 Qing-Hua Liu1
Qing-Hua Liu1 Xi Qiao1Li-Jun Zhang1Jin Wang2Li Zhang3Xu-Wen Zhai4Xiao-Ze Ren2Yu Li5Xiao-Na Cao5Qi-Long Feng2Ji-Min Cao2*Bo-Wei Wu2*
Xi Qiao1Li-Jun Zhang1Jin Wang2Li Zhang3Xu-Wen Zhai4Xiao-Ze Ren2Yu Li5Xiao-Na Cao5Qi-Long Feng2Ji-Min Cao2*Bo-Wei Wu2*- 1Department of Pathophysiology, Shanxi Medical University, Taiyuan, China
- 2Key Laboratory of Cellular Physiology at Shanxi Medical University, Ministry of Education, and the Department of Physiology, Shanxi Medical University, Taiyuan, China
- 3Clinical Laboratory, Children’s Hospital of Shanxi, Taiyuan, China
- 4Clinical Skills Teaching Simulation Hospital, Shanxi Medical University, Taiyuan, China
- 5Department of Internal Medicine, The Hospital of Beijing Sports University, Beijing, China
Intracellular Ca2+ overload, prolongation of the action potential duration (APD), and downregulation of inward rectifier potassium (IK1) channel are hallmarks of electrical remodeling in cardiac hypertrophy and heart failure (HF). We hypothesized that enhancement of IK1 currents is a compensation for IK1 deficit and a novel modulation for cardiac Ca2+ homeostasis and pathological remodeling. In adult Sprague-Dawley (SD) rats in vivo, cardiac hypertrophy was induced by isoproterenol (Iso) injection (i.p., 3 mg/kg/d) for 3, 10, and 30 days. Neonatal rat ventricular myocytes (NRVMs) were isolated from 1 to 3 days SD rat pups and treated with 1 μmol/L Iso for 24 h in vitro. The effects of zacopride, a selective IK1/Kir2.1 channel agonist, on cardiac remodeling/hypertrophy were observed in the settings of 15 μg/kg in vivo and 1 μmol/L in vitro. After exposing to Iso for 3 days and 10 days, rat hearts showed distinct concentric hypertrophy and fibrosis and enhanced pumping function (P < 0.01 or P < 0.05), then progressed to dilatation and dysfunction post 30 days. Compared with the age-matched control, cardiomyocytes exhibited higher cytosolic Ca2+ (P < 0.01 or P < 0.05) and lower SR Ca2+ content (P < 0.01 or P < 0.05) all through 3, 10, and 30 days of Iso infusion. The expressions of Kir2.1 and SERCA2 were downregulated, while p-CaMKII, p-RyR2, and cleaved caspase-3 were upregulated. Iso-induced electrophysiological abnormalities were also manifested with resting potential (RP) depolarization (P < 0.01), APD prolongation (P < 0.01) in adult cardiomyocytes, and calcium overload in cultured NRVMs (P < 0.01). Zacopride treatment effectively retarded myocardial hypertrophy and fibrosis, preserved the expression of Kir2.1 and some key players in Ca2+ homeostasis, normalized the RP (P < 0.05), and abbreviated APD (P < 0.01), thus lowered cytosolic [Ca2 +]i (P < 0.01 or P < 0.05). IK1channel blocker BaCl2 or chloroquine largely reversed the cardioprotection of zacopride. We conclude that cardiac electrical remodeling is concurrent with structural remodeling. By enhancing cardiac IK1, zacopride prevents Iso-induced electrical remodeling around intracellular Ca2+ overload, thereby attenuates cardiac structural disorder and dysfunction. Early electrical interventions may provide protection on cardiac remodeling.
Introduction
Ventricular remodeling is characterized by myocardial hypertrophy and interstitial fibrosis in response to exercise or damage. It is a dynamic and time-dependent process. Physiological remodeling could improve pumping function by increasing the amount of contractile units and reducing the wall stress (Fedak et al., 2005). While maladaptive remodeling may lead to progressive ventricular dilatation, dysfunction, and even malignant arrhythmias (St John Sutton et al., 2003). Cardiac remodeling generally encompasses two components, structural remodeling, and electrical remodeling. The former exhibits hypertrophy, necrosis, apoptosis, as well as interstitial fibrosis, resulting in changes in heart size, shape, and mass (Tsukamoto et al., 2006; Ryan et al., 2007; Zhu et al., 2007; Li et al., 2009; Stewart et al., 2010). Electrical remodeling involves alterations in cardiac ion channels, exchangers, or pumps such as L-type calcium channels (LTCC), transient outward potassium channel, ATP-sensitive potassium channel (KATP), inward rectifier potassium channel (IK1), sodium-calcium exchanger (NCX), and sodium-potassium pump (Aimond et al., 1999; Long et al., 2015). Large-scale animal and clinical trials have confirmed that β-blockers, angiotensin-converting enzyme inhibitors (ACEI), angiotensin II receptor blockers (ARB), aldosterone antagonists, and endothelin receptor antagonists avail to limit ventricular dysfunction and remodeling (reviewed by Burchfield et al., 2013). However, the mortality associated with cardiac remodeling, heart failure (HF), and malignant arrhythmias remains high. It is crucial to identify new targets and develop effective therapies.
In some cases of cardiac diseases, electrical remodeling, such as alterations in ion channels or Ca2+ cycling, precedes the observed depression of mechanical performance, suggesting that amelioration of electrical remodeling might be an effective therapeutic strategy against HF (Houser and Margulies, 2003; Mueller et al., 2011). KATP is reportedly involved in ventricular remodeling, and KATP channel agonists exert beneficial effects on cardiac structural remodeling and dysfunction (Lee et al., 2008; Sun et al., 2015). IK1 and KATP channels are both members of inward rectifier potassium (Kir) channel family and are respectively constituted by Kir2.x and Kir6.x subunits (Hibino et al., 2010). Prolongation of the action potential duration (APD) and downregulation of IK1 channel are well documented hallmarks of electrical remodeling in HF (Janse, 2004). Inhibition of IK1 also contributes to APD prolongation. Besides, IK1 is reduced by elevated diastolic Ca2+ in HF (Fauconnier et al., 2005). Therefore, IK1 channel is probably involved in cardiac remodeling, and IK1 channel agonism or up-regulation may improve cardiac structure and dysfunction.
We previously reported a selective IK1/Kir2.1 channel agonist, namely, zacopride. In rat ventricular myocytes, zacopride significantly enhanced IK1 while with no effect on other ion channels, transporters, or pumps (Liu et al., 2012, Zhai et al., 2017). Liu et al. (2016) showed that zacopride inhibited maladaptive cardiac repair following myocardial infarction (MI), and this effect was mediated by the activation of IK1 channel. The present study was designed to demonstrate the potential effect of zacopride on isoproterenol (Iso)-induced ventricular remodeling and to clarify the interplay between electrical remodeling and structural remodeling around Ca2+ dyshomeostasis.
Materials and Methods
Animal and Ethical Approval
Sprague-Dawley (SD) rat pups (1–3 days old, both male and female) or adult male rats (2 months old) were provided by Laboratory Animal Research Center of Shanxi Medical University (Taiyuan, China). The adult rats were housed under standard conditions, room temperature 20–24°C, humidity 40–60%, 12:12 h light dark (LD) cycles with light intensity up to 200 lux and fed standard chow and water ad libitum. This study was carried out in accordance with the recommendations of the guidelines for the Care and Use of Laboratory Animals (NIH, revised 2011), Ethics Committee of Shanxi Medical University. The protocol was approved by the Ethics Committee of Shanxi Medical University.
Induction of Cardiac Hypertrophy and Failure by Isoproterenol
Cardiac hypertrophy and failure were induced by daily injection of isoproterenol (3mg/kg/d) for 3–30 days in rats in vivo and were evaluated by calculating the heart mass index (the ratio of heart weight/body weight or left ventricle (LV) weight/body weight), and by echocardiography, histology, confocal microscopy, patch clamp, and western blotting.
Experimental Protocol
Isoproterenol (Iso, Sigma) was administered by intraperitoneal injection (i.p.) once a day for 3, 10, and 30 days, respectively, to establish temporal cardiac remodeling. An experimental protocol scheme including grouping and treatments is shown in Figure 1, and more information about the experiments including treatments and animal numbers is shown in Table S1. Pharmacological treatments were as follows: Iso (3 mg/kg/day, i.p.), zacopride (IK1 agonist, 15 µg/kg/day, i.p.) (Tocris, England), chloroquine (IK1 antagonist, 7.5 µg/kg/day, i.p.) (Sigma, USA), RS23597-190 (5-HT4 receptor antagonist, 0.27 mg/kg/day, i.p.) (Tocris, England), and m-chlorophenylbiguanide (m-CPBG, 5-HT3 receptor agonist, 0.19 mg/kg/day, i.p.) (Tocris, England). Age-matched control rats were administered with the same volume of saline. The dose of zacopride and chloroquine were applied according to our previous study (Liu et al., 2016) and preliminary experiment.
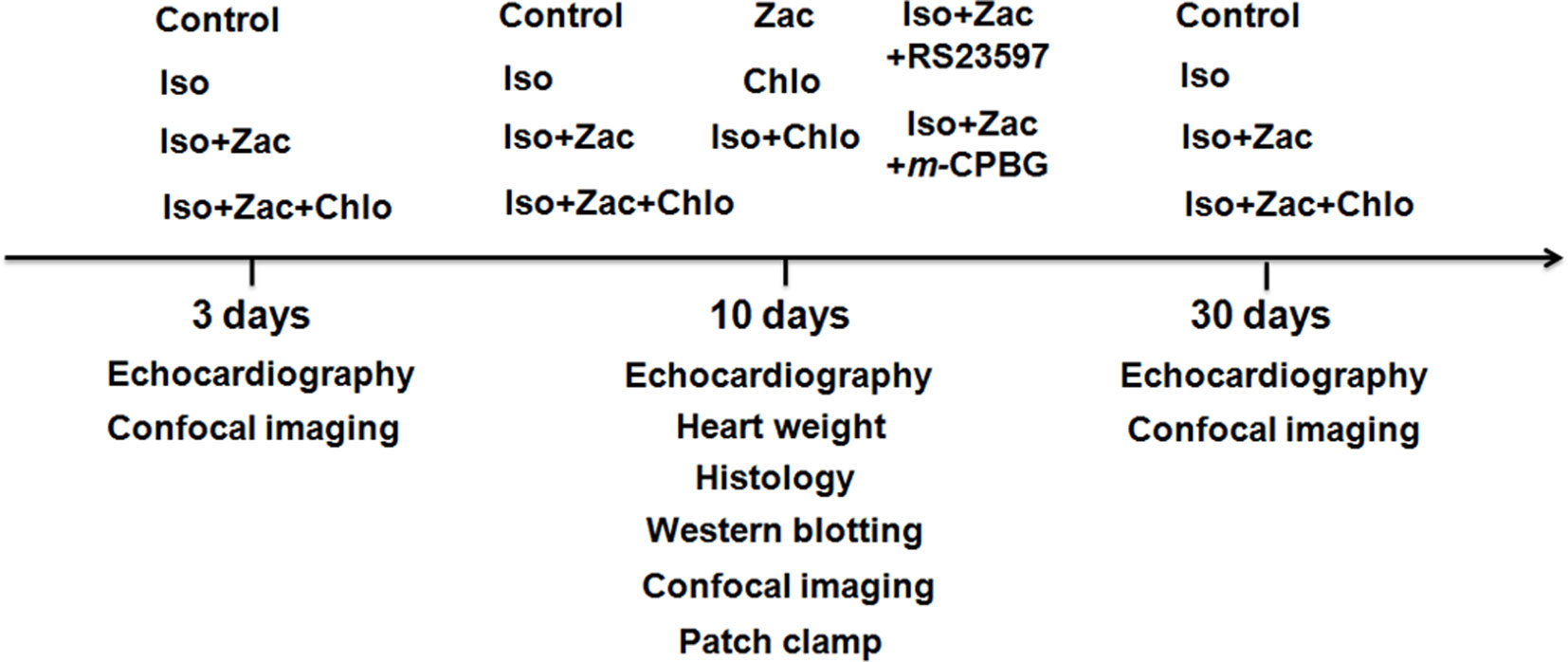
Figure 1 Schematic protocol of in vivo experiments. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine; RS23597, RS23597-190, an antagonist of 5-HT4 receptor. m-CPBG, m-chlorophenylbiguanide, an agonist of 5-HT3 receptor.
Echocardiography
The GE Vivid 7 Pro Ultrasound System (10 S probe, probe frequency 8.0 MHz, equipped with 2D strain imaging software and EchoPAC workstation) was used in M-mode for rodent hearts. Approximate exploration angle was at 15−30°, depth at 2−3 cm, frame rate > 250/s, and maximum frame rate up to 400/s. The positioning criterion was LV long-axis section. The measured parameters included LV dimensions at end diastole (LVIDd) and end systole (LVIDs), interventricular septum thickness at end diastole (IVSd) and end systole (IVSs), LV posterior wall thickness at end diastole (LVPWd) and end systole (LVPWs), and LV ejection fraction (EF) and LV short-axis fractional shortening (FS).
Histology
Samples of LV from all groups were fixed in 10% phosphate-buffered formalin and subjected to routine histological processing. Transverse LV sections (5 μm thick) were cut using a cryostat microtome (Leica, Wetzlar, Germany). After hematoxylin and eosin (HE) staining, the cross-sectional area of myofibers was measured using a microscope (Olympus, Tokyo, Japan) under a high-powered field (HPF) (×250 magnification). Fibrosis was evaluated by Masson’s trichrome staining, and the collagen content in the interstitial space was estimated by analyzing the images of each group. Total collagen area was calculated and expressed as percent of total ventricular area (Benjamin et al., 1989).
Western Blotting
Proteins from LV samples were loaded (40 μg per lane) on 5−15% acrylamide gels. After electrophoretic transfer and incubation with 5% non-fat milk in Tris-buffered saline (TBS), the nitrocellulose membranes were incubated overnight at 4°C with target-protein antibodies. Standard western blotting were performed using respective antibodies to quantify the relative levels of Kir2.1 (mouse monoclonal anti-Kir2.1, dilution 1:1,000, Sigma; or rabbit monoclonal anti-Kir2.1, 1:1,000, Abcam), CaMKII and phosphorylated CaMKII (rabbit monoclonal anti-CaMKII, 1:1,000, Cell Signaling), SERCA2 (rabbit polyclonal anti-SERCA2, 1:1,000, Cell Signaling), RyR2-phospho S2808 or total RyR2 (rabbit polyclonal anti-(p)-RyR2, 1:1,000, Abcam), cleaved caspase 3 (rabbit monoclonal anti-c-caspase 3, 1:1,000, Cell Signaling). The GAPDH (rabbit monoclonal anti-GAPDH, 1: 2,000, Sigma) was used as the loading control in each case. Quantification of bands was executed by ImageJ and Image Lab.
Isolation of Adult Rat Ventricular Myocytes (ARVMs)
LV myocytes from adult rats in control, Iso, Iso+Zac, and Iso+Zac+Chlo groups were respectively isolated using an enzymatic dissociation procedure. In brief, after anesthesia (sodium pentobarbital, 65 mg/kg, i.p.), the heart was quickly harvested and placed into chilled (4°C), oxygenated (100% O2), and Ca2+-free Tyrode’s solution and then was mounted onto a Langendorff retrograde perfusion apparatus via the aorta with a perfusion pressure of 80-cm H2O. The composition of Tyrode’s solution was (in mmol/L): NaCl 135.0, KCl 5.4, CaCl2 1.8, MgCl2 1.0, NaH2PO4 0.33, HEPES 10.0, and glucose 10.0 (pH 7.3−7.4 adjusted with NaOH). The heart was perfused first with oxygenated (100% O2) and Ca2+-free Tyrode’s solution at 37°C for 10 min, and then perfused with enzyme-containing Tyrode’s solution for about 20 min until the tissue was adequately digested. The enzyme-containing Tyrode’s solution was composed of (in mmol/L) NaCl 125.0, KCl 5.4, MgCl2 1.0, NaH2PO4 0.33, HEPES 10.0, glucose 10.0, taurine 20.0, and 5.0−8.0 mg/50 ml collagenase P (Roche, Switzerland). LV myocytes was then separated and stored in Krebs buffer (KB) solution at room temperature (25°C) at least 4 hours before use. The KB solution contained (in mmol/L): KOH 85.0, L-glutamic acid 50.0, KCl 30.0, MgCl2 1.0, KH2PO4 30.0, glucose 10.0, taurine 20.0, HEPES 10.0, and EGTA 0.5. The pH was adjusted to 7.4 with KOH.
Measurements of Cytosolic Ca2+ and SR Ca2+ Levels in ARVMs
The extracellular Ca2+ of ARVMs was recalcificated gradiently to 1.0 mmol/L with modified Tyrode’s solution. Cells from different groups were incubated with 5 μmol/L Fluo-4 AM (cytosolic Ca2+ indicator, Dojindo, Japan) and 5 μmol/L Fluo-5N/AM (SR Ca2+ indicator, Invitrogen, USA) respectively in fresh Tyrode’s solution (1.0 mmol/L Ca2+) supplemented with BSA (0.5%) at 37°C for 45 min. Unincorporated Fluo-4 or Fluo-5N was removed by washing myocytes thrice in modified Tyrode’s solution. The average intensity of Ca2+ fluorescence in cardiomyocytes was recorded using FV1000 laser confocal scanning microscope (Olympus, Japan).
Patch Clamp to Record Transmembrane Potential of Cardiomyocytes
To measure the resting potential (RP) and action potential (AP) of LV myocytes, Tyrode’s solution was used as the bath solution. The pipette solution contained (in mmol/L) KCl 150.0, MgCl2 1.0, EGTA 5.0, HEPES 5.0, and ATP-K2 3.0; pH was adjusted to 7.3 with KOH. Cells were superfused with bath solution at 36°C, and the perfusion flow rate was at 2 ml/min. Current clamp mode of whole-cell configuration was performed using Axopatch-200B patch clamp amplifier (Axon Instrument, USA). Patch electrodes were made by a two-stage vertical microelectrode puller (PP-83, Narishge Scientific Instrument, Japan) with resistance of 2−5 MΩ. The pClampex 8.2 program (Axon Instrument, USA) was utilized to produce clamping commands. The RP results were corrected for the calculated junction potential (–8 mV).
Isolation of Neonatal Rat Ventricular Myocytes (NRVMs)
NRVMs from 80 neonatal SD rats were isolated and cultured as previously described (Chlopcíková et al., 2001). Briefly, a combination of trypsin (0.08%, Sigma) and collagenase II (0.04%, Sigma) was used to dissociate the dissected pieces of ventricular tissues into single cells. The tissue pieces in enzyme solution were stirred gently for 6 min. The cell suspension was collected in 20% fetal bovine serum (FBS, Gibco), and the remaining tissue fragments were further digested by fresh enzyme solution. After 5−8 digestion cycles, all the supernatants containing isolated cells were collected and centrifuged at 4°C, 600 rpm for 6 min; washed once; and resuspended in DMEM culture medium containing 15% FBS. Non-myocytes were removed by differential adhesiveness, and cardiomyocytes were plated at a density of 2 × 105 viable cells in culture medium supplemented with 5-bromo-2-deoxyuridine (0.1 mmol/L, Sigma). Cultured neonatal cardiomyocytes were randomly separated into six groups: control, Iso (1 μmol/L), zacopride (1 μmol/L), Iso+zacopride, Iso+zacopride+BaCl2 (1 μmol/L), and Iso+zacopride +chloroquine (0.3 μmol/L). All the NRVMs were incubated for 24 h after drug treatment for further study.
Confocal Microscopy to Measure Intracellular [Ca2+] of NRVMs
NRVMs were incubated with 5 μmol/L Fluo-4 AM (Dojindo, Japan) in DMEM solution containing 0.5% BSA and 1 mM CaCl2 at 37°C for 0.5 h. Unincorporated Fluo-4 AM was removed by washing myocytes twice in PBS. The cell surface area and the intensity of [Ca2+]i fluorescence in cardiomyocytes were recorded using FV1000 laser confocal scanning microscope (Olympus, Japan).
Flow Cytometry to Measure Apoptosis of NRVMs
Flow cytometry was performed with propidium iodide (PI) and fluorescein isothiocyanate (FITC)–labeled Annexin V (KeyGEN Biotech, Nanjing, China). In brief, the NRVMs were treated for 24 h with different drugs as described in the grouping, then were harvested, rinsed twice with cold PBS, resuspended in binding buffer at the density of 1 × 106 cells/ml, and incubated with 5 μmol/L FITC-Annexin V and 5 μmol/L PI. Cells were gently vortexed and incubated in the dark for 15 min at room temperature. Flow cytometry was performed within 1 h using a FC500 Flow Cytometer (Coulter, Beckman, Palo Alto, CA, USA).
Statistical Analysis
All statistical analyses were performed using SPSS statistics software version 17.0 (IBM Corp, Chicago, IL, United States). Data were expressed as the mean ± standard error (SEM). Quantitative data were analyzed by one-way ANOVA. Multiple comparisons were performed using the least significant different test. Differences in mortality among groups were analyzed using chi-square test. P < 0.05 was considered statistically significant.
Results
The Mortality Rate of Iso-Modeled Rats
As shown in Table 1, daily Iso infusion for 3 d, 10 d, and 30 d led to sudden death in some rats. The mortalities were 40%, 41.7% (P < 0.01 vs. control), and 50% (P < 0.05 vs. control), respectively. In zacopride-treated groups, the mortalities were 16.7%, 23.8%, and 30.0% for 3, 10, and 30 days, respectively, but did not reach a statistical significance compared with the Iso group. Co-application of zacopride and chloroquine (IK1 channel blocker) increased the mortalities respectively to 30% (3 days), 37.5% (10 days, P < 0.01 vs. control), and 58.3% (30 days, P < 0.05 vs. control). All control rats, rats treated with zacopride alone or chloroquine alone, survived in the entire period of the experiments. These results indicate that 15 µg/kg zacopride and 7.5 µg/kg chloroquine per se had no significant toxicity to the rats.
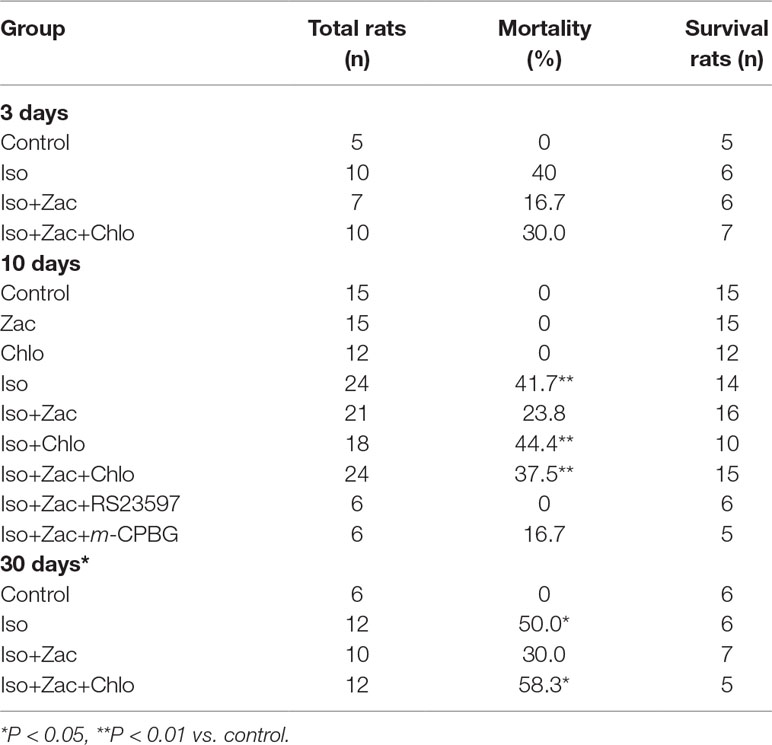
Table 1 The mortalities of rats in a time-course study in vivo.
Morphological Features of Iso-Induced Cardiac Hypertrophy in Vivo
The gross morphology of the whole heart (Figure 2A) revealed cardiac enlargement 10 days after daily Iso exposure. Echocardiographic detection demonstrated distinct characteristics of concentric hypertrophy and enhanced pumping function (Figure 2B and Table 2). To conform the results obtained from echocardiography, histological examination was performed by HE (Figure 2C) and Masson’s trichrome staining (Figure 2D). As shown in Figures 2C and E, compared with controls, the cardiac myofibers in Iso-treated rats are disorganized and hypertrophic, with certain degree of cell necrosis and relatively light staining of the cytoplasm. In zacopride-treated rats, cardiac myofibers are better arranged and with normal size. IK1 channel blocker chloroquine abolished these protective effects from zacopride as shown by worsened manifestation, indicating that the anti-remodeling effect of zacopride is mediated by the activation of IK1 channels. After 10 days of Iso infusion, rat hearts exhibited significant fibrosis, validated by increased collagen deposition (Figures 2D, F P < 0.01 vs. control). Zacopride treatment dramatically attenuated the fibrosis (P < 0.01), and this effect was largely abolished by chloroquine (P < 0.01). Iso-induced cardiac hypertrophy was further measured by heart mass index. The whole-heart weight and LV weight, normalized to body weights (Figure 2G), were greater in Iso-treated rats than controls (P < 0.01 vs. control). Zacopride treatment effectively prevented Iso-induced cardiac hypertrophy (P < 0.01), and the effect was attenuated by chloroquine (P < 0.01).
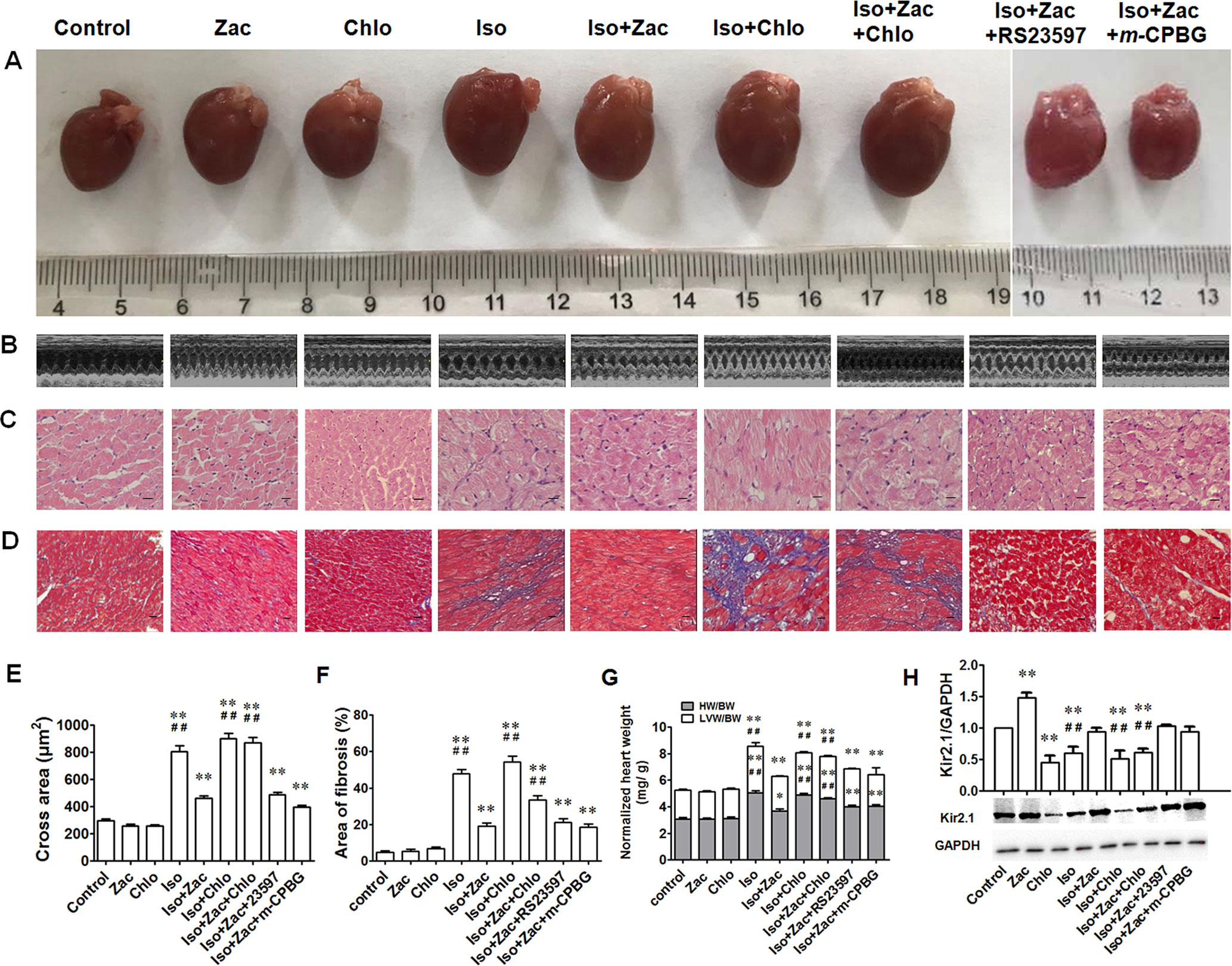
Figure 2 Cardiac remodeling induced by 10 days of Iso exposure in rats in vivo. (A) The gross morphology of the whole hearts. (B) Representative echocardiographic images from each of the corresponding hearts shown in (A). (C) HE staining of transverse LV sections (250×). Scale bars = 50 µm. (D) Masson’s trichrome staining showing collagen deposition in rat LV (100 ×). Cardiomyocytes and collagen fibers were stained as red and blue, respectively. Scale bars = 100 µm. (E) Statistical results of the cross sectional area of myofiber in different groups. N = 50 in each group. (F) Statistical results of fibrotic area which was expressed as the percentage of total area in each field, n = 5 in each group. (G) The heart/LV mass index. (H) The expression levels of Kir2.1 channel protein. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine; RS23597, RS23597-190; m-CPBG, m-chlorophenylbiguanide. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 compared with control. ##P < 0.01 compared with Iso+Zac.
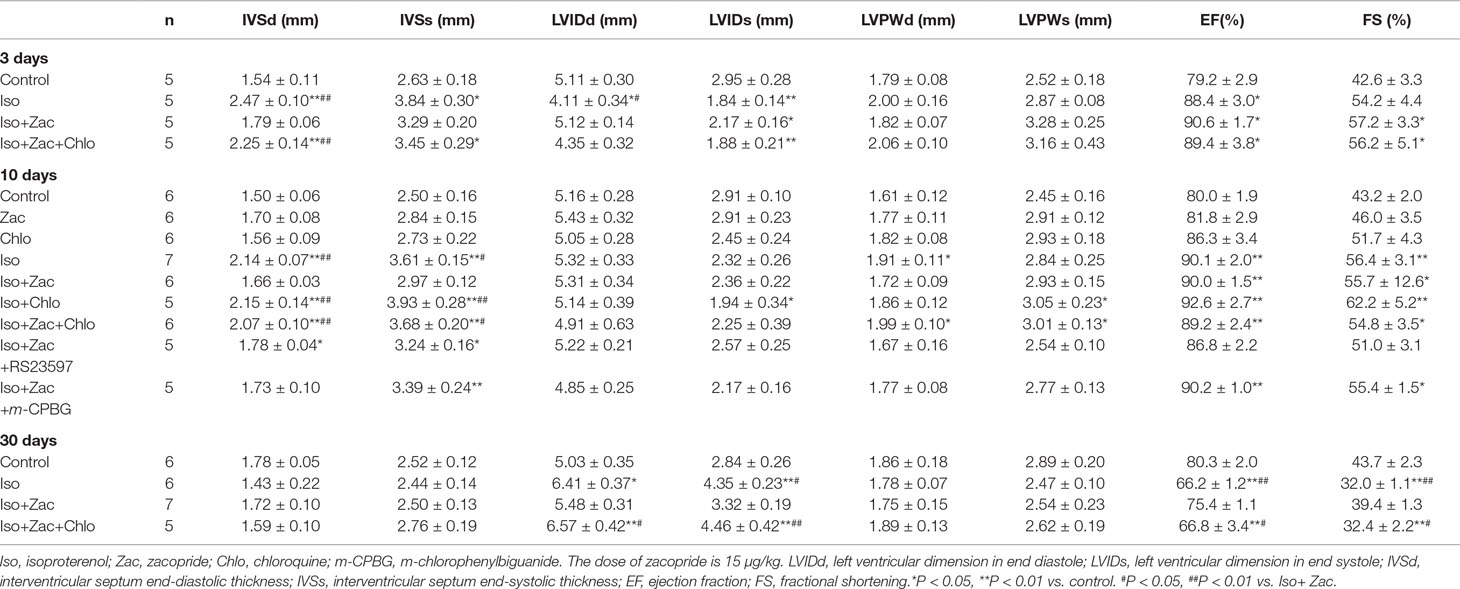
Table 2 Echocardiographic parameters in Iso-induced cardiac remodeling (mean ± SEM).
Zacopride (15 µg/kg) or chloroquine (7.5 µg/kg) per se had no significant effects on cardiac structure or function. Because zacopride is also a known 5-HT4 receptor agonist and 5-HT3 receptor antagonist, we examined whether 5-HT receptors are involved in the anti-remodeling effect using pharmacological tools, 5-HT4 receptor antagonist RS23597-190, and 5-HT3 receptor agonist m-CPBG. Results showed that RS23597-190 and m-CPBG could not counteract the effects of zacopride on cardiac remodeling including changes of hypertrophy and fibrosis (Table 2, Figures 2A–G), suggesting that the protective effects of zacopride on LV remodeling is mediated by IK1 channel but not by 5-HT receptors.
The Expression of Kir2.1 Channel Protein in Iso-Induced Hypertrophic LV
The native IK1 channels in the heart are assembled by Kir2.1 (KCNJ2), Kir2.2 (KCNJ12), and Kir2.3 (KCNJ4) channels. We have proven that Kir2.1 is the dominant isoform in rat ventricles and zacopride is a selective Kir2.1 channel agonist (Zhang et al., 2013). Compared with control, zacopride treatment (15 µg/kg/d) alone for 10 days upregulated Kir2.1 channel protein by 48.6% (P < 0.01), while chloroquine (7.5 µg/kg/d) treatment alone for 10 days decreased Kir2.1 channel protein level by 44.4% (P < 0.01) (Figure 2H). Consistently, zacopride prevented Iso-induced IK1 inhibition (P < 0.01), and this effect could be reversed by chloroquine (P < 0.01). Coapplication of RS23597-190 (5-HT4R antagonist) or m-CPBG (5-HT3R agonist) could not counteract the effect of zacopride on Kir2.1 expression (Figure 2H).
The Temporal Significance of Zacopride on Iso-Induced Cardiac Remodeling, Dysfunction, and Ca2+ Dyshomeostasis
Echocardiography Revealed the Protective Effects of Zacopride on Iso-Induced Cardiac Structural Remodeling and Dysfunction in Vivo
To elucidate the precise relationship between electrical remodeling and structural remodeling in a temporal sense, we observed the development of cardiac remodeling in the period of 3, 10, and 30 days of Iso infusion in vivo. Post 3 days of Iso infusion, IVSd, and IVSs were increased (P < 0.01); LVIDd (P < 0.05) and LVIDs (P < 0.01) were reduced compared with age-matched control rats (Table 2 and Figures 3A−B). Ten days after Iso treatment, IVSd (P < 0.01), IVSs (P < 0.01), and LVPWd (P < 0.05) were increased, while LVIDs and LVIDd had no significant differences compared with age-matched control. LVEF and LVFS were increased both in 3 days (P < 0.05) and 10 days (P < 0.01) of Iso groups compared with age-matched controls. These results indicated that Iso-induced cardiac remodeling occurred much early and was characterized by concentric hypertrophy and enhanced pumping function. Zacopride treatment prevented the thickening of interventricular septum and the decrease of LV volume (P < 0.01 or P < 0.05). 30 days after Iso treatment, LVIDd (P < 0.05) and LVIDs (P < 0.01) were significantly increased; LVEF and LVFS (P < 0.01) were decreased compared with age-matched control. The thickness of IVS was decreased compared with that in 3 and 10 days of Iso groups (P < 0.01). Collectively, these results indicated that longer (30 days) Iso exposure led to progression of cardiac pathological remodeling, dysfunctions, and even failure. Zacopride treatment prevented LV chamber dilatation (P < 0.05) and preserved the systolic function (P < 0.01). These effects were largely reversed by IK1 channel antagonist chloroquine (P < 0.01 or P < 0.05).
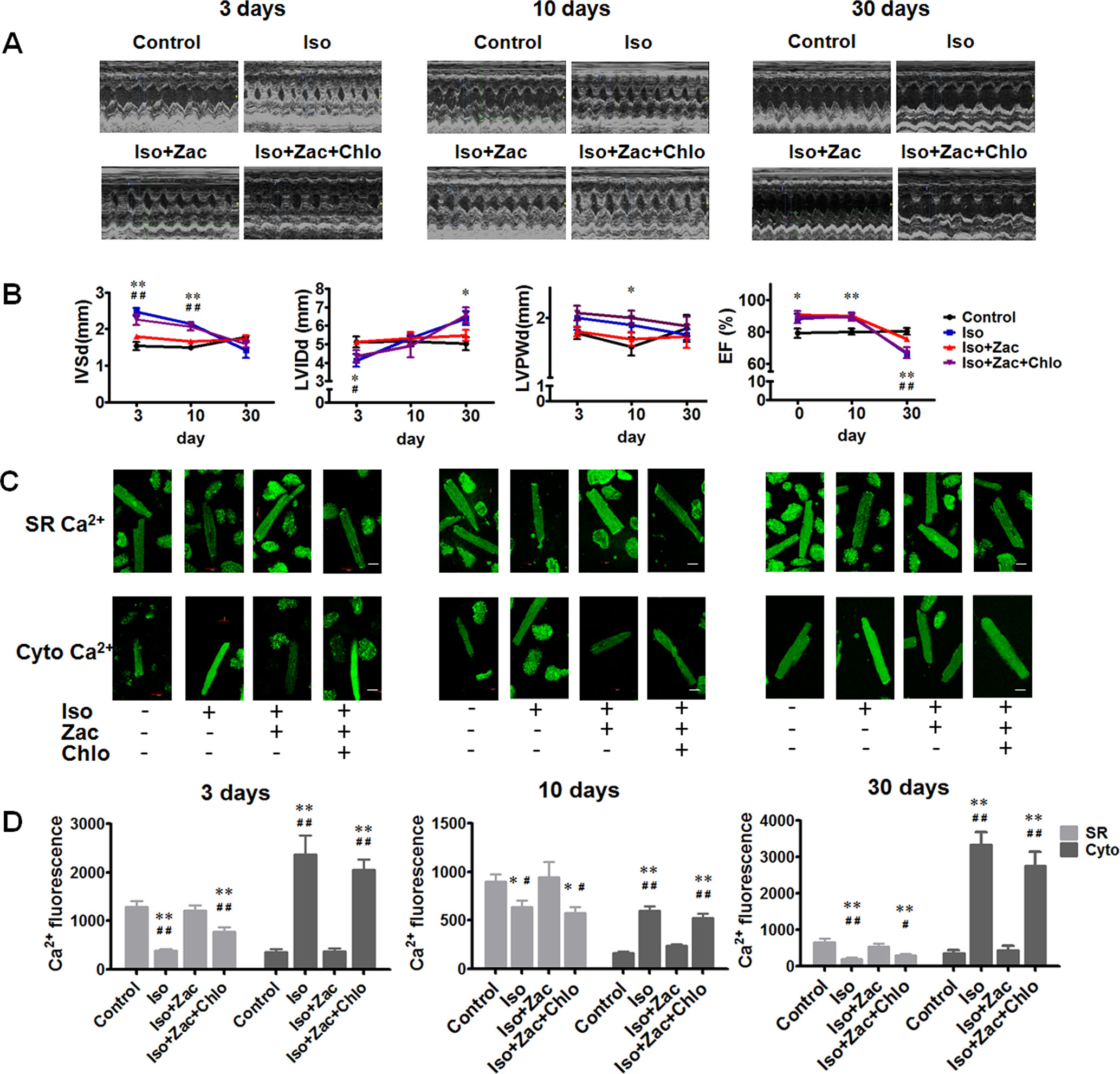
Figure 3 Time courses of structural remodeling and electrical remodeling 3 days, 10 days, and 30 days post-Iso infusion. (A) Representative echocardiographic images of the corresponding hearts. (B) Time courses of IVSd, LVIDd, LVPWd, and EF changes post-Iso toxication. *P < 0.05, **P < 0.01 for Iso compared with age-matched control. #P < 0.05, ##P < 0.01 for Iso compared with age-matched Iso+Zac group. (C) Cytosolic Ca2+ and SR Ca2+ fluorescences measured using laser scanning confocal microscopy. Upper row, Fluo-5N cellular distribution indicating SR Ca2+. Lower row, Fluo-4 cellular distribution indicating cytosolic Ca2+. Scale bars = 20 µm. (D) Statistical summary of cytosolic Ca2+ and SR Ca2+ levels of 3 days, 10 days, and 30 days post-Iso exposure, respectively. SR, sarcoplasmic reticulum. LVIDd, left ventricular dimension in end diastole. IVSd, interventricular septum end-diastolic thickness. LVPWd, LV posterior wall thickness at end diastole; EF, ejection fraction; Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine; RS23597, RS23597-190; m-CPBG, m-chlorophenylbiguanide. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 compared with age-matched control. #P < 0.05, ##P < 0.01 compared with age-matched Iso+Zac.
Confocal Microscopy Exhibited the Beneficial Effect of Zacopride on Iso-Induced Ca2+ Dyshomeostasis in Cardiomyocytes in Vitro
It is known that more than 90% of the Ca2+ is cycled between the cytosol and the SR in the rat (Bers, 2002). To observe the Ca2+ homeostasis in cardiomyocytes, we used Fluo-4 AM to quantify the cytosolic [Ca2+] and Fluo-5N AM as SR Ca2+ indicator. Figure 3C shows representative confocal images for cytosolic Ca2+ and SR Ca2+ fluorescence. Compared with the age-matched control, cardiomyocytes from the Iso-treated rat hearts exhibited higher cytosolic Ca2+ (P < 0.01) and lower SR Ca2+ content (P < 0.01 or P < 0.05) at all three observing time points (3, 10, and 30 days post-Iso daily infusion) (Figure 3D), and the Ca2+ dyshomeostasis was concurrent with cardiac structural and functional remodeling as shown above. Zacopride pretreatment prevented Iso-induced intracellular calcium overload and the decrease of SR Ca2+ content, and the effects were largely reversed by chloroquine (P < 0.05 or P < 0.01).
Western Blotting Demonstrated the Protective Effect of Zacopride on Iso-Induced Cardiomyocyte Ca2+ Dyshomeostasis in Vivo
In vivo experiments show that treatment with Iso for 3, 10, and 30 days all decreased the expression of Kir2.1 in cardiomyocytes (Figures 4A, B) (P < 0.01 vs. control). Concurrently, the phosphorylated CaMKII (Figure 4A, C) and RyR2 protein levels (Figure 4A, D) were progressively elevated (P < 0.05 or P < 0.01 vs. control). SERCA 2 protein (Figure 4A, E) did not change during short-term (3 days) Iso challenge, but was downregulated in 10 days and 30 days of Iso groups (P < 0.01). Iso also activated caspase 3 (Figure 4A, F) as indicated by elevation of cleaved caspase 3 (P < 0.05 or P < 0.01 vs. control) along with the progression of cardiac remodeling. These alterations were largely normalized by zacopride treatment (P < 0.05 or P < 0.01), and lower dose chloroquine reversed the effects of zacopride (P < 0.05 or P < 0.01).
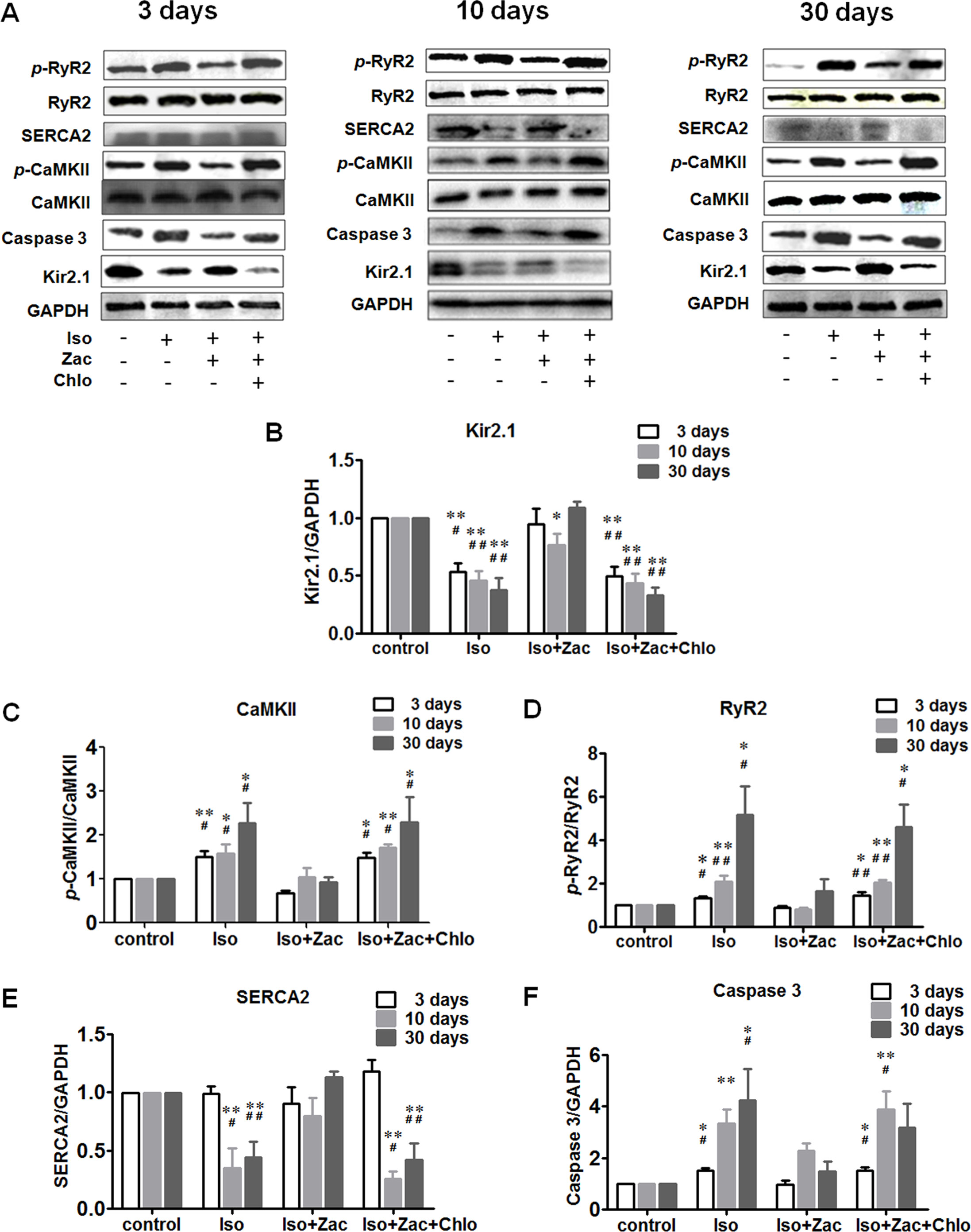
Figure 4 Zacopride improved Iso-induced maladaptive protein alterations in rat hearts. (A) Representative western blotting images 3 days, 10 days, and 30 days after the onset of Iso infusion. (B) Dynamic changes of Kir2.1 protein expression levels. (C) Dynamic changes of p-CaMKII protein expression levels relative to total CaMKII. (D) Dynamic changes in p-RyR2 protein expression levels relative to total RyR2. (E) Dynamic changes in SERCA2 protein expression. (F) Dynamic changes of cleaved caspase 3 protein expression. All data were normalized to control. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine. Data were presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01 compared with control. #P < 0.05, ##P < 0.01 compared with Iso+Zac.
Patch Clamp Experiments Showed the Suppressing Effect of Zacopride on Iso-Induced Changes of Transmembrane Potentials in ARVMs in Vitro
Iso treatment for 10 days induced significant depolarization of RP (P < 0.01 vs. control) and prolongations of APD50 (P < 0.01 vs. control) and APD90 (P < 0.01 vs. control). Zacopride treatment restored the RP depolarization (P < 0.05) and prolongations of APD50 (P < 0.01) and APD90 (P < 0.01) to normal or near normal levels. These effects were largely abolished by chloroquine (P < 0.01) (Figure 5, Table 3).
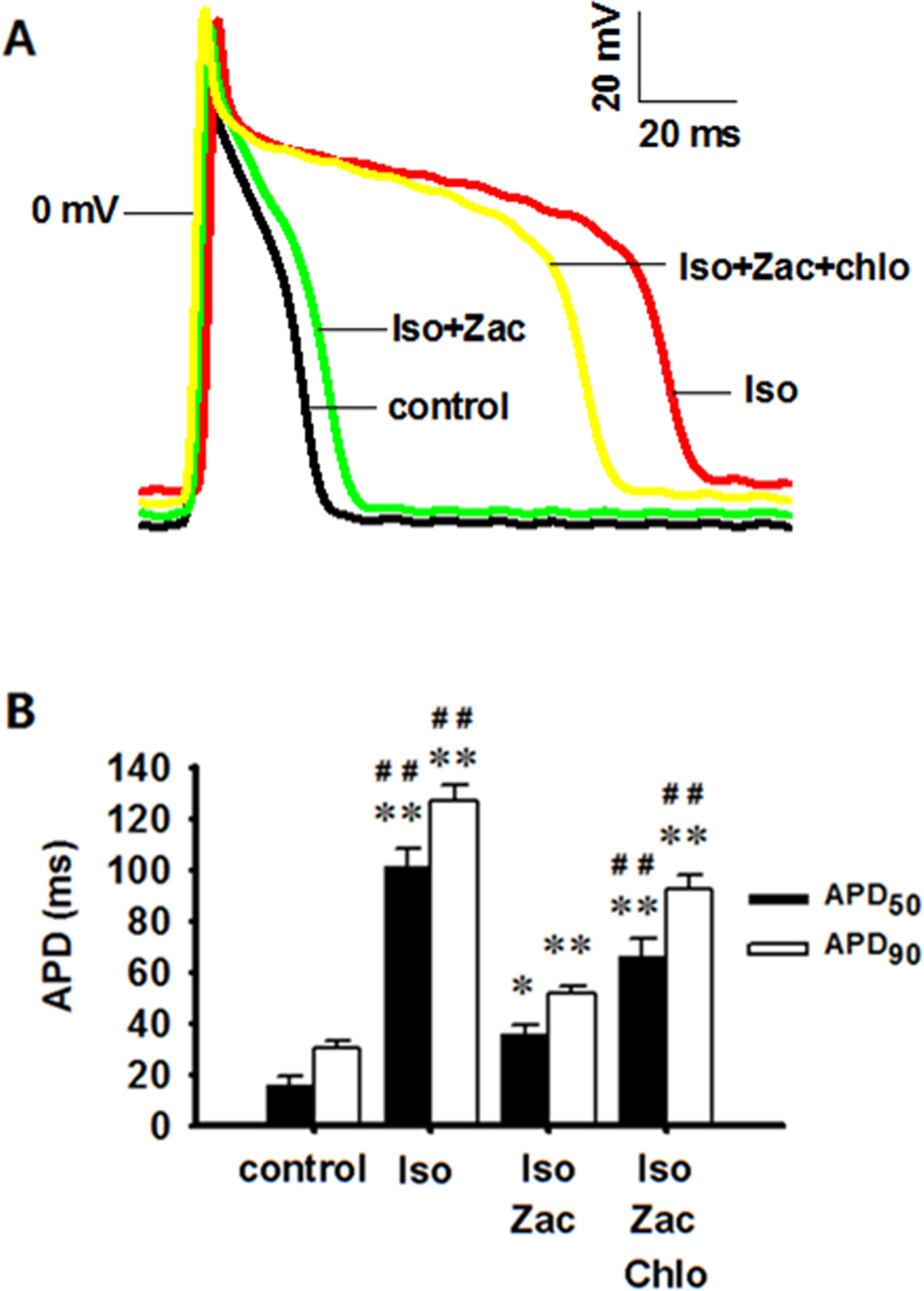
Figure 5 Zacopride restored RP depolarization and APD prolongation in isolated rat ventricular myocytes 10 days post-Iso infusion. These effects could be partially reversed by chloroquine. (A) Representative AP recording. (B) APD50 and APD90 in different groups. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine. N = 6 cells. Data were presented as mean ± SEM. *P < 0.05, **P < 0.01 compared with control. ##P < 0.01 compared with Iso+Zac.

Table 3 The effects of zacopride on the morphology of action potential (AP).
Zacopride Prevented Iso-Induced Hypertrophy and Intracellular Ca2+ Overload in NRVMs in Vitro
Iso at 1 μmol/L induced hypertrophy of cultured neonatal cardiomyocytes as evidenced by enlarged cell area and higher [Ca2 +]i (P < 0.01) (Figure 6). Zacopride treatment restored cell morphology to normal or near normal levels (Figure 6B). This in vitro result was consistent with that from ARVMs. Confocal detection indicates that zacopride significantly attenuated Iso-induced calcium overload (P < 0.01) (Figure 6C). This effect was reversed by IK1 channel blockers BaCl2 or chloroquine (P < 0.01). Notably, zacopride had no effect on [Ca2 +]i in normal NRVMs, suggesting that suppression of Ca2+ overload by zacopride may contribute to the protective effect of zacopride on cardiac remodeling.
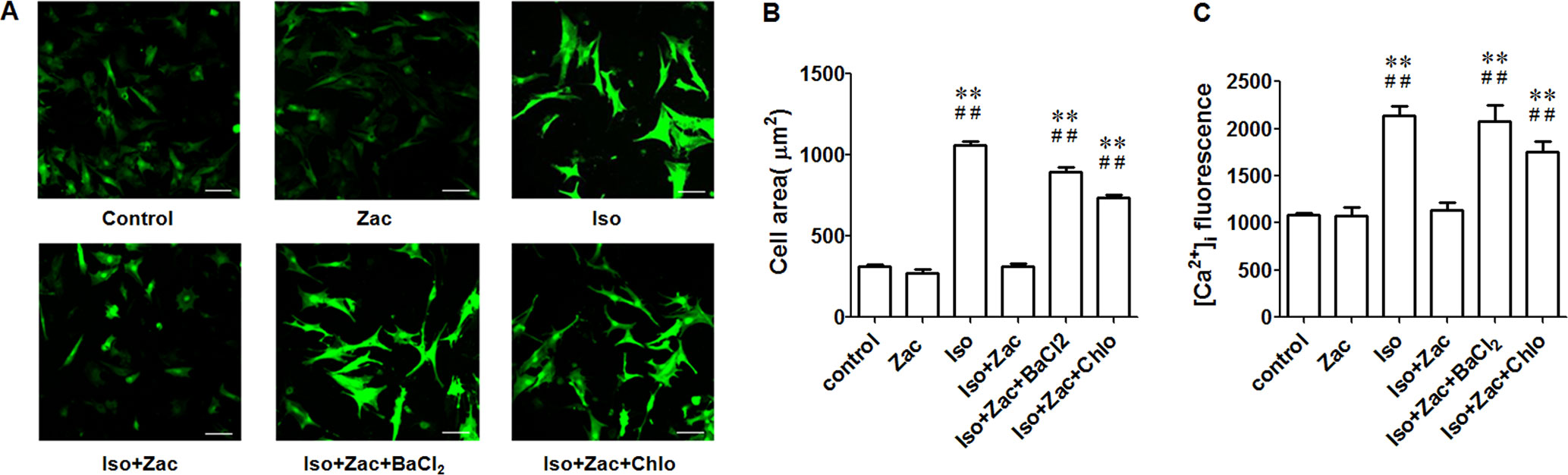
Figure 6 Zacopride inhibited Iso-induced hypertrophy and intracellular calcium overload in cultured NRVMs. (A) Fluorescent images of Fluo-4/AM loaded neonatal cardiomyocytes. Bar = 50 μm. (B) Zacopride treatment normalized the cell size of NRVMs. (C) Zacopride treatment attenuated Iso-induced intracellular calcium overload. BaCl2 or chloroquine reversed the effects of zacopride on cell sizes (B) and [Ca2+]i(C). Zacopride had no effect on [Ca2+]i in normal NRVMs. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine. N = 6 cells. Data were presented as mean ± SEM. **P < 0.01 compared with control; ##P < 0.01 compared with Iso+Zac.
Zacopride Inhibited Iso-Induced Apoptosis of NRVMs
Annexin V-FITC/PI staining was performed to detect early-stage apoptosis of cells. The apoptosis in Iso-incubated NRVMs was significantly higher than that in controls (13.7 ± 1.0% vs. 3.8 ± 0.6%, P < 0.01) (Figure 7). Zacopride decreased the apoptosis to 5.6 ± 0.2% (P < 0.01). IK1 channel blocker BaCl2 (P < 0.01) or chloroquine (P < 0.05) reversed the effects of zacopride, indicating that the anti-apoptotic effect of zacopride was mediated by IK1 channel activation.
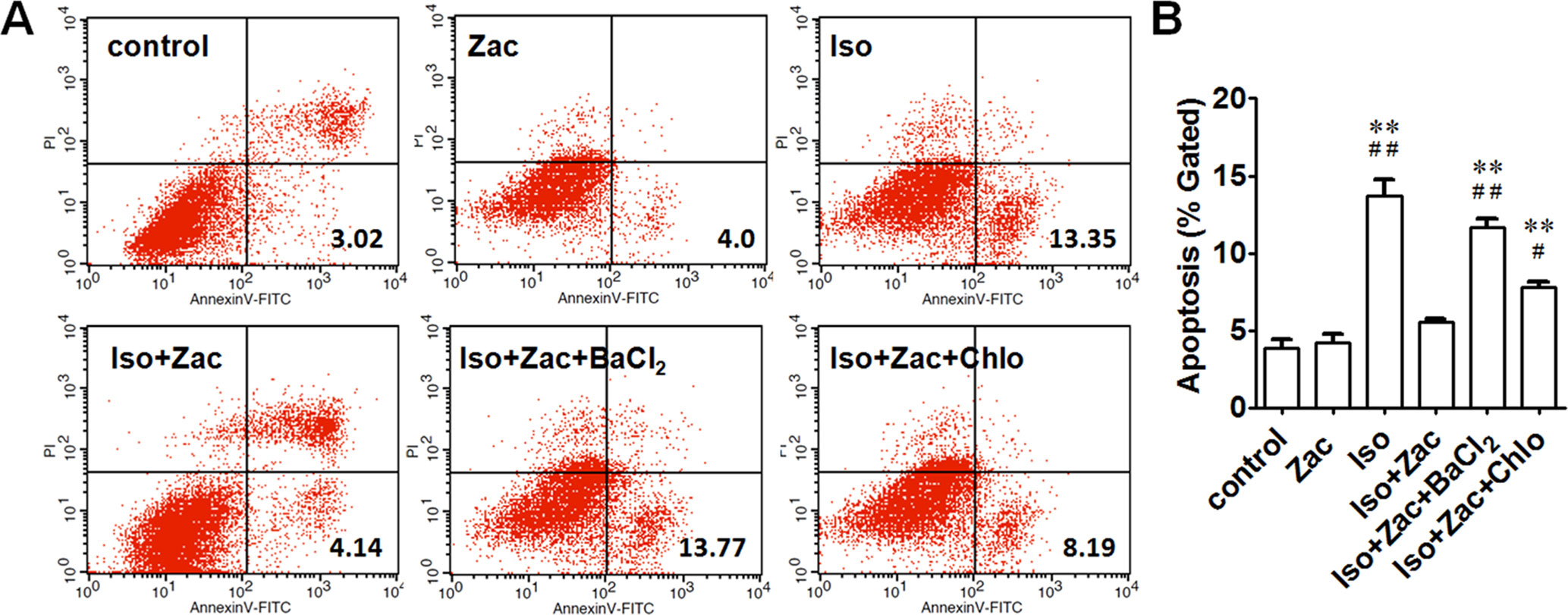
Figure 7 Apoptosis of NRVMs following exposure to 1 μmol/L Iso for 24 h. (A) Cells in the lower right (LR) quarter represents early apoptosis. (B) Zacopride protected cardiomyocytes from Iso-induced apoptosis. IK1 channel blocker BaCl2 or chloroquine reversed the effects of Zac. Iso, isoproterenol; Zac, zacopride; Chlo, chloroquine. N = 5 parallel samples in each group. Data were presented as means ± SEM. **P < 0.01, compared with control. #P < 0.05, ##P < 0.01, compared with Iso+Zac.
Discussion
A variety of diseases, including hypertension, coronary heart disease, hereditary defect, and toxic insults may all cause cardiac remodeling. End-stage remodeling is a major contributor in the development of HF. Currently, the principles for improving cardiac remodeling mainly involve the reduction in cardiac workload, improvement of myocardial systolic and diastolic functions, and inhibition of certain gene expression and release of humoral factors that induce cardiac hypertrophy and fibrosis (Hellawell and Margulies, 2012). Although significant progress has been achieved in recent years, the mortality among patients with HF remains high. Therefore, elucidating the molecular mechanisms underlying cardiac remodeling and HF and identifying novel therapeutic strategies to prevent or reverse cardiac remodeling are still important issues in cardiovascular research. The main findings of the present study are that 1) electrical remodeling is concurrent with structural remodeling, and they may not be two independent processes, but are two circumstances of the same scenario. Consequently, reversing electrical disorder might facilitate the improvement of cardiac structure and function; 2) modulation of the function and expression of IK1/Kir2.1 channel might be a novel strategy for handling intracellular Ca2+; 3) IK1 channel is a promising target for the treatment of cardiac remodeling in future clinical interventions.
Enhancing IK1 Prevented Iso-Induced Intracellular Calcium Overload
Ca2+ plays pivotal roles in myocardial excitation-contraction coupling, substance metabolism, cell cycle regulation, cell–cell communication, and gene expression (Aiba and Tomaselli, 2010). Intracellular Ca2+ homeostasis is maintained by coordination between ATP-dependent ion pumps and transporters located in plasma membranes or organelles, as well as Ca2+-binding proteins (Carafoli et al., 2001; Muth et al., 2001). Calcium dyshomeostasis, especially pathologic elevation of intracellular Ca2+ (Ca2+ overload), is a central event during the development of hypertrophy and HF (Molkentin et al., 1998; Zhang and Brown, 2004). Beta-adrenergic receptor (β-AR) stimulation (such as by Iso) activates the cAMP-dependent kinase (protein kinase A, PKA), consequently leads to the phosphorylation of several Ca2+ handling proteins. LTCCs allow Ca2+ influx (McDonald et al., 1994), ryanodine receptors (RyRs) are responsible for Ca2+ release from sarcoplasmic reticulum (SR), and phospholamban (PLN) reduces inhibition of SR Ca2+-ATPase (SERCA) which uptakes cytosolic Ca2+. From the view of electrophysiology, depolarized RP and prolonged APD facilitate the opening of LTCCs thus promote intracellular Ca2+ accumulation (Swynghedauw, 1999), while IK1 downregulation delays AP repolarization and in turn further prolongs APD (Miake et al., 2003). In the ventricle, LTCC is the main Ca2+ influx pathway and plays a key role in the excitation-contraction coupling. Overactivation of LTCC elevates intracellular Ca2+ and correlates with the genesis of hypertrophy (Muth et al., 2001; Song et al., 2002; Bodi et al., 2003; Viola et al., 2009). LTCC antagonists have been expected to retard Ca2+ influx and prevent hypertrophy (Sugiura et al., 2001). However, as Ca2+ influx through LTCC is important in initiating and maintaining contraction, LTCC blockers might be limited in the clinical application because of the potential risk of pumping dysfunction.
Elevation of cardiomyocyte [Ca2+]i elicits a series of biochemical signals through multifaceted Ca2+-regulated enzymes including Ca2+-/calmodulin-dependent protein kinase II (CaMKII). Iso is also a definite stimulation for the autophosphorylation of CaMKII in cardiomyocytes (Zhu et al., 2003; Pereira et al., 2007; Mangmool et al., 2010). Activated CaMKII phosphorylates multiple ion channels and Ca2+ handling proteins such as voltage-gated Na+ channel, LTCC, Cl− channel, and RyR receptor (Grandi et al., 2007; Bers and Grandi, 2009; Wagner et al., 2009). We show here that hyperphosphorylated CaMKII is a key player for intracellular Ca2+ dyshomeostasis. Reduction of SR Ca2+ content in hypertrophic or failing cardiomyocytes were largely resulted from reduced SERCA2 and increased RyR leak, and in turn, facilitated cytosolic Ca2+ overload. If considering the ineffectiveness of zacopride on ICa-L or INCX (Liu et al., 2012), the inhibition of Ca2+ overload by zacopride is probably secondary and mediated by the activation of IK1 channels. By enhancing IK1 or upregulating Kir2.1, zacopride reversed RP depolarization and APD prolongation, restored some key Ca2+ handling proteins, consequently decreased cardiac Ca2+ overload. Of note, zacopride inhibited Ca2+ overload in hypertrophic cardiomyocytes but not affected [Ca2 +]i in normal cardiomyocytes (Figure 6).
PKA Signaling may be Involved in IK1 Channel–Mediated Calcium Homeostasis
Kir2.1, Kir2.2, and Kir2.3 are the substrates of PKA and protein kinase C (PKC). Phosphorylation of these pore-forming channel proteins thus may play important roles in regulating IK1 channel function although the mechanism remains a matter of debate (Fakler et al., 1994; Koumi et al., 1995; Sonoyama et al., 2006; Kiesecker et al., 2006). Fakler et al. demonstrate that IK1 channels expressed in Xenopus oocytes is upregulated by addition of the catalytic subunit of PKA and is downregulated following application of a specific activator of PKC (Fakler et al., 1994). While Koumi et al. found that native IK1 channels in guinea-pig ventricular myocytes are inhibited by exposure of the cytosolic side of the membrane to purified catalytic subunit of PKA. In human cardiomyocytes, ET-1 induced a marked inhibition of IK1 which could be suppressed by a PKC inhibitor staurosporine but not be altered by PKA inhibitor KT5720 (Kiesecker et al., 2006). In our previous work, zacopride selectively activated heterologously expressed Kir2.1 channels in HEK 293 cells, and the activation could be reversed by PKA inhibitor KT5720 but not by PKC inhibitor GF109203X or PKG inhibitor KT5823. Further mutation of the PKA phosphorylation site S425L completely abolished the agonizing effect of zacopride on Kir2.1. These data suggest that zacopride selectively activates Kir2.1 channel via a PKA-mediated signaling pathway (Zhang et al., 2013).
As a heterotetrameric threonine/serine kinase, PKA is also involved in the regulation of intracellular calcium homeostasis. Activated cyclic-AMP/PKA triggers Ca2+ influx through LTCC and Ca2+ release from SR, resulting in RP depolarization, then Ca2+ extrusion from the cardiomyocyte by NCX (Roe et al., 2015). Considering that zacopride has no direct effect on INCX, ICa-L (Liu et al., 2012), and IKATP in rat LV myocytes (Zhai et al., 2017), enhancing IK1 may be an negative feedback which limits the PKA-mediated depolarization and calcium overload.
The Interplay Between Electrical and Structural Remodelings Around Calcium Overload
In present study, chronic exposure to β-AR agonist Iso induced both structural and electrical remodelings in the rat ventricles. The structural remodeling manifested with increase of ventricle mass, cardiac hypertrophy, apoptosis, abnormalities of proteins expression, and interstitial fibrosis. The electrical remodeling is featured with depolarization of RP, prolongation of APD, downregulation of IK1 (Kir2.1) channel, elevation of cytosolic free Ca2+, and reduction of SR Ca2+ content. Among these events, Ca2+ overload is a key factor linking the electrical remodeling and structural remodeling. For example, secondary to Ca2+ overload, Ca2+-activated CaMKII phosphorylates various transcription factors such as class II HDACs (Backs et al., 2006; Zhang et al., 2007), transcription factor 1(ATF-1) (Sun et al., 1996), cAMP response element–binding protein (CREB) (Sheng et al., 1991), and myocyte enhancer factor 2 (MEF2) (Passier et al., 2000; Zhang et al., 2007). These events promote the expression of cardiac specific genes involved in the structural remodeling. Chronic CaMKII overexpression also caused downregulation of IK1 channels (Wagner et al., 2009), whereas inhibition of CaMKII increased IK1 density which partially accounted for the shortening of APD (Li et al., 2006). These findings agree well with the observations in the present study albeit the disputable causality. From the in vivo time-course study, cardiac electrical remodeling, such as inhibition of IK1 and Ca2+ dyshomeostasis, is concurrent with the structural remodeling and dysfunction. This connection was also supported by the genesis of cardiac apoptosis. An inappropriate rise in intracellular Ca2+ activates Ca2+-/Mg2+-dependent endonucleases and glutamine transferases which degrade DNA and cytoskeletal proteins. Apoptosis may lead to cardiac fibrosis (Zhang et al., 2011). Therefore, reduction of intracellular calcium overload has been an important focus in the prevention of maladaptive cardiac remodeling. By upregulating cardiac IK1, zacopride prevented Iso-induced electrical remodeling, preserved Ca2+ homeostasis, and thus inhibited calcium-activated remodeling signaling and apoptosis, thereafter improved structural remodeling. The most convincing data shown here were that BaCl2 in vitro or chloroquine in vitro/vivo could blunt the beneficial effects of zacopride on both electrical and structural remodelings. IK1 channel might be a novel target for the regulation of cardiac calcium homeostasis and remodeling.
It is worth mentioning that in the in vivo experiment, about 50% rats suffered from sudden death in the follow-up period after Iso infusion. There are two major causes of death in HF: pumping function decline and arrhythmias. These two causes both link to Ca2+ dyshomeostasis in cardiomyocytes (Bers, 2006). Cardiac pumping dysfunction may not sufficiently account for the sudden death in the present study, because the rats subjected to 3 days and 10 days of Iso infusion showed enhanced pumping function. Acute Iso challenging induced DADs which facilitate arrhythmogenesis (Zhai et al., 2017). Ventricular tachyarrhythmia and LV hypertrophy both increase the risk of sudden cardiac death (SCD) up to 10-fold (Aronow et al., 1988), and resolution of LV hypertrophy reduces the risk of SCD (Wachtell et al., 2007). As an IK1 channel agonist, zacopride has been recognized as a new antiarrhythmic agent on triggered arrhythmias (Liu et al., 2012; Zhai et al., 2017). Here, we further show that zacopride was also effective in reducing cardiac hypertrophy. The dual actions of zacopride might protect animals from SCD. We expect that targeting myocardial IK1 channel and Ca2+ homeostasis may have great potential for the prevention of triggered arrhythmias in HF patients.
Data Availability
All datasets generated for this study are included in the manuscript and the supplementary files.
Ethics Statement
This study was carried out in accordance with the recommendations of the guidelines for the Care and Use of Laboratory Animals (NIH, revised 2011), Ethics Committee of Shanxi Medical University. The protocol was approved by the Ethics Committee of Shanxi Medical University.
Author Contributions
Q-HL, designed the study, performed the experiments, and drafted the manuscript. XQ, L-JZ, YL, X-NC and X-ZR acquired and analyzed the data. Q-LF, analyzed the data. JW performed studies evaluating cardiac function. LZ and X-WZ carried out the patch clamp experiments. B-WW and J-MC participated in the protocol design and critically revised the manuscript, and gave the final proof for the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 31200864 to Q-HL, No. 81670313 to J-MC), a grant from Shanxi Scholarship Council of China (No. 2016-059 to Q-HL), grants from the Key Laboratory of Medical Electrophysiology (Southwest Medical University), Ministry of Education of China (No. 201704 to Q-HL), and a fund for Shanxi “1331 Project” Key Subjects Construction (1331KSC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
APD, action potential duration; IK1, inward rectifier potassium channel or current; ARVM, adult rat ventricular myocyte; NRVM, neonatal rat ventricular myocyte; Iso, isoproterenol; Chlo, chloroquine; Zac, zacopride; LTCC/ICa-L, L-type calcium channel; Ito, transient outward K+ current; IKATP, ATP-sensitive potassium current; INCX, Na+-Ca2+ exchanger (NCX) current; RP, resting potential; SERCA, sarcoplasmic reticulum Ca2+-ATPase; CaMKII, Ca2+-/calmodulin-dependent protein kinase II; DAD, delayed afterdepolarization; m-CPBG, m-chlorophenylbiguanide.
Acknowledgments
The authors would like to thank Prof. Rui-Ling Xu (Shanxi Medical University) for critical reviews, and Wei-Fang Zhang and Xue-Fen Pang (Shanxi Medical University) for their helpful technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00929/full#supplementary-material
References
Aiba, T., Tomaselli, G. F. (2010). Electrical remodeling in the failing heart. Curr. Opin. Cardiol. 25, 29–36. doi: 10.1097/HCO.0b013e328333d3d6
Aimond, F., Alvarez, J. L., Rauzier, J. M., Lorente, P., Vassort, G. (1999). Ionic basis of ventricular arrhythmias in remodeled rat heart during long-term myocardial infarction. Cardiovasc. Res. 42, 402–415. doi: 10.1016/S0008-6363(99)00070-X
Aronow, W. S., Epstein, S., Koenigsberg, M., Schwartz, K. S. (1988). Usefulness of echocardiographic left ventricular hypertrophy, ventricular tachycardia and complex ventricular arrhythmias in predicting ventricular fibrillation or sudden cardiac death in elderly patients. Am. J. Cardiol. 62, 1124–1125. doi: 10.1016/0002-9149(88)90562-0
Backs, J., Song, K., Bezprozvannaya, S., Chang, S., Olson, E. N. (2006). CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 116, 1853–1864. doi: 10.1172/JCI27438
Benjamin, I. J., Jalil, J. E., Tan, L. B., Cho, K., Weber, K. T., Clark, W. A. (1989). Isoproterenol induced-myocardial fibrosis in relation to myocyte necrosis. Circ. Res. 65, 657–670. doi: 10.1161/01.RES.65.3.657
Berridge, M. J., Bootman, M. D., Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiol. (Bethesda) 21, 380–387. doi: 10.1152/physiol.00019.2006
Bers, D. M., Grandi, E. (2009). Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cadriovasc. Res. 54, 180–187. doi: 10.1097/FJC.0b013e3181a25078
Bodi, I., Muth, J. N., Hahn, H. S., Petrashevskaya, N. N., Rubio, M., Koch, S. E., et al. (2003). Electrical remodeling in hearts from a calcium-dependent mouse model of hypertrophy and failure: complex nature of K+ current changes and action potential duration. J. Am. Coll. Cardiol. 41, 1611–1622. doi: 10.1016/S0735-1097(03)00244-4
Burchfield, J. S., Xie, M., Hill, J. A. (2013). Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 128, 388–400. doi: 10.1161/CIRCULATIONAHA.113.001878
Carafoli, E., Santella, L., Branca, D., Brini, M. (2001). Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 36, 107–260. doi: 10.1080/20014091074183
Chlopcíková, S., Psotová, J., Miketová, P. (2001). Neonatal rat cardiomyocytes-a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 145, 49–55. doi: 10.5507/bp.2001.011
Fakler, B., Brändle, U., Glowatzki, E., Zenner, H. P., Ruppersberg, J. P. (1994). Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron 13, 1413–1420. doi: 10.1016/0896-6273(94)90426-X
Fauconnier, J., Lacampagne, A., Rauzier, J. M., Vassort, G., Richard, S. (2005). Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc. Res. 68, 204–212. doi: 10.1016/j.cardiores.2005.05.024
Fedak, P. W., Verma, S., Weisel, R. D., Li, R. K. (2005). Cardiac remodeling and failure: from molecules to man (part I). Cardiovasc. Pathol. 14, 1–11. doi: 10.1016/j.carpath.2004.12.002
Grandi, E., Puglisi, J. L., Wagner, S., Maier, L. S., Severi, S., Bers, D. M. (2007). Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 93 (11), 835–347. doi: 10.1529/biophysj.107.114868
Grueter, C. E., Colbran, R. J., Anderson, M. E. (2007). CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J. Mol. Med. (Berl.) 85, 5–14. doi: 10.1007/s00109-006-0125-6
Hellawell, J. L., Margulies, K. B. (2012). Myocardial reverse remodeling. Cardiovasc. Ther. 30, 172–181. doi: 10.1111/j.1755-5922.2010.00247.x
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Houser, S. R., Margulies, K. B. (2003). Is depressed myocyte contractility centrally involved in heart failure? Circ. Res. 92, 350–358. doi: 10.1161/01.RES.0000060027.40275.A6
Janse, M. J. (2004). Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc. Res. 61, 208–217. doi: 10.1016/j.cardiores.2003.11.018
Kiesecker, C., Zitron, E., Scherer, D., Lueck, S., Bloehs, R., Scholz, E. P., et al. (2006). Regulation of cardiac inwardly rectifying potassium current IK1 and Kir2.x channels by endothelin-1. J. Mol. Med. 84, 46–56. doi: 10.1007/s00109-005-0707-8
Koumi, S., Wasserstrom, J. A., Ten Eick, R. E. (1995). Beta-adrenergic and cholinergic modulation of inward rectifier K+ channel function and phosphorylation in guinea-pig ventricle. J. Physiol. 486, 661–678. doi: 10.1113/jphysiol.1995.sp020842
Lee, T. M., Linm, M. S., Chang, N. C. (2008). Effect of ATP-sensitive potassium channel agonists on ventricular remodeling in healed rat infarcts. J. Am. Coll. Cardiol. 51, 1309–1318. doi: 10.1016/j.jacc.2007.11.067
Li, J., Marionneau, C., Zhang, R., Shah, V., Hell, J. W., Nerbonne, J. M., et al. (2006). Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ. Res. 99, 1092–1099. doi: 10.1161/01.RES.0000249369.71709.5c
Li, L., Okada, H., Takemura, G., Kosai, K., Kanamori, H., Esaki, M., et al. (2009). Postinfarction gene therapy with adenoviral vector expressing decorin mitigates cardiac remodeling and dysfunction. Am. J. Physiol. Heart Circ. Physiol. 297, H1504–H1513. doi: 10.1152/ajpheart.00194.2009
Liu, C. F., Liu, E. L., Luo, T. E., Zhang, W. F., He, R. L. (2016). Opening of the inward rectifier potassium channel alleviates maladaptive tissue repair following myocardial infarction. Acta. Biochim. Biophys. Sin. 48, 687–695. doi: 10.1093/abbs/gmw060
Liu, Q. H., Li, X. L., Xu, Y. W., Lin, Y. Y., Cao, J. M., Wu, B. W. (2012). A novel discovery of IK1 channel agonist: zacopride selectively enhances IK1 current and suppresses triggered arrhythmias in the rat. J. Cardiovasc. Pharmacol. 59, 37–48. doi: 10.1097/FJC.0b013e3182350bcc
Long, V. P., 3rd, Bonilla, I. M., Vargas-Pinto, P., Nishijima, Y., Sridhar, A., Li, C., et al. (2015). Heart failure duration progressively modulates the arrhythmia substrate through structural and electrical remodeling. Life Sci. 123, 61–71. doi: 10.1016/j.lfs.2014.12.024
Mangmool, S., Shukla, A. K., Rockman, H. A. (2010). Beta-arrestin-dependent activation of Ca (2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J. Cell Biol. 189, 573–587. doi: 10.1083/jcb.200911047
McDonald, T. F., Pelzer, S., Trautwein, W., Pelzer, D. J. (1994). Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 74, 365–507. doi: 10.1152/physrev.1994.74.2.365
Miake, J., Marbán, E., Nuss, H. B. (2003). Functional role of inward rectifier current in heart probed by Kir2.1 overexpression and dominant-negative suppression. J. Clin. Invest. 111, 1529–1536. doi: 10.1172/JCI17959
Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., et al. (1998). A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228. doi: 10.1016/S0092-8674(00)81573-1
Mueller, E. E., Momen, A., Massé, S., Zhou, Y. Q., Liu, J., Backx, P. H., et al. (2011). Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc. Res. 89, 623–633. doi: 10.1093/cvr/cvq351
Muth, J. N., Bodi, I., Lewis, W., Varad, G., Schwartz, A. (2001). A Ca(2+) -dependent transgenic model of cardiac hypertrophy: a role for protein kinase Calpha. Circulation 103, 140–147. doi: 10.1161/01.CIR.103.1.140
Passier, R., Zeng, H., Frey, N., Naya, F. J., Nicol, R. L., McKinsey, T. A., et al. (2000). CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 105, 1395–1406. doi: 10.1172/JCI8551
Pereira, L., Metrich, M., Fernandez-Velasco, M., Lucas, A., Leroy, J., Perrier, R., et al. (2007). The cAMP binding protein Epac modulates Ca sparks by a Ca/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 583 (Pt 2), 685–694. doi: 10.1113/jphysiol.2007.133066
Roe, A. T., Frisk, M., Louch, W. E. (2015). Targeting cardiomyocyte Ca2+ homeostasis in ostasis in heart failure. Curr. Pharm. Des. 21, 431–448. doi: 10.2174/138161282104141204124129
Ryan, T. D., Rothstein, E. C., Aban, I., Tallaj, J. A., Husain, A., Lucchesi, P. A., et al. (2007). Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J. Am. Coll. Cardiol. 49, 811–821. doi: 10.1016/j.jacc.2006.06.083
Sheng, M., Thompson, M. A., Greenberg, M. E. (1991). CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252, 1427–1430. doi: 10.1126/science.1646483
Song, L. S., Guia, A., Muth, J. N., Rubio, M., Wang, S. Q., Xiao, R. P., et al. (2002). Ca(2+) signaling in cardiac myocytes overexpressing the alpha(1) subunit of L-type Ca(2+) channel. Circ. Res. 90, 174–181. doi: 10.1161/hh0202.103230
Sonoyama, K., Ninomiya, H., Igawa, O., Kaetsu, Y., Furuse, Y., Hamada, T., et al. (2006). Inhibition of inward rectifier K+ currents by angiotensin II in rat atrial myocytes: lack of effects in cells from spontaneously hypertensive rats. Hypertens. Res. 29, 923–934. doi: 10.1291/hypres.29.923
St John Sutton, M., Lee, D., Rouleau, J. L., Goldman, S., Plappert, T., Braunwald, E., et al. (2003). Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation 107, 2577–2582. doi: 10.1161/01.CIR.0000070420.51787.A8
Stewart, J. A., Jr., Massey, E. P., Fix, C., Zhu, J., Goldsmith, E. C., Carver, W. (2010). Temporal alterations in cardiac fibroblast function following induction of pressure overload. Cell Tissue Res. 340, 117–126. doi: 10.1007/s00441-010-0943-2
Sugiura, R., Sio, S. O., Shuntoh, H., Kuno, T. (2001). Molecular genetic analysis of the calcineurin signaling pathways. Cell. Mol. Life Sci. 58, 278–288. doi: 10.1007/PL00000855
Sun, J. M., Wang, C. M., Guo, Z., Hao, Y. Y., Xie, Y. J., Gu, J., et al. (2015). Reduction of isoproterenol-induced cardiac hypertrophy and modulation of myocardial connexin43 by a KATP channel agonist. Mol. Med. Rep. 11, 1845–1850. doi: 10.3892/mmr.2014.2988
Sun, P., Lou, L., Maurer, R. A. (1996). Regulation of activating transcription factor-1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II and IV. J. Biol. Chem. 271, 3066–3073. doi: 10.1074/jbc.271.6.3066
Swynghedauw, B. (1999). Molecular mechanisms of myocardial remodeling. Physiol. Rev. 79, 215–262. doi: 10.1152/physrev.1999.79.1.215
Tsukamoto, O., Minamino, T., Okada, K., Shintani, Y., Takashima, S., Kato, H., et al. (2006). Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem. Biophys. Res. Commun. 340, 1125–1133. doi: 10.1016/j.bbrc.2005.12.120
Viola, H. M., Macdonald, W. A., Tang, H., Hool, L. C. (2009). The L-type Ca(2+) channel as a therapeutic target in heart disease. Curr. Med. Chem. 16, 3341–3358. doi: 10.2174/092986709789057671
Wachtell, K., Okin, P. M., Olsen, M. H., Dahlöf, B., Devereux, R. B., Ibsen, H., et al. (2007). Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: the life study. Circulation 116, 700–705. doi: 10.1161/CIRCULATIONAHA.106.666594
Wagner, S., Hacker, E., Grandi, E., Weber, S. L., Dybkova, N., Sossalla, S., et al. (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294. doi: 10.1161/CIRCEP.108.842799
Zhai, X., Zhang, L., Guo, Y., Yang, Y., Wang, D., Zhang, Y., et al. (2017). The IK1/Kir2.1 channel agonist zacopride prevents and cures acute ischemic arrhythmias in the rat. PLoS One 12, e0177600. doi: 10.1371/journal.pone.0177600
Zhang, L., Liu, Q., Liu, C., Zhai, X., Feng, Q., Xu, R., et al. (2013). Zacopride selectively activates the Kir2.1 channel via a PKA signaling pathway in rat cardiomyocyte. Sci. China Life Sci. 56, 788–796. doi: 10.1007/s11427-013-4531-z
Zhang, T., Brown, J. H. (2004). Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 63, 476–486. doi: 10.1016/j.cardiores.2004.04.026
Zhang, T., Kohlhaas, M., Backs, J., Mishra, S., Phillips, W., Dybkova, N., et al. (2007). CaMKIIδ isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 282, 35078–35087. doi: 10.1074/jbc.M707083200
Zhang, T., Yong, S. L., Drinko, J. K., Popović, Z. B., Shryock, J. C., Belardinelli, L., et al. (2011). LQTS mutation N1325S in cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis, cardiac fibrosis and contractile dysfunction in mice. Int. J. Cardiol. 147, 39–45. doi: 10.1016/j.ijcard.2009.08.047
Zhou, X., Huang, J., Ideker, R. E. (2002). Transmural recording of monophasic action potentials. Am. J. Physiol. Heart Circ. Physiol. 282, H855–H861. doi: 10.1152/ajpheart.01172.2000
Zhu, H., Tannous, P., Johnstone, J. L., Kong, Y., Shelton, J. M., Richardson, J. A., et al. (2007). Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793. doi: 10.1172/JCI27523
Keywords: inward rectifier potassium channel, isoproterenol, calcium overload, cardiac remodeling, zacopride
Citation: Liu Q-H, Qiao X, Zhang L-J, Wang J, Zhang L, Zhai X-W, Ren X-Z, Li Y, Cao X-N, Feng Q-L, Cao J-M and Wu B-W (2019) IK1 Channel Agonist Zacopride Alleviates Cardiac Hypertrophy and Failure via Alterations in Calcium Dyshomeostasis and Electrical Remodeling in Rats. Front. Pharmacol. 10:929. doi: 10.3389/fphar.2019.00929
Received: 21 February 2019; Accepted: 22 July 2019;
Published: 26 August 2019.
Edited by:
Eleonora Grandi, University of California, United StatesReviewed by:
Na Li, Baylor College of Medicine, United StatesDaniel M. Johnson, University of Birmingham, United Kingdom
Copyright © 2019 Liu, Qiao, Zhang, Wang, Zhang, Zhai, Ren, Li, Cao, Feng, Cao and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Min Cao, Y2FvamltaW5Ac3htdS5lZHUuY24=; Bo-Wei Wu, cXdlYXNkMjk5QDE2My5jb20=