 Yang Yang1†
Yang Yang1† Yu Cao
Yu Cao Hao Li
Hao Li- 1Institute of Geriatrics, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Beijing Key Laboratory of Pharmacology of Chinese Materia Region, Institute of Basic Medical Sciences of Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Alzheimer’s disease (AD) is the most common type of dementia worldwide. The deposition of amyloid β (Aβ) is one of the most important pathological changes in AD. Autophagy, which mediates degradation of toxic proteins and maintains normal neuronal function, is dysfunctional in AD; dysfunctional autophagy is believed to be a critical pathological feature of AD. Here, we evaluated the in vitro and in vivo effects of a traditional Chinese medicinal formula called Yizhiqingxin formula (YQF) on autophagy. We determined that treatment with a high dose of YQF improved spatial memory and decreased the hippocampal Aβ burden in APP/PS1 mice, an early onset AD model. Transmission electron microscopy and immunohistochemical data revealed that YQF enhanced autophagosome formation and also increased the levels of LC3II/LC3I and Beclin1. Further, we found that YQF treatment promoted autophagic activity by inhibiting the phosphorylation of the Mammalian target of rapamycin (mTOR) at the Ser2448 site. Moreover, the level of 4EBP1 increased after YQF intervention, indicating a suppression of mTOR signaling. YQF was also found to promote autophagosome degradation, as indicated by the decreased p62 levels and increased cathepsin D and V-ATPase levels. Taken together, YQF could improve spatial learning in APP/PS1 mice and ameliorate the accumulation of Aβ while promoting autophagy via mTOR pathway modulation.
Introduction
Alzheimer’s disease (AD), manifested by core clinical features of progressive memory loss and executive dysfunction, is the most common type of dementia worldwide. In China, the current prevalence of AD in people over 65 is 3.21% (Jia et al., 2014). The aggravating trend of aging population also drives the increasing number of AD patients. According to the World Alzheimer Report 2018, it is estimated that there are about 47 million Alzheimer’s patients worldwide, with 10 million new cases each year (Alzheimer’s Association, 2018). As the disease progresses, most patients eventually need constant care and help with most basic daily activities, exacting a heavy burden on families and society (Jia et al., 2018). AD therapy is reported to be the most expensive treatment in the U.S. (Souchet et al., 2018).
Autophagy is an evolutionarily conserved intracellular process responsible for the lysosomal degradation and removal of degraded and aggregated proteins, and damaged intracellular organelles to maintain cellular homeostasis (Ryter et al., 2013). This robust quality control mechanism is critical in neurons to support their long-term viability and functionality. Multiple studies have identified defective autophagic activity in several neurodegenerative diseases, especially in AD, which is characterized by amyloid β (Aβ) peptide aggregation. Moreover, both in vitro and in vivo studies have shown that autophagy retards neuronal degeneration and age-associated cognitive disorders by the clearance of accumulated toxic proteins. An abnormal alteration of autophagy and Aβ deposition has been observed in AD patients, along with a hyperactivation of the mammalian target of rapamycin (mTOR) pathway (Sun et al., 2014; Tramutola et al., 2017). Consistent with these clinical findings, overwhelming data suggest a vicious cycle between Aβ deposition and mTOR hyperactivity (Oddo, 2012; Talboom et al., 2015).
Yizhiqingxin formula (YQF; previously called Fuzheng Quxie Decoction Supplementary Data Sheet 1) is a traditional Chinese formula composed of Panax ginseng (radix), Coptis chinensis (rhizome), and Conioselinum anthriscoides ‘Chuanxiong’ (rhizome). Our previous work found that YQF could alleviate cognitive deficits and tau hyperphosphorylation in SAMP8 mice (Yang et al., 2017); vascular endothelial growth factor (VEGF) and VEGF receptor were the other molecular targets of YQF (Wang et al., 2018). However, the effects of YQF on autophagy and the underlying mechanisms are not clear. In the present study, we investigate the potential roles of YQF on autophagy and mTOR signaling in APP/PS1 mice and APP/PS1 double transgenic HEK293 cells. Data revealed that YQF could ameliorate cognitive deficits and decrease Aβ deposition, suggesting that YQF is a potential candidate for the treatment of AD.
Materials and Methods
Animals
Sixty 3-month-old male APP/PS1 transgenic mice were randomly divided into five groups: Model group, Rapa group (2.24 mg·kg−1·d−1), YQF high dose group (YQF-H, 10.4 g·kg−1·d−1 raw drugs), YQF medium dose group (YQF-M, 5.2 g·kg−1·d−1 raw drugs), and YQF low dose group (YQF-L, 2.6 g·kg−1·d−1 raw drugs). Twelve male C57BL/6 mice with the same genetic background at 3 months of age were used as control. Each group was given 24 weeks of gavage intervention. Drugs used in Rapa and YQF-L groups were equivalent doses suggested in the clinic. The Control and Model groups were given equal volumes of distilled water. The other groups were given corresponding drugs. Mice were all housed in groups of three to four per cage and maintained in a specific pathogen-free conditions at 22 ± 2°C with automatic light cycles (12 h light/dark) and relative humidity of 50–70%. Animals received food and water ad libitum and were allowed to adapt to the environment for 1 week before experiment. All experiments were carried out in accordance with the guidelines for animal care and were approved by the animal ethics committee of the Ethics Committee of Xiyuan Hospital, China Academy of Chinese Medical Sciences (NO. 2018XLC004-1).
Reagents
Rabbit monoclonal anti-beta amyloid 1–42 (ab201060) antibody and rabbit polyclonal anti-LC3 II (ab48394), anti-p62 (ab91526), anti-mTOR (ab2732), and anti-cathepsin D (ab75852) antibodies were procured from Abcam (UK). Rabbit polyclonal anti-ATPase (sc-28801) antibody was procured from Santa Cruz (USA). Rabbit polyclonal anti-p-mTOR (5536S), anti-4EBP1 (9644S), anti-P70S6K (2708S), anti-Beclin 1 (3738S), and anti-LAMP1 (3243S) antibodies were procured from Cell Signaling Technology (USA). Mouse monoclonal anti-GAPDH (YM3029) antibody was purchased from Immunoway (USA). Mouse monoclonal anti-β-actin (sc-69879) antibody was purchased from Santa Cruz (USA). Aβ ELISA kit (DAB142) was purchased from RD (USA).
Drug Preparation
YQF is composed of Panax ginseng (radix), Coptis chinensis (rhizome), and Conioselinum anthriscoides ‘Chuanxiong’ (rhizome), with a weight ratio of 9:5:6. Raw drugs were purchased from Beijing Kangrentang Pharmaceutical Co. Ltd. They were deposited at the Herbarium of National Resource Center for Chinese Materia Medica (CMMI), China Academy of Chinese Medical Sciences. The codes for Panax ginseng (radix), Coptis chinensis (rhizome), and Conioselinum anthriscoides ‘Chuanxiong’ (rhizome) specimens were 0220582LY0015, 420528LY0075, and 331127LY0805, respectively. The extraction was done by the Department of Pharmaceutics of Xiyuan Hospital, China Academy of Chinese Medical Sciences. Per gram extraction was obtained from 3.71 grams raw drug mixture. Seven major bioactive compounds (gginsenoside Rg1, ginsenoside Re, ginsenoside Rb1, coptisine, berberine, ligustilide, and ferulic acid) were identified using HPLC (high performance liquid chromatography) (Yang et al., 2017).
Cell Culture, Gene Transfection, and Drug Intervention Protocols
The human embryonic kidney cell line (HEK293 cells) was purchased from National Infrastructure of Cell Line Resource (NO: 3111C0001CCC000128). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Hyclone, USA) supplemented with penicillin–streptomycin (100 IU/ml and 100 µg/ml, Gibco, USA) and 10% fetal bovine serum (Gibco, USA) in a humidified atmosphere of 5% CO2 at 37°C. Cells were passaged using 0.25% trypsin with 0.02% EDTA every 3–4 days. During transfection, HEK293 cells were cultured in Opti-MEM I Serum Medium (Gibco, USA) and transfected with APP695 plasmid (Addgene, USA) with Lipofectamine 2000 (Invitrogen, USA) following manufacturers’ instructions. The APP695 gene HEK293 (APP-HEK293) cell clones were selected using puromycin (4 μg/ml, Amresco, USA). PSEN1dE9 plasmids (Beijing Borui Technology Co., Ltd., China) were transfected into the APP-HEK293 cells [clones were selected using G418 (1 mg/ml, Amresco, USA)], and then the APP/PS1 double gene transfected HEK293 cells stably expressing APP695 and PSEN1dE9 genes (APP/PS1 double transgenic HEK293 cells) were obtained.
After gene transfection, HEK293 cells were divided into the Control group (treated with Control group serum), and APP/PS1 double transgenic HEK293 cells were divided into the Model group, YQF-H group, YQF-M group, and YQF-L group and were treated with serum containing YQF.
Cell Viability Assay
Cell viabilities were measured by the Cell Counting Kit-8/WST-8 kit (CCK8, Dojindo, Japan) following the manufacturer’s instruction. Briefly, cells were plated in a 96-well plate at a density of 1 × 105/ml. CCK8 mixed with DMEM without phenol red at a ratio of 1:9 was added to the 96-well plate, and then the plate was incubated in an incubator for 1–2 h. Optical density readings were measured with a Microplate Reader (Bio Tek, USA) at 450 nm wavelength.
Preparation of Serum Containing YQF (YQF-Serum)
Forty 3-month-old male Sprague Dawley rats purchased from Huafukang Biological Technologies [Beijing, China, NO: SCXK (Beijing) 2014-0004] were divided into four groups: YQF high dose group (YQF-H, 18 g·kg−1·d−1, herbal medicine), YQF medium dose group (YQF-M, 9 g·kg−1·d−1, herbal medicine), YQF low dose group (YQF-L, 1.8 g·kg−1·d−1, herbal medicine), and Control group (control, equal volume of distilled water) with 10 rats in each group. Rats in the four groups received a daily gavage with the respective treatment for 1 week. Subsequently, the rats were anaesthetized by intraperitoneal injection with pentobarbital sodium. Blood was drawn from the abdominal aorta and centrifuged at 3000 rpm for 10 min. The YQF-Serum so obtained was used for further experiments.
Morris Water Maze Tests
Spatial memory was assessed using the Morris water maze (MWM) test, as described previously (Yang et al., 2017). Briefly, a tank (diameter 120 cm, height 50 cm, painted white) filled with water (22°C) to the depth of 40 cm was used. The water was stained with nontoxic white dye. A white platform (diameter, 10 cm; height, 15 cm) was placed in the center of the target quadrant 1 cm beneath the water. The place navigation trial consisted of five training days and four trials per day with a 20-min inter-trial interval. Mice were placed into the water from four different starting positions facing the wall, and each trial lasted for 60 s. If a mouse did not reach the platform within 60 s, it was guided to the platform where it had to remain for 10 s. The swimming distance and escape latencies were recorded. One day after the task acquisition (day 5), a probe trial was performed in order to assess the spatial memory. The platform was removed. Animals were put into the tank from the position opposite to the platform and allowed to swim freely for 60 s. The percentage of total time spent in each quadrant and the number of crossings where the platform had been previously located were recorded.
Analysis of Blood and Brain Tissue for Active Components of YQF
After the MWM experiments, blood samples were collected from rats being transcardially perfused with ice-cold physiological saline for brain tissue isolation. Brains were quickly removed on ice; cerebral cortices were then separated and homogenized in ice-cold physiological saline (2 ml saline per 1 g cortex). Plasma was isolated from the blood samples of each group. Subsequently, liquid chromatography tandem mass spectrometry (LC-MS/MS) was used to detect the active components of YQF present in the blood plasma and brain tissue homogenate.
Transmission Electron Microscopy Analysis
Cells from in vitro experiments and hippocampal tissues obtained from the sacrificed mice, post MWM tests, were fixed with 2.5% glutaraldehyde and washed three times in phosphate-buffered saline (PBS) (pH 7.2), following which they were post fixed in 1% osmium tetroxide, dehydrated in graded ethanol and acetone solution, and embedded in epoxy resin. Sectioning and staining were subsequently performed under general electron microscopy supervision. Autophagosomes in cells of different samples were observed and imaged under a transmission electron microscope at 4000× magnification.
Immunohistochemistry Assays
After the behavior test, five mice from each group were anesthetized with pentobarbital sodium (30 mg/kg) and were perfused by intracardiac puncture with PBS (4°C) and 4% paraformaldehyde. Immunohistochemistry staining was conducted on 20 mm thick coronal sections as previously reported (Wang et al., 2010). Briefly, the sections were dewaxed, rehydrated, and incubated in PBS containing 3% H2O2 for 10 min. After washing with tris-buffered saline (TBS), sections were boiled in citric acid buffer for 5 min in a microwave oven and then incubated overnight at 4°C with rabbit antibody against Aβ1-42 antibody (1:1000 dilutions). After rinsing, sections were incubated with biotinylated secondary antibody for 1 h, followed by streptavidin peroxidase for 1 h at room temperature. Subsequently, the sections were visualized with 3, 3-diaminobenzidine and analyzed using an inverted microscope (OLYMPUS CKX41, Tokyo, Japan).
To assess the Aβ burden in the cortex and hippocampus, five sections with the same reference position were selected from each mouse (n = 5). The percentage of the total area of Aβ-positive areas compared with the total area of the hippocampus was quantified (in square micrometers). The data were analyzed with the image analysis system (Image-Pro Plus 6.0).
ELISA
Cell cultures were treated with YQF-serum at the indicated concentrations for the specified durations. The conditioned medium was then collected and subjected to a sandwich ELISA for the Aβ42 levels according to the manufacturer’s guidelines.
Western Blot Analysis
The hippocampus was lysed on ice in lysis buffer and then centrifuged at 12,000g for 10 min at 4°C. The protein content was determined using a BCA protein assay kit (Pierce, Rockford, IL). Proteins (40 mg) were separated by SDS-PAGE electrophoresis and then transferred to 0.45 μm polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were blocked for 2 h in 5% BCA and were then incubated overnight at 4°C with the primary antibody (1:1000 dilutions) followed by a horseradish peroxidase-conjugated secondary antibody (1:2000 dilutions). The blots of proteins of interest were visualized using an ECL advanced Western blotting detection kit (Thermo Fisher Scientific, MA, USA). Quantitative analyses of the protein bands were performed with a Molecular Imager Carestream MI SE system (Rochester, NY, USA).
Statistical Analysis
Data are expressed as mean ± SEM and were analyzed by GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). MWM latencies were analyzed using two-way analysis of variance (ANOVA) with repeated measures. All the other data were analyzed using one-way ANOVA. Fisher’s least significant difference post hoc test was used to test the differences between two groups. Significance level was set at p < 0.05.
Results
Analysis of Blood and Brain Tissue for Active Components of YQF
Our previous HPLC study revealed that the important active components of YQF are ginsenoside Rg1, ginsenoside Re, ginsenoside Rb1, coptisine, berberine, ligustilide, and ferulic acid (Yang et al., 2017). Here, the LC-MS/MS analyses of active components of YQF in the plasma and brain tissue (Table 1 and Supplementary DataTable 1) showed that many alkaloids like berberine, palmatine, worenine, and protopine, and ginsenosides like Rg1, Re, Rb1, Rd, and Rc were found in plasma, while berberine, palmatine, epiberberine, coptisine, and ginsenoside Rb1 were found in brain tissue and ferulic acid of Conioselinum anthriscoides ‘Chuanxiong’ (rhizome) was found in the plasma but not brain tissue. Since many of the active components of Conioselinum anthriscoides ‘Chuanxiong’ (rhizome) are volatile, we believe that not all of its components were detected by this method.
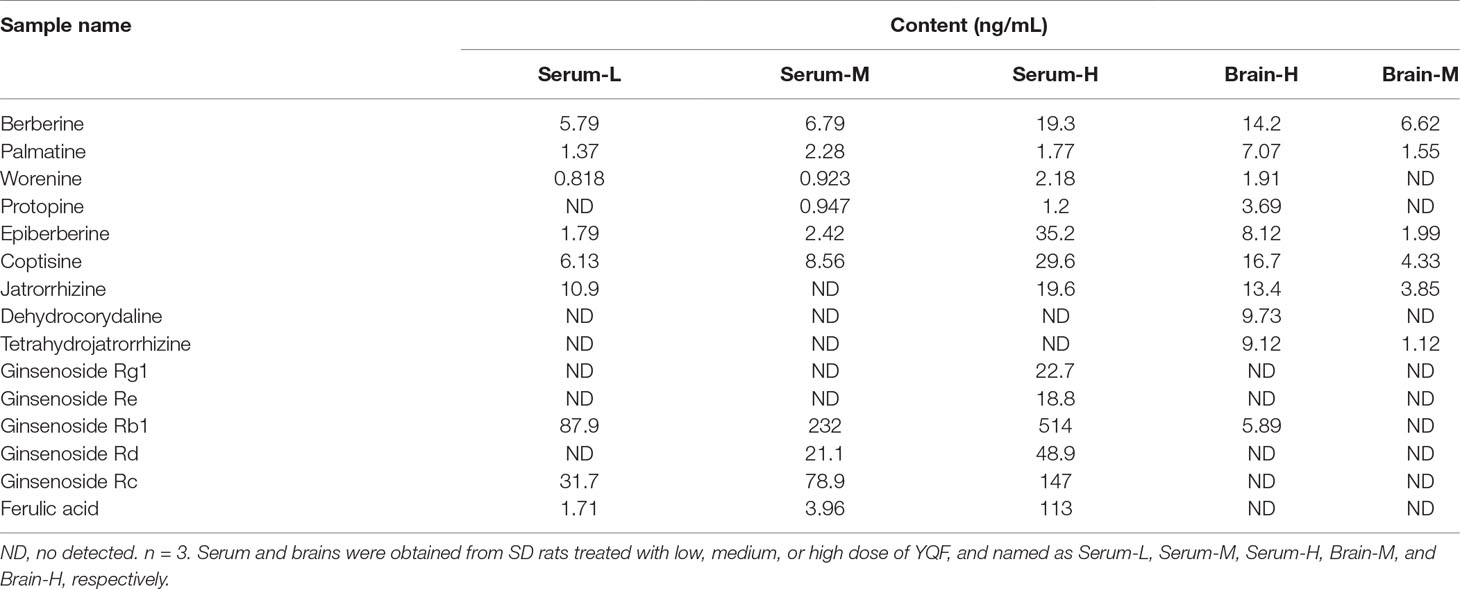
Table 1 Active components of YQF in the serum and brain tissue of SD rats.
YQF Treatment Alleviates Spatial Memory Deficits in APP/PS1 Mice
APP/PS1 mice cognitive–behavioral changes occur at 3 months of age (Garcia-Alloza et al., 2006), APP/PS1 mice have a substantial number of amyloid deposits by 6 months of age. The plaque burden increases progressively with time (Jankowsky et al., 2004). Therefore, APP/PS1 mice have well simulated the main pathological changes of AD and are currently recognized as the international animal model of AD. To investigate whether a long-term YQF treatment could ameliorate spatial learning deficits in transgenic APP/PS1 mice, MWM tests were performed. As shown in Figure 1A, all mice exhibited a significant decrease in escape latency over time. Two-way ANOVA reported a significant treatment effect on the escape latency [F(5, 53) = 3.237, p = 0.013]. However, APP/PS1 mice spent more time searching for the hidden platform than controls (p < 0.01 model group vs. control group), indicating a significant cognitive decline. After high dose YQF treatment, the escape latency was shortened remarkably (p < 0.01 YQF-H group vs. model group). Rapamycin also reduced the mean latency significantly (p < 0.05 Rapa group vs. model group) (Figure 1A).
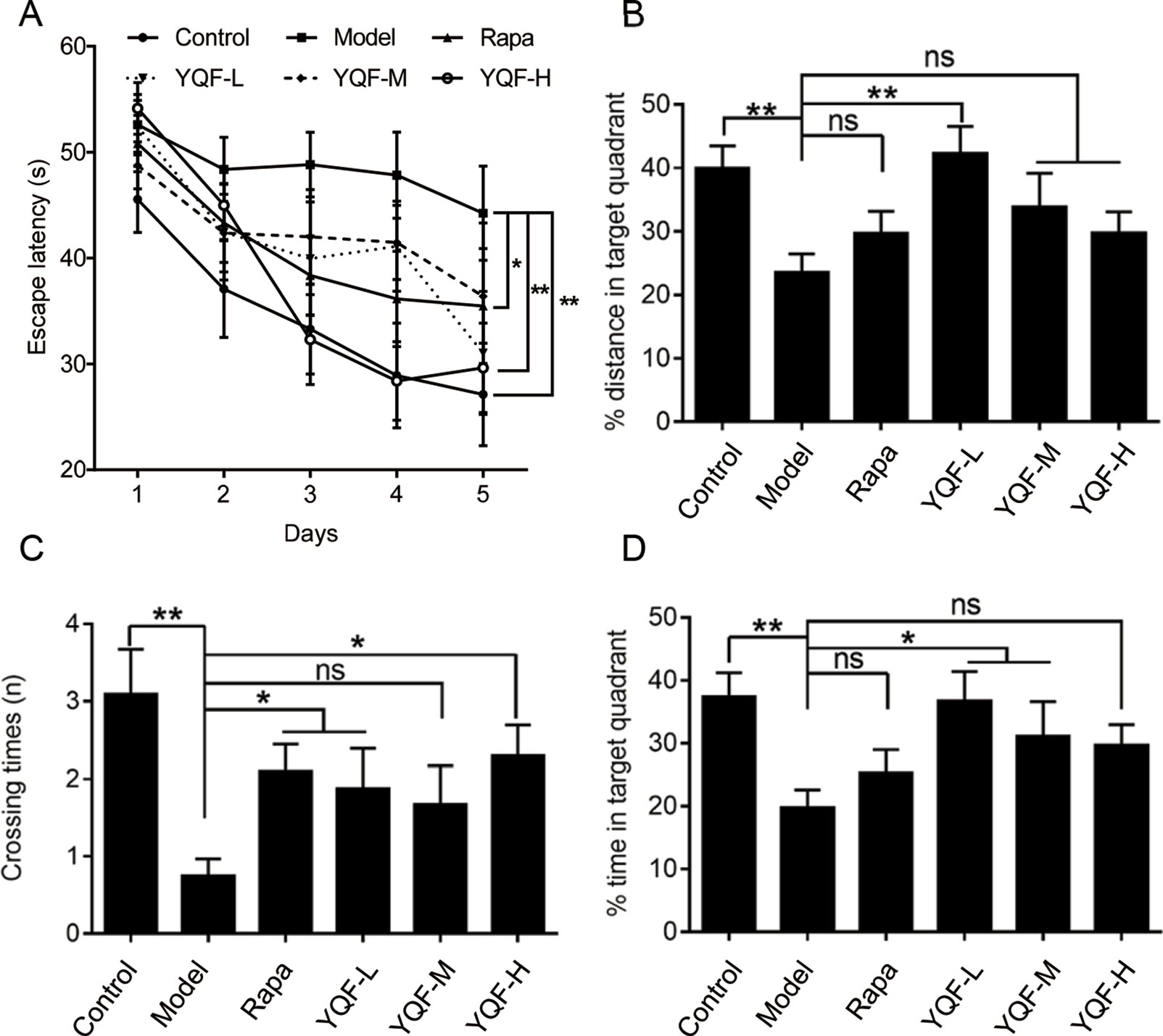
Figure 1 YQF alleviates spatial memory deficits of APP/PS1 mice (A) Escape latency, (B) Distance in the target quadrant, (C) Crossing times, (D) Time spent in the target quadrant, n = 8–12; *p < 0.05; **p < 0.01; ns, no significant difference.
Moreover, in the probe trial, APP/PS1 mice failed to remember the accurate location of the platform, and the crossover times were remarkably less than the Control group [F(5, 54) = 3.615 p = 0.007, p < 0.01 model group vs. control group]. Administration of rapamycin or high dose YQF significantly mitigated the deficits (p < 0.05 Rapa group vs. model group and p < 0.05 YQF-H group vs. model group). Besides, the mice treated with medium dose of YQF spent significantly more time in the target quadrant [F(5, 52) = 3.359 p = 0.011, p < 0.05 YQF-M group vs. model group]. The low dose-treated group also showed a higher percentage of the swimming distance and time in the target quadrant than the Model group [F(5, 52) = 3.712 p = 0.006, p < 0.01 YQF-L group vs. model group and F(5, 52) = 3.359 p = 0.011, p < 0.05 YQF-L group vs. model group]. These results indicate that YQF treatment improves the spatial learning and memory function in APP/PS1 mice (Figures 1B–D).
YQF Reduces Aβ Levels in the Hippocampus of APP/PS1 Mice and APP/PS1 Double Transgenic HEK293 Cells
To investigate the effect of YQF treatment on Aβ deposition and senile plaque formation, brain slices were stained with a monoclonal antibody against Aβ1-42 and quantitative image analysis was performed (Figures 2A, B). Results show that in 9-month-old APP/PS1 mice, immuno-positive diffuse plaques occupied a great deal of the hippocampal areas compared to non-transgenic mice [F(5, 24) = 11.618 p = 0.000, p < 0.01 model group vs. control group]. After YQF treatment, all YQF groups had significantly reduced Aβ burden (p < 0.05 YQF-L group vs. model group and p < 0.01 YQF-M/H group vs. model group). Rapamycin group also showed a remarkable reduction in Aβ burden (p < 0.01 Rapa group vs. model group).
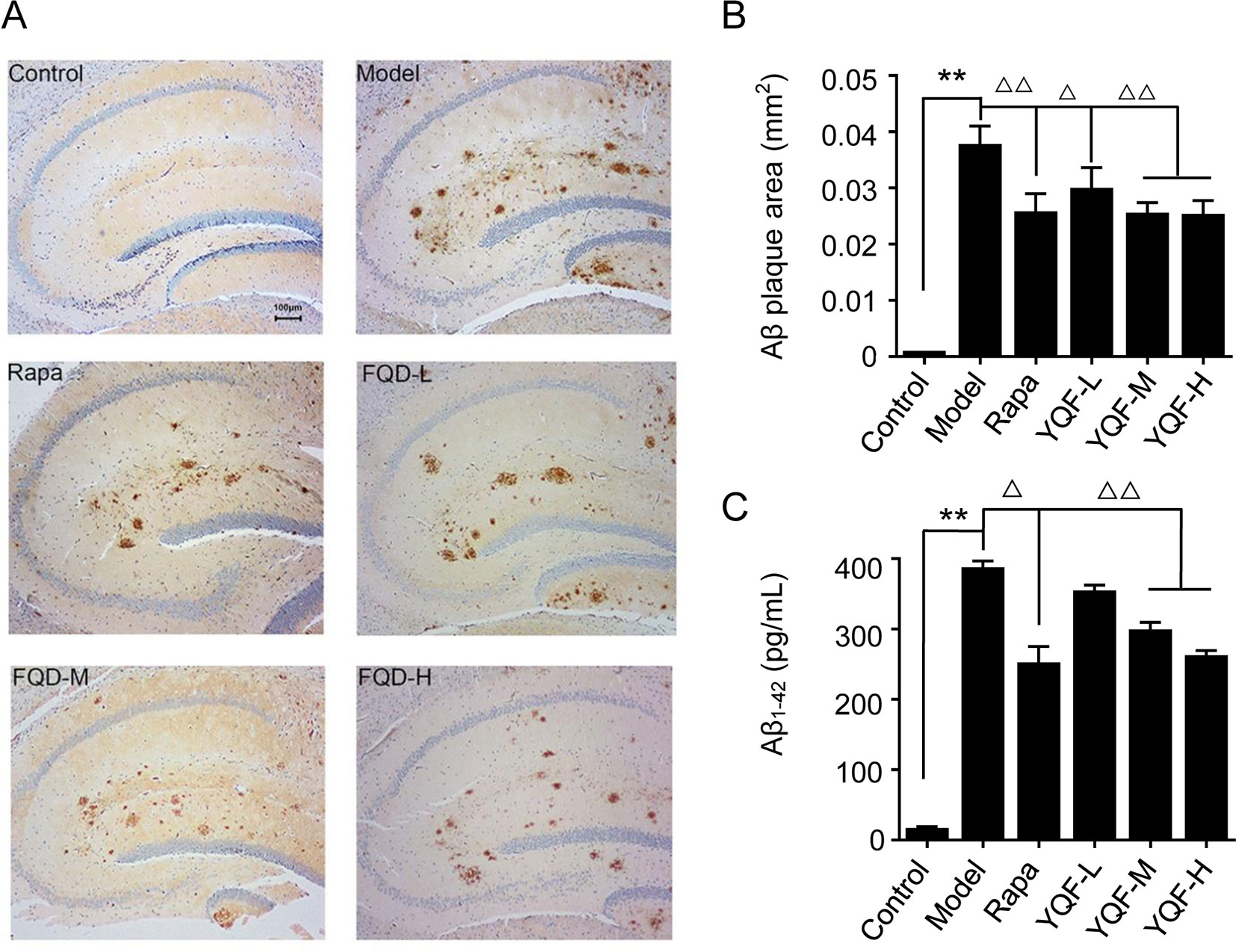
Figure 2 Expression of Aβ1-42 in hippocampus of mice (A and B) and HEK293 cells (C). Data are expressed as mean ± SEM, n = 5, **p < 0.01 compared with the control group, △p < 0.05 and △△p < 0.01 compared with the model group.
In vitro (Figure 2C), we also found that APP/PS1 double transgenic HEK293 cells expressed the highest level of Aβ1-42 compared with the other groups [F(5,18) = 130.531 p = 0.000, p < 0.01 model group vs. control group]; YQF could reduce the expression of Aβ1-42, especially in the YQF-M and YQF-H groups (p < 0.01 YQF-M/H group vs. model group).
YQF Promotes the Formation and Degradation of Autophagosome In Vivo and In Vitro
By transmission electron microscopy (Figure 3), we found using in vivo and vitro experiments that many large autophagosomes are assembled in APP/PS1 mice and cells compared with non-transgenic animals and cells. Mitochondria and other cytoplasmic components could be found in the autophagosomes. Furthermore, rapamycin could assemble large number of autophagosomes in neuronal cells and transgenic HEK293 cells as compared to the Model group. Every cell in the YQF groups exhibited autophagosomes more than that in Model groups but less than in Rapamycin groups. Moreover, we could also find that contents in the autophagic vacuoles of YQF groups were partly degraded, especially in the YQF-H group; apparently, a portion of the autophagic vacuoles were autophagolysosomes.
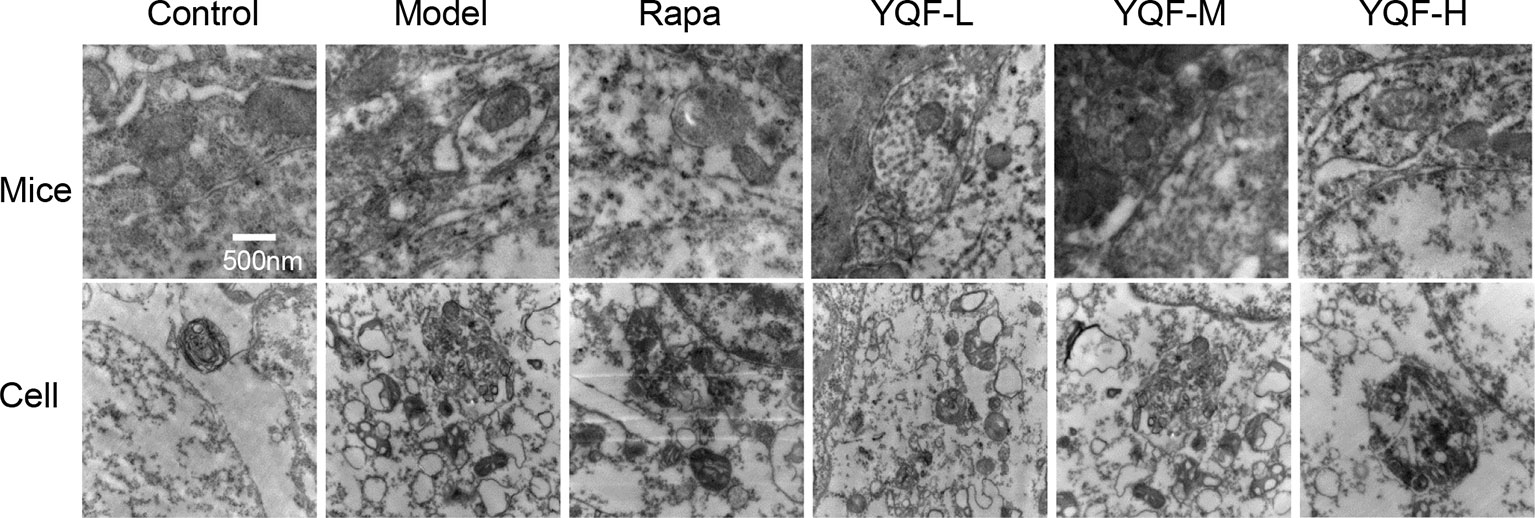
Figure 3 Ultrastructure of hippocampus of 9-month-old APP/PS1 transgenic mice, C57BL/6J mice, HEK293 cells, and APP/PS1 HEK293 cells. Autophagy vesicles were detected by transmission electron microscope after the treatment of rapamycin and YQF (500nm).
YQF Treatment Promotes Autophagy Activity In Vivo and In Vitro
Beclin 1, LC3, and P62 are protein markers of autophagy activity. In vitro and in vivo experiments revealed that Beclin 1 levels decreased significantly in Model group mice and cell lines [χ2 = 15.565 p = 0.008, p < 0.05 model group vs. control group in vivo and F(5,12) = 5.142, p = 0.009, p < 0.01 model group vs. control group in vitro]. With the formation and maturation of the autophagosome, LC3I converts to LC3II, and here, we take the LC3II/LC3I ratio as the sign of the formation of autophagosome. Although we did not find any differences in vivo between Model group mice and Control group mice, the LC3II/LC3I ratio of in vitro model cell lines decreased significantly [F(5,12) = 10.263, p = 0.001, p < 0.01 model group vs. control group]. Rapamycin could promote the expression of Beclin 1 (χ2 = 15.565 p = 0.008, p < 0.05 Rapa group vs. model group in vivo) and LC3II [F(5,12) = 10.263, p = 0.001, p < 0.05 Rapa group vs. model group in vitro], similar to the in vivo and in vitro effects of YQF (Beclin 1 p < 0.05 YQF-H group vs. model group in vivo, p < 0.01 YQF-L/H group vs. model group in vitro and LC3II, p < 0.01 YQF-L/H group vs. model group in vivo, p < 0.05 YQF-M/H group vs. model group in vitro) (Figure 4 and Figures 5A–C). P62 expresses rapidly with the formation and maturation of the autophagosome but decreases with the formation and degradation of autolysosomes. In this study, we found that P62 levels increased in Model groups [χ2 = 16.298 p = 0.006, p < 0.01 Rapa group vs. model group in vivo and F(5, 12) = 11.783 p = 0.000, p < 0.05 Rapa group vs. model group in vitro] but decreased in Rapamycin (p < 0.01 in vivo, p < 0.05 in vitro) and YQF groups (p < 0.01 YQF-L/M/H group vs. model group in vivo, p < 0.01 YQF-M/H group vs. model group in vitro) (Figure 4 and Figures 5A, D).
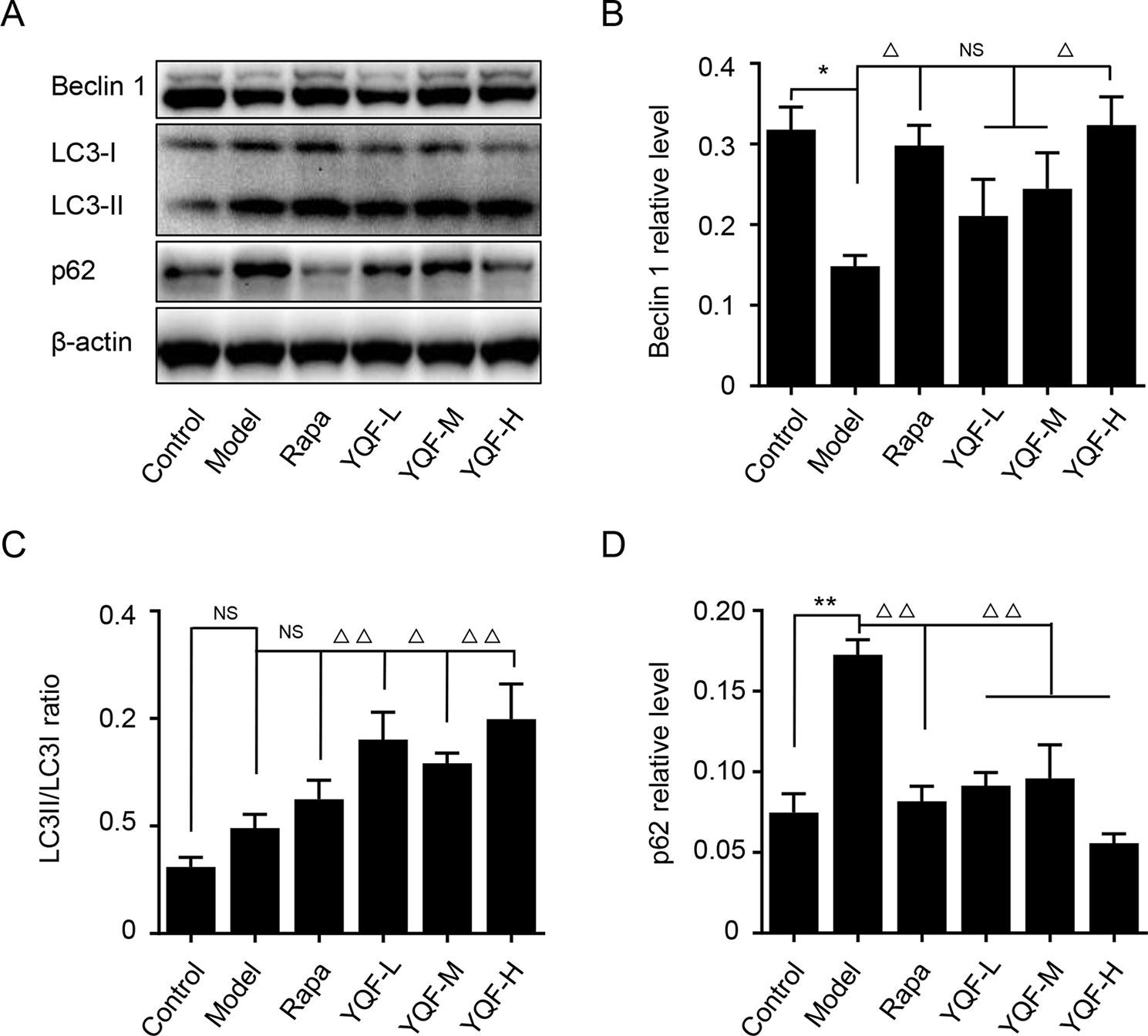
Figure 4 The expression of autophagy related proteins in APP/PS1 and C57BL/6J mice hippocampus intervened with YQF. (A) Western blot bands, (B) Beclin 1, (C) LC3/LC3, (D) P62. Data are expressed as mean ± SEM, n = 5, *p < 0.05 and **p < 0.01 compared with the control group, △p < 0.05 and △△p < 0.01 compared with the model group, NS, no significant difference.
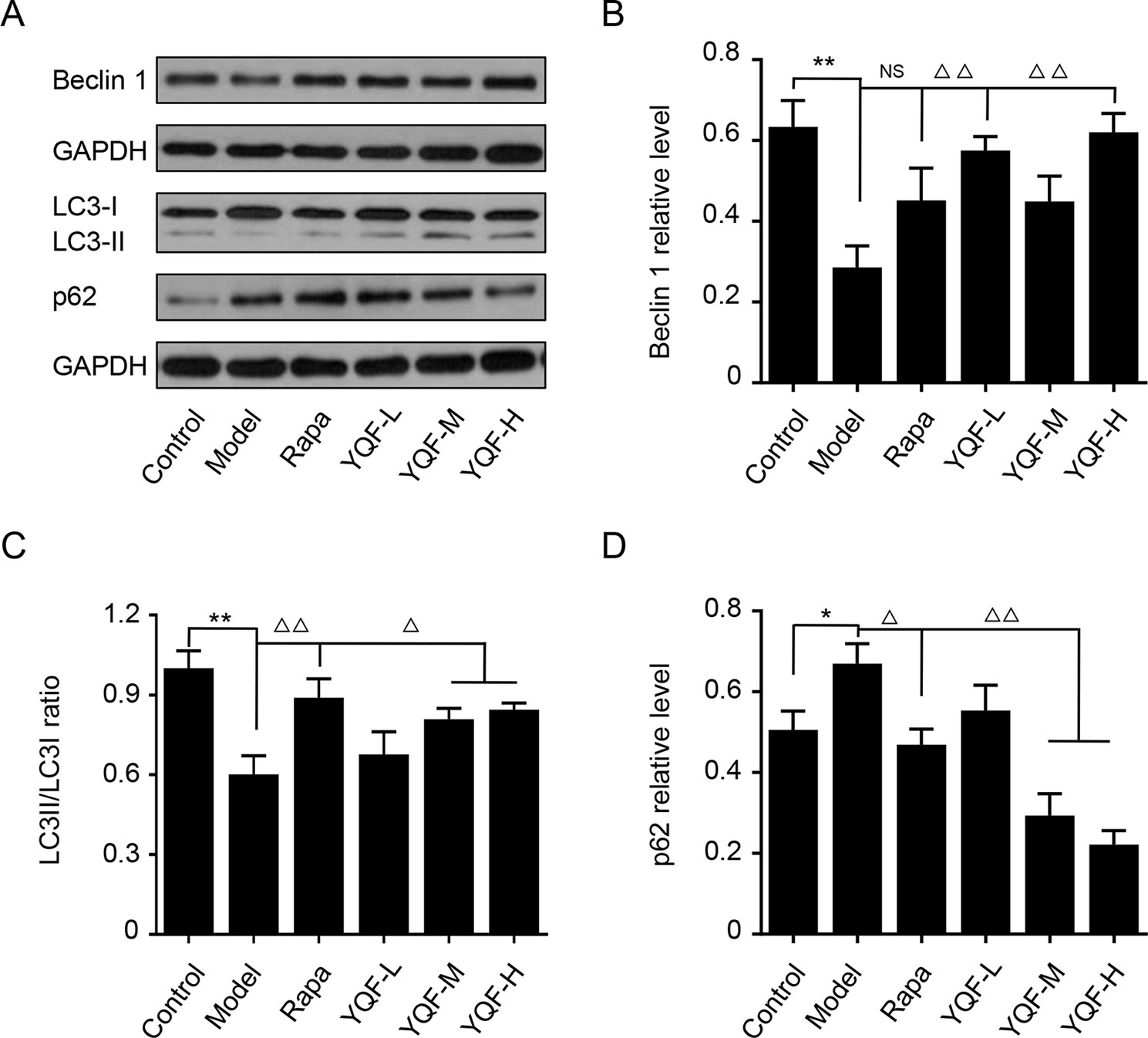
Figure 5 The expression of autophagy related proteins in APP/PS1 and HEk293 cells intervened with YQF. (A) Western blot bands, (B) Beclin 1, (C) LC3II/LC3I, (D) P62. Data are expressed as mean ± SEM, n = 3, *p < 0.05 and **p < 0.01 compared with the control group, △p < 0.05 and △△p < 0.01 compared with the model group, NS, no significant difference.
YQF Treatment Suppresses the Activity of mTOR Signaling Pathway
Autophagy is regulated by the mTOR signaling pathway, so we studied how it is affected by YQF. Our results showed that YQF could significantly suppress the activity of this pathway (Figure 6 and Figure 7). We considered p-mTOR/mTOR ratio as the indicator of mTOR activity, and we found significantly elevated mTOR activity in model mice and cells [F(5,24) = 7.950 p = 0.000, p < 0.05 model group vs. control group in vivo and F(5,12) = 1.990 p = 0.153, p < 0.05 model group vs. control group in vitro]. Rapamycin and YQF could reverse this condition (p < 0.05 Rapa group vs. model group in vivo and in vitro, p < 0.05 YQF-H group vs. model group in vivo and in vitro), in effect suppressing the activity of mTOR. P70S6K and 4EBP1 are two major downstream signaling molecules of mTOR; with the activation of mTOR, P70S6K is activated and 4EBP1 is suppressed. Here, we found that in each group, P70S6K varied directly with pmTOR/mTOR ratio and had an indirect relationship with 4EBP1 levels. However, we did not find significant differences in P70S6K levels between the different study groups. Although rapamycin could significantly promote the expression of 4EBP1, we did not find any difference between YQF groups and the Model group.
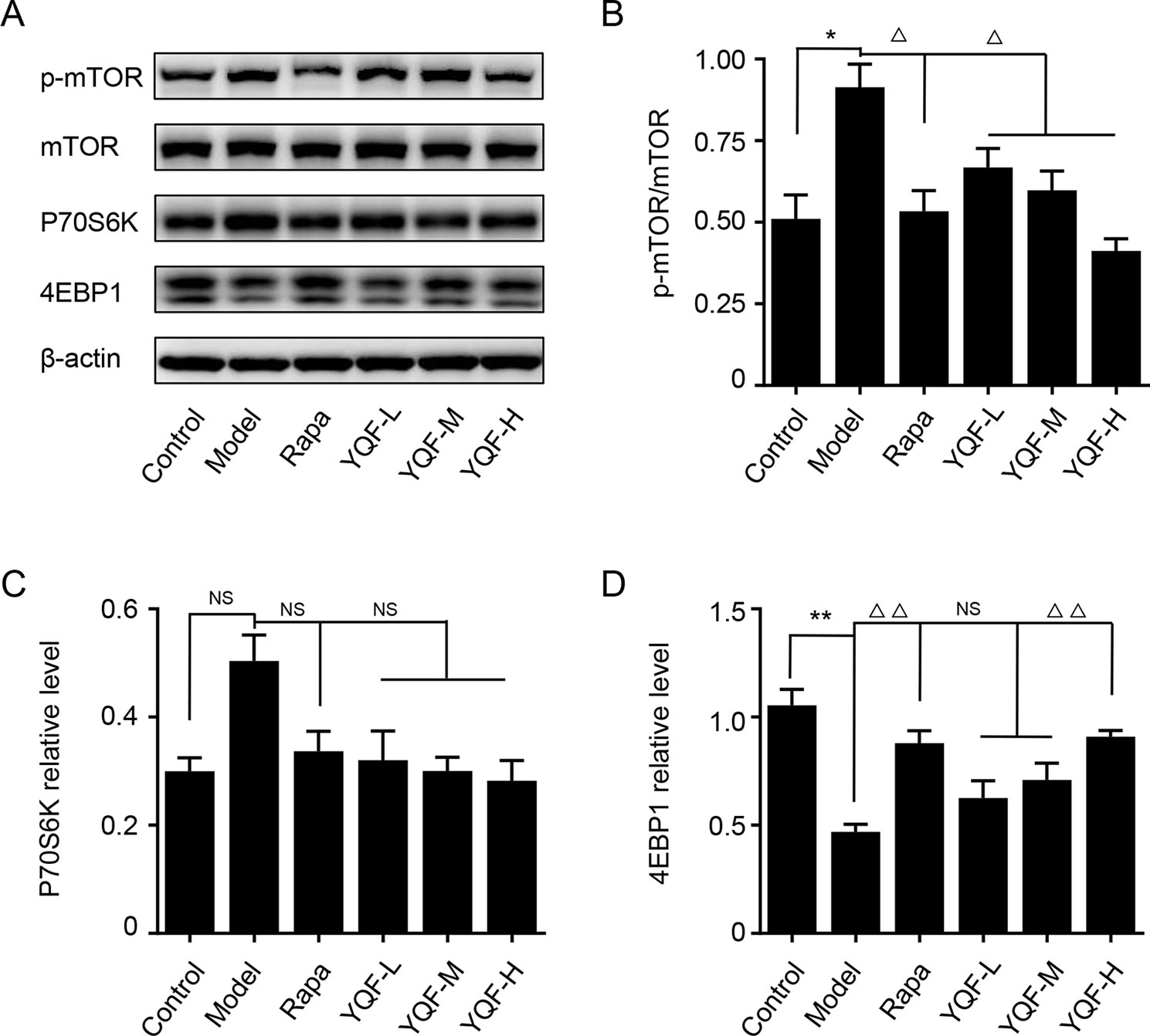
Figure 6 The expression of mTOR, 4EBP1, and P70S6K in APP/PS1 mice intervened with YQF. (A) Western blot bands, (B) p-mTOR/mTOR, (C) P70S6K, (D) 4EBP1. Data are expressed as mean ± SEM, n = 3, *p < 0.05 and **p < 0.01 compared with the control group, △p < 0.05 and △△p < 0.01 compared with the model group, NS, no significant difference.
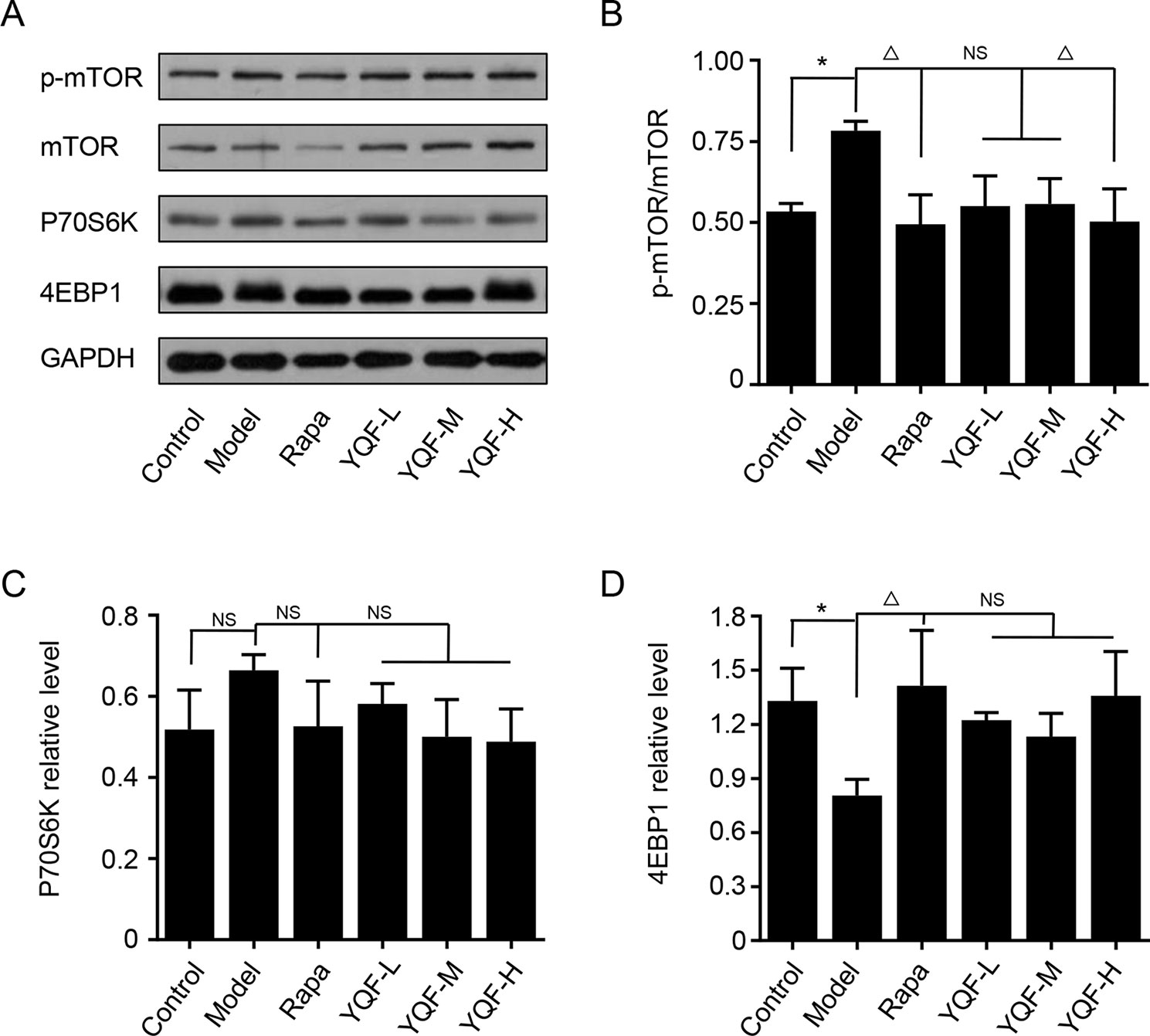
Figure 7 The expression of mTOR, 4EBP1, and P70S6K in HEk293 cells intervened with YQF. (A) Western blot bands, (B) p-mTOR/mTOR, (C) P70S6K, (D) 4EBP1. Data are expressed as mean ± SEM, n = 3, *p < 0.05 compared with the control group, △p < 0.05 compared with the model group, NS, no significant difference.
YQF Treatment Promotes the Degradation of Autolysosome In Vitro
To further explore the effect of YQF on the whole process of autophagy, we analyzed the levels of autolysosome related proteins like lysosomal-associated membrane protein 1 (LAMP-1), cathepsin D, and vacuolar-type H+-ATPase (V-ATPase) (Figure 8). We found that the levels of LAMP-1 in the Model group and the Control group were not significantly different [F(5,12) = 4.257 p = 0.019, p > 0.05 model group vs. control group]. Rapamycin could promote LAMP-1 expression (p < 0.05 Rapa group vs. model group). Although YQF could promote LAMP-1 expression, non-significant differences were found. Cathepsin D and V-ATPase were found to be significantly decreased in the Model group compared with the Control group [F(5,12) = 4.322 p = 0.018, p < 0.05 model group vs. control group and F(5,12) = 2.237 p = 0.113, p < 0.05 model group vs. control group]. Rapamycin could increase the cathepsin D and V-ATPase levels; however, the differences were non-significant. YQF could significantly enhance the expression of cathepsin D (p < 0.05 YQF-H group vs. model group) and V-ATPase (p < 0.05 YQF-L group vs. model group and p < 0.01 YQF-M/H group vs. model group).
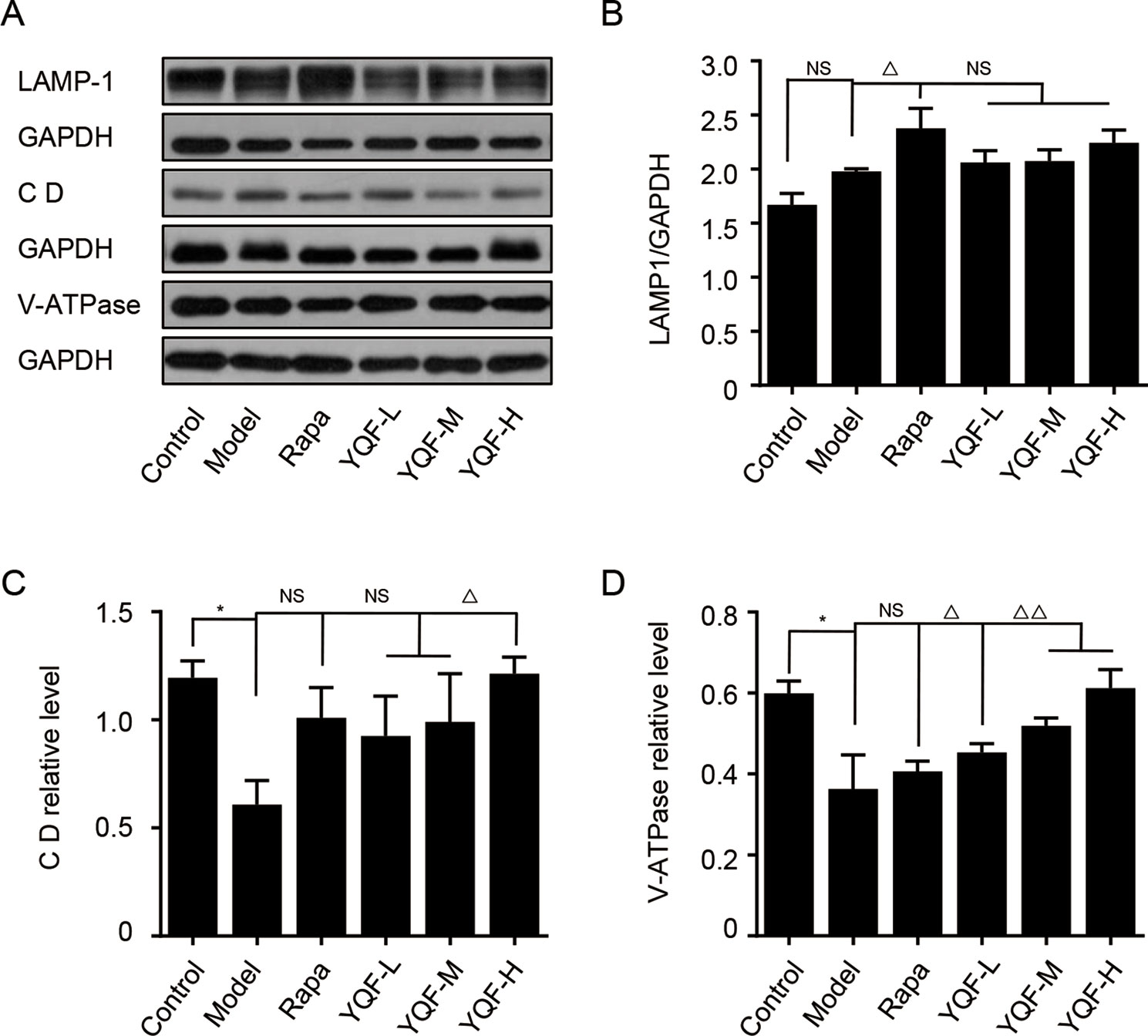
Figure 8 The expression of LAMP-1, Cathepsin D CD and V-ATPase in HEK293 cells intervened with YQF. (A) Western blot bands, (B) LAMP-1, (C) Cathepsin D, (D) V-ATPase. Data are expressed as mean ± SEM, n = 3, *p < 0.05 compared with the control group, △p < 0.05 and △△p < 0.01 compared with the model group, NS, no significant difference.
Discussion
Under physiological conditions, autophagy, by recycling damaged organelle and defective proteins, is crucial for the homeostasis of energy metabolism in neural cells. During normal aging or the progression of AD, autophagy activity gradually declines, attributing to the aggregation of Aβ (Pickford et al., 2008; Uchiyama et al., 2008), which is now widely considered as a prominent feature found in AD brain. The impairment of autophagy in AD has been substantiated by the accumulation of autophagosomes in dystrophic neurites (Tanida et al., 2005). Consistent with these results, activation of the autophagy pathway by rapamycin retards Aβ and Tau pathology and enhances cognitive functions (Caccamo et al., 2010; Majumder et al., 2011). Apart from these data, previous study also reported that autophagy participates in the extracellular Aβ secretion, thereby directly affecting Aβ plaque formation (Nilsson et al., 2013). Therefore, considerable efforts have been focusing on autophagy to discover potential targets for AD therapeutic interventions (Zhu et al., 2013; Jiang et al., 2014). Beclin 1, the mammalian orthologue of the yeast Vps30/Apg6 gene, functioning as a scaffold, is necessary for the formation of autophagosome. Previous study showed a reduction of Beclin 1 level in the entorhinal cortex of AD patients (Pickford et al., 2008). In APP transgenic mice, Beclin 1 deficiency exacerbates Aβ deposition and promotes synaptodendritic degeneration. On the contrary, Beclin 1 overexpression reduces amyloid pathology (Pickford et al., 2008). Our results suggest a significant reduction of Beclin 1, accompanied with an increased Aβ accumulation in the brain of APP/PS1 mice and APP/PS1 HEK293 cells. Furthermore, results demonstrate that YQF or rapamycin treatment elevates the level of Beclin 1 and reduces the formation of Aβ and amyloid plaques.
Microtubule-associated protein 1 light chain 3 (LC3 I) plays a key role in autophagosome biogenesis/maturation. During autophagy, a cytosolic form of LC3 (LC3 I) is converted from a nonlipidated form (LC3 I) to a phosphatidylethanolamine (PE)-conjugated form (LC3 II), which is recruited to autophagosomal membranes. LC3 II specifically localizes in autophagosome membranes, and it is therefore considered a specific autophagy marker (Lai and Devenish, 2012). Sequestosome-1 (SQSTM1)/p62 is a ubiquitin-LC3-binding protein and also an adaptor protein for the autophagic degradation of misfolded proteins or organelles. p62 itself is a selective autophagy substrate targeted by the autophagy–lysosome degradation system. Hence, the level of p62 inversely correlates with autophagy activity and is considered as an important biomarker for autophagy degradation (Tanida et al., 2005). In accordance with previous data (Lee et al., 2014; Guo et al., 2015), here we show that an impaired autophagic flux is present, as indicated by an increase of LC3 II/LC3 I and p62 level in vivo in model mice, whereas with the in vitro study, LC3 II/LC3 I decreased but the level of p62 increased in APP/PS1 double transgenic HEK293 cells, indicating that both the formation and degradation of autophagosome are obstructed in AD. Importantly, in this study, treatment with different doses of YQF rectified the aberrant autophagy activity. Our results show that YQF treatment increases the LC3 II/LC3 I ratio and reduces p62 levels.
In order to explore the underlying mechanism, we evaluated the phosphorylation of mTOR and the lysosomal function. mTOR is a critical negative regulator of autophagy, which senses and integrates signals under various conditions including growth factors, amino acids, hypoxia, and energy levels (Liang, 2010; Kim and Guan, 2015). In the brain, it is primarily expressed in neurons and is involved in synaptic plasticity and cognition (Talboom et al., 2015). In AD, there is ample evidence suggesting that hyperactive mTOR contributes to the accumulation of Aβ and cognitive impairment. mTOR inhibitors reduce AD hallmarks and protect cognitive function (Godoy et al., 2014; Tramutola et al., 2017). In this study, we found that 9-month-old APP/PS1 mice and APP/PS1 double transgenic HEK293 cells exhibit hyperphosphorylated mTOR compared with age-matched wild-type control C57BL/6J mice and non-transfected HEK293 cells, which is in accordance with previous study (Zhou et al., 2008). Our study found that YQF and rapamycin treatments retard the abnormal phosphorylation, and thus inhibit the activity of mTOR and promoted autophagy. By phosphorylating the downstream signaling molecules, mTOR activates P70S6K and inactivates 4EBP1, thereby exerting intracellular signal transduction (Perluigi et al., 2015). Although we did not find significant changes in P70S6K activity in each group, we found that the levels of 4EBP1 decreased significantly in Model groups and increased after the in vivo YQF treatment, further proof that YQF inhibits the activity of mTOR.
Formation of autolysosomes by the fusion of autophagosome and lysosome is needed for the subsequent completion of substrate clearance. Lysosome function is crucial in this step. Studies found that lysosome dysfunction may be the most important point in the autophagy disorder in AD (Bordi et al., 2016; Ma et al., 2017). LAMP-1 is a lysosome-associated membrane glycoprotein, cathepsin D is an aspartic endo-protease that is ubiquitously distributed in lysosomes, and V-ATPase is an ATP-dependent proton pump (transporting H+ ions into the lysosome) that maintains the acidity of lysosomes. Our in vitro studies revealed that LAMP-1 levels were largely the same between different groups, but the levels of cathepsin D and V-ATPase decreased significantly in the Model groups, while YQF could increase their levels. This phenomenon indicates that while the levels of lysosomes are not significantly changed in different groups, their functions changed; lysosome function was impaired in Model groups, and YQF could alleviate this defect.
In conclusion, in the present study, we show that long-term administration of YQF ameliorates learning and memory deficits in the APP/PS1 mice. The presence of Aβ plaques is a key pathological feature of AD. Here, immunohistochemistry results demonstrate that YQF treatment mitigates the formation of amyloid plaques and decreases the levels of Aβ. The results from ELISA assays show that YQF could up-regulate Beclin-1 and LC3 II, down-regulate p62, and inhibit the activity of the mTOR signaling pathway, in vivo and in vitro. Thus, we conclude that YQF can improve cognitive function and promote Aβ clearance by activating autophagy function via suppressing the mTOR pathway, which may provide new insight in the treatment of AD. We have detected several active components of YQF, but we cannot sure which one play the critical role. We will do some more work to research which active components play the key role in YQF.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the animal ethics committee of the Ethics Committee of Xiyuan Hospital, China Academy of Chinese Medical Sciences.
Author Contributions
YY and ZW contributed equally to this work. YY and ZW performed experiments and data analysis and wrote the manuscript. YC assisted in the in vivo experiments and manuscript writing. PL assisted in the in vitro experiments. HL and JL supervised the experiments. HL and ML conceived the study and designed the experiments. All the authors reviewed and approved the manuscript.
Funding
The study was funded by the National Natural Science Foundation of China (No. 81503628, 81573819, and 81873350).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors HL, YY, ZW, YC, JL, PL, ML at the time of the review.
Acknowledgments
The authors thank the National Natural Science Foundation of China for the financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.01041/full#supplementary-material
References
Alzheimer’s Association (2018). Alzheimer’s Association Report 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 14, 367–429. doi: 10.1016/j.jalz.2018.02.001
Bordi, M., Berg, M. J., Mohan, P. S., Peterhoff, C. M., Alldred, M. J., Che, S., et al. (2016). Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483. doi: 10.1080/15548627.2016.1239003
Caccamo, A., Majumder, S., Richardson, A., Strong, R., Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.M110.100420
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., et al. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 24 (3), 516–524. doi: 10.1016/j.nbd.2006.08.017
Godoy, J. A., Rios, J. A., Zolezzi, J. M., Braidy, N., Inestrosa, N. C. (2014). Signaling pathway cross talk in Alzheimer’s disease. Cell Commun. Signal. 12, 23. doi: 10.1186/1478-811X-12-23
Guo, Z., Sun, H., Zhang, H., Zhang, Y. (2015). Anti-hypertensive and renoprotective effects of berberine in spontaneously hypertensive rats. Clin. Exp. Hypertens. 37, 332–339. doi: 10.3109/10641963.2014.972560
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170. doi: 10.1093/hmg/ddh019
Jia, J., Wang, F., Wei, C., Zhou, A., Jia, X., Li, F., et al. (2014). The prevalence of dementia in urban and rural areas of China. Alzheimers Dement. 10, 1–9. doi: 10.1016/j.jalz.2013.01.012
Jia, J., Wei, C., Chen, S., Li, F., Tang, Y., Qin, W., et al. (2018). The cost of Alzheimer’s disease in China and re-estimation of costs worldwide. Alzheimers Dement. 14, 483–491. doi: 10.1016/j.jalz.2017.12.006
Jiang, T., Yu, J. T., Zhu, X. C., Tans, M. S., Wang, H. F., Cao, L., et al. (2014). Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 81, 54–63. doi: 10.1016/j.phrs.2014.02.008
Kim, Y. C., Guan, K. L. (2015). MTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25–32. doi: 10.1172/JCI73939
Lai, S. C., Devenish, R. J. (2012). LC3-associated phagocytosis (LAP): connections with host autophagy. Cells 1, 396–408. doi: 10.3390/cells1030396
Lee, J. K., Jin, H. K., Park, M. H., Kim, B. R., Lee, P. H., Nakauchi, H., et al. (2014). Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 211, 1551–1570. doi: 10.1084/jem.20132451
Liang, C. (2010). Negative regulation of autophagy. Cell Death Differ. 17, 1807–1815. doi: 10.1038/cdd.2010.115
Ma, L. Y., Lv, Y. L., Huo, K., Liu, J., Shang, S. H., Fei, Y. L., et al. (2017). Autophagy-lysosome dysfunction is involved in Abeta deposition in STZ-induced diabetic rats. Behav. Brain Res. 320, 484–493. doi: 10.1016/j.bbr.2016.10.031
Majumder, S., Richardson, A., Strong, R., Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6, e25416. doi: 10.1371/journal.pone.0025416
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69. doi: 10.1016/j.celrep.2013.08.042
Oddo, S. (2012). The role of mTOR signaling in Alzheimer disease. Front. Biosci. (Schol Ed) 4, 941–952. doi: 10.2741/s310
Perluigi, M., Di Domenico, F., Butterfield, D. A. (2015). mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 84, 39–49. doi: 10.1016/j.nbd.2015.03.014
Pickford, F., Masliah, E., Britschqi, M., Lucin, K., Narsimhan, R., Jaegar, P. A., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Invest. 118, 2190–2199. doi: 10.1172/JCI33585
Ryter, S. W., Cloonan, S. M., Choi, A. M. (2013). Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol. Cells 36, 7–16. doi: 10.1007/s10059-013-0140-8
Souchet, B., Audrain, M., Billoir, B., Lecanu, L., Tada, S., Braudeau, J., et al. (2018). Is it time to rethink the Alzheimer’s disease drug development strategy by targeting its silent phase? Neural Regen Res. 13, 224–225. doi: 10.4103/1673-5374.226389
Sun, Y. X., Ji, X., Mao, X., Xie, L., Jia, J., Galvan, V., et al. (2014). Differential activation of mTOR complex 1 signaling in human brain with mild to severe Alzheimer’s disease. J. Alzheimers Dis. 38, 437–444. doi: 10.3233/JAD-131124
Talboom, J. S., Velazquez, R., Oddo, S. (2015). The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease. NPJ Aging Mech. Dis. 1, 15008. doi: 10.1038/npjamd.2015.8
Tanida, I., Minematsu-Ikeguchi, N., Ueno, T., Kominami, E. (2005). Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1, 84–91. doi: 10.4161/auto.1.2.1697
Tramutola, A., Lanzillotta, C., Di Domenico, F. (2017). Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev. Neurother. 17, 33–45. doi: 10.1111/jnc.13037
Uchiyama, Y., Shibata, M., Koike, M., Yoshimura, K., Sasaki, M. (2008). Autophagy–physiology and pathophysiology. Histochem. Cell Biol. 129, 407–420. doi: 10.1007/s00418-008-0406-y
Wang, F., Feng, J., Yang, Y., Liu, J., Liu, M., Wang, Z., et al. (2018). The Chinese herbal formula Fuzheng Quxie decoction attenuates cognitive impairment and protects cerebrovascular function in SAMP8 mice. Neuropsychiatr Dis. Treat. 14, 3037–3051. doi: 10.2147/NDT
Wang, X., Zheng, W., Xie, J. W., Wang, T., Wang, S. L., Teng, W. P., et al. (2010). Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol. Neurodegener. 5, 46. doi: 10.1186/1750-1326-5-46
Yang, Y., Jia, X., Feng, J., Wang, Z., Cao, Y., Liu, J., et al. (2017). Fuzheng Quxie decoction ameliorates learning and memory impairment in SAMP8 mice by decreasing tau hyperphosphorylation. Evid. Based Complement. Alternat. Med. 2017, 5934254. doi: 10.1155/2017/5934254
Zhou, X. W., Tanila, H., Pei, J. J. (2008). Parallel increase in p70 kinase activation and tau phosphorylation (S262) with Aβ overproduction. FEBS Lett. 582, 159–164. doi: 10.1016/j.febslet.2007.11.078
Keywords: Alzheimer’s disease, Yizhiqingxin formula, autophagy, mTOR, APP/PS1 mice
Citation: Yang Y, Wang Z, Cao Y, Liu J, Li P, Li H and Liu M (2019) Yizhiqingxin Formula Alleviates Cognitive Deficits and Enhances Autophagy via mTOR Signaling Pathway Modulation in Early Onset Alzheimer’s Disease Mice. Front. Pharmacol. 10:1041. doi: 10.3389/fphar.2019.01041
Received: 07 June 2019; Accepted: 16 August 2019;
Published: 17 September 2019.
Edited by:
Yue Liu, Xiyuan Hospital, ChinaReviewed by:
Jun Xu, Capital Medical University, ChinaYan-Fang Xian, The Chinese University of Hong Kong, Hong Kong
Copyright © 2019 Yang, Wang, Cao, Liu, Li, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Li, xyhplihao1965@126.com; Meixia Liu, MTg3MDE1MjIzNjJAMTI2LmNvbQ==
†These authors have contributed equally to this work