 Shuangjun Shen1,2
Shuangjun Shen1,2 Bin Li1,2
Bin Li1,2 Juanjuan Dai1,2Zengkai Wu1,2Yan He1,2
Juanjuan Dai1,2Zengkai Wu1,2Yan He1,2 Li Wen1,2*Xingpeng Wang1,2*
Li Wen1,2*Xingpeng Wang1,2* Guoyong Hu1,2*
Guoyong Hu1,2*- 1Department of Gastroenterology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Shanghai Key Laboratory of Pancreatic Disease, Institute of Pancreatic Disease, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Impaired autophagy has been shown to play a critical role in experimental and human acute pancreatitis (AP). However, the mechanism for transcriptional regulation of autophagy remains largely unknown. In this study, we aim to explore the role of BRD4 (bromodomain-containing protein 4), a transcriptional repressor of autophagy, during AP. Changes in pancreatic BRD4 expression and the effect of BRD4 inhibition were measured in mice with AP (induced by caerulein and ethanol and palmitoleic acid) and in isolated pancreatic acinar cells stimulated with cholecystokinin (CCK). Pancreatitis severity was evaluated by serum amylase and pancreatic histopathology. The autophagic flux, the fusion of autophagosome and lysosome, and lysosomal degradation were evaluated. Sirtuin 1 (SIRT1) expression and the effect of SIRT1 inhibition were assessed. We found that pancreatic BRD4 expression was upregulated during various models of AP. BRD4 inhibition reduced CCK-stimulated pancreatic acinar cell injury and pro-inflammatory expression in vitro and protected against two models of experimental AP. Mechanistically, BRD4 inhibition restored impaired autophagic flux via promoting autophagosome-lysosome fusion and lysosomal degradation. BRD4 inhibition also upregulated SIRT1 and inhibition of SIRT1 reversed the effects of BRD4 inhibition on autophagic flux. Our data suggest that BRD4 is a potential therapeutic target for treating AP.
Introduction
Acute pancreatitis (AP) is an inflammatory disease of the exocrine pancreas, which is closely related to high morbidity and mortality, and has an increasing incidence in recent years worldwide (Pandol et al., 2007; Habtezion et al., 2019). Currently, there is no effective treatment that can change the pathological process of the disease (Vege et al., 2018). Autophagy is an essential cellular process which can degrade defective cellular proteins and recycle useful components (Parzych and Klionsky, 2014). In experimental pancreatitis, impaired autophagy is due to the disability of lysosomes to degrade cargo and a consequent increase in the formation of autophagosomes (Gukovskaya et al., 2017; Habtezion et al., 2019). Studies focusing on autophagy related genes knockout mice models revealed the mechanistic role of autophagy in maintaining the homeostasis of pancreas and the pathogenesis of AP. Autophagy genes (Atg) 5 and Atg7 knockout mice developed pancreatitis and extensive fibrosis (Antonucci et al., 2015; Diakopoulos et al., 2015). In addition, the impaired lysosomal function, for example in lysosomal-associated membrane protein 2 (LAMP2) deficient mice, leading to pancreatic inflammation and acinar cell degeneration. Furthermore, LAMP2 deficiency also increased the severity of experimental pancreatitis (Mareninova et al., 2015). All these findings showed the essential role of impaired autophagy in the development of AP, suggesting pharmacologic approaches to enhance autophagy efficiency may be of great importance to treat AP.
Bromodomain-containing protein 4 (BRD4) is a member of the Bromo- and Extra-Terminal domain (BET) family proteins with two N-terminal bromodomains and an extra-terminal domain (Shi and Vakoc, 2014; Hogg et al., 2017). BRD4 plays a crucial role in regulating gene transcription during cellular proliferation and differentiation processes (Shi and Vakoc, 2014; Jiang et al., 2017). Moreover, BRD4 has been recently identified to repress autophagy and lysosomal genes transcription by binding to the promoter regions (Sakamaki et al., 2017). BRD4 inhibition can enhance autophagic flux and lysosomal function, thus promoting the degradation of pathogenic proteins (Sakamaki et al., 2017). However, the role of BRD4 in the pathogenesis of AP remains undetermined.
Therefore, we aim to explore the role of BRD4 using experimental models of pancreatitis. Firstly, we found that BRD4 is upregulated in various models of AP. Inhibition of BRD4 by JQ1 protected against pancreatic acinar cell injury induced by cholecystokinin (CCK). Furthermore, inhibition or knockdown of BRD4 restored impaired autophagic flux via enhancing autophagosome-lysosome fusion and lysosomal degradation. Interestingly, BRD4 did not alter the initiation of autophagy in pancreatic acinar cells. In addition, BRD4 inhibition upregulated SIRT1 and inhibition of SIRT1 reversed the effects of BRD4 inhibition on autophagic flux, suggesting that inhibition of BRD4 via upregulating SIRT1 exerted its effects on autophagy. Finally, we showed that BRD4 inhibition also protected against two clinically representative models of AP through restoring impaired autophagy in vivo.
Materials and Methods
Chemicals and Reagents
Caerulein (CER) (#HY-A0190) was purchased from MedChemExpress (Monmouth Junction, NJ). Palmitoleic acid (POA), CCK, L-Arginine (L-Arg), and Na taurocholate (NaT) were purchased from Sigma-Aldrich Chemical (St. Louis, MO, USA). BSA was purchased from Yeasen (Shanghai, China). JQ1, EX527 were purchased from Selleck Chemicals (Houston, TX, USA). Antibodies against microtubule associated protein 1 light chain 3 beta (LC3B) (#3868S), p62 (#23214S), ATG14 (#96752S), cathepsin B (#31718S), AMP activated protein kinase (AMPK) (#5831T), phosphorylated AMPK (#2535T), mammalian target of rapamycin (mTOR) (#2983T), phosphorylated mTOR (#5536T), β-actin (#3700s) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against Amylase (#sc46657) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against BRD4 (#ab128874), LAMP2 (#ab203224), syntaxin 17 (STX17) (#ab229646), cathepsin L (#ab6314) were purchased from Abcam (Cambridge, MA, USA). Antibody against CD45 (#550539) were purchased from BD Biosciences (Franklin, NJ, USA).
Animals
All experiments involving animals were approved by the Institutional Animal Care and Research Ethics Committee of Shanghai Jiao Tong University School of Medicine (SYXK 2013–0050, Shanghai, China) and carried out in accordance with the guidelines of the National Institute of Health for the Care and Use of Laboratory Animals. Male C57BL/6 mice (6–8 weeks, 20–22 g) were purchased from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, China). All mice were housed under specific pathogen-free environment with controlled temperature (23 ± 1°C), 12 h light-dark cycle, humidity of 40–70% and free access to water and standard rodent diet. Mice were randomly allocated into groups for all the in vivo studies (n = 5 per group).
Isolation and Treatments of Mouse Pancreatic Acinar Cells
Pancreatic acinar cells were prepared by collagenase digestion, as described previously (Wen et al., 2018). Isolated pancreatic acinar cells were incubated at 37°C in DMEM/F-12 medium containing 10% fetal bovine serum with or without CCK or BRD4 inhibitor (JQ1) or chloroquine (CQ) (Sigma, #C6628) or SIRT1 inhibitor (EX527). For viral transduction, cells were infected with 107 plaque forming unit per ml adenovirus 24 h before stimulation. The siRNA sequence used for viral transduction is CCATGGATATGGGAACAAT (#1), GCCTCCAAAGAAGGATGTA (#2), GCCTGAAGAGCCAGTTGTT (#3), and TTCTCCGAACGTGTCACGT (Negative Control).
ATP Measurement
ATP levels in acinar cells were detected by using the Cell Titer Glo Luminescent Cell Viability Assay kit (Promega, Madison, WI) according to the manufacturer’s instructions, as previously described (Han et al., 2017). In brief, cells (3.0 × 106/ml) were treated with JQ1 (500 nmol/L) for 1 h, prior to CCK (200 nmol/L) treatment for 4 h. After the treatment, add 100 ul cell suspension into 96-well culture plate. Then add the ATP depletion reagents, and detect the level of bioluminescence using a Synergy multifunctional Microplate Reader (Gene Company Ltd, China). Data were normalized to protein concentration for each sample, then normalized to the untreated controls as 100%.
Assessment of PI Uptake
Isolated pancreatic acinar cells (3.0 × 106 per ml) were treated with JQ1 (500 nmol/L) for 1 h, prior to CCK (200 nmol/L) treatment for 4 h. Then cells were treated with propidium iodide (PI; 1 μmol/ml) for 5 min and the fluorescent intensity (excitation 536, emission 617), as PI uptake by the cells, was detected using a Synergy multifunctional Microplate Reader. Then 10 µl of 25% Triton-X100 (Sigma, #T8787) was added into the cells, and shake for 10 min and the fluorescent intensity (excitation 536, emission 617) was measured, as total amount of the cells. The percentage of PI uptake was calculated by Read 1 dividing Read 2 (% PI uptake = Read 1/Read 2 ×100).
Measurement of LDH Release
Detecting necrosis in pancreatic acinar cells was used the method of determining LDH released into the cultured medium, as reported earlier (Gukovskaya et al., 1997; Mareninova et al., 2006; Sung et al., 2009). In brief, cells (3.0 × 106/ml) were treated with JQ1 (500 nmol/L) for 1 h, prior to CCK (200 nmol/L) treatment for 4 h. LDH release was measured using LDH Cytotoxicity Assay Kit (Beyotime, Shanghai, China. C0017) according to the manufacturer’s instructions. The absorbance at 490 nm was detected by using Microplate Reader (BioTek Instruments, USA).
LysoTarcker Red Staining
Isolated pancreatic acinar cells (3.0 × 106/ml) were treated with JQ1 (500 nmol/L) for 1 h, prior to CCK (200 nmol/L) treatment for 4 h and then were harvested. After incubating with 500 μl of pre-warmed medium containing 75 nmol/L LysoTracker Red DND-99 dye (excitation 577 nm, emission 590 nm; 40739ES50, Yeasen, China) for 1 h, cells were washed and resuspended with Hoechst 33528 (40730ES10, Yeasen, China) for 15 min at 37°C. Lysosomal function was imaged by confocal imaging (Leica, Wetzlar, Germany).
Measurement the Activities of Cathepsin B and Cathepsin L
The activities of cathepsin B and cathepsin L were measured by the Cathepsin B Assay Kit (Abcam, #ab65300) and Cathepsin L Activity Assay Kit (Abcam, #ab65306), respectively, according to the manufacturer’s instructions. Briefly, isolated pancreatic acinar cells (3.0 × 106/ml) were pre-treated with JQ1 (500 nmol/L) with or without 10 μmol/L EX527 for 1 h, prior to CCK (200 nmol/L) stimulation for 4 h. The cell lysate and reaction buffer were added to a 96‐well black plate (Block Plate, WHB, Shanghai, China). The fluorescent intensity (excitation 400, emission 505) for both was detected using a Synergy multifunctional Microplate Reader.
Induction of Experimental Acute Pancreatitis
CER hyperstimulation pancreatitis was induced by ten hourly intraperitoneal injections of CER (100 μg/kg) (Lerch and Gorelick, 2013). Controls received similar injections of physiologic saline. JQ1 (20 mg/kg) was injected via tail vein 1 h before the first injection of CER. Mice were anesthetized by pentobarbital sodium (1.5%, w/v) 12 h after the first injection of CER, then blood and pancreas were collected.
Fatty acid ethyl ester pancreatitis model was induced by two hourly intraperitoneal injections of POA (150 mg/kg) and ethanol (1.35 g/kg) (Huang et al., 2014; Wen et al., 2015). The control animals were injected with equal volumes of ethanol. JQ1 (20 mg/kg) was injected via tail vein 1 h before the first injection of POA and ethanol. Mice were anesthetized by pentobarbital sodium (1.5%, w/v) 24 h after the first injection of POA and ethanol, then blood and pancreas were harvested.
L-Arg pancreatitis model was induced by two hourly intraperitoneal injections of 8% L-Arg (pH = 7.0), at a dose of 4 g/kg body weight (Dawra et al., 2007). Control group were injected with equal volume of saline. Mice were killed humanely by cervical dislocation 72 h after the first L-Arg injection and pancreas was harvested.
NaT pancreatitis was induced by pancreatic duct retrograde injection of 2% NaT (5 μl/min by infusion pump for 10 min), which is described previously (Perides et al., 2010). Control mice received the laparotomy only. After 24 h, mice were humanely killed by cervical dislocation pancreas were collected.
Serum Amylase
Blood samples were collected and centrifuged for 10 min at 3,000 rpm in 4°C. Serum amylase levels were detected by enzyme dynamics chemistry using commercial kits according to the manufacturer’s protocols (Roche, Basel, Switzerland).
Hematoxylin–Eosin Staining and Immunohistochemistry
H&E staining was performed after being fixed in 4% paraformaldehyde and embedded in paraffin, tissues were cut into 4 μm sections. Pancreatic sections were scored by two pathologists in a blind manner for edema, inflammatory infiltration, and necrosis, ranging from 0 to 3 (Wildi et al., 2007). CD45 antibody (1:100) was used for immunohistochemistry to evaluate pancreatic inflammatory cell infiltration (Wen et al., 2018). Briefly, after deparaffinization and antigen retrieval by proteinase K, non-specific bindings were blocked by 5% bovine serum albumin, and then primary antibody was incubated overnight at 4°C. Sections were treated with alkaline phosphatase labeled secondary antibody for 1 h and then imaged by substrate tablets (Sigma-Aldrich, St. Louis, MO, USA).
Immunofluorescence
The pancreas samples were embedded in paraffin and then deparaffinized. Antigen retrieval was performed by sodium citrate buffer (pH= 6), and then sections were blocked by 5% bovine serum albumin for 1 h. Sections were stained with monoclonal antibody against BRD4 (1:200) and amylase (1:300) overnight at 4°C; Alexa Fluor 488-labeled secondary antibody (1:200) and Alexa Fluor 488-labeled secondary antibody (1:300) for 1 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Sections were imaged by confocal microscope.
Western Blot
Total protein of pancreatic tissue and pancreatic acinar cells were lysed with RIPA which contained protease and phosphatase inhibitors, as previously described (Han et al., 2017). Proteins were loaded on a 15% or 10% polyacrylamide gel. Primary antibodies LC3B (1:1,000), p62 (1:1,000), AMPK (1:1,000), phosphorylated AMPK (1:1,000), mTOR (1:1,000), phosphorylated mTOR (1:1,000), ATG14 (1:1,000), STX17 (1:1,000), cathepsin B (1:1,000), cathepsin L (1:1,000), LAMP2 (1:1,000), BRD4 (1:800), and β-actin (1:1000) were used. The protein bands were detected by chemiluminescence (Millipore, USA) using Amersham Imager 600 (GE Healthcare, USA) and quantified by using Image J software.
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA from the pancreatic acinar cells or pancreatic tissues was isolated using Trizol reagent (Takara, Japan). Reverse transcription was used PrimeScript RT Master Mix (Perfect Real Time) (Takara, Japan). Gene expression was detected by real-time PCR using TB Green chemistry (Takara, Japan) on a QuantStudio 6 Flex System using gene-specific, intron-spanning primers (Table 1). The results were normalized to β-actin and expressed as fold changes over control group.

Table 1 Primer sequences used for qRT-PCR.
Statistical Analysis
Data were showed as mean ± SEM and analyzed by GraphPad Prism 4.0c (GraphPad Software, Inc.). The comparison between two groups was determined by Student’s un-paired, two-tailed t-test. P value <0.05 was considered statistically significant.
Results
BRD4 Is Upregulated in Mouse Models of Acute Pancreatitis
To examine the role of BRD4 during experimental AP, we firstly evaluated the expression of BRD4 in various clinically representative mouse models of AP. We found that the mRNA and protein expression of BRD4 were markedly upregulated in caerulein hyperstimulation pancreatitis, L-arginine-induced pancreatitis representing severe form of AP (Dawra et al., 2007), fatty acid ethyl ester pancreatitis mimicking alcohol associated acute pancreatitis (Huang et al., 2014), and NaT pancreatitis mimicking biliary acute pancreatitis (Perides et al., 2010) (Figures 1A–C). Consistently, immunofluorescent co-staining for BRD4 and amylase revealed that there was virtually undetectable expression of BRD4 in the exocrine of the pancreas in normal mice (Figure 1D). In contrast, BRD4 expression was markedly increased in the pancreas during four models of AP (Figure 1D). These results suggest that BRD4 may play a role in the pathogenesis of AP.
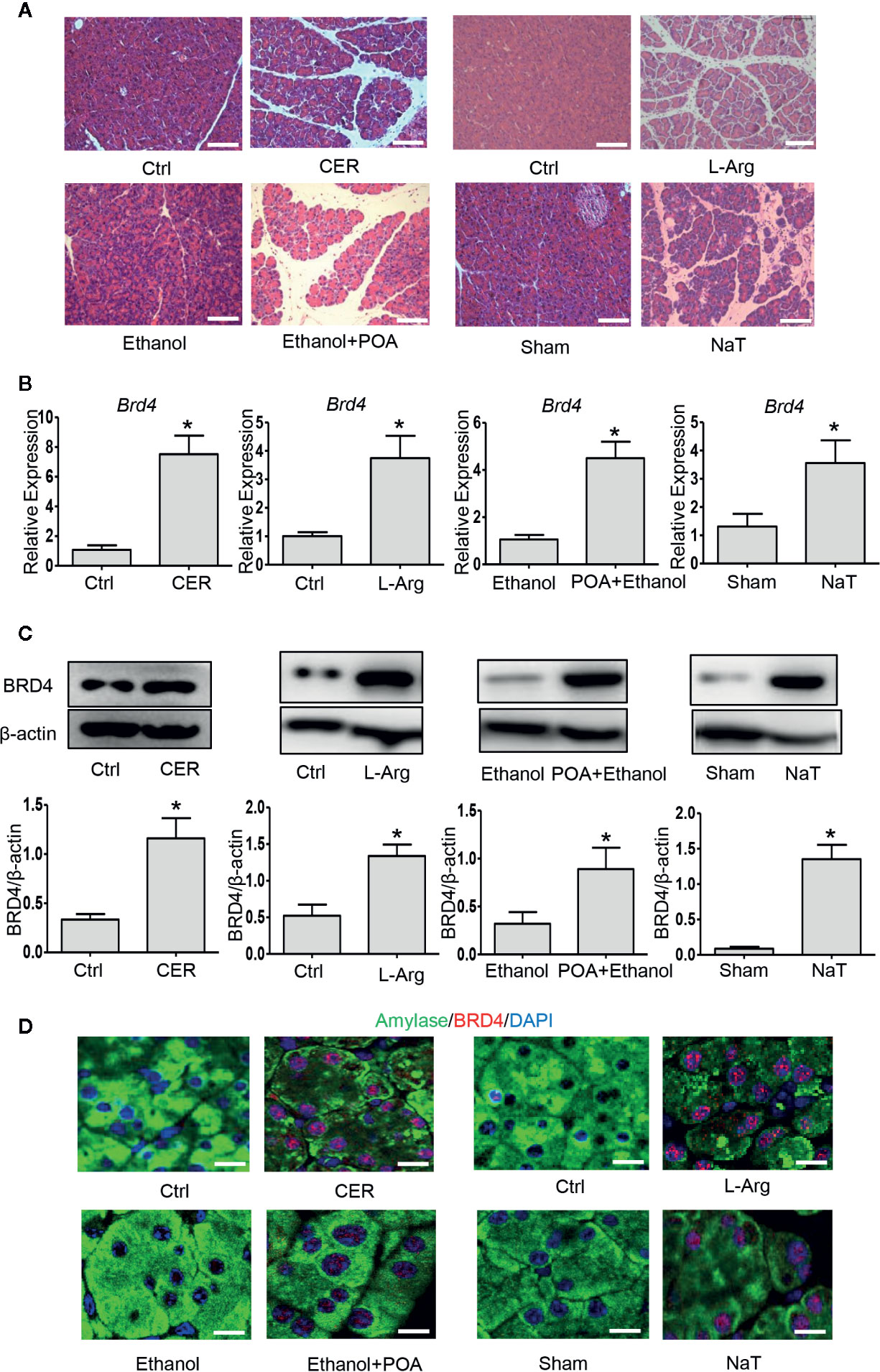
Figure 1 BRD4 is upregulated during various models of experimental acute pancreatitis. (A) Representative H&E images of pancreatic sections in CER, L-Arg, Ethanol- and POA, and NaT (200×), scale bar: 100 μm. (B) mRNA levels of Brd4. (C) Immunoblot analysis of BRD4 from pancreatic tissue. (D) Double immunofluorescent staining of BRD4 (red) and amylase (green) from pancreatic tissues (400×). 4′,6-Diamidino-2-phenylindole (DAPI; blue) was used to counterstain nuclei. Scale bar: 20 μm. Data represent the mean values ± SEM (n = 5). Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05 versus control.
BRD4 Inhibition Protects Against Pancreatic Acinar Cell Injury
In order to evaluate the therapeutic benefit of BRD4 in AP, we measured the effect of BRD4 inhibition on CCK-induced pancreatic acinar cell injury in vitro. In isolated primary pancreatic acinar cells, stimulation with CCK resulted in a marked reduction in the intracellular ATP levels and treatment with BRD4 inhibitor (JQ1) prevented CCK-induced loss of intracellular ATP (Figure 2A). To further confirm the effect of BRD4 inhibition on pancreatic acinar cell necrosis, we quantified acinar cell necrosis by LDH release and PI uptake. Inhibition of BRD4 by JQ1 markedly inhibited CCK-induced LDH release and the percentage of PI uptake, suggesting that BRD4 inhibition protected against necrotic cell death pathway activation in primary pancreatic acinar cells (Figures 2B, C). Moreover, we found that BRD4 inhibition also evidently inhibited the expression of several pro-inflammatory mediators, including Tnf, Il1b, and Il6 (Figure 2D). These results demonstrate that BRD4 inhibition protects against CCK-induced pancreatic acinar cell injury and inflammation.
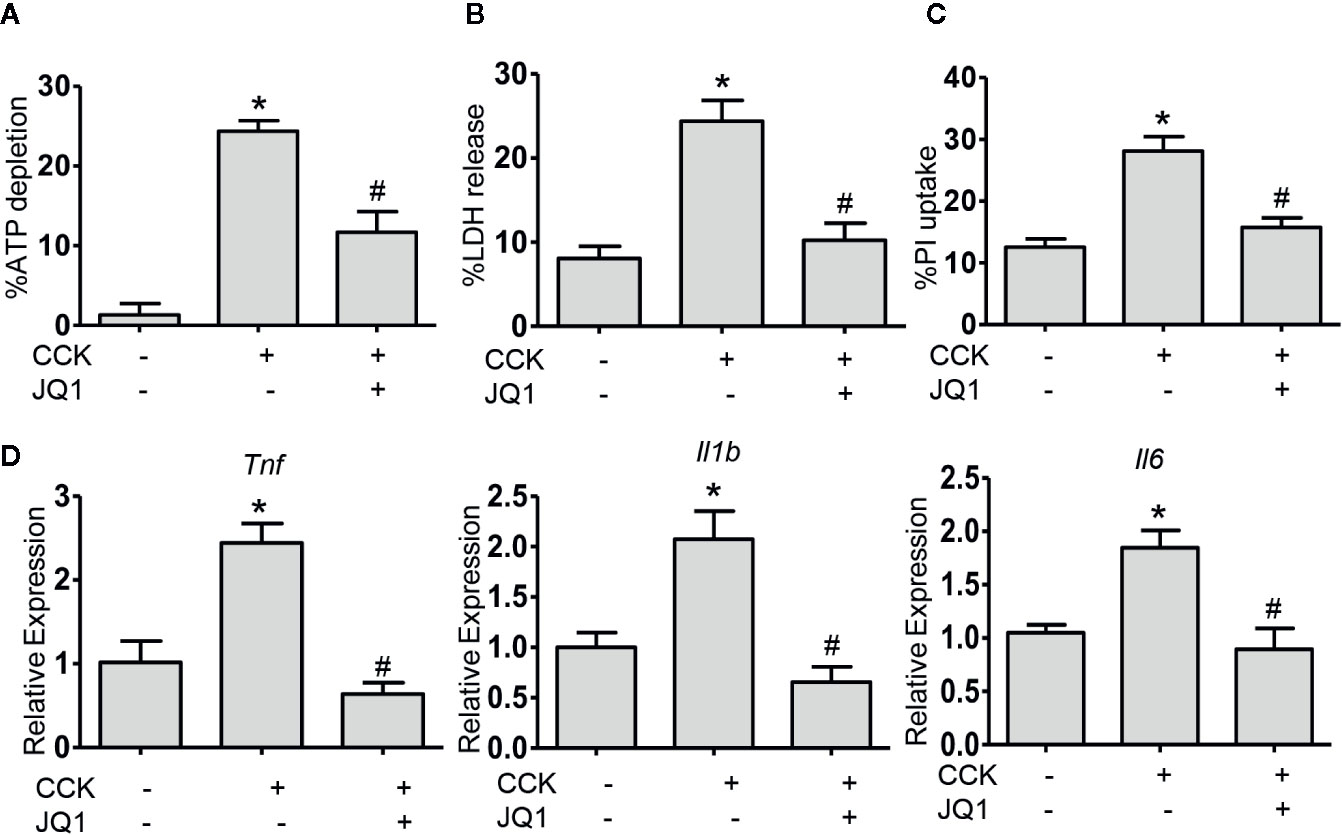
Figure 2 BRD4 inhibition prevents the activation of cell death pathway and downregulation of proinflammatory gene expression in vitro. (A) ATP levels were measured by luminescence in isolated pancreatic acinar cells. Data were normalized to untreated control as 100% (n = 5). (B) The percentage of total cellular lactate dehydrogenase (LDH) released into the extracellular medium in isolated pancreatic acinar cells stimulated with CCK (200 nmol·L-1) in the absence or presence of JQ1 (500 nmol·L-1) (n = 5). (C) PI uptake induced by CCK (200 nmol·L-1) with or without JQ1 (500 nmol·L-1) in isolated pancreatic acinar cells (n = 5). (D) mRNA levels of Tnf, Il1b, and Il6 in CCK-stimulated pancreatic acinar cells with or without JQ1 (n = 3). Data represent the mean values ± SEM. Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05, compared to the control; #P < 0.05, compared to CCK-stimulated group.
BRD4 Inhibition Restores Impaired Autophagic Flux In Vitro and the Effect of BRD4 Inhibition Is Mediated by Upregulating SIRT1
Impaired autophagy plays an important role in the pathogenesis of AP (Piplani et al., 2019). It has been shown that a series of autophagy-associated genes, including induction of autophagy, the fusion of autophagosome with lysosome and lysosomal degradation are repressed by BRD4 (Sakamaki et al., 2017), we next sought to determine whether BRD4 influences autophagic flux in AP. Firstly, we found that the protein expression of the LC3B-II and p62 was significantly elevated in pancreatic acinar cells after CCK stimulation. Treatment with chloroquine (CQ) in the presence or absence of CCK, an well-known inhibitor of autophagy that blocks lysosomal degradation, similarly led to an increase in the protein expression of the LC3B-II. Compared to CCK-stimulated and CQ-treated cells, inhibition or knockdown of BRD4 decreased the accumulation of LC3B-II and more markedly decreased the accumulation of p62, indicating that BRD4 inhibition may enhance autophagic flux (Figure 3A; Figures S1A, B). Next, we assessed the effect of BRD4 inhibition on AMPK and mTOR, which regulate autophagy induction through Ulk1 phosphorylation (Kim et al., 2011). Interestingly, we found that BRD4 inhibition had no significant effect on the activation of AMPK and mTOR, suggesting that inhibition of BRD4 did not affect the induction of autophagy in isolated pancreatic acinar cells (Figure 3B). We also found BRD4 inhibition did not affect the expression of the genes that were related to autophagosomes formation, including Becn1, Vmp1, Atg2a, and Map1lc3b (Figure 3C). Since the histone deacetylase, SIRT1 has been reported to regulate autophagy-lysosomal pathway (Hariharan et al., 2010; Huang et al., 2019) and can be upregulated by BRD4 inhibition (Kokkola et al., 2015). We examined whether BRD4 inhibition restores impaired autophagy by upregulating SIRT1. As expected, the expression of SIRT1 was upregulated by JQ1 treatment or BRD4 knockdown in isolated pancreatic acinar cells (Figures 4A, B; Figure S4). Furthermore, compared with JQ1-treated group, treatment with the SIRT1 inhibitor EX527 resulted in an increased expression of the autophagic markers such as LC3B-II and p62 (Figure 4C).
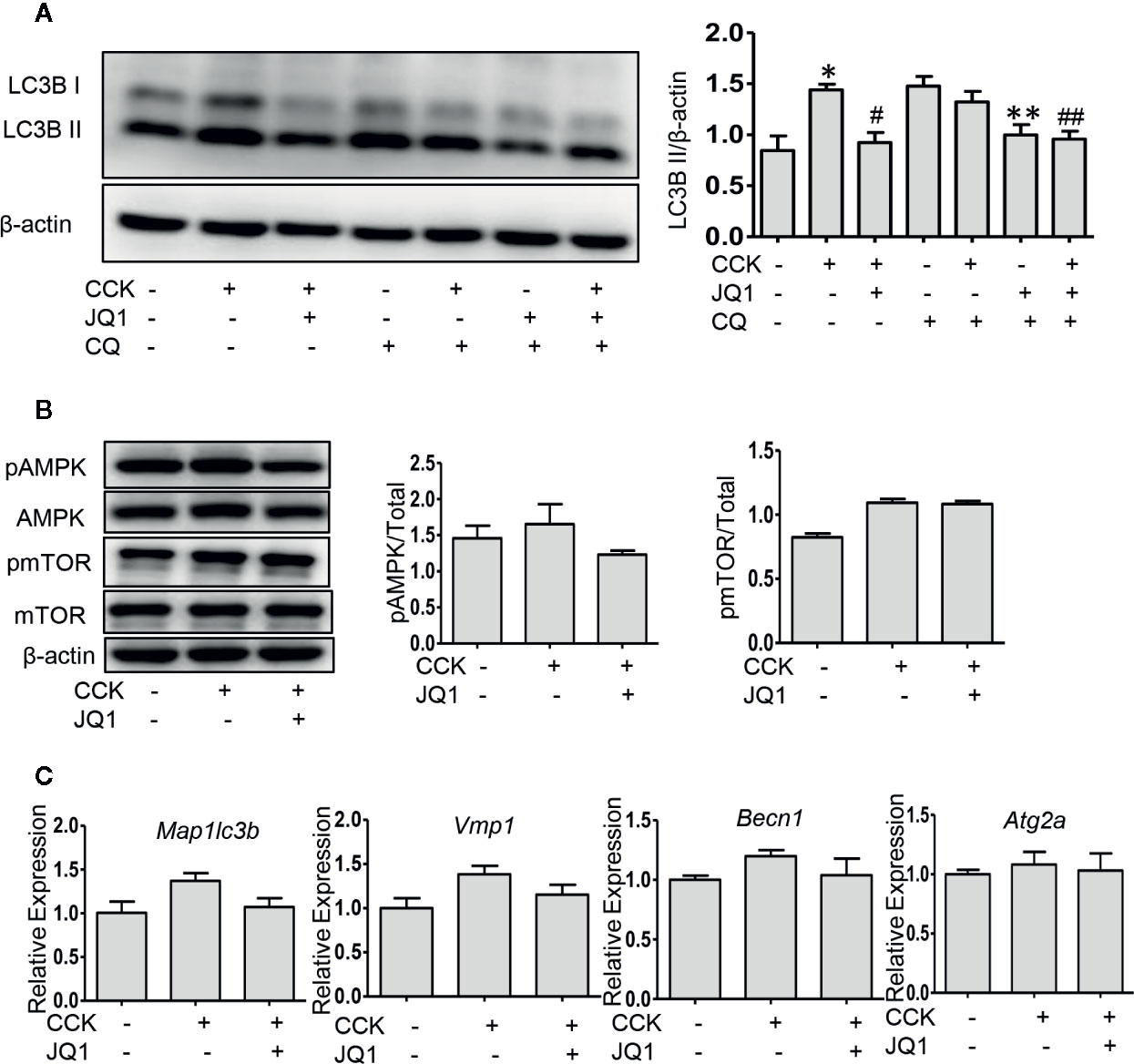
Figure 3 BRD4 inhibition restores impaired autophagic influx, but has no effect on autophagy initiation. (A) Immunoblot analysis of LC3B level at 4 h after CCK or CQ stimulation in isolated pancreatic acinar cells (n = 3). (B) Immunoblot analysis of AMPK, mTOR phosphorylation levels at 1 h after CCK stimulation in isolated pancreatic acinar cells (n = 3). (C) mRNA levels of Map1lc3b, Vmp1, Becn1, and Atg2a at 4 h after CCK stimulation (n = 3). Data represent the mean values ± SEM. Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05, compared to the control; #P < 0.05, compared to CCK-stimulated group;##P < 0.05, compared to the CQ-stimulated group; #P < 0.05, compared to CCK+CQ-stimulated group.
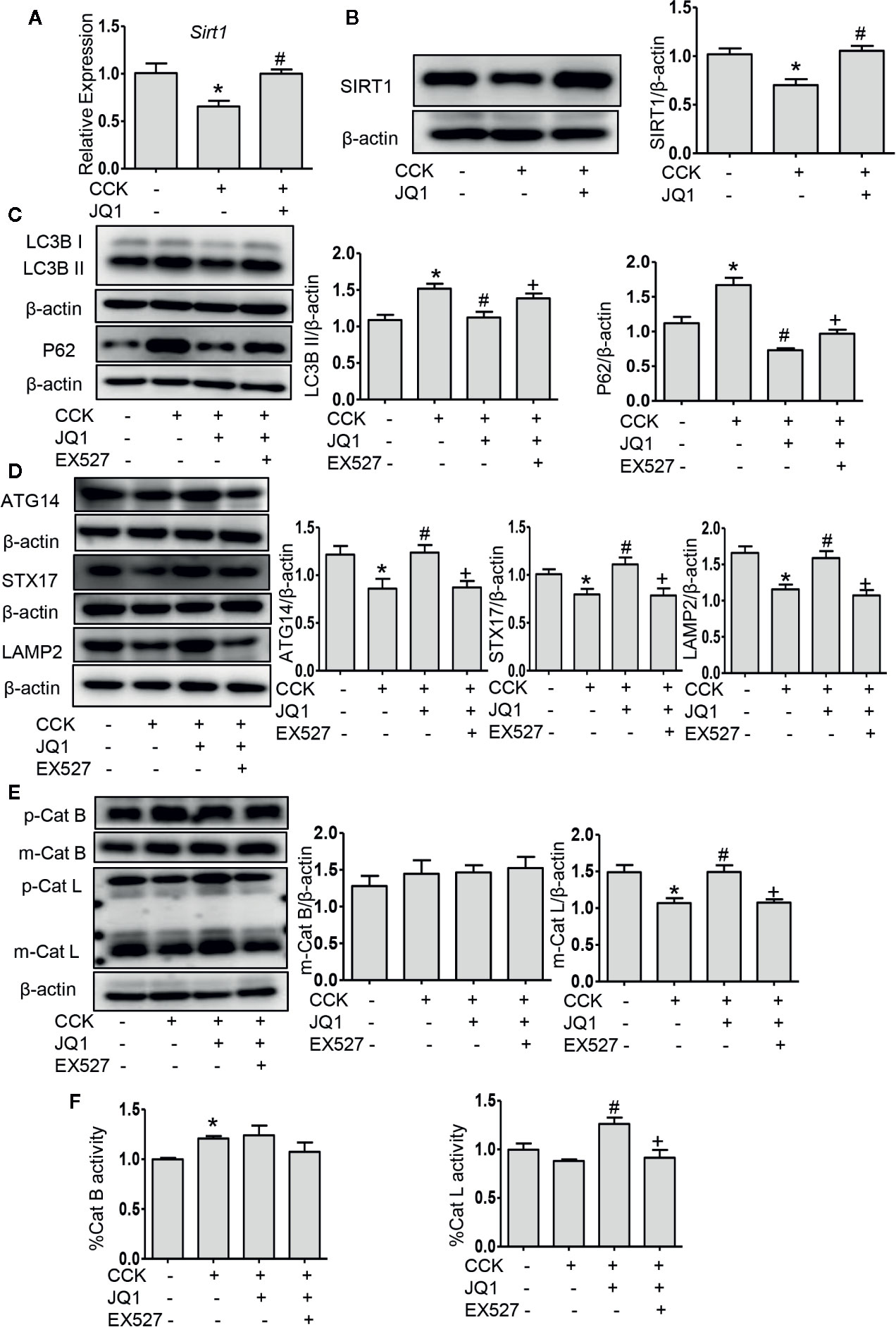
Figure 4 BRD4 inhibition upregulated SIRT1 and inhibition of SIRT1 reversed the effects of BRD4 inhibition on autophagic flux. (A) mRNA level of Sirt1 at 4 h after CCK stimulation in isolated pancreatic acinar cells (n = 3). (B) Immunoblot analysis of SIRT1 level at 4 h after CCK stimulation (n = 5). (C) Immunoblot analysis for LC3B and p62 expression in isolated pancreatic acinar cells pretreated with 500 nmol·L-1 JQ1 or 10 μmol·L-1 EX527 followed by stimulation with 200 nmol·L-1 CCK for 4 h (n = 4). (D) Immunoblot analysis for ATG14, STX17, and LAMP2 expression (n = 5). (E) Immunoblot analysis for cathepsin B (n = 3) and cathepsin L expression (n = 5). (F) The activities of cathepsin B (n = 3) and cathepsin L (n = 3). Data represent the mean values ± SEM. Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05, compared to the control; #P < 0.05, compared to CCK-stimulated group; +P < 0.05, compared to JQ1-treated group.
Autophagosome fusion with lysosome is a key step in autophagy progression, which is regulated by ATG14 and the complex formed by STX17, synaptosome associated protein 29, and vesicle associated membrane protein 8 (Itakura and Mizushima, 2013). We measured the expression of ATG14, STX17, and LAMP2 and found that the protein levels of ATG14, STX17, and LAMP2 were downregulated in pancreatic acinar cells after CCK stimulation and inhibition of BRD4 by JQ1 or knockdown of BRD4 by transfecting shRNA restored their expression (Figure 4D; Figures S2A, B), suggesting that BRD4 inhibition enhances the fusion of autophagosomes with lysosomes. Furthermore, using LysoTracker staining, we found that BRD4 inhibition maintained lysosomal pH (Figure S3). Interestingly, we also observed that treatment with the SITR1 inhibitor, EX527 reversed the effects of BRD4 inhibition on the expression of autophagosome and lysosome fusion markers, including ATG14, STX17, and LAMP2 (Figure 4D), indicating the influence of BRD4 inhibition on autophagosome fusion with lysosome was mediated by SIRT1.
It has been shown that during AP, impaired autophagy is related to an imbalance between cathepsin L and cathepsin B as the former degrades trypsinogen and trypsin into amino acid and the latter converts trypsinogen into trypsin (Mareninova et al., 2009). We next measured the expression of cathepsin L and cathepsin B and found that inhibition or knockdown BRD4 significantly increased the level of the pro-enzyme and cleaved form of cathepsin L. Interestingly, BRD4 inhibition did not alter the expression of cathepsin B, but BRD4 knockdown markedly decreased the expression of cathepsin B (Figure 4E; Figure S2C), suggesting that BRD4 mediates the balance between cathepsin L and cathepsin B, therefore, resulting in reduced pancreatic acinar cell injury. And with the SITR1 inhibitor treatment, the pro-enzyme and the cleaved form of cathepsin L was decreased, but the level of cathepsin B was not changed (Figure 4E). To further assess the effect of JQ1 on lysosomal function, we measured the activity of cathepsin B and L. BRD4 inhibition increased the activity of cathepsin L and SIRT1 inhibitor reversed its effect, but there was no effect on the activity of cathepsin B (Figure 4F). Taken together, these data suggest that BRD4 inhibition enhances the fusion of autophagosome with lysosome and improves lysosomal degradation, but has no influence on autophagy initiation and autophagosome formation. Secondly, the influence of BRD4 inhibition on autophagy was likely mediated by upregulating SIRT1.
BRD4 Inhibition Alleviates Two Acute Pancreatitis Models
Then, we examined the influence of BRD4 inhibition on pancreatitis severity during two experimental models of AP in vivo. Firstly, we tested JQ1, the inhibitor of BRD4 in caerulein hyperstimulation pancreatitis, a widely used and highly reproducible AP model (Lerch and Gorelick, 2013). We showed that JQ1 markedly reduced pancreatic edema, inflammatory infiltration, and necrosis (Figures 5A, B). Furthermore, leukocyte infiltration assessed by CD45 immunostaining and pro-inflammatory cytokines such as Tnf, Il1b, and Il6 in the pancreas were decreased with JQ1 treatment (Figures 5C, D). The second AP model was induced by administrations of POA and ethanol, which is associated with alcohol-induced pancreatitis (Huang et al., 2014). Similarly, we found that JQ1 significantly reduced histological scores in the pancreas, including edema, inflammation, and necrosis (Figures 5A, B). Furthermore, JQ1 decreased leukocyte infiltration and inflammatory factors in the pancreas and serum amylase levels in this model (Figures 5C, D). Consistently with our in vitro findings, these data showed that BRD4 inhibition protects against two clinically relevant models of AP.
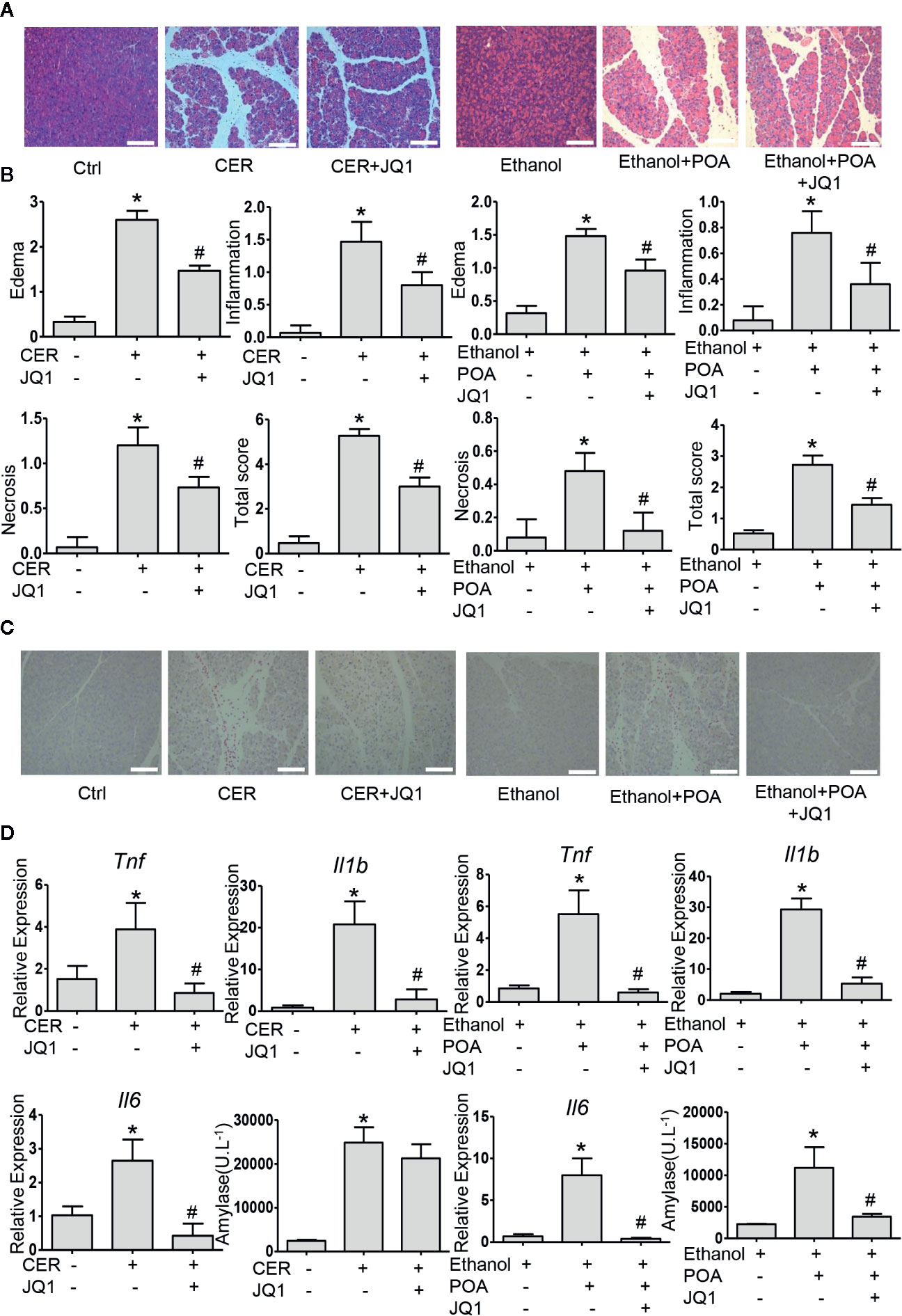
Figure 5 BRD4 inhibition protects against caerulein- and ethanol plus POA-induced pancreatitis in vivo. (A) Representative H&E images of pancreatic sections in CER and ethanol plus POA-induced pancreatitis (200×) (n = 5). (B) Histopathological scores for edema, inflammation, necrosis, and the total histology score (n = 5). (C) Representative micrographs of leukocyte marker CD45 immunohistochemical staining for the pancreas (200×). (D) mRNA levels of Tnf, Il1b, and Il6 from pancreatic tissue (n = 3) and serum amylase levels (n = 5). Scale bar = 100 μm. Data represent the mean values ± SEM. Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05, compared to the control; #P < 0.05, compared to AP group.
BRD4 Inhibition Restores Impaired Autophagy In Vivo
Finally, we evaluated the effect of BRD4 inhibition on autophagy in vivo. In these two AP models, JQ1 treatment significantly downregulated p62 levels (Figure 6A). BRD4 inhibition downregulated LC3B-II expression in fatty acid ethyl ester-induced pancreatitis, while had no effect on LC3B-II expression CER model (Figure 6A). These data suggest that BRD4 inhibition restores impaired autophagy in vivo. Furthermore, we found that BRD4 inhibition upregulated the expression of ATG14 and LAMP2, but had no effect on STX17 in both models (Figures 6B, C), indicating BRD4 inhibition enhances the fusion of the autophagosome with lysosome in vivo. Finally, we detected the expression of SIRT1 in vivo and found that BRD4 inhibition significantly upregulated SIRT1 levels (Figure S5). Collectively, our data suggest that BRD4 inhibition via upregulating SIRT1 restores impaired autophagy in vivo.
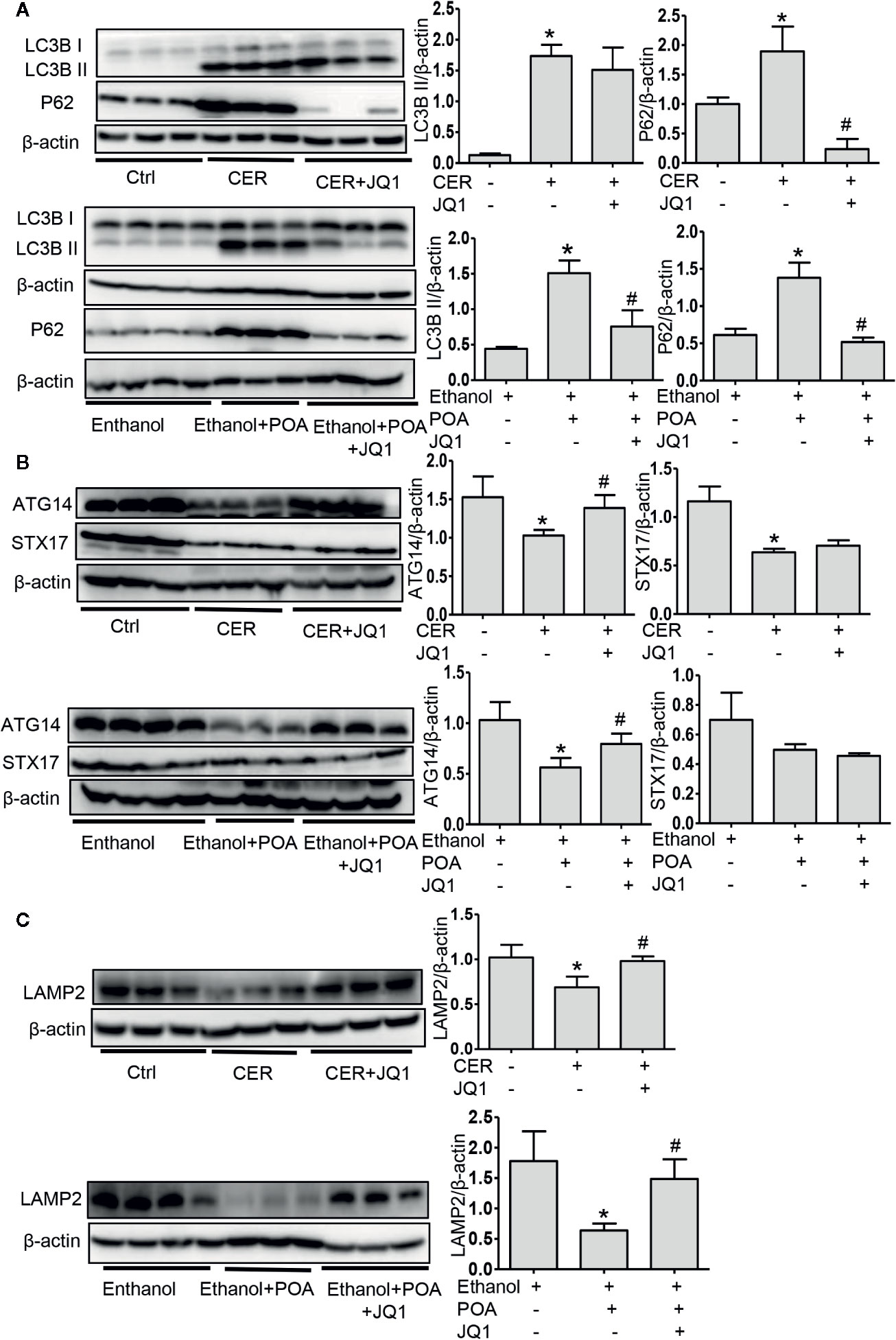
Figure 6 BRD4 inhibition restores impaired autophagic influx in vivo via promoting autophagosome-lysosome fusion and lysosomal degradation. (A) Immunoblot analysis for LC3 and p62 in the pancreas from CER (upper) or ethanol plus POA (lower). (B) Immunoblot analysis for ATG14 and STX17 in the pancreas from CER (upper) or ethanol plus POA (lower). (C) Immunoblot analysis for LAMP2 in the pancreas from CER (upper) or ethanol plus POA (lower). Data represent the mean values ± SEM (n = 3). Statistical analysis was performed by Student’s un-paired, two-tailed t-test between two groups, *P < 0.05, compared to the control; #P < 0.05, compared to AP group.
Discussion
The BET protein family includes four subtypes: Brd2, Brd3, Brd4, and Brd6/t which is testis specific. (Shi and Vakoc, 2014; Sakamaki et al., 2017). It has been known that among these isoforms only BRD4 knockdown mediates autophagy and lysosomal function (Sakamaki et al., 2017). Therefore, we focused our study on the role of BRD4 in AP. Among BRD4 inhibitors, we choose JQ1 since it is widely used pharmacological BRD4 inhibitor in various other diseases (Jiang et al., 2017; Rudman et al., 2018; Song et al., 2019). We found that BRD4 inhibition reduced CCK-induced pancreatic acinar cell injury and pro-inflammatory expression in vitro, and protected against two AP models. Interestingly, Huang et al. reported that treatment with I-BET-762, another BET inhibitor, markedly alleviated taurolithocholic acid-induced pancreatitis and POA plus ethanol AP, but not caerulein hyperstimulation pancreatitis (Huang et al., 2017), suggesting that different BET inhibitor may exert their anti-inflammatory effects through different mechanisms. As shown in our study, BRD4 inhibition by JQ1 protects against AP mainly through restoring impaired autophagic flux, which has been reported to be pivotal in the pathogenesis of AP.
Autophagy is a critical catabolic process which can degrade and recycle damaged organelles, lipids, and proteins (Habtezion et al., 2019). SQSTM1, also known as p62, is an autophagy receptor. As degraded along with cargos, it can be used as a marker of autophagic degradation (Klionsky et al., 2016). The excessive accumulation of p62 is a marker of impaired autophagy (Gukovskaya et al., 2017). We found that BRD4 inhibition and knockdown had a pronounced effect on reducing the accumulation of p62, indicating that BRD4 inhibition enhances autophagic degradation. It has been reported that BRD4 inhibition and knockdown promotes autophagy induction in early stages, the formation of autophagosomes, and subsequent fusion with lysosomes (Sakamaki et al., 2017). In our study, we found that BRD4 inhibition had no effects on the phosphorylation of mTOR and AMPK, two markers of autophagic induction, nor on the genes that were related to autophagosomes formation, suggesting that BRD4 inhibition did not affect autophagy induction and autophagosome formation. This may partly be explained by that pancreatic basal autophagy is high and is crucial for pancreatic hemostasis (Antonucci et al., 2015), but during experimental AP, activated autophagy is protective while incomplete autophagic process might be detrimental (Gukovskaya et al., 2017). Accumulating evidence demonstrated that autophagy in acute pancreatitis models is impaired (Mareninova et al., 2009; Gukovskaya et al., 2017; Biczo et al., 2018). The fusion of autophagosome with lysosome is critical for efficient lysosomal degradation and is regulated by a series of proteins, such as ATG14, Rab7, LAMP2, and STX17 (Itakura and Mizushima, 2013; Zhang et al., 2019). We found that BRD4 inhibition or knockdown increased the expression of ATG14, STX17, and LAMP2 in isolated pancreatic acinar cells and in two clinically relevant models of AP, suggesting that BRD4 inhibition restores impaired autophagy by improving the fusion of autophagosome with lysosome in vitro and in vivo.
In addition to the fusion of autophagosome with lysosome, efficient lysosomal degradation depends on the integrity of lysosome membrane and the activities of acidic hydrolases (Saftig and Klumperman, 2009). Cathepsin B and cathepsin L are involved in this process, cathepsin B converts trypsinogen to trypsin while cathepsin L degrades both trypsin and trypsinogen into amino acids (Halangk et al., 2000; Wartmann et al., 2010; Gukovskaya and Gukovsky, 2012). Previous studies have shown that in pancreatitis level of fully processed (mature) forms of these cathepsins decreased and accumulation of intermediate forms increased (Mareninova et al., 2009; Gukovskaya and Gukovsky, 2012; Biczo et al., 2018). Moreover, acidic environment in the lysosome plays a critical role in maintaining the normal activities of these enzymes (Mindell, 2012). In this study, we found that BRD4 inhibition maintained lysosomal pH, increased the expression of mature form of cathepsin L and the activity of cathepsin L in isolated pancreatic acinar cells, suggesting that BRD4 inhibition restores impaired autophagy during AP also by enhancing lysosomal degradation.
SIRT1 is a member of class III histone deacetylase, which is involved in the regulation of cell metabolism, apoptosis, and autophagy (Oellerich and Potente, 2012). It has been showed that BRD4 inhibitor JQ1 upregulated SIRT1 and alleviated inflammatory responses in a cellular model of lung disease (Kokkola et al., 2015). In addition, BRD4 inhibition induced ferritinophagy and downregulated the expression of genes that are related to ferroptosis by enhancing the expression of SIRT1 or suppressing the expression of the histone methyltransferase G9a (Sui et al., 2019). Consistently, we found that BRD4 inhibition or knockdown upregulated SIRT1 in pancreatic acinar cells and in experimental models of AP. It has been reported that SIRT1 regulates autophagy by interacting with autophagy related genes and deacetylating them. For example, SIRT1 deacetylate autophagy genes such as Atg5, Atg7, and Atg8, which is critical for the activation of autophagy induced by starvation (Lee et al., 2008). Moreover, SIRT1 deacetylated FOXO1 (forkhead box O1), enhancing autophagosome-lysosome fusion (Hariharan et al., 2010; Huang et al., 2019). SIRT1 also deacetylates FOXO3 (forkhead box O3), leading Bnip3‐mediated autophagy (Kume et al., 2010). In addition, SIRT1 deacetylates TFEB (transcription factor EB), enhancing the expression of autophagy/lysosome-associated genes (Bao et al., 2016). In this study, we observed that the inhibition of SIRT1 deacetylation with EX527 reversed the effects of BRD4 inhibition on autophagic flux. Specifically, inhibition of SIRT1 reversed the increased expression of LC3B-II and p62 with BRD4 inhibition, downregulated the increased expression of ATG14, STX17, and LAMP2 with BRD4 inhibition, and decreased the pro-enzyme and the cleaved form and the activity of cathepsin L, but the level and the activity of cathepsin B was not changed. These data suggest that SIRT1 is a crucial mediator that is responsible for BRD4-mediated autophagy during AP.
Conclusion
In summary, in this study, we showed that BRD4 expression is upregulated in various experimental models of pancreatitis. BRD4 inhibition alleviated pancreatic acinar cell injury and two clinically relevant mouse models of experimental AP. These protective effects were mediated by restoring impaired autophagic flux, primarily through enhancing autophagosome fusion with lysosome and lysosomal degradation. The regulation of impaired autophagic flux by BRD4 inhibition is through upregulating SIRT1 (Figure 7).
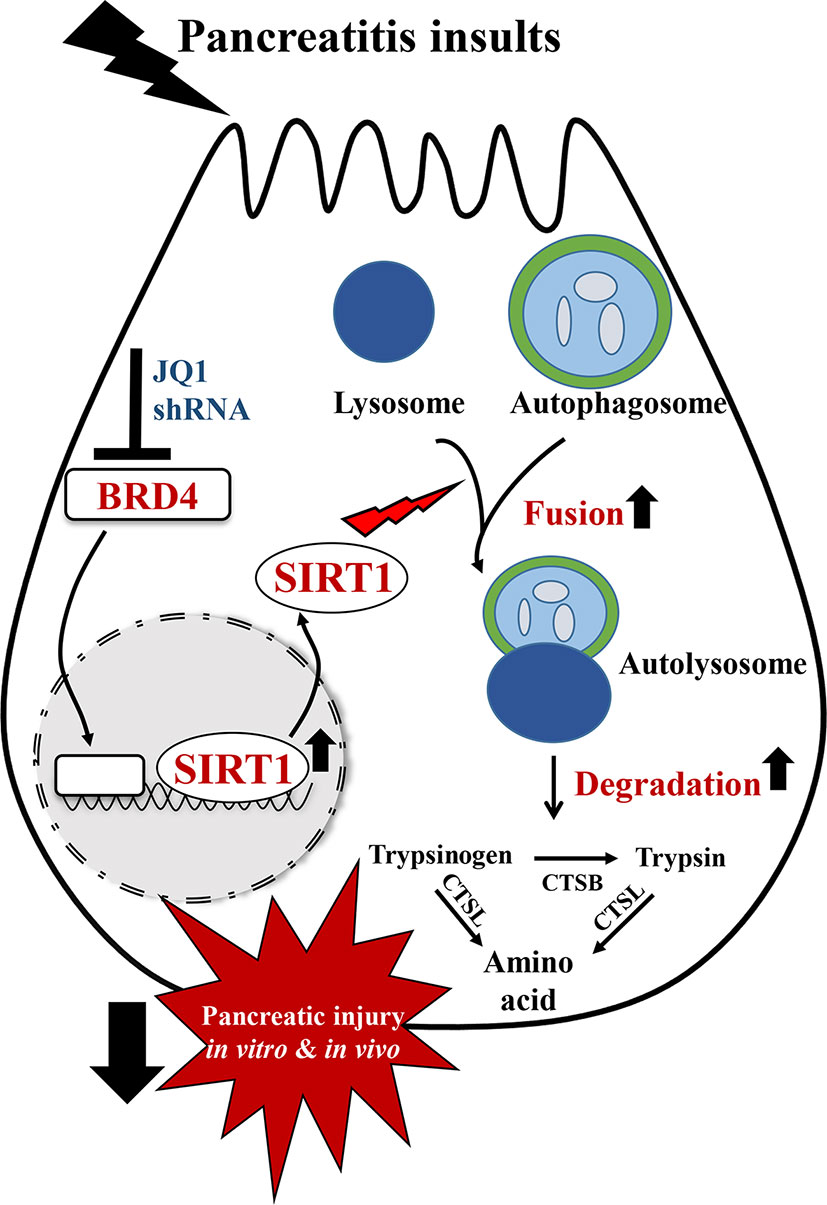
Figure 7 Schematic diagram for the mechanism of BRD4 inhibition on impaired autophagy during acute pancreatitis. BRD4 inhibition protects against pancreatic injury in the in vitro and in vivo settings of AP. Inhibition or knockdown of BRD4 upregulated SIRT1, leading to enhanced autophagosome fusion with lysosome and lysosomal degradation, therefore restored impaired autophagic flux by pancreatitis insults.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
All experiments involving animals were approved by the Institutional Animal Care and Research Ethics Committee of Shanghai Jiao Tong University School of Medicine (SYXK 2013–0050, Shanghai, China).
Author Contributions
SS, BL, and JD performed the experiments and drafted the manuscript. SS, ZW, and YH performed the statistical analysis. LW, GH, and XW designed, conceived the study, revised the manuscript, and provided funding to support the study. All authors read and approved the submitted version.
Funding
This work was sponsored by Natural Science Foundation of China to GH (81670584 and 81970556), XW (81570580), and LW (81900585), Shanghai Pujiang Program to GH (18PJD041) and LW (19PJ1408400), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant to LW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflicts of interest.
The reviewer OM declared a past co-authorship with one of the authors LW to the handling editor.
Acknowledgments
The abstract entitled “BRD4 inhibition protects against experimental acute pancreatitis through restoring impaired autophagy” was selected for poster presentation (PW-56) at the annual meeting of the American Pancreatic Association 2019 in Maui, Hawaii.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00618/full#supplementary-material.
Abbreviations
AP, acute pancreatitis; BRD4, Bromodomain-containing protein 4; BET, bromodomain and extraterminal; CCK, cholecystokinin; CQ, chloroquine; SIRT1, sirtuin 1; Atg, autophagy genes; CER, caerulein; POA, palmitoleic acid; L-Arg, L-Arginine; NaT, Na taurocholate; LAMP2, lysosomal-associated membrane protein 2; AMPK, AMP activated protein kinase; LC3B, microtubule associated protein 1 light chain 3 beta; STX17, syntaxin 17; mTOR, mammalian target of rapamycin; FOXO1, forkhead box O1; FOXO3, forkhead box O1; TFEB, transcription factor EB.
References
Antonucci, L., Fagman, J. B., Kim, J. Y., Todoric, J., Gukovsky, I., Mackey, M., et al. (2015). Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. U. S. A 112 (45), E6166–E6174. doi: 10.1073/pnas.1519384112
Bao, J., Zheng, L., Zhang, Q., Li, X., Zhang, X., Li, Z., et al. (2016). Deacetylation of TFEB promotes fibrillar Abeta degradation by upregulating lysosomal biogenesis in microglia. Protein Cell 7 (6), 417–433. doi: 10.1007/s13238-016-0269-2
Biczo, G., Vegh, E. T., Shalbueva, N., Mareninova, O. A., Elperin, J., Lotshaw, E., et al. (2018). Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 154 (3), 689–703. doi: 10.1053/j.gastro.2017.10.012
Dawra, R., Sharif, R., Phillips, P., Dudeja, V., Dhaulakhandi, D., Saluja, A. K. (2007). Development of a new mouse model of acute pancreatitis induced by administration of L-arginine. Am. J. Physiol. Gastrointest. Liver Physiol. 292 (4), G1009–G1018. doi: 10.1152/ajpgi.00167.2006
Diakopoulos, K. N., Lesina, M., Wormann, S., Song, L., Aichler, M., Schild, L., et al. (2015). Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 148 (3), 626–638 e617. doi: 10.1053/j.gastro.2014.12.003
Gukovskaya, A. S., Gukovsky, I. (2012). Autophagy and pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 303 (9), G993–G1003. doi: 10.1152/ajpgi.00122.2012
Gukovskaya, A. S., Gukovsky, I., Zaninovic, V., Song, M., Sandoval, D., Gukovsky, S., et al. (1997). Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J. Clin. Invest. 100 (7), 1853–1862. doi: 10.1172/JCI119714
Gukovskaya, A. S., Gukovsky, I., Algul, H., Habtezion, A. (2017). Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 153 (5), 1212–1226. doi: 10.1053/j.gastro.2017.08.071
Habtezion, A., Gukovskaya, A. S., Pandol, S. J. (2019). Acute Pancreatitis: A Multifaceted Set of Organelle and Cellular Interactions. Gastroenterology 156 (7), 1941–1950. doi: 10.1053/j.gastro.2018.11.082
Halangk, W., Lerch, M. M., Brandt-Nedelev, B., Roth, W., Ruthenbuerger, M., Reinheckel, T., et al. (2000). Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106 (6), 773–781. doi: 10.1172/JCI9411
Han, X., Li, B., Ye, X., Mulatibieke, T., Wu, J., Dai, J., et al. (2017). Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-kappaB signalling pathway. Br. J. Pharmacol. 174 (24), 4751–4770. doi: 10.1111/bph.14057
Hariharan, N., Maejima, Y., Nakae, J., Paik, J., Depinho, R. A., Sadoshima, J. (2010). Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 107 (12), 1470–1482. doi: 10.1161/CIRCRESAHA.110.227371
Hogg, S. J., Vervoort, S. J., Deswal, S., Ott, C. J., Li, J., Cluse, L. A., et al. (2017). BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 18 (9), 2162–2174. doi: 10.1016/j.celrep.2017.02.011
Huang, W., Booth, D. M., Cane, M. C., Chvanov, M., Javed, M. A., Elliott, V. L., et al. (2014). Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63 (8), 1313–1324. doi: 10.1136/gutjnl-2012-304058
Huang, W., Haynes, A. C., Mukherjee, R., Wen, L., Latawiec, D., Tepikin, A. V., et al. (2017). Selective inhibition of BET proteins reduces pancreatic damage and systemic inflammation in bile acid- and fatty acid ethyl ester- but not caerulein-induced acute pancreatitis. Pancreatology 17 (5), 689–697. doi: 10.1016/j.pan.2017.06.005
Huang, J., Wang, X., Zhu, Y., Li, Z., Zhu, Y. T., Wu, J. C., et al. (2019). Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci. Ther. 25 (6), 796–807. doi: 10.1111/cns.13114
Itakura, E., Mizushima, N. (2013). Syntaxin 17: the autophagosomal SNARE. Autophagy 9 (6), 917–919. doi: 10.4161/auto.24109
Jiang, Y., Zhu, L., Zhang, T., Lu, H., Wang, C., Xue, B., et al. (2017). BRD4 has dual effects on the HMGB1 and NF-kappaB signalling pathways and is a potential therapeutic target for osteoarthritis. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (12), 3001–3015. doi: 10.1016/j.bbadis.2017.08.009
Kim, J., Kundu, M., Viollet, B., Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13 (2), 132–141. doi: 10.1038/ncb2152
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12 (1), 1–222. doi: 10.1080/15548627.2015.1100356
Kokkola, T., Suuronen, T., Pesonen, M., Filippakopoulos, P., Salminen, A., Jarho, E. M., et al. (2015). BET Inhibition Upregulates SIRT1 and Alleviates Inflammatory Responses. Chembiochem 16 (14), 1997–2001. doi: 10.1002/cbic.201500272
Kume, S., Uzu, T., Horiike, K., Chin-Kanasaki, M., Isshiki, K., Araki, S., et al. (2010). Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 120 (4), 1043–1055. doi: 10.1172/JCI41376
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U. S. A 105 (9), 3374–3379. doi: 10.1073/pnas.0712145105
Lerch, M. M., Gorelick, F. S. (2013). Models of acute and chronic pancreatitis. Gastroenterology 144 (6), 1180–1193. doi: 10.1053/j.gastro.2012.12.043
Mareninova, O. A., Sung, K. F., Hong, P., Lugea, A., Pandol, S. J., Gukovsky, I., et al. (2006). Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J. Biol. Chem. 281 (6), 3370–3381. doi: 10.1074/jbc.M511276200
Mareninova, O. A., Hermann, K., French, S. W., O’Konski, M. S., Pandol, S. J., Webster, P., et al. (2009). Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Invest. 119 (11), 3340–3355. doi: 10.1172/JCI38674
Mareninova, O. A., Sendler, M., Malla, S. R., Yakubov, I., French, S. W., Tokhtaeva, E., et al. (2015). Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol. Gastroenterol. Hepatol. 1 (6), 678–694. doi: 10.1016/j.jcmgh.2015.07.006
Mindell, J. A. (2012). Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86. doi: 10.1146/annurev-physiol-012110-142317
Oellerich, M. F., Potente, M. (2012). FOXOs and sirtuins in vascular growth, maintenance, and aging. Circ. Res. 110 (9), 1238–1251. doi: 10.1161/CIRCRESAHA.111.246488
Pandol, S. J., Saluja, A. K., Imrie, C. W., Banks, P. A. (2007). Acute pancreatitis: bench to the bedside. Gastroenterology 132 (3), 1127–1151. doi: 10.1053/j.gastro.2007.01.055
Parzych, K. R., Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxidants Redox Signaling 20 (3), 460–473. doi: 10.1089/ars.2013.5371
Perides, G., van Acker, G. J., Laukkarinen, J. M., Steer, M. L. (2010). Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat. Protoc. 5 (2), 335–341. doi: 10.1038/nprot.2009.243
Piplani, H., Marek-Iannucci, S., Sin, J., Hou, J., Takahashi, T., Sharma, A., et al. (2019). Simvastatin induces autophagic flux to restore cerulein-impaired phagosome-lysosome fusion in acute pancreatitis. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (11), 165530. doi: 10.1016/j.bbadis.2019.08.006
Rudman, M. D., Choi, J. S., Lee, H. E., Tan, S. K., Ayad, N. G., Lee, J. K. (2018). Bromodomain and extraterminal domain-containing protein inhibition attenuates acute inflammation after spinal cord injury. Exp. Neurol. 309, 181–192. doi: 10.1016/j.expneurol.2018.08.005
Saftig, P., Klumperman, J. (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10 (9), 623–635. doi: 10.1038/nrm2745
Sakamaki, J. I., Wilkinson, S., Hahn, M., Tasdemir, N., O’Prey, J., Clark, W., et al. (2017). Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 66 (4), 517–532 e519. doi: 10.1016/j.molcel.2017.04.027
Shi, J., Vakoc, C. R. (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54 (5), 728–736. doi: 10.1016/j.molcel.2014.05.016
Song, S., Liu, L., Yu, Y., Zhang, R., Li, Y., Cao, W., et al. (2019). Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-beta-induced endothelial-mesenchymal transition and cardiac fibrosis. J. Mol. Cell Cardiol. 127, 83–96. doi: 10.1016/j.yjmcc.2018.12.002
Sui, S., Zhang, J., Xu, S., Wang, Q., Wang, P., Pang, D. (2019). Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 10 (5), 331. doi: 10.1038/s41419-019-1564-7
Sung, K. F., Odinokova, I. V., Mareninova, O. A., Rakonczay, Z., Jr., Hegyi, P., Pandol, S. J., et al. (2009). Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp. Cell Res. 315 (11), 1975–1989. doi: 10.1016/j.yexcr.2009.01.009
Vege, S. S., DiMagno, M. J., Forsmark, C. E., Martel, M., Barkun, A. N. (2018). Initial Medical Treatment of Acute Pancreatitis: American Gastroenterological Association Institute Technical Review. Gastroenterology 154 (4), 1103–1139. doi: 10.1053/j.gastro.2018.01.031
Wartmann, T., Mayerle, J., Kahne, T., Sahin-Toth, M., Ruthenburger, M., Matthias, R., et al. (2010). Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 138 (2), 726–737. doi: 10.1053/j.gastro.2009.10.048
Wen, L., Voronina, S., Javed, M. A., Awais, M., Szatmary, P., Latawiec, D., et al. (2015). Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149481-492 (2), e487. doi: 10.1053/j.gastro.2015.04.015
Wen, L., Javed, T. A., Yimlamai, D., Mukherjee, A., Xiao, X., Husain, S. Z. (2018). Transient High Pressure in Pancreatic Ducts Promotes Inflammation and Alters Tight Junctions via Calcineurin Signaling in Mice. Gastroenterology 155 (4), 1250–1263 e1255. doi: 10.1053/j.gastro.2018.06.036
Wildi, S., Kleeff, J., Mayerle, J., Zimmermann, A., Bottinger, E. P., Wakefield, L., et al. (2007). Suppression of transforming growth factor beta signalling aborts caerulein induced pancreatitis and eliminates restricted stimulation at high caerulein concentrations. Gut 56 (5), 685–692. doi: 10.1136/gut.2006.105833
Keywords: BRD4, acute pancreatitis, autophagic flux, SIRT1, lysosomal degradation
Citation: Shen S, Li B, Dai J, Wu Z, He Y, Wen L, Wang X and Hu G (2020) BRD4 Inhibition Protects Against Acute Pancreatitis Through Restoring Impaired Autophagic Flux. Front. Pharmacol. 11:618. doi: 10.3389/fphar.2020.00618
Received: 19 December 2019; Accepted: 20 April 2020;
Published: 08 May 2020.
Edited by:
Nabi Eid Moustafa, Osaka Medical College, JapanReviewed by:
Olga A. Mareninova, University of California, Los Angeles, United StatesBinan Lu, Minzu University of China, China
Manoj B. Menon, Indian Institute of Technology Delhi, India
Copyright © 2020 Shen, Li, Dai, Wu, He, Wen, Wang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Wen, d2VubGk3MDA3QGdtYWlsLmNvbQ==; Xingpeng Wang, cmljaGFyZHdhbmd4cEAxNjMuY29t; Guoyong Hu, aHVndW95b25nc2hAc2luYS5jb20=