 Fen Yang
Fen Yang Min Jiang1†
Min Jiang1† Pei Hu
Pei Hu- 1Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Center of Drug Clinical Trial, Peking University Cancer Hospital and Institute, Beijing, China
- 2Clinical Pharmacology Research Center, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
- 3Beijing Key Laboratory of Clinical PK and PD Investigation for Innovative Drugs, Beijing, China
Objective: This phase Ia study was designed to assess the pharmacokinetic (PK) characters of free vincristine (F-VCR, refer to as non-liposomal VCR and VCR released from liposome) and total vincristine (T-VCR, the sum of both liposomal VCR and F-VCR), urinary excretion and safety of intravenous administration of vincristine sulfate liposomes injection (VSLI) in Chinese patients with malignant lymphoma and compare the results with those for conventional vincristine sulfate injection (VSI).
Methods: In the phase Ia, randomized, open-label, two sequence cross-over study, patients from one group were exposed to treatment 1 including cytoxan (cyclophosphamide power injection), hydroxyrubicin (adriamycin power injection), oncovin (VSI), and prednisone tablets (standard CHOP scheme) before crossed over to treatment 2 (modified CHOP scheme in which VSI was replaced with VSLI). Patients from another group received treatments in reverse order.
Results: In this phase Ia study, a total of eight subjects participated. VCR elimination from the circulation after injection of VSLI was characterized by a significantly increased maximum concentration (Cmax, 86.6 ng/mL) and plasma area under the plasma concentration-time curve from zero to infinity (AUC0-Inf, 222.1 ng/mL h), markedly decreased distribution volume (Vz, 224.1 L) and plasma clearance (CL, 8.9 L/h) compared to lower Cmax (26.6 ng/mL) and AUC0-Inf (95.1 ng/mL h), larger Vz (688.8 L) and CL (22.1 L/h) for VSI. The small proportion of F-VCR following infusion of VSLI in circulation was reflected by very low Cmax (1.8 ng/mL) and AUC0-Inf (50.5 ng/mL h). Less than 3% of the administered dose of VSLI was excreted in urine and the extent was similar to that for VSI. The elimination percentage of 40–21–14% for VSI changed to 6.2–24–39% for VSLI at intervals of 0–5, 5–13 and 13–25 h, respectively. Significant difference of toxicity between VSLI and VSI was not observed.
Conclusion: VSLI exhibits higher AUC0-Inf of T-VCR, lower CL and Vz compared with VSI. VSLI was well tolerated, maybe due to the markedly decreasing AUC0-Inf of F-VCR. The majority of VCR was enveloped in liposome and VCR was released gradually from liposome following injection of VSLI. Liposomal encapsulation of VCR does not alter the route and extent of VCR excretion in urine.
Introduction
Vincristine sulfate (VCR) remains a potent and widely used antitumor agent for more than 50 years (Johnson et al., 1963) and it has significant activity against a non-Hodgkin lymphoma (NHL) subtypes and acute lymphoblastic leukemia (ALL) (Gidding et al., 1999). VCR exhibits cell cycle-specific cytotoxic activity by binding to tubulin, resulting in microtubule depolymerization, metaphase arrest and apoptosis in cells (Schou et al., 1968; Owellen et al., 1972, 1976). Thus, the antitumor efficiency of VCR is dependent on the drug concentration and duration of exposure at the tumor site (Horton et al., 1988). However, the routinely individual standard VCR dose is limited to 1.4 mg/m2 or a maximum 2 mg (i.e., dose capping) in most VCR-containing cancer treatment regimens in order to reduce the risk of severe peripheral and central nervous system (CNS) neurotoxicity (Hildebrand et al., 1972; Legha, 1986), which may limit its optimal clinic benefit.
Liposomes are versatile drug carriers that allow effective delivery of drug to target tissue, prolong the circulation time of encapsulated drug and slowly release the drug, resulting in high levels of encapsulated drug in target tissues and a long duration of exposure of tumor cells to therapeutic drug concentrations (Drummond et al., 1999; Allen et al., 2006). Vincristine sulfate liposome injection (VSLI) is an encapsulated preparation of standard VCR in liposomes, which was designed to overcome the dosing and PK limitations of conventional vincristine sulfate injection (VSI). VSLI has been studied extensively both in laboratory (Kanter et al., 1994; Webb et al., 1995, 1998; Krishna et al., 2001; Zhong et al., 2014; Shah et al., 2016a) and in the clinic (Embree et al., 1998; Gelmon et al., 1999; Bedikian et al., 2006, 2011; Rodriguez et al., 2009; Thomas et al., 2009; Yan et al., 2012; Silverman et al., 2013; Douer, 2016; Shah et al., 2016b) to show the superiority over standard VCR. Marqibo® (Hana Biosciences, Inc.) was the first listed VSLI approved by FDA at 2012 (Silverman and Deitcher, 2013). Although higher antitumor activity with good tolerance was observed for VSLI, a clear understanding of concentration-effect/toxicity relationship, the release properties of VCR from liposome and the contribution of F-VCR to the overall PK profile of T-VCR for VSLI in human being are still limited.
In the present study, a new VSLI (developed by Shanghai Fudan-zhangjiang Bio-Pharmaceutical Co.) was introduced in the phase Ia trial, which was conducted to study the PK characters of F-VCR and T-VCR, the VCR urinary excretion and the safety in Chinese subjects with malignant lymphoma after VSLI injection.
Materials and Methods
Materials
Vincristine sulfate liposome injection (0.16 mg/mL) was provided by Shanghai Fudan-zhangjiang Bio-Pharmaceutical Co., Ltd., China, which is a three-part formulation containing empty liposomes, disodium hydrogen phosphate and VCR sulfate for injection. After the standard procedure, the encapsulation efficiency of liposomal VCR was up to 95% and liposomal VCR remains stable for at least 24 h when stored at 4°C. VSI, cytoxan (cyclophosphamide power injection), hydroxyrubicin (adriamycin power injection), oncovin (VSI) and prednisone tablets were purchased through Nanjing Pharmaceutical Hefei Tianxing Co., Ltd., China.
Study Subjects
Eligible patients aged from 18 to 65 years had histologically or cytologically confirmed malignant lymphoma, which were refractory to conventional forms of cancer therapy. They should not receive other therapy including surgery, chemotherapy or radiotherapy within at least previous 4 weeks. In addition, it was required for eligible patients to have life expectancy of at least 12 weeks, and Eastern Cooperative Oncology Group performance status of 0 to 2, and adequate bone marrow function (leukocyte count ≥4.0 × 109/L, an absolute granulocyte count ≥1.5 × 109/L, a platelet count 75.0 × 109/L and a hemoglobin count ≥80g/L), and adequate hepatic function (alanine aminotransferase [ALT], aspartate aminotransferase [AST] and total bilirubin ≤3 × the upper limit of normal [ULN], and total bilirubin ≤1.5 × ULN), and renal function (serum creatinine ≤1.5 × ULN).
Patients with brain metastases, or CNS disorder, or severe cardiac and cerebral vascular diseases, or active infection were excluded. Patients who were allergic to the test drug and accessories were ineligible.
Study Design
The study design was shown in Figure 1. In the Phase Ia study with multi-center, randomized, open-label and cross-over design, two treatments were administered under fasted conditions. The treatment sequence for each participant was assigned by a computer-generated randomization list. Treatment 1 was standard CHOP scheme including cytoxan (cyclophosphamide power injection, 750 mg/m2), hydroxyrubicin (adriamycin power injection, 50 mg/m2), oncovin (VSI, 1.4 mg/m2) and prednisone tablets (100 mg). The drugs for injection were administrated intravenously (I.V.) on study day 1 and the tablets were swallowed whole for concessive 5 days from day 1 to 5 of each 21-day circle. Treatment 2 was modified CHOP scheme in which VSLI (1.0 mg/m2) replaced VSI and others remained unchanged. VSI and VSLI should be administrated within 60 ± 5 min.
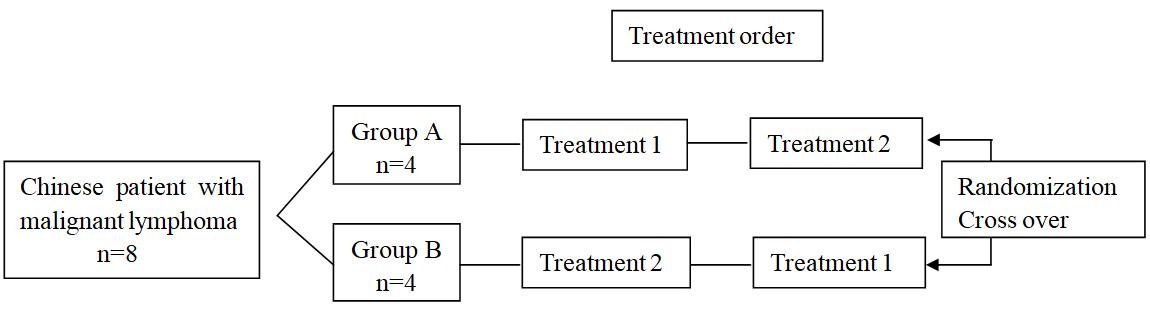
FIGURE 1. Study design. Treatment 1, standard CHOP scheme including cytoxan (cyclophosphamide power injection, 750 mg/m2), hydroxyrubicin (adriamycin power injection, 50 mg/m2), oncovin (VSI, 1.4 mg/m2) and prednisone tablets (100 mg); Treatment 2, modified CHOP scheme in which VSLI (1.0mg/m2) replaced VSI. The drugs for injection were administrated intravenously (I.V.) on study day 1 and the tablets were swallowed whole for concessive 5 days from day 1 to 5 of each 21-day circle.
Subjects fasted overnight for at least 10 h prior to drug administration and for at least 2 h post-dose, after which controlled food intake was allowed. Cigarette, alcohol, tea, coffee, grapefruit-, or drug-containing food or drink, and non-investigator-approved prescription medications or over-the-counter products were to be avoided at least 24 h post-dose and during the study.
Blood and Urine Sampling for PK Analyses
Blood samples were collected at pre-dosing, 15, 30, and 60 min after infusion and 0.25, 0.5, 2, 4, 8, 12, 24, 48, 72, and 96 h after the end of infusion. The whole blood was centrifuged (450 ×g at 4°C for 10 min) and obtained plasma then was divided into three parts. One part was used to separate F-VCR from liposomes using solid phase extraction (SPE) method immediately and the remaining two parts (one part was used as the backup) were frozen for assay of the T-VCR concentration.
Urine samples were collected from four patients of group A (one female and three males) before and after drug infusion up to 97 h at six intervals: 0–5, 5–13, 13–25, 25–49, 49–73, and 73–97 h. The samples from each interval were mixed and the volume was measured. Then 10 mL urine samples were remained and frozen at -80°C.
Bioanalytical Methods
The plasma concentration of F-VCR and T-VCR and the urine VCR concentration was determined using the ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) system (Waters Corporation, Milford, MA, United States) with vinblastine sulfate used as the internal standard (IS) as previously described by our team (Yang et al., 2013, 2015). The compounds were detected as doubly charged ions and the multiple reaction monitor (MRM) transitions of VCR and the IS were m/z 413.2 → 353.2 and m/z 406.2 → 271.6, respectively. F-VCR was separated from liposomal form in plasma using SPE method; plasma T-VCR and urine VCR were extracted using liquid-liquid extraction method. F-VCR and T-VCR were identified and quantified over a theoretical concentration range of 0.2–50 ng/ml for F-VCR in plasma, 0.5–400 ng/ml for T-VCR in plasma and 0.5–100 ng/ml for VCR in urine, respectively. Assay specificity was assessed using blank sample from six different lots to verify the absence of interference at retention time. Quantitation was made using peak area ratios of analyte/IS, and back-calculated concentrations were determined using a weighted (1/ × 2) linear regression (y = ax + b).
Data and Statistical Analyses
The measured plasma concentration was used to obtain Cmax and the time to reach Cmax (Tmax) directly. The main PK parameters, calculated from the plasma concentration-time data using a non-compartmental analysis method (WinNonlin Professional Network Edition, Version 7.0, Pharsight Corp., Palo Alto, CA, United States), were elimination half-life (t1/2), the area under plasma concentration-time curve from zero to last time (AUC0-t) and from zero to infinity (AUC0-Inf), the mean retain time from zero to last time (MRT0-t) and from zero to infinity (MRT0-Inf), clearance (CL) and apparent volume of distribution (Vd). Mann-Whitney test was used to compare PK parameters (Cmax, AUC0-t, t1/2, Vz and CL) between the two groups using GraphPad Prism software (Version 7.0). P-value less than 0.05 was considered to be significant.
Safety Assessments
The safety of the subjects was monitored by evaluation of physical examinations, vital signs, electrocardiograms, and clinical laboratory tests and adverse events (AEs) reporting. AEs were classified by system organ class and were graded according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4. Safety results were analyzed using descriptive statistics.
Results
Subjects
There were eight patients (four males and four females) enrolled in this study. Other demographic data are shown in Table 1. Nobody withdrew/were withdrawn before the end of the study. All patients were assessable for safety. All subjects provided measurable PK data and were therefore included in the PK evaluation and statistical analysis. Table 2 is a summary of dose level, number of the patients and actual dose delivered.
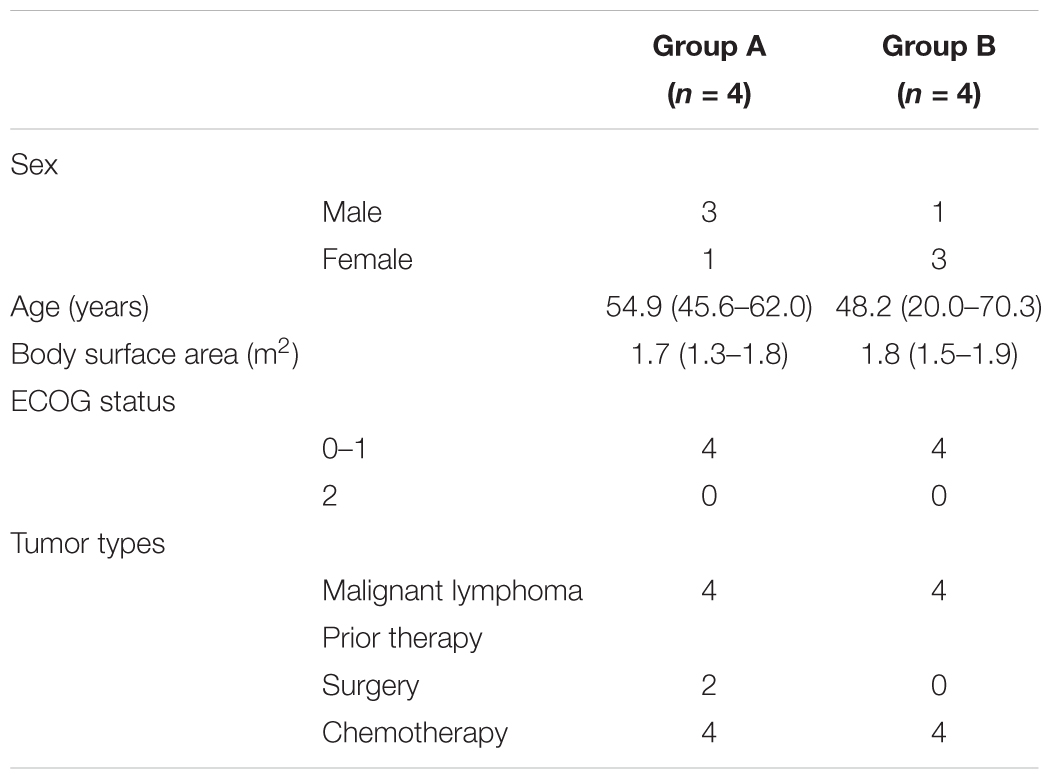
TABLE 1. Demographic characteristics of the patients.
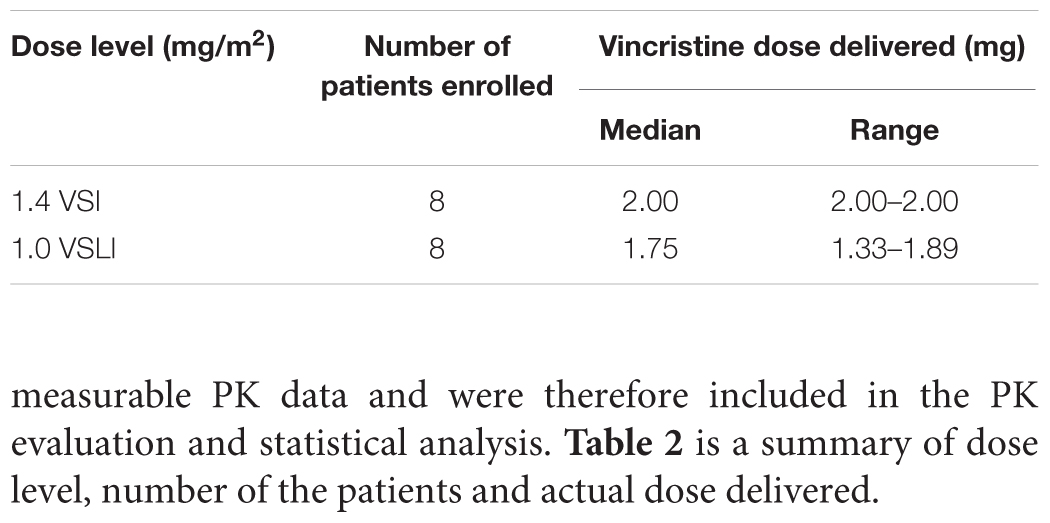
TABLE 2. Planned dose levels and doses delivered.
PKs of T-VCR
The mean plasma T-VCR concentration vs. time curves of patients who received VSLI (1.0 mg/m2) or VSI (1.4 mg/m2) are shown in Figure 2. The corresponding PKs of T-VCR were calculated from these data and are presented in Table 3. The maximum plasma T-VCR concentrations (Cmax) were observed at the end of the VSLI infusion, subsequently a rapid distribution phase occurred, in which concentration rapidly declined within 1 h, followed by a slow distribution phase and a terminal elimination phase. Therefore, the T-VCR plasma concentrations for all subjects were characterized by a triexponential decline after treated with VSLI. Although the profiles for VSI are very similar to those for VSLI, VSI exhibited significant lower Cmax and AUC0-Inf of VCR, as well as markedly higher Vz and CL than those of T-VCR for VSLI. After administration of VSLI, significantly elevated systematic exposure (Cmax was 86.6 ng/mL and AUC0-Inf was 222.1 ng/mL h), associated with decreased CL (8.9 L/h) and Vz (224.2 L) of T-VCR were obtained, compared with those of VCR for VSI (P < 0.01). There was no significant difference for half life (t1/2) between two groups (VSLI, 18.4 h vs. VSI, 22.5 h, P = 0.065).
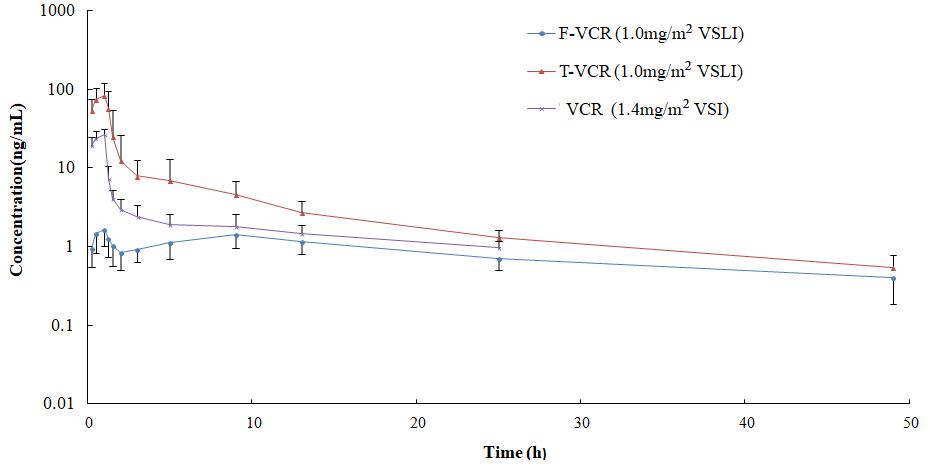
FIGURE 2. Mean logarithmic concentration vs. time plot of T-VCR and F-VCR following infusion of VSLI (1.0 mg/m2) and VCR post injection of VSI (1.4 mg/m2).
PKs of F-VCR
The mean plasma F-VCR concentration-time plots of patients after administration with VSLI at dose level of 1.0 mg/m2 is shown in Figure 2. Key PK parameters of plasma F-VCR are shown in Table 3.
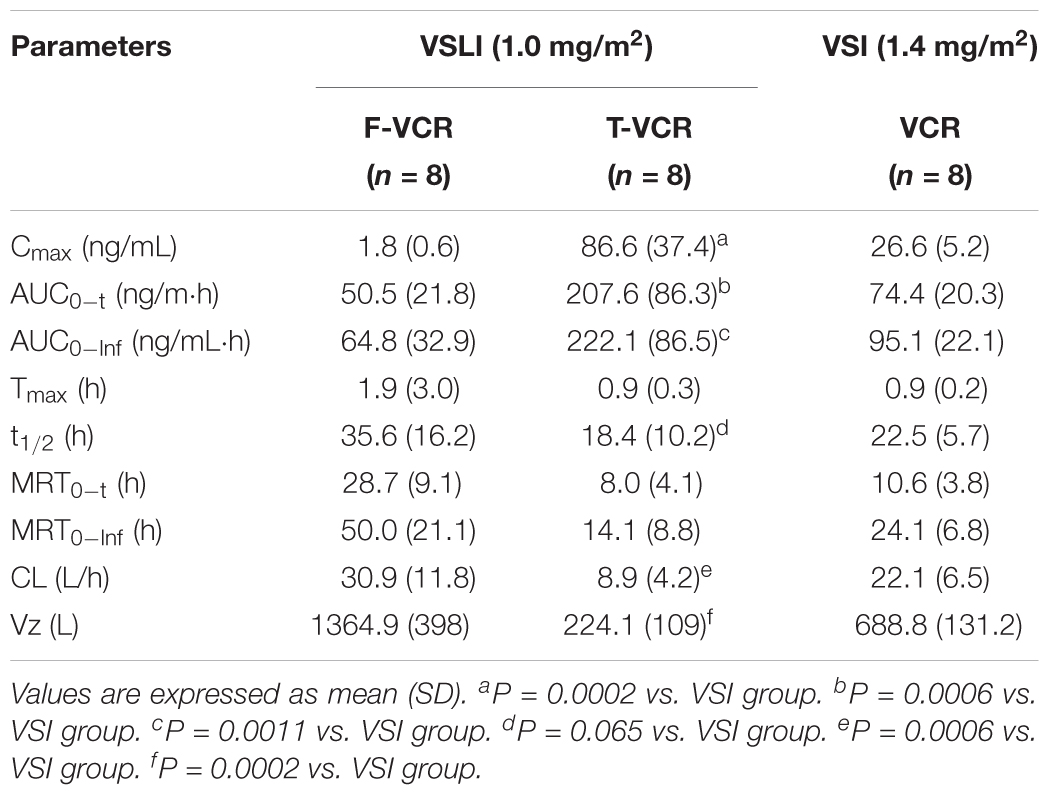
TABLE 3. Main PK parameters of F-VCR and T-VCR following infusion of VSLI (1.0 mg/m2) and VCR post injection of VSI (1.4 mg/m2).
Multiple peaks were apparently observed for the individual F-VCR concentration-time curve (Supplementary Material). The peak concentration (Cmax) of F-VCR was obtained at the end of VSLI infusion (1 h) in most patients (7 of 8 patients). Another one or two lower concentration peaks appeared at 9 or 3/9 h after the start of infusion. Then, the F-VCR concentration gradually declined. At 1 h after i.v. administration of VSLI, the mean plasma concentration of F-VCR was 1.8 ng/ml, compared with 86.6 ng/ml for T-VCR. This indicated that F-VCR represented 2% of the T-VCR in the plasma at this time point. The proportion of F-VCR in the plasma was evaluated by comparing the AUC values. The AUC0-Inf value for F-VCR after the administration of VSLI was 64.8 ng/mL h, which represent 29.2% of the AUC0-Inf for T-VCR. In addition, systemic exposure of F-VCR for VSLI was compared with that of VCR for VSI. Although the similar AUC0-Inf value of F-VCR to that of VCR was obtained after dose calibration, the lower F-VCR concentration at every time point from 0.25 to 25 h after administration of VSLI than that of VCR for VSI was observed. It is important to note that the significant decreased Cmax of F-VCR contrasts the increased plasma VCR concentration observed after VSI administration where peak value was 26.3 ng/mL, 16-fold greater than that of F-VCR.
Urinary Excretion
The cumulative excretion percentage of VCR in urine from four patients treated with VSLI (1.0 mg/m2) and VSI (1.4 mg/m2) are shown in Figure 3, respectively. Less than 3% of the injected VSLI dose was eliminated in the urine over the 97-h period in the form of unchanged VCR and the extent of urinary excretion was similar to that after treated with VSI. Notably, though about 70% of the total amount of VCR excreted in the urine was recovered within 25 h after both VSLI and VSI administration, time course of excretion had large difference. The majority of VCR was excreted during the first few hours (40% of total VCR excretion within 0–5 h followed by 21% within 5–13 h and 14% within 13–25 h) post infusion of VSI. In contrast, the VCR excretion rate gradually increased over the 25-h period (6.2, 24, and 39% of the total amount of VCR excreted in the urine within 0–5, 5–13, and 13–25 h period, respectively) after VSLI administration.
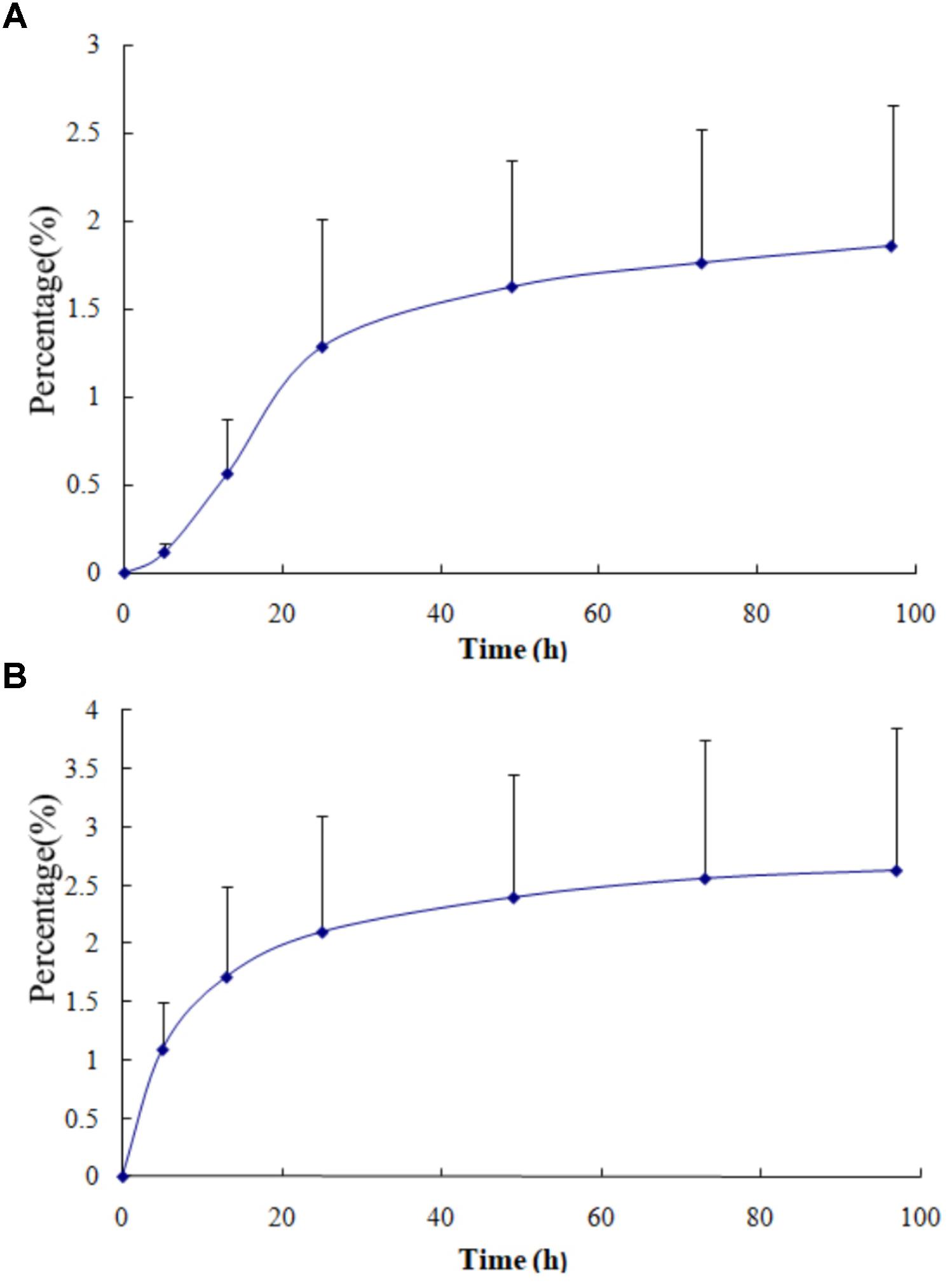
FIGURE 3. The cumulative urinary excretion of VCR in urine from four patients after dosing with VSLI [1.0 mg/m2, (A)] and VSI [1.4 mg/m2, (B)], respectively.
Safety
Adverse events (AEs) were graded according to NCI CTCAE for Adverse Events (version 4.0). AEs mentioned in this paper refer to AEs associated with VALI or VSI (shown in Table 4). Eight patients (100%) experienced AEs both in treatment 1and 2, seven (87.5%) and four (50.0%) of whom experienced grade ≥3 AE in treatment 1 and 2, respectively. Hematologic toxicity were the most frequently reported AEs (seven patients, 87.5% and eight patients, 100% in treatment 1 and 2, respectively) followed by metabolic disorder (five patients, 62.5% both in treatment 1and 2) and gastrointestinal reaction (four patients, 50% both in treatment 1and 2). Eight patients showed good tolerance using treatment 1and 2 and there was no significant difference of AEs between two treatments, showing good safety of VSLI at 1.0 mg/m2 dose level. No serious AEs occurred in this study.
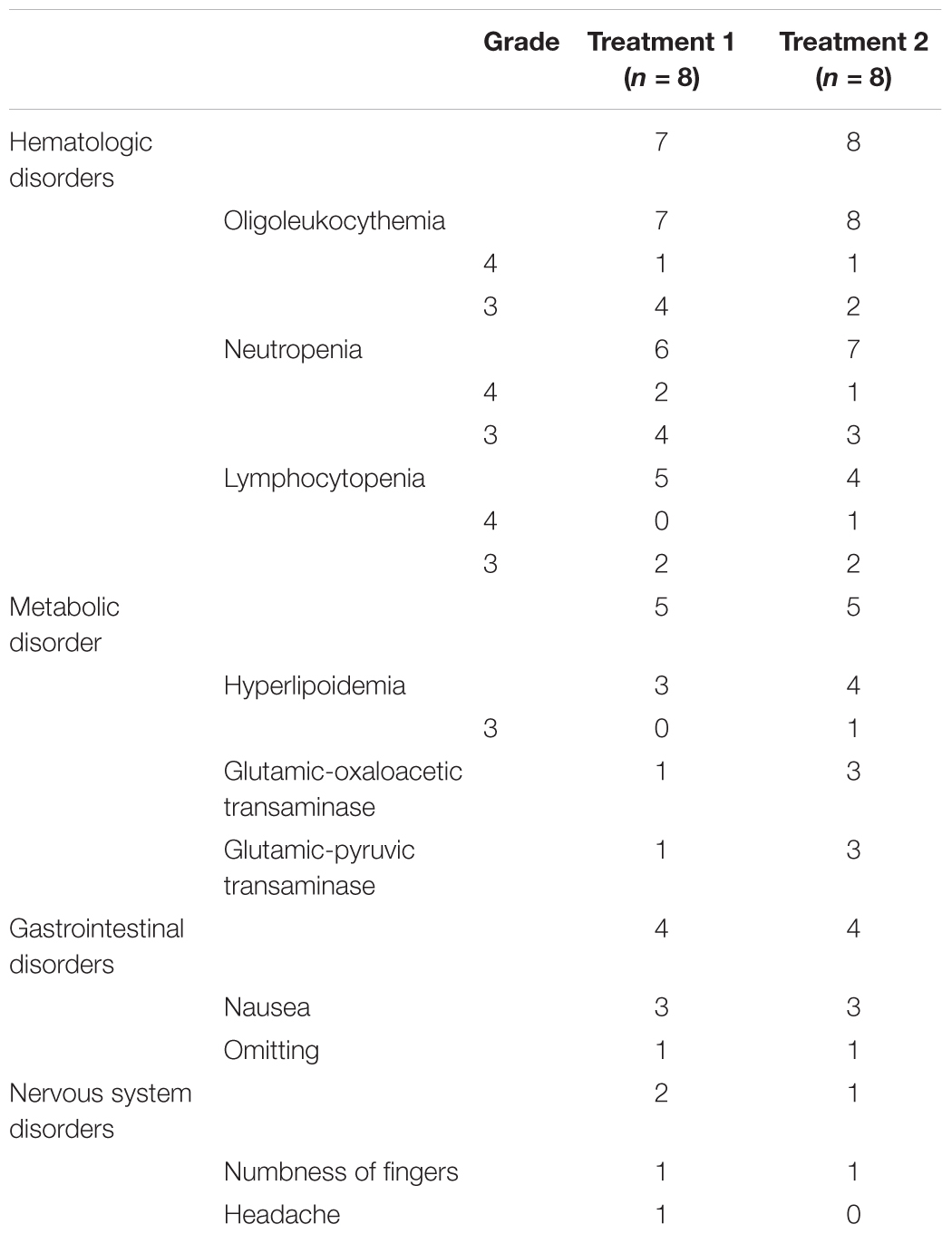
TABLE 4. Summary of adverse events (AEs) associated with VSLI or VSI reported by frequency.
Discussion
Cross-over design was used in order to minimize the variability between the groups. CHOP or modified CHOP scheme, instead of single-agent VSLI or VSI, was applied in this study considering the patient benefit. A conservative dose of 1.0 mg/m2 for VSLI was selected in this study, which is approximately 1.4-fold lower than the recommended dose for traditional VCR for safety reasons, because a new VSLI may lead to unpredictable toxicity owing to different pharmaceutical characteristics, though liposomal VCR seemed to exhibit reduced toxicity in previous clinical study.
The PK characteristic of F-VCR and the release properties of VCR from the liposome in human being were studied. The significant lower Cmax and AUC0-Inf of F-VCR demonstrated that majority of the VCR was encapsulated in the liposome and the VCR was released slowly from the liposome though the sudden release was observed reflected by multiple peaks of plasma-concentration profiles.
In this study, the higher Cmax values, longer circulation half-lives, and longer mean residence times observed with the T-VCR of VSLI, compared with VSI, were associated with a significantly higher plasma AUC values for T-VCR. This could be explained that VSLI decreases in plasma clearance rates and the volume of distribution compared with VSI. These data were very similar to those obtained from another study in China (Yan et al., 2012). In addition, the PK parameter values of T-VCR including Cmax, AUC0-Inf, CL and Vz from the present study were in consistent with those from the study mentioned above (Yan et al., 2012). However, the main PK parameters of T-VCR in this study had large difference from those reported in previous studies from United States (Bedikian et al., 2011) and Canada (Embree et al., 1998; Gelmon et al., 1999). The data, summarized in Table 5, showed that the dose-normalized Cmax and AUC0-Inf value from three international studies were similar and exceeded those from this current study by approximately 5- and 30-fold, respectively. Besides, compared to the current study, the Vz of T-VCR in two studies from United States (Bedikian et al., 2011) and Canada (Embree et al., 1998) reduced by 80- and 50-fold, respectively. The CL of T-VCR reduced by 24-fold in study from United States vs. this present study. The differences in pharmaceutical property of VSLI including the ingredient and particle size uniformity of liposome, or in the patients’ characteristics (race, cancer types and disease states) in the three international studies vs. the current study may be responsible for these inconsistencies. In addition, methodological differences used in different studies may contribute to the inconsistencies.
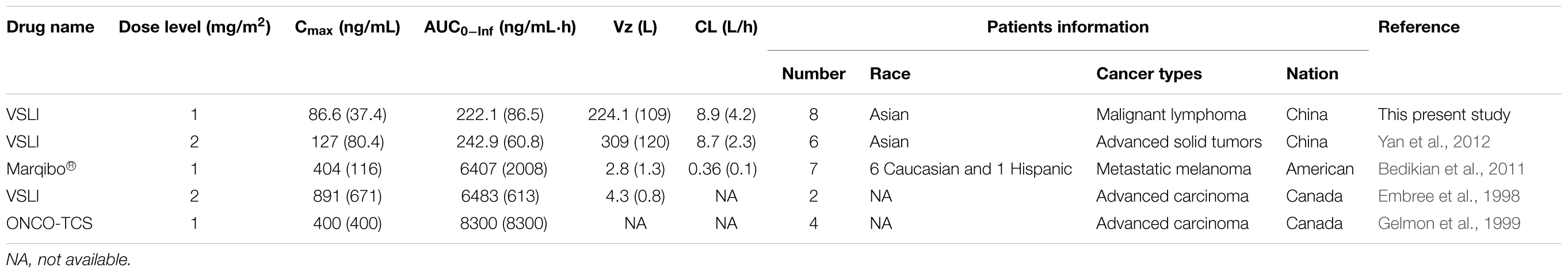
TABLE 5. PK parameters of T-VCR from different studies.
Two physical states of T-VCR exist in the circulation after VSLI administration including F-VCR (unentrapped and released from liposome) and liposome-entrapped VCR. It is theoretically agreed that F-VCR is responsible for causing toxicity and liposomal VCR accounts for providing the activity. From the safety perspective, it is essential to ensure that the plasma levels of F-VCR post injection of VSLI are comparable with or below those of VCR after treatment with VSI. Thus, it was anticipated that VCR is released from the liposome to increase the concentration of VCR in tumor site and to decrease the concentration of free drug in plasma, then the vast majority of drug present in circulation remains entrapped with liposomes. In order to confirm this hypothesis and better understand the release character of VCR from liposome of VSLI in human being, we developed an SPE method to separate the F-VCR from the liposomal VCR in plasma and the concentration of F-VCR was determined (Yang et al., 2013). Based on these data, we observed that the VCR was not released at constant speed because multiple peaks appeared in the plasma F-VCR concentration-time profile. While, the lower Cmax showed that the release rate of VCR from lipid carrier was slow and controllable in general, even though the sudden release existed to some degree. When comparing the AUC0-Inf value of F-VCR to that of T-VCR after administration with VSLI, the ratio at 29.2% indicated that there are only a small fraction of VCR released from liposome in the plasma. Furthermore, the lower F-VCR concentration in plasma for VSLI than that of VCR for VSI supplied the theoretical basis for the fact that VSLI lead to decreased toxicity compared to VSI, and this is consistent with the hypothesis.
It is expected that the excretion route of VCR released from liposome was similar to that of traditional VCR. The biliary system is the principal excretion route for traditional VCR (Jackson et al., 1978). To confirm it, we developed a simple method to determine the VCR concentration in urine and calculate the cumulative excretion percentage of VCR in urine (Yang et al., 2015). Less than 3% of the administered dose of VSLI was excreted from urinary post injection and the extent was similar to that for VSI. Nevertheless, both the time course and the elimination rate were very different. The elimination percentage of 40–21–14% for VSI changed to 6.2–24–39% for VSLI at intervals of 0–5, 5–13, and 13–25 h, respectively. Considering the fact that it is impossible for liposomes to be cleared via glomerular filtration due to the size, the elimination rate of VCR from urine should entirely be determined by the release rate of VCR from liposome. Hence, the gradual increasing of excretion rate for VSLI indicated that VCR was slowly released from liposome. In a word, the above data declared that the liposomal VCR did not change the elimination route of VCR, but the elimination rate was different because of the slow release of VCR from liposome.
Vincristine sulfate liposome injection was well tolerated, no serious AEs were reported and no patient was withdrawn due to an AE during the study. The profile and frequency of AE, as well as the numbers of the patients who experienced the grade ≥3 AE for VSLI were consistent with that for conventional VSI. No additional toxicity was observed even though the 2.8-times higher plasma AUC0-Inf of T-VCR for VSLI than that of VCR for VSI was obtained.
Conclusion
In summary, the high exposure of T-VCR and low exposure of F-VCR demonstrated that the proportion of F-VCR was very small and most of VCR was entrapped in liposome in circulation following injection of VSLI, which explained the fact that VSLI showed higher efficacy and lower toxicity than that for VSI. VCR was released from liposome slowly, which was reflected by the low plasma concentration of F-VCR and the delayed excretion of VCR in urine. Liposomal encapsulation of VCR did not alter the extent of VCR excretion in urine, but time course changed.
Ethics Statement
The phase Ia period (Clinical Trials.gov Identifier: 2009L01014) was conducted at three sites in China. It was approved by the Ethics Committee of Peking Union Medical College Hospital (Site 1, Permission Number: 1254), Nanjing First Hospital Affiliated of Nanjing Medical University (Site 2, Permission Number: 2011-MD- I22) and The Second Affiliated Hospital of Soochow University (Site 3, Permission Number: 2012-3), respectively. The study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The written Informed Consent Form was obtained from each subject prior to any study procedure.
Author Contributions
FY and MJ contributed equally to the study design, process of samples, data analysis, and article writing. ML assisted with the process of samples and data acquisition. PH was involved in the design of the study and article writing. JJ and HW were the principal investigators of this trial and were involved in the study design and analysis of data. All authors revised the article critically and gave final approval of the version to be published.
Funding
This work was supported by National Natural Science Foundation of China (No. 81602655) and “13th Five-Year” National Major New Drug Projects (No. 2018ZX09734006-001).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Shanghai Fudan-zhangjiang Bio-Pharmaceutical Co., Ltd., China for supplying VSLI.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00991/full#supplementary-material
FIGURE S1 | F-VCR concentration-time curve for subject 1.
FIGURE S2 | F-VCR concentration-time curve for subject 2.
FIGURE S3 | F-VCR concentration-time curve for subject 3.
FIGURE S4 | F-VCR concentration-time curve for subject 4.
FIGURE S5 | F-VCR concentration-time curve for subject 5.
FIGURE S6 | F-VCR concentration-time curve for subject 6.
FIGURE S7 | F-VCR concentration-time curve for subject 7.
FIGURE S8 | F-VCR concentration-time curve for subject 8.
Abbreviations
AE, adverse event; AUC0-Inf, area under plasma concentration-time curve from zero to infinity; Cmax, maximum concentration; CHOP, cytoxan (cyclophosphamide power injection), hydroxyrubicin (adriamycin power injection), oncovin (vincristine sulfate injection) and prednisone tablets; CL, clearance; CTCAE, Common Terminology Criteria for Adverse Events; F-VCR, free vincristine; IS, internal standard; I.V., intravenously; NCI, National Cancer Institute; PK, pharmacokinetic; SPE, solid phase extraction; T-VCR, total vincristine; UPLC-MS/MS, ultra-performance liquid chromatography-tandem mass spectrometry; VSI, vincristine sulfate injection; VSLI, vincristine sulfate liposomes injection; Vz, distribution volume; t1/2, elimination half-life.
References
Allen, T. M., Cheng, W. W., Hare, J. I., and Laginha, K. M. (2006). Pharmacokinetics and pharmacodynamics of lipidic nano-particles in cancer. Anticancer Agents Med. Chem. 6, 513–523. doi: 10.2174/187152006778699121
Bedikian, A. Y., Silverman, J. A., Papadopoulos, N. E., Kim, K. B., Hagey, A. E., Vardeleon, A., et al. (2011). Pharmacokinetics and safety of marqibo® (vincristine sulfate liposomes injection) in cancer patients with impaired liver function. J. Clin. Pharmacol. 51, 1205–1212. doi: 10.1177/0091270010381499
Bedikian, A. Y., Vardeleon, A., Smith, T., Campbell, S., and Namdari, R. (2006). Pharmacokinetics and urinary excretion of vincristine sulfate liposomes injection in metastatic melanoma patients. J. Clin. Pharmacol. 46, 727–737. doi: 10.1177/0091270006288953
Douer, D. (2016). Efficacy and safety of vincristine sulfate liposome injection in the treatment of adult acute lymphocytic leukemia. Oncologist 21, 840–847. doi: 10.1634/theoncologist.2015-0391
Drummond, D. C., Meyer, O., Hong, K., Kirpotin, D. B., and Papahadjopoulos, D. (1999). Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 51, 691–743.
Embree, L., Gelmon, K., Tolcher, A., Hudon, N., Heggie, J., Dedhar, C., et al. (1998). Pharmacokinetic behavior of vincristine sulfate following administration of vincristine sulfate liposome injection. Cancer Chemother. Pharmacol. 41, 347–352. doi: 10.1007/s002800050750
Gelmon, K. A., Tolcher, A., Diab, A. R., Bally, M. B., Embree, L., Hudon, N., et al. (1999). Phase I study of liposomal vincristine. J. Clin. Oncol. 17, 679–705. doi: 10.1200/JCO.1999.17.2.697
Gidding, C. E., Kellie, S. J., Kamps, W. A., and de Graaf, S. S. (1999). Vincristine revisited. Crit. Rev. Oncol. Hematol. 29, 267–287. doi: 10.1016/S1040-8428(98)00023-7
Hildebrand, J., Kenis, Y., Mubashir, B. A., and Bart, J. B. (1972). Vincristine neurotoxicity. N. Engl. J. Med. 287:517. doi: 10.1056/NEJM197209072871014
Horton, J. K., Houghton, P. J., and Houghton, J. A. (1988). Relationships between tumor responsiveness, vincristine pharmacokinetics and arrest of mitosis in human tumor xenografts. Biochem. Pharmacol. 37, 3995–4000. doi: 10.1016/0006-2952(88)90085-8
Jackson, D. V. Jr., Castle, M. C., and Bender, R. A. (1978). Biliary excretion of vincristine. Clin. Pharmacol. Ther. 24, 101–107. doi: 10.1002/cpt1978241101
Johnson, I. S., Armstrong, J. G., Gorman, M., and Burnett, J. P. Jr. (1963). The vinca alkaloids: a new class of oncolytic agents. Cancer Res. 23, 1390–1427.
Kanter, P. M., Klaich, G. M., Bullard, G. A., King, J. M., Bally, M. B., and Mayer, L. D. (1994). Liposome encapsulated vincristine: preclinical toxicologic and pharmacologic comparison with free vincristine and empty liposomes in mice, rats and dogs. Anticancer Drugs 5, 579–590. doi: 10.1097/00001813-199410000-00010
Krishna, R., Webb, M. S., St Onge, G., and Mayer, L. D. (2001). Liposomal and nonliposomal drug pharmacokinetics after administration of liposome-encapsulated vincristine and their contribution to drug tissue distribution properties. J. Pharmacol. Exp. Ther. 298, 1206–1212.
Legha, S. S. (1986). Vincristine neurotoxicity. pathophysiology and management. Med. Toxicol. 1, 421–427. doi: 10.1007/BF03259853
Owellen, R. J., Hartke, C. A., Dickerson, R. M., and Hains, F. O. (1976). Inhibition of tubulin-microtubule polymerization by drugs of the Vinca alkaloid class. Cancer Res. 36, 1499–1502.
Owellen, R. J., Owens, A. H. Jr., and Donigian, D. W. (1972). The binding of vincristine, vinblastine and colchicine to tubulin. Biochem. Biophys. Res. Commun. 47, 685–691. doi: 10.1016/0006-291X(72)90546-3
Rodriguez, M. A., Pytlik, R., Kozak, T., Chhanabhai, M., Gascoyne, R., Lu, B., et al. (2009). Vincristine sulfate liposomes injection (Marqibo®) in heavily pretreated patients with refractory aggressive non-hodgkin lymphoma: report of the pivotal phase 2 study. Cancer 115, 3475–3482. doi: 10.1002/cncr.24359
Schou, M., Amdisen, A., and Thomsen, K. (1968). Clinical and experimental observations concerning the absorption and elimination of lithium and on lithium poisoning. Acta Psychiatr. Scand. 203(Suppl. ), 153–155. doi: 10.1111/j.1600-0447.1968.tb01983.x
Shah, N. N., Cole, D. E., Lester-McCully, C. M., Wayne, A. S., Warren, K. E., and Widemann, B. C. (2016a). Plasma and cerebrospinal fluid pharmacokinetics of vincristine and vincristine sulfate liposomes injection (VSLI, Marqibo®(R)) after intravenous administration in non-human primates. Invest. New Drugs 34, 61–65. doi: 10.1007/s10637-015-0311-x
Shah, N. N., Merchant, M. S., Cole, D. E., Jayaprakash, N., Bernstein, D., Delbrook, C., et al. (2016b). Vincristine sulfate liposomes injection (VSLI, Marqibo®(R)): results from a phase I study in children, adolescents, and young adults with refractory solid tumors or leukemias. Pediatr. Blood Cancer 63, 997–1005. doi: 10.1002/pbc.25937
Silverman, J. A., and Deitcher, S. R. (2013). Marqibo®(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 71, 555–564. doi: 10.1007/s00280-012-2042-4
Silverman, J. A., Reynolds, L., and Deitcher, S. R. (2013). Pharmacokinetics and pharmacodynamics of vincristine sulfate liposome injection (VSLI) in adults with acute lymphoblastic leukemia. J. Clin. Pharmacol. 53, 1139–1145. doi: 10.1002/jcph.155
Thomas, D. A., Kantarjian, H. M., Stock, W., Heffner, L. T., Faderl, S., Garcia-Manero, G., et al. (2009). Phase 1 multicenter study of vincristine sulfate liposomes injection and dexamethasone in adults with relapsed or refractory acute lymphoblastic leukemia. Cancer 115, 5490–5498. doi: 10.1002/cncr.24632
Webb, M. S., Harasym, T. O., Masin, D., Bally, M. B., and Mayer, L. D. (1995). Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br. J. Cancer 72, 896–904. doi: 10.1038/bjc.1995.430
Webb, M. S., Logan, P., Kanter, P. M., St-Onge, G., Gelmon, K., Harasym, T., et al. (1998). Preclinical pharmacology, toxicology and efficacy of sphingomyelin/cholesterol liposomal vincristine for therapeutic treatment of cancer. Cancer Chemother. Pharmacol. 42, 461–470. doi: 10.1007/s002800050846
Yan, Z., Zhu, Z. L., Qian, Z. Z., Hu, G., Wang, H. Q., Liu, W. H., et al. (2012). Pharmacokinetic characteristics of vincristine sulfate liposomes in patients with advanced solid tumors. Acta Pharmacol. Sin. 33, 852–858. doi: 10.1038/aps.2012.44
Yang, F., Wang, H., Hu, P., and Jiang, J. (2015). Validation of an UPLC-MS-MS method for quantitative analysis of vincristine in human urine after intravenous administration of vincristine sulfate liposome injection. J. Chromatogr. Sci. 53, 974–978. doi: 10.1093/chromsci/bmu164
Yang, F., Wang, H., Liu, M., Hu, P., and Jiang, J. (2013). Determination of free and total vincristine in human plasma after intravenous administration of vincristine sulfate liposome injection using ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 1275, 61–69. doi: 10.1016/j.chroma.2012.12.026
Keywords: free vincristine, total vincristine, pharmacokinetics, vincristine sulfate liposome injection, urinary excretion, safety
Citation: Yang F, Jiang M, Lu M, Hu P, Wang H and Jiang J (2018) Pharmacokinetic Behavior of Vincristine and Safety Following Intravenous Administration of Vincristine Sulfate Liposome Injection in Chinese Patients With Malignant Lymphoma. Front. Pharmacol. 9:991. doi: 10.3389/fphar.2018.00991
Received: 04 May 2018; Accepted: 13 August 2018;
Published: 29 August 2018.
Edited by:
Yurong Lai, Gilead, United StatesReviewed by:
Cengiz Gokbulut, Balíkesir University, TurkeyZhihao Liu, University of Illinois at Chicago, United States
Copyright © 2018 Yang, Jiang, Lu, Hu, Wang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyun Wang, d2FuZ2h5QHB1bWNoLmNu Ji Jiang, cGsuZnJvc2hAZ21haWwuY29t
†These authors have contributed equally to this work