 Vincent M. Lam1
Vincent M. Lam1 Catharine A. Mielnik1Corey Baimel2Pieter Beerepoot1,3
Catharine A. Mielnik1Corey Baimel2Pieter Beerepoot1,3 Stefano Espinoza4
Stefano Espinoza4 Ilya Sukhanov4,5Wendy Horsfall1
Ilya Sukhanov4,5Wendy Horsfall1 Raul R. Gainetdinov6Stephanie L. Borgland2
Raul R. Gainetdinov6Stephanie L. Borgland2 Amy J. Ramsey1
Amy J. Ramsey1 Ali Salahpour1*
Ali Salahpour1*- 1Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada
- 2Department of Physiology and Pharmacology, University of Calgary, Calgary, AB, Canada
- 3Boston Children’s Hospital, F.M. Kirby Center for Neurobiology, Harvard Medical School, Boston, MA, United States
- 4Department of Neuroscience and Brain Technologies, Fondazione Istituto Italiano di Tecnologia, Genoa, Italy
- 5Pavlov First Saint Petersburg State Medical University, Valdman Institute of Pharmacology, Saint Petersburg, Russia
- 6Institute of Translational Biomedicine, Saint Petersburg State University, Saint Petersburg, Russia
The trace amine associated receptor 1 (TAAR1) is a G-protein coupled receptor expressed in the monoaminergic regions of the brain, and represents a potential novel therapeutic target for the treatment of neurological disorders. While selective agonists for TAAR1 have been successfully identified, only one high affinity TAAR1 antagonist has been described thus far. We previously identified four potential low potency TAAR1 antagonists through an in silico screen on a TAAR1 homology model. One of the identified antagonists (compound 22) was predicted to have favorable physicochemical properties, which would allow the drug to cross the blood brain barrier. In vivo studies were therefore carried out and showed that compound 22 potentiates amphetamine- and cocaine-mediated locomotor activity. Furthermore, electrophysiology experiments demonstrated that compound 22 increased firing of dopamine neurons similar to EPPTB, the only known TAAR1 antagonist. In order to assess whether the effects of compound 22 were mediated through TAAR1, experiments were carried out on TAAR1-KO mice. The results showed that compound 22 is able to enhance amphetamine- and cocaine-mediated locomotor activity, even in TAAR1-KO mice, suggesting that the in vivo effects of this compound are not mediated by TAAR1. In collaboration with Psychoactive Drug Screening Program, we attempted to determine the targets for compound 22. Psychoactive Drug Screening Program (PDSP) results suggested several potential targets for compound 22 including, the dopamine, norepinephrine and serotonin transporters; as well as sigma 1 and 2 receptors. Our follow-up studies using heterologous cell systems showed that the dopamine transporter is not a target of compound 22. Therefore, the biological target of compound 22 mediating its psychoactive effects still remains unknown.
Introduction
The trace amine associated receptor 1 (TAAR1) is a GPCR that in part acts as an autoreceptor in presynaptic monoamine neurons, where TAAR1 signaling decreases the firing rate of dopaminergic neurons and dopamine release from terminals (Bradaia et al., 2009; Revel et al., 2011; Leo et al., 2014; Lam et al., 2015a). In addition, TAAR1 has also been shown to interact with the dopamine D2 receptors both pre- and post-synaptically (Leo et al., 2014; Espinoza et al., 2015); as well as in heterologous cell systems (Espinoza et al., 2011). Indeed, functionally it has been proposed that the TAAR1-D2 heteromer negatively modulates GSK3β signaling (Harmeier et al., 2015). Due to these mechanisms of TAAR1 action, there has been much focus on TAAR1 as a potential target for the treatment of neurological and psychiatric diseases, which can arise from the dysregulation of the brain dopamine system.
Selective TAAR1 agonists based on either the 2-benzyl-imidazoline (Galley et al., 2012) or 2-aminooxazole backbones (Galley et al., 2016) have been shown to decrease the firing rate of dopaminergic neurons. These observations led to the testing of TAAR1 agonists as potential treatments for schizophrenia, a disease characterized by hyper-dopaminergia (Brisch et al., 2014). In a series of recent studies, TAAR1 agonists demonstrated antipsychotic activity (Revel et al., 2011, 2012, 2013) in the animal models of schizophrenia. Interestingly, TAAR1 agonists are shown to have similar efficacy in improving both positive and negative symptoms of schizophrenia and also were able to improve few cognitive deficits. Moreover, RO5263397, a partial TAAR1 agonist, does not have the same adverse metabolic side effects as olanzapine, and co-treatment of RO5263397 with olanzapine reduced the metabolic side effects observed with olanzapine alone (Revel et al., 2013). Although still no TAAR1 ligand has been approved for clinical use, TAAR1 remains an intriguing and novel drug target for schizophrenia.
In disease states of hypo-dopaminergic dysregulation such as Parkinson’s disease, a recent study has shown that TAAR1 signaling is also involved and that TAAR1 antagonism could potentially slow the progression of the disease (Alvarsson et al., 2015). Unlike TAAR1 agonists which display antipsychotic activity, a TAAR1 antagonist should enhance dopamine signaling and be useful for the treatment of diseases arising from hypo-dopaminergia such as Parkinsons’s disease. In contrast to the several selective synthetic TAAR1 agonists available, there only exists one selective high affinity TAAR1 antagonist, EPPTB (Bradaia et al., 2009). Unlike TAAR1 agonists, the potential for TAAR1 antagonists in the treatment of disorders arising from hypo-dopaminergia has not been explored in vivo; due to poor in vivo pharmacokinetic properties of EPPTB (Stalder et al., 2011). To identify novel TAAR1 antagonists, we have recently used in silico screening of commercially available compounds on a TAAR1 homology model (Cichero et al., 2014; Lam et al., 2015b). These studies allowed for the identification of low affinity TAAR1 antagonists, which were validated in vitro. In the present study, the behavioral characterization of a previously discovered novel antagonist (compound 22) was performed in vivo. Compound 22 is predicted to have good pharmacokinetic properties that allow the drug to cross the blood brain barrier (BBB). Our data indicated that this compound is able to regulate dopamine transmission by potentiating the locomotor stimulating effects of the psychostimulants cocaine and amphetamine, however, these effects are found to be independent of TAAR1.
Materials and Methods
Cocaine hydrochloride (Medisca, New York, NY; Batch: 0723-06) and amphetamine (Tocris Bioscience, Bristol, United Kingdom; Batch: 4A/137502) were handled and stored according to regulations set by Health Canada. Compound 22 was purchased from Enamine Ltd. (Kiev, Ukraine). Cell culture reagents and buffers were obtained from Sigma-Aldrich (St. Louis, MO, United States) and Life Technologies (Carlsbad, CA, United States). HEK293 (CRL-1573) cells were purchased from American Type Culture Collection (Hopkinton, United States). Poly-D-lysine was purchased from Sigma-Aldrich and prepared by dissolving the powder to a concentration of 1 mg/mL in ddH2O. Polyethylenimine (PEI) was purchased from Polyscience Inc. (Warminster, PA, United States) and dissolved to a concentration of 1 mg/mL. Aliquots of PEI were stored at -80°C.
The human HA-DAT construct was provided by Sorkina et al. (2006). The backbone of this construct is the peYFP-c1 vector, where the YFP is located on the N-terminus of DAT. In addition, an HA epitope was added onto the second extracellular loop replacing residues 193–203.
All animals were housed in the Division of Comparative Medicine at the University of Toronto. Procedures were conducted in accordance with the Canadian Council for Animal Care and the University of Toronto Faculty of Medicine and Pharmacy Animal Care Committee. Mice were housed 1–4 per cage with 12 h light/dark cycles (7:00–19:00), with ad libitum access to food (Teklad, Envigo, IN, United States) and water.
Cell Culture
HEK293 cells were cultured in Dulbecco’s Modified Eagle Serum (DMEM), supplemented with 10% fetal bovine serum (Sigma-Aldrich), and maintained at 37°C with 5% CO2 in a humidified atmosphere. Cells were passaged 24 h prior to transfection at 50% confluency (∼2 × 106 cells in a 10 cm plate). Transfections were carried out using the PEI method as described previously (Ehrhardt et al., 2006; Lam et al., 2013; Beerepoot et al., 2016). PEI and plasmid DNA (3 μl:1 μg PEI:DNA ratio) were added into separate tubes (tube 1: PEI, tube 2: DNA) followed by 200 μL of DMEM into each tube, containing no supplements. Tubes were allowed to incubate for 5 min before the two tubes were combined (PEI with DNA). The PEI:DNA mixture was then further incubated for 30 min at room temperature and subsequently added drop wise to a 10 cm plate containing HEK293 cells at 50% confluency. For stable cell line generation with the HA-DAT construct, 24 h after transfection, media was replaced with selection media containing G418 (500 μg/mL, Bioshop, Burlington, ON, Canada). Clonal cell lines were generated by picking individual colonies ∼2 weeks post-transfection. Expression was confirmed by western blot and fluorescence microscopy.
Fluorescent Dopamine Uptake Assay
Fluorescent dopamine uptake assay kits were purchased from Molecular Devices (Sunnyvale, CA; catalog #: R6138). Stable cells expressing human HA-DAT were seeded on poly-D-lysine treated, black clear-bottom 96-well plates (Corning Catalog #: 3603) at a density of 1 × 105 cells/well, and incubated for 24 h prior to the start of the uptake experiment. The media was removed and replaced with 80 μL of assay buffer (20 mM HEPES, 1× HBSS, pH 7.4), followed by 10 μL of either 2× concentrated compound 22, 10 μL of 2× concentrated cocaine, or vehicle solutions, previously dissolved in assay buffer. The plates were then incubated for 30 min at 37°C. Following incubation, 100 μL of dye solution was added and fluorescence intensity was measured for 30 min at 37°C using the SpectraMax M3 (Molecular Devices, excitation: 440 nm, emission: 520 nm). The rate of reaction (slope of the curve in the linear range) was taken as the readout for the assay.
Electrophysiology
All protocols were in accordance with the ethical guidelines established by the Canadian Council for Animal Care and were approved by the University of Calgary Animal Care Committees. All mice were housed in groups of 2–5 and were maintained on a 12-h light: dark schedule and were given food and water ad libitum. Experiments were performed during the animal’s light cycle.
All electrophysiological recordings were performed in slice preparations from C57Bl/6J mice (P21-P30). Briefly, mice were anaesthetized with isoflurane and transcardially perfused with an ice-cold sucrose solution containing (in mM): 50 sucrose, 26.2 NaHCO3, 1.25 glucose, 4.9 MgCl2, 3 kynurenic acid, 0.1 CaCl2, and 1.32 ascorbic acid in bicarbonate-buffered solution (aCSF, described below). Mice were then decapitated and brains were extracted. Horizontal sections (180 μm) containing the VTA were cut on a vibratome (Leica, Nussloch, Germany) and incubated in a holding chamber for at least 45 min before being transferred to a recording chamber and superfused with aCSF containing (in mM): 126 NaCl, 1.6 KCl, 1.1 NaH2PO4, 1.4 MgCl2, 2.4 CaCl2, 26 NaHCO3, 11 glucose (32-34oC), and saturated with 95% O2/5% CO2. Cells were visualized on an upright microscope using “Dodt-type” gradient contrast infrared optics (Dodt et al., 2002) and whole-cell recordings were made using a MultiClamp 700B amplifier (Axon Instruments, Union City, CA, United States). Recording electrodes (3–5 MΩ) were filled with (in mM): 136 potassium-D-gluconate, 4 MgCl2, 1.1 HEPES, 5, EGTA, 10 sodium creatine phosphate, 3.4 Mg-ATP, and 0.1 Na2GTP. Putative VTA dopamine neurons were identified by the presence of a large hyperpolarization-activated, cyclic nucleotide-regulated cation (Ih) current. Spontaneous firing activity was recorded in current-clamp mode. Compound 22 and EPPTB were both dissolved in DMSO and diluted to their final concentration in aCSF and bath applied to slices for 5 min. Firing data for all neurons was analyzed with the MiniAnalysis program (Synaptosoft) using the same criteria. Drug-induced changes in firing are expressed as a percentage of baseline. Drug effects were calculated by comparing the response during the baseline/pre-drug period to the response 5 min after onset of drug administration.
Experimental Mice
The Taarl-/- (TAAR1-KO) mice were obtained from Lundbeck (Wolinsky et al., 2007). All wild type (WT) and TAAR1-KO mice used for experiments were generated from TAAR1-KO heterozygous mice in a C57BL/6J x 129S1/Sv mixed background.
Behavioral Experiments
Experimentally naïve mice, of at least 12 weeks of age, were used for all behavioral experiments. The mice were randomly assigned to treatment or control groups, balanced by sex and weight. Locomotor activity was assessed using the automated locomotor analysis monitors (Omnitech Electronics, Columbus, OH, United States). The apparatus included four open field monitors. Each Open Field monitor consisted of sets of 16 light beams arrayed in the horizontal X and Y axes. The hardware detected beams broken by the animal, which allowed the software to determine the location of the mouse in the cage. Total distance covered by mice was used to characterize locomotor activity of the animals. The monitors were divided into four compartments (20 cm × 20 cm). Animals were tested individually for defined periods with 5-min intervals. The mice were first weighed and then placed into the apparatus, allowing for 30 min habituation. Following the habituation, the mice were removed from the al apparatus and injected with drugs (see below for administration) or vehicle and returned immediately to the locomotor chamber. The locomotor activity was then measured for additional 1 h. After the experiments, the animals were euthanized by cervical dislocation.
Drug Administration
In all behavioral studies, compound 22 was co-injected with saline, cocaine or amphetamine. All drug solutions were prepared freshly on the day of the experiment and injected i.p. at the volume of 10 ml/kg. Cocaine hydrochloride was dissolved in 0.9% saline at a concentration of 1 mg/mL. Amphetamine was dissolved in 0.9% saline at a concentration of 0.2 mg/mL. Compound 22 was then dissolved into the cocaine or amphetamine solutions, respectively, to the correct dose for the experiment.
Statistical Analysis
Data analyses were performed with Graphpad Prism 5.01 (GraphPad Software, Inc., La Jolla, CA). Linear regression analysis was used to quantify fluorescent dopamine transporter uptake activity. Dose response curves were fitted with non-linear curve fitting. Two-tailed Student’s t-tests or one-way ANOVA analysis with Dunnett’s post hoc correction was used where appropriate to determine differences between data sets.
Results
Predicted Chemical Properties
Previously, we discovered four potential low potency antagonists of TAAR1 (Lam et al., 2015b). These four identified compounds were assessed for their potential suitability for in vivo use, along with their ability to cross the blood brain barrier. To assess a compound’s permeability of BBB, we followed the criteria outlined by an extensive review of marketed drugs for CNS targets, which yielded a series of chemical properties that could predict BBB penetration (Pajouhesh and Lenz, 2005). The following six criteria were used to evaluate our compounds: (1) liquid water partition coefficient (logP), (2) total polar surface area (tPSA), (3) hydrogen bond donor, (4) hydrogen bond acceptor, (5) rotatable bonds, and (6) molecular weight (Pajouhesh and Lenz, 2005). The physical properties of the antagonist hits from Lam et al. (2015b) (compound 9, 16, 22, and 24) were estimated (Table 1). These four compounds shared similar chemical properties, with the largest differences seen in the logP and tPSA. Based on these predicted values, compound 22 and 24 had the most favorable logP values at 3.30 and 2.98, respectively, whereas compound 9 and 16 had logP values of 1.95 and 1.18, respectively. Therefore, compound 22 and 24 had superior predicted chemical properties for crossing the blood brain barrier. However, due to the constraints of commercial availability, compound 22 was chosen for use in the in vivo studies described here.
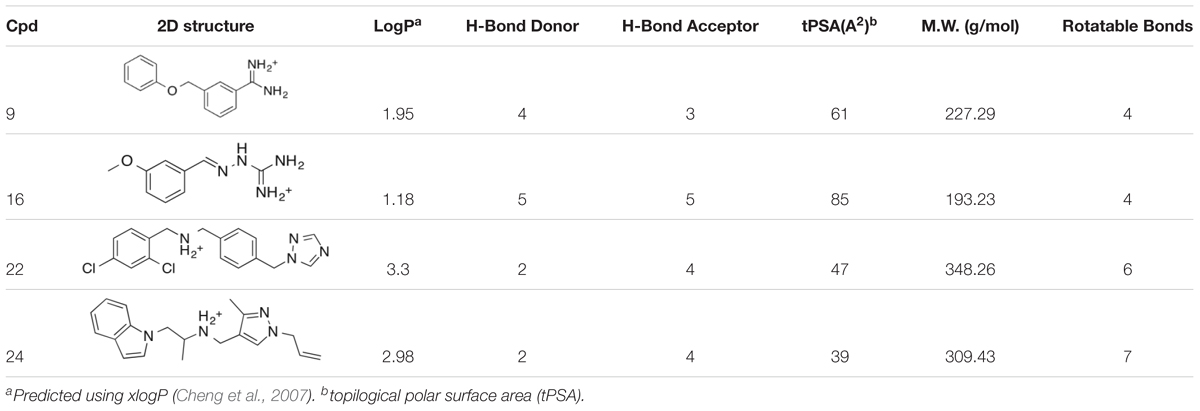
TABLE 1. Comparison of predicted physical properties of antagonist hits from the PubChem database (Wang et al., 2012).
Behavioral Experiments in WT C57BL/6J and TAAR1-KO Mice
It has been previously shown that the TAAR1-KO mice have a potentiated response to the psychostimulant locomotor inducing effects of amphetamine and cocaine (Wolinsky et al., 2007; Lindemann et al., 2008; Di Cara et al., 2011). Therefore, we used locomotor activity as our in vivo readout for the testing of compound 22. We hypothesized that a functional TAAR1 antagonist in WT mice should mimic the phenotypes that are seen in the TAAR1-KO mice, and potentiate their locomotor response to amphetamine and cocaine. Behavioral experiments with compound 22 were carried out in C57BL/6J mice, as well as in TAAR1-KO mice.
Compound 22 Effects on Basal Locomotor Activity in C57BL/6J Mice
In order to assess the effects of compound 22 on basal locomotor activity, C57BL/6 mice were injected with doses of 5 and 30 mg/kg of compound 22 or vehicle (Figure 1). At the dose of 5 mg/kg, compound 22 inhibited basal locomotor activity by 58% (∗p = 0.02). Although not significant, there was a trend toward a decrease in locomotor activity at the dose of 30 mg/kg as well (26% decrease, p = 0.15). Based on these results, compound 22, when administered alone, did not stimulate locomotor activity.
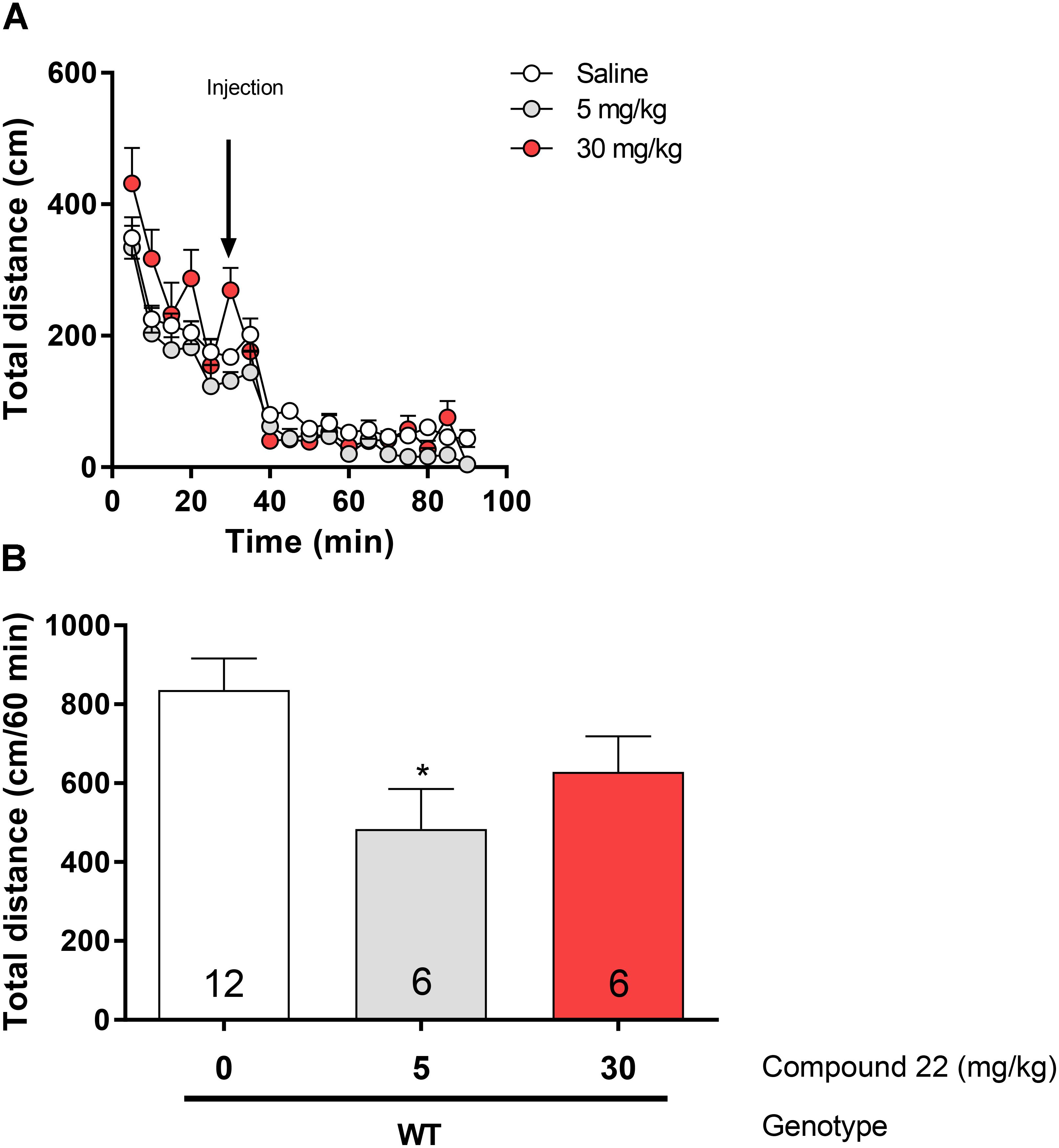
FIGURE 1. In vivo studies with compound 22 on basal locomotor activity. Wild type C57BL/6J mice were first habituated for 30 min followed by co-injection of saline or compound 22 at a dose of 5 or 30 mg/kg. The locomotor activity was assessed for 60 min after injection. (A) Locomotor activity over time for saline only or co-injected with 5 or 30 mg/kg compound 22. (B) Sum of locomotor activity over 60 min after the injection of saline or compound 22. Data are means ± SEM; N = 6 for compound 22 treated alone and N = 12 for saline treated mice. One-way ANOVA was performed [F(3, 32) = 3.49, p = 0.027] followed by Dunnett’s post hoc analyses (∗p < 0.05). Data are means ± SEM.
Amphetamine Co-injection With Compound 22 in C57BL/6J Mice
The effect of compound 22 (5, 15, 20, 30, and 50 mg/kg) on locomotor activity, in mice, in combination to a single, sub-maximal dose of amphetamine (2 mg/kg) was carried out (Figure 2). Treatment of WT C57BL/6J mice with 15 mg/kg of compound 22 showed enhanced amphetamine locomotor response by 44% (∗p = 0.04). At doses of 20 or 30 mg/kg of compound 22, mice exhibited 57% (∗p = 0.02) and 77% (∗∗∗p = 0.0009) increases in amphetamine-stimulate motor activity, respectively. Although not significant, there was a trend toward an increase in locomotor activity at the dose of 5 mg/kg as well (28% increase, p = 0.32). There was no difference in locomotor activity at the dose of 50 mg/kg of compound 22.
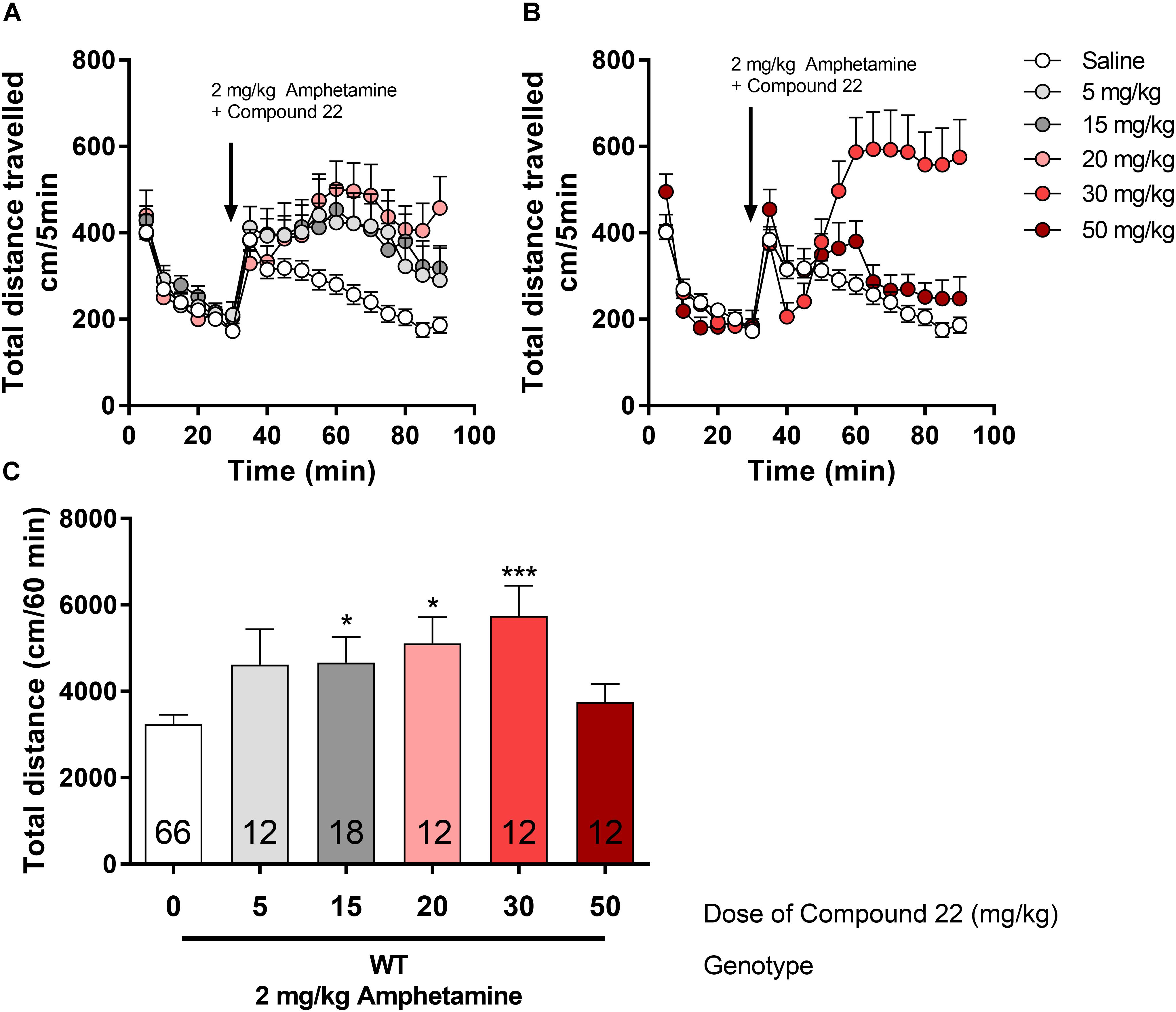
FIGURE 2. In vivo studies with compound 22 co-injected with amphetamine in WT mice. Wild type C57BL/6J mice were first habituated for 30 min followed by the co-injection of amphetamine (2 mg/kg) and saline or compound 22 at doses 5, 15, 20, 25, 30, and 50 mg/kg. The locomotor activity was assessed for 60 min after the injection. (A) Locomotor activity over time for 2 mg/kg amphetamine only or co-injected with 5, 15, or 20 mg/kg compound 22; (B) locomotor activity over time for 2 mg/kg amphetamine only or co-injected with 30 or 50 mg/kg compound 22; (C) sum of locomotor activity over 60 min after the injection of amphetamine and compound 22. Data are means ± SEM; N = 12–18 for compound 22 treated alone and N = 66 for amphetamine treated alone. A one way ANOVA was performed [F(5,126) = 4.788, p = 0.0005] followed by Dunnett’s post hoc analyses (∗p < 0.05, ∗∗∗p < 0.001).
Cocaine Co-injection With Compound 22 in C57BL/6J Mice
Since compound 22 enhanced amphetamine-induced locomotor response, we assessed if compound 22 could also enhance cocaine-induced locomotion in WT C57BL/6J mice (Figure 3). Using a single sub-maximal dose of cocaine (10 mg/kg, Orsini et al., 2005), three doses of compound 22 were tested (5, 15, and 25 mg/kg). 5 and 15 mg/kg of compound 22 increased cocaine locomotor activity by 77% (∗p = 0.03) and 84% (∗p = 0.02), respectively. At a dose of 25 mg/kg of compound 22, the mice had a 124% (∗∗∗p = 0.003) increase in cocaine-induced locomotor activity, clearly indicating that compound 22 enhanced cocaine-induced locomotor activity.
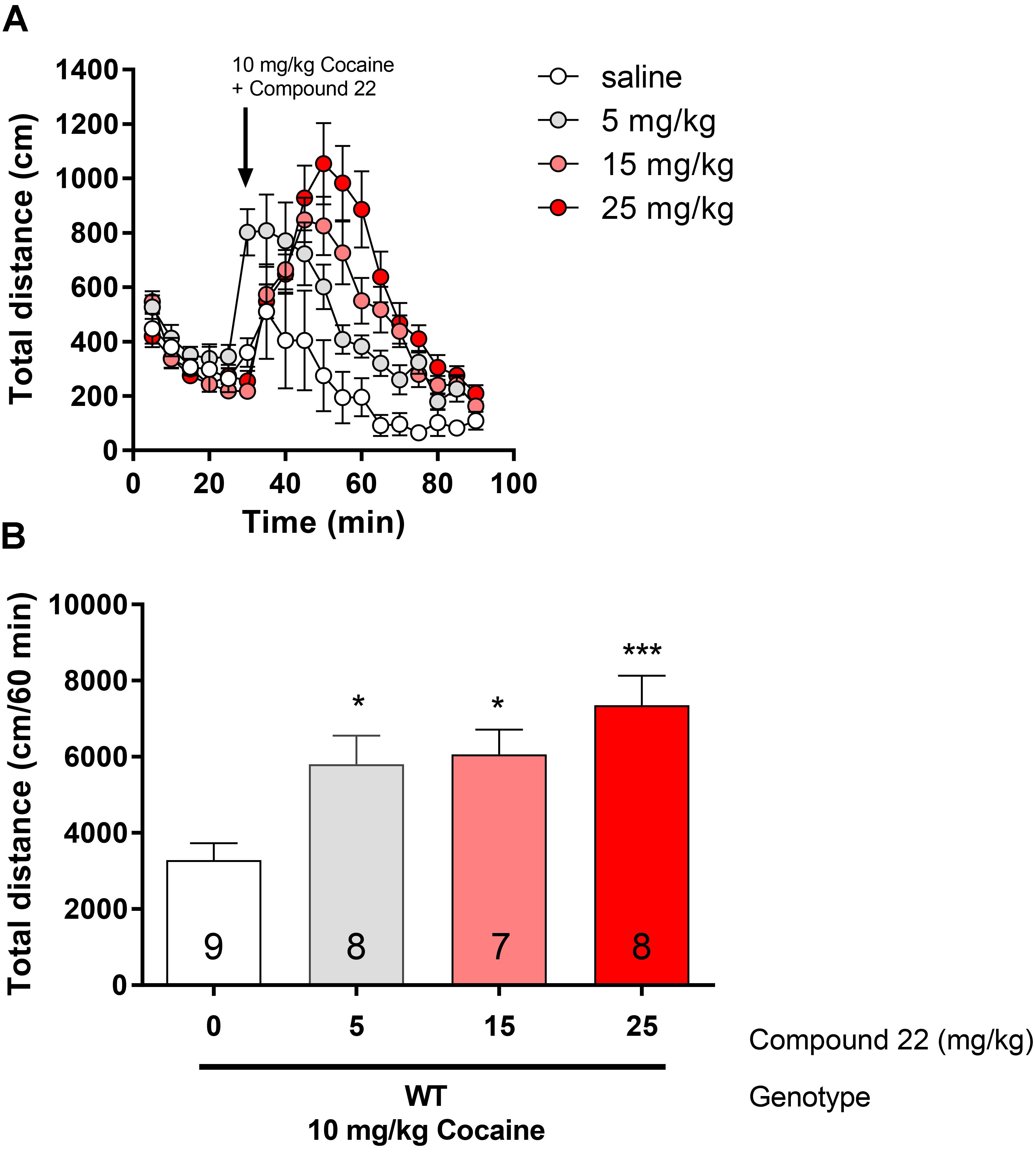
FIGURE 3. In vivo studies with compound 22 co-injected with cocaine. Wild type C57BL/6J mice were first habituated for 30 min followed by co-injection of cocaine (10 mg/kg) and saline or compound 22 at doses 5, 15, and 25 mg/kg. The locomotor activity was assessed for 60 min after the injection. (A) Locomotor activity over time for 10 mg/kg cocaine only or co-injected with 5, 15, or 25 mg/kg compound 22; (B) sum of locomotor activity over 60 min after injection of cocaine and compound 22. Data are means ± SEM; N = 7–9. One way ANOVA was performed [F(3,28) = 7.138, p = 0.001] followed by Dunnett’s post hoc analyses (∗p < 0.05, ∗∗∗p < 0.001).
Effects of Compound 22 Co-injection With Amphetamine and Cocaine on Stereotypic Counts in C57BL/6J Mice
When compound 22 was co-administered with amphetamine we found that there is a statistically significant increase in stereotypic counts at doses of 5 and 20 mg/kg of compound 22 (Supplementary Figure 1A). Furthermore, when compound was co-injected with cocaine, there was an increase in stereotypic counts at doses of 5 and 25 mg/kg of compound 22 (Supplementary Figure 1B). These results indicate that in addition to locomotor activity, compound 22 can enhance stereotypic counts.
Effect of Compound 22 on Firing of Dopamine Neurons
Previous studies have shown that EPPTB increases the firing rate of dopamine neurons (Bradaia et al., 2009; De Gregorio et al., 2016). We therefore assessed the effects of compound 22 on firing rate of dopamine neurons. At a dose of 100 μM, compound 22 increased the firing rate of dopaminergic neurons from VTA by 88% (Figure 4). As a positive control we show that EPPTB caused a 74% increase in firing of dopamine neurons at a dose of 10 nM (Figure 4). These observations showing that compound 22 increases the firing rate of dopamine neurons could partially explain its ability to enhance amphetamine and cocaine locomotor stimulating effects.
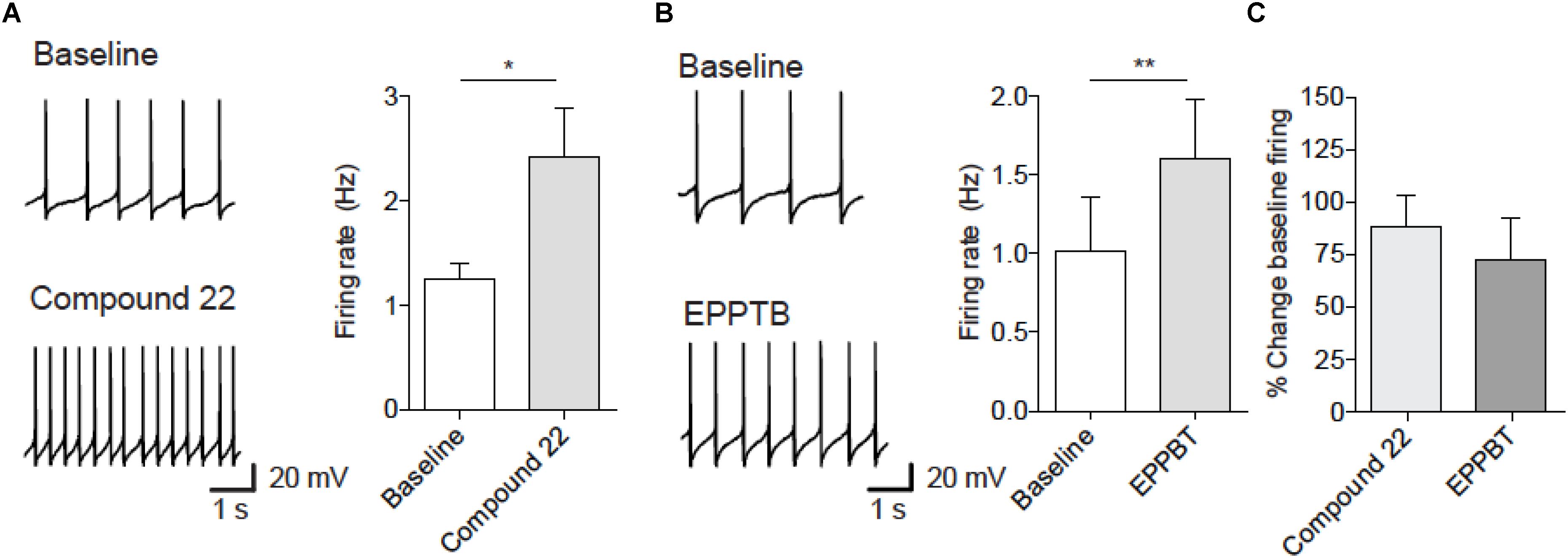
FIGURE 4. Compound 22 and EPPTB increase spontaneous firing of VTA dopamine neurons. (A) Left, example traces of firing before (upper) or after compound 22 (lower). Right, firing rate significantly increased 5 min after onset of bath application of compound 22 (100 μM) [paired t-test; t(4) = 3.67, ∗p = 0.02, N = 5]. (B) Left, example traces of firing before (upper) or after EPPTB (lower). Right, firing rate significantly increased 5 min after onset of bath application of EPPTB (10 nM) [paired t-test; t(3) = 8.74, ∗∗p = 0.003, N = 4]. (C) Effect size of compound 22 or EPPTB. Bars are mean ± SEM.
Compound 22 Effects on Basal Locomotor Activity in TAAR1-KO Mice
To investigate the response of the TAAR1-KO mice to compound 22, the locomotor activity of TAAR1-KO and WT littermates were tested with different doses of compound 22 alone. Previous experiments in C57BL/6J mice indicated that compound 22 did not stimulate basal locomotor activity. Two doses of compound 22 (5 and 25 mg/kg) were used in TAAR1-KO mice or their WT littermates (Figure 5). For both doses tested, compound 22 did not significantly alter the basal locomotor activity of the TAAR1-KO mice or their WT littermates.
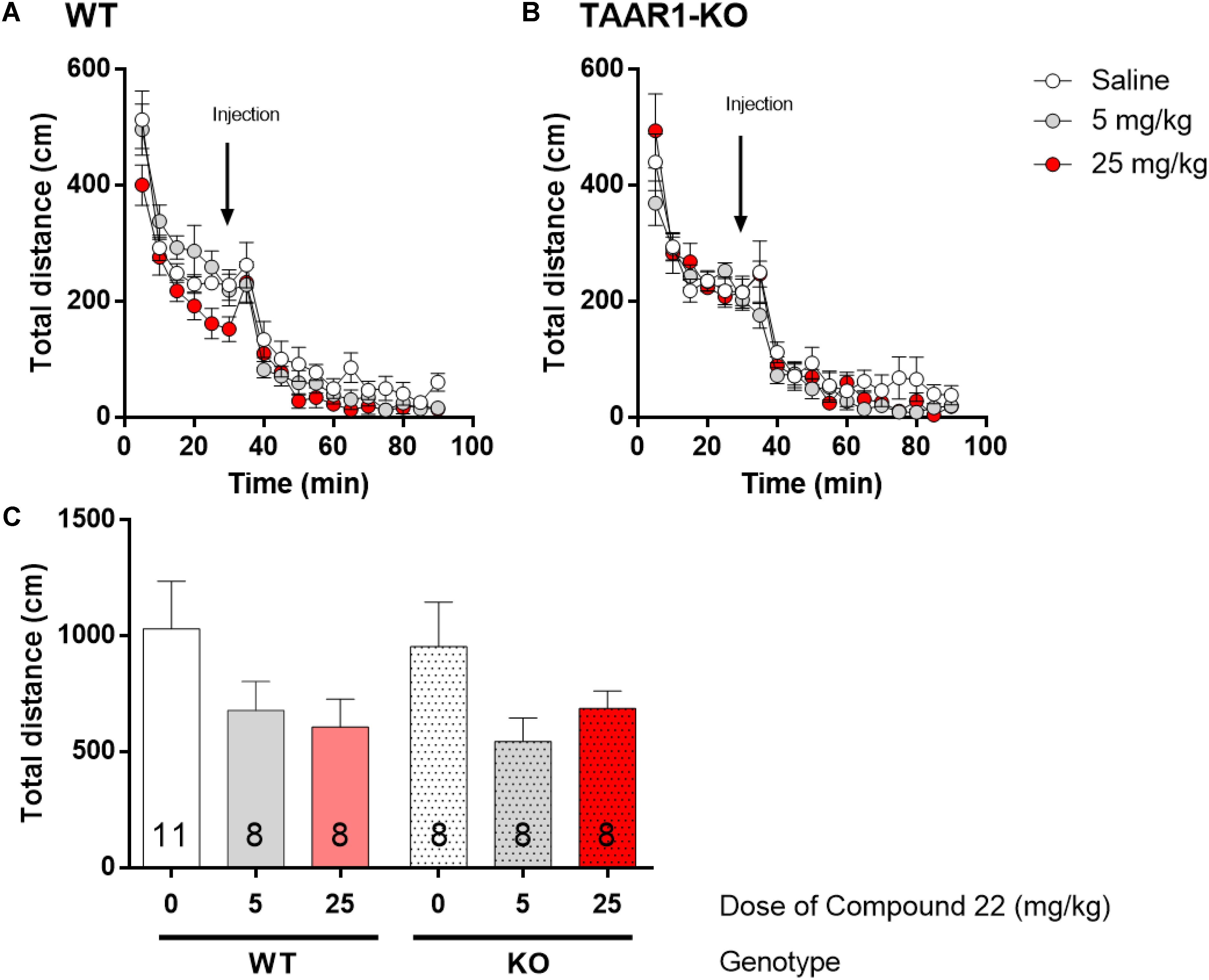
FIGURE 5. In vivo studies with TAAR1-KO mice and compound 22 on basal locomotor activity. TAAR1-KO mice and WT littermates (C57BL/6J × 129S2/Sv) were first habituated for 30 min followed by saline or compound 22 (5 and 25 mg/kg). The locomotor activity was assessed for 60 min following the injection. (A) WT locomotor activity over time for saline or compound 22 (5 and 25 mg/kg); (B) TAAR1-KO locomotor activity over time for saline only or compound 22 (5 and 25 mg/kg); (C) sum of locomotor activity over 60 min after injection of compound 22 in WT (solid bars) or TAAR1-KO mice (dotted bars). Data are means ± SEM; N = 8–11. One-way ANOVA was performed for each genotype; WT [F(2,24) = 1.915, p = 0.1691] and TAAR1-KO [F(2,21) = 2.431, p = 0.1123] followed by Dunnett’s post hoc analyses.
Amphetamine Co-injection with Compound 22 in the TAAR1-KO Mice
In order to assess if the in vivo effects we observed with compound 22 in WT C57BL/6 mice were due to the antagonism of TAAR1, we repeated our co-injection experiments of compound 22 with amphetamine in TAAR1-KO mice. As with the previous in vivo experiments with compound 22, a single submaximal dose of amphetamine (2 mg/kg) was used. In these experiments, doses of 2.5, 5, and 15 mg/kg of compound 22 were tested in TAAR1-KO mice and their WT littermates. As shown in Figure 6, in TAAR1-KO mice, the locomotor stimulating effects of amphetamine were potentiated by 84% with 15 mg/kg of compound 22 (∗∗p = 0.004). Interestingly, in the WT littermates of TAAR1-KO mice, significant potentiation of amphetamine response was seen at 5 mg/kg (44%, ∗p = 0.049). Taken together, these results show that enhancement of amphetamine-induced locomotor response by compound 22 is not through a TAAR1 selective mechanism.
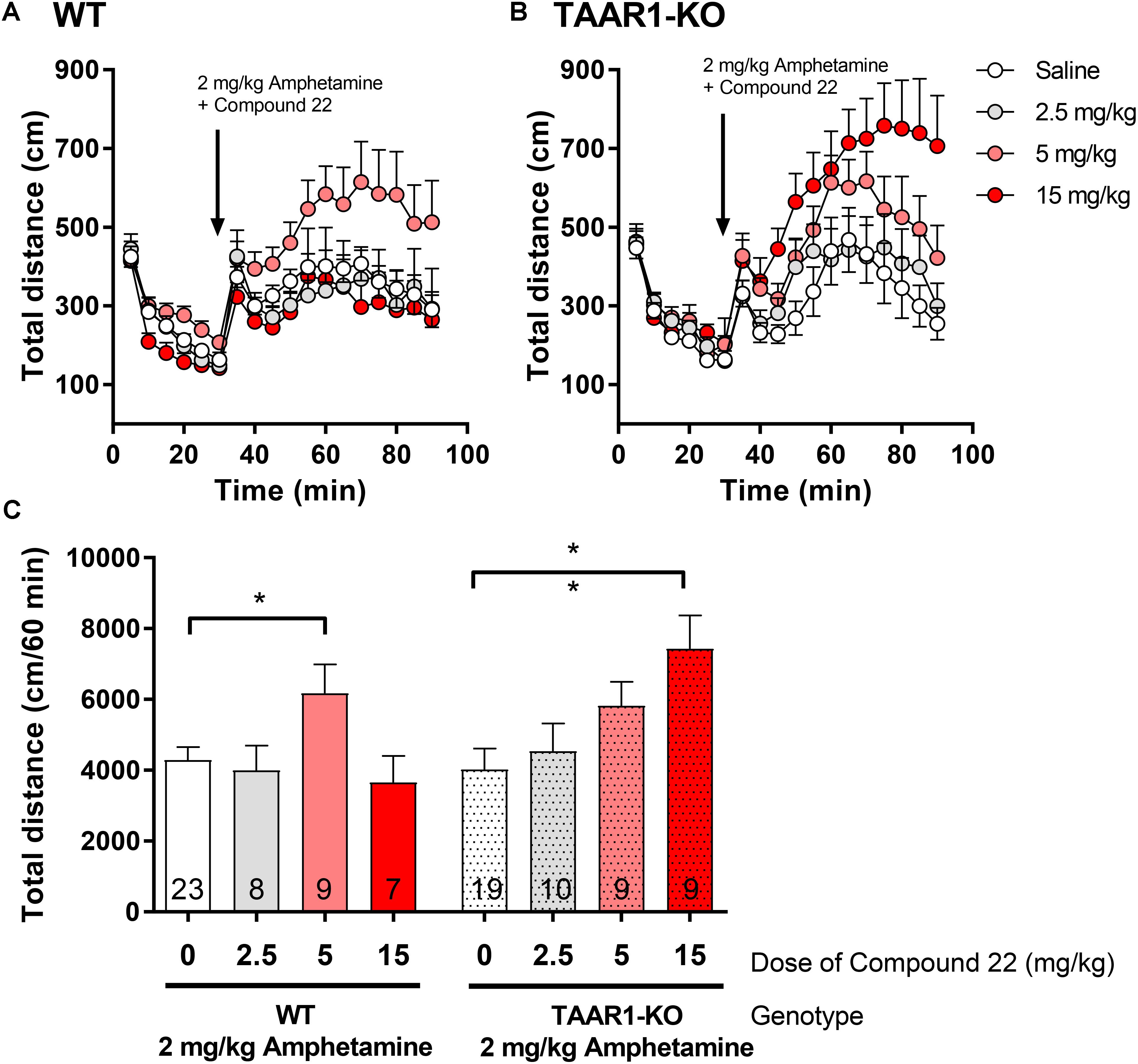
FIGURE 6. In vivo studies with TAAR1-KO mice co-injected with compound 22 and amphetamine. TAAR1-KO mice and their WT littermates (C57BL/6J × 129S2/Sv) were first habituated for 30 min followed by co-injection of amphetamine (2 mg/kg) and saline or compound 22 at doses 2.5, 5, and 15 mg/kg. The locomotor activity was assessed for 60 min after the injection. (A) WT locomotor activity over time for 2 mg/kg amphetamine only or co-injected with 2.5, 5, and 15 mg/kg of compound 22; (B) TAAR1-KO locomotor activity over time for 2 mg/kg amphetamine only or co-injected with 2.5, 5, and 15 mg/kg of compound 22; (C) sum of locomotor activity over 60 min after the injection of amphetamine and compound 22 in WT (solid bars) or TAAR1-KO mice (spotted bars). Data are means ± SEM; N = 7–23. One-way ANOVA was performed for each genotype; WT [F(3,43) = 2.862, p = 0.048] and TAAR1-KO [F(3,43) = 4.159, p = 0.011] followed by Dunnett’s post hoc analyses (∗p < 0.05, ∗∗p < 0.01).
Cocaine Co-injection With Compound 22 in the TAAR1-KO Mice
Since we showed that compound 22 potentiated amphetamine locomotor response did not act through a TAAR1 specific mechanism, next, we investigated whether the same was true for compound 22-potentiated cocaine locomotor response. Doses of 5, 15, and 25 mg/kg of compound 22 were assessed with cocaine co-injection in the TAAR1-KO mice and their WT littermates. As in previous studies, a submaximal dose of 10 mg/kg of cocaine was used for all locomotor assays (Figure 7). All three doses of compound 22 were able to significantly potentiate the locomotor stimulating effects of cocaine in both WT and TAAR1-KO mice. These data further supported that compound 22 is able to potentiate the locomotor stimulant action of amphetamine (Figure 6) and cocaine (Figure 7) in a TAAR1-independent manner.
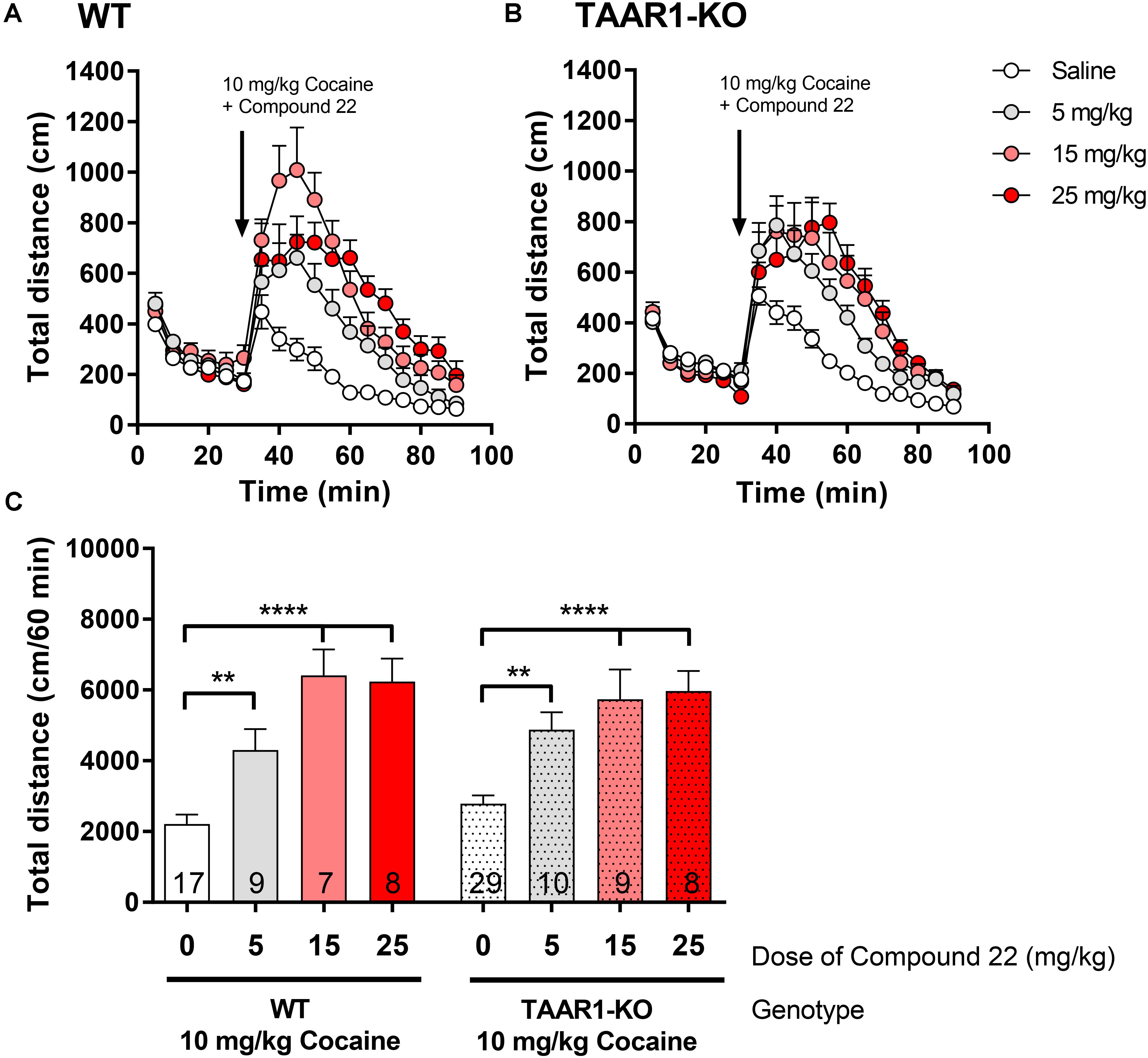
FIGURE 7. In vivo studies with TAAR1-KO mice co-injected with compound 22 and cocaine. TAAR1-KO mice and WT littermates (C57BL/6J × 129S2/Sv) were first habituated for 30 min, followed by co-injection of cocaine (10 mg/kg) with saline or compound 22 (5, 15, and 25 mg/kg). The locomotor activity was assessed for 60 min following the injection. (A) WT locomotor activity over time for cocaine (10 mg/kg) only or co-injected with compound 22 (5, 15, and 25 mg/kg), (B) TAAR1-KO locomotor activity over time for cocaine (10 mg/kg) only, or co-injected with compound 22 (2.5, 5, and 15 mg/kg). (C) Sum of locomotor activity over 60 min following the injection of cocaine and compound 22 in WT (solid bars) or TAAR1-KO mice (dotted bars). Data are means ± SEM; N = 7–29. A one-way ANOVA was performed for each genotype; WT [F(3,38) = 17.97, p < 0.0001] and TAAR1-KO [F(3,52) = 13.93, p < 0.0001] followed by Dunnett’s post hoc analyses (∗∗p < 0.01, ∗∗∗∗p < 0.0001).
Effects of Compound 22 Co-injection With Amphetamine and Cocaine on Stereotypic Counts in the TAAR1-KO Mice
In the TAAR1-KO mice and their WT littermates, we found a similar trend for increased stereotypic counts compared to locomotor activity. For amphetamine treated mice, the WT animals did not have statistically enhanced stereotypic counts in any dose of compound 22. In the TAAR1-KO mice, only the dose of 15 mg/kg produced a statistically significant increase in stereotypic counts (Supplementary Figure 2A). When compound 22 was co-injected with cocaine, compound 22 enhanced stereotypic counts in WT animals at doses of 15 and 25 mg/kg of compound 22. Lastly the TAAR1-KO mice showed significant increases in stereotypic counts after cocaine injection for all three doses tested of compound 22 (5, 15, and 25 mg/kg; Supplementary Figure 2B).
Elucidating the Mechanism of Compound 22
Since our data in TAAR1-KO mice indicated that compound 22 modulated the locomotor response to psychostimulants in a TAAR1-independent manner, we next aimed to identify potential targets for compound 22 that would mediate this effect. This was achieved through the identification of potential pharmacological targets of compound 22 using the Psychoactive Drug Screening Program (PDSP) at University of North Carolina-Chapel-Hill. PDSP provides a platform for screening novel psychoactive compounds on human proteins expressed in the central nervous system in order to identify their biological target(s) (Besnard et al., 2012). Binding studies were done on 47 targets at a single dose of compound 22 (10 μM; Supplementary Table S1). This primary screen yielded a total of 5 hits for compound 22 (Table 2). These hits were the serotonin, dopamine, and norepinephrine transporters, as well as the sigma 1 and sigma 2 receptors. The affinities of compound 22 for the serotonin, dopamine, and norepinephrine transporters were relatively low with Ki = 1800, 1053, and 1902 nM, respectively. In addition to these monoamine transporters, compound 22 was found to have moderate affinity for the sigma 1 and 2 receptors with Ki = 276 and 412 nM, respectively.

TABLE 2. Compound 22 binding studies from PDSP: secondary screen for compound 22 and subsequent Ki values (list of compound 22 potential targets).
Since the PDSP screen showed that compound 22 was able to bind to the dopamine transporter (Ki = 1053 nM), which is also the drug target of amphetamine and cocaine, we hypothesized that the psychostimulant potentiating effects of compound 22 could be mediated by modulation of the dopamine transporter. Therefore, we carried out experiments to directly assess the ability of compound 22 to modulate dopamine transporter activity.
Cells stably expressing the human dopamine transporter were pre-treated with increasing doses of compound 22 or cocaine which was used as a positive control. The ability of compound 22 to directly disrupt uptake activity was assessed with a fluorescent uptake assay. As shown in Figure 7, cocaine dose-dependently inhibited dopamine uptake by DAT (IC50 = 0.95 ± 0.02 μM), while compound 22 had no effect on dopamine uptake activity (Figure 8A). Next, we assessed whether compound 22 could modulate cocaine inhibitory effects on DAT. As shown in Figure 7B, compound 22 at three doses (1, 10, and 100 μM) did not alter the cocaine dose response inhibition of dopamine uptake (Figure 8B). These data are in contrast to the results obtained from PDSP, and indicate that if compound 22 binds to the dopamine transporter, it does not block dopamine uptake via the transporter.
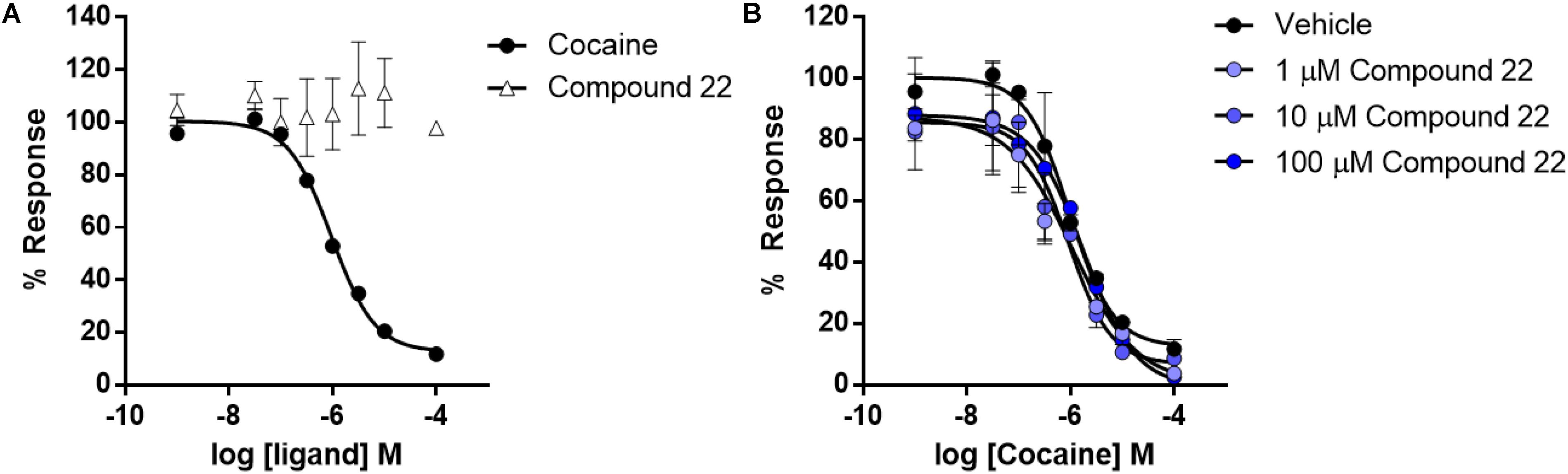
FIGURE 8. Dose response curves for cocaine and compound 22 effects on dopamine transporter uptake activity. Dose response curves were generated for compound 22 or cocaine by the addition of a range of doses to HEK293 cells stably expressing human HA-DAT. (A) Dose response of cocaine and compound 22 on inhibition of uptake; (B) cocaine co-treatment with: vehicle, 1, 10, or 100 μM of compound 22. Error bars represent standard error of mean at N = 3.
Discussion
In our previous study, compound 22 appeared to be a potential weak antagonist (IC50 > 100 μM) for TAAR1 (Lam et al., 2015b). The favorable predicted chemical properties of compound 22 (Table 1) led to the hypothesis that compound 22 would cross BBB and have effects in vivo. Indeed, our experiments in this study confirmed that compound 22 was able to cross BBB and potentiate the locomotor stimulating effects and stereotypic counts when co-injected with amphetamine and cocaine in WT animals.
Since TAAR1-KO mice have a potentiated locomotor response to amphetamine and cocaine when compared to WT mice (Wolinsky et al., 2007; Lindemann et al., 2008; Di Cara et al., 2011), we hypothesized that the locomotor and stereotypic effects of compound 22 seen in WT mice were consistent with TAAR1-based antagonism. However, we observed that compound 22 could also potentiate cocaine- and amphetamine-mediated locomotor activity and stereotypic counts in TAAR1-KO mice, showing that the in vivo locomotor effects of compound 22 are, in fact, TAAR1-independent. Further attempts at elucidating the compound 22 mechanism of action using PDSP suggested that the effects of compound 22 could be mediated by the dopamine transporter. However, our follow up experiments in heterologous cells excluded this possibility. In addition, PDSP also showed the sigma 1 receptor as a hit for compound 22. The sigma 1 receptor is a single transmembrane protein that is primarily localized in the endoplasmic reticulum. The sigma 1 receptor is expressed in peripheral tissues (Stone et al., 2006), as well as highly expressed in the brain (Alonso et al., 2000; Hayashi and Su, 2005). Previous studies with sigma 1 ligands showed that sigma 1 agonists potentiated cocaine mediated locomotor activity (Menkel et al., 1991; Matsumoto et al., 2001; Rodvelt et al., 2011a,b; Lever et al., 2014; Hong et al., 2017), however, agonists inhibited amphetamine mediated locomotor activity (Poncelet et al., 1993; Rückert and Schmidt, 1993; Guitart et al., 1998; Skuza and Rogóz, 2006). Therefore, it is unlikely that compound 22 is a sigma 1 ligand as we have shown that compound 22 potentiates both cocaine and amphetamine mediated locomotor activity. In sum, our data suggest that compound 22, which is a low potency TAAR1 antagonist, is able to enhance amphetamine- and cocaine-mediated locomotor activity through a currently unknown mechanism.
Within the basal ganglia circuitry, there are multiple receptor systems that could explain the in vivo results we observed with compound 22. While it is rare for a compound to potentiate both amphetamine- and cocaine-induced locomotor activity and not stimulate locomotor activity alone, several other compounds have been previously discovered that act in a similar mode. In general, such compounds fit into three distinct mechanisms of action: 1) enhanced firing rate of dopaminergic neurons and 2) enhanced stimulation of D2 dopamine receptor expressing medium spiny neurons (MSN).
Given that compound 22 was found to enhance the firing rate of dopamine neurons, we hypothesize the target of compound 22 to be critical for modulating the firing rate of dopamine neurons. There are several presynaptic receptors that regulate this phenomenon. Such receptors include, but are not limited to, the D2 dopamine receptor, TAAR1, and 5HT2C receptors. For example, the 5HT2C receptor antagonist SB232082 potentiates the locomotor stimulating effects of MDMA, amphetamine, fenfluramine, cocaine, methylphenidate, nicotine, and morphine (Fletcher et al., 2006). Mechanistically, the antagonism of the 5HT2C receptors increases the firing rate of dopaminergic neurons in the VTA, resulting in enhanced dopamine release (Millan et al., 1998; Di Giovanni et al., 1999; Di Matteo et al., 1999, 2002). However, it is unlikely that compound 22 is acting as a 5HT2C receptor antagonist since this receptor was not a hit in the PDSP screen. It is possible that compound 22 could bind to another pre or postsynaptic receptor to enhance the firing rate of dopaminergic neurons as a mechanism of action.
Lastly, regulation of locomotor activity can also occur at the level of postsynaptic MSN expressing the dopamine D2 receptor. One potential target of compound 22 could be the adenosine A2A receptor, which is expressed in D2-expressing MSN (Schiffmann et al., 2007; Fredholm et al., 2011). Mechanistically, it has been shown that the A2A receptor has mutual antagonistic activities with the D2 dopamine receptor. Both the A2A receptor and the D2 dopamine receptor have been shown to dimerize in vitro, as well as in striatal membrane preparations from rats (Ferre et al., 1991; Yang et al., 1995; Dasgupta et al., 1996; Kamiya et al., 2003). The activation of the A2A receptor via A2A agonists inhibits amphetamine- and cocaine-mediated behaviors (Turgeon et al., 1996; Ferré et al., 1997; Rimondini et al., 1997; Baldo et al., 1999; Chen et al., 2001; Knapp et al., 2001; Filip et al., 2006). Conversely, antagonism of the A2A receptor potentiates amphetamine- and cocaine-mediated behaviors (Casas et al., 1989; Turgeon et al., 1996; Ferré et al., 1997; Fredholm et al., 1999; Shiozaki et al., 1999). Since the adenosine A2A receptor was not tested in the original PDSP screen, it is possible that compound 22 is acting as an adenosine A2A receptor antagonist.
One of the limitations of our study is the species differences between the receptors used for our in vivo and in vitro experiments. For instance, the original TAAR1 homology model used to identify compound 22 was generated using the human TAAR1 primary amino sequence (Lam et al., 2015b). In addition, the PDSP assays were performed on human proteins. However, our in vivo studies were carried out in mice. While the human and mouse TAAR1 receptors share 76% sequence homology (Miller et al., 2005), there are important differences in the affinities for known compounds between these receptors from humans and mice. For instance, EPPTB has a 0.9 nM affinity for the mouse TAAR1; while EPPTB does not bind to the human TAAR1 receptor (Bradaia et al., 2009). Therefore, it is possible that compound 22 could be more selective for a mouse receptor (over a human receptor) that would explain mechanistically the in vivo results. Despite species differences, compound 22 still elicits a potentiation of psychoactive-induced locomotor responses in the absence of TAAR1 (TAAR1-KO mice).
Conclusion
In conclusion, our in vivo studies have shown compound 22 to potentiate the locomotor stimulating effects of both amphetamine and cocaine. Our original hypothesis was that compound 22 mediated these effects through the antagonism of TAAR1. However, these findings were also observed in the TAAR1-KO mice, suggesting that compound 22 is not mediating potentiation of amphetamine- and cocaine-induced locomotor response through TAAR1. In collaboration with PDSP, we attempted to determine the target for compound 22; however, the target for compound 22 remains unknown. Therefore, compound 22 appears to be a potent modulator of dopamine signaling within the brain, through a yet unknown mechanism.
Author Contributions
VL, AR, AS, SE, IS, and RG contributed conception and design of the study. VL, CM, PB, and WH designed and contributed to experimental data found in the figures of the manuscript. CB and SB contributed to electrophysiology experiments in Figure 4. VL wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) to AS and Natural Sciences and Engineering Research Council (NSERC) of Canada (DG-372517) to SB. VL was supported by a Doctoral Fellowship from NSERC. PB was supported a Doctoral Fellowship from Canadian Institutes of Health Research (CIHR). RG was supported by the Russian Science Foundation grant 14-50-00069. CB was supported by Brain Canada Doctoral Studentship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with one of the authors PB.
Acknowledgments
We thank Thomas Zhang for preliminary experiments. We thank Dr. Alexander Sorkin for the YFP-HA-DAT DNA. We also thank PDSP services and Dr. Bryan Roth (UNC Chapel Hill) for profiling compound 22 against 47 CNS targets. We are grateful to LundbeckA/G and Lundbeck United States for generously providing TAAR1 knockout mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00953/full#supplementary-material
FIGURE S1 | Stereotypic counts of C57BL/6J mice co-injected with compound 22 and cocaine. (A) Wild type C57BL/6J mice were first habituated for 30 min followed by the co-injection of amphetamine (2 mg/kg) and saline or compound 22 at doses 5, 15, 20, 25, 30, and 50 mg/kg. The locomotor activity was assessed for 60 min after the injection. Sum of stereotypic counts assessed over 60 min after the injection of amphetamine and compound 22. Data are means ± SEM; N = 12–18 for compound 22 treated alone and N = 66 for amphetamine treated alone. A one way ANOVA was performed [F(5,126) = 4.482, p = 0.0009] followed by Dunnett’s post hoc analyses (∗p < 0.05, ∗∗∗p < 0.001). (B) Wild type C57BL/6J mice were first habituated for 30 min followed by co-injection of cocaine (10 mg/kg) and saline or compound 22 at doses 5, 15, and 25 mg/kg. The locomotor activity was assessed for 60 min after the injection. Sum of stereotypic counts assessed over 60 min after injection of cocaine and compound 22. Data are means ± SEM; N = 7–9. One way ANOVA was performed [F(3,29) = 4.365, p = 0.0118] followed by Dunnett’s post hoc analyses (∗p < 0.05, ∗∗∗p < 0.001).
FIGURE S2 |In vivo studies with TAAR1-KO mice and compound 22 on stereotypic counts activity. (A) TAAR1-KO mice and their WT littermates (C57BL/6J x 129S2/Sv) were first habituated for 30 min followed by co-injection of amphetamine (2 mg/kg) and saline or compound 22 at doses 2.5, 5, and 15 mg/kg. The sum of stereotypic counts was assessed over 60 min after the injection of amphetamine and compound 22 in WT (solid bars) or TAAR1-KO mice (spotted bars). Data are means ± SEM; N = 7–23. One-way ANOVA was performed for each genotype; WT [F(3,43) = 1.843, p = 0.1536] and TAAR1-KO [F(3,43) = 4.303, p = 0.0097] followed by Dunnett’s post hoc analyses ∗∗p < 0.01). (B) TAAR1-KO mice and WT littermates (C57BL/6J x 129S2/Sv) were first habituated for 30 min, followed by co-injection of cocaine (10 mg/kg) with saline or compound 22 (5, 15, and 25 mg/kg). The sum of stereotypic counts was assessed over 60 min following the injection in WT (solid bars) or TAAR1-KO mice (dotted bars). Data are means ± SEM; N = 7–29. A one-way ANOVA was performed for each genotype; WT [F(3,37) = 6.696, p = 0.001) and TAAR1-KO [F(3,51) = 13.57, p < 0.0001] followed by Dunnett’s post hoc analyses (∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
TABLE S1 | Compound 22 binding studies from PDSP: primary screen for compound 22 and subsequent hits (50% cut-off, hits highlighted in red). The primary screen was performed by PDSP as described previously (Besnard et al., 2012).
References
Alonso, G., Phan, V.-L., Guillemain, I., Saunier, M., Legrand, A., Anoal, M., et al. (2000). Immunocytochemical localization of the sigma1 receptor in the adult rat central nervous system. Neuroscience 97, 155–170. doi: 10.1016/S0306-4522(00)00014-2
Alvarsson, A., Zhang, X., Stan, T. L., Schintu, N., Kadkhodaei, B., Millan, M. J., et al. (2015). Modulation by trace amine-associated receptor 1 of experimental parkinsonism, L-DOPA Responsivity, and glutamatergic neurotransmission. J. Neurosci. 35, 14057–14069. doi: 10.1523/JNEUROSCI.1312-15.2015
Baldo, B. A., Koob, G. F., and Markou, A. (1999). Role of adenosine A2 receptors in brain stimulation reward under baseline conditions and during cocaine withdrawal in rats. J. Neurosci. 19, 11017–11026.
Beerepoot, P., Lam, V. M., and Salahpour, A. (2016). Pharmacological chaperones of the dopamine transporter rescue dopamine transporter deficiency syndrome mutations in heterologous cells. J. Biol. Chem. 291, 22053–22062. doi: 10.1074/jbc.M116.749119
Besnard, J., Ruda, G. F., Setola, V., Abecassis, K., Rodriguiz, R. M., Huang, X.-P., et al. (2012). Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. doi: 10.1038/nature11691
Bradaia, A., Trube, G., Stalder, H., Norcross, R. D., Ozmen, L., Wettstein, J. G., et al. (2009). The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. U.S.A. 106, 20081–20086. doi: 10.1073/pnas.0906522106
Brisch, R., Saniotis, A., Wolf, R., Bielau, H., Bernstein, H.-G., Steiner, J., et al. (2014). The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front. Psychiatry 5:47. doi: 10.3389/fpsyt.2014.00047
Casas, M., Ferré, S., Cobos, A., Grau, J. M., and Jané, F. (1989). Relationship between rotational behaviour induced by apomorphine and caffeine in rats with unilateral lesion of the nigrostriatal pathway. Neuropharmacology 28, 407–409. doi: 10.1016/0028-3908(89)90037-3
Chen, J. F., Moratalla, R., Impagnatiello, F., Grandy, D. K., Cuellar, B., Rubinstein, M., et al. (2001). The role of the D(2) dopamine receptor (D(2)R) in A(2A) adenosine receptor (A(2A)R)-mediated behavioral and cellular responses as revealed by A(2A) and D(2) receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1970–1975. doi: 10.1073/pnas.98.4.1970
Cheng, T., Zhao, Y., Li, X., Lin, F., Xu, Y., Zhang, X., et al. (2007). Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 47, 2140–2148. doi: 10.1021/ci700257y
Cichero, E., Espinoza, S., Franchini, S., Guariento, S., Brasili, L., Gainetdinov, R. R., et al. (2014). Further insights into the pharmacology of the human trace amine-associated receptors: discovery of novel ligands for TAAR1 by a virtual screening approach. Chem. Biol. Drug Des. 84, 712–720. doi: 10.1111/cbdd.12367
Dasgupta, S., Ferré, S., Kull, B., Hedlund, P. B., Finnman, U.-B., Ahlberg, S., et al. (1996). Adenosine A2A receptors modulate the binding characteristics of dopamine D2 receptors in stably cotransfected fibroblast cells. Eur. J. Pharmacol. 316, 325–331. doi: 10.1016/S0014-2999(96)00665-6
De Gregorio, D., Posa, L., Ochoa-Sanchez, R., McLaughlin, R., Maione, S., Comai, S., et al. (2016). The hallucinogen D-lysergic diethylamide (LSD) decreases dopamine firing activity through 5-HT1A, D2 and TAAR1 receptors. Pharmacol. Res. 113, 81–91. doi: 10.1016/j.phrs.2016.08.022
Di Cara, B., Maggio, R., Aloisi, G., Rivet, J.-M., Lundius, E. G., Yoshitake, T., et al. (2011). Genetic deletion of trace amine 1 receptors reveals their role in auto-inhibiting the actions of ecstasy (MDMA). J. Neurosci. 31, 16928–16940. doi: 10.1523/JNEUROSCI.2502-11.2011
Di Giovanni, G., De Deurwaerdére, P., Di Mascio, M., Di Matteo, V., Esposito, E., and Spampinato, U. (1999). Selective blockade of serotonin-2C/2B receptors enhances mesolimbic and mesostriatal dopaminergic function: a combined in vivo electrophysiological and microdialysis study. Neuroscience 91, 587–597. doi: 10.1016/S0306-4522(98)00655-1
Di Matteo, V., Cacchio, M., Di Giulio, C., and Esposito, E. (2002). Role of serotonin2C receptors in the control of brain dopaminergic function. Pharmacol. Biochem. Behav. 71, 727–734. doi: 10.1016/S0091-3057(01)00705-5
Di Matteo, V., Di Giovanni, G., Di Mascio, M., and Esposito, E. (1999). SB 242 084, a selective serotonin(2C) receptor antagonist, increases dopaminergic transmission in the mesolimbic system. Neuropharmacology 38, 1195–1205. doi: 10.1016/S0028-3908(99)00047-7
Dodt, H.-U., Eder, M., Schierloh, A., and Zieglgänsberger, W. (2002). Infrared-guided laser stimulation of neurons in brain slices. Sci. STKE 2002:pl2. doi: 10.1126/stke.2002.120.pl2
Ehrhardt, C., Schmolke, M., Matzke, A., Knoblauch, A., Will, C., Wixler, V., et al. (2006). Polyethylenimine, a cost-effective transfection reagent. Signal Transduct. 6, 179–184. doi: 10.1002/sita.200500073
Espinoza, S., Ghisi, V., Emanuele, M., Leo, D., Sukhanov, I., Sotnikova, T. D., et al. (2015). Postsynaptic D2 dopamine receptor supersensitivity in the striatum of mice lacking TAAR1. Neuropharmacology 93, 308–313. doi: 10.1016/j.neuropharm.2015.02.010
Espinoza, S., Salahpour, A., Masri, B., Sotnikova, T. D., Messa, M., Barak, L. S., et al. (2011). Functional Interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol. Pharmacol. 80, 416–425. doi: 10.1124/mol.111.073304
Ferré, S., Fredholm, B. B., Morelli, M., Popoli, P., and Fuxe, K. (1997). Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 20, 482–487. doi: 10.1016/S0166-2236(97)01096-5
Ferre, S., von Euler, G., Johansson, B., Fredholm, B. B., and Fuxe, K. (1991). Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc. Natl. Acad. Sci. U.S.A. 88, 7238–7241. doi: 10.1073/pnas.88.16.7238
Filip, M., Frankowska, M., Zaniewska, M., Przegaliński, E., Muller, C. E., Agnati, L., et al. (2006). Involvement of adenosine A2A and dopamine receptors in the locomotor and sensitizing effects of cocaine. Brain Res. 1077, 67–80. doi: 10.1016/j.brainres.2006.01.038
Fletcher, P. J., Sinyard, J., and Higgins, G. A. (2006). The effects of the 5-HT2C receptor antagonist SB242084 on locomotor activity induced by selective, or mixed, indirect serotonergic and dopaminergic agonists. Psychopharmacology 187, 515–525. doi: 10.1007/s00213-006-0453-9
Fredholm, B. B., Bättig, K., Holmén, J., Nehlig, A., and Zvartau, E. E. (1999). Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 51, 83–133.
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Linden, J., and Müller, C. E. (2011). International union of basic and clinical pharmacology. lXXXI. nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 63, 1–34. doi: 10.1124/pr.110.003285
Galley, G., Beurier, A., Décoret, G., Goergler, A., Hutter, R., Mohr, S., et al. (2016). Discovery and characterization of 2-aminooxazolines as highly potent, selective, and orally active TAAR1 agonists. ACS Med. Chem. Lett. 7, 192–197. doi: 10.1021/acsmedchemlett.5b00449
Galley, G., Stalder, H., Goergler, A., Hoener, M. C., and Norcross, R. D. (2012). Optimisation of imidazole compounds as selective TAAR1 agonists: discovery of RO5073012. Bioorg. Med. Chem. Lett. 22, 5244–5248. doi: 10.1016/j.bmcl.2012.06.060
Guitart, X., Codony, X., Ballarín, M., Dordal, A., and Farré, A. J. (1998). E-5842: a new potent and preferential sigma ligand. preclinical pharmacological profile. CNS Drug Rev. 4, 201–224. doi: 10.1111/j.1527-3458.1998.tb00065.x
Harmeier, A., Obermueller, S., Meyer, C. A., Revel, F. G., Buchy, D., Chaboz, S., et al. (2015). Trace amine-associated receptor 1 activation silences GSK3 β signaling of TAAR1 and D2R heteromers. Eur. Neuropsychopharmacol. 25, 2049–2061. doi: 10.1016/j.euroneuro.2015.08.011
Hayashi, T., and Su, T. (2005). The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr. Neuropharmacol. 3, 267–280. doi: 10.2174/157015905774322516
Hong, W. C., Yano, H., Hiranita, T., Chin, F. T., McCurdy, C. R., Su, T.-P., et al. (2017). The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J. Biol. Chem. 292, 11250–11261. doi: 10.1074/jbc.M116.774075
Kamiya, T., Saitoh, O., Yoshioka, K., and Nakata, H. (2003). Oligomerization of adenosine A2A and dopamine D2 receptors in living cells. Biochem. Biophys. Res. Commun. 306, 544–549. doi: 10.1016/S0006-291X(03)00991-4
Knapp, C. M., Foye, M. M., Cottam, N., Ciraulo, D. A., and Kornetsky, C. (2001). Adenosine agonists CGS 21680 and NECA inhibit the initiation of cocaine self-administration. Pharmacol. Biochem. Behav. 68, 797–803. doi: 10.1016/S0091-3057(01)00486-5
Lam, V. M., Beerepoot, P., Angers, S., and Salahpour, A. (2013). A novel assay for measurement of membrane-protein surface expression using a β-lactamase. Traffic 14, 778–784. doi: 10.1111/tra.12073
Lam, V. M., Espinoza, S., Gerasimov, A. S., Gainetdinov, R. R., and Salahpour, A. (2015a). In-vivo pharmacology of trace-amine associated receptor 1. Eur. J. Pharmacol. 763, 136–142. doi: 10.1016/j.ejphar.2015.06.026
Lam, V. M., Rodríguez, D., Zhang, T., Koh, E. J., Carlsson, J., and Salahpour, A. (2015b). Discovery of trace amine-associated receptor 1 ligands by molecular docking screening against a homology model. Med. Chem. Commun. 6, 2216–2223. doi: 10.1039/C5MD00400D
Leo, D., Mus, L., Espinoza, S., Hoener, M. C., Sotnikova, T. D., and Gainetdinov, R. R. (2014). Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: role of D2 dopamine autoreceptors. Neuropharmacology 81, 283–291. doi: 10.1016/j.neuropharm.2014.02.007
Lever, J. R., Miller, D. K., Fergason-Cantrell, E. A., Green, C. L., Watkinson, L. D., Carmack, T. L., et al. (2014). Relationship between cerebral sigma-1 receptor occupancy and attenuation of cocaine’s motor stimulatory effects in mice by PD144418. J. Pharmacol. Exp. Ther. 351, 153–163. doi: 10.1124/jpet.114.216671
Lindemann, L., Meyer, C. A., Jeanneau, K., Bradaia, A., Ozmen, L., Bluethmann, H., et al. (2008). Trace amine-associated receptor 1 modulates dopaminergic activity. J. Pharmacol. Exp. Ther. 324, 948–956. doi: 10.1124/jpet.107.132647
Matsumoto, R. R., McCracken, K. A., Friedman, M. J., Pouw, B., De Costa, B. R., and Bowen, W. D. (2001). Conformationally restricted analogs of BD1008 and an antisense oligodeoxynucleotide targeting σ1 receptors produce anti-cocaine effects in mice. Eur. J. Pharmacol. 419, 163–174. doi: 10.1016/S0014-2999(01)00968-2
Menkel, M., Terry, P., Pontecorvo, M., Katz, J. L., and Witkin, J. M. (1991). Selective σ ligands block stimulant effects of cocaine. Eur. J. Pharmacol. 201, 251–252. doi: 10.1016/0014-2999(91)90355-T
Millan, M. J., Dekeyne, A., and Gobert, A. (1998). Serotonin (5-HT)(2C) receptors tonically inhibit dopamine (DA) and noradrenaline (NA), but not 5-HT, release in the frontal cortex in vivo. Neuropharmacology 37, 953–955. doi: 10.1016/S0028-3908(98)00078-1
Miller, G. M., Verrico, C. D., Jassen, A., Konar, M., Yang, H., Panas, H., et al. (2005). Primate trace amine receptor 1 modulation by the dopamine transporter. J. Pharmacol. Exp. Ther. 313, 983–994. doi: 10.1124/jpet.105.084459
Orsini, C., Bonito-Oliva, A., Conversi, D., and Cabib, S. (2005). Susceptibility to conditioned place preference induced by addictive drugs in mice of the C57BL/6 and DBA/2 inbred strains. Psychopharmacology (Berl). 181, 327–336. doi: 10.1007/s00213-005-2259-6
Pajouhesh, H., and Lenz, G. R. (2005). Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2, 541–553. doi: 10.1602/neurorx.2.4.541
Poncelet, M., Santucci, V., Paul, R., Gueudet, C., Lavastre, S., Guitard, J., et al. (1993). Neuropharmacological profile of a novel and selective ligand of the sigma site: SR 31742A. Neuropharmacology 32, 605–615. doi: 10.1016/0028-3908(93)90057-A
Revel, F. G., Moreau, J., Pouzet, B., Mory, R., Bradaia, A., Buchy, D., et al. (2013). A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 18, 543–556. doi: 10.1038/mp.2012.57
Revel, F. G., Moreau, J.-L., Gainetdinov, R. R., Bradaia, A., Sotnikova, T. D., Mory, R., et al. (2011). TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. U.S.A. 108, 8485–8490. doi: 10.1073/pnas.1103029108
Revel, F. G., Moreau, J. L., Gainetdinov, R. R., Ferragud, A., Velázquez-Sánchez, C., Sotnikova, T. D., et al. (2012). Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics. Biol. Psychiatry 72, 934–942. doi: 10.1016/j.biopsych.2012.05.014
Rimondini, R., Ferré, S., Ogren, S. O., and Fuxe, K. (1997). Adenosine A2A agonists: a potential new type of atypical antipsychotic. Neuropsychopharmacology 17, 82–91. doi: 10.1016/S0893-133X(97)00033-X
Rodvelt, K. R., Lever, S. Z., Lever, J. R., Blount, L. R., Fan, K.-H., and Miller, D. K. (2011a). SA 4503 attenuates cocaine-induced hyperactivity and enhances methamphetamine substitution for a cocaine discriminative stimulus. Pharmacol. Biochem. Behav. 97, 676–682. doi: 10.1016/j.pbb.2010.11.016
Rodvelt, K. R., Oelrichs, C. E., Blount, L. R., Fan, K.-H., Lever, S. Z., Lever, J. R., et al. (2011b). The sigma receptor agonist SA4503 both attenuates and enhances the effects of methamphetamine. Drug Alcohol Depend. 116, 203–210. doi: 10.1016/j.drugalcdep.2010.12.018
Rückert, N. G. H., and Schmidt, W. J. (1993). The σ receptor ligand 1,3-di-(2-tolyl) guanidine in animal models of schizophrenia. Eur. J. Pharmacol. 233, 261–267. doi: 10.1016/0014-2999(93)90059-Q
Schiffmann, S. N., Fisone, G., Moresco, R., Cunha, R. A., and Ferré, S. (2007). Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 83, 277–292. doi: 10.1016/j.pneurobio.2007.05.001
Shiozaki, S., Ichikawa, S., Nakamura, J., Kitamura, S., Yamada, K., and Kuwana, Y. (1999). Actions of adenosine A(2A) receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology 147, 90–95. doi: 10.1007/s002130051146
Skuza, G., and Rogóz, Z. (2006). Effect of BD 1047, a sigma1 receptor antagonist, in the animal models predictive of antipsychotic activity. Pharmacol. Rep. 58, 626–635.
Sorkina, T., Miranda, M., Dionne, K. R., Hoover, B. R., Zahniser, N. R., and Sorkin, A. (2006). RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J. Neurosci. 26, 8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006
Stalder, H., Hoener, M. C., and Norcross, R. D. (2011). Selective antagonists of mouse trace amine-associated receptor 1 (mTAAR1): discovery of EPPTB (RO5212773). Bioorg. Med. Chem. Lett. 21, 1227–1231. doi: 10.1016/j.bmcl.2010.12.075
Stone, J. M., Årstad, E., Erlandsson, K., Waterhouse, R. N., Ell, P. J., and Pilowsky, L. S. (2006). [123I]TPCNE—A novel SPET tracer for the sigma-1 receptor: first human studies and in vivo haloperidol challenge. Synapse 60, 109–117. doi: 10.1002/syn.20281
Turgeon, S. M., Pollack, A. E., Schusheim, L., and Fink, J. S. (1996). Effects of selective adenosine A1 and A2a agonists on amphetamine-induced locomotion and c-Fos in striatum and nucleus accumbens. Brain Res. 707, 75–80.
Wang, Y., Xiao, J., Suzek, T. O., Zhang, J., Wang, J., Zhou, Z., et al. (2012). PubChem’s bioassay database. Nucleic Acids Res. 40, D400–D412. doi: 10.1093/nar/gkr1132
Wolinsky, T. D., Swanson, C. J., Smith, K. E., Zhong, H., Borowsky, B., Seeman, P., et al. (2007). The Trace Amine 1 receptor knockout mouse: an animal model with relevance to schizophrenia. Genes Brain Behav. 6, 628–639. doi: 10.1111/j.1601-183X.2006.00292.x
Yang, S.-N., Dasgupta, S., Lledo, P.-M., Vincent, J.-D., and Fuxe, K. (1995). Reduction of dopamine D2 receptor transduction by activation of adenosine A2a receptors in stablyA2a/D2 (long-form) receptor co-transfected mouse fibroblast cell lines: studies on intracellular calcium levels. Neuroscience 68, 729–736. doi: 10.1016/0306-4522(95)00171-E
Keywords: TAAR1, dopamine transporter (DAT), cocaine, amphetamine, locomotor activity, electrophysiology
Citation: Lam VM, Mielnik CA, Baimel C, Beerepoot P, Espinoza S, Sukhanov I, Horsfall W, Gainetdinov RR, Borgland SL, Ramsey AJ and Salahpour A (2018) Behavioral Effects of a Potential Novel TAAR1 Antagonist. Front. Pharmacol. 9:953. doi: 10.3389/fphar.2018.00953
Received: 03 February 2018; Accepted: 03 August 2018;
Published: 04 September 2018.
Edited by:
Nick Andrews, Harvard Medical School, United StatesReviewed by:
Wladyslaw Lason, Institute of Pharmacology of the Polish Academy of Sciences, PolandGabriella Gobbi, McGill University, Canada
Copyright © 2018 Lam, Mielnik, Baimel, Beerepoot, Espinoza, Sukhanov, Horsfall, Gainetdinov, Borgland, Ramsey and Salahpour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Salahpour, YWxpLnNhbGFocG91ckB1dG9yb250by5jYQ==