 Zhonglian Zhang
Zhonglian Zhang Yue Zhang
Yue Zhang Meifang Song2
Meifang Song2 Xiaojun Ma
Xiaojun Ma- 1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 2Yunnan Branch of Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Jinghong, China
The taxonomy and nomenclature of Dracaena plants are much disputed, particularly for several Dracaena species in Asia. However, neither morphological features nor common DNA regions are ideal for identification of Dracaena spp. Meanwhile, although multiple Dracaena spp. are sources of the rare traditional medicine dragon’s blood, the Pharmacopoeia of the People’s Republic of China has defined Dracaena cochinchinensis as the only source plant. The inaccurate identification of Dracaena spp. will inevitably affect the clinical efficacy of dragon’s blood. It is therefore important to find a better method to distinguish these species. Here, we report the complete chloroplast (CP) genomes of six Dracaena spp., D. cochinchinensis, D. cambodiana, D. angustifolia, D. terniflora, D. hokouensis, and D. elliptica, obtained through high-throughput Illumina sequencing. These CP genomes exhibited typical circular tetramerous structure, and their sizes ranged from 155,055 (D. elliptica) to 155,449 bp (D. cochinchinensis). The GC content of each CP genome was 37.5%. Furthermore, each CP genome contained 130 genes, including 84 protein-coding genes, 38 tRNA genes, and 8 rRNA genes. There were no potential coding or non-coding regions to distinguish these six species, but the maximum likelihood tree of the six Dracaena spp. and other related species revealed that the whole CP genome can be used as a super-barcode to identify these Dracaena spp. This study provides not only invaluable data for species identification and safe medical application of Dracaena but also an important reference and foundation for species identification and phylogeny of Liliaceae plants.
Introduction
Dracaena Vand. ex L., belonging to the Dracaeneae tribe of Liliaceae, comprises approximately 50 species that are mainly distributed in tropical Africa, Australia, and Asia (Chen and Turland, 2000). In China, there are six species of Dracaena, mainly distributed in Yunnan, Guangxi, and Hainan Provinces (Chen and Turland, 2000). At present, there are many disputes over the taxonomy of the Dracaena plant, especially with regard to several Dracaena spp. in Asia that has been studied very little for several decades (Chen and Turland, 2000; Paul et al, 2012), consequently there are many taxononomic and nomenclatural problems and large numbers of unidentified specimens in herbaria (Chen and Turland, 2000; Yan, 2005). In addition, the traditional classification of the genus Dracaena is mainly based on leaf sheath, leaf blade central costa, tepals, and flaments (Wilkin et al.,2013). However, the diversity of natural environment in Southeast Asia area results in significant morphological variations of the Dracaena species, making it difficult to accurately identify the Dracaena species according to the above identification characteristics. As for the molecular identification, previous studies have demonstrated that barcoding sequences (e.g., ITS, trnL, matK, psbA-trnH, and rbcL) are also not ideal for accurate identification of Dracaena spp. (Li, 2012; Zheng, 2013; Wang et al., 2015).
In application, Dracaena species are usually used as ornamental horticultural plants or as source plants of rare medicinal material dragon’s blood (Adolt and Pavlis, 2004; Aslam et al., 2013). Dragon’s blood is a deep red resin that is widely used throughout the world; it is known to enhance immune function, promote skin repair, stop bleeding, and enhance blood circulation (Fan et al., 2014), and it has been utilized as a traditional medicine for wounds, fractures, piles, leucorrhea, diarrhea, stomach and intestinal ulcers, and even some types of cancer in the histories of many cultures (Gupta et al., 2007; Tapondjou et al., 2008; Kougan et al., 2010; Xu et al., 2010). Modern chemical and pharmacological studies have indicated that the flavonoids, stilbene, saponins, and terpenes in dragon’s blood are the main effective compounds (Hu et al., 2001; González et al., 2004; Chen et al., 2012; Mei et al., 2013; Fan et al., 2014). Among the Dracaena plant, multiple Dracaena species are the source plants of dragon’s blood (Gupta et al., 2007; Lyons, 1974) such as D. cinnabari Balf.f, D. draco L, Dracaena cochinchinensis (Lour.) S.C.Chen and D. cambodiana Pierre ex Gagnp (Gupta et al., 2007; Pearson and Prendergast, 2001), but the Pharmacopoeia of the People’s Republic of China has defined D. cochinchinensis as the only source plant. At present, dragon’s blood resources in China mainly depend on imported supplies from Thailand, Laos, Myanmar, and other countries. However, the Dracaena species in these areas are more abundant, the inaccurate identification of the Dracaena species will inevitably affect the clinical application effect of dragon’s blood. Therefore, the identification of molecular markers that can unambiguously distinguish the Dracaena spp. is critical for ensuring the beneficial effects of medicinal products.
The chloroplast (CP), an organelle that converts solar energy to carbohydrates through photosynthesis, has important roles in the biosynthesis of starch, fatty acids, amino acids, and pigments (Jansen and Ruhlman, 2012; Zhao et al., 2015; Daniell et al., 2016). The CP genome is a circular DNA molecule that includes a small single-copy (SSC) region, a large single-copy (LSC) region, and two inverted repeats (IRa and IRb) (Sato et al., 1999). It is highly conserved (Tonti-Filippini et al., 2017) in plants and therefore ideal for ecological, evolutionary, and diversity studies (Wicke et al., 2011). Recently, researchers have used the CP genome as a super-barcode to distinguish species (Xia et al., 2016; Chen et al., 2018b), or have screened sequences from the whole CP genome for species identification (Hu et al., 2016; Thode and Lohmann, 2019; Zhang et al., 2017a). At present, several CP genomes from Liliaceae have previously been reported and deposited in the National Center for Biotechnology Information (NCBI) database. However, from the largest genus of tribe Dracaeneae, Dracaena, only one CP genome (D. cambodiana) has been published previously (Zhu et al., 2018). In this study, we report the CP genomes of six Dracaena spp., including D. cochinchinensis (Lour.) S.C.Chen, D. cambodiana Pierre ex Gagnep, D. angustifolia (Medik.) Roxb, D. terniflora Roxb., D. hokouensis G.Z.Ye, and D. elliptica Thunb. & Dalm., obtained via high-throughput Illumina sequencing technology. Our aim is to utilize the CP genome as a super-barcode for the identification of Dracaena spp. and to provide invaluable genetic information for understanding their phylogenetic relationships and other future studies.
Materials and Methods
Plant Materials, DNA Extraction, and Illumina Sequencing
Plant materials from six Dracaena spp. were collected from the Yunnan Branch of the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences; the voucher specimens were deposited in the Institute of Medicinal Plant Development herbarium. Total genomic DNA was extracted from clean leaves from samples frozen at −80°C using the TaKaRa MiniBEST Universal Genomic DNA Extraction Kit (TaKaRa, Kusatsu, Japan) with a standard protocol, DNA quality was assessed using a Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA) and by electrophoresis in a 1% (w/v) agarose gel. The OD260/280 values ranged from 1.8 to 2.0, and >2 µg of DNA was equally pooled from individuals of the six species to generate shotgun libraries. cpDNA samples were randomly sheared, incubated with fragmentation buffer, and broken into 300–500-bp fragments in an M220 focused ultrasonicator (Covaris, Woburn, MA, USA). Connect the A & B connector at both ends of the DNA fragment, Screen the segments, then remove the self-connecting segments of the connector. Fragment screening by electrophoresis in agarose gel, keep the fragment with A connector at one end and B connector at the other end, which subsequent addition of NaOH to denatures, produce single-stranded DNA fragments. The library was sequenced using Illumina HiSeq 4000 sequencing platform at the Major Company (Shanghai, China).
Genome Assembly and Annotation
Raw data for the six Dracaena spp. were generated with a paired-end read length of 150 bp. The low-quality reads (Q < 20) were filtered out and the clean reads were used for CP genome assembly. A reference database was created by downloading all plant CP sequences from NCBI. The high-quality reads were mapped to the database, and the mapped reads were extracted based on sequence coverage and similarity. The extracted reads were assembled into contigs using SOAPdenovo2 (Luo et al., 2012), and the resulting contigs were combined and extended to obtain complete CP genome sequence. Finally, the positions of the LSC, SSC, and two IR regions of the CP genomes were determined by localization, and complete CP genomes were obtained. Annotation of the six complete Dracaena CP genomes were executed using the online program Dual Organellar GenoMe Annotator (DOGMA, http://dogma.ccbb.utexas.edu/) (Wyman et al., 2004) coupled with manual correction. The tRNAscan-SE software (v2.0, University of California, Santa Cruz, CA, USA) (Schattner et al., 2005) and DOGMA (Wyman et al., 2004) were used to identify the tRNA genes. The BLAST versus reference sequences was used for verifying boundaries genes, intron/exon and coding regions. Moreover, MEGA 6.0 was used to analyze the GC content (Tamura et al., 2013). The Organellar Genome DRAW (OGDRAW) (v1.2, Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany) (Lohse et al., 2007) was used with default settings to draw the gene map, which was then checked manually. Finally, a.sqn file was generated to submit the sequences to NCBI. The complete and correct CP genome sequences of the six Dracaena species were deposited in GenBank, accession numbers are MN200193 (D. angustifolia), MN200194 (D. cambodiana), MN200195 (D. cochinchinensis), MN200196 (D. elliptica), MN200197 (D. hokouensis), and MN200198 (D. terniflora) respectively.
Genome Structure and Comparative Genome Analysis
The CodonW software (University of Texas, Houston, TX, USA) was used to investigate the distribution of codons based on the relative synonymous codon usage (RSCU) ratio (Sharp and Li, 1987). Conserved sequences were identified between the CP genomes of D. cochinchinensis and those of D. cambodiana, D. angustifolia, D. terniflora, D. hokouensis, and D. elliptica by BLASTN analysis with an E-value cutoff of 1e−10. The mVISTA (Frazer et al., 2004) program was used in Shuffle-LAGAN mode to compare the six Dracaena CP genomes using the D. cochinchinensis CP genome as a reference.
Repeat Sequence Analysis
RSCU value, which is the ratio between the frequency of use and the expected frequency of a particular codon, was used to detected non-uniform synonymous codon usage within a coding sequence (Sharp and Li, 1987). Moreover, simple sequence repeats (SSRs) were detected using the MISA software (http://pgrc.ipk-gatersleben.de/misa/), with parameters set to encompass the number of repeat units of a mononucleotide SSR ≥10 nucleotides in length, followed by ≥5 and ≥4 repeat units for di- and tri-nucleotide SSRs, respectively, and ≥3 repeat units for tetra-, penta-, and hexa-nucleotide SSRs. The sizes and locations of repeat sequences in the CP genomes of the six Dracaena spp. were identified using REPuter (University of Bielefeld, Bielefeld, Germany) (Kurtz et al., 2001) with the parameters set to a similarity percentage of scattered repeat copies ≥90% and a minimal repeat size of 30 bp.
Phylogenetic Analysis
To determine phylogenetic positions of the six Dracaena species within Liliaceae, we analyzed the CP genomes of 37 species, encompassing 31 additional taxa within this lineage. The CP genome sequences of 31 species were downloaded from NCBI. The sequences were initially compared using MAFFT (Katoh et al., 2005), followed by multiple sequence visual analysis and manual adjustment using BioEdit (Hall, 1999). We used the CP genomes of Stemona japonica (Blume) Miq. (NC_039675.1) as outgroups and constructed phylogenetic trees employing 37 CP genomes sequences using the maximum parsimony (MP) and neighbor-joining (NJ) methods in MEGA 6.0 (Tamura et al., 2013) with 1000 bootstrap replicates.
Results and Discussion
CP Genomes of Six Dracaena spp.
At present, complete CP genome sequencing is feasible with the genome skimming approach as a result of the wide application of next-generation DNA sequencing technology and bioinformatics software tools (Twyford and Ness, 2016). Illumina technology can be used for assembling the complete CP genome without the tedious of separating CP DNA from nuclear DNAs (Wang et al., 2016a). The raw data from the six Dracaena species is 7.04 Gb for D. cochinchinensis, 7.02 Gb for D. cambodiana, 6.59 Gb for D. angustifolia, 6.24 Gb for D. terniflora, 7.74 Gb for D. hokouensis, and 6.88 Gb for D. elliptica. The genome sequences assembled using the reads obtained from the Illumina sequencing platform ranged from 155,055 bp for D. elliptica to 155,449 bp for D. cochinchinensis (Table 1), which are similar to other Liliaceae CP genomes (Kim et al., 2016; Park et al., 2017; Liu et al., 2018). The genome exhibited typical cyclic tetramer structure, including the SSC and LSC regions separated by two IR regions (Figure 1), and the four regions from the six species had similar lengths (Table S1). The SSC lengths ranged from 18,456 to 18,494 bp, which is larger than those from the six Ipomoea species (Park et al., 2018) and Strobilanthes cusia (Nees) Kuntze (Chen et al., 2018a). Meanwhile, the LSC lengths ranged from 83,621 to 83,907 bp, which is shorter than those from the six Ipomoea species and S. cusia. In addition, the IR lengths each ranged from 26,489 bp to 26,530 bp, which is larger than those obtained from S. cusia (Chen et al., 2018a), Taxillus chinensis (DC.) Danser (Li et al., 2017), and T. sutchuenensis (Lecomte) Danser (Li et al., 2017). Moreover, the GC contents of the six Dracaena CP genomes were similar, and the IR regions (42.9%) had higher GC contents than the single-copy regions (LSC: 35.4%–35.6% and SSC: 31.1%–31.2%). This result is consistent with previous reports (Park et al., 2018; Li and Zheng, 2018). The high GC contents in the IR regions may be attributable to the presence of four rRNA genes (rrn16, rrn23, rrn4.5, and rrn5) (Chen et al., 2018b).
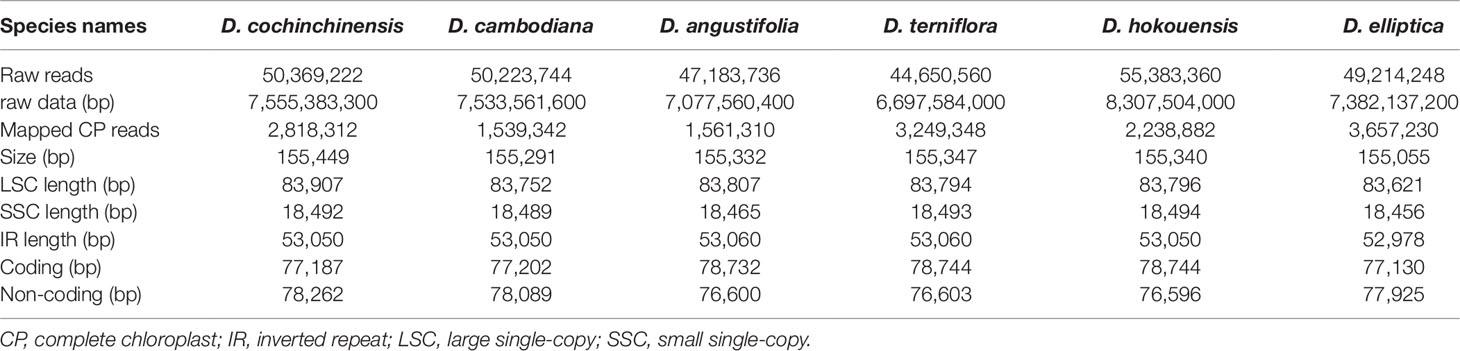
Table 1 Summary statistics for assembly of the six complete chloroplast (CP) genomes of Dracaena species.
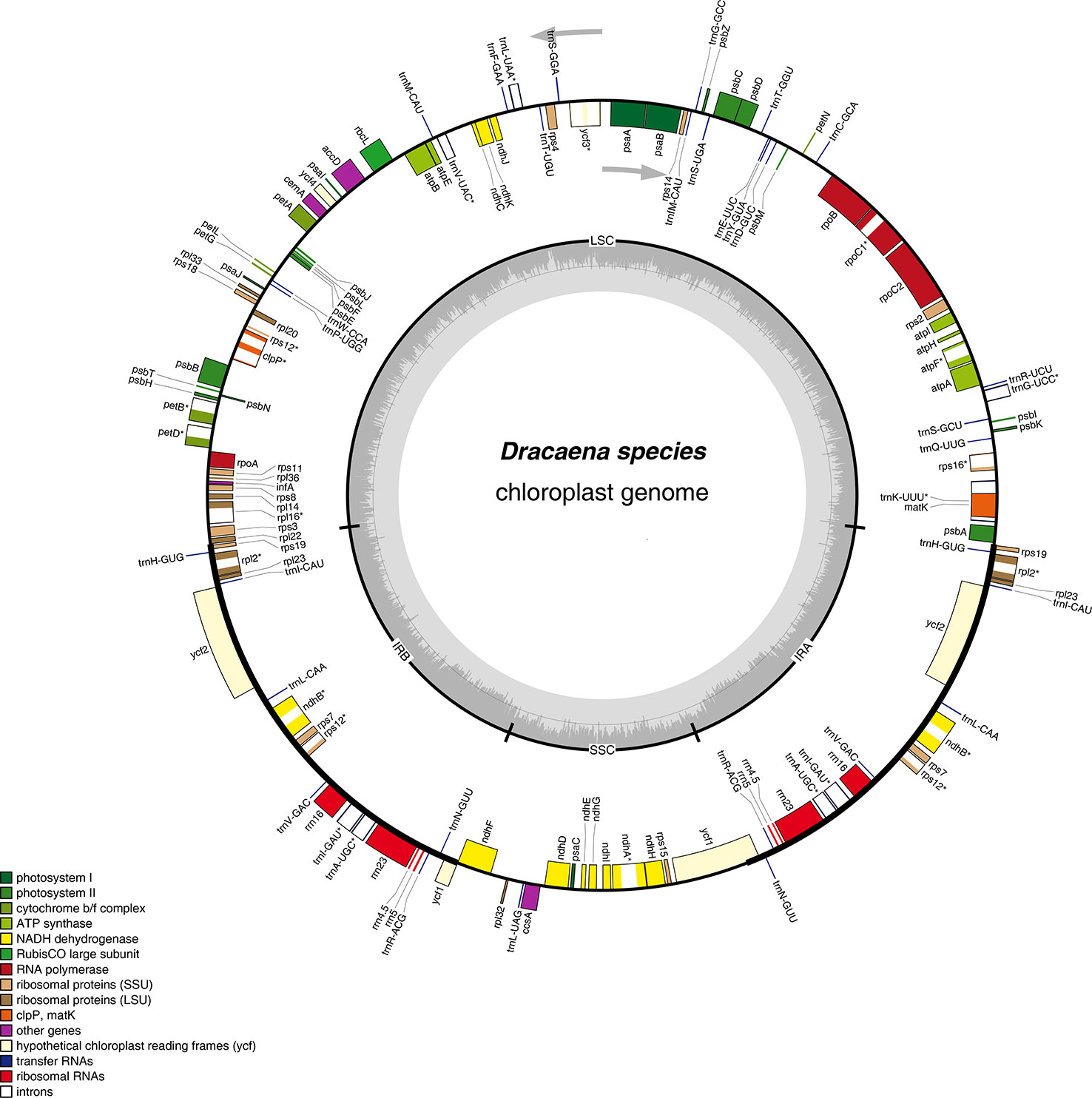
Figure 1 Gene map of the complete chloroplast genomes of Dracaena species. Genes on the inside of the circle are transcribed clockwise, and those on the outside are transcribed counter-clockwise. The darker gray area in the inner circle corresponds to GC content, whereas the lighter gray corresponds to AT content.
Furthermore, the six Dracaena CP genomes have similar gene contents, sequences, and orientations, which are the typical characteristics of higher plant CP genomes (Wicke et al., 2011; Qian et al., 2013). The genes in the six CP genomes consisted of 84 protein-coding genes, 38 tRNA genes, and 8 rRNA genes. Among them, seven protein-coding genes (rps19, rpl2, rpl23, ycf2, ndhB, rps7, and rps12), four rRNA genes (rrn4.5×2, rrn5×2, rrn16×2, and rrn23×2), and eight tRNA genes (trnR-ACG, trnA-UGC, trnI-GAU, trnV-GAC, trnL-CAA, trnI-CAU, trnH-GUG, and trnN-GUU), as well as the IR regions, contained two repeat units, similar to Ligularia intermedia Nakai (Chen et al., 2018b) and Artemisia annua L. (Shen et al., 2017). Moreover, 18 of the genes contained introns, and among them, 10 protein-coding genes (petB, petD, atpF, ndhA, ndhB, rpoC1, rps12, rps16, rpl16, and rpl2) and 6 tRNA genes contained a single intron, and 2 protein-coding genes (clpP and ycf3) contained two introns (Table 2). Introns play an important role in the regulation of gene expression by enhancing the expression of exogenous genes at specific sites and at specific times in plants (Xu et al., 2003). In total, the coding regions including protein-coding genes, tRNAs, and rRNAs occupied 57.35%–58.37% of the six Dracaena CP genomes, while the non-coding regions constituted 41.63%–42.68% of the genomes.

Table 2 List of genes found in the six CP genomes of Dracaena species.
Codon Usage
RSCU values reveal biases in synonymous codon usage. The codon usage and anti-codon recognition patterns of the six Dracaena CP genomes are shown in Table S2. The CP protein-coding genes of these six species contained 61 codons encoding 20 amino acids. The coding regions (CDS) comprised 25,710 codons in D. elliptica to 26,248 codons in D. terniflora and D. hokouensis. Codons for isoleucine (5.63%∼5.89%) and lysine (5.41%∼5.66%) were the most abundant in the six Dracaena CP genomes, whereas those for cysteine (0.37∼∼ 0.39%) and arginine (0.44∼∼0.48%) were observed least often, at lower frequencies than in artichoke (Curci et al., 2015) and the six Ligularia species (Chen et al., 2018b).
An RSCU value <1.00 indicates that the codon usage frequency is lower than expected, whereas an RSCU value >1.00 indicates that the codon usage frequency is higher than expected (Sharp and Li, 1987). In this study, other than leucine and isoleucine, amino acid codons in the CP genomes of the six Dracaena spp. preferentially ended with A or U (RSCU >1). Codons ending in A and/or U accounted for 69.76% (D. cochinchinensis) to 72.87% (D. terniflora) of all CDS codons in the six CP genomes. These results are similar to those observed for Papaver rhoeas L.and Papaver orientale L. (Zhou et al., 2018). The codon usage pattern may be determined by the high proportion of A/T composition bias. In other terrestrial higher plant CP genomes, high codon preference in codon usage, especially A/T bias, is very common (Kim and Lee, 2004; Qian et al., 2013). This phenomenon indicates that stable CP evolution is beneficial for protecting important CP genes from harmful mutations and adaptation to selection stress (Wang et al., 2016b; Ivanova et al., 2017; Zuo et al., 2017).
Meanwhile, codons ending in A and/or T (U) usually had high RSCU values in the six CP genomes, e.g., GCU (1.79) for alanine, UCU (1.69) for serine, and GGA (1.64) for glycine. In addition, the use of the start codon (ATG) and TGG encoding Trp showed no bias in the six CP genomes (RSCU = 1), and the remaining amino acids have preferred codons. Furthermore, protein-coding genes accounted for 49.65%∼50.69% of the whole genome sequence, and tRNA genes accounted for 1.849%∼1.853% (Table S3). A total of 18 genes contained introns in the six Dracaena CP genomes, including 12 protein-coding genes (rps16, atpF, rpoC1, ycf3, rps12, clpP, petB, petD, rpl16, rpl2, ndhB, and ndhA), and 6 tRNA genes (trnK-UUU, trnG-UCC, trnL-UAA, trnV-UAC, trnl-GAU, and trnA-UGC; Table S4) were identified in this research. TrnK-UUU has the longest intron (857 bp).
SSRs in the CP Genomes of the Six Dracaena spp.
SSRs are widely present in CP genomes, consisting of 1–6-nucleotide repeat units, and are valuable molecular markers of high variation within the same species (Powell et al., 1995). The repeats were divided into tandem repeats and dispersed repeats, and the dispersed repeats can be further divided into four repetition types: complement, forward, reverse, and palindromic repeats (Kurtz et al., 2001). In this study, we analyzed repeats using the software tools Tandem Repeats Finder and REPuter, and the distributions of repeated sequences and SSRs in the CP genomes of the six species were analyzed. The repeat structure analysis is shown in Figure 2. The results showed that the numbers of repeat types in the six Dracaena spp. were extremely similar. Among the repeat types, palindromes (29∼33) were the most abundant, followed by forward (21∼25) and reverse repeats (0∼2), and there were no complement repeats in the six Dracaena CP genomes. Furthermore, totals of 69, 69, 67, 64, 70, and 71 SSRs were identified using the microsatellite identification tool (MISA) from the CP genomes of D. cochinchinensis, D. cambodiana, D. angustifolia, D. terniflora, D. hokouensis, and D. elliptica, respectively (Table 3).
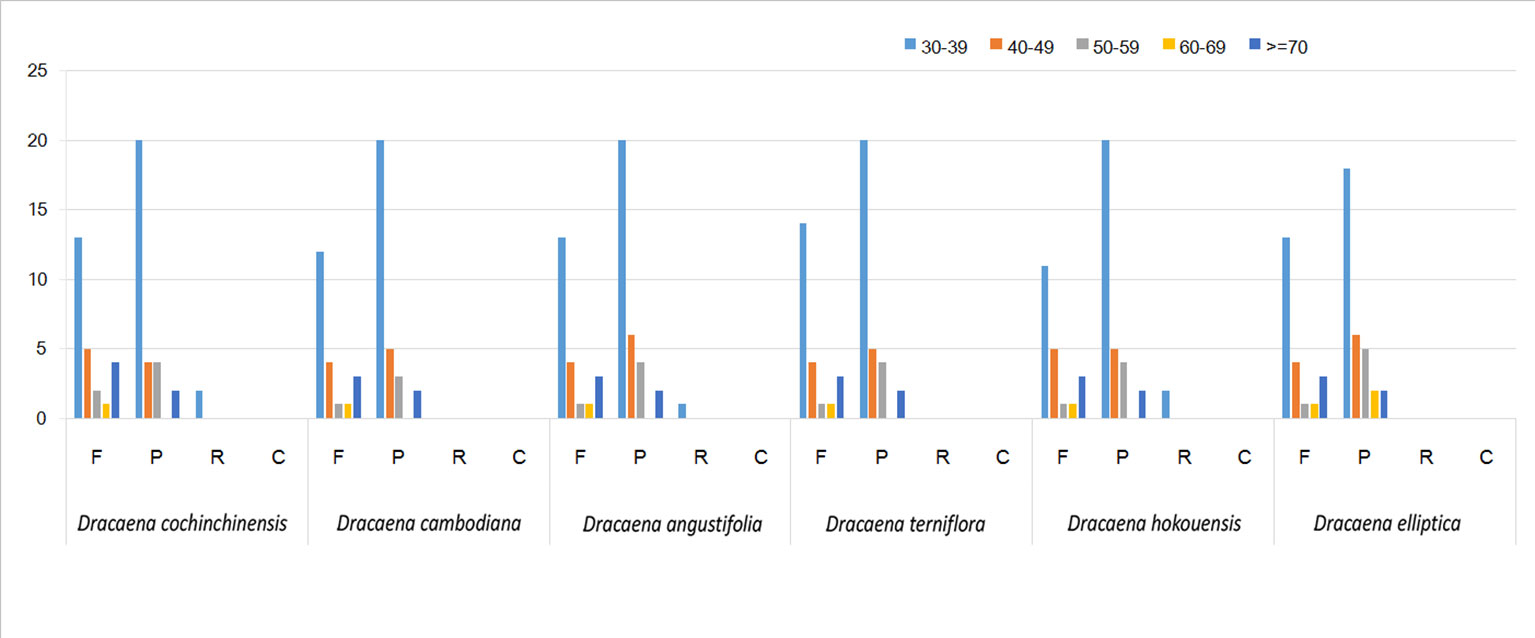
Figure 2 Repeat analysis in six Dracaena complete chloroplast (CP) genomes. REPuter was used to identify repeat sequences with length ≥30 bp and sequence identified ≥90% in the CP genomes. F, P, R, and C indicate the repeat types F (forward), P (palindrome), R (reverse), and C (complement), respectively. Repeats with different lengths are indicated in different colors.
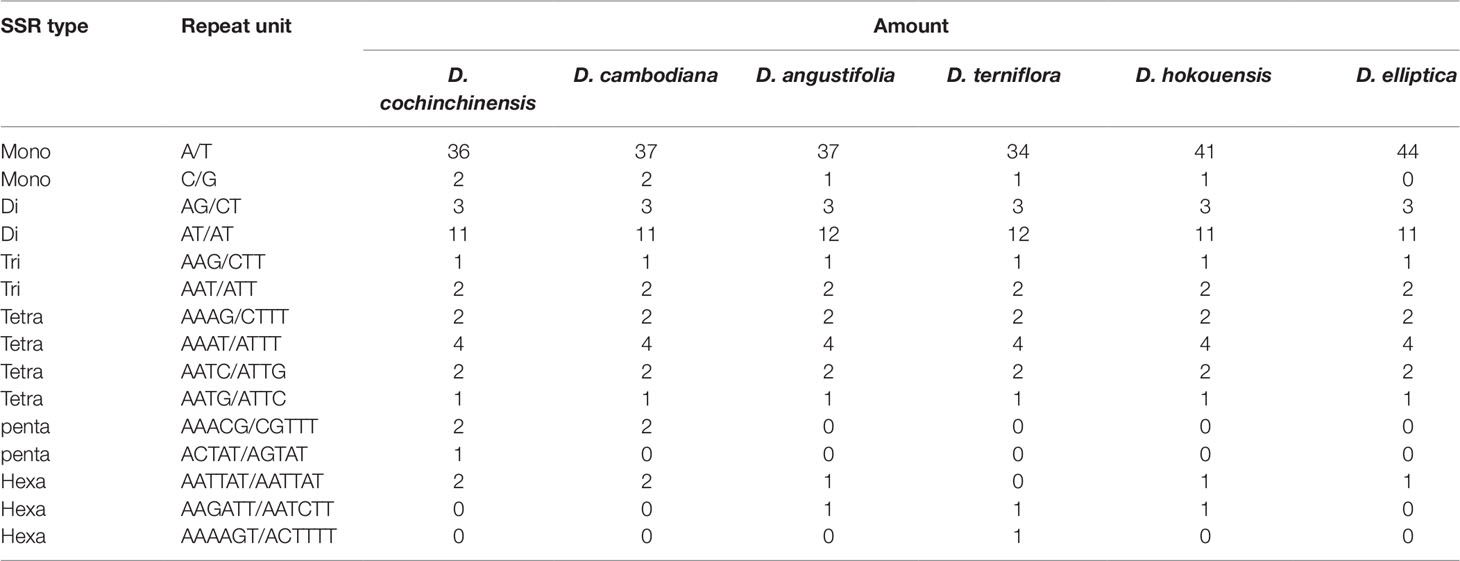
Table 3 The simple sequence repeat (SSR) types of the six CP genomes of Dracaena species.
It is well known that the repeat sequences are associated with plastome tissues, mostly in the intergenic and intron regions, and only a small portion is distributed in the genetic region (Salih et al., 2017; Zhou et al., 2017). In the present study, SSRs were less abundant in the CDS (15∼16 SSRs) of the six Dracaena CP genomes than in the non-coding region. In addition, the types and distributions of the potential SSRs were investigated. The most abundant type was repeated mononucleotide (54.69%∼61.97%), which were found 14∼15 times in the six Dracaena species. These were followed by dinucleotide (19.72%∼23.44%), trinucleotide (4.23%∼4.69%), tetranucleotide (12.68%∼14.06%), pentanucleotide (0%∼4.35%), and hexanucleotide repeats (1.41%∼2.99%). Among these repeats, the A/T repeat was the most abundant motif, followed by AT/TA dinucleotide repeats, then AAAT/ATTT tetranucleotide repeats. These results were consistent with previous reports that CP SSRs usually consist of short poly-A or poly-T repeats and rarely contain tandem G or C repeats in many plants (Kuang et al., 2011). In addition, with the exceptions of D. cochinchinensis and D. cambodiana, the other four species had no pentanucleotide SSRs (Table 3). At present, SSR markers are widely used in genetic diversity and population structure assessments, comparative genomics, development of genetic maps, and marker-assisted selective breeding (Chen et al., 2015; Zhou et al., 2018; Zhang et al., 2017b). The repeats identified in this study will provide valuable resources for species identification and population studies of Dracaena spp.
Comparative Genome Analysis
Comparative analysis of CP genome is advantageous for understanding the genetic diversity and evolutionary relationships of plants in different environments (Daniell et al., 2016; Zhao et al., 2018). The sequence homology of the six Dracaena CP genomes was compared and analyzed using the mVISTA software, and D. cochinchinensis was used as a reference sequence (Figure 3). The comparison revealed that some differences between D. cochinchinensis and the other five Dracaena spp. existed within intergenic spacers (IGS), such as rps12-trnV and psaI-ycf4; and the CP genomes of the other five Dracaena spp. were not significantly different., only some differences existed in the ycf1 gene and intergenic regions, such as trnS-trnG, rpoB-trnC, rpl16-rps3, psbE-petL, and ndhF-rpl32.
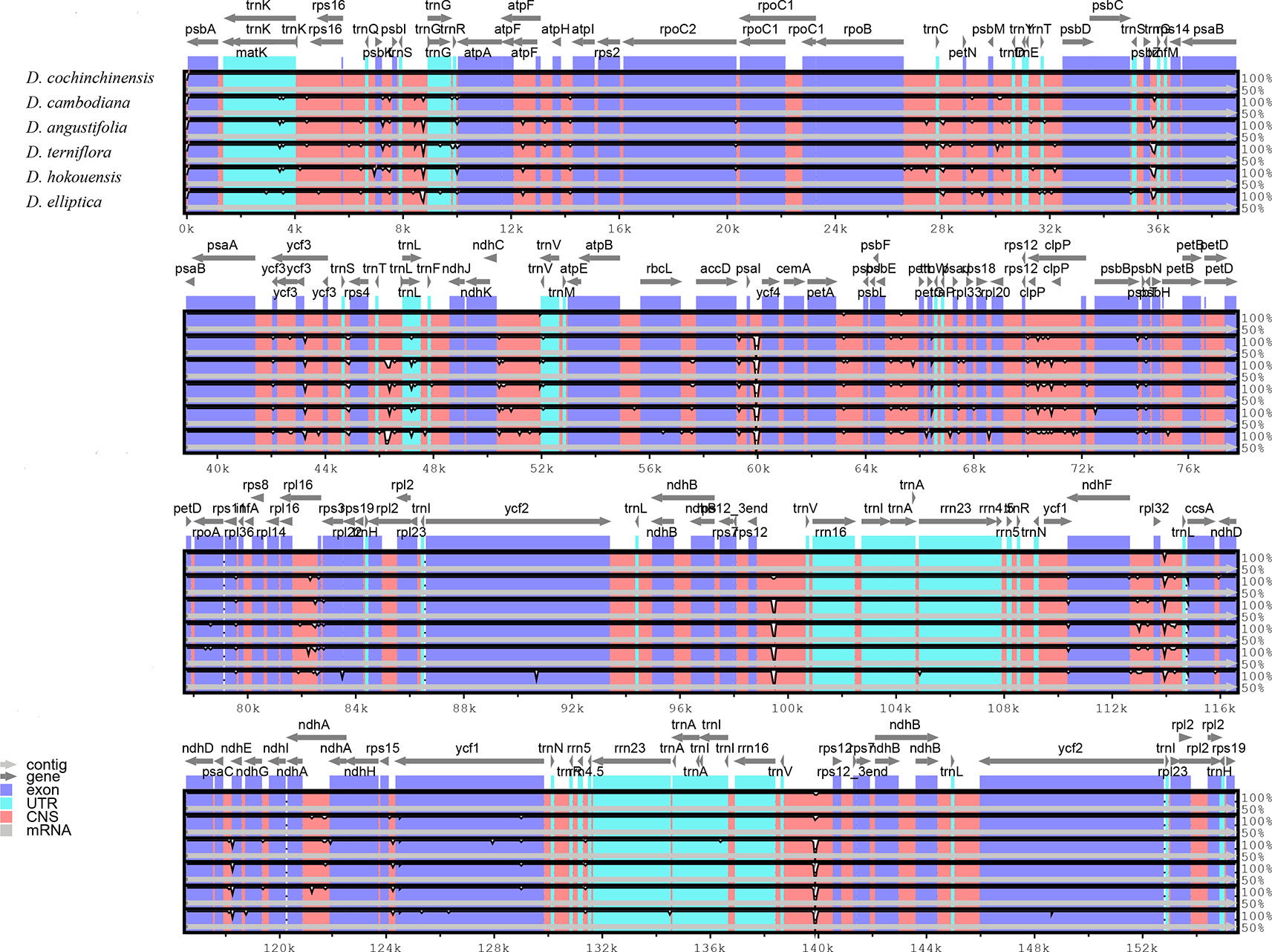
Figure 3 Structure comparison of the six Dracaena CP genomes by using the mVISTA program. Gray arrows and thick black lines above the alignment indicate genes with their orientation and the position of the IRs, respectively. A cut-off value of 70% identity was used for the plots, and the Y-scale represents the percent identity between 50% and 100%.
As shown in Table 4, the gene with the most variable sites in coding regions is ycf1, which has 73 variable sites and 35 indels within the 5,466 bp alignment. the intergenic regions with the most variable sites is rps7 → trnV-GAC, which has 59 variable sites and 16 indels within the 2,716 bp length. On the whole, the variable sites of the six Dracaena CP genomes are more than some other species (Zhang et al., 2017c). Furthermore, we found that the majority of sequence variations were within the LSC and SSC regions, whereas the IR regions had relatively fewer sequence variations. This result further supports the findings of previous studies: the two IR regions are more conserved than the LSC and SSC regions (Nazareno et al., 2015; Li et al., 2017; Lu et al., 2017). This may be because mutations in the IR sequences are corrected by gene conversion (Khakhlova and Bock, 2006). In addition, the sequence variation in the non-coding regions was higher than in the gene coding regions in the CP genomes of the six Dracaena spp., which is consistent with previous findings for most gymnosperms (Zhang et al., 2011; Lu et al., 2017; Zhao et al., 2018).
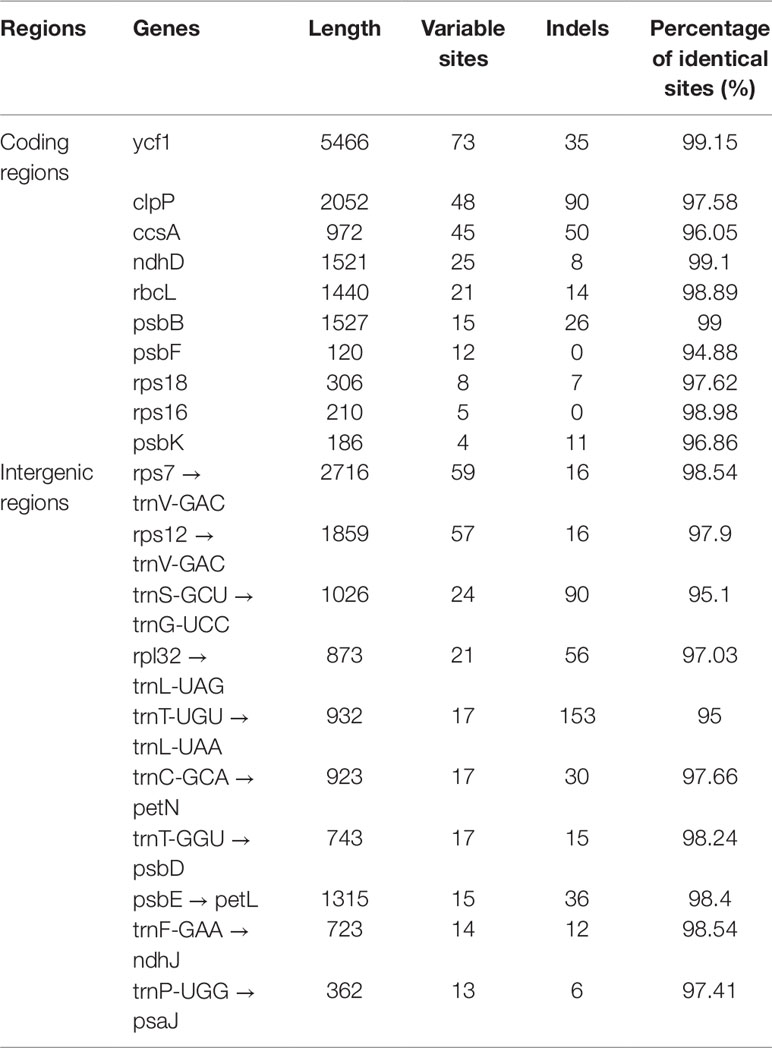
Table 4 The 10 most-divergent coding regions and intergenic regions in the six Dracaena species.
Identification and Phylogenetic Analysis of Dracaena Species
The highly variable regions of CP genomes can be used as potential DNA barcodes for species identification and phylogenetic analyses, such as in Fritillaria species (Li et al., 2016). Our results showed high similarity among the six sequences. The largest change in gene length occurred in ndhF, with 2,211 bp in D. hokouensis, 2,214 bp in D. elliptica, 2,220 bp in D. cochinchinensis, 2,223 bp in D. angustifolia and D. terniflora, and 2,244 bp in D. cambodiana. Furthermore, the most divergent regions between the six Dracaena species localized in the IGS among the CP genomes, but these variable regions is not enough to distinguish the six Dracaena species. As there are no significant difference in structure and size of the six CP genome sequence of Dracaena species, we thought that the complete CP genome could be used as a barcode marker to distinguish Dracaena species.
The CP sequence is crucial for studying phylogenetic relationships and determining taxonomic status among angiosperms (Jansen et al., 2007). To identify the phylogenetic positions of the six Dracaena spp. within the Liliaceae, we obtained 30 complete CP genome sequences belonging to 20 genera of the family Liliaceae (including two species each from Aletris, Allium, Aloe, Asparagus, Hosta, Yucca, Polygonatum, Fritillaria, Lilium, and Paris, and one species each from Anemarrhena, Chlorophytum, Maianthemum, Rohdea, Cordyline, Tricyrtis, Chionographis, Trillium, Veratrum, and Gloriosa) and one species belonging to Stemonaceae from the RefSeq database (Table S5). In the MP (Figure 4) and NJ (Figure S1) tree, the six species of the genus Dracaena are clustered together and separated from the other 20 genera of the family Liliaceae, indicating the close relationship of the six Dracaena species. Meanwhile, the six Dracaena spp. were distinct from one another, with support values ≥85%; D. angustifolia and D. terniflora were clustered together, D. hokouensis exhibited a sister relationship, and D. cambodiana and D. cochinchinensis were clustered together. The results showed that the CP genomes can be used to identify the six Dracaena species. In addition, we explored the construction of a phylogenetic tree based on 36 complete Liliaceae CP genomes sequence multiple alignments in present study. Our results will provide an important reference and foundation for species identification and phylogeny of Liliaceae plants.
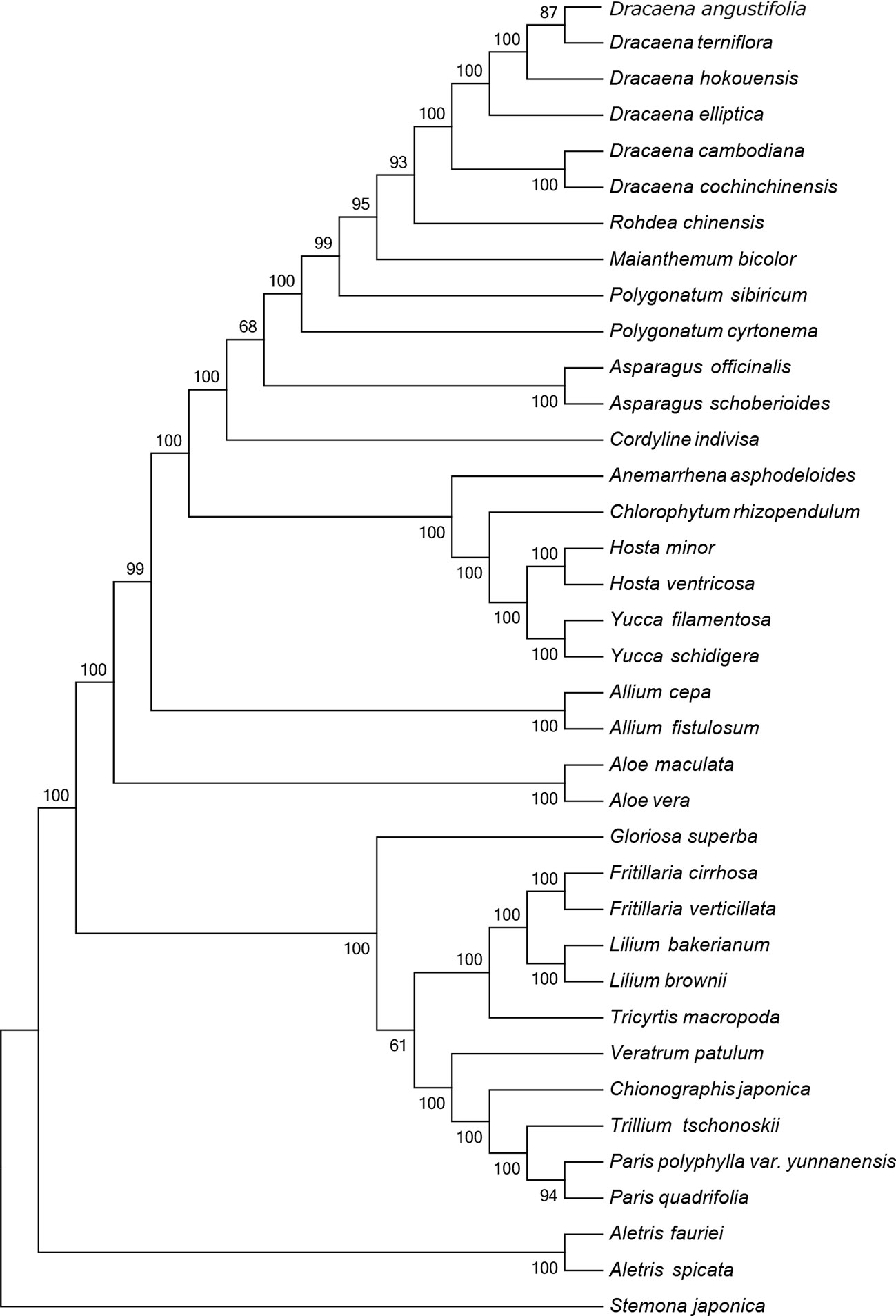
Figure 4 Phylogenetic tree constructed using maximum parsimony (MP) based on complete CP genomes of six Dracaena and other 31 species. Numbers above the branches are the bootstrap support values.
Conclusion
This study reported the CP genomes from six Dracana species, range from 155,055 bp (D. elliptica) to 155,449 bp (D. cochinchinensis), and the structure and composition of the CP genomes are highly similar. The CP genomes genes consisted of 84 protein-coding genes and 38 tRNAs, with 8 rRNA genes in the six genomes. Among the six Dracana species, D. angustifolia had the closest relationship with D. terniflora, D. cochinchinensis had the closest relationship with D. cambodiana respectively. The MP and NJ tree showed that the CP genome can be used to identify the six Dracana species and is expected to become a super-barcode for the identification of Dracana species.
Data Availability Statement
The datasets generated for this study can be found in GenBank, accession numbers are MN200193 (D. angustifolia), MN200194 (D. cambodiana), MN200195 (D. cochinchinensis), MN200196 (D. elliptica), MN200197 (D. hokouensis) and MN200198 (D. terniflora).
Author Contributions
ZZ and XM conceived and design the study. XM acquired the funding. ZZ and YG collected samples and determined the species. YZ performed the genome assembly and analysis on the data. ZZ wrote the manuscript. XM supervised the manuscript. All authors have read and approved the final manuscript.
Funding
This research was funded by CAMS Initiative for Innovative Medicine (No. 2017-12M-B&R-09) and Quality guarantee system of Chinese herbal medicines (No. 201507002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.01441/full#supplementary-material
References
Adolt, R., Pavlis, J. (2004). Age structure and growth of Dracaena cinnabari populations on Socotra. Trees 18, 43–53. doi: 10.1007/s00468-003-0279-6
Aslam, J., Mujib, A., Sharma, M. P. (2013). In vitro micropropagation of Dracaena sanderiana Sander ex Mast: an important indoor ornamental plant. Saudi. J. Biol. Sci. 20, 63–68. doi: 10.1016/j.sjbs.2012.11.005
Chen, X., Turland, N. (2000). Dracaena Vandelli ex Linnaeus Vol. 24. Eds. Wu, Z.-Y., Raven, P. H. (Flora of China. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press), 215–217.
Chen, H. Q., Zuo, W. J., Wang, H., Shen, H. Y., Luo, Y., Dai, H. F., et al. (2012). Two new antimicrobial flavanes from dragon’s blood of Dracaena cambodiana. J. Asian Nat. Prod. Res. 14, 436–440. doi: 10.1080/10286020.2012.668534
Chen, L. Y., Cao, Y. N., Yuan, N., Nakamura, K., Wang, G. M., Qiu, Y. X. (2015). Characterization of transcription and development of novel ESTSSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to Asian land-bridge islands. Mol. Breed. 35, 187. doi: 10.1007/s11032-015-0379-1
Chen, H. M., Shao, J. J., Zhang, H., Jiang, M., Huang, L. F., et al. (2018a). Sequencing and analysis of Strobilanthes cusia (Nees) Kuntze chloroplast genome revealed the rare simultaneous contraction and expansion of the inverted repeat region in angiosperm. Front. Plant Sci. 9, 324. doi: 10.3389/fpls.2018.00324
Chen, X., Zhou, J., Cui, Y., Wang, Y., Duan, B., Yao, H. (2018b). Identification of Ligularia herbs susing the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9, 695. doi: 10.3389/fphar.2018.00695
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G., Sonnante, G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PloS One 10, e0120589. doi: 10.1371/journal.pone.0120589
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Fan, J. Y., Yi, T., Sze-To, C. M., Zhu, L., Peng, W. L., Zhang, Y. Z., et al. (2014). A systematic review of the botanical, phytochemical and pharmacological profile of Dracaena cochinchinensis, a plant source of the ethnomedicine “dragon’s blood”. Molecules 19, 10650–10669. doi: 10.3390/molecules190710650
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
González, A. G., León, F., Hernández, J. C., Padrón, J. I., Sánchez-Pinto, L., Barrera, J. B. (2004). Flavans of dragon’s blood from Dracaena draco and Dracaena tamaranae. Biochem. Syst. Ecol. 32, 179–184. doi: 10.1016/S0305-1978(03)00133-9
Gupta, D., Bleakley, B., Gupta, R. K. (2007). Dragon’s blood: botany, chemistry and therapeutic uses. J. Ethnopharmacol. 115, 361–380. doi: 10.1016/j.jep.2007.10.018
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hu, Y. Q., Tu, P. F., Li, R. Y., Wan, J., Wang, D. L. (2001). Studies on stilbene derivatives from Dracaena cochinchinensis and their antifungal activities. Chin. Traditional Herb. Drugs 32, 104–106. doi: 10.3321/j.issn:0253-2670.2001.02.004
Hu, Y., Woeste, K. E., Zhao, P. (2016). Completion of the chloroplast genomes of five Chinese juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7, 1955. doi: 10.3389/fpls.2016.01955
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., Apostolova, E., Yahubyan, G., et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8, 204. doi: 10.3389/fpls.2017.00204
Jansen, R. K., Ruhlman, T. A. (2012). “Genomics of chloroplasts and mitochondria: advances in photosynthesis and respiration (including bioenergy and related processes),” in Plastid genomes of seed plants, vol. 35. Eds. Bock, R., Knoop, V. (Dordrecht: Springer).
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Nat. Acad. Sci. U. S. A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., Kuma, K., Toh, H., Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313x.2006.02673.x
Kim, K. J., Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, S. C., Kim, J. S., Kim, J. H. (2016). Insight into infrageneric circumscription through complete chloroplast genome sequences of two Trillium species. AoB Plants 8, plw015. doi: 10.1093/aobpla/plw015
Kougan, G. B., Miyamoto, T., Tanaka, C., Paululat, T., Mirjolet, J. F., Duchamp, O., et al. (2010). Steroidal saponins from two species of Dracaena. J. Nat. Prod. 73, 1266–1270. doi: 10.1021/np100153m
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/G11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, B., Zheng, Y. Q. (2018). Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 8, 9285. doi: 10.1038/s41598-018-27453-7
Li, Y., Yao, H., Song, J., Ren, F., Li, X., Sun, C. (2016). Screening Fritillaria genus-specific DNA barcodes based on complete chloroplast genome sequences. Mod. Tradit. Chin. Med. Mater. Med. World Sci. Technol. 18, 24–28.
Li, Y., Zhou, J. G., Chen, X. L., Cui, Y. X., Xu, Z. C., et al. (2017). Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 7, 12834. doi: 10.1038/s41598-017-13401-4
Li, Y. (2012). Study of DNA barcoding and SRAP markers of Dracaena. Master’s dissertation. University of Hainan, Haikou.
Liu, H.-Y., Yu, Y., Deng, Y.-Q., Li, J., Huang, Z-X., Zhou, S-D. (2018). The chloroplast genome of Lilium henrici: genome structure and comparative analysis. Molecules 23, E1276. doi: 10.3390/molecules23061276
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lu, R. S., Li, P., Qiu, Y. X. (2017). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7, 2054. doi: 10.3389/fpls.2016.02054
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18–23. doi: 10.1186/2047-217X-1-18
Mei, W. L., Luo, Y., Wang, H., Shen, H. Y., Zeng, Y. B., Dai, H. F. (2013). Two new flavonoids from dragon’s blood of Dracaena cambodiana. B. Korea Chem. Soc 34, 1791–1794. doi: 10.5012/bkcs.2013.34.6.1791
Nazareno, A. G., Carlsen, M., Lohmann, L. G. (2015). Complete chloroplast genome of Tanaecium tetragonolobum: the first Bignoniaceae plastome. PloS One 10, e0129930. doi: 10.1371/journal.pone.0129930
Park, I., Kim, W. J., Yeo, S. M., Choi, G., Kang, Y. M., Piao, R., et al. (2017). The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 22, E982. doi: 10.3390/molecules22060982
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., Moon, B. C. (2018). The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic pharbitidis semen (seeds of I. nil or I. purpurea). Front. Plant Sci. 9, 965. doi: 10.3389/fpls.2018.00965
Paul, W., Piyakaset, S., Kaweesak, K., Peter, V., Justyna, W. (2012). A new threatened endemic species from central and northeastern Thailand, Dracaena jayniana (Asparagaceae: tribe Nolinoideae). Kew Bull. 67, 697–705. doi: 10.2307/23489231
Pearson, J., Prendergast, H. D. V. (2001). Daemonorops, Dracaena and other dragon’s blood. Econ. Bot. 55, 474–477. doi: 10.1007/bf02871711
Powell, W., Morgante, M., Mcdevitt, R., Vendramin, G. G., Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U. S. A. 92, 7759–7763. doi: 10.2307/2368128
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS One 8, e57607. doi: 10.1371/journal.pone.0057607
Salih, R. H. M., Majeský, L’., Schwarzacher, T., Gornall, R., Heslop-Harrison, P. (2017). Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): identity and variation between three microspecies. PloS One 12, e0168008. doi: 10.1371/journal.pone.0168008
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Tabata, S. (1999). Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290. doi: 10.1093/dnares/6.5.283
Schattner, P., Brooks, A. N., Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sharp, P. M., Li, W. H. (1987). The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Shen, X., Wu, M., Liao, B., Liu, Z., Bai, R., Xiao, S., et al. (2017). Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia Annua. Molecules 22, 1330–1343. doi: 10.3390/molecules22081330
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0 . Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tapondjou, L. A., Ponou, K. B., Teponno, R. B., Mbiantcha, M., Djoukeng, J. D., Nguelefack, T. B., et al. (2008). In vivo antiinflammatory effect of a new steroidal saponin, mannioside A, and its derivatives isolated from Dracaena mannii. Arch. Pharm. Res. 31, 653–658. doi: 10.1007/s12272-001-1208-3
Thode, V. A., Lohmann, L. G. (2019). Comparative chloroplast genomics at low taxonomic levels: a case study using Amphilophium (Bignonieae, Bignoniaceae). Front. Plant Sci. 10, 796. doi: 10.3389/fpls.2019.00796
Tonti-Filippini, J., Nevill, P. G., Dixon, K., Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. 90, 808–818. doi: 10.1111/tpj.13491
Twyford, A. D., Ness, R. W. (2016). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
Wang, J., Liu, X., Sun, W., Wang, B., Li, D. P., Fan, J. J., et al. (2015). DNA barcodes identification of one rare traditional chinese medicine Draconis Sanguis. Chin. Pharmaceut. J. 50, 1261–1265. doi: 10.11669/cpj.2015.15.001
Wang, B., Chen, H., Ma, H., Zhang, H., Lei, W., Wu, W., et al. (2016a). Complete plastid genome of Astragalus membranaceus (Fisch.) Bunge var. membranaceus. Mitochondrial. DNA B. Resour. 1, 517–519. doi: 10.1038/srep21669
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016b). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the malvales order. Front. Plant Sci. 7, 280. doi: 10.3389/fpls.2016.00280
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wilkin, P., Suksathan, P., Keeratikiat, K., Van-Welzen, P., Wiland-Szymanska, J. (2013). A new species from Thailand and Burma, Dracaena kaweesakii Wilkin & Suksathan (Asparagaceae subfamily Nolinoideae). PhytoKeys 26, 101–112. doi: 10.3897/phytokeys.26.5335
Wyman, S. K., Jansen, R. K., Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xia, Y., Hu, Z., Li, X., Wang, P., Zhang, X., Li, Q., et al. (2016). The complete chloroplast genome sequence of Chrysanthemum indicum. Mitochondrial. DNA A. DNA Mapp. Seq. Anal. 27, 4668–4669. doi: 10.3109/19401736.2015.1106494
Xu, J., Feng, D., Song, G., Wei, X., Chen, L., Wu, X., et al. (2003). The first intron of rice EPSP synthase enhances expression of foreign gene. Sci. China Life Sci. 46, 561–569. doi: 10.1360/02yc0120
Xu, M., Zhang, Y. J., Li, X. C., Jacob, M. R., Yang, C. R. (2010). Steroidal saponins from fresh stems of Dracaena angustifolia. J. Nat. Prod. 73, 1524–1528. doi: org/10.1021/np100351p
Yan, Y. (2005). A phytoxonomical study on Dracaena and its status in plant community in yunnan. Master’s dissertation. . Academy of Sciences, Beijing.
Zhang, Y. J., Ma, P. F., Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PloS One 6, e20596. doi: 10.1371/journal.pone.0020596
Zhang, N., Erickson, D. L., Ramachandran, P., Ottesen, A. R., Timme, R. E., Funk, V. A., et al. (2017a). An analysis of Echinacea chloroplast genomes: implications for future botanical identification. Sci. Rep. 7, 216. doi: 10.1038/s41598-017-00321-6
Zhang, Y., Iaffaldano, B. J., Zhuang, X., Cardina, J., Cornish, K. (2017b). Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biol. 17, 34. doi: 10.1186/s12870-016-0967-1
Zhang, Y., Zhang, Y., Wang, Y. H., Shen, S. K. (2017c). De novo assembly of transcriptome and development of novel EST-SSR markers in Rhododendron rex Lévl. through illumina sequencing. Front. Plant Sci. 8, 1664. doi: 10.3389/fpls.2017.01664
Zhao, Y., Yin, J., Guo, H., Zhang, Y., Xiao, W., Sun, C., et al. (2015). The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front. Plant Sci. 5, 696. doi: 10.3389/fpls.2014.00696
Zhao, J. T., Xu, Y., Xi, L. J., Yang, J. W., Chen, H. W., Zhang, J. (2018). Characterization of the chloroplast genome sequence of Acer miaotaiense: comparative and phylogenetic analyses. Molecules 23, 1740. doi: 10.3390/molecules23071740
Zheng, S. (2013). The preliminiary studies of induction mechanism of Dragon’s Blood and molecular taxonomy of several Dracaena species. Master’s dissertation. Yunnan University, Kunming.
Zhou, J. G., Chen, X. L., Cui, Y. X., Sun, W., Li, Y. H., Wang, Y., et al. (2017). Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 18, 1839. doi: 10.3390/ijms18091839
Zhou, J. G., Cui, Y. X., Chen, X. L., Li, Y., Xu, Z. C., et al. (2018). Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: molecular structures, comparative analysis, and phylogenetic analysis. Molecules 23, 437. doi: 10.3390/molecules23020437
Zhu, Z. X., Mu, W. X., Wang, J. H., Zhang, J. R., Zhao, K. K., Ross Friedman, C., et al. (2018). Complete plastome sequence of Dracaena cambodiana (Asparagaceae): a species considered ‘Vulnerable’ in Southeast Asia. Mitochondrial. DNA B. 3, 620–621. doi: 10.1080/23802359.2018.1473740
Keywords: Dracaena Vand. ex L., chloroplast genome, identification, super-barcode, Liliaceae
Citation: Zhang Z, Zhang Y, Song M, Guan Y and Ma X (2019) Species Identification of Dracaena Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 10:1441. doi: 10.3389/fphar.2019.01441
Received: 27 July 2019; Accepted: 12 November 2019;
Published: 29 November 2019.
Edited by:
Hugo J. De Boer, University of Oslo, NorwayCopyright © 2019 Zhang, Zhang, Song, Guan and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojun Ma, bWF5aXh1YW4xMEAxNjMuY29t