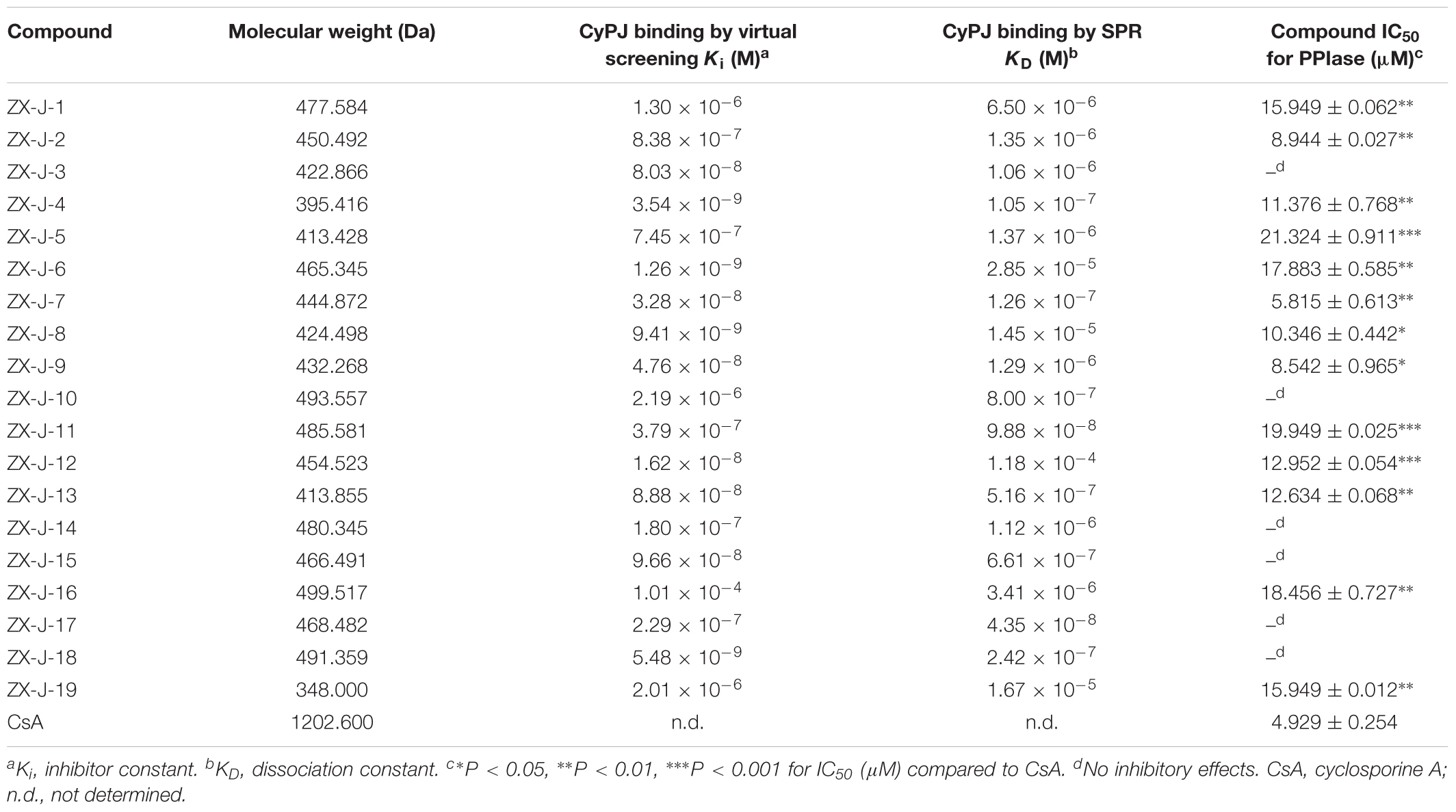
TABLE 1. Affinity of inhibitors to CyPJ, and their respective IC50 for CyPJ PPIase inhibition.
Inhibition of the PPIase Activity by 13 Identified Compounds
To investigate whether the selected compounds inhibit the PPIase activity of CyPJ, we performed a PPIase assay as described previously (Chen et al., 2015). The inhibitory effect of each compounds was determined with a standard spectrophotometric method in chymotrypsin-coupled assays with different concentrations of peptide substrate. The rate constants for the cis–trans conversion were evaluated by fitting the data to the integrated first-order rate equation through nonlinear least-square analysis. Since CyPJ belongs to the cyclophilin protein family, the inhibitory effect of these compounds appears to follow Michaelis–Menten kinetics (Chen et al., 2015). There were 13 compounds including ZX-J-1, ZX-J-2, ZX-J-4, ZX-J-5, ZX-J-6, ZX-J-7, ZX-J-8, ZX-J-9, ZX-J-11, ZX-J-12, ZX-J-13, ZX-J-16, and ZX-J-19 that had IC50 values ranging from 5 to ∼20 μM, slightly higher than that of CsA (Table 1). Meanwhile, no inhibition of PPIase activity of CyPJ was detectable in the remaining six compounds, ZX-J-3, ZX-J-10, ZX-J-14, ZX-J-15, ZX-J-17, and ZX-J-18 (Table 1). We concluded that 13 out of 19 compounds may be regarded as the inhibitors of CyPJ.
HCC Tumor Cell Growth Inhibition by Quinoxaline-Containing Compound ZX-J-19
Among the 13 inhibitory compounds identified previously, ZX-J-19 is a quinoxaline derivative composed of a quinoxaline nucleus with a furanyl group at positions 2 and 3 and a urea group at position 6 (Figure 2). Since the quinoxaline nucleus may potentially have antitumor activity (Supplementary Table S1), together with our previous observation that siRNA-based CyPJ targeting inhibited tumor growth of HCC (Chen et al., 2015), we wanted to know whether the quinoxaline-containing ZX-J-19 can inhibit HCC tumor cell growth. First of all, we verified the alterations of CyPJ in HCC in a large cohort from The Cancer Genome Atlas (TCGA). We found a higher CyPJ gene copy number in about 15% of HCC samples (n = 370) (Supplementary Figure S1A). Also, the CyPJ alterations were positively correlated with poor disease-free survival in HCC patients (P = 0.0287) (Supplementary Figure S1B), suggesting that CyPJ is of importance in HCC.
Next, the inhibitory effects of ZX-J-19 were determined using MTT-based in vitro cell proliferation assay along with CsA (positive control for inhibiting CyPJ) and 5-FU (positive control for antitumor activity) on HCC cells SK-HEP1 and QGY. As shown in Table 2, the IC50 of ZX-J-19 on SK-HEP1 and QGY HCC cells was 40.440 and 52.438 μM, respectively. Although these IC50 values were slightly higher than those of CsA (10.243 and 7.902 μM, respectively), as the positive control inhibitor of CyPJ, they were strikingly lower than those of 5-FU (177.238 and 238.528 μM, respectively), a conventional clinical drug (Table 2), suggesting that ZX-J-19 potently inhibits HCC tumor cell growth in vitro.
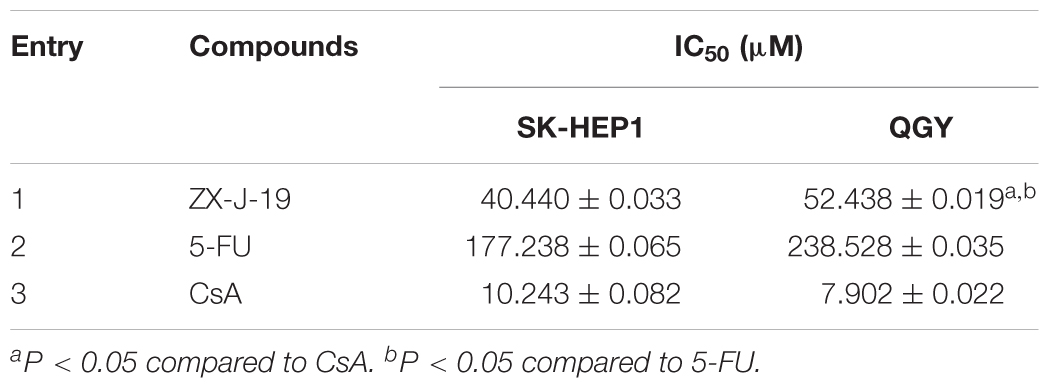
TABLE 2. IC50 of selected compounds on HCC cells.
Modifications of 2,3-Substituted-Quinoxaline-6-Amine Derivatives with Synthetic Chemistry
To identify more potent quinoxaline-containing compounds and further optimize them for tumor cell inhibition, we modified the quinoxaline nucleus at positions 2, 3, and 6 based on the structure of ZX-J-19 using the rational-design strategy. First, we synthesized the ethanediones (compound 1, e.g., 1a, 1b, and 1c) as presented in Figure 3A, which provided the necessary diversity of the R1 residues at positions 2 and 3. Benzaldehyde and furaldehyde reactions were catalyzed by thiamine to generate 2-hydroxy-1,2-diphenylethanone and 1,2-di (furan-2-yl)-2-hydroxyethanone, respectively (yields: 88 and 84%, respectively). The resulting hydroxyethanone derivatives were then oxidized by Cu(NO3)2 to generate the diones 1a and 1b in HOAc/H2O (1:1, V/V) at 70°C in 6 h (yields: 91 and 87%, respectively). For the synthesis of 1c, selective acylation of pyrrole was catalyzed with oxalyl chloride in carbon disulfide at -70°C (yield: 81%).
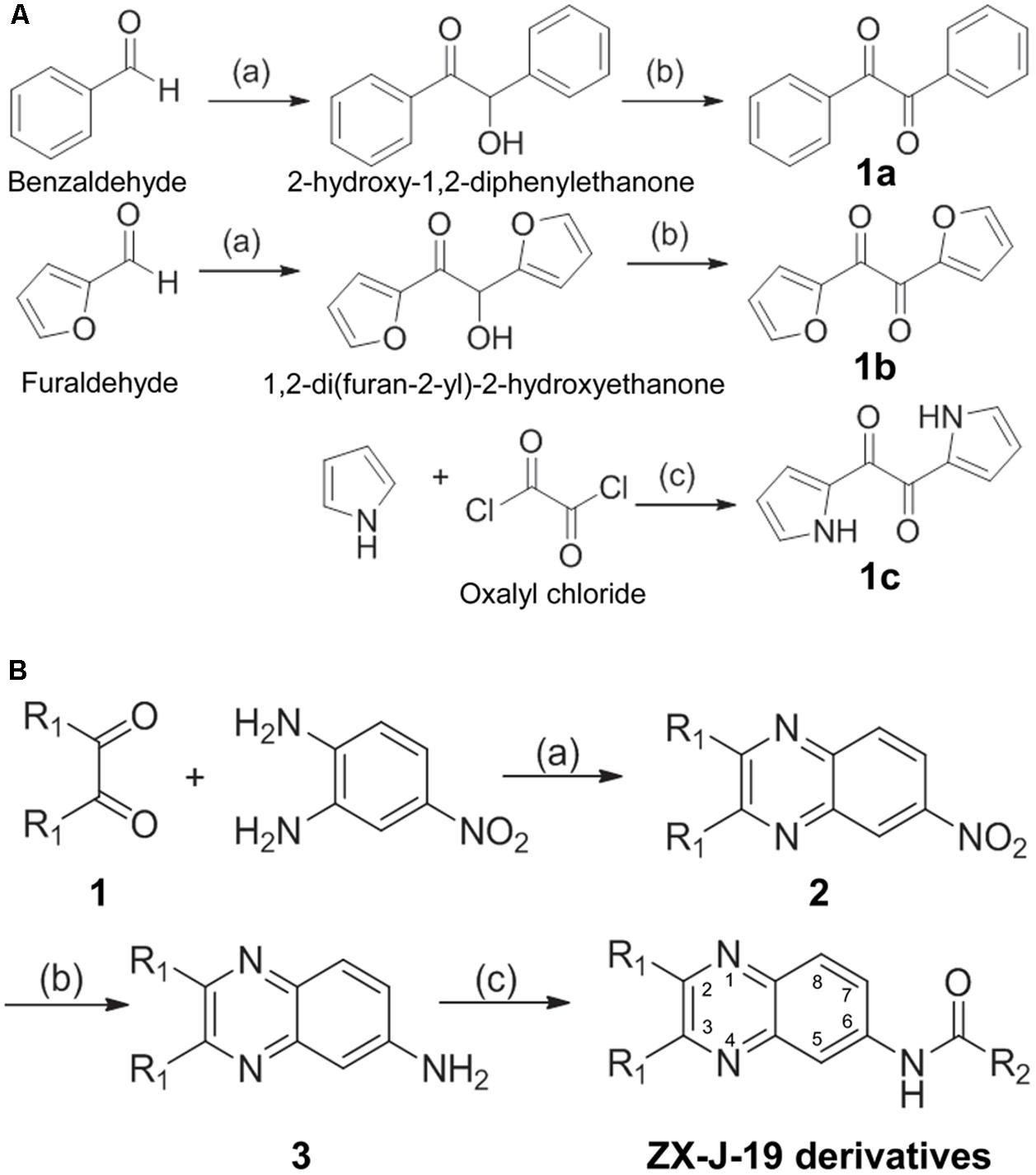
FIGURE 3. The synthesis route of 2,3-substituted-quinoxaline-6-amine derivatives. (A) The synthetic production of ethanediones (including intermediate chemicals 1a, 1b, 1c). Reagents and conditions: (a) Vitamin B1 (VB1), EtOH/H2O 2:1, NaOH, room temperature, 12 h; (b) Cu(NO3)2, HOAc/H2O 1:1, 70°C, 6 h; (c) carbon disulfide, –70°C, pyridine, 4 h. (B) The synthetic process of 2,3-substituted-quinoxaline-6-amine derivatives (i.e., ZX-J-19 derivatives). Compound 1 was referred to as 1a, 1b, or 1c obtained from (A). Reagents and conditions: (a) HOAc, reflux, 8 h; (b) Na2Sx, x = 2–6, EtOH, reflux, 12 h; (c) RCOCl, pyridine, acetone, reflux, 4 h. The numbers in the ZX-J-19 derivatives represent the position of the carbons in the quinoxaline ring. R1 refers to the side residuals at the positions 2 and 3 of the quinoxalines. R2 refers to the side residual linked to the amide group at the position 6 of the quinoxalines.
Next, 2,3-substituted-6-amine derivatives of quinoxaline were synthesized, and the synthesis route is illustrated in Figure 3B. The compounds 1a, 1b, or 1c were condensed with 4-nitro-o-phenylene diamine in HOAc under reflux for about 8 h to generate 6-nitro-2,3-diphenyl quinoxaline, 2,3-di(furan-2-yl)-6-nitroquinoxaline, or 6-nitro-2,3-di(1H-pyrrol-2-yl) quinoxaline (compound 2), respectively. The resulting nitro-quinoxalines (2) (e.g., 6-nitro-2,3-diphenyl quinoxaline) was reduced by Na2Sx (1.5 equiv) under reflux in ethanol to furnish the 2,3-diphenylquinoxaline-6-amine (compound 3). Compound 3 (1 equiv) was then treated with acyl chloride (1.3 equiv) in acetone at room temperature to produce the 22 2,3-quinoxaline-6-amine (ZX-J-19) derivatives, as shown in Table 3 (ZX-J-19a–ZX-J-19v; yield: 63–81%). All synthesized compounds were structurally characterized by 1H NMR, elemental analysis, and mass spectral analysis (see the Supplementary Structure Data listed in the Supplementary Materials). These compounds were categorized into three groups according to the property of the residues at the R1 position: the phenyl type (ZX-J-19a–ZX-J-19l), the furanyl type (ZX-J-19m–ZX-J-19s), and the pyrrole type (ZX-J-19t–ZX-J-19v), and the compounds with the furanyl type were more closely related to compound ZX-J-19.
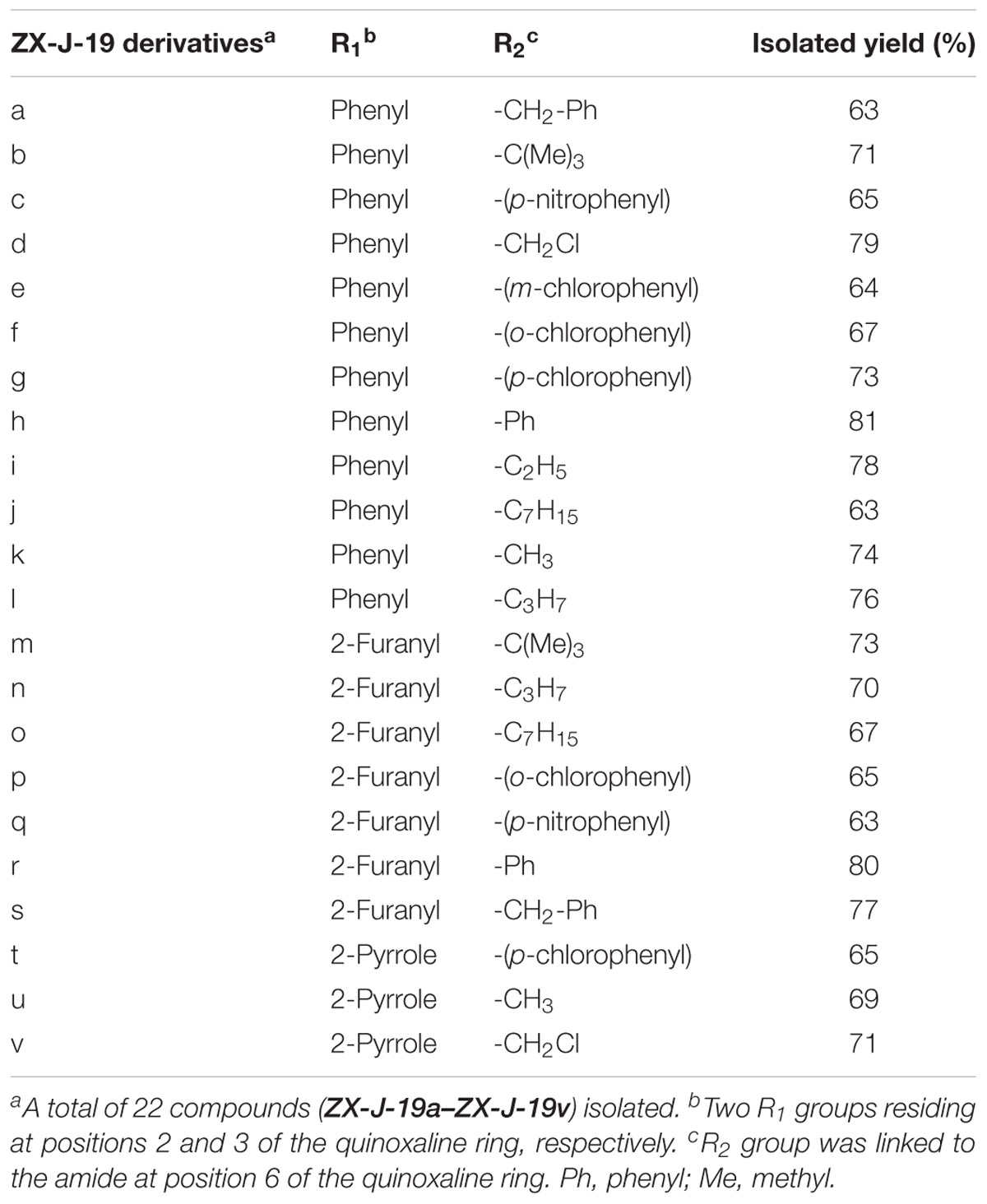
TABLE 3. Isolated yields of 2,3-substituted quinoxaline-6-amine derivatives.
Remarkable Inhibition of Tumor Cell Growth by ZX-J-19 Derivatives
To determine the effects of the 22 ZX-J-19 derivatives (a–v) on tumor cell growth, we first examined their activity on HCC cells as detected by the MTT-based in vitro cell proliferation assay. As shown in Table 4, four compounds ZX-J-19e, ZX-J-19g, ZX-J-19j, and ZX-J-19l, all belonging to the phenyl type of 2,3-substituted quinoxaline-6-amine derivatives, exhibited remarkable inhibitory effects on HCC cell growth. Their potency was comparable to the potency of the positive CyPJ inhibitor control, CsA, shown in Table 2. Among these four compounds, ZX-J-19j and ZX-J-19l exhibited the most potency. This result is evident by their lower IC50 on SK-HEP1 cells (6.725 and 3.512 μM, respectively) (Table 4), suggesting that ZX-J-19j and ZX-J-19l may be better than ZX-J-19 itself and other ZX-J-19 derivatives in HCC cell growth inhibition.
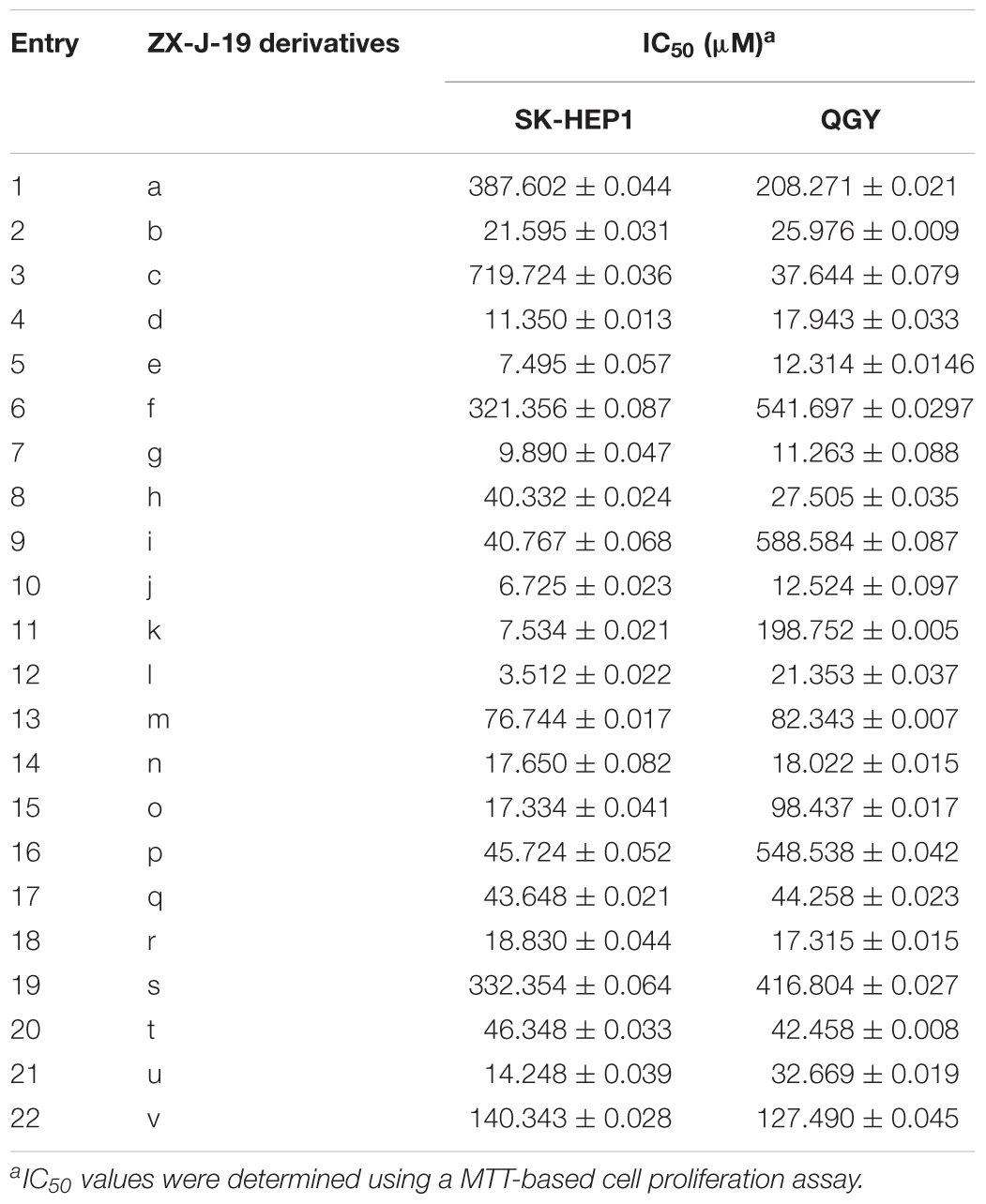
TABLE 4. IC50 of ZX-J-19 derivatives on HCC cells.
Besides HCC, the CyPJ gene is also aberrantly dysregulated in several other malignancies such as breast, prostate, and ovarian cancers (Supplementary Figure S2) (Qi et al., 2005; Couch et al., 2016; Gong et al., 2017). With this fact in mind, we further determined if ZX-J-19j and ZX-J-19l could inhibit growth of tumor cells originating from other cancers, including breast cancer cells HCC1954, BT474, and MDA-MB468; ovarian cancer cells SKOV3; and prostate cancer cells PC3 and LNCaP. As shown in Table 5, both ZX-J-19j and ZX-J-19l showed significant growth inhibition in these tumor cells, suggesting ZX-J-19j and ZX-J-19l might inhibit tumor cell growth in a wide spectrum of malignancies.
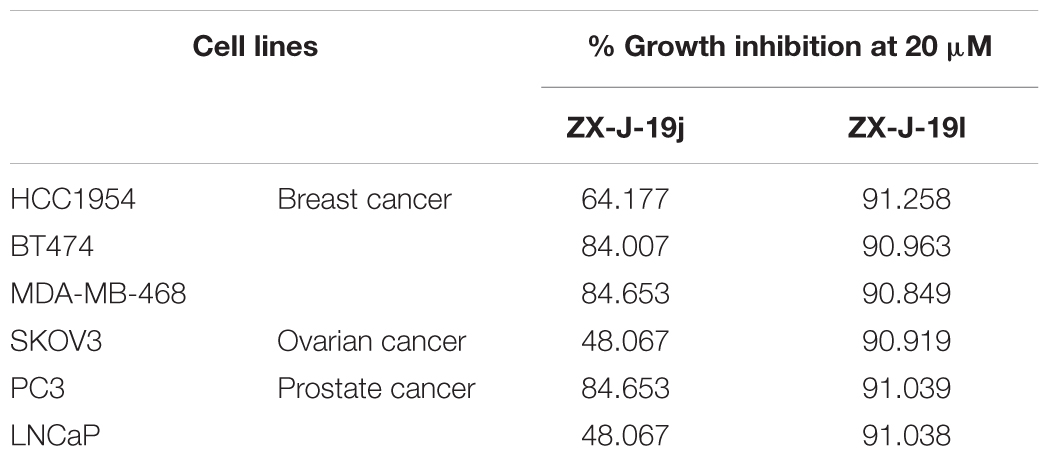
TABLE 5. Growth inhibition of multiple cancer cell lines with compounds ZX-J-19j and ZX-J-19l.
It should be pointed out that although ZX-J-19j and ZX-J-19l demonstrated convincing inhibitory effects on tumor cell growth by using in vitro cell models, these two compounds still retain similar lipophilicity to ZX-J-19. This characteristic may contribute to poor antitumor activity of ZX-J-19 in an in vivo xenograft mouse tumor model due to its poor pharmacokinetics (unpublished data). For example, this compound was absorbed and accumulated in blood serum within 10 min after intraperitoneal administration, peaked within 30–60 min, and then declined and diminished to control levels after 8 h. These results suggest that these 2,3-substituted quinoxaline-6 amine compounds need to be further optimized for in vivo application, a finding which is under investigation.
Taken together, our data suggest that these quinoxaline-containing derivatives (e.g., ZX-J-19j and ZX-J-19l) are lead compounds for novel CyPJ-targeting antitumor drugs.
Discussion
Based on the three-dimensional structure of CyPJ reported in our previous research (Hu et al., 2005; Huang et al., 2005; Chen et al., 2015), we virtually screened the chemical libraries of SPECS and CNPD in silico, and were able to identify 13 inhibitors of CyPJ (Table 1). Because of the well-characterized potential antitumor activity of quinoxaline derivatives and their relative ease of synthesis, we focused on the quinoxaline-containing compound ZX-J-19 and quinoxaline-based modifications of that template. A total of 22 2,3-substituted-quinoxaline-6-amine derivatives were successfully synthesized using a structure–activity relationship (SAR) strategy (Table 3). Moreover, we were able to provide convincing evidence that the growth in several tumor cell lines was inhibited by ZX-J-19j and ZX-J-19l (Tables 4, 5). These representatives of strong antitumor activity may act as lead compounds to facilitate the development of promising drugs targeting CyPJ-associated cancers.
In this study, we confirmed aberrations of the CyPJ gene previously reported in HCC (Chen et al., 2015) and demonstrated that its expression was associated with HCC progression (Supplementary Figure S1). Although CyPJ was relatively uninvestigated previously, a few reports suggested that it might play an important role in tumorigenesis and/or progression in HCC (Chen et al., 2015) and other cancers (Qi et al., 2005; Couch et al., 2016; Gong et al., 2017). For example, we have previously shown the importance of CyPJ expression in HCC; CyPJ inactivation by CsA- or siRNA-based CyPJ knockdown diminished HCC tumor cell growth in vitro and in vivo, demonstrating CyPJ was capable of initiating tumorigenesis of HCC (Chen et al., 2015). Our observations in this study are consistent with these previous findings (Supplementary Figure S1). Previous research provided support for the further investigation of CpPJ inhibitors ZX-J-19j and ZX-J-19l as potent inhibitors of tumor cell growth.
Cyclosporine A is a well-established potent inhibitor of cyclophilins that can also block the PPIase catalytic sites of CyPJ (Chen et al., 2015). CsA has profound implications in immunosuppression for organ transplantation (Colombo and Ammirati, 2011; Ziaei et al., 2016), however, as an antitumor agent, CsA may not be suitable due to potent side effects related to secondary skin cancers and other malignancies (Behnam et al., 2005; Gallagher et al., 2010; Norman et al., 2010; Han et al., 2012; Muellenhoff and Koo, 2012). It is therefore necessary to identify novel small molecule antitumor compounds that target cyclophilins including CyPJ. In this report, we identified at least 13 compounds, including the quinoxaline-containing compound ZX-J-19, that exhibited notable inhibitory activities of PPIase in CyPJ (Table 1). Intriguingly, based on the quinoxaline nucleus scaffold of ZX-J-19, we rationally designed and ultimately synthesized 22 ZX-J-19-like quinoxaline derivatives with 2,3-substituted-6-amine modifications. Two of the resulting compounds ZX-J-19j and ZX-J-19l had notably enhanced antitumor activities (Tables 4, 5).
Consistent with these results, the quinoxaline nucleus is known to exhibit antitumor activity, and quinoxaline-derived compounds have been reported to have potential in cancer treatment (Zarranz et al., 2004; Amin et al., 2006; Weng et al., 2008; Pereira et al., 2015). For example, several quinoxaline-1,4-di-N-oxide derivatives exhibited considerable antitumor activity in MCF-7 (breast), NCI-H460 (lung), and SF-268 (CNS) cells; such antitumor activity was dependent on the substituents at the carbonyl group (Zarranz et al., 2004). With regard to ZX-J-19j and ZX-J-19l, the phenyl at position R1 and the heptyl (ZX-J-19j) or propyl (ZX-J-19l) at position R2 were considered as the residues that contribute to the amelioration of antitumor capability. Similarly, our data indicated that at least ZX-J-19j and ZX-J-19l are optimal lead candidate compounds for further development of novel CyPJ inhibitors and the potential antitumor drugs for CyPJ-based targeting.
As discussed previously, the quinoxaline derivative structures were determined based on CyPJ protein structure. However, we cannot rule out the possibility that these derivatives may impair the PPIase activity of other human cyclophilins. There are at least 15 members of the cyclophilin protein family (Fruman et al., 1994; Galat and Metcalfe, 1995). Due to high degree of similarity between members, cyclophilins inhibitors may have overlapping inhibitory action against many cyclophilins. For instance, a potent, well-characterized cyclophilin inhibitor, CsA, can inhibit the PPIase activities of almost all cyclophilins including CyPJ (Handschumacher et al., 1984; Chen et al., 2015). Cyclophilin CyPA has similarly been reported to be involved in many diseases; CyPA PPIase-based inhibitor Debio-025 (Alisporivr) has been shown to inhibit several other cyclophilins including CyPB and CyPD (Flisiak et al., 2008; Quarato et al., 2012; Lee, 2013). Specifically for quinoxaline-derived compounds, it has been reported that they may inhibit at least CyPA and CyPD (Guo et al., 2005; Li et al., 2006a). In this respect, it may be not unexpected that our CyPJ inhibitors may also inhibit other members of cyclophilins at some extent, and this issue will be the focus of future work.
In summary, we carried out a computer-aided virtual inhibitor screening of chemical databases based on the crystal structure of CyPJ/CsA complex, and identified 13 potential inhibitors of CyPJ. The quinoxaline-containing ZX-J-19, and its derivatives ZX-J-19j and ZX-J-19l showed potent antitumor activity. To the best of our knowledge, this study is the first to identify the inhibitors of CyPJ; these compounds are potential leads for rational design of novel CyPJ-based antitumor drugs.
Author Contributions
XZ and LY designed the project. XZ, CX, XW, HW, MX, and YL performed the experiments and data analysis. XZ, CX, and YL wrote the manuscript. All authors discussed the results and revised the manuscript.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (Grant Nos. 81172959 to XZ and 81372589 to YL), the Shandong Science and Technology Development Planning of China (Grant Nos. BS2009SW059 and 2007GG20002017, both to XZ), and the Shandong Medical and Health Science and Technology Development Planning of China (Grant No. 2011HZ104 to XZ).
Conflict of Interest Statement
Two patents related to this work have been granted in China: one for the compound 1,2-di(1H-pyrrol-2-yl)ethane-1,2-dione in 2011 (Patent No. ZL200910019140.2, inventors: XZ and CX) and the other for ZX-J-19j [N-(2,3-diphenylquinoxalin-6-yl)octanamide] in 2013 (Patent No. ZL201110178176.2, inventors: XZ and CX).
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all members in the laboratory of Prof. Long Yu who provided technical assistance with SPR assay and Dr. Jinzhi Tan at the State Key Laboratory of Drug Research at the Chinese Academy of Sciences (Shanghai, China) for assisting in virtual screening.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00126/full#supplementary-material
Footnotes
References
Amin, K. M., Ismail, M. M., Noaman, E., Soliman, D. H., and Ammar, Y. A. (2006). New quinoxaline 1,4-di-N-oxides. Part 1: Hypoxia-selective cytotoxins and anticancer agents derived from quinoxaline 1,4-di-N-oxides. Bioorg. Med. Chem. 14, 6917–6923. doi: 10.1016/j.bmc.2006.06.038
Behnam, S. M., Behnam, S. E., and Koo, J. Y. (2005). Review of cyclosporine immunosuppressive safety data in dermatology patients after two decades of use. J. Drugs Dermatol. 4, 189–194.
Butler, K. V., Ma, A., Yu, W., Li, F., Tempel, W., Babault, N., et al. (2016). Structure-based design of a covalent inhibitor of the set domain-containing protein 8 (SETD8) lysine methyltransferase. J. Med. Chem. 59, 9881–9889. doi: 10.1021/acs.jmedchem.6b01244
Campagna-Slater, V., Therrien, E., Weill, N., and Moitessier, N. (2014). Methods for docking small molecules to macromolecules: a user’s perspective. 2. applications. Curr. Pharm. Des. 20, 3360–3372. doi: 10.2174/13816128113199990606
Chen, J., Chen, S., Wang, J., Zhang, M., Gong, Z., Wei, Y., et al. (2015). Cyclophilin J is a novel peptidyl-prolyl isomerase and target for repressing the growth of hepatocellular carcinoma. PLoS One 10:e0127668. doi: 10.1371/journal.pone.0127668
Chen, Q., Bryant, V. C., Lopez, H., Kelly, D. L., Luo, X., and Natarajan, A. (2011). 2,3-Substituted quinoxalin-6-amine analogs as antiproliferatives: a structure-activity relationship study. Bioorg. Med. Chem. Lett. 21, 1929–1932. doi: 10.1016/j.bmcl.2011.02.055
Chen, S., Zhao, X., Tan, J., Lu, H., Qi, Z., Huang, Q., et al. (2007). Structure-based identification of small molecule compounds targeting cell cyclophilin A with anti-HIV-1 activity. Eur. J. Pharmacol. 565, 54–59. doi: 10.1016/j.ejphar.2007.03.023
Choi, J. Y., Fuerst, R., Knapinska, A. M., Taylor, A. B., Smith, L., Cao, X., et al. (2017). Structure-Based Design and Synthesis of Potent and Selective Matrix Metalloproteinase 13 Inhibitors. J. Med. Chem. 60, 5816–5825. doi: 10.1021/acs.jmedchem.7b00514
Colombo, D., and Ammirati, E. (2011). Cyclosporine in transplantation - a history of converging timelines. J. Biol. Regul. Homeost. Agents 25, 493–504.
Couch, F. J., Kuchenbaecker, K. B., Michailidou, K., Mendoza-Fandino, G. A., Nord, S., Lilyquist, J., et al. (2016). Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat. Commun. 7:11375. doi: 10.1038/ncomms11375
Dawar, F. U., Tu, J., Khattak, M. N., Mei, J., and Lin, L. (2017). Cyclophilin A: a key factor in virus replication and potential target for anti-viral therapy. Curr. Issues Mol. Biol. 21, 1–20.
Dunyak, B. M., and Gestwicki, J. E. (2016). Peptidyl-proline isomerases (PPIases): targets for natural products and natural product-inspired compounds. J. Med. Chem. 59, 9622–9644. doi: 10.1021/acs.jmedchem.6b00411
El Aissi, R., Liu, J., Besse, S., Canitrot, D., Chavignon, O., Chezal, J. M., et al. (2014). Synthesis and biological evaluation of new quinoxaline derivatives of ICF01012 as melanoma-targeting probes. ACS Med. Chem. Lett. 5, 468–473. doi: 10.1021/ml400468x
Flisiak, R., Horban, A., Gallay, P., Bobardt, M., Selvarajah, S., Wiercinska-Drapalo, A., et al. (2008). The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47, 817–826. doi: 10.1002/hep.22131
Fruman, D. A., Burakoff, S. J., and Bierer, B. E. (1994). Immunophilins in protein folding and immunosuppression. FASEB J. 8, 391–400. doi: 10.1096/fasebj.8.6.7513288
Galat, A., and Metcalfe, S. M. (1995). Peptidylproline cis/trans isomerases. Prog. Biophys. Mol. Biol. 63, 67–118. doi: 10.1016/0079-6107(94)00009-X
Gallagher, M. P., Kelly, P. J., Jardine, M., Perkovic, V., Cass, A., Craig, J. C., et al. (2010). Long-term cancer risk of immunosuppressive regimens after kidney transplantation. J. Am. Soc. Nephrol. 21, 852–858. doi: 10.1681/ASN.2009101043
Gong, Z., Mu, Y., Chen, J., Chu, H., Lian, P., Wang, C., et al. (2017). Expression and significance of cyclophilin J in primary gastric adenocarcinoma. Anticancer Res. 37, 4475–4481.
Gothel, S. F., and Marahiel, M. A. (1999). Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol. Life. Sci. 55, 423–436.
Gu, W., Wang, S., Jin, X., Zhang, Y., Hua, D., Miao, T., et al. (2017). Synthesis and evaluation of new quinoxaline derivatives of dehydroabietic acid as potential antitumor agents. Molecules 22:E1154. doi: 10.3390/molecules22071154
Guo, H. X., Wang, F., Yu, K. Q., Chen, J., Bai, D. L., Chen, K. X., et al. (2005). Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/ release. Acta Pharmacol. Sin. 26, 1201–1211. doi: 10.1111/j.1745-7254.2005.00189.x
Han, W., Soltani, K., Ming, M., and He, Y. Y. (2012). Deregulation of XPC and CypA by cyclosporin A: an immunosuppression-independent mechanism of skin carcinogenesis. Cancer Prev. Res. 5, 1155–1162. doi: 10.1158/1940-6207.CAPR-12-0185-T
Handschumacher, R. E., Harding, M. W., Rice, J., Drugge, R. J., and Speicher, D. W. (1984). Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226, 544–547. doi: 10.1126/science.6238408
Harikishore, A., and Yoon, H. S. (2015). Immunophilins: structures, mechanisms and ligands. Curr. Mol. Pharmacol. 9, 37–47. doi: 10.2174/1874467208666150519113427
Hopkins, S., and Gallay, P. A. (2015). The role of immunophilins in viral infection. Biochim. Biophys. Acta 1850, 2103–2110. doi: 10.1016/j.bbagen.2014.11.011
Houston, D. R., and Walkinshaw, M. D. (2013). Consensus docking: improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 53, 384–390. doi: 10.1021/ci300399w
Hu, H., Huang, C. Q., Liu, H. L., Han, Y., Yu, L., and Bi, R. C. (2005). Crystallization and preliminary X-ray crystallographic studies of human cyclophilin J. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 216–218. doi: 10.1107/S1744309105000643
Huang, L. L., Zhao, X. M., Huang, C. Q., Yu, L., and Xia, Z. X. (2005). Structure of recombinant human cyclophilin J, a novel member of the cyclophilin family. Acta Crystallogr. D Biol. Crystallogr. 61, 316–321. doi: 10.1107/S0907444904033189
Hui, X., Desrivot, J., Bories, C., Loiseau, P. M., Franck, X., Hocquemiller, R., et al. (2006). Synthesis and antiprotozoal activity of some new synthetic substituted quinoxalines. Bioorg. Med. Chem. Lett. 16, 815–820. doi: 10.1016/j.bmcl.2005.11.025
Kim, H., Kim, W. J., Jeon, S. T., Koh, E. M., Cha, H. S., Ahn, K. S., et al. (2005). Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin. Immunol. 116, 217–224. doi: 10.1016/j.clim.2005.05.004
Kofron, J. L., Kuzmic, P., Kishore, V., Colon-Bonilla, E., and Rich, D. H. (1991). Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30, 6127–6134. doi: 10.1021/bi00239a007
Kuntz, I. D. (1992). Structure-based strategies for drug design and discovery. Science 257, 1078–1082. doi: 10.1126/science.257.5073.1078
Lee, J. (2013). Cyclophilin A as a new therapeutic target for hepatitis C virus-induced hepatocellular carcinoma. Korean J. Physiol. Pharmacol. 17, 375–383. doi: 10.4196/kjpp.2013.17.5.375
Lee, J., and Kim, S. S. (2010). An overview of cyclophilins in human cancers. J. Int. Med. Res. 38, 1561–1574. doi: 10.1177/147323001003800501
Li, J., Chen, J., Zhang, L., Wang, F., Gui, C., Zhang, L., et al. (2006a). One novel quinoxaline derivative as a potent human cyclophilin A inhibitor shows highly inhibitory activity against mouse spleen cell proliferation. Bioorg. Med. Chem. 14, 5527–5534.
Li, J., Tan, J. Z., Chen, L. L., Zhang, J., Shen, X., Mei, C. L., et al. (2006b). Design, synthesis and antitumor evaluation of a new series of N-substituted-thiourea derivatives. Acta Pharmacol. Sin. 27, 1259–1271.
Lucke, C., and Weiwad, M. (2011). Insights into immunophilin structure and function. Curr. Med. Chem. 18, 5333–5354. doi: 10.2174/092986711798194324
Miller, E. M., Xia, Q., Cella, M. E., Nenninger, A. W., Mruzik, M. N., Brillos-Monia, K. A., et al. (2017). Voltammetric study of some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives with anti-tumor activities. Molecules 22:E1442. doi: 10.3390/molecules22091442
Muellenhoff, M. W., and Koo, J. Y. (2012). Cyclosporine and skin cancer: an international dermatologic perspective over 25 years of experience. A comprehensive review and pursuit to define safe use of cyclosporine in dermatology. J. Dermatolog. Treat. 23, 290–304. doi: 10.3109/09546634.2011.590792
Nigro, P., Pompilio, G., and Capogrossi, M. C. (2013). Cyclophilin A: a key player for human disease. Cell Death Dis. 4:e888. doi: 10.1038/cddis.2013.410
Nigro, P., Satoh, K., O’dell, M. R., Soe, N. N., Cui, Z., Mohan, A., et al. (2011). Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 208, 53–66. doi: 10.1084/jem.20101174
Norman, K. G., Canter, J. A., Shi, M., Milne, G. L., Morrow, J. D., and Sligh, J. E. (2010). Cyclosporine A suppresses keratinocyte cell death through MPTP inhibition in a model for skin cancer in organ transplant recipients. Mitochondrion 10, 94–101. doi: 10.1016/j.mito.2009.10.001
Pap, T. (2005). Cyclophilins in rheumatoid arthritis–stepping into an undiscovered country? Clin. Immunol. 116, 199–201. doi: 10.1016/j.clim.2005.06.013
Pereira, J. A., Pessoa, A. M., Cordeiro, M. N., Fernandes, R., Prudencio, C., Noronha, J. P., et al. (2015). Quinoxaline, its derivatives and applications: a state of the art review. Eur. J. Med. Chem. 97, 664–672. doi: 10.1016/j.ejmech.2014.06.058
Qi, Z. Y., Hui, G. Z., Li, Y., Zhou, Z. X., Gu, S. H., Ying, K., et al. (2005). cDNA microarray in isolation of novel differentially expressed genes related to human glioma and clone of a novel full-length gene. Chin. Med. J. 118, 799–805.
Quarato, G., D’aprile, A., Gavillet, B., Vuagniaux, G., Moradpour, D., Capitanio, N., et al. (2012). The cyclophilin inhibitor alisporivir prevents hepatitis C virus-mediated mitochondrial dysfunction. Hepatology 55, 1333–1343. doi: 10.1002/hep.25514
Satoh, K., Nigro, P., Matoba, T., O’dell, M. R., Cui, Z., Shi, X., et al. (2009). Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat. Med. 15, 649–656. doi: 10.1038/nm.1958
Sridevi, C., Balaji, K., and Naidu, A. (2011). Synthesis and pharmacological evaluation of some phenylpyrazolo indoquinoxaline derivatives. E-J. Chem. 8, 924–930. doi: 10.1155/2011/584817
Towers, G. J., Hatziioannou, T., Cowan, S., Goff, S. P., Luban, J., and Bieniasz, P. D. (2003). Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9, 1138–1143. doi: 10.1038/nm910
Walsh, C. T., Zydowsky, L. D., and Mckeon, F. D. (1992). Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J. Biol. Chem. 267, 13115–13118.
Wang, G., Shen, J., Sun, J., Jiang, Z., Fan, J., Wang, H., et al. (2017). Cyclophilin A maintains glioma-initiating cell stemness by regulating Wnt/beta-catenin signaling. Clin. Cancer Res. 23, 6640–6649. doi: 10.1158/1078-0432.CCR-17-0774
Weill, N., Therrien, E., Campagna-Slater, V., and Moitessier, N. (2014). Methods for docking small molecules to macromolecules: a user’s perspective. 1. The theory. Curr. Pharm. Des. 20, 3338–3359. doi: 10.2174/13816128113199990605
Weng, Q., Wang, D., Guo, P., Fang, L., Hu, Y., He, Q., et al. (2008). Q39, a novel synthetic Quinoxaline 1,4-Di-N-oxide compound with anti-cancer activity in hypoxia. Eur. J. Pharmacol. 581, 262–269. doi: 10.1016/j.ejphar.2007.12.006
Xue, C., Sowden, M., and Berk, B. C. (2017). Extracellular cyclophilin A, especially acetylated, causes pulmonary hypertension by stimulating endothelial apoptosis, redox stress, and inflammation. Arterioscler. Thromb. Vasc. Biol. 37, 1138–1146. doi: 10.1161/ATVBAHA.117.309212
Zarranz, B., Jaso, A., Aldana, I., and Monge, A. (2004). Synthesis and anticancer activity evaluation of new 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives. Bioorg. Med. Chem. 12, 3711–3721. doi: 10.1016/j.bmc.2004.04.013
Zhang, L., Qiu, B., Xiong, B., Li, X., Li, J., Wang, X., et al. (2007). Quinoxalinylurea derivatives as a novel class of JSP-1 inhibitors. Bioorg. Med. Chem. Lett. 17, 2118–2122. doi: 10.1016/j.bmcl.2007.01.094
Zhou, Z., Ying, K., Dai, J., Tang, R., Wang, W., Huang, Y., et al. (2001). Molecular cloning and characterization of a novel peptidylprolyl isomerase (cyclophilin)-like gene (PPIL3) from human fetal brain. Cytogenet. Cell Genet. 92, 231–236. doi: 10.1159/000056909
Keywords: PPIL3, cyclophilin J, quinoxaline derivative, hepatocellular carcinoma, cyclophilin J inhibitor, peptidylprolyl isomerase, CyPJ, PPIase
Citation: Zhao X, Xia C, Wang X, Wang H, Xin M, Yu L and Liang Y (2018) Cyclophilin J PPIase Inhibitors Derived from 2,3-Quinoxaline-6 Amine Exhibit Antitumor Activity. Front. Pharmacol. 9:126. doi: 10.3389/fphar.2018.00126
Received: 16 October 2017; Accepted: 05 February 2018;
Published: 21 February 2018.
Edited by:
Chiranjib Chakraborty, Galgotias University, IndiaReviewed by:
Joel D. A. Tyndall, University of Otago, New ZealandShiqiao Ye, University of Arkansas for Medical Sciences, United States
Copyright © 2018 Zhao, Xia, Wang, Wang, Xin, Yu and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulong Liang, eXVsb25nQGJjbS5lZHU= Long Yu, bG9uZ3l1QGZ1ZGFuLmVkdS5jbg==
† These authors have contributed equally to this work.