 Masatsugu Ohara1
Masatsugu Ohara1 Shunsuke Ohnishi1*Hidetaka Hosono1Koji Yamamoto1Qingjie Fu1Osamu Maehara2Goki Suda1Naoya Sakamoto1
Shunsuke Ohnishi1*Hidetaka Hosono1Koji Yamamoto1Qingjie Fu1Osamu Maehara2Goki Suda1Naoya Sakamoto1- 1Department of Gastroenterology and Hepatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
- 2Department of Pathophysiology and Therapeutics, Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan
Background: Liver fibrosis is a complex inflammatory and fibrogenic process, and the progression of fibrosis leads to cirrhosis. The only therapeutic approaches are the removal of injurious stimuli and liver transplantation. Therefore, the development of anti-fibrotic therapies is desired. Palmitoylethanolamide (PEA) is an endogenous fatty acid amide belonging to the N-acylethanolamines family and contained in foods such as egg yolks and peanuts. PEA has therapeutic anti-inflammatory, analgesic, and neuroprotective effects. However, the effects and roles of PEA in liver fibrosis remain unknown. Here we investigated the therapeutic effects of PEA in rats with liver fibrosis.
Methods: We conducted in vitro experiments to investigate the effects of PEA on the activation of hepatic stellate cells (HSCs, LX-2). Liver fibrosis was induced by an intraperitoneal injection of 1.5 mL/kg of 50% carbon tetrachloride twice a week for 4 weeks. Beginning at 3 weeks, PEA (20 mg/kg) was intraperitoneally injected thrice a week for 2 weeks. Then rats were sacrificed and we performed histological and quantitative reverse-transcription polymerase chain reaction analyses.
Results: The expression of α-smooth muscle actin (SMA) induced by transforming growth factor (TGF)-β1 in HSCs was significantly downregulated by PEA. PEA treatment inhibited the TGF-β1-induced phosphorylation of SMAD2 in a dose-dependent manner, and upregulated the expression of SMAD7. The reporter gene assay demonstrated that PEA downregulated the transcriptional activity of the SMAD complex upregulated by TGF-β1. Administration of PEA significantly reduced the fibrotic area, deposition of type I collagen, and activation of HSCs and Kupffer cells in rats with liver fibrosis.
Conclusion: Activation of HSCs was significantly decreased by PEA through suppression of the TGF-β1/SMAD signaling pathway. Administration of PEA produced significant improvement in a rat model of liver fibrosis, possibly by inhibiting the activation of HSCs and Kupffer cells. PEA may be a potential new treatment for liver fibrosis.
Introduction
Liver fibrosis is a complex inflammatory and fibrogenic process that results from chronic liver injury (Zhang et al., 2016). It is characterized by the excessive deposition of extracellular matrix proteins including collagen, and by activation of HSCs and Kupffer cells (Friedman, 2008; Tacke and Zimmermann, 2014; van Dijk et al., 2015). HSCs, once activated, are responsible for liver fibrosis (Xu et al., 2005). A significant increase in TGF-β expression is observed in the activated HSCs (Yoshida et al., 2014). TGF-β family members (TGF-β1, -β2, and -β3) are induced and activated in a variety of fibrotic diseases (Govinden and Bhoola, 2003; Xu et al., 2014). The progression of liver fibrosis leads to cirrhosis, a condition characterized by distortion of the normal liver architecture, the formation of septae/nodules, portal hypertension, and eventually tumor formation (Han et al., 2004; van Dijk et al., 2015). The only therapeutic approaches are removal of the injurious stimuli and liver transplantation, which is limited by organ shortages, high associated expenses, and the need for lifelong immunosuppressive medications (Campana and Iredale, 2017; Wang et al., 2017). Therefore, development of anti-fibrotic therapies is a major area of unmet clinical need.
Palmitoylethanolamide is an endogenous fatty acid amide belonging to the N-acylethanolamines (NAEs) family, and contained in foods such as egg yolks, peanuts, and soy seeds (Kilaru et al., 2007; Esposito and Cuzzocrea, 2013; Impellizzeri et al., 2015). PEA is synthesized “on-demand” during a variety of inflammatory disease states and produces marked protective properties for inflammatory responses and pruritus (Re et al., 2007; Wang et al., 2014). Furthermore, PEA has therapeutic analgesic and neuroprotective effects, acting at different molecular targets such as tumor necrosis factor (TNF)-α, vascular endothelial growth factor (VEGF), and intercellular adhesion molecules (ICAM)-1 (Hoareau et al., 2006; Esposito and Cuzzocrea, 2013; Wang et al., 2014; Paterniti et al., 2015). PEA reduces lung inflammation in mice with pulmonary fibrosis (Di Paola et al., 2016), and is able to modulate activation of one or more members of the PPAR family of nuclear receptors and/or a cannabinoid CB2-like receptor (Di Paola et al., 2016). In addition, PPARα is required for the anti-inflammatory actions of PEA (Lo Verme et al., 2005), and presence of PPARα mRNA in HSCs (Miyahara et al., 2000) and suppressive effect of PPARα agonist Wy-14,643 on liver fibrosis (Ip et al., 2004) have been reported. However, the effects and roles of PEA in liver fibrosis remain unknown. The aim of this study was to examine the effects of PEA on liver fibrosis in rats and to investigate its underlying mechanisms.
Materials and Methods
Reagents
Palmitoylethanolamide, MK886, a PPARα antagonist, and GW6471, a PPARα antagonist, were purchased from Cayman Chemical (Ann Arbor, MI, United States). PEA was dissolved in dimethyl sulfoxide (Wako Pure Chemical Industries, Osaka, Japan) for the in vitro experiments, and dissolved in a vehicle composed of Tween 80 (Kanto Chemical, Tokyo, Japan), dimethyl sulfoxide, and phosphate-buffered saline (PBS, Life Technologies, Carlsbad, CA, United States) (1:0.5:18.5 by volume) for the in vivo experiments.
Cell Culture
LX-2 cells, immortalized human HSCs, were purchased from Merck Millipore (Billerica, MA, United States). In all experiments, the cells were subjected to no more than 15 cell passages. The cells were cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA, United States) containing 2% fetal bovine serum (Moregate Biotech, Bulimba, QLD, Australia), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Wako Pure Chemical Industries), and maintained at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every other day. Human embryonic kidney cells (HEK293, provided by the RIKEN BioResource Center, Tsukuba, Japan) or their derivatives, which were stably transfected with the human Toll-like receptor (TLR) 4a, MD2, and CD14 genes (293/hTLR4A-MD2-CD14; InvivoGen, San Diego, CA, United States), were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 U/mL of penicillin, and 100 μg/mL of streptomycin. 293/hTLR4A-MD2-CD14 cells were activated by lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO, United States) treatment.
Western Blot Analysis
To investigate the phosphorylation of SMAD2 and expression of α-SMA, LX-2 cells were plated into six-well plates (2 × 105 cells/well, Corning, Corning, NY, United States) and cultured. The next day, the culture medium was changed to a medium containing PEA or dimethyl sulfoxide, and the cells were incubated for an additional 30 min or 1 h. Then cells were treated with 2.0 ng/mL of TGF-β1 (PeproTech, Rocky Hill, NJ, United States) for 30 min, 24 h, and 72 h, and washed with ice-cold PBS. Cell lysates were prepared using a radio-immunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl (pH8.0), 150 mM NaCl, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulfate (SDS), 1.0% (w/v) NP-40 substitute, and Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology, Beverly, MA, United States). Equal amounts of cellular protein extracts were diluted in a 4 × Laemmli sample buffer (Bio-Rad, Hercules, CA, United States). The samples were heated at 95°C for 5 min and then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE, Bio-Rad). The separate proteins were transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Merck Millipore), which were subsequently incubated in tris buffered saline with 0.05% Tween 20 (Wako Pure Chemical Industries) consisting of a 5% PhosphoBLOCKER blocking reagent (Cell Biolabs, San Diego, CA, United States) at room temperature for 60 min. The membranes were probed with primary antibodies for phospho-SMAD2 (1:2000; Cell Signaling Technology), SMAD2/3 (1:2000; Cell Signaling Technology), α-SMA (1:1000; Abcam, Cambridge, United Kingdom) and bound antibodies were detected with peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (1:10,000; Jackson ImmunoResearch, West Grove, PA, United States) or peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) (1:10,000; Jackson ImmunoResearch), and visualized and photographed using ECL Prime detection reagent (GE Healthcare, Chicago, IL, United States). The blots were analyzed using ImageQuant LAS-4000 (Fujifilm, Tokyo, Japan).
Plasmids
cDNAs of TGF-β receptor 1 (TGFβR-1) and SMAD2 were obtained by RT-PCR and the resulting fragments were cloned into pCI-neo-HA(c) and pCMV-3 × FLAG. Active mutants of TGFβR-1 (T204D) (Wieser et al., 1995) and SMAD2-2D (S465D/S467D) (Souchelnytskyi et al., 1997) were consecutively generated using a PCR-based method.
Transient Transfection and Reporter Gene Assay
LX-2 cells and HEK293 cells were plated into 24-well plates (Corning) containing 500 μL of culture medium (4.0 × 104 cells/well and 1.25 × 105 cells/well, respectively). After incubation for 24 h at 37°C, cells were transfected with 25 ng of luciferase plasmid DNA with 25 ng of Renilla pGL4.74(hRluc/TK) vector (Promega, Madison, WI, United States) as an internal control, and 500 ng of plasmid DNA containing three copies of a SMAD binding element that drove the transcription of the luciferase reporter gene [pGL4.48(luc2P/SBE/Hygro), Promega], using Lipofectamine® LTX (Life Technologies). After 24 h of incubation at 37°C, the cells were treated with 1.0 ng/mL of TGF-β1 for 3 h; a reporter gene assay was performed using the Dual Luciferase Reporter Assay System (Promega). Luminescence intensity was measured using a GloMax®-Multi Detection System (Promega) according to manufacturer’s instructions. Transcription activity was normalized according to the Renilla luciferase activity. These experiments were performed in triplicate.
Immunofluorescent Staining
LX-2 cells were plated into a eight-well coverglass chamber (Asahi Glass, Tokyo, Japan), and pretreated with PEA for 1 h. Then, the cells were incubated with 2.0 ng/mL of TGF-β1 for 24 h. After incubation, the cells were washed with PBS, and then fixed in 100% methanol (Wako Pure Chemical Industries) for 15 min at 4°C. The cells were blocked with 2% human serum albumin (The Chemo-Sero-Therapeutic Research Institute, Kumamoto, Japan) in PBS for 1 h at 4°C. A primary antibody against α-SMA (1:250; Abcam) was incubated for 2 h at 4°C, and then the cells were incubated with a fluorescein-labeled secondary antibody (1:2000; Cell Signaling Technology) for 1 h. Samples were imaged using a fluorescence microscopy (Olympus, Tokyo, Japan). Fluorescent intensities were analyzed using ImageJ (NIH, Bethesda, MD, United States). At least 10 cells were included in the analysis for each condition.
Animals
This study was carried out in accordance with the recommendations of the Animal Care Unit and Use Committees of Hokkaido University. The protocol was approved by the Animal Care Unit and Use Committees of Hokkaido University. Six-week-old male Sprague-Dawley rats were procured from Japan SLC (Hamamatsu, Japan), and three rats were housed per cage in a temperature-controlled room (24°C) on a 12-h/12-h light/dark cycle. All rats had ad libitum access to standard chow and water.
Induction of Liver Fibrosis and PEA Treatment
Liver fibrosis was induced by an intraperitoneal injection of 1.5 mL/kg of 50% carbon tetrachloride (CCl4, Wako Pure Chemical Industries) in olive oil twice a week for 4 weeks (Figure 1). The control group rats were injected with olive oil alone. PEA (20 mg/kg) was injected intraperitoneally thrice a week after 2 weeks of CCl4 treatment. The dose of PEA was based on a previously reported study (Richter et al., 2016).
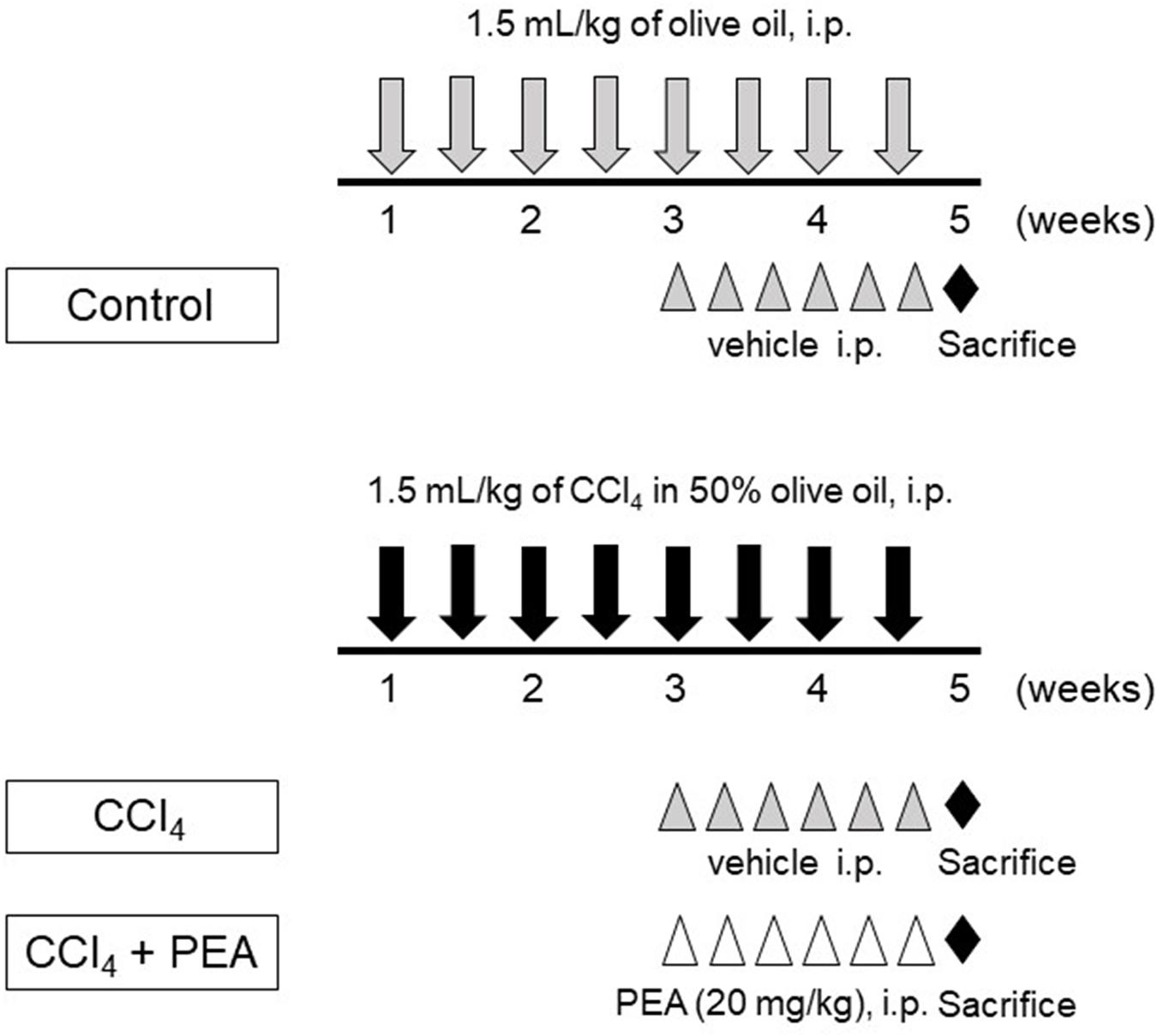
FIGURE 1. Experimental protocol for carbon tetrachlroide (CCl4)-induced liver fibrosis. Rats received intraperitoneal (i.p.) injection of 1.5 mL/kg of CCl4 in 50% olive oil twice a week for 4 weeks. Palmitoylethanolamide (PEA, 20 mg/kg) was i.p. injected thrice a week beginning at 3 weeks. All rats were sacrificed at 5 weeks.
Histological Examination
The rats were sacrificed after 4 weeks of CCl4 treatment. The left lobe of the liver was removed, fixed in 40 g/L of formaldehyde saline, embedded in paraffin, and cut into 5-μm sections. Tissue sections were stained with Hematoxylin and Eosin (H&E) and Masson’s trichrome. We photographed 10 random fields on a section from each rat, and calculated the blue-stained area from the entire liver cross-sectional area (%, ×100) with a digital image analyzer (WinROOF; Mitani, Co., Fukui, Japan).
Immunohistochemical Examination
The tissue sections were stained with anti-rat type I collagen antibody (1:100,000; LSL, Tokyo, Japan) for 60 min at room temperature. To assess HSC activation, the tissue sections were stained with anti-rat α-SMA antibody (1:800, Thermo Fisher Scientific) for 30 min at room temperature. To assess the infiltration of Kupffer cells, the tissue sections were stained with anti-rat CD68 monoclonal antibody (1:50; AbD Serotec, Kidlington, United Kingdom) for 40 min at room temperature. We photographed 10 random fields on a section from each rat, and measured the stained areas from the entire liver cross-sectional area using a digital image analyzer (WinROOF).
RNA Isolation and Quantitative RT-PCR
Total RNA of cultured cells or the rat liver was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany), and 1 μg of the total RNA was reverse-transcribed into cDNA using the PrimeScript RT reagent Kit (Takara Bio, Kusatsu, Japan). PCR was performed using a 25-μL reaction mixture that contained 1 μL of cDNA and 12.5 μL Platinum SYBR Green PCR Mix (Life Technologies). β-actin messenger RNA that was amplified from the same samples served as an internal control. After initial denaturation at 95°C for 2 min, we used a two-step cycle procedure (denaturation at 95°C for 15 s, annealing and extension at 60°C for 1 min) for 40 cycles in a 7700 Sequence Detector (Applied Biosystems, Foster City, CA, United States). Gene expression levels were determined using the comparative threshold cycle (ΔΔCt) method with β-actin used as an endogenous control. Data were analyzed with Sequences Detection Systems software (Applied Biosystems). Primer sequences are shown in Table 1.

TABLE 1. Primer sequences.
Statistical Analysis
Data are expressed as mean ± standard deviation (SD). Parameters among the groups were compared by one-way ANOVA, followed by Tukey’s test. The difference was considered significant at p < 0.05. All analyses were performed using the GraphPad Prism, version 7 (GraphPad software, San Diego, CA, United States).
Results
PEA Suppresses LPS-Induced Inflammatory Reaction and TGF-β1-Induced Activation of HSCs in Vitro
We first investigated the anti-inflammatory effect of PEA in 293/hTLR4A-MD2-CD14 cells. Treatment with LPS significantly upregulated the expression of TNF-α, and PEA significantly and dose-dependently decreased the expression of TNF-α (Figure 2A). We next examined whether PEA suppresses TGF-β1-induced activation of LX-2 cells. Treatment with TGF-β1 significantly upregulated the expression of α-SMA, and PEA significantly downregulated the expression of α-SMA in a dose-dependent manner (Figure 2B). Similarly, Western blotting and immunofluorescent staining demonstrated that PEA decreased the protein levels of α-SMA that were induced by TGF-β1 (Figures 2C,D, respectively). Furthermore, treatment with TGF-β1 significantly increased the expression of collagen1a1 (COL1A1) in LX-2 cells, and PEA significantly decreased the expression of COL1A1 (Figure 2E). However, the inhibitory effect of PEA on activation of LX-2 cells were not canceled by PPARα antagonist MK886 and GW6471 (Figures 2F,G, respectively). These results suggest that PEA suppresses LPS-induced inflammatory reaction and TGF-β1-induced fibrogenic reaction. In addition, the mechanisms of the inhibitory effect on HSC activation are independent of PPARα.
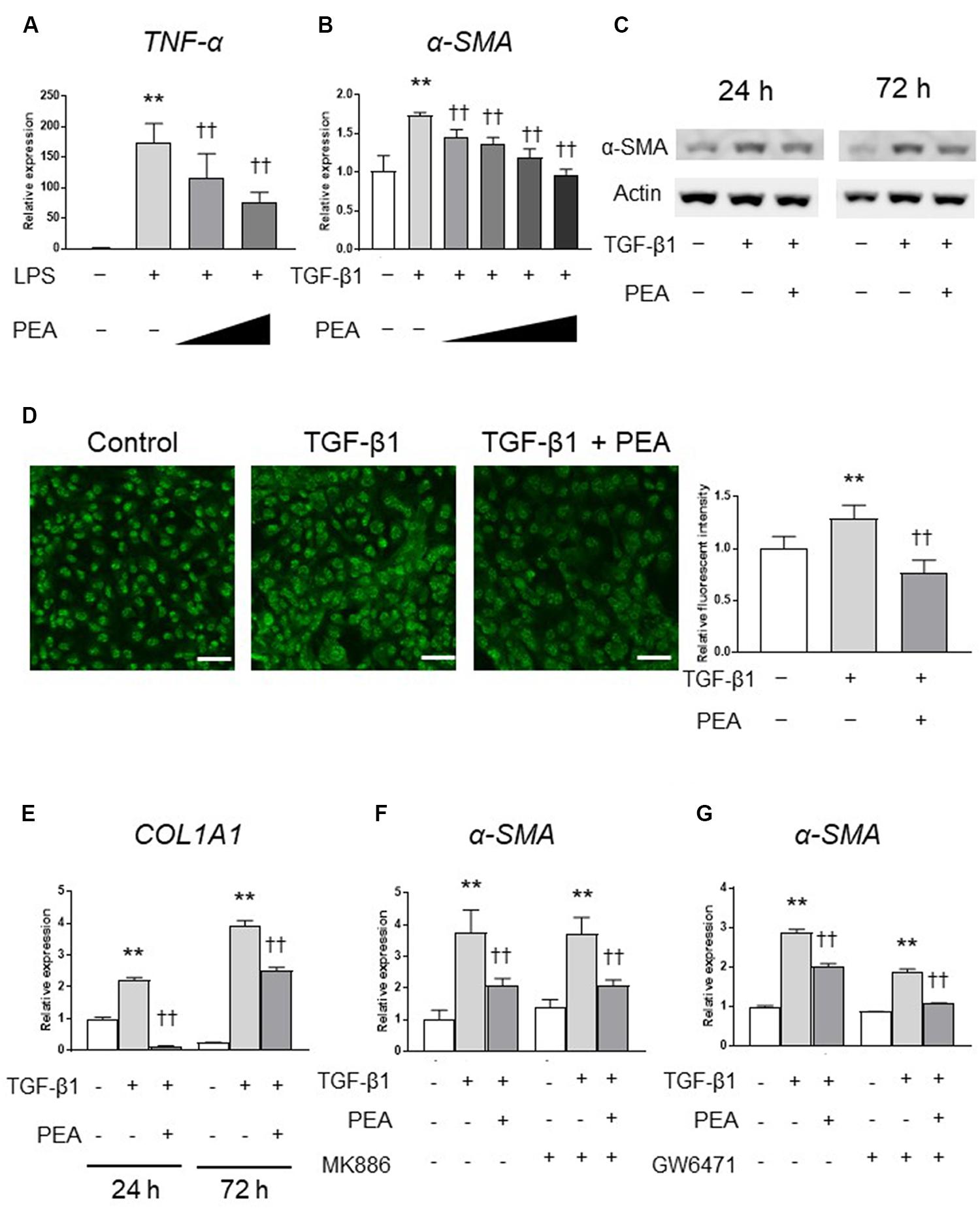
FIGURE 2. Effect of palmitoylethanolamide (PEA) on inflammatory reactions and fibrogenic responses in vitro. (A) PEA (0.1, 1.0 μM) was added to cultured 293/hTLR4A-MD2-CD14 cells with 2.5 ng/mL lipopolysaccharide (LPS). Total RNA was isolated 3 h after LPS administration, and the expression of tumor necrosis factor (TNF)-α was investigated by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). (B) PEA (1.0, 5.0, 10.0, and 20.0 μM) was added to LX-2 cells with 2.0 ng/mL transforming growth factor (TGF)-β1. Total RNA was isolated 24 h after TGF-β1 administration, and the expression of α-smooth muscle actin (SMA) was investigated by qRT-PCR. (C) PEA (10 μM) was added to LX-2 cells with 2.0 ng/mL TGF-β1. Total protein was isolated 24 or 72 h after TGF-β1 administration and the expression of α-SMA was investigated by Western blotting. (D) Immunofluorescent staining with α-SMA (green) in LX-2 cells 24 h after TGF-β1 administration. Scale bars: 20 μm. (E) PEA (10 μM) was added to LX-2 cells with 2.0 ng/mL TGF-β1. Total RNA was isolated 24 and 72 h after TGF-β1 administration and the expression of collagen1a1 (COL1A1) was investigated by qRT-PCR. (F) LX-2 cells were treated with PEA (10 μM) followed by TGF-β1 for 24 h with or without MK886 (10 μM) treatment. (G) LX-2 cells were treated with PEA (10 μM) followed by TGF-β1 for 24 h with or without GW6471 (2 μM) treatment. The values are the mean ± standard deviation (n = 3). ∗∗p < 0.01 versus the Control. ††p < 0.01 versus TGF-β1.
PEA Suppresses Phosphorylation of SMAD2 and Upregulates SMAD7
We next investigated whether PEA affects the TGF-β/SMAD pathway. Western blotting demonstrated that TGF-β1 treatment induced the phosphorylation of SMAD2, and PEA treatment inhibited the TGF-β1-induced phosphorylation of SMAD2 in a dose-dependent manner (Figure 3A). In addition, treatment with TGF-β1 significantly increased the expression of SMAD7, an inhibitor of the SMAD pathway, and PEA treatment further increased the expression of SMAD7 (Figure 3B). The reporter gene assay demonstrated that TGF-β1 markedly upregulated the transcription activity of the SMAD complex, and it was significantly downregulated by PEA (Figure 3C). Overexpression of TGFβR-1 (T204D), a constitutively active form of TGFβR-1, significantly upregulated the transcription activity of the SMAD complex, but PEA did not suppress luciferase activity (Figure 3D). Furthermore, overexpression of SMAD2-2D (S465D/S467D), a constitutively active form of SMAD2, also significantly elevated the transcription activity of the SMAD complex, but PEA did not suppress the luciferase activity (Figure 3E). These results suggest that PEA suppresses an early step of TGF-β/SMAD pathway in LX-2 cells.
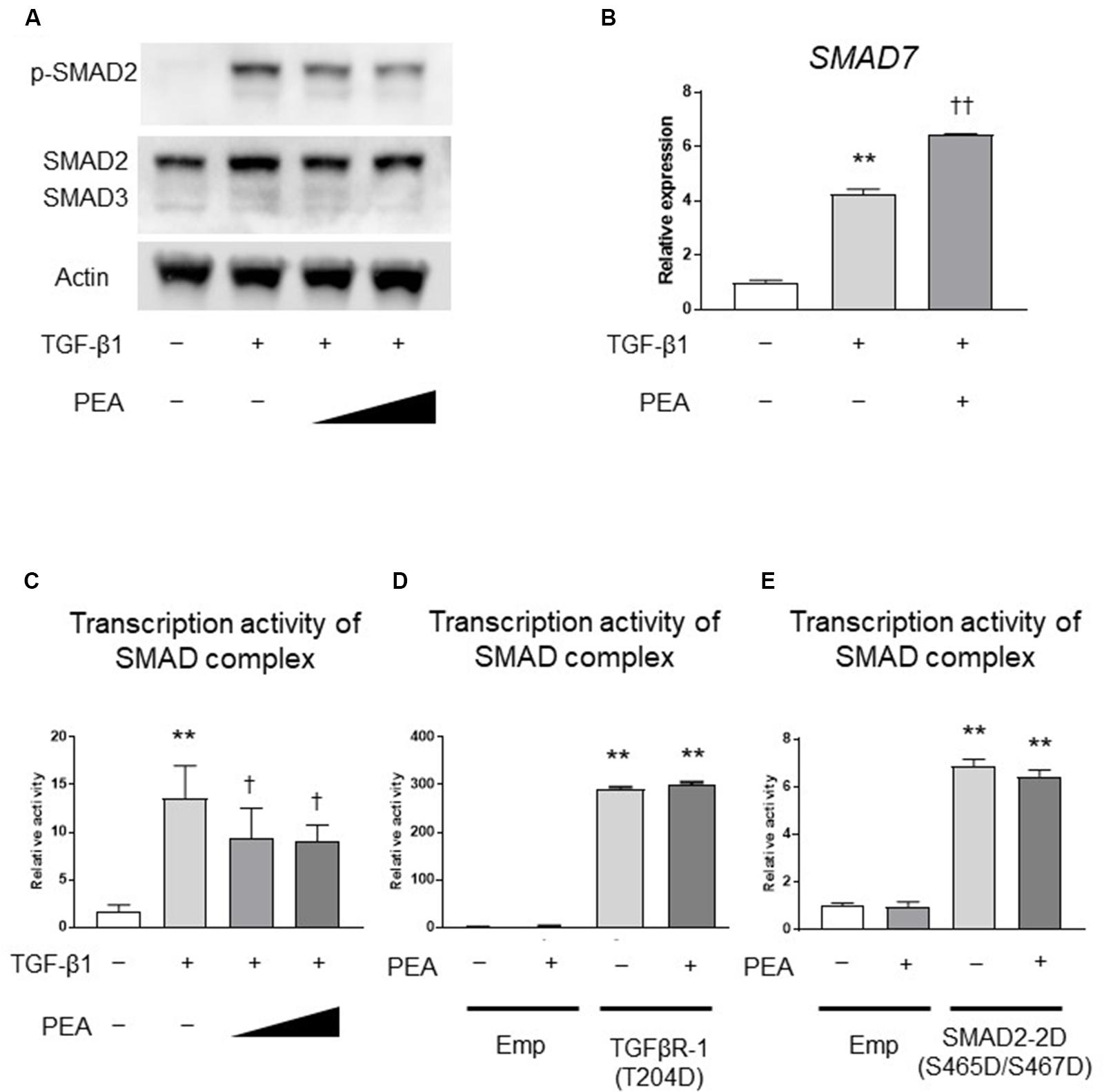
FIGURE 3. Effect of PEA on TGF-β/SMAD signaling pathways. (A) LX-2 cells were treated with TGF-β1 (2.0 ng/mL) for 30 min after pretreatment with PEA (10 and 20 μM) for 30 min, and the expression of phosphorylated SMAD2 (p-SMAD2) was investigated by Western blotting. (B) Effect of PEA (10 μM) on mRNA expression of SMAD7 in LX-2 cells treated with TGF-β1 (2.0 ng/mL) for 30 min. (C) Effect of PEA (10 and 20 μM) on transcription activity of SMAD complex in LX-2 cells treated with TGF-β1 (1.0 ng/mL) for 3 h. (D) Effect of PEA (20 μM) on transcription activity of SMAD complex in LX-2 cells with overexpression of TGFβR-1 (T204D). (E) Effect of PEA (20 μM) on transcription activity of SMAD complex in LX-2 cells with overexpression of SMAD2-2D (S465D/S467D). The values are the mean ± standard deviation (n = 3). ∗∗p < 0.01 versus the Control. †p < 0.05, ††p < 0.01 versus TGF-β1.
PEA Suppresses Liver Fibrosis in Rats
We investigated the effects of PEA on liver fibrosis in rats. In H&E staining, the thick fibrotic septa and pseudolobular formation were more extensive in the CCl4 group compared to the PEA group (Figure 4A). Masson’s trichrome staining demonstrated that fiber accumulation induced by CCl4 was significantly attenuated by administration of PEA for 2 weeks (Figure 4B). Consistent with this finding, the upregulated expression of type I collagen by CCl4 treatment was also attenuated by PEA administration (Figure 4C). The expression of α-SMA, a marker for HSC activation, was significantly increased in the CCl4 group; however, PEA administration significantly suppressed this increase (Figure 4D). The expression of CD68, a marker for Kupffer cells, was significantly increased in the CCl4 group; however, PEA administration decreased the infiltration of CD68-positive Kupffer cells (Figure 4E).
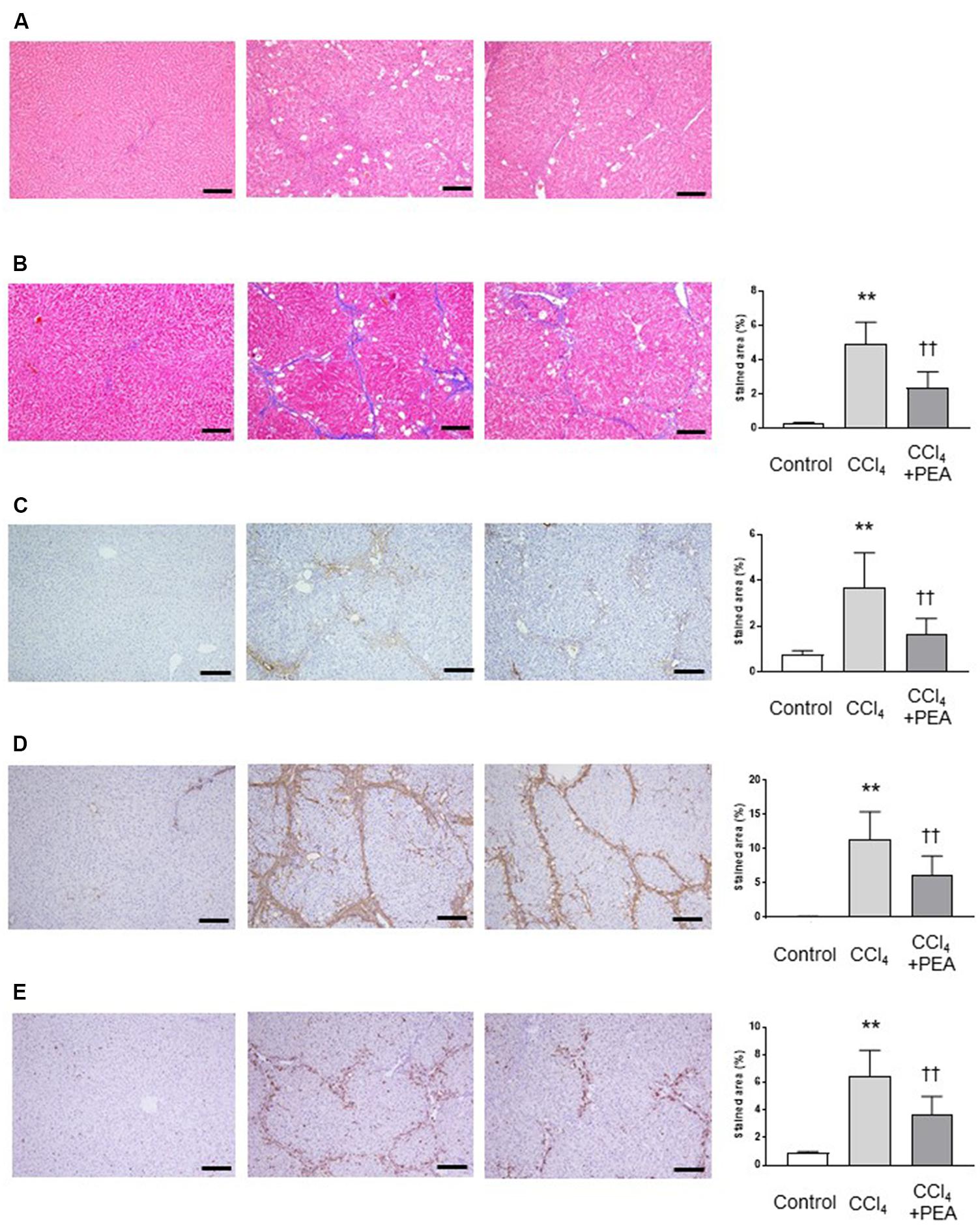
FIGURE 4. Effect of PEA on liver fibrosis in rats. (A) Hematoxylin and eosin staining. (B) Masson’s trichrome staining. Fibrotic area was stained blue and measured from the entire liver cross-sectional area. (C) Expression of type I collagen. (D) α-SMA expression (E) CD68 expression. The stained areas were measured from the entire liver cross-sectional area. Scale bars: 200 μm. The values are the mean ± standard deviation of (n = 6 in Control group, n = 14 in CCl4 group, and n = 14 in CCl4 + PEA group). ∗∗p < 0.01 versus Control group. ††p < 0.01 versus CCl4 group.
Effects of PEA Administration on Gene Expressions in the Liver
We next examined the expression of fibrosis-related genes in the liver. CCl4 treatment significantly increased the expression of α-Sma, collagen1a1 (Col1a1), Tgf-β, and tissue inhibitor of metalloproteinases (Timp)-1 (Figures 5A–C,E, respectively), and the expression of α-Sma, Col1a1 and Tgf-β were significantly decreased by administration of PEA (Figures 5A–C). The expression of Cd68 was significantly increased by CCl4; however, PEA significantly decreased the expression of Cd68 (Figure 5H). The expressions of monocyte chemoattractant protein (Mcp)-1 and Lbp tended to decrease following administration of PEA (Figures 5I,J, respectively). The expressions of matrix metalloproteinase (Mmp)-9, Tnf-α and Pparα were not affected by CCl4 treatment and PEA administration (Figures 5D,F,G, respectively).
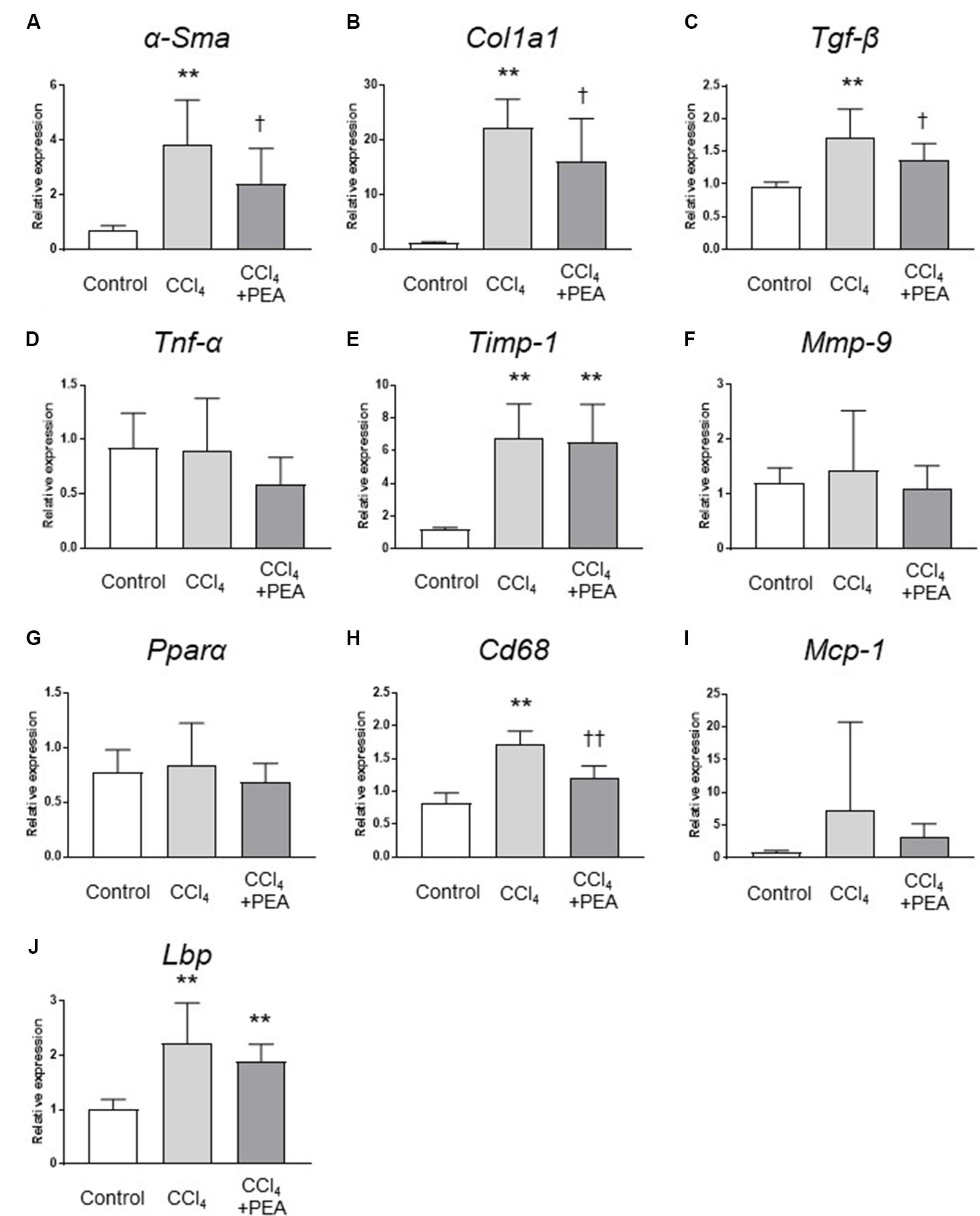
FIGURE 5. Gene expression analyses of the liver. qRT-PCR analyses of (A) α-smooth muscle actin (α-Sma), (B) collagen1a1 (Col1a1), (C) Tgf-β, (D) Tnf-α, (E) tissue inhibitor of metalloproteinases (Timp)-1, (F) matrix metalloproteinase (Mmp)-9, (G) peroxisome proliferator-activated receptor (Ppar)α, (H) Cd68, (I) monocyte chemoattractant protein (Mcp)-1, (J) lipopolysaccharide-binding protein (Lbp). The values are the mean ± standard deviation (n = 6 in Control group, n = 14 in CCl4 group, and n = 14 in CCl4 + PEA group). ∗∗p < 0.01 versus Control group. †p < 0.05 versus CCl4 group. ††p < 0.01 versus CCl4 group.
Discussion
In this study, we investigated the anti-fibrotic effects of PEA in vitro and in vivo. We found that (i) PEA suppressed LPS-induced inflammatory reaction in vitro, (ii) PEA suppressed TGF-β1-induced activation of cultured HSCs, and (iii) PEA administration ameliorated liver fibrosis in rats.
The therapeutic effects of PEA were previously reported in small animals with intestinal radiation injuries (Wang et al., 2014), uveitis (Impellizzeri et al., 2015), and cortical spreading depression (Richter et al., 2016). Proposed targets for PEA actions included a number of receptors, namely, cannabinoid receptors (Farquhar-Smith et al., 2002), transient receptor potential vanilloid type-1 (TRPV1) ion channels (Ambrosino et al., 2013), the orphan G protein-coupled receptor 55 (GPR55) (Pertwee, 2007), and PPARα (Lo Verme et al., 2005). PPARα is one of the main pharmacological targets of PEA action (LoVerme et al., 2005). On the other hand, PPARα is prominently expressed in hepatocytes (Ying-Ying and Lin, 2010), and PPARα ligands exert antifibrotic effects in rats with thioacetamide-induced liver cirrhosis (Toyama et al., 2004). Furthermore, PPARα agonists contrast inflammation and fibrosis in experimental models of steatohepatitis (Ip et al., 2004). However, in the present study, PEA-induced inactivation of LX-2 cells was not canceled by PPARα antagonists, suggesting that PEA attenuates activation of HSCs, independent of PPARα. Furthermore, Paterniti et al. (2015) demonstrated that PEA attenuates the degree of inflammation while preserving the blood–retinal barrier in rats with experimental diabetic retinopathy. This study revealed that PEA treatment reduces VEGF levels of the retina tissues in diabetic rats, and suggested that PEA may directly inhibit VEGF or may affect VEGF expression through TNF-α via its effects on VEGFR-2.
The molecular mechanisms of HSC activation were recently investigated (Huang et al., 2017; Senoo et al., 2017). TGF-β1 is known to be the most potent pro-fibrogenic cytokine in HSCs’ activation (Liu et al., 2006). We demonstrated that PEA inhibited the TGFβ-1-induced expression of fibrogenic genes, such as α-SMA and COL1A1 in LX-2 cells. TGF-β regulates numerous cell signaling pathways (Doerks et al., 2002), and the TGF-β/SMAD signaling pathway is one of the key fibrogenic and inflammatory pathways in the liver (Gressner and Weiskirchen, 2006). It is well-known that TGF-β activates downstream mediators SMAD2 and SMAD3, and the TGF-β/SMAD pathway is negatively regulated by SMAD7 (Xu et al., 2016). Our in vitro study demonstrated that PEA inhibited phosphorylation of SMAD2 and upregulated SMAD7 in LX-2 cells. In addition, PEA suppressed transcription activity of the SMAD complex, and expression of their downstream target genes and proteins. TGF-β1 binds TGFβR-2 with high affinity, which then recruits the lower-affinity TGFβR-1 (Hata and Chen, 2016). This induces the assembly of a complex that includes these receptors (Macias et al., 2015). Within the complex, TGFβR-2 subunits phosphorylate TGFβR-1 (Weiss and Attisano, 2013). Activated TGFβR-1 kinases recruit and phosphorylate SMAD proteins for signal transduction. Our in vitro study demonstrated that PEA did not suppress the transcription activity of the SMAD complex in HEK293 cells with overexpression of TGFβR-1 (T204D). Therefore, PEA may have suppressed the early steps of the TGF-β1/SMAD signaling pathway by interacting with TGFβR, or at the cell membrane.
In the present study, CCl4 was used to induce liver fibrosis. Liver fibrosis can be induced by one of the following approaches: (1) Chemical compounds and toxins. These agents cause direct injury to hepatocytes and trigger secondary inflammatory reaction in the liver, which in turn activate HSCs and result in fibrosis. Commonly used chemical agents include CCl4, thioacetamide, dimethyl nitrosamine, dioxin, sodium arsenate, and ethanol; (2) Special diet, such as choline-deficient, L-amino acid-defined, methionine-deficient diet, and high-fat diet; (3) Physical methods. Bile duct ligation creates obstruction of the extrahepatic bile duct; (4) Fibrosis induced by immune reaction; (5) Genetic modification (Zhou et al., 2014). Among above methods, bile duct ligation and CCl4 are well-validated models for fibrosis progression and resolution (Hernandez-Gea and Friedman, 2011). Therefore, we chose CCl4 to induce liver fibrosis in rats. This chemical changes the membrane permeability in liver mitochondria and plasm in various animals, and forms highly toxic free radicals, probably mediated by cytochrome p450 2E1 (Khan et al., 2015). Its repeated and prolonged treatment (≥4 weeks) causes severe liver disease, such as fibrosis and cirrhosis (Campana and Iredale, 2017). In liver fibrosis, the accumulation of extracellular matrix is a hallmark feature, and the activation of HSCs is a precursor of this phenomenon. In the normal liver, HSCs are quiescent and not fibrogenic (Senoo et al., 2017); however, the cells are activated by inflammatory processes of liver injury, such as the production of toxic cytokines and the recruitment of inflammatory cells (Bataller and Brenner, 2005). In the present study, we measured liver weight/body weight, and it was significantly increased by CCl4, and PEA treatment tended decrease the liver weight/body weight. The reason for this may be that it was the early stage of fibrosis with administration of CCl4 for 4 weeks (data are shown in Supplementary Materials). However, PEA administration suppressed CCl4-induced collagen deposition and activation of HSCs. In addition, PEA administration inhibited the expression of TGF-β in the liver. During liver injury, activated Kupffer cells secrete a large number of proinflammatory and fibrogenic mediators, which can drive HSC activation (Li et al., 2017) and are main targets of LPS, through TLR4 (Hernandez-Gea and Friedman, 2011). In the present study, PEA suppressed activation of 293/hTLR4A-MD2-CD14 cells, and inhibited the infiltration of Kupffer cells in rats with CCl4-induced liver injury. It has been reported that PEA has anti-inflammatory effect (Re et al., 2007), and PEA can attenuate LPS-induced inflammatory responses in the murine macrophage cell line RAW264.7 (Ross et al., 2000). Our in vitro experiment demonstrated that PEA has the anti-inflammatory effect in 293/hTLR4A-MD2-CD14 through LPS/TLR4 signal. Furthermore, macrophages are key-immune cells in the inflammatory process of liver fibrosis (Li et al., 2017), and responsible for liver cirrhosis induced by CCl4 (Muriel and Escobar, 2003). For other representative immune cells, it has been reported that neutrophils did not infiltrate in CCl4-induced liver fibrosis model (Campana and Iredale, 2017). Therefore, we evaluated the expression of CD68 in our in vivo experiment, and PEA would attenuate liver fibrosis by inhibiting activation of HSCs as well as Kupffer cells.
Conclusion
PEA administration ameliorated fibrogenesis in a rat model of CCl4-induced liver fibrosis, possibly by attenuating the activation of HSCs and Kupffer cells. PEA administration may be a new therapeutic strategy for treating liver fibrosis, and should be investigated in other liver fibrosis models as well as human pathogenesis.
Data Availability
The raw data supporting the conclusion of this manuscript will be made available by the authors, without undue reservation.
Author Contributions
MO contributed to all experiments, data analysis, and manuscript writing. SO and NS contributed to conception and design, and final approval of the manuscript. HH, KY, QF, OM, and GS contributed to assembly of data and data analysis. All authors have read and approved the manuscript.
Funding
This study was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS, 16H05282).
Conflict of Interest Statement
GS has received honoraria from Bristol-Myers Squibb, MSD KK, and AbbVie. Naoya Sakamoto has received honoraria and research funding from Gilead Sciences, Bristol-Myers Squibb, MSD KK, Otsuka Pharm, Abbvie, Shionogi, and Takeda.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Megumi Kimura, Tomoe Shimazaki, and Akiko Hirano for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00709/full#supplementary-material
Abbreviations
α-SMA, α-smooth muscle actin; CCl4, carbon tetrachloride; Col1a1, collagen1a1; HSC, hepatic stellate cell; LBP, lipopolysaccharide-binding protein; MMP, matrix metalloproteinase; PBS, phosphate-buffered saline; PEA, palmitoylethanolamide; PPAR, peroxisome proliferator-activated receptor; RT-PCR, reverse transcription-polymerase chain reaction; TGF-β, transforming growth factor-β; TGFβR, transforming growth factor-β receptor; TIMPs, tissue inhibitor of metalloproteinases; TNF-α, tumor necrosis factor-α.
References
Ambrosino, P., Soldovieri, M. V., Russo, C., and Taglialatela, M. (2013). Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br. J. Pharmacol. 168, 1430–1444. doi: 10.1111/bph.12029
Bataller, R., and Brenner, D. A. (2005). Liver fibrosis. J. Clin. Investig. 115, 209–218. doi: 10.1172/jci24282
Campana, L., and Iredale, J. P. (2017). Regression of liver fibrosis. Semin. Liver Dis. 37, 1–10. doi: 10.1055/s-0036-1597816
Di Paola, R., Impellizzeri, D., Fusco, R., Cordaro, M., Siracusa, R., Crupi, R., et al. (2016). Ultramicronized palmitoylethanolamide (PEA-um) in the treatment of idiopathic pulmonary fibrosis. Pharmacol. Res. 111, 405–412. doi: 10.1016/j.phrs.2016.07.010
Doerks, T., Copley, R. R., Schultz, J., Ponting, C. P., and Bork, P. (2002). Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 12, 47–56. doi: 10.1101/gr.203201
Esposito, E., and Cuzzocrea, S. (2013). Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol. Disord. Drug Targets 12, 55–61. doi: 10.2174/1871527311312010010
Farquhar-Smith, W. P., Jaggar, S. I., and Rice, A. S. (2002). Attenuation of nerve growth factor-induced visceral hyperalgesia via cannabinoid CB1 and CB2-like receptors. Pain 97, 11–21. doi: 10.1016/S0304-3959(01)00419-5
Friedman, S. L. (2008). Hepatic fibrosis – overview. Toxicology 254, 120–129. doi: 10.1016/j.tox.2008.06.013
Govinden, R., and Bhoola, K. D. (2003). Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacol. Ther. 98, 257–265. doi: 10.1016/s0163-7258(03)00035-4
Gressner, A. M., and Weiskirchen, R. (2006). Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell Mol. Med. 10, 76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x
Han, Y. P., Zhou, L., Wang, J., Xiong, S., Garner, W. L., French, S. W., et al. (2004). Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J. Biol. Chem. 279, 4820–4828. doi: 10.1074/jbc.M310999200
Hata, A., and Chen, Y. G. (2016). TGF-β signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 8:a022061. doi: 10.1101/cshperspect.a022061
Hernandez-Gea, V., and Friedman, S. L. (2011). Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6, 425–456. doi: 10.1146/annurev-pathol-011110-130246
Hoareau, L., Ravanan, P., Gonthier, M. P., Delarue, P., Goncalves, J., Cesari, M., et al. (2006). Effect of PEA on LPS inflammatory action in human adipocytes. Cytokine 34, 291–296. doi: 10.1016/j.cyto.2006.06.005
Huang, Y., Deng, X., and Liang, J. (2017). Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp. Cell Res. 352, 420–426. doi: 10.1016/j.yexcr.2017.02.038
Impellizzeri, D., Ahmad, A., Bruschetta, G., Di Paola, R., Crupi, R., Paterniti, I., et al. (2015). The anti-inflammatory effects of palmitoylethanolamide (PEA) on endotoxin-induced uveitis in rats. Eur. J. Pharmacol. 761, 28–35. doi: 10.1016/j.ejphar.2015.04.025
Ip, E., Farrell, G., Hall, P., Robertson, G., and Leclercq, I. (2004). Administration of the potent PPARα agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 39, 1286–1296. doi: 10.1002/hep.20170
Khan, R. A., Khan, M. R., Sahreen, S., Ahmed, M., and Shah, N. A. (2015). Carbon tetrachloride-induced lipid peroxidation and hyperglycemia in rat: a novel study. Toxicol. Ind. Health 31, 546–553. doi: 10.1177/0748233713475503
Kilaru, A., Blancaflor, E. B., Venables, B. J., Tripathy, S., Mysore, K. S., and Chapman, K. D. (2007). The N-acylethanolamine-mediated regulatory pathway in plants. Chem. Biodivers. 4, 1933–1955. doi: 10.1002/cbdv.200790161
Li, P., He, K., Li, J., Liu, Z., and Gong, J. (2017). The role of Kupffer cells in hepatic diseases. Mol. Immunol. 85, 222–229. doi: 10.1016/j.molimm.2017.02.018
Liu, X., Hu, H., and Yin, J. Q. (2006). Therapeutic strategies against TGF-β signaling pathway in hepatic fibrosis. Liver Int. 26, 8–22. doi: 10.1111/j.1478-3231.2005.01192.x
Lo Verme, J., Fu, J., Astarita, G., La Rana, G., Russo, R., Calignano, A., et al. (2005). The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 67, 15–19. doi: 10.1124/mol.104.006353
LoVerme, J., La Rana, G., Russo, R., Calignano, A., and Piomelli, D. (2005). The search for the palmitoylethanolamide receptor. Life Sci. 77, 1685–1698. doi: 10.1016/j.lfs.2005.05.012
Macias, M. J., Martin-Malpartida, P., and Massague, J. (2015). Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 40, 296–308. doi: 10.1016/j.tibs.2015.03.012
Miyahara, T., Schrum, L., Rippe, R., Xiong, S., Yee, HF Jr, Motomura, K., et al. (2000). Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 275, 35715–35722. doi: 10.1074/jbc.M006577200
Muriel, P., and Escobar, Y. (2003). Kupffer cells are responsible for liver cirrhosis induced by carbon tetrachloride. J. Appl. Toxicol. 23, 103–108. doi: 10.1002/jat.892
Paterniti, I., Di Paola, R., Campolo, M., Siracusa, R., Cordaro, M., Bruschetta, G., et al. (2015). Palmitoylethanolamide treatment reduces retinal inflammation in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 769, 313–323. doi: 10.1016/j.ejphar.2015.11.035
Pertwee, R. G. (2007). GPR55: a new member of the cannabinoid receptor clan? Br. J. Pharmacol. 152, 984–986. doi: 10.1038/sj.bjp.0707464
Re, G., Barbero, R., Miolo, A., and Di Marzo, V. (2007). Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: potential use in companion animals. Vet. J. 173, 21–30. doi: 10.1016/j.tvjl.2005.10.003
Richter, F., Koulen, P., and Kaja, S. (2016). N-palmitoylethanolamine prevents the run-down of amplitudes in cortical spreading depression possibly implicating proinflammatory cytokine release. Sci. Rep. 6:23481. doi: 10.1038/srep23481
Ross, R. A., Brockie, H. C., and Pertwee, R. G. (2000). Inhibition of nitric oxide production in RAW264.7 macrophages by cannabinoids and palmitoylethanolamide. Eur. J. Pharmacol. 401, 121–130. doi: 10.1016/S0014-2999(00)00437-4
Senoo, H., Mezaki, Y., and Fujiwara, M. (2017). The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 92, 387–455. doi: 10.1007/s12565-017-0395-9
Souchelnytskyi, S., Tamaki, K., Engstrom, U., Wernstedt, C., ten Dijke, P., and Heldin, C. H. (1997). Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 272, 28107–28115. doi: 10.1074/jbc.272.44.28107
Tacke, F., and Zimmermann, H. W. (2014). Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60, 1090–1096. doi: 10.1016/j.jhep.2013.12.025
Toyama, T., Nakamura, H., Harano, Y., Yamauchi, N., Morita, A., Kirishima, T., et al. (2004). PPARα ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 324, 697–704. doi: 10.1016/j.bbrc.2004.09.110
van Dijk, F., Olinga, P., Poelstra, K., and Beljaars, L. (2015). Targeted therapies in liver fibrosis: combining the best parts of platelet-derived growth factor BB and interferon gamma. Front. Med. 2:72. doi: 10.3389/fmed.2015.00072
Wang, J., Cen, P., Chen, J., Fan, L., Li, J., Cao, H., et al. (2017). Role of mesenchymal stem cells, their derived factors, and extracellular vesicles in liver failure. Stem Cell Res. Ther. 8:137. doi: 10.1186/s13287-017-0576-4
Wang, J., Zheng, J., Kulkarni, A., Wang, W., Garg, S., Prather, P. L., et al. (2014). Palmitoylethanolamide regulates development of intestinal radiation injury in a mast cell-dependent manner. Dig. Dis. Sci. 59, 2693–2703. doi: 10.1007/s10620-014-3212-5
Weiss, A., and Attisano, L. (2013). The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2, 47–63. doi: 10.1002/wdev.86
Wieser, R., Wrana, J. L., and Massague, J. (1995). GS domain mutations that constitutively activate T βR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 14, 2199–2208.
Xu, F., Liu, C., Zhou, D., and Zhang, L. (2016). TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem. Cytochem. 64, 157–167. doi: 10.1369/0022155415627681
Xu, J., Liu, X., Koyama, Y., Wang, P., Lan, T., Kim, I. G., et al. (2014). The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 5:167. doi: 10.3389/fphar.2014.00167
Xu, L., Hui, A. Y., Albanis, E., Arthur, M. J., O’Byrne, S. M., Blaner, W. S., et al. (2005). Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54, 142–151. doi: 10.1136/gut.2004.042127
Ying-Ying, Y., and Lin, H. C. (2010). Endocannabinoids and non-endocannabinoid fatty acid amides in cirrhosis. Liver Int. 30, 780–781. doi: 10.1111/j.1478-3231.2010.02244.x
Yoshida, K., Murata, M., Yamaguchi, T., and Matsuzaki, K. (2014). TGF-β/Smad signaling during hepatic fibro-carcinogenesis (review). Int. J. Oncol. 45, 1363–1371. doi: 10.3892/ijo.2014.2552
Zhang, C. Y., Yuan, W. G., He, P., Lei, J. H., and Wang, C. X. (2016). Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 22, 10512–10522. doi: 10.3748/wjg.v22.i48.10512
Keywords: palmitoylethanolamide, carbon tetrachloride, liver fibrosis, hepatic stellate cells (HSCs), transforming growth factor-β, smad
Citation: Ohara M, Ohnishi S, Hosono H, Yamamoto K, Fu Q, Maehara O, Suda G and Sakamoto N (2018) Palmitoylethanolamide Ameliorates Carbon Tetrachloride-Induced Liver Fibrosis in Rats. Front. Pharmacol. 9:709. doi: 10.3389/fphar.2018.00709
Received: 31 March 2018; Accepted: 12 June 2018;
Published: 13 July 2018.
Edited by:
Ralf Weiskirchen, RWTH Aachen Universität, GermanyReviewed by:
Ana Cristina Simões E. Silva, Universidade Federal de Minas Gerais, BrazilJosef Van Helden, Labor Mönchengladbach MVZ Dr. Stein and Kollegen GbR, Germany
Copyright © 2018 Ohara, Ohnishi, Hosono, Yamamoto, Fu, Maehara, Suda and Sakamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shunsuke Ohnishi, c29uaXNoaUBwb3AubWVkLmhva3VkYWkuYWMuanA=