 Huimin Zhong
Huimin Zhong Huan Yu
Huan Yu Junjue Chen2
Junjue Chen2 Jun Sun
Jun Sun Yisheng Zhong
Yisheng Zhong- 1Shanghai Key Laboratory for Prevention and Treatment of Bone and Joint Diseases, Shanghai Institute of Traumatology and Orthopaedics, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Ophthalmology, Ruijin Hospital Affiliated Medical School, Shanghai Jiaotong University, Shanghai, China
There are three members of the endogenous gas transmitter family. The first two are nitric oxide and carbon monoxide, and the third newly added member is hydrogen sulfide (H2S). They all have similar functions: relaxing blood vessels, smoothing muscles, and getting involved in the regulation of neuronal excitation, learning, and memory. The cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfur transferase acts together with cysteine aminotransferase (3-MST/CAT), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase with D-amino acid oxidase (3-MST/DAO) pathways are involved in the enzymatic production of H2S. More and more researches focus on the role of H2S in the central nervous system (CNS), and H2S plays a significant function in neuroprotection processes, regulating the function of the nervous system as a signaling molecule in the CNS. Endoplasmic reticulum stress (ERS) and protein misfolding in its mechanism are related to neurodegenerative diseases. H2S exhibits a wide variety of cytoprotective and physiological functions in the CNS degenerative diseases by regulating ERS. This review summarized on the neuroprotective effect of H2S for ERS played in several CNS diseases including Alzheimer’s disease, Parkinson’s disease, and depression disorder, and discussed the corresponding possible signaling pathways or mechanisms as well.
Introduction
Hydrogen sulfide (H2S) plays an important role in terms of cell signal transduction and modulation in the central nerve system (CNS), the cardiovascular system, and many organs like hepatic function (Olas, 2015; Wu et al., 2019). H2S shows its function on inflammatory cells, endoplasmic reticulum, and mitochondria so that H2S may promote resolution of inflammation, energy metabolism, mitochondrial function and misfold protein (Sivarajah et al., 2009; Polhemus and Lefer, 2014; Wallace et al., 2018). Moreover, neuronal signaling mediated by H2S contributes to neuromodulation properties and neuroprotection because H2S concentration distribution in the brain is up to threefold higher than in serum (Zhao et al., 2001; Hogg, 2009; Paul and Snyder, 2018). Many different donors of H2S to cells are widely used in scientific research. Endogenous H2S is difficult to accurately survey due to low concentration in the body. Therefore, the biological effects of H2S can be simulated by injecting exogenous gas into cells or donors in vitro (Powell et al., 2018). Sodium hydrosulfide (NaHS) and sodium sulfide have been exerted to give a burst of H2S but short duration (Cheung et al., 2007). GYY4137, AP97, and AP105 can trigger the corresponding release of H2S in vivo. Another advantage is that they can target organelles (Li and Moore, 2007; Li et al., 2011; Hancock and Whiteman, 2014). The article reviews the effects of H2S on the endoplasmic reticulum stress (ERS) pathway in the pathogenesis of neurodegenerative and psychiatric diseases.
H2S in CNS
As is known to all, H2S is a colorless, water-soluble, highly toxic acidic gas with a depressing smell of rotten eggs (Xiao et al., 2018). H2S is slightly soluble at the physiological condition of 37°C , pH 7.4 and pK1 = 6.76; four-fifths of H2S is dissociated form (HS- and S2-), and less than one-fifths is undissociated form (H2S). At PH 6.0, H2S mainly exists as gas (Ishigami et al., 2009). However, H2S, an endogenously produced gas, also has been qualified as the new third gas transmitter, signaling molecule, antioxidants, antiapoptotic agents and nerve cell protectant (Kumar and Sandhir, 2018). H2S exerts its function in maintaining a balance between oxidation and antioxidant to protect neurons from oxidative stress (Shefa et al., 2018). The role of H2S in ischemic brain depends on the concentration; low concentrations have a protective effect, and high concentrations do the opposite (Chan and Wong, 2017).
H2S is produced from cysteine by enzymes. There are four enzymes responsible for endogenous H2S generation: Cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfur transferase acts together with cysteine aminotransferase (3-MST/CAT), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase with D-amino acid oxidase (3-MST/DAO) pathways (Chen et al., 2015). The first three utilize L-cysteine or homocysteine as substrates. While the 3-MST/DAO pathway is a novel source of endogenous H2S; the substrate is the less toxic D-cysteine (Shibuya et al., 2013). D-cysteine would be more advantageous than L-cysteine as a neuroprotectant against cerebellar ataxias. Furthermore, their distribution is highly tissue specific. The three enzymes CBS, 3-MST/CAT, and 3-MST/DAO are mainly localized in the brain, while the fourth enzyme CSE produces H2S in other organs. Additionally, the 3-MST pathway functions as the main producer to release H2S in the brain. Additionally, the 3-MST pathway functions as the main producer of H2S and polysulfides (H2Sn) in the brain. 3-Mercaptopyruvate (3-MP), the substrate of 3-MST, can produce protein-bound polysulfides so that H2Sn generated by 3-MST exists in the brain (Hylin and Wood, 1959; Kimura, 2019). In the meantime, H2S in CNS parts such as the hippocampus, brain stem, cerebellum, and brain is commonly generated by CBS as well (Chen et al., 2015). CSE is largely associated with peripheral or nonnervous tissues. Thus, H2S produced by CBS and 3-MST/CAT is mainly discussed.
Production of H2S and H2Sn
There are two possibilities for the mechanism of H2S releasing. One is a nonenzymatic pathway that is released immediately once H2S is produced. The other possibility is the enzymatic pathway that releases H2S storage produced by enzyme under specific conditions (Ishigami et al., 2009; Zaorska et al., 2020). As mentioned above, H2S production is closely related to enzymes. CBS and CSE comprise the transsulfuration pathway and also have the ability to catalyze the desulfhydration of cysteine. Relatively, 3-MST that is mostly located in the mitochondria gets involved in the cysteine catabolic pathway (Banerjee et al., 2015). Distribution of enzymes is highly tissue specific although all of them can be detected in many organs. Representative enzyme in the brain is CBS followed by 3-MST, whereas CSE is the most active in the cardiovascular system (Bao et al., 1998; Yang et al., 2008).
The amino acids cysteine and homocysteine have been qualified as the sulfur source. Under the catalysis of CBS, homocysteine and serine undergo a replacement reaction, and the products are water and cystathionine. Next is the elimination reaction dominated by CSE, which produces cysteine, α-ketobutyrate and NH3+. This series of reactions completes the conversion of the harmful substance homocysteine to cysteine. Then CSE and CBS achieve condensation of homocysteine and cysteine in common. Furthermore, homocysteine itself can also generate H2S under the catalysis of CSE enzyme. Cysteine plays a crucial role in diverse H2S-producing reactions, which can complete replacement and cracking reaction of different objects under the catalysis of CBS and CSE. CBS catalyzes the β-replacement of cysteine + homocysteine or cysteine + H2O to liberate H2S. Similarly, in addition to participating in the cracking reaction of cysteine + homocysteine and the γ-replacement reaction of two moles of homocysteine, CSE also takes part in cysteine’s condensation reaction with itself to produce H2S (Banerjee et al., 2015) (Figure 1).
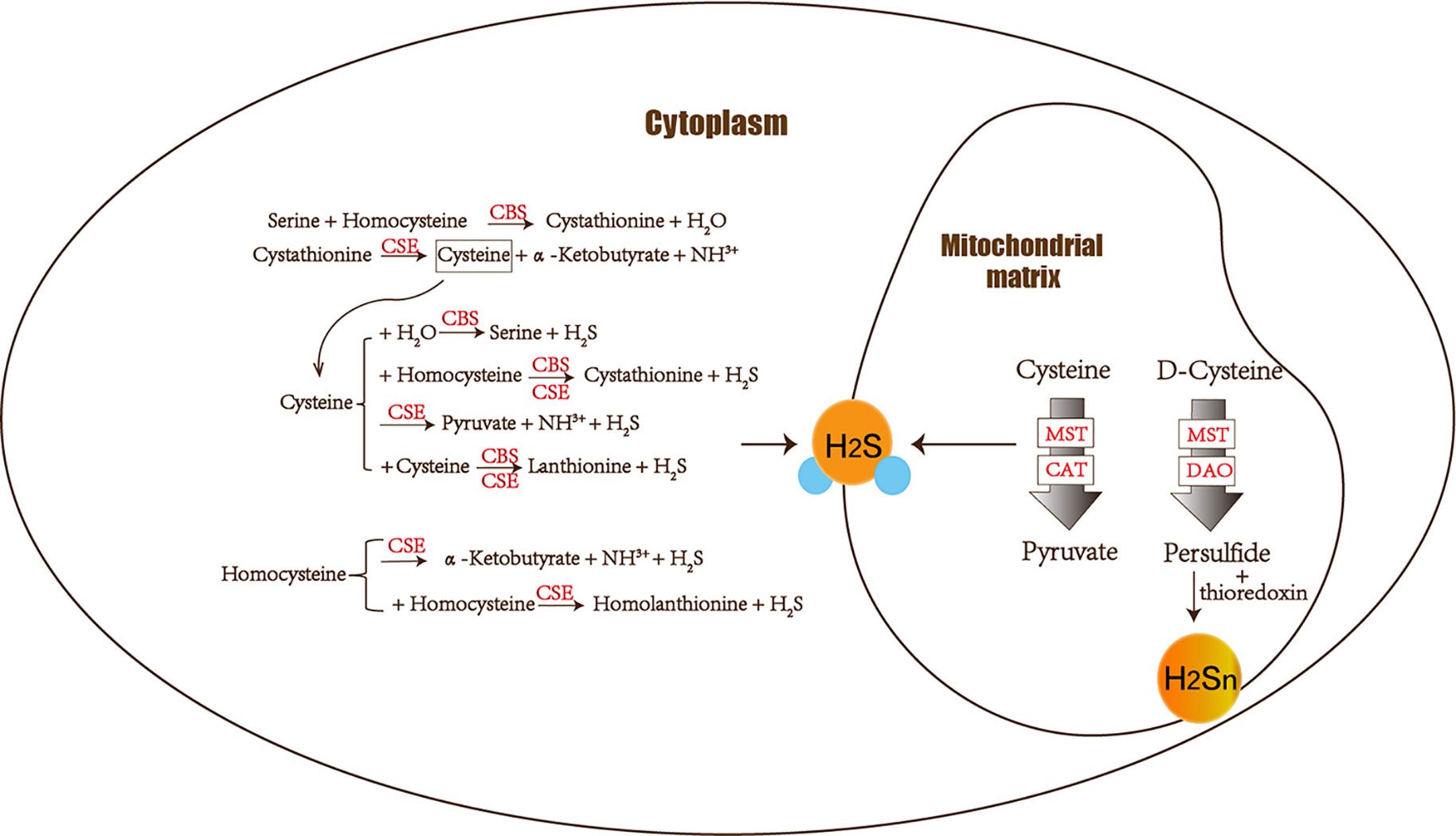
Figure 1 The figure shows the enzymes involved in H2S biogenesis. Although enzymes can be present in the cytoplasm and mitochondria, H2S produced by MST dominates the mitochondria. CBS, CSE, MST/CAT, and MST/DAO represent cystathionine β-synthase, cystathionine γ-lyase ; mercaptopyruvate sulfur transferase acts together with cysteine aminotransferase and mercaptopyruvate sulfur transferase with D-amino acid oxidase. The 3-MST/DAO pathway also can produce persulfide, which interacts with thioredoxin to release H2Sn.
CAT also catalyzes the transamination between cysteine and α-ketoglutarate, but the products are mercaptopyruvate and glutamate. Subsequently MST forms persulfide and pyruvate to liberate H2S under reducing conditions (Yadav et al., 2013). MST is an enzyme that can transfer sulfur from mercaptopyruvate to make MST-SH into MST-SSH. MST-SSH is a persulfide intermediate that releases H2S in the presence of a reductant (Banerjee et al., 2015). H2S is produced from 3-MP by 3-MST. When synthesizing 3-MP in the mitochondria from cytoplasmic D-cysteine, it needs participation of the enzyme DAO (Ubuka et al., 1978; Shibuya et al., 2013) (Figure 1).
Hideo Kimura identified H2S2 and H2S3 produced by 3MST in the brain. The intermediate products persulfide or polysulfide generated after 3-MST catalysis interact with thioredoxin to release H2Sn (mainly H2S2) (Kimura, 2019). Most H2Sn-related enzymes are distinct from H2S. Copper/zinc superoxide dismutase can be utilized to produce H2S2, H2S3, and H2S5 (Searcy et al., 1995; Olson et al., 2018). Peroxidases such as lactoperoxidase and myeloperoxidase can oxidize H2S to polysulfides (Nakamura et al., 1984; Garai et al., 2017). Two gas signal molecules NO and H2S will also interact and produce H2Sn (Eberhardt et al., 2014; Moustafa and Habara, 2016). H2Sn (n ≥ 2) induces Ca2+ influx in astrocytes more effectively than H2S (Kimura et al., 2013). Neurons can be protected by H2S and H2Sn (such as H2S4) to reduce the damage of oxidative stress (Kimura et al., 2019). As a result, both H2S and H2Sn play a role in neurodegenerative diseases.
H2S Is Produced by CBS
CBS is an enzyme that depended on pyridoxal-5′-phosphate, and CBS has been found in the hippocampus, brainstem, cerebellum, cerebrum. CBS can produce efficient H2S through the β displacement condensation reaction of cysteine and homocysteine β substitution (Chen et al., 2004). CBS is of great importance to regulate homocysteine levels in vivo because mice lacking CBS behave hyperhomocysteinemia and hypermethioninemia (Ishii et al., 2010). Endogenous H2S in the brain is generated mainly by CBS so that the change of H2S level depends on altering CBS expression. CBS mRNA expression or CBS transcription can be increased under the regulation of epidermal growth factor, transforming growth factor-α, and cyclic adenosine monophosphate (CAMP). Activation of astrocytes and microglia inflammatory pathways in neuronal cells will reduce CBS expression, leading to downgrade H2S levels in the brain (Schicho et al., 2006; Kimura et al., 2010).
Under local oxidizing conditions, the ferrous form of CBS is less active than the ferric form (Taoka et al., 1998). Due to the very low chemical reaction potential of Fe3+/Fe2+ in CBS (−350mV), the availability of ferrous states in CBS is unclear (Kabil et al., 2011). The CO system will interfere with the H2S system, a novel member of the gas-transmitter family, because CO in the ferrous state will combine with CBS to inhibit the activity of CBS (Shintani et al., 2009). S-adenosylmethionine is the precursor of homocysteine, which can activate CBS by binding to the carboxy-terminal domain of CBS, thus H2S also increases (Sen et al., 2012). CBS expression is abnormal in several diseases. CBS level is found to be threefold higher in the brains of Down’s syndrome patients than in the normal people, but CBS allele expression is lower in children with high intelligence quotient (Kimura, 2010). Experiments have shown that L-glutamic acid and calcium (Ca2+)/calmodulin in hippocampal slices of rats can promote and maintain the production of H2S by CBS, respectively. N-Methyl-D-aspartate (NMDA) is involved in this process as well because CBS functions as an antioxidant inhibitory via triggering NMDA receptors. One of the reasons for the high H2S concentration in the cerebrospinal fluid of patients with Down’s syndrome may be that the addition of NMDA receptors has altered the long-term enhancement of the hippocampus. Encoded and overexpressed CBS on chromosome 21 in these patients causes this increased H2S concentration. What’s more, H2S level of the patients with Alzheimer disease reduces 55%. Studies show that continuous leaks of H2S can impair fetal neuronal development and monoamine neurochemistry in experimental rats (Cheung et al., 2007).
Thus, H2S is very relevant to neurodegenerative diseases. In a word, overexpression and deficiency of CBS both lead to serious diseases such as cognitive dysfunction. If we can understand how to balance the expression of CBS in the brain, the potential therapeutic pathways for many central nervous system diseases can be expanded.
H2S Is Produced by 3-MST/CAT
It has been reported that 3-MST/CAT induces H2S in the brain and mostly get involved in the neuronal generation of H2S (Kimura et al., 2010; Panthi et al., 2016). CBS is mainly localized in the cytosolic of cells, while 3-MST is found in the mitochondrial matrix of neurons in the brain and retina (Nagahara et al., 1998; Shibuya et al., 2009a; Shibuya et al., 2009b; Mikami et al., 2011a; Mikami et al., 2011b).
It has recently been shown that substrates containing L-cysteine and D-cysteine in the brain can produce H2S through the 3-MST/CAT and 3-MST/DAO pathways (Panthi et al., 2016). Thioredoxin and dihydrolipoic acid (DHLA) integrate with 3-MST to release H2S. The concentration of DHLA in the brain is approximately 40 μM, which enhances the H2S production effectively (Mikami et al., 2011a; Mikami et al., 2011b). Actually, 3-MP is the substrate of the mitochondrial enzyme 3-MST, which itself also acts as an endogenous H2S donor (Mitidieri et al., 2018). The mitochondria are the intracellular storage form of H2S. 3-MP stimulated mitochondrial H2S production and enhanced mitochondrial electron transport at low concentrations (Modis et al., 2013). L-cysteine and α-ketoglutarate provide 3-MP by the CAT pathway. When synthesizing 3-MP in the mitochondria from cytoplasmic D-cysteine, it needs participation of enzyme DAO (Ubuka et al., 1978; Shibuya et al., 2013). DAO is rich in cerebellar tissues which can convert D-cysteine to 3MP, thereby effectively producing H2S. H2S generated by D-cysteine exerts its function in promoting dendritic development of cerebella Purkinje cells (Seki et al., 2018). H2S produces from 3-MP by 3-MST, so unstable molecule 3-MP is also the intermediate of CAT catalysis that affects the formation of 3-MST (Shibuya et al., 2009b; Mikami et al., 2011a). Under the presence of dithiothreitol (DTT), persulfide can be produced to release H2S through the way of providing sulfur by 3-MP at the active site of 3-MST (Shibuya et al., 2009b; Kabil and Banerjee, 2010).
On the other hand, Ca2+ concentration is closely related to the production of H2S. The production of H2S is the highest when the Ca2+ concentration is zero and minimum under the condition of 2.9 μM Ca2+ so that the activity of CAT is inhibited by the Ca2+ concentration as well (Mikami et al., 2011b). Mikami et al. also found that 3-MST produced H2S from thiosulfate. In the presence of high concentrations of DHLA, H2S can be produced from both 3MP and thiosulfate. They also concluded that DHLA was detected to release H2S effectively from the brain post-nuclear supernatant containing bound sulfane sulfur (Mikami et al., 2011a).
There are three primary biological forms of H2S including free, acid-labile, and bound sulfur. The acid-labile sulfur is another form of the sulfur pool to release H2S, which is primarily located at the iron–sulfur cluster of enzymes in the mitochondria. Experiments have shown that acid-labile sulfur can be discovered in the brains of rats and humans by detecting the shape of different sulfides. H2S can be released from acid-labile sulfur at acidic conditions while from bound sulfur in alkaline microenvironment. The highest PH for H2S to release from acid-labile sulfur is 5.4. In fact, the acid-labile sulfur releasing H2S pathway may be difficult because the mitochondria are usually not in the acidic environment (Ishigami et al., 2009). Bound sulfur is localized to the cytoplasm and acts as an intracellular storage of H2S. Because 3-MST cell expression mutation lacks H2S-producing activity, the concentration of bound sulfur is low (Shibuya et al., 2009b). 1,500 nmol/g protein concentrations of bound sulfur can release enough H2S to stimulate the target molecules in the brain. When neurons are excited, the increase of extracellular potassium ion concentration causes the intracellular pH of astrocytes to increase to release H2S from the bound sulfur (Ishigami et al., 2009). Moreover, another substrate for CAT, aspartate can combine competitively with CAT to suppress the production of H2S. 3-MST generates bound sulfur more efficiently than CBS. 3-MST is more active than CBS to transfer bound sulfur from H2S (Kimura, 2010).
However, specific proteins that can be used to identify whether H2S releases physiological or pathological signals from the storage form are still unknown.
H2S as a Signaling Molecule in the CNS
H2S plays a significant role in regulating the function of the CNS as a signaling molecule. It has been found that H2S is involved in neuroprotection processes and neurotransmission in various models (Nagai et al., 2004; Hu et al., 2008; Qu et al., 2008). The potential mechanism of neuroprotection of H2S contains anti-inflammation and upregulation of antioxidative enzymes. H2S may protect neurons from apoptosis and degeneration as well (Popov, 2013). H2S also plays a neuroprotective function by regulating the intracellular pH of microglia and restricting the injury of activated microglia in the damaged site (Lu et al., 2010). H2S results in immoderate NMDA receptor stimulation through the transmitter cAMP. One of the endogenous ligands of the NMDA receptors is glutamic acid. The function of protein kinase A (PKA) is to make various intracellular proteins phosphorylation and to get involved in maintaining brain activity. After the addition of the glutamate receptor subunit, phosphorylation happens in the NMDA receptor 1 ion channel by activation of cAMP-dependent PKA (Zhao et al., 2016). Thus, H2S may influence the behaviors of NMDA receptors and second messenger systems through changing intracellular cAMP levels and increases intracellular Ca2+ by activating voltage-gated sodium channels in neuronal cells (Zhang and Bian, 2014). Generation of cAMP stimulates PKA, thereby phosphorylating various intracellular proteins, and an influx of Ca2+ ions is observed in this process (Tan et al., 2010). Studies have shown that CAMP of the primary cultures of the cerebral cortex, cerebellar neurons, and glial cells increases because of the enhanced concentration of NaHS sustained release H2S (Kimura, 2000). Therefore, H2S may modulate intracellular cAMP levels and thereby activate PKA to regulate the activity of NMDA receptors. At the same time, H2S may increase intracellular Ca2+ concentration by activating sodium channels on neuronal cell membranes (Kimura, 2011).
Research shows that H2S protects neurons from oxidative stress. Hypochlorous acid (HOCl) or peroxynitrite (ONOO−) interacts with tyrosine to form 3-chlorotyrosine, which is toxic to neurons. H2S has the function of restraining the activity of reactive oxygen species (ROS), reactive nitrogen species (RNS) or chlorine (such as HOCl), so it can significantly inhibit HOCl toxicity by eliminating HOCl before neurodegenerative changes occur (Olas, 2017). H2S protects neuroblastoma cells from oxidative/nitrative stress induced by ONOO− or HOCl (Whiteman et al., 2004). In addition, H2S may protect retinal neurons from light-stimulated degeneration (Mikami et al., 2011b; Kimura, 2014).
ONOO− can nitrate phenolic groups of tryptophan and tyrosine in proteins, and 3-nitrotyrosine is formed consequently. Due to the toxicity of ONOO− itself and its products to neuronal cells, it plays a role in neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease or amyotrophic lateral sclerosis. Reduced H2S levels in these diseases lead to increasing ONOO− activity and neuronal degeneration. H2S exerts a protective function by inhibiting the interaction between ONOO− and tyrosine (Cheung et al., 2007). Rho-associated protein kinase 2, a key factor that promotes neurodegeneration in Parkinson’s disease, can be reduced by H2S through microRNA-mediated protection of nerve cells (Liu et al., 2016). Persulfidation of H2S signals actually modifies cysteine residues on target protein and converts SH group to SSH group. Dysregulation of the transsulfuration pathway which generates H2S occurs in several neurodegenerative diseases (Paul and Snyder, 2018). Sulfhydration might be impaired in protein misfolding diseases such as Parkinson’s disease. H2S inhalation has been studied for its neuroprotective action in a tested mouse model of Parkinson’s disease (Kida et al., 2011). Administration of H2S donor in the APP/PS1 mouse model can attenuate cognitive dysfunction caused by homocysteine. H2S can inhibit the oxidative stress pathways that affect AD. The generation of oxidative stress markers homocysteine-induced malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) can be reduced by H2S. Nrf2 plays a key role in maintaining redox balance so that NaHS (one of H2S donors) can increase the expression of Nrf2. As a result, H2S mediates neuroprotection via multiple pathways (Paul and Snyder, 2018).
Everything has two sides. Steady intracellular concentrations of H2S depend on enzymatic generation and clearance rates (Kabil and Banerjee, 2014). H2S is also a two-edged sword that excessive H2S can have an adverse result. The inflammatory factor interleukin-1β (IL-1β) causes memory loss through stimulating the CBS enzyme. H2S-mediated IL-1β results in degradation of postsynaptic density 95 which is an important scaffold protein to promote synapse maturation. The loss of postsynaptic density 95 implicates in brain diseases such as consequent neuronal spine retraction (Mir et al., 2014). CBS level is found to be threefold higher in the brains of Down’s syndrome patients than in the normal people but CBS allele expression is lower in children with high intelligence quotient. Overexpression of CBS is present on the trisomy of chromosome 21 (Kimura, 2010). CBS is localized to astrocytes adjacent to the senile plaques in the brains of Down’s syndrome patients (Paul and Snyder, 2015).
H2S production and metabolism should be balanced and regulated in the nervous system. It should be focused on uncovering the exact role and function of H2S in the CNS with the aims of dissecting the involved signaling pathways.
ERS of Neurons in CNS
In eukaryotic cells, the endoplasmic reticulum (ER) is an organelle that synthesizes, modifies, and folds proteins into the correct structure. Only correctly folded proteins can be transported to the Golgi apparatus for further processing. When the protein load capacity of ER is insufficient to deal with unfolded or misfolded proteins accumulated in the ER, the ER balance will be broken and the Ca2+ balance will be disturbed, resulting in endoplasmic reticulum stress (ERS). To alleviate this stress state, the cell firstly initiates an unfolded protein response (UPR), a self-protection mechanism, to eliminate unfolded protein stacks and facilitate cell survival. UPR is mediated by glucose-regulated protein 78 (GPR78)/immunoglobulin heavy chain binding protein (Bip), and three ERS-sensing proteins located on the ER membrane. The three ERS-sensing proteins are double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK) and type-1 ER transmembrane protein kinase (IRE-1) and activating transcription factor 6, ATF6 (Saito, 2014). Although ERS can activate UPR, a self-protection mechanism, severe ERS can still induce apoptosis and death (Cabral Miranda et al., 2014). Short-term activation of UPR is protective, but sustained UPR stimulation caused by prolonged ERS duration can promote neurotoxicity. Protein misfolding and trafficking in the ER lumen initiate UPR and cause the toxic accumulation in energy-starved neurons. Chorionic activation of ERS plays an important role in various neurological diseases, mainly neurodegenerative diseases, followed by spinal cord injury, sclerosis and diabetic nerves (Zhang et al., 2015).
Under normal circumstances, the three inactive proteins are bound to GPR78/Bip, respectively. When the ER homeostasis is broken and a large amount of unfolded proteins accumulate in the ER, GPR78/Bip dissociates from the three sensory proteins in order to bind the accumulated unfolded proteins, releasing and activating the three sensory proteins, thereby activating UPR (Almanza et al., 2019). The role of GRP78 is mainly to regulate the initiation of UPR through direct interaction with each sensory protein (Bertolotti et al., 2000; Shen et al., 2002). After dissociation of PERK and GPR78/Bip, the downstream eukaryotic initiation factor 2α (eIF2α) is subsequently phosphorylated. Phosphorylated eIF2α restricts unfolded proteins from entering the ER, which is beneficial to cell survival; in addition, phosphorylated eIF2α can regulate the expression of activated transcription factor 4 (ATF4) and participate in the recovery of protein synthesis. After IRE-1 is activated, X-box binding protein-1 (XPB-1) is further activated. Activated XBP-1 accelerates the degradation of ER-related proteins, repairs the steady state of endoplasmic reticulum, and plays a role in cell protection. Dissociation of ATF6 from GPR78/Bip can activate the molecular chaperone Bip, the ER protein target gene and upregulate XBP-1, helping the protein to fold, modify, and transport correctly, thereby maintaining cell homeostasis. However, under the stimulation of continuous or excessively strong ERS, UPR cannot continue to maintain homeostasis in the ER, leading to CCAAT-enhancer-binding protein homologous protein (CHOP) activation. Activated CHOP disrupts the balance between downstream apoptotic genes and finally indirectly induces apoptosis (Hetz and Papa, 2018).
Features of pathophysiological stress induced by ERS like protein aggregates, inflammatory signals, metabolic alterations trigger UPR. The changes in the UPR pathway due to ERS may work in the pathogenesis of diabetic neuropathy (Sims-Robinson et al., 2012). Data indicated that icariin reduced neuronal apoptosis and suppressed ERS signaling including decreased the level of GRP78, phosphorylated ER-regulated kinase and phosphorylated eIF2α, as well ATF4, CHOP, DNA damage inducible protein 34 and tribbles homologous protein 3 to protect against Alzheimer’s disease animal model (Li et al., 2019). Carboxyl-terminus of the Hsp70 interacting protein (CHIP) prevents severe ERS-induced hippocampal neuron death. Experiments have shown that overexpression of CHIP prevents upregulation of both ERS-induced CHOP and p53 pro-apoptotic pathways and does not prevent growth of UPR-induced GRP78/Bip. Therefore, it is shown that overexpression of CHIP can weaken the ERS-induced apoptotic response while maintaining ERS adaptive changes in CNS (Cabral Miranda et al., 2014).
ROS and RNS disrupt correct protein folding structure in the ER lumen. Often, cells respond to oxidative stress by initiating ERS response. To human immunodeficiency virus (HIV)-associated neurocognitive impairment, Bip expression in HIV-positive cortex significantly increases and the cell specificity of the Bip level significantly increases in neurons and astrocytes. For the same group of patients, the expression of ATF-6β also upregulates (Lindl et al., 2007). Additionally, ERS is closely relevant to cell death and inflammatory signals. ERS induces astrocytes and neurons to secrete molecules with lipid characteristics. This molecule is a cascade reaction, which in turn regulates other astrocytes and neurons’ inflammation and ERS responses. Methylmercury enhances ERS levels to exert its toxicity through the inactivated Akt pathway mediated by ROS, thereby inducing neuronal apoptosis and eventually leading to death (Chung et al., 2019). Trace metals such as zinc (Zn), copper (Cu), and nickel (Ni) are essential in various physiological functions and have powerful biological functions. They are involved in the metabolic processes of enzymes, hormones, vitamins, and nucleic acids, but their excessive amounts cause disorders in various tissues of the CNS. Cu2+ markedly enhances Zn2+-induced neuronal cell death by activating ERS response. Excess Ni2+ can trigger the ERS response, which significantly enhances Zn2+-induced neuronal cell death, especially the expression of CHOP (Tanaka et al., 2019). H2S donor may be beneficial not only for the brain but also for spinal cord injury recovery via the ERS pathway. Cell autophagy induced by spinal cord injury can be remarkably blocked by the ERS inhibitor. From this, whether H2S can help ERS pathway against autophagy in CNS or not would be an interesting research (Wang et al., 2018). By targeting ERS molecular signaling responses, there will be more new perspectives on the protection and function maintenance of CNS neurons.
The Effect of H2S on ERS of Neurons in CNS
Neuroprotective Effects of H2S in Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most common destructive and progressive neurodegenerative diseases in the elderly. It mainly affects CNS, cognition, memory, and optic nerve abnormalities. The pathological manifestation is the presence of β amyloid deposits in the brain. Endogenous H2S may be closely related to the pathogenesis of AD because of the disordered H2S levels in the serum of AD patients. Proper H2S concentration protects neurons by inhibiting ROS generation and preserving the mitochondrial membrane potential (MMP) pathway (Tang et al., 2008). Hyperhomocysteinemia is a closely independent risk factor for AD because homocysteine can increase neuronal cell apoptosis and inhibit the production of endogenous H2S. Moreover, homocysteine causes the upregulation of ERS-related GRP78, CHOP, and cleaved caspase-12 (Wei et al., 2014). Studies have shown that NaHS interference in animal models of hyperhomocysteinemia can attenuate DNA damage and death of apoptotic cells to prevent neurodegeneration (Kumar et al., 2018). H2S also enhances the expression of anti-apoptotic Bcl-2 or reduce cellular ROS toxicity to protect homocysteine-induced cytotoxicity and apoptosis. Some evidence shows that H2S plays a key role in ERS pathology of AD. Experiments have demonstrated that H2S can attenuate the learning and memory decline in AD and inhibit the hippocampal ERS in homocysteine-exposed rats by reducing the expression of GRP78, CHOP, and cleaved caspase-12 (Zou et al., 2017). H2S can also restrain homocysteine-induced ERS and hippocampal neuronal apoptosis by upregulating the brain-derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB) pathway in AD rat models (Tang et al., 2010). NaHS releases endogenous H2S in vivo and increases the expression of BDNF in a dose-dependent manner, thereby significantly reducing homocysteine-induced apoptosis in ERS and hippocampal neurons. The protective effects of NaHS against homocysteine-induced ERS disappear when using k252a, a specific antagonist of TrkB (Wei et al., 2014). In brief, it is valued that H2S has a neuroprotective effect in AD, and ERS with its related pathways as shown in Figure 2 should be referenced.
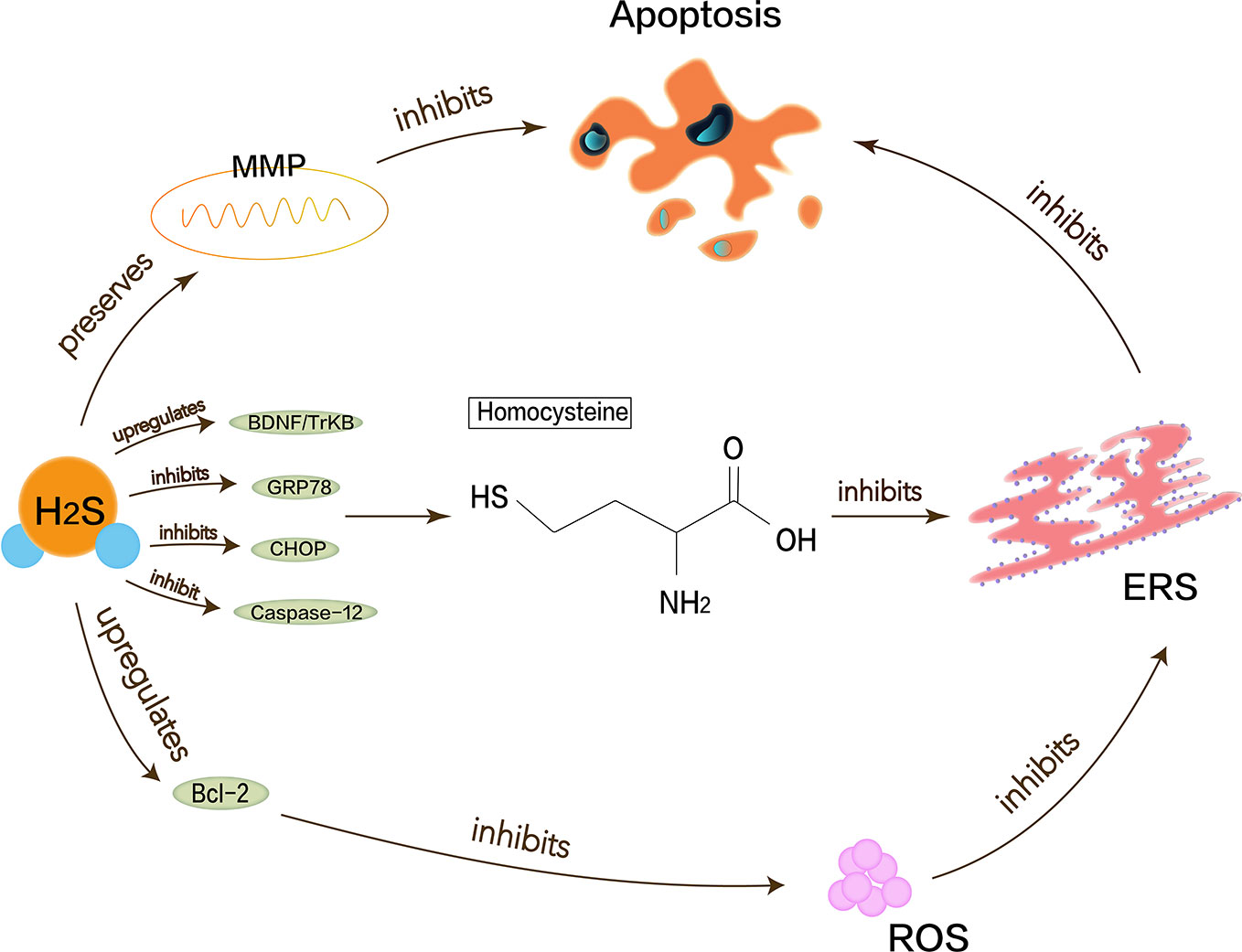
Figure 2 Neuroprotective effects of H2S in Alzheimer’s disease. Homocysteine can increase neuronal cell apoptosis and inhibit the production of endogenous H2S and cause the upregulation of ERS-related GRP78, CHOP, and cleaved caspase-12. Proper H2S concentration protects neurons by inhibiting ROS generation and preserving MMP pathway to reduce Alzheimer’s disease symptoms. H2S can increase expression of anti-apoptotic Bcl-2 or reduce cellular ROS toxicity and also restrain homocysteine-induced ERS and hippocampal neuronal apoptosis by upregulating BDNF/TrkB pathway in Alzheimer’s disease rat models.
Neuroprotective Effects of H2S in Parkinson’s Disease
Parkinson’s disease (PD) is the second most devastating progressive neurodegenerative disease after AD, with characteristic motor symptoms such as resting tremor and muscle stiffness. Broad and complex changes in neurons lead to Parkinsonian symptoms (Obeso et al., 2008).
Oxidative stress, mitochondrial dysfunction, neurotoxicity, neuroinflammation, and apoptosis have been considered as possible mechanisms that cause PD (Hirsch and Hunot, 2009). H2S is also closely relevant to PD. Neurotoxins such as 6-hydroxy-dopamine (6-OHDA) and 1-methyl-4-phenylpyridinium (MPP+) are used to simulate PD models in vitro and in vivo. MPP+ is the active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which functions to stimulate the generation of superoxide radicals in vitro and induce cell apoptosis (Xiao et al., 2016). In a 6-OHDA-induced PD rat model, the endogenous H2S level of the primary lesion site of PD, substantia nigra (SN), is significantly reduced (Xue and Bian, 2015). Symptoms of PD may reflect compromised ubiquitylation. Neuroprotective ubiquitin E3 ligase of parkin in the brain of PD patients inactivates. and sulfhydration of parkin diminishes, while the persulfidation of parkin protein promotes the activity of ubiquitin E3 ligase, thereby mediating cell protection. H2S may upregulate the expression of deubiquitinating enzymes USP8 to antagonize the degradation of Parkin protein (Sun et al., 2020). This implies that H2S donors may be potentially therapeutic (Vandiver et al., 2013).
It turns out that the accumulation of misfolded or damaged proteins is related to the development mechanism of PD. The growth of mitochondrial dysfunction and the imbalance of oxidation and antioxidant will lead to the ER overload response that is the generation of ERS (Sarkar et al., 2016). Mitochondrial dysfunction and the imbalance of oxidation and antioxidant activate the pro-apoptotic pathway and inhibit the anti-apoptotic pathway. It is also proposed that mitochondrial dysfunction changes the energy-dependent cell membrane potential to generate free radicals and has toxic damage to cells (Sarkar et al., 2016). Excessive ROS can cause oxidative stress.
Lu et al. have demonstrated that H2S can attenuate the loss of SN-dense dopamine neurons and the MPTP-induced accumulation of ROS, thereby reducing oxidative stress and ERS. Mitochondrial uncoupling protein 2 (UCP2) can serve as a mechanism for H2S to reduce ROS generation. Mitochondrial uncoupling protein 2 (UCP2), which is associated with dopamine neurons, can function for H2S to reduce ROS production, acting upstream of KATP channels. In addition, H2S can directly and indirectly reduce ROS accumulation through the KATP/PI3K/AKT/Bcl-2 pathway (Lu et al., 2012). Chen’s work shows that appropriate concentrations of H2S can protect neurons by maintaining MMP and weakening ROS generation (Chen et al., 2015). MPP+ inhibits the production of endogenous H2S, so H2S not only needs to maintain MMP but also to resist MPP+-induced cytotoxicity and apoptosis by reducing ROS accumulation (Tang et al., 2011; Xiao et al., 2016). The pathway that described the above content is shown in Figure 3. Therefore, the neuroprotective therapy for PD can exert H2S to prevent ERS.
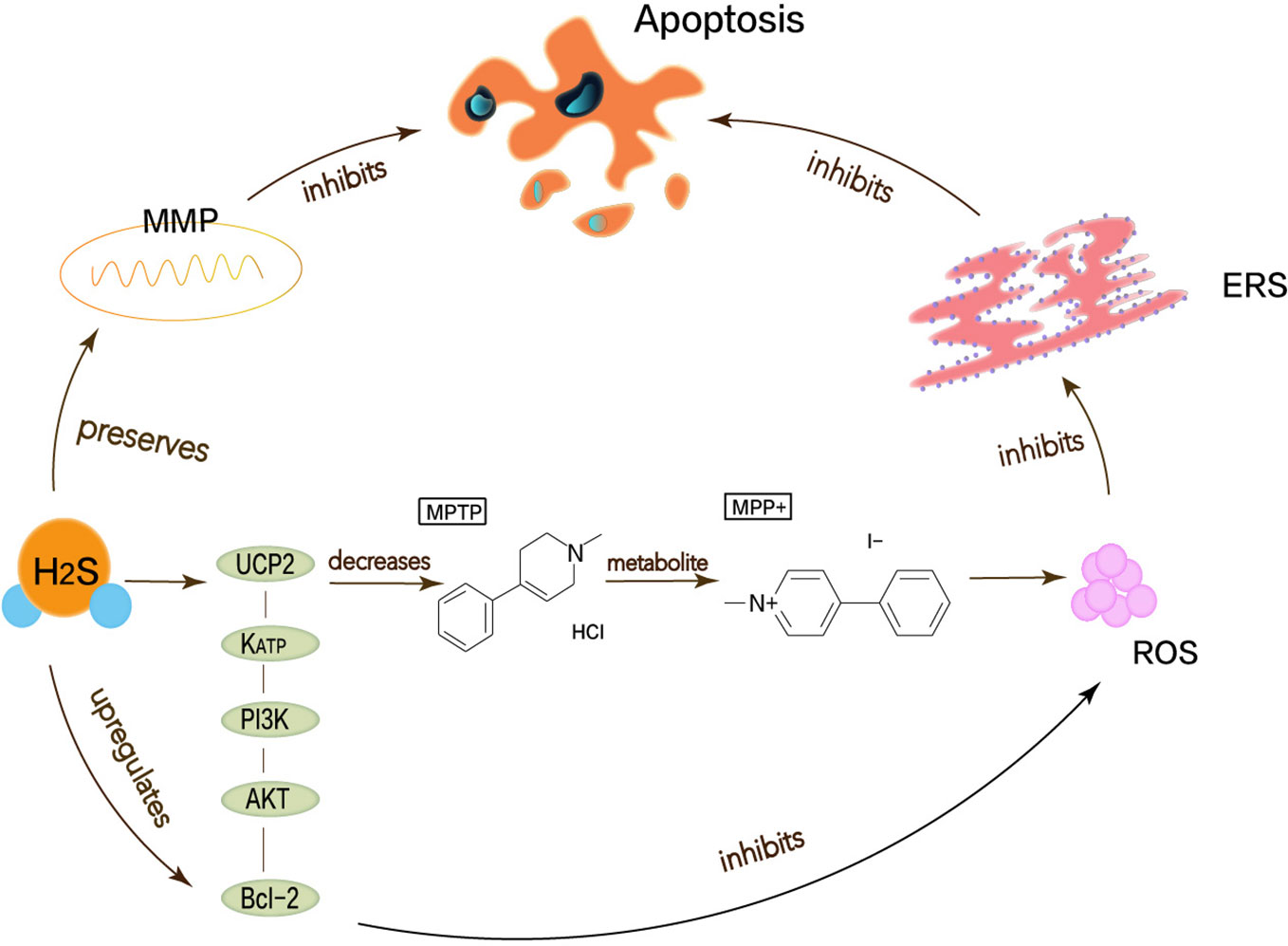
Figure 3 Neuroprotective effects of H2S in Parkinson’s disease. Oxidative stress, mitochondrial dysfunction, apoptosis has been considered as possible mechanisms to cause Parkinson’s disease, thereby leading to ERS. MPP+ is the active metabolite of MPTP, which functions to stimulate the generation of superoxide radicals in vitro and induce cell apoptosis. H2S can attenuate the MPTP-induced accumulation of ROS, thereby reducing oxidative stress and ERS. UCP2 can function for H2S to reduce ROS production, acting upstream of KATP channels. In addition, H2S can directly and indirectly reduce ROS accumulation through the KATP/PI3K/AKT/Bcl-2 pathway.
Neuroprotective Effects of H2S in Major Depression Disorder
As people’s psychological pressure gradually increases, the incidence of depression increases year by year. Depression is a common mental disorder. Its clinical features are emotional disorders, discomfort, and despair. Severe cases even have suicide attempts. The etiology of major depressive disorder (MDD) is a combination of multiple factors (Chirita et al., 2015). Neurochemical mechanisms of depression mainly involve the synergistic action of three major neurotransmitter systems: 5-hydroxytryptamine (5-HT), Noradrenaline (NE), and dopamine (DA). According to the World Health Organization estimates, depression places a huge social burden on nonfatal health consequences (Chirita et al., 2015). Therefore, the treatment of MDD is very important. Unfortunately, existing treatment methods cannot prevent the high recurrence of symptoms (Liu et al., 2011). New targets for depression still need to be studied.
The pathogenesis of depression is not yet complete, but some major hypotheses have been proposed: hippocampal neurogenesis and the BDNF/TrkB pathway may be involved in the pathophysiology of depression (Castren and Rantamaki, 2010; Hanson et al., 2011). The neuro-plasticity of MDD is abnormal. BDNF maintains the development of neurons. Under the circumstances of stress, the expression of BDNF may be suppressed, which interrupts the supply of BDNF in the hippocampus. Atrophy or apoptosis of susceptible neurons causes depression and recurrent episodes. Therefore, abnormalities in synaptic plasticity can make MDD worse (Duman, 2002). H2S has been reported to enhance neuronal synaptic transmission and promote its long-term enhanced induction (Du et al., 2004). H2S can play a positive role in MDD based on its protection of hippocampus and nerve cells. Tan et al. have shown that H2S interferes with the process of hippocampal neuron volume reduction and impaired function under stress-induced MDD (Tan et al., 2015). ERS refers to overload caused by ER dysfunction which is a key step in the pathogenesis of neurodegenerative diseases (Stefani et al., 2012). ERS links to the pathogenesis of depression caused by chronic unpredictable mild stress (CUMS). Amphetamine and inhibition of rat brain striatum stress can activate transcription of ERS transcription factors ATF3 and ATF4 (Pavlovsky et al., 2013). H2S not only attenuates homocysteine-induced apoptosis in hippocampal neurons and ERS by upregulating the expression of BDNF-TrkB in the MDD model but also improves CUMS-induced depression and inhibits hippocampus by promoting the expression of hippocampal Sirt-1 ERS (Wei et al., 2014; Liu et al., 2017).
Taken together, as shown in Figure 4, H2S signaling molecules in the brain can understand new antidepressant pathways and mechanisms through ERS.
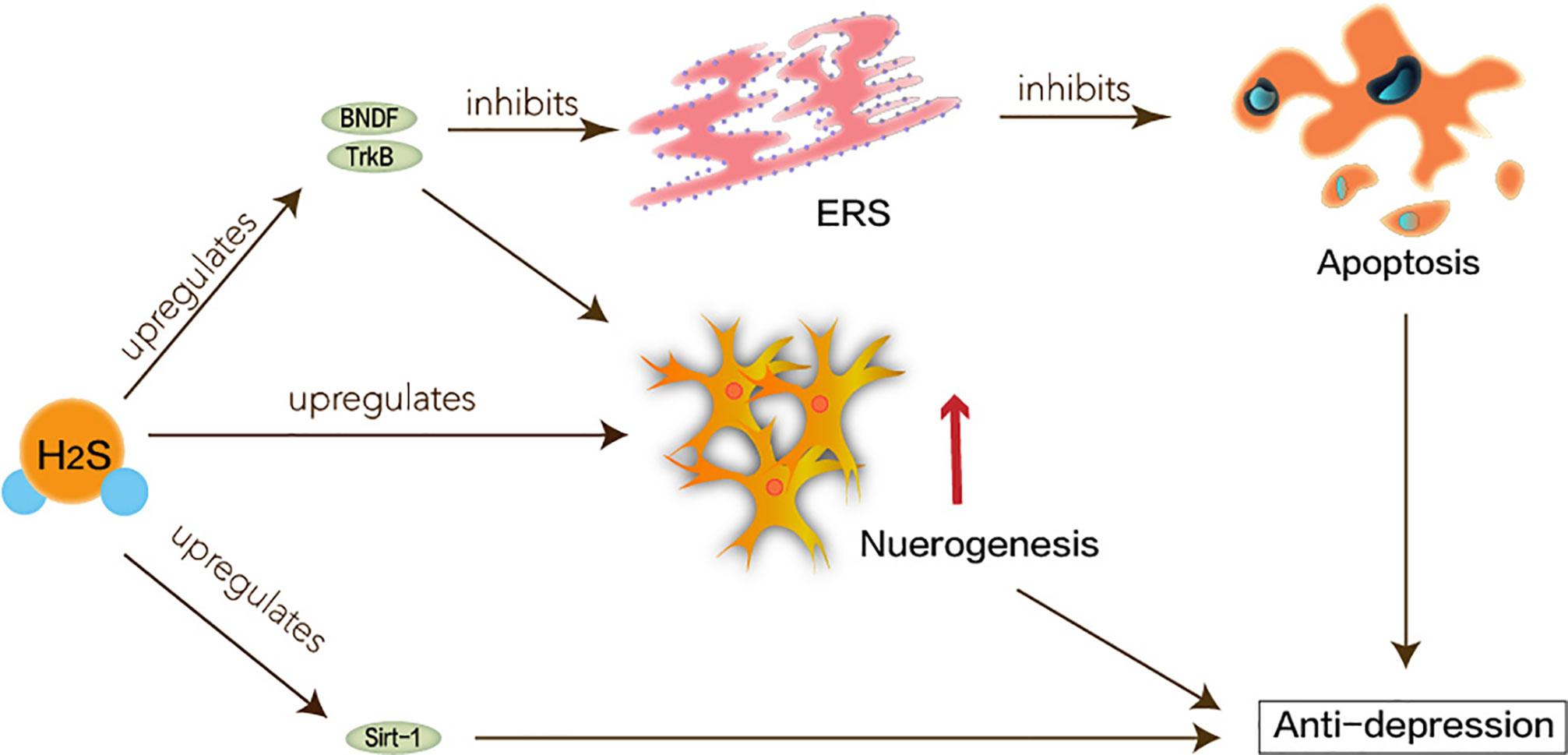
Figure 4 Neuroprotective effects of H2S in major depression disorder. ERS linked to the pathogenesis of depression caused by CUMS. Hippocampal neurogenesis and the BDNF/TrkB pathway may be involved in the pathophysiology of depression. H2S interferes with the process of hippocampal neuron volume reduction and impaired function. H2S not only attenuates homocysteine-induced apoptosis in hippocampal neurons and ERS by upregulating the expression of BDNF-TrkB in the MDD model, but also improves CUMS-induced depression and inhibits hippocampus by promoting the expression of hippocampal Sirt-1 ERS.
Conclusion and Perspectives
Different studies have recognized that H2S plays an important role in physiological and pathological conditions in the body. Appropriate concentration of H2S has a protective and regulatory role in the CNS, existing in cells in free, acid-labile, and bound sulfane sulfur form. Free exogenous H2S is able to exert a physiologic function in neurotransmission and cell survival. Although the neuroprotective effects of H2S are mainly emphasized in this article, either excessive or insufficient H2S still has pathogenic effects on various systems. Excessive H2S initiates neuro-cytotoxic mechanism in the brain. Cells and tissues need to maintain appropriate concentrations of H2S to prevent potential toxicity. There is no doubt that the misfolding of proteins and the accumulation of unfolded proteins in the ER cavity trigger neurotoxic effects. The overload of ER activates the ERS pathway and plays a role in the pathogenesis of a series of neurological diseases. Potential mechanisms that trigger the ERS response may be closely related to toxic levels of homocysteine, oxidative stress, and abnormal epigenetic modification. H2S can regulate the expression of various proteins and genes under the condition of ERS,and maintain the homeostasis of cells in vivo. It is considered using H2S to directly or indirectly target drug-mediated treatment of CNS diseases by modulating the ERS mechanism. One challenge of H2S-based therapeutics is its delivery development. H2S has attractive applications in neurological diseases and psychiatry. H2S will be a promising agent for neurodegenerative diseases.
Author Contributions
HZ and HY discussed the concepts and wrote the manuscript. JC, JS, LG, PH, and YZ reviewed the literature and provided critical revision of the manuscript for important content.
Funding
This study received support from the Shanghai Municipal Health and Family Planning Commission Traditional Chinese Medicine Research Project Foundation (No.2018JP008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Almanza, A., Carlesso, A., Chintha, C., Creedican, S., Doultsinos, D., Leuzzi, B., et al. (2019). Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 286, 241–278. doi: 10.1111/febs.14608
Banerjee, R., Chiku, T., Kabil, O., Libiad, M., Motl, N., Yadav, P. K. (2015). Assay methods for H2S biogenesis and catabolism enzymes. Methods Enzymol. 554, 189–200. doi: 10.1016/bs.mie.2014.11.016
Bao, L., Vlcek, C., Paces, V., Kraus, J. P. (1998). Identification and tissue distribution of human cystathionine beta-synthase mRNA isoforms. Arch. Biochem. Biophys. 350, 95–103. doi: 10.1006/abbi.1997.0486
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Cabral Miranda, F., Adao-Novaes, J., Hauswirth, W. W., Linden, R., Petrs-Silva, H., Chiarini, L. B. (2014). CHIP, a carboxy terminus HSP-70 interacting protein, prevents cell death induced by endoplasmic reticulum stress in the central nervous system. Front. Cell Neurosci. 8, 438. doi: 10.3389/fncel.2014.00438
Castren, E., Rantamaki, T. (2010). The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 70, 289–297. doi: 10.1002/dneu.20758
Chan, S. J., Wong, P. T. (2017). Hydrogen sulfide in stroke: Protective or deleterious? Neurochem. Int. 105, 1–10. doi: 10.1016/j.neuint.2016.11.015
Chen, X., Jhee, K. H., Kruger, W. D. (2004). Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem. 279, 52082–52086. doi: 10.1074/jbc.C400481200
Chen, W. L., Niu, Y. Y., Jiang, W. Z., Tang, H. L., Zhang, C., Xia, Q. M., et al. (2015). Neuroprotective effects of hydrogen sulfide and the underlying signaling pathways. Rev. Neurosci. 26, 129–142. doi: 10.1515/revneuro-2014-0051
Cheung, N. S., Peng, Z. F., Chen, M. J., Moore, P. K., Whiteman, M. (2007). Hydrogen sulfide induced neuronal death occurs via glutamate receptor and is associated with calpain activation and lysosomal rupture in mouse primary cortical neurons. Neuropharmacology 53, 505–514. doi: 10.1016/j.neuropharm.2007.06.014
Chirita, A. L., Gheorman, V., Bondari, D., Rogoveanu, I. (2015). Current understanding of the neurobiology of major depressive disorder. Rom. J. Morphol. Embryol. 56, 651–658.
Chung, Y. P., Yen, C. C., Tang, F. C., Lee, K. I., Liu, S. H., Wu, C. C., et al. (2019). Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology 425, 152245. doi: 10.1016/j.tox.2019.152245
Du, J., Hui, Y., Cheung, Y., Bin, G., Jiang, H., Chen, X., et al. (2004). The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels 19, 75–80. doi: 10.1007/s00380-003-0743-7
Duman, R. S. (2002). Pathophysiology of depression: the concept of synaptic plasticity. Eur. Psychiatry 17 Suppl 3, 306–310. doi: 10.1016/s0924-9338(02)00654-5
Eberhardt, M., Dux, M., Namer, B., Miljkovic, J., Cordasic, N., Will, C., et al. (2014). H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 5, 4381. doi: 10.1038/ncomms5381
Garai, D., Rios-Gonzalez, B. B., Furtmuller, P. G., Fukuto, J. M., Xian, M., Lopez-Garriga, J., et al. (2017). Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 113, 551–563. doi: 10.1016/j.freeradbiomed.2017.10.384
Hancock, J. T., Whiteman, M. (2014). Hydrogen sulfide and cell signaling: team player or referee? Plant Physiol. Biochem. 78, 37–42. doi: 10.1016/j.plaphy.2014.02.012
Hanson, N. D., Owens, M. J., Nemeroff, C. B. (2011). Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology 36, 2589–2602. doi: 10.1038/npp.2011.220
Hetz, C., Papa, F. R. (2018). The Unfolded Protein Response and Cell Fate Control. Mol. Cell. 69, 169–181. doi: 10.1016/j.molcel.2017.06.017
Hirsch, E. C., Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/S1474-4422(09)70062-6
Hogg, P. J. (2009). Contribution of allosteric disulfide bonds to regulation of hemostasis. J. Thromb. Haemost. 7 Suppl 1, 13–16. doi: 10.1111/j.1538-7836.2009.03364.x
Hu, Y., Chen, X., Pan, T. T., Neo, K. L., Lee, S. W., Khin, E. S., et al. (2008). Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 455, 607–616. doi: 10.1007/s00424-007-0321-4
Hylin, J. W., Wood, J. L. (1959). Enzymatic formation of polysulfides from mercaptopyruvate. J. Biol. Chem. 234, 2141–2144. doi: 10.1002/jbmte.390010408
Ishigami, M., Hiraki, K., Umemura, K., Ogasawara, Y., Ishii, K., Kimura, H. (2009). A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 11, 205–214. doi: 10.1089/ARS.2008.2132
Ishii, I., Akahoshi, N., Yamada, H., Nakano, S., Izumi, T., Suematsu, M. (2010). Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 285, 26358–26368. doi: 10.1074/jbc.M110.147439
Kabil, O., Banerjee, R. (2010). Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907. doi: 10.1074/jbc.R110.128363
Kabil, O., Banerjee, R. (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 20, 770–782. doi: 10.1089/ars.2013.5339
Kabil, O., Weeks, C. L., Carballal, S., Gherasim, C., Alvarez, B., Spiro, T. G., et al. (2011). Reversible heme-dependent regulation of human cystathionine beta-synthase by a flavoprotein oxidoreductase. Biochemistry 50, 8261–8263. doi: 10.1021/bi201270q
Kida, K., Yamada, M., Tokuda, K., Marutani, E., Kakinohana, M., Kaneki, M., et al. (2011). Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid. Redox Signal. 15, 343–352. doi: 10.1089/ars.2010.3671
Kimura, Y., Goto, Y., Kimura, H. (2010). Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 12, 1–13. doi: 10.1089/ars.2008.2282
Kimura, Y., Mikami, Y., Osumi, K., Tsugane, M., Oka, J., Kimura, H. (2013). Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 27, 2451–2457. doi: 10.1096/fj.12-226415
Kimura, Y., Shibuya, N., Kimura, H. (2019). Sulfite protects neurons from oxidative stress. Br. J. Pharmacol. 176, 571–582. doi: 10.1111/bph.14373
Kimura, H. (2000). Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267, 129–133. doi: 10.1006/bbrc.1999.1915
Kimura, H. (2010). Hydrogen sulfide: from brain to gut. Antioxid. Redox Signal. 12, 1111–1123. doi: 10.1089/ars.2009.2919
Kimura, H. (2011). Hydrogen sulfide: its production, release and functions. Amino Acids 41, 113–121. doi: 10.1007/s00726-010-0510-x
Kimura, H. (2014). The physiological role of hydrogen sulfide and beyond. Nitric. Oxide 41, 4–10. doi: 10.1016/j.niox.2014.01.002
Kimura, H. (2019). Signaling by hydrogen sulfide (H2S) and polysulfides (H2Sn) in the central nervous system. Neurochem. Int. 126, 118–125. doi: 10.1016/j.neuint.2019.01.027
Kumar, M., Sandhir, R. (2018). Hydrogen Sulfide in Physiological and Pathological Mechanisms in Brain. CNS Neurol. Disord. Drug Targets. 17, 654–670. doi: 10.2174/1871527317666180605072018
Kumar, M., Ray, R. S., Sandhir, R. (2018). Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies. Neurochem. Int. 120, 87–98. doi: 10.1016/j.neuint.2018.07.010
Li, L., Moore, P. K. (2007). An overview of the biological significance of endogenous gases: new roles for old molecules. Biochem. Soc. Trans. 35, 1138–1141. doi: 10.1042/BST0351138
Li, L., Rose, P., Moore, P. K. (2011). Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 51, 169–187. doi: 10.1146/annurev-pharmtox-010510-100505
Li, F., Zhang, Y., Lu, X., Shi, J., Gong, Q. (2019). Icariin improves the cognitive function of APP/PS1 mice via suppressing endoplasmic reticulum stress. Life Sci. 234, 116739. doi: 10.1016/j.lfs.2019.116739
Lindl, K. A., Akay, C., Wang, Y., White, M. G., Jordan-Sciutto, K. L. (2007). Expression of the endoplasmic reticulum stress response marker, BiP, in the central nervous system of HIV-positive individuals. Neuropathol. Appl. Neurobiol. 33, 658–669. doi: 10.1111/j.1365-2990.2007.00866.x
Liu, Y., Yang, N., Hao, W., Zhao, Q., Ying, T., Liu, S., et al. (2011). Dynamic proteomic analysis of protein expression profiles in whole brain of Balb/C mice subjected to unpredictable chronic mild stress: implications for depressive disorders and future therapies. Neurochem. Int. 58, 904–913. doi: 10.1016/j.neuint.2011.02.019
Liu, Y., Liao, S., Quan, H., Lin, Y., Li, J., Yang, Q. (2016). Involvement of microRNA-135a-5p in the Protective Effects of Hydrogen Sulfide Against Parkinson’s Disease. Cell Physiol. Biochem. 40, 18–26. doi: 10.1159/000452521
Liu, S. Y., Li, D., Zeng, H. Y., Kan, L. Y., Zou, W., Zhang, P., et al. (2017). Hydrogen Sulfide Inhibits Chronic Unpredictable Mild Stress-Induced Depressive-Like Behavior by Upregulation of Sirt-1: Involvement in Suppression of Hippocampal Endoplasmic Reticulum Stress. Int. J. Neuropsychopharmacol. 20, 867–876. doi: 10.1093/ijnp/pyx030
Lu, M., Choo, C. H., Hu, L. F., Tan, B. H., Hu, G., Bian, J. S. (2010). Hydrogen sulfide regulates intracellular pH in rat primary cultured glia cells. Neurosci. Res. 66, 92–98. doi: 10.1016/j.neures.2009.09.1713
Lu, M., Zhao, F. F., Tang, J. J., Su, C. J., Fan, Y., Ding, J. H., et al. (2012). The neuroprotection of hydrogen sulfide against MPTP-induced dopaminergic neuron degeneration involves uncoupling protein 2 rather than ATP-sensitive potassium channels. Antioxid. Redox Signal. 17, 849–859. doi: 10.1089/ars.2011.4507
Mikami, Y., Shibuya, N., Kimura, Y., Nagahara, N., Ogasawara, Y., Kimura, H. (2011a). Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 439, 479–485. doi: 10.1042/BJ20110841
Mikami, Y., Shibuya, N., Kimura, Y., Nagahara, N., Yamada, M., Kimura, H. (2011b). Hydrogen sulfide protects the retina from light-induced degeneration by the modulation of Ca2+ influx. J. Biol. Chem. 286, 39379–39386. doi: 10.1074/jbc.M111.298208
Mir, S., Sen, T., Sen, N. (2014). Cytokine-induced GAPDH sulfhydration affects PSD95 degradation and memory. Mol. Cell. 56, 786–795. doi: 10.1016/j.molcel.2014.10.019
Mitidieri, E., Tramontano, T., Gurgone, D., Citi, V., Calderone, V., Brancaleone, V., et al. (2018). Mercaptopyruvate acts as endogenous vasodilator independently of 3-mercaptopyruvate sulfurtransferase activity. Nitric. Oxide 75, 53–59. doi: 10.1016/j.niox.2018.02.003
Modis, K., Coletta, C., Erdelyi, K., Papapetropoulos, A., Szabo, C. (2013). Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611. doi: 10.1096/fj.12-216507
Moustafa, A., Habara, Y. (2016). Cross talk between polysulfide and nitric oxide in rat peritoneal mast cells. Am. J. Physiol. Cell Physiol. 310, C894–C902. doi: 10.1152/ajpcell.00028.2016
Nagahara, N., Ito, T., Kitamura, H., Nishino, T. (1998). Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 110, 243–250. doi: 10.1007/s004180050286
Nagai, Y., Tsugane, M., Oka, J., Kimura, H. (2004). Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 18, 557–559. doi: 10.1096/fj.03-1052fje
Nakamura, S., Nakamura, M., Yamazaki, I., Morrison, M. (1984). Reactions of ferryl lactoperoxidase (compound II) with sulfide and sulfhydryl compounds. J. Biol. Chem. 259, 7080–7085.
Obeso, J. A., Rodriguez-Oroz, M. C., Benitez-Temino, B., Blesa, F. J., Guridi, J., Marin, C., et al. (2008). Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 23 Suppl 3, S548–S559. doi: 10.1002/mds.22062
Olas, B. (2015). Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 439, 212–218. doi: 10.1016/j.cca.2014.10.037
Olas, B. (2017). Hydrogen Sulfide as a “Double-Faced” Compound: One with Pro- and Antioxidant Effect. Adv. Clin. Chem. 78, 187–196. doi: 10.1016/bs.acc.2016.07.005
Olson, K. R., Gao, Y., Arif, F., Arora, K., Patel, S., DeLeon, E. R., et al. (2018). Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 15, 74–85. doi: 10.1016/j.redox.2017.11.009
Panthi, S., Chung, H. J., Jung, J., Jeong, N. Y. (2016). Physiological Importance of Hydrogen Sulfide: Emerging Potent Neuroprotector and Neuromodulator. Oxid. Med. Cell Longev. 2016, 9049782. doi: 10.1155/2016/9049782
Paul, B. D., Snyder, S. H. (2015). H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 40, 687–700. doi: 10.1016/j.tibs.2015.08.007
Paul, B. D., Snyder, S. H. (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 149, 101–109. doi: 10.1016/j.bcp.2017.11.019
Pavlovsky, A. A., Boehning, D., Li, D., Zhang, Y., Fan, X., Green, T. A. (2013). Psychological stress, cocaine and natural reward each induce endoplasmic reticulum stress genes in rat brain. Neuroscience 246, 160–169. doi: 10.1016/j.neuroscience.2013.04.057
Polhemus, D. J., Lefer, D. J. (2014). Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 114, 730–737. doi: 10.1161/CIRCRESAHA.114.300505
Popov, D. (2013). An outlook on vascular hydrogen sulphide effects, signalling, and therapeutic potential. Arch. Physiol. Biochem. 119, 189–194. doi: 10.3109/13813455.2013.803578
Powell, C. R., Dillon, K. M., Matson, J. B. (2018). A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 149, 110–123. doi: 10.1016/j.bcp.2017.11.014
Qu, K., Lee, S. W., Bian, J. S., Low, C. M., Wong, P. T. (2008). Hydrogen sulfide: neurochemistry and neurobiology. Neurochem. Int. 52, 155–165. doi: 10.1016/j.neuint.2007.05.016
Saito, A. (2014). Physiological functions of endoplasmic reticulum stress transducer OASIS in central nervous system. Anat. Sci. Int. 89, 11–20. doi: 10.1007/s12565-013-0214-x
Sarkar, S., Raymick, J., Imam, S. (2016). Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 17, 904. doi: 10.3390/ijms17060904
Schicho, R., Krueger, D., Zeller, F., Von Weyhern, C. W., Frieling, T., Kimura, H., et al. (2006). Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology 131, 1542–1552. doi: 10.1053/j.gastro.2006.08.035
Searcy, D. G., Whitehead, J. P., Maroney, M. J. (1995). Interaction of Cu,Zn superoxide dismutase with hydrogen sulfide. Arch. Biochem. Biophys. 318, 251–263. doi: 10.1006/abbi.1995.1228
Seki, T., Sato, M., Konno, A., Hirai, H., Kurauchi, Y., Hisatsune, A., et al. (2018). d-Cysteine promotes dendritic development in primary cultured cerebellar Purkinje cells via hydrogen sulfide production. Mol. Cell Neurosci. 93, 36–47. doi: 10.1016/j.mcn.2018.10.002
Sen, U., Sathnur, P. B., Kundu, S., Givvimani, S., Coley, D. M., Mishra, P. K., et al. (2012). Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Cell Physiol. 303, C41–C51. doi: 10.1152/ajpcell.00398.2011
Shefa, U., Kim, M. S., Jeong, N. Y., Jung, J. (2018). Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid. Med. Cell Longev. 2018, 1873962. doi: 10.1155/2018/1873962
Shen, J., Chen, X., Hendershot, L., Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 3, 99–111. doi: 10.1016/s1534-5807(02)00203-4
Shibuya, N., Mikami, Y., Kimura, Y., Nagahara, N., Kimura, H. (2009a). Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 146, 623–626. doi: 10.1093/jb/mvp111
Shibuya, N., Tanaka, M., Yoshida, M., Ogasawara, Y., Togawa, T., Ishii, K., et al. (2009b). 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714. doi: 10.1089/ARS.2008.2253
Shibuya, N., Koike, S., Tanaka, M., Ishigami-Yuasa, M., Kimura, Y., Ogasawara, Y., et al. (2013). A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 4, 1366. doi: 10.1038/ncomms2371
Shintani, T., Iwabuchi, T., Soga, T., Kato, Y., Yamamoto, T., Takano, N., et al. (2009). Cystathionine beta-synthase as a carbon monoxide-sensitive regulator of bile excretion. Hepatology 49, 141–150. doi: 10.1002/hep.22604
Sims-Robinson, C., Zhao, S., Hur, J., Feldman, E. L. (2012). Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia 55, 2276–2284. doi: 10.1007/s00125-012-2573-6
Sivarajah, A., Collino, M., Yasin, M., Benetti, E., Gallicchio, M., Mazzon, E., et al. (2009). Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 31, 267–274. doi: 10.1097/SHK.0b013e318180ff89
Stefani, I. C., Wright, D., Polizzi, K. M., Kontoravdi, C. (2012). The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimer Res. 9, 373–387. doi: 10.2174/156720512800107618
Sun, Y., Lu, F., Yu, X., Wang, B., Chen, J., Lu, F., et al. (2020). Exogenous H2S Promoted USP8 Sulfhydration to Regulate Mitophagy in the Hearts of db/db Mice. Aging Dis. 11, 269–285. doi: 10.14336/AD.2019.0524
Tan, B. H., Wong, P. T., Bian, J. S. (2010). Hydrogen sulfide: a novel signaling molecule in the central nervous system. Neurochem. Int. 56, 3–10. doi: 10.1016/j.neuint.2009.08.008
Tan, H., Zou, W., Jiang, J., Tian, Y., Xiao, Z., Bi, L., et al. (2015). Disturbance of hippocampal H2S generation contributes to CUMS-induced depression-like behavior: involvement in endoplasmic reticulum stress of hippocampus. Acta Biochim. Biophys. Sin. (Shanghai) 47, 285–291. doi: 10.1093/abbs/gmv009
Tanaka, K. I., Kasai, M., Shimoda, M., Shimizu, A., Kubota, M., Kawahara, M. (2019). Nickel Enhances Zinc-Induced Neuronal Cell Death by Priming the Endoplasmic Reticulum Stress Response. Oxid. Med. Cell Longev. 2019, 9693726. doi: 10.1155/2019/9693726
Tang, X. Q., Yang, C. T., Chen, J., Yin, W. L., Tian, S. W., Hu, B., et al. (2008). Effect of hydrogen sulphide on beta-amyloid-induced damage in PC12 cells. Clin. Exp. Pharmacol. Physiol. 35, 180–186. doi: 10.1111/j.1440-1681.2007.04799.x
Tang, X. Q., Shen, X. T., Huang, Y. E., Ren, Y. K., Chen, R. Q., Hu, B., et al. (2010). Hydrogen sulfide antagonizes homocysteine-induced neurotoxicity in PC12 cells. Neurosci. Res. 68, 241–249. doi: 10.1016/j.neures.2010.07.2039
Tang, X. Q., Fan, L. L., Li, Y. J., Shen, X. T., Zhuan, Y. Y., He, J. Q., et al. (2011). Inhibition of hydrogen sulfide generation contributes to 1-methy-4-phenylpyridinium ion-induced neurotoxicity. Neurotox Res. 19, 403–411. doi: 10.1007/s12640-010-9180-4
Taoka, S., Ohja, S., Shan, X., Kruger, W. D., Banerjee, R. (1998). Evidence for heme-mediated redox regulation of human cystathionine beta-synthase activity. J. Biol. Chem. 273, 25179–25184. doi: 10.1074/jbc.273.39.25179
Ubuka, T., Umemura, S., Yuasa, S., Kinuta, M., Watanabe, K. (1978). Purification and characterization of mitochondrial cysteine aminotransferase from rat liver. Physiol. Chem. Phys. 10, 483–500. doi: 10.1007/BF00580836
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4, 1626. doi: 10.1038/ncomms2623
Wallace, J. L., Motta, J. P., Buret, A. G. (2018). Hydrogen sulfide: an agent of stability at the microbiome-mucosa interface. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G143–G149. doi: 10.1152/ajpgi.00249.2017
Wang, H., Wu, Y., Han, W., Li, J., Xu, K., Li, Z., et al. (2018). Hydrogen Sulfide Ameliorates Blood-Spinal Cord Barrier Disruption and Improves Functional Recovery by Inhibiting Endoplasmic Reticulum Stress-Dependent Autophagy. Front. Pharmacol. 9, 858. doi: 10.3389/fphar.2018.00858
Wei, H. J., Xu, J. H., Li, M. H., Tang, J. P., Zou, W., Zhang, P., et al. (2014). Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 35, 707–715. doi: 10.1038/aps.2013.197
Whiteman, M., Armstrong, J. S., Chu, S. H., Jia-Ling, S., Wong, B. S., Cheung, N. S., et al. (2004). The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J. Neurochem. 90, 765–768. doi: 10.1111/j.1471-4159.2004.02617.x
Wu, D. D., Wang, D. Y., Li, H. M., Guo, J. C., Duan, S. F., Ji, X. Y. (2019). Hydrogen Sulfide as a Novel Regulatory Factor in Liver Health and Disease. Oxid. Med. Cell Longev. 2019, 3831713. doi: 10.1155/2019/3831713
Xiao, F., Zhang, P., Chen, A. H., Wang, C. Y., Zou, W., Gu, H. F., et al. (2016). Hydrogen sulfide inhibits MPP(+)-induced aldehyde stress and endoplasmic reticulum stress in PC12 cells: involving upregulation of BDNF. Exp. Cell Res. 348, 106–114. doi: 10.1016/j.yexcr.2016.09.006
Xiao, Q., Ying, J., Xiang, L., Zhang, C. (2018). The biologic effect of hydrogen sulfide and its function in various diseases. Med. (Baltimore) 97, e13065. doi: 10.1097/MD.0000000000013065
Xue, X., Bian, J. S. (2015). Neuroprotective effects of hydrogen sulfide in Parkinson’s disease animal models: methods and protocols. Methods Enzymol. 554, 169–186. doi: 10.1016/bs.mie.2014.11.015
Yadav, P. K., Yamada, K., Chiku, T., Koutmos, M., Banerjee, R. (2013). Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013. doi: 10.1074/jbc.M113.466177
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Zaorska, E., Tomasova, L., Koszelewski, D., Ostaszewski, R., Ufnal, M. (2020). Hydrogen Sulfide in Pharmacotherapy, Beyond the Hydrogen Sulfide-Donors. Biomolecules 10, 323. doi: 10.3390/biom10020323
Zhang, X., Bian, J. S. (2014). Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chem. Neurosci. 5, 876–883. doi: 10.1021/cn500185g
Zhang, H. Y., Wang, Z. G., Lu, X. H., Kong, X. X., Wu, F. Z., Lin, L., et al. (2015). Endoplasmic reticulum stress: relevance and therapeutics in central nervous system diseases. Mol. Neurobiol. 51, 1343–1352. doi: 10.1007/s12035-014-8813-7
Zhao, W., Zhang, J., Lu, Y., Wang, R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20, 6008–6016. doi: 10.1093/emboj/20.21.6008
Zhao, S., Liu, F. F., Wu, Y. M., Jiang, Y. Q., Guo, Y. X., Wang, X. L. (2016). Upregulation of spinal NMDA receptors mediates hydrogen sulfide-induced hyperalgesia. J. Neurol. Sci. 363, 176–181. doi: 10.1016/j.jns.2016.02.058
Keywords: hydrogen sulphide, central nervous system, degeneration disease, endoplasmic reticulum stress, neuroprotection
Citation: Zhong H, Yu H, Chen J, Sun J, Guo L, Huang P and Zhong Y (2020) Hydrogen Sulfide and Endoplasmic Reticulum Stress: A Potential Therapeutic Target for Central Nervous System Degeneration Diseases. Front. Pharmacol. 11:702. doi: 10.3389/fphar.2020.00702
Received: 12 February 2020; Accepted: 29 April 2020;
Published: 14 May 2020.
Edited by:
Nikolaos Pitsikas, University of Thessaly, GreeceReviewed by:
Vincenzo Brancaleone, University of Basilicata, ItalyDongbao Chen, University of California, Irvine, United States
Norihiro Shibuya, Sanyo-Onoda City University, Japan
Copyright © 2020 Zhong, Yu, Chen, Sun, Guo, Huang and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Guo, Z3VvbGVpNjA3QDEyNi5jb20=; Ping Huang, cGluZ2hwaW5naEAxMjYuY29t; Yisheng Zhong, eXN6aG9uZzY4QDEyNi5jb20=
†These authors have contributed equally to this work