Abstract
Misfolding and extracellular deposition of proteins is the hallmark of a heterogeneous group of conditions collectively termed protein misfolding and deposition diseases or amyloidoses. These include both localized (e.g. Alzheimer’s disease, prion diseases, type 2 diabetes mellitus) and systemic amyloidoses. Historically regarded as a group of maladies with limited, even inexistent, therapeutic options, some forms of systemic amyloidoses have recently witnessed a series of unparalleled therapeutic successes, positively impacting on their natural history and sometimes even on their incidence. In this review article we will revisit the most relevant of these accomplishments. Collectively, current evidence converges towards a crucial role of an early and conspicuous reduction or stabilization of the amyloid-forming protein in its native conformation. Such an approach can reduce disease incidence in at risk individuals, limit organ function deterioration, promote organ function recovery, improve quality of life and extend survival in diseased subjects. Therapeutic success achieved in these forms of systemic amyloidoses may guide the research on other protein misfolding and deposition diseases for which effective etiologic therapeutic options are still absent.
Protein Misfolding Diseases
Protein misfolding and deposition diseases arise when one of an ever growing list of proteins (the amyloid-forming protein in its native conformation, also referred to as the amyloidogenic precursor) acquires an alternative folding state (the misfolded state), starts to aggregate and to form oligomers, then protofibrils and finally fibrillar structures (termed amyloid fibrils) which eventually deposit within tissues (forming the amyloid deposits) (Benson et al., 2018a). This process can occur at the site of protein production, leading to localized amyloidoses, as it is the case of Alzheimer’s disease and the majority of prion diseases affecting the central nervous system (CNS) and type 2 diabetes mellitus affecting the islets of Langerhans within the pancreas (Aguzzi et al., 2013; Westermark and Westermark, 2013; Selkoe and Hardy, 2016). Conversely, amyloid deposition can affect multiple body sites (typically sparing the central nervous system parenchyma) when the amyloid-forming protein is a circulating protein, as in the case of systemic amyloidoses (Nuvolone and Merlini, 2017b) (Table 1). The prion protein can form amyloid deposits outside of the CNS in the presence of selected mutations of the PRNP gene, leading to PrP systemic amyloidosis (Mead et al., 2013; Matsuzono et al., 2016; Capellari et al., 2018), or in the context of variant Creutzfeldt-Jakob disease (Will et al., 1996; Bruce et al., 2001). Rarely, iatrogenic amyloid deposits can be formed at the site of drug injection, as reported for insulin and enfuvirtide (Storkel et al., 1983; Morilla et al., 2009).
Table 1
| Localized amyloidoses | Systemic amyloidoses | ||||||
|---|---|---|---|---|---|---|---|
| Alzheimer’s disease | Prion diseases | Type 2 diabetes | Localized AL amyloidosis | Systemic AL amyloidosis | AA amyloidosis | ATTR amyloidosis | |
| Source | CNS | CNS | Pancreas | Extra-medullary plasma cell clone | BM plasma cell clone | Liver | Liver Choroid plexus Retina |
| Amyloid-forming protein | Aβ | PrP | IAPP | Locally secreted monoclonal LC | Circulating monoclonal LC | SAA | TTR |
| Site of amyloid deposit | CNS | CNS# | Pancreas | One site: • Urogenital tract • Larynx • Skin • Lung • GI tract | • Heart • Kidney • Liver • GI tract • Peripheral nerves • Soft tissue | • Kidney • Liver • GI tract • Spleen • Thyroid | • Heart • Peripheral nerves • Carpal tunnel • Meninges • Eye |
Localized versus systemic forms of amyloidosis.
#Systemic amyloid deposits can be observed in some forms of prion diseases, including variant Creutzfeldt-Jacob disease and in PrP systemic amyloidosis. Aβ, amyloid β; BM, bone marrow; CNS, central nervous system; GI tract, gastrointestinal tract; IAPP, islet amyloid polypeptide; LC, immunoglobulin light chain; PrP, prion protein; SAA, serum amyloid A; TTR, transthyretin.
The complex process of protein misfolding, aggregation, amyloid oligomer and fibril formation and deposition can lead to cytotoxicity, subversion of tissue architecture, progressive organ damage, and eventually death (Merlini and Bellotti, 2003). Notwithstanding, amyloid itself may not be necessarily toxic, as witnessed by the existence of so-called functional amyloids within the body (Chiti and Dobson, 2006).
Sometimes amyloidogenesis occurs when a protein concentration is persistently elevated, like for the acute phase reactant serum amyloid A (SAA) in systemic amyloidosis associated to chronic inflammation (AA amyloidosis) or wild-type β2-microglobulin in systemic amyloidosis associated to dialysis (Westermark et al., 2015; Kaneko and Yamagata, 2018). In other cases, amyloidosis can be caused by genetic mutations which destabilize an otherwise non-amyloidogenic or only mildly amyloidogenic protein, as in familial prion diseases and hereditary systemic amyloidoses (including ATTRv amyloidosis associated to variant transthyretin, TTR) (Connors et al., 2003; Rowczenio et al., 2019), or by mutations which increase the synthesis or the proteolytic release of the amyloid-forming protein, as in the case of AA amyloidosis associated to hereditary autoinflammatory syndromes or familial Alzheimer’s disease, respectively (De Strooper et al., 2012; Obici and Merlini, 2012). In systemic amyloidosis associated to immunoglobulin light chains (AL amyloidosis), amyloidogenesis is the result of the presence of elevated concentrations of a patient’s unique, unstable light chain produced by an underlying B-cell clonal disorder (Merlini et al., 2018).
In some other instances, amyloid formation occurs in the absence of other apparent predisposing conditions, as a process associated to aging, presumably due to the intrinsic amyloidogenicity of a specific protein, as in the case of sporadic forms of Alzheimer’s and prion diseases, as well as in the case of systemic amyloidosis associated to wild-type TTR (ATTRwt amyloidosis) (Prusiner, 2001; Gertz et al., 2015; Long and Holtzman, 2019). Historically regarded as a group of conditions with limited, even inexistent, therapeutic options, some forms of systemic amyloidoses have recently witnessed a series of unparalleled therapeutic successes, positively impacting on their natural history and sometimes even on their incidence. Here we will review the most relevant of these accomplishments in order to draw some lessons which may guide research on other protein misfolding diseases for which effective etiologic therapeutic options are still absent.
Elimination or Reduction of the Amyloid-Forming Protein Translates into Clinical Benefit
Substantial therapeutic success has been achieved by interventions aimed at eliminating or substantially reducing the supply of the amyloid-forming protein, thus interfering with the process of misfolding, oligomerization and amyloid formation (Nuvolone and Merlini, 2017b) (Table 2).
Table 2
| Strategy | AL amyloidosis | AA amyloidosis | ATTR amyloidosis |
|---|---|---|---|
| Eliminate or reduce the amyloid-forming protein | Anti-PC (or B cells) drugs: • Chemotherapy/ASCT • Immunomodulatory drugs • Steroids • Monoclonal antibodies | Treatment of underlying flogosis: • Steroids • DMARDs • Monoclonal antibodies • Antibiotics • Surgery • … | TTR directed interventions: • Liver transplantation (hATTR)° • TTR tetramer stabilizers @ • TTR silencing agents # |
| Monitor the concentration of the amyloid-forming protein | Amyloid-forming protein: monoclonal LCGoal: disappearance Monitoring: s+uEP+IFIX serum free LC levels | Amyloid-forming protein: SAA Goal: SAA normalization Monitoring: serum SAA levels | Amyloid-forming protein: TTR Goal: reduced mutant TTR° tetramer stabilization @ reduced TTR levels # Monitoring: tetramer stability/dissociation @ or serum TTR levels #@ |
| Early diagnosis | At risk individuals: MGUS, MM, other PC dyscrasia Proposed screening: • Proteinuria • NT-proBNP • ALP | At risk individuals: Chronic inflammation, morbid obesity (?) Proposed screening: • Proteinuria | At risk individuals: pre-symptomatic mutation carriers (hATTR) Proposed screening (based on TTR mutation): • NT-proBNP • Nerve MRI |
Therapeutic interventions against the most common forms of systemic amyloidosis.
ALP, alkaline phosphatase; ASCT, autologous stem cell transplantation; BM, bone marrow; DMARDs, disease-modifying anti-rheumatic drugs; hATTR, hereditary ATTR amyloidosis; LC, immunoglobulin light chains; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; MRD, minimal residual disease; MRI, magnetic resonance imaging; NT-proBNP, amino-terminal pro brain natriuretic peptide; PC, plasma cell; SAA, serum amyloid A; s&uEP+IFIX, serum and urine electrophoresis with immunofixation; TTR, transthyretin. °, @ and # denote different therapeutic strategies in ATTR amyloidosis, with different goals, and whose efficacy can be monitored by different modalities.
The first known examples of successful therapeutic approaches against systemic amyloidoses have been described in the context of AA amyloidosis (Figure 1). This is a form of systemic amyloidosis complicating longstanding chronic inflammation and most commonly arises in association with chronic arthritides, hereditary autoinflammatory syndromes, chronic inflammatory bowel diseases or chronic infections (Table 3). Amyloid deposits derive from the proteolytic processing of the liver-derived acute phase reactant SAA and almost invariably affect kidneys.
Figure 1
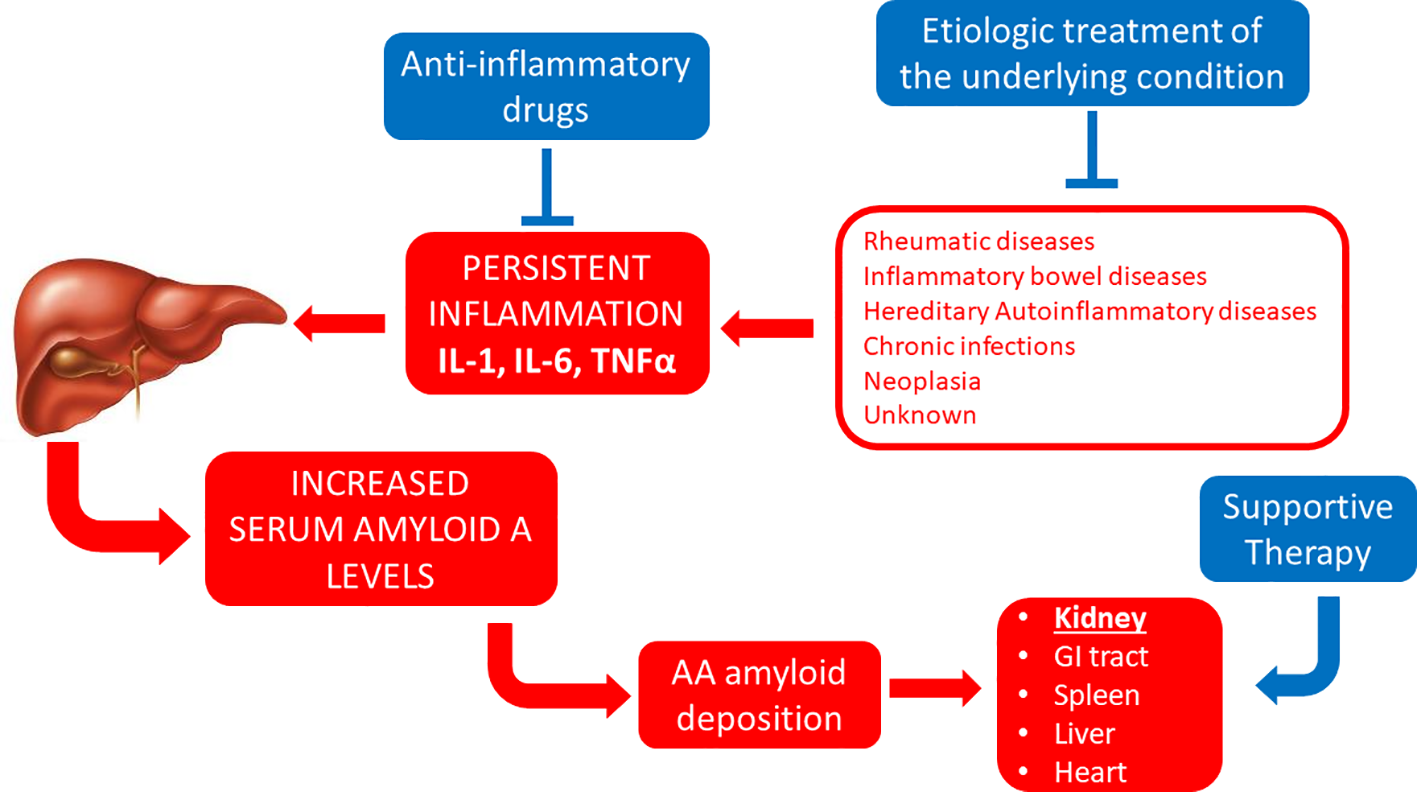
Established therapeutic interventions against AA amyloidosis. Red blocks indicate steps in the pathogenesis of AA amyloidosis; blue blocks indicate established therapeutic interventions against AA amyloidosis. IL-1, interleukin 1; IL-6, interleukin 6; GI tract, gastro-intestinal tract; TNFα, tumor necrosis factor α.
Table 3
| Class of underlying disorder | |
|---|---|
| Chronic infections | Tuberculosis |
| Leprosy | |
| Osteomyelitis | |
| Whipple disease | |
| Chronic infections as a consequence of: | |
| Cystic fibrosis | |
| Bronchiectasis | |
| Paraplegia | |
| Injection-drug abuse | |
| Epidermolysis bullosa | |
| Immunodeficiencies | |
| Others | |
| Rheumatologic diseases | Ankylosing spondylitis |
| Juvenile idiopathic arthritis | |
| Psoriatic arthropathy Rheumatoid arthritis | |
| Systemic vasculitis | |
| Others | |
| Inflammatory bowel diseases | Crohn’s disease Ulcerative colitis |
| Hereditary Auto-inflammatory Disorders | Familial Mediterranean fever (FMF) Mevalonate kinase deficiency (MKD) |
| Cryopyrin-associated periodic fever syndrome (CAPS) | |
| Tumor necrosis factor receptor associated periodic syndrome (TRAPS) | |
| Others | |
| Neoplasia | Castelman’s disease |
| Lymphoma | |
| Sarcoma | |
| Adenocarcinoma | |
| Mesothelioma | |
| Others | |
| Unknown | Morbid obesity (?) |
Chronic inflammatory conditions underlying AA amyloidosis.
In 1928, Henning Waldenström reported the rapid resolution of hepatomegaly and liver amyloid deposits after successful surgical treatment of “lymphoid TBC fistulae” in a child presumably affected by AA amyloidosis (Waldenstrom, 1928). Similar successful regression of AA amyloid deposits has been described after surgical excision of localized Castleman’s disease leading to the resolution of the associated chronic inflammation (Perfetti et al., 1994; Lachmann et al., 2002; Mandreoli et al., 2002; Verbrugghe et al., 2005; Androulaki et al., 2007).
Studies of experimental murine AA amyloidosis show that AA amyloid can be indeed spontaneously cleared upon cessation of inflammation through redundant innate immune mechanisms (Nystrom and Westermark, 2012; Lundmark et al., 2013; Sponarova et al., 2013). They also demonstrate that, after apparent resolution of AA amyloid deposits, a relapse of inflammation can lead to a dramatic, rapid exacerbation of amyloid deposits, presumably due to residual AA amyloid fibrils resisting clearing mechanisms and serving as nucleating seeds, further stressing the importance of maintaining reduced SAA levels throughout the disease (Hawkins and Pepys, 1990; Simons et al., 2013).
To which extent SAA levels influence the natural history of AA amyloidosis has been substantiated by work of Lachmann and collaborators based on the largest series of patients with AA amyloidosis reported so far. This study shows that amyloid burden, renal outcome and overall survival all correlate with SAA levels in the course of the disease (Lachmann et al., 2007). In this series, amyloid regression was seen in 60% of patients in which therapeutic interventions against the underlying cause of systemic inflammation succeeded at lowering SAA levels below the threshold of 10 mg/L (Lachmann et al., 2007). This should be therefore regarded as the therapeutic goal when treating these patients.
For a subset of patients with AA amyloidosis, which has proportionally increased in more recent case series, the cause of the underlying chronic inflammatory status cannot be identified despite an intensive diagnostic workout (Lane et al., 2017a). Recent work has shown that some of these patients may benefit from empirical treatment with interleukin-1 inhibition (Lane et al., 2017b). Whether these patients may benefit also from silencing agents against SAA, which have been studied in the murine model, is presently unknown (Kluve-Beckerman et al., 2011).
Even more satisfactory therapeutic results have been achieved in another form of systemic amyloidosis that is AL amyloidosis (Figure 2). This is caused by an underlying plasma cell (or B cell) clonal disorder resulting in the production of an unstable immunoglobulin light chain (Table 4), which can form amyloid in the heart, kidneys, liver, gastrointestinal tract, peripheral nerves, soft tissues and elsewhere, commonly in various combinations (Merlini et al., 2018). The obvious therapeutic strategy in this case has been to use drugs developed against other, more prevalent plasma cell (or B cell) disorders, including alkylating agents, steroids, proteasome inhibitors, immunomodulatory drugs and monoclonal antibodies (Merlini et al., 2018).
Figure 2
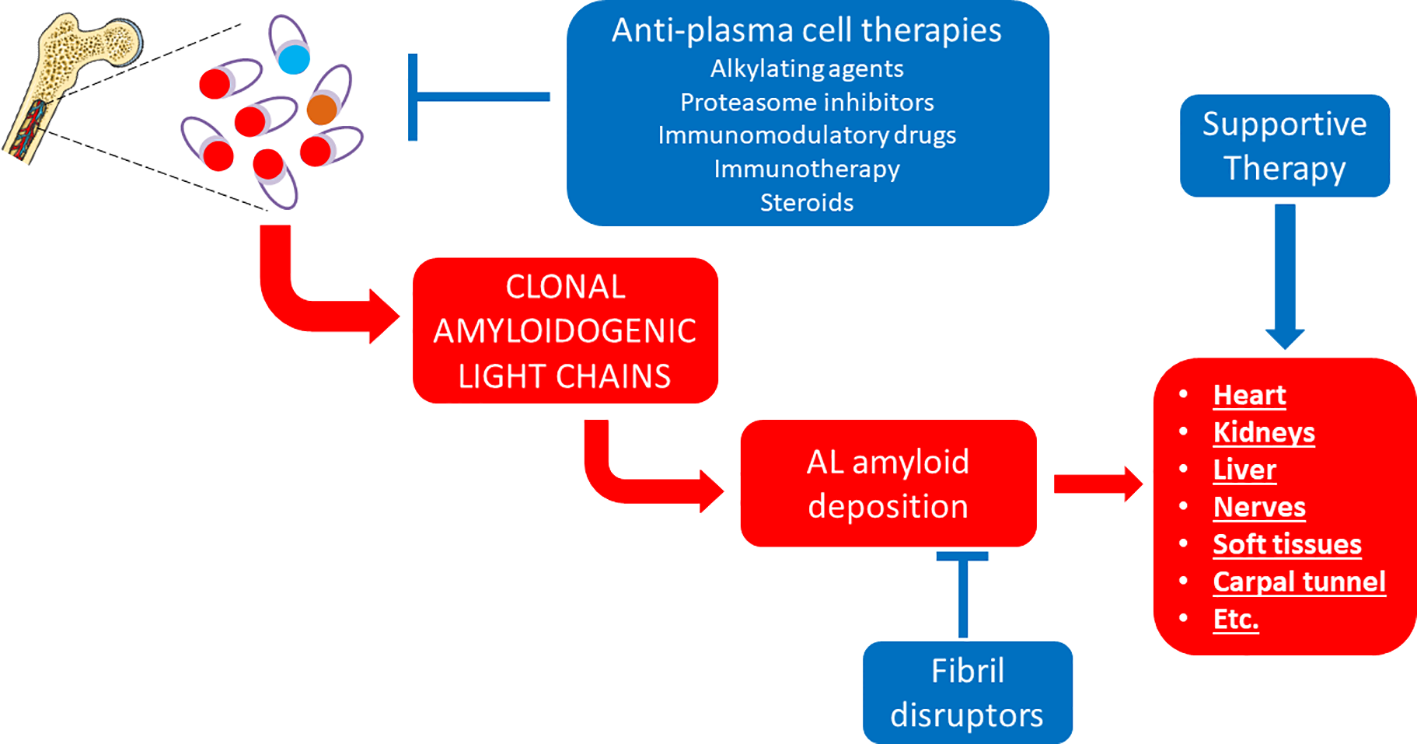
Established therapeutic interventions against AL amyloidosis. Red blocks indicate steps in the pathogenesis of AL amyloidosis; blue blocks indicate established therapeutic interventions against AL amyloidosis.
Table 4
| M protein type | Clonal cell | Related conditions |
|---|---|---|
| Non-IgM | Plasma cell | AL |
| MM + AL | ||
| PCL + AL | ||
| B-cell CLL + AL | ||
| B-cell NHL + AL | ||
| Other | ||
| IgM | B cell (exceptionally plasma cell) | AL with IgMκ or IgMλ clone |
| WM + AL | ||
| B-cell CLL + AL | ||
| B-cell NHL + AL | ||
| (exceptionally IgM MM + AL) | ||
| Other |
Main clinical conditions associated with AL amyloidosis based on type of M protein and underlying clonal cell.
AL, immunoglobulin light chain amyloidosis; CLL, chronic lymphocytic leukemia; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; PCL, plasma cell leukemia; WM, Waldenström macroglobulinemia.
High dose melphalan followed by rescue therapy with autologous stem cell transplantation (ASCT) for the fittest, transplant-eligible subset of AL amyloidosis patients grants the most durable responses (Sanchorawala et al., 2015; Sidiqi et al., 2018; Sidana et al., 2019). Scrupulous selection of transplant eligible patients has dramatically reduced transplant-related mortality in more recent patients’ series (Sidiqi et al., 2018). When full dose melphalan (200 mg/m2) is employed, the median overall survival achieved is superior to 10 years (Sanchorawala et al., 2015; Sidiqi et al., 2018). In patients with bone marrow plasma cell infiltration above 10%, induction therapy before ASCT to expedite lowering of toxic light chains may improve rates of hematologic response and overall survival (Hwa et al., 2016; Scharman et al., 2017), stressing the importance of swiftly turning off the production of the amyloid-forming protein (Mikhael, 2018).
Transplant-ineligible patients due to advanced disease or significant comorbidity are treated with standard or attenuated doses of chemotherapy against the plasma cell clone, using alkylating agents, proteasome inhibitors, immunomodulatory drugs, monoclonal antibodies and steroids (Merlini et al., 2018). Several drug combinations are available and they are associated with median survivals exceeding 5 years, with the exception of patients with very advanced cardiac involvement at diagnosis, who show dismal prognosis irrespectively of the employed regimen (Merlini et al., 2018).
The extent of reduction of the amyloidogenic light chains with respect to pre-therapy levels can be used to grade the depth of hematologic response to therapy into four main response categories, that is no response, partial response, very good partial response and complete response, which are characterized by a progressively better prognosis (Palladini et al., 2012). Indeed, drop of the amyloidogenic, clonal light chain is paralleled by reduction of markers of heart dysfunction (Palladini et al., 2006) and hematologic response to therapy is the prerequisite to halt organ function deterioration or even promote organ function recovery (i.e. organ response to therapy) (Dispenzieri et al., 2006; Palladini et al., 2012; Palladini et al., 2014; Muchtar et al., 2018). Of note, the extent and rapidity of amyloidogenic light chain reduction is associated with higher rates of organ response and longer event-free survival (Palladini et al., 2020).
Among patients achieving the hematologic complete response status, that is the disappearance of M protein/monoclonal light chains and the normalization of serum free light chain levels, next generation flow cytometry or next generation sequencing can be employed to identify residual clonal plasma cells (defined as minimal residual disease, MRD) within the bone marrow with high (10−5–10−6) sensitivity (Kastritis et al., 2018). Recent studies are indicating that AL patients with MRD persistence have lower rates of organ response (Sidana et al., 2020; Staron et al., 2020). While the biologic bases of this observation are not clear, one possibility is that low levels of toxic, amyloidogenic light chains, below detection limits of routine diagnostic tests, may be enough to prevent organ function restoration. This highlights the importance of completely eradicating the underlying plasma cell clone, to arrest further damage and recover organ function.
More recently, the elucidation of the molecular mechanisms underlying the pathogenesis of ATTR amyloidosis has enabled the development of novel targeted therapies against this disease (Ruberg et al., 2019) (Figure 3).
Figure 3
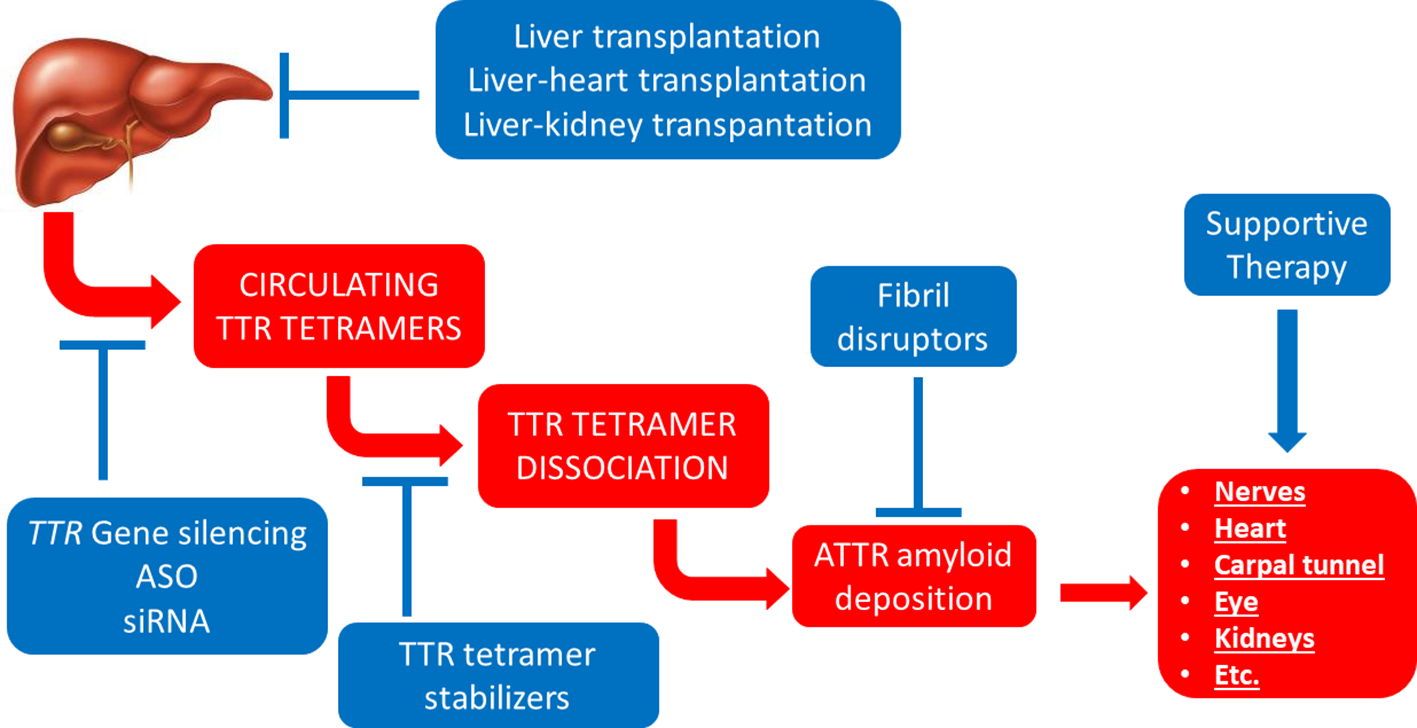
Established therapeutic interventions against ATTR amyloidosis. Red blocks indicate steps in the pathogenesis of ATTR amyloidosis; blue blocks indicate established therapeutic interventions against ATTR amyloidosis. ASO, anti-sense oligonucleotides; siRNA, short interfering RNA.
Circulating TTR is a homo-tetrameric protein mainly synthetized by the liver. Transthyretin is intrinsically amyloidogenic and can form amyloid also in its wild-type form, mainly within the heart, leading to ATTRwt amyloidosis (formerly senile systemic amyloidosis or senile cardiac amyloidosis) affecting predominantly elderly men. This is the result of a presumably very slow process which can become clinically manifest in elderly subjects or remain unappreciated and be detected as a post-mortem finding. Indeed, cardiac amyloid deposits containing TTR can be detected in 12–25% of subjects older than 80 years at autopsy (Cornwell et al., 1983; Tanskanen et al., 2008; Ueda et al., 2011). ATTRwt amyloidosis is an increasingly recognized cause of amyloid cardiomyopathy whose clinical detection has been conspicuously boosted by scintigraphy with bone tracers and increased awareness (Ravichandran et al., 2020). Of note, scintigraphy with bone tracers enabled to detect ATTRwt amyloidosis in 14–16% of patients with severe symptomatic aortic stenosis listed for transcatheter aortic valve replacement (Castano et al., 2017; Cavalcante et al., 2017; Scully et al., 2018; Scully et al., 2020).
Tetramer dissociation is currently regarded as the rate limiting factor for ATTR amyloidogenesis, as it releases TTR monomers which can then misfold and aggregate. A parallel mechano-enzymatic amyloidogenic mechanism has also been proposed (Marcoux et al., 2015). The presence of one of an ever-growing list of TTR tetramer-destabilizing mutations is the cause of hereditary or variant ATTR amyloidosis (ATTRv), which can affect peripheral nerves (hereditary ATTR amyloidosis with polyneuropathy, also known as familial amyloid polyneuropathy), the heart (hereditary ATTR amyloid cardiomyopathy, also known as familial amyloid cardiomyopathy) or both (at various degrees), based on the underlying TTR gene mutation (Connors et al., 2003; Rowczenio et al., 2019), with the neurotropic Val30Met (p.Val50Met) and the cardiotropic Val122Ile (p.Val142Ile) mutations being at the two extremities of the disease spectrum (Rapezzi et al., 2010). ATTRv amyloidosis associated to the Val30Met mutation mainly affects the peripheral and autonomous nervous systems (Andrade, 1952; Saraiva, 2002), but may involve the heart and the kidney. The disease is endemic in some areas of Portugal, Japan and Sweden, even though substantial differences in disease penetrance, age of onset (early or late onset) and other biochemical and clinical features exist among these geographic areas (Araki et al., 1968; Andersson, 1976; Araki, 1984; Alves et al., 1997). The Val122Ile TTR mutation is present in approximately 4% of African Americans and can lead to late-onset restrictive amyloid cardiomyopathy (Quarta et al., 2015).
For long time, liver transplantation to abolish variant TTR production as a sort of surgical gene therapy has been the only available therapeutic option in selected ATTRv patients. The best results are seen in patients with early-onset hereditary ATTR amyloidosis with polyneuropathy associated with the Val30Metmutation transplanted at early disease stages (Carvalho et al., 2015).
While most of the mutations in the TTR gene destabilize TTR tetramer and favor amyloid formation, the Thr119Met, or p.Thr139Met, mutation stood out for its unprecedented properties. Indeed, this mutation was identified in compound heterozygous subjects who were protected from the development of amyloid deposits despite the presence of a highly prevalent amyloidogenic mutation (the Val30Met mutation in the endemic Portuguese area) (Coelho et al., 1993). Subsequent biochemical and biophysical studies clarified that the protective effect of the Thr119Met mutation was due to the stabilization of TTR hetero-tetramer formed by both Val30Met and Thr119Met monomers (Hammarstrom et al., 2001). Collectively, these clinical and laboratory observations paved the way for a novel therapeutic approach against TTR-related amyloidosis, that is TTR tetramer stabilization (Hammarstrom et al., 2003). Both structure-based drug design and drug repurposing were pursued to identify TTR stabilizers with therapeutic effects.
The non-steroidal anti-inflammatory drug diflunisal was identified as a TTR stabilizer, with a >1 stoichiometry to serum TTR when orally administered thanks to its high bioavailability (Hammarstrom et al., 2003; Sekijima et al., 2006). An international randomized, double-blind, placebo-controlled study on patients with hereditary ATTR amyloidosis with polyneuropathy showed that diflunisal administration for 2 years reduced the rate of progression of neurological impairment and preserved quality of life (Berk et al., 2013).
The first drug to obtain approval for the treatment of ATTRv amyloidosis, tafamidis, was the result of structure-based drug design. This molecule was indeed designed to bind to one of the two T4-binding pockets of the TTR tetramer and to slow tetramer dissociation (Razavi et al., 2003; Bulawa et al., 2012). Its efficacy was tested in the context of a multicenter, international, randomized, placebo-controlled clinical trial. Daily treatment of patients affected by hereditary ATTR amyloidosis with polyneuropathy (TTR Val30Met mutation) with tafamidis led to circulating TTR tetramer stabilization and, in the efficacy-evaluable cohort of patients, to a significant delay in peripheral neurologic impairment over a 18-month treatment period compared to placebo, even though the coprimary endpoints of the trial were not met (Coelho et al., 2012). Subsequent case series confirmed tafamidis efficacy also in additional patients’ cohorts, including in patients with non-Val30MetTTR mutations and in patients with ATTRv cardiomyopathy (Coelho et al., 2013b; Merlini et al., 2013; Damy et al., 2015; Cortese et al., 2016; Ishii et al., 2019). Another clinical trial assessed tafamidis efficacy against ATTR cardiomyopathy (both ATTRv and ATTRwt types). Daily treatment with tafamidis was associated with reductions in all-cause mortality and cardiovascular-related hospitalizations, along with reduced decline in functional capacity and quality of life as compared with placebo (Maurer et al., 2018). The outcome of this trial led to the FDA and EMA approval of tafamidis as the first drug for the treatment of ATTR amyloidosis cardiomyopathy.
An additional TTR tetramer stabilizer developed based on structural data is represented by AG10. This small molecule stabilizes both wild-type and Val122Ile-containing TTR tetramers from patients’ sera (Penchala et al., 2013; Miller et al., 2018). Twice daily treatment of patients with ATTR cardiomyopathy (both ATTRv and ATTRwt) with AG10 led to a dose-dependent stabilization of TTR tetramers and to increased circulating TTR levels (Judge et al., 2019). An ongoing phase 3 trial is investigating the efficacy and safety of AG10 compared to placebo administered on a background of stable heart failure therapy in patients with ATTRv or ATTRwt cardiomyopathy (NCT03860935).
Other rationally designed small molecule TTR tetramer binders, including the palindromic molecule mds84, which simultaneously occupies both T4-binding pockets of TTR tetramers (Kolstoe et al., 2010; Corazza et al., 2019), are in preclinical development.
Screening of molecules under clinical development or already in use in humans led to the discovery that tolcapone, a drug used to treat patients with Parkinson’s disease, is a potent TTR tetramer binder, which can bind TTR in human plasma, stabilize TTR tetramers in vivo and inhibit TTR cytotoxicity (Sant’Anna et al., 2016; Gamez et al., 2019). As tolcapone is capable of passing the blood-brain-barrier, this molecule is particularly promising for the treatment of the clinical manifestations associated with the deposition of amyloid within the central nervous system, which are seen especially in the presence of selected TTR mutations as a result of local TTR production at the choroid plexus. A clinical trial assessing the TTR tetramer stabilizing effect of tolcapone in plasma and cerebrospinal fluid of patients with symptomatic ATTRv leptomeningeal amyloidosis and asymptomatic TTR mutation carriers has recently completed recruitment (NCT03591757).
More recently, an alternative therapeutic approach against TTR-related amyloidosis has been explored, one which is based on gene silencing. This has been achieved through either anti-sense oligonucleotides (ASOs) or silencing RNA (siRNA), which ultimately lower mature TTR mRNA levels within the liver, thus reducing hepatic TTR protein synthesis and secretion. As both wild-type and mutant TTR are found in the amyloid deposits, gene silencing agents have been designed to target highly conserved portions of the 3’-untranslated region of both wild-type and mutant TTR transcripts (Coelho et al., 2013a; Ackermann et al., 2016). Two large international randomized, double-blind, placebo-controlled studies demonstrated the efficacy of gene silencing agents for hereditary ATTR amyloidosis with polyneuropathy. Subcutaneous injection of the antisense oligonucleotide inotersen (three injections in the first week followed by weekly injections) over a 15-month period led to a median reduction in serum TTR concentration of 79% and improved the course of neurologic disease and health-related quality of life (Benson et al., 2018b). Intravenous administration of the siRNA agent patisiran once every 3 weeks over an 18-month period led to a median reduction in serum TTR concentration of 81% and improved several clinical manifestations of the disease compared to placebo (Adams et al., 2018). Remarkable findings were observed in the subset of enrolled patients displaying concomitant amyloid cardiomyopathy. In these patients, patisiran treatment was associated with echocardiographic features of better cardiac structure and function, reduced levels of the cardiac biomarker NT-proBNP and decreased adverse cardiac outcome, suggesting that this drug may halt or even reverse the progression of cardiac manifestations of hereditary ATTR amyloidosis (Adams et al., 2018; Minamisawa et al., 2019; Solomon et al., 2019).
As a result of the unprecedented successes seen in recent years, several alternative effective drugs are now available which can potentially reduce the availability of the amyloid-forming protein and interfere with the process of amyloid formation and organ damage for both AL and ATTR amyloidosis. Considering the paucity (or even the absence) of controlled clinical trials directly comparing different treatment regimens, now the question is how to select the best drug for each patient.
In AL amyloidosis, retrospective data from large series of homogenously treated patients have highlighted a potential role of molecular cytogenetics in predicting sensitivity of the underlying plasma cell clone to some anti-plasma cell drugs. Gain of chromosome 1q21 is an independent adverse prognostic factor in patients treated with melphalan and dexamethasone (Bochtler et al., 2014). Conversely, translocation t(11;14) is associated with adverse outcome in newly diagnosed and transplant-ineligible patients who are treated with bortezomib-based regimens (Bochtler et al., 2015). Interestingly, the survival disadvantage wrought by the presence of the t(11;14) translocation can be abrogated by exposure to melphalan (Muchtar et al., 2017). Based on these observations, the combination of bortezomib, melphalan and dexamethasone, which has been compared to treatment with melphalan and dexamethasone alone in the context of a clinical trial (NCT01277016), has the potential to by-pass the reduced sensitivity of gain 1q21- or t(11;14)-positive plasma cell clones towards melphalan or bortezomib, respectively.
The t(11;14) translocation, which is present in up to 60% of AL plasma cell clones, results in the overexpression of the anti-apoptotic BCL-2 protein. The selective BCL-2 inhibitor venetoclax showed therapeutic effect in a few cases of relapsed/refractory AL amyloidosis (Leung et al., 2018; Ghilardi et al., 2019; Premkumar et al., 2019), in line with positive findings against relapsed/refractory t(11;14)-positive multiple myeloma cases, both as a single agent or in combination with bortezomib (Kumar et al., 2017; Moreau et al., 2017).
In ATTR amyloidosis, retrospective data from a large series of patients with hereditary ATTR amyloidosis and polyneuropathy treated with tafamidis were analyzed to identify possible predictors of tafamidis response. This analysis enabled the definition of a prognostic model based on patient’s sex, disease severity and native TTR concentration at the outset of treatment to predict response to this TTR tetramer stabilizer (Monteiro et al., 2019).
Monitoring the Concentration of the Amyloid-Forming Protein Guides Therapeutic Interventions
Experimental and clinical evidence point towards a crucial pathogenetic role of soluble oligomeric intermediates of fibril assembly as the main mediators of cytotoxicity and organ damage (Comenzo et al., 1996; Lambert et al., 1998; Hartley et al., 1999; Dember et al., 2001; Sousa et al., 2001; Andersson et al., 2002; Walsh et al., 2002). However, such oligomeric species remain difficult to be measured through routine methods. In the impossibility of directly assessing levels of oligomeric species, the effect of etiologic interventions against AA, AL and ATTR amyloidosis can be assessed by closely monitoring circulating levels of the amyloid-forming protein (that is, SAA, circulating free light chains, and TTR, respectively).
In AA amyloidosis, SAA levels above 10 mg/L should justify a potentiation of the anti-inflammatory therapy, as levels above this threshold value are associated to a significantly increased risk of organ damage progression or amyloid recurrence in renal graft for transplanted patients (Pinney et al., 2013).
In AL amyloidosis, the amount of the amyloid-forming protein, as indicated by baseline immunoglobulin free light chain (FLC) levels, bears a negative prognostic effect (Dispenzieri et al., 2006). Also, the difference between involved and uninvolved FLC (denoted as dFLC, which is indicative of the burden of amyloidogenic light chains in patients with AL amyloidosis) is a negative risk factor independent of the extent of cardiac involvement (Kumar et al., 2012). In this context, the aim of anti-plasma cell therapy is to profoundly reduce or possibly eliminate the amyloidogenic light chain. Reaching adFLC of <40 mg/L or less was considered the aim of therapy (Palladini et al., 2012). However, recent evidence has emerged indicating that a concentration of dFLC of 10 mg/L or less after therapy (termed stringent dFLC response) is associated with improved outcome (Manwani et al., 2019). Also in patients with low levels of dFLC at diagnosis, who have less frequent and less severe heart involvement and a better prognosis, achievement of dFLC below 10 mg/L after therapy is associated with a better overall survival and organ response (Dittrich et al., 2017; Milani et al., 2017; Sidana et al., 2018).
As for TTR, it should be noted that tetramer stabilizers and gene silencers lead to opposing changes in serum TTR levels, with the former leading to increased levels due to tetramer stabilization and the latter leading to reduced levels due to suppressed synthesis. Of interest, in the clinical trial investigating the therapeutic effect of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy, a correlation between the degree of the reduction in TTR levels from baseline and neurological manifestations was observed (Adams et al., 2018). In line, analysis of serum TTR levels from a large series of patients after 12 months of treatment with tafamidis showed higher levels of circulating TTR in patients classified as responders to tafamidis, compared to patients defined as partial responders or non-responders based on clinical evaluations (Monteiro et al., 2019).
For those cases in which experimental therapeutic approaches against the amyloid-forming protein are planned, the availability of reliable diagnostic assays to accurately monitor the levels if the amyloid-forming protein in the relevant compartment represents an essential prerequisite for clinical trial design and patient management. In the case of prion diseases, where neurodegeneration is due to replication of the endogenous prion protein in the presence of the infectious agents, the prion, there are efforts at developing silencing agents to lower PRNP expression and reduce cellular prion protein expression, with the hope to attenuate prion-induced neurodegeneration or even prevent it in at risk individuals (e.g. pre-symptomatic carriers of highly penetrant mutations) (Nuvolone et al., 2009). The validity of this approach is corroborated by observations in animal models (Mallucci et al., 2003; Mallucci et al., 2007). In this context, the development of a robust diagnostic assay to measure prion protein levels in cerebrospinal fluids (Vallabh et al., 2019), together with ongoing animal studies on the therapeutic effect of PRNP silencing agents, are welcomed achievements in the pathway leading to clinical trials against these as of yet orphan diseases.
Reduction/Elimination of the Amyloid-Forming Protein may Delay/Prevent Disease Onset
Whenever possible, prevention can be highly effective and can significantly impact on the epidemiology of the disease and modify incidence and/or prevalence. This has been achieved so far—even though only for a few forms of systemic amyloidosis—by avoiding increased concentrations of the amyloid-forming protein in at risk individuals.
Amyloidosis associated with wild-type β2-microglobulin (Aβ2Mwt amyloidosis, also known as dialysis-related amyloidosis) can occur in patients undergoing long-term hemodialysis (occasionally also in patients in peritoneal dialysis or in patients with end stage renal disease not yet on dialysis) (Zingraff et al., 1990). Because of lack of efficient renal catabolism and clearance of β2-microglobulin, circulating levels of this protein are chronically increased in these patients, and this can result in the formation of amyloid deposits, most commonly within the osteo-articular system (Scarpioni et al., 2016). Post-mortem studies showed that this condition can be detected histologically in almost 50% of hemodialysis patients at autopsy, even though the disease becomes clinically or radiologically evident only in a subset of these cases (Jadoul et al., 1997). Over the last two decades, significant advances in dialysis technology, including the use of high flux membranes, convective regimens, adsorptive columns/membranes and biocompatible materials have succeeded at reducing β2-microglobulin levels in dialysis patients (Figure 4). In line, these improvements have resulted in a significant delay of the occurrence of Aβ2Mwt amyloidosis, leading to overall reduced prevalence and severity of this invalidating complication of long-term dialysis (Schiffl, 2014; Hoshino et al., 2016; Morris et al., 2019).
Figure 4
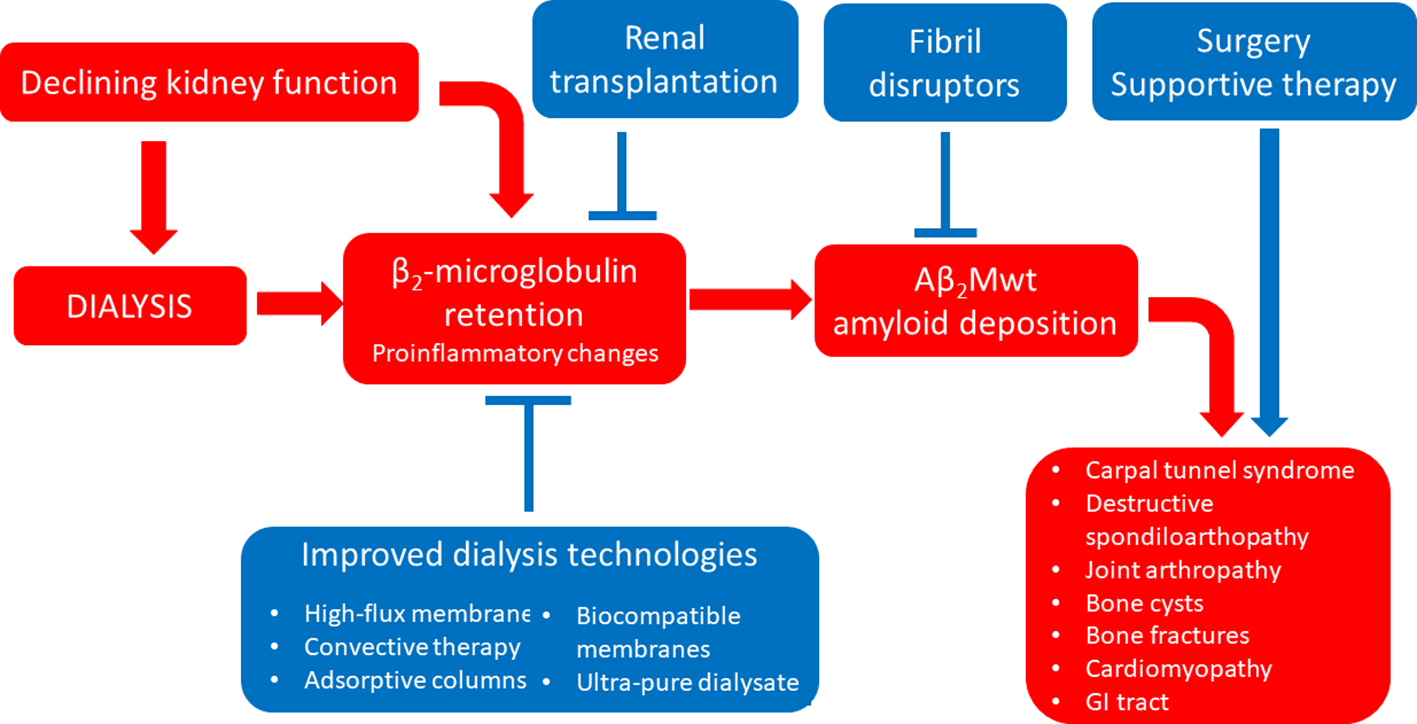
Established therapeutic interventions against Aβ2Mwt amyloidosis. Red blocks indicate steps in the pathogenesis of Aβ2Mwt amyloidosis; blue blocks indicate established therapeutic interventions against Aβ2Mwt amyloidosis. GI tract, gastro-intestinal tract.
In the case of AA amyloidosis, a better control of the inflammatory status through more effective drugs, including disease-modifying anti-rheumatic drugs and monoclonal antibodies, is the most plausible explanation for the reduced incidence of this type of amyloidosis in high income Countries (Lane et al., 2017a).
In principle, this approach could be extended to other protein misfolding and deposition diseases. A promising clinical setting for preemptive interventions is represented by asymptomatic carriers of amyloidogenic TTR mutations, especially in the era of effective silencing agents. There are currently no data to support the potential benefit of preemptive interventions in pre-symptomatic mutation carriers. Because of the existence of incomplete penetrance, the first setting where to test this approach may be TTR Val30Met carriers from endemic areas with high disease penetrance. In this context, the elucidation of the bases of susceptibility, including the study of potential modifier genes (Santos et al., 2016; Santos et al., 2018; Dias et al., 2019; Santos et al., 2019), along with the development of high-sensitivity imaging modalities to detect minute amount of amyloid deposits at the pre-symptomatic stage (Kollmer et al., 2015; Kollmer et al., 2017) would help to identify individuals who could benefit most from such a preemptive intervention.
For ATTRwt amyloidosis, no known predisposing factors have been identified so far beyond aging and male sex, even though an association with hip and knee arthroplasty, carpal tunnel syndrome, spinal canal stenosis and aortic stenosis has been described (Rubin et al., 2017; Aus dem Siepen et al., 2019; Scully et al., 2020).
In AL amyloidosis, at risk individuals are represented by subjects with a known monoclonal gammopathy. However, only a small proportion of patients with MGUS, MM or other plasma cell dyscrasia eventually develop AL amyloidosis.
There are current research efforts aiming at identifying individuals at higher risk of developing AL amyloidosis among subjects with monoclonal gammopathy. One such effort exploits the known association of AL amyloidosis with a few germ-line genes encoding for the variable light chain gene, VL, possibly due to the high amyloidogenicity of these germ-line genes. In particular, patients with a monoclonal protein expressing the IGLV6-57, IGLV1-44, IGLV2-14 or IGLV1-51 have a relative risk of developing AL amyloidosis vs myeloma ranging from 7.3, 2.5, 1.6 to 1.2, respectively (Zhou et al., 2019) (NCT02741999). Based on this, the impact of ascertaining the VL gene family through sequencing approaches in bone marrow specimens in patients with MGUS/monoclonal gammopathy is currently being explored (Zhou et al., 2019).
Another approach is based on the functional characterization of patient-derived monoclonal light chains. Indeed, a functional competition assay based on in vitro fibrillization has been established. The test succeeded at differentiating amyloidogenic from control light chains, based on the superior capacity of the formers to inhibit in vitro recruitment of an amyloidogenic variable light chain domain by homologous amyloid-like fibrils (Martin et al., 2017; Martin et al., 2018).
Recent interesting data have emerged from the study of post-translational modifications of monoclonal light chains from the serum of patients with AL amyloidosis or other plasma cell disorders. In particular, the use of mass spectrometry on immunopurified light chains from patients’ serum has identified distinctive M protein mass distributions patterns possibly indicating N-glycosylation of monoclonal light chains in 33 and 10.2% of patients with AL-κ and AL-λ amyloidosis, compared to 3.7 and 4.9% in patients with non-AL monoclonal gammopathies of κ and λ type, respectively (Kumar et al., 2019). Subsequent analyses in a subset of cases involving enzymatic deglycosylation and mass spectrometric analysis confirmed the presence of N-glycosylation in the analysed monoclonal light chains (Kumar et al., 2019). These observations are in line with previous studies on a limited number of amyloidogenic light chains showing N-glycosylation as a post-translational modification detected in a subset of cases (Stevens, 2000). As mass spectrometry analysis is an emerging area for the screening of M proteins, the identification of potential N-glycosylation in monoclonal light chain may help identifying subjects at higher risk of developing AL amyloidosis (Kumar et al., 2019).
Additional studies are needed to show the potential clinical application of these different approaches and their impact on patients’ outcome.
Early Diagnosis is Vital
As systemic amyloidoses are characterized by the progressive organ dysfunction of affected organs, early diagnosis is of utmost importance in order to intercept patients before the development of irreversible organ damage.
A delayed diagnosis, in the presence of advanced organ damage and possibly of multi-systemic involvement, unavoidably results in increased frailty, bears negative prognostic impact and represents a significant limitation for therapy.
In the general population of patients with AA amyloidosis, more than 10% showed end-stage renal disease at diagnosis (Lachmann et al., 2007; Palladini et al., 2017b). This figure increases up to ≈25% in patients developing AA amyloidosis as a complication of hereditary autoinflammatory syndromes (Lane et al., 2013). Proteinuria-based screening should be considered to promote early recognition of AA amyloidosis in at risk-patients with chronic inflammatory diseases (Papa and Lachmann, 2018).
In a series of 108 patients with ATTRwt amyloidosis, approximately one third of subjects had severe heart failure (New York Heart Association, NYHA, class III or IV) at diagnosis (Gonzalez-Lopez et al., 2017). Of note, in the clinical trial testing the therapeutic effect of tafamidis-mediated TTR tetramer stabilization in patients with ATTR amyloid cardiomyopathy, tafamidis reduced the rate of cardiovascular-related hospitalization in all patients’ subgroups, except patients with NYHA class III (patients with NYHA IV class were excluded from the trial). This again stresses the importance of an earlier diagnosis and a prompt initiation of therapy. Luckily, the increased awareness of this disease, along with the increased role of cardiac imaging and the possibility of a biopsy-free diagnosis through scintigraphy with radiotracers (Gillmore et al., 2016) facilitate the diagnosis of ATTR amyloid cardiomyopathy at earlier disease stages. Indeed, among ATTRwt amyloidosis patients evaluated at a single institution, patients diagnosed after the introduction of a scintigraphy-based non-invasive diagnosis showed lower rate of advanced disease stage compared to patients diagnosed earlier (Lane et al., 2019). In line, recent real-world case series of newly diagnosed ATTRwt amyloidosis patients show milder disease stages compared to cohorts of patients enrolled in clinical trials in terms of NYHA functional class, hemodynamic parameters and cardiac biomarkers (Canepa et al., 2019).
As carpal tunnel syndrome, often bilateral, is present in 20–50% of ATTRwt amyloidosis patients at diagnosis (Nakagawa et al., 2016; Gonzalez-Lopez et al., 2017; Milandri et al., 2020) as well as in 10–20% of AL amyloidosis patients (Prokaeva et al., 2007; Rapezzi et al., 2009), and in most cases this condition precedes cardiac (or other) manifestations of the diseases by many years, different screening approaches have been applied in this clinical setting. Congo red staining of tenosynovial tissue has been employed to detect amyloid deposits at this site in patients with carpal tunnel syndrome (males aged ≥50 years and females ≥60 years) undergoing carpal tunnel release surgery. Amyloid deposits were found in 10% of evaluated patients, enabling the diagnosis of previously unrecognized amyloidosis (ATTRwt, ATTRv and AL), including symptomatic patients requiring therapy (Sperry et al., 2018). In another study, patients aged 60 years or older who underwent carpal tunnel release surgery were evaluated by echocardiography and, in the presence of increased left ventricular hypertrophy, by additional tests to ascertain the presence of cardiac amyloidosis. Cardiac amyloidosis (ATTRwt and AL) was identified in 1% of screened patients and this figure increased to >10% when focusing on patients with bilateral carpal tunnel syndrome and left ventricular hypertrophy (Zegri-Reiriz et al., 2019).
Also in the case of hereditary ATTR amyloidosis with polyneuropathy, administration of tafamidis at earlier disease stages led to a greater preservation of neurologic function compared to patients in which tafamidis was initiated later (Coelho et al., 2013b; Plante-Bordeneuve et al., 2017).
In AL amyloidosis, a delayed diagnosis is the rule rather than the exception. According to a recent patient-initiated survey, patients received a diagnosis after an average of 3 years from clinical onset and after consulting ≥4 specialists (McCausland et al., 2018). Not surprisingly, about 85% of AL patients are not eligible for ASCT at diagnosis due to advanced organ involvement. Noteworthy, a subset of these patients may eventually become transplant-eligible and undergo successful deferred ASCT upon response to standard dose, induction chemotherapy, as a result of reversal of organ dysfunction present at diagnosis, with excellent outcome (Manwani et al., 2018). Fifteen to 20% of AL amyloidosis patients show advanced cardiac involvement at diagnosis. These patients are particularly frail, have limited therapeutic options and have a dismal prognosis (Wechalekar et al., 2013).
Recently, a strategy to promote early diagnosis of AL amyloidosis, before overt symptomatic organ damage has been promoted. At diagnosis, virtually all (95%) AL amyloidosis patients have an abnormal serum immunoglobulin free light chain ratio and >80% of cases have increased NT-proBNP levels and/or proteinuria due to heart and/or kidney involvement. Based on these figures, the addition of NT-proBNP and proteinuria within the battery of biomarkers employed for the regular follow up of patients with MGUS has been advocated for those MGUS cases with an abnormal free light chain ratio (Merlini and Palladini, 2012). Such a strategy has indeed succeeded at identifying pre-symptomatic patients with AL amyloidosis, at early disease stages, resulting in excellent outcomes (Palladini et al., 2017a). As AL amyloidosis is often diagnosed late also in patients with a known monoclonal gammopathy under hematologic follow up (Kourelis et al., 2014), this approach has the potential to significantly reduce the diagnostic delay. In general, increasing the awareness of AL amyloidosis among physicians may reduce the patients’ journey before achieving a diagnosis (McCausland et al., 2018).
Prompt reduction of the amyloid-forming protein is necessary but not sufficient to guarantee restoration of organ function, improvement of quality of life and prolonged survival. Organ function recovery may depend on the type and severity of organ damage and possibly, also on the type of amyloid and on as of yet poorly identified inter-individual factors.
Lack of organ function restoration may be explained, at least in part, by the persistence of amyloid deposits within affected organs. Of note, among AA amyloidosis patients where anti-inflammatory therapies successfully lowered circulating SAA levels below 10 ng/L, amyloid regression was seen in 60% of cases and survival among these patients was superior to survival among those in whom amyloid deposits did not regress (Lachmann et al., 2007).
In this context, therapeutics able to promote amyloid clearance may facilitate restoration of organ function. This approach has attracted much attention in recent years, with three monoclonal antibodies targeting amyloid deposits being tested in clinical trials, and more molecules in the pipeline (Nuvolone and Merlini, 2017a). Despite promising preliminary results of NEOD001, a monoclonal antibody targeting misfolded amyloid proteins (Gertz et al., 2016), and of miridesap, a monoclonal antibody targeting amyloid-bound SAP after pharmacological depletion of circulating SAP (Richards et al., 2015; Richards et al., 2018), several clinical trials investigating these biologicals were recently interrupted due to futility analysis (for NEOD001, NCT02312206; NCT03168906) or to a change in benefit/risk profile (for miridesap, NCT03044353). A third chimeric fibril reactive monoclonal antibody, CAEL-101, formerly 11-1F4, is currently under clinical evaluation (Edwards et al., 2019) (NCT02245867).
An alternative strategy to promote amyloid clearance is based on the amyloid fibril disrupting activity of 4ʹ-iodo-4ʹ-deoxydoxorubicin and of the structurally related molecule doxycycline (Merlini et al., 1995; Cardoso et al., 2010). Several clinical observations support a potential benefit of doxycycline treatment against ATTR, AL and Aβ2Mwt amyloidosis (Obici et al., 2012; Montagna et al., 2013; Piccoli et al., 2017; Wechalekar and Whelan, 2017; Karlstedt et al., 2019) and several ongoing clinical trials are currently investigating the potential therapeutic effect of doxycycline against different forms of systemic amyloidosis.
Conclusive Remarks
Recent, unprecedented progress in the treatment of different forms of systemic amyloidoses is rapidly changing the natural history of these diseases, significantly prolonging patients’ survival and even leading to effective preemptive interventions which result in reduced disease incidence in some instances. Still, systemic amyloidoses remain serious illnesses, which may lead to deterioration of organ function, poor quality of life and reduced survival, and only subsets of patients benefit from currently available therapies. Thus, there is still much space for improvements in the treatment of these diseases.
Clinical observations stemming from the therapeutic success achieved in the more common forms of systemic amyloidoses all converge at indicating the early and conspicuous reduction of the amyloid-forming protein as the presently most effective treatment strategy. Such observations also stress the importance of identifying these diseases early, to enable timely therapeutic interventions, to prevent advanced organ damage and to increase the likelihood of organ function restoration and improvement in quality of life and disease-free survival.
While the few tested interventions aimed at promoting amyloid clearance did not show clear clinical benefit against systemic amyloidoses so far, this therapeutic approach should be further pursued, as it could represent an important complementation to halt or reverse organ dysfunction.
Most of the success obtained in the treatment of AL amyloidosis is the result of the impressive progress made in recent years by academic and industrial research at developing drugs against the more prevalent plasma cell tumor multiple myeloma. This has enabled the use of novel, potent anti-plasma cell drugs to treat also patients with AL amyloidosis. Similarly, the changing paradigm seen in the context of AA amyloidosis, with reduced incidence of this type of amyloidosis and a relative increase of cases in which the underlying inflammatory condition cannot be unambiguously identified, reflects recent improvements in the treatment of inflammatory arthritides, chronic inflammatory bowel diseases and infections. While one could argue that most of the progress made in the treatment of AL and AA amyloidosis was serendipitous and just reflects improvements in the treatment of myeloma and chronic inflammatory conditions, on a more positive note the scenario is totally different in the case of TTR-related amyloidosis. Here, relentless, dedicated efforts at uncovering the molecular mechanisms of this disease and at identifying or designing novel effective drugs have eventually succeeded at providing tailored, targeted therapies against this disorder. This unprecedented therapeutic success was the result of the use of different approaches for drug discovery, including high-throughput screens, rational structure-based drug design and gene silencing. The common denominator of these approaches is to reduce the supply or to stabilize transthyretin, again stressing the importance of rapidly and profoundly turning off the supply of the amyloid-forming protein to treat amyloid diseases.
There is hope that the example of ATTR amyloidosis will not remain isolated, and that this will be replicated for the many forms of amyloidosis, either systemic or localized, for which etiologic therapies are still eagerly awaited.
Funding
This work was supported by grants from the Italian Ministry of Health (Ricerca Finalizzata, grants RF-2013-02355259 and GR-2018-12368387), the CARIPLO Foundation (grants 2013-0964 and 2018-0257), the Amyloidosis Foundation, the “Associazione Italiana per la Ricerca sul Cancro—Special Program Molecular Clinical Oncology 5 per mille” (grant 9965) and by an Accelerator Award from the Cancer Research UK, the Fundación Científica—Asociación Española Contra el Cáncer and the Associazione Italiana Ricerca sul Cancro.
Statements
Author contributions
AN, GM, and MN wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
GM: consultant for Millennium Pharmaceuticals, Inc., Pfizer, Janssen, Prothena, and IONIS. MN: speaker honoraria from Janssen-Cilag.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AckermannE. J.GuoS.BensonM. D.BootenS.FreierS.HughesS. G.et al. (2016). Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid23 (3), 148–157. doi: 10.1080/13506129.2016.1191458
2
AdamsD.Gonzalez-DuarteA.O’RiordanW. D.YangC. C.UedaM.KristenA. V.et al. (2018). Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med.379 (1), 11–21. doi: 10.1056/NEJMoa1716153
3
AguzziA.NuvoloneM.ZhuC. (2013). The immunobiology of prion diseases. Nat. Rev. Immunol.13 (12), 888–902. doi: 10.1038/nri3553
4
AlvesI. L.AltlandK.AlmeidaM. R.WinterP.SaraivaM. J. (1997). Screening and biochemical characterization of transthyretin variants in the Portuguese population. Hum. Mutat.9 (3), 226–233. doi: 10.1002/(SICI)1098-1004(1997)9:3<226::AID-HUMU3>3.0.CO;2-5
5
AnderssonK.OlofssonA.NielsenE. H.SvehagS. E.LundgrenE. (2002). Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem. Biophys. Res. Commun.294 (2), 309–314. doi: 10.1016/S0006-291X(02)00465-5
6
AnderssonR. (1976). Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden. Acta Med. Scand. Suppl.590, 1–64.
7
AndradeC. (1952). A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain75 (3), 408–427. doi: 10.1093/brain/75.3.408
8
AndroulakiA.GiaslakiotisK.GiakoumiX.AessoposA.LazarisA. C. (2007). Localized Castleman’s disease associated with systemic AA amyloidosis. Regression of amyloid deposits after tumor removal. Ann. Hematol.86 (1), 55–57. doi: 10.1007/s00277-006-0187-0
9
ArakiS.MawatariS.OhtaM.NakajimaA.KuroiwaY. (1968). Polyneuritic amyloidosis in a Japanese family. Arch. Neurol.18 (6), 593–602. doi: 10.1001/archneur.1968.00470360015001
10
ArakiS. (1984). Type I familial amyloidotic polyneuropathy (Japanese type). Brain Dev.6 (2), 128–133. doi: 10.1016/s0387-7604(84)80061-3
11
Aus dem SiepenF.HeinS.PrestelS.BaumgartnerC.SchonlandS.HegenbartU.et al. (2019). Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin. Res. Cardiol.108 (12), 1324–1330. doi: 10.1007/s00392-019-01467-1
12
BensonM. D.BuxbaumJ. N.EisenbergD. S.MerliniG.SaraivaM. J. M.SekijimaY.et al. (2018a). Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid25 (4), 215–219. doi: 10.1080/13506129.2018.1549825
13
BensonM. D.Waddington-CruzM.BerkJ. L.PolydefkisM.DyckP. J.WangA. K.et al. (2018b). Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med.379 (1), 22–31. doi: 10.1056/NEJMoa1716793
14
BerkJ. L.SuhrO. B.ObiciL.SekijimaY.ZeldenrustS. R.YamashitaT.et al. (2013). Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA310 (24), 2658–2667. doi: 10.1001/jama.2013.283815
15
BochtlerT.HegenbartU.KunzC.BennerA.SeckingerA.DietrichS.et al. (2014). Gain of chromosome 1q21 is an independent adverse prognostic factor in light chain amyloidosis patients treated with melphalan/dexamethasone. Amyloid21 (1), 9–17. doi: 10.3109/13506129.2013.854766
16
BochtlerT.HegenbartU.KunzC.GranzowM.BennerA.SeckingerA.et al. (2015). Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J. Clin. Oncol.33 (12), 1371–1378. doi: 10.1200/JCO.2014.57.4947
17
BruceM. E.McConnellI.WillR. G.IronsideJ. W. (2001). Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet358 (9277), 208–209. doi: 10.1016/s0140-6736(01)05411-3
18
BulawaC. E.ConnellyS.DevitM.WangL.WeigelC.FlemingJ. A.et al. (2012). Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A.109 (24), 9629–9634. doi: 10.1073/pnas.1121005109
19
CanepaM.TiniG.MusumeciB.CappelliF.MilandriA.MussinelliR.et al. (2019). Real-world versus trial patients with transthyretin amyloid cardiomyopathy. Eur. J. Heart Fail.21 (11), 1479–1481. doi: 10.1002/ejhf.1563
20
CapellariS.BaiardiS.RinaldiR.Bartoletti-StellaA.GrazianoC.PirasS.et al. (2018). Two novel PRNP truncating mutations broaden the spectrum of prion amyloidosis. Ann. Clin. Transl. Neurol.5 (6), 777–783. doi: 10.1002/acn3.568
21
CardosoI.MartinsD.RibeiroT.MerliniG.SaraivaM. J. (2010). Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J. Transl. Med.8, 74. doi: 10.1186/1479-5876-8-74
22
CarvalhoA.RochaA.LobatoL. (2015). Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl.21 (3), 282–292. doi: 10.1002/lt.24058
23
CastanoA.NarotskyD. L.HamidN.KhaliqueO. K.MorgensternR.DeLucaA.et al. (2017). Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J.38 (38), 2879–2887. doi: 10.1093/eurheartj/ehx350
24
CavalcanteJ. L.RijalS.AbdelkarimI.AlthouseA. D.SharbaughM. S.FridmanY.et al. (2017). Cardiac amyloidosis is prevalent in older patients with aortic stenosis and carries worse prognosis. J. Cardiovasc. Magn. Reson.19 (1), 98. doi: 10.1186/s12968-017-0415-x
25
ChitiF.DobsonC. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem.75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
26
CoelhoT.da Gloria CarvalhoM.SaraivaM.AlvesC.AlmeidaM.CostaP.et al. (1993). A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET. J. Rheumatol.20, 179.
27
CoelhoT.MaiaL. F.Martins da SilvaA.Waddington CruzM.Plante-BordeneuveV.LozeronP.et al. (2012). Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology79 (8), 785–792. doi: 10.1212/WNL.0b013e3182661eb1
28
CoelhoT.AdamsD.SilvaA.LozeronP.HawkinsP. N.MantT.et al. (2013a). Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med.369 (9), 819–829. doi: 10.1056/NEJMoa1208760
29
CoelhoT.MaiaL. F.da SilvaA. M.CruzM. W.Plante-BordeneuveV.SuhrO. B.et al. (2013b). Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol.260 (11), 2802–2814. doi: 10.1007/s00415-013-7051-7
30
ComenzoR. L.VosburghE.SimmsR. W.BergethonP.SarnackiD.FinnK.et al. (1996). Dose-intensive melphalan with blood stem cell support for the treatment of AL amyloidosis: one-year follow-up in five patients. Blood88 (7), 2801–2806. doi: 10.1182/blood.V88.7.2801.bloodjournal8872801
31
ConnorsL. H.LimA.ProkaevaT.RoskensV. A.CostelloC. E. (2003). Tabulation of human transthyretin (TTR) variants 2003. Amyloid10 (3), 160–184. doi: 10.3109/13506120308998998
32
CorazzaA.VeronaG.WaudbyC. A.MangioneP. P.BinghamR.UingsI.et al. (2019). Binding of Monovalent and Bivalent Ligands by Transthyretin Causes Different Short- and Long-Distance Conformational Changes. J. Med. Chem.62 (17), 8274–8283. doi: 10.1021/acs.jmedchem.9b01037
33
CornwellG.MurdochW. L.KyleR. A.WestermarkP.PitkanenP. (1983). Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med.75 (4), 618–623. doi: 10.1016/0002-9343(83)90443-6
34
CorteseA.VitaG.LuigettiM.RussoM.BisogniG.SabatelliM.et al. (2016). Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J. Neurol.263 (5), 916–924. doi: 10.1007/s00415-016-8064-9
35
DamyT.JudgeD. P.KristenA. V.BerthetK.LiH.AartsJ. (2015). Cardiac findings and events observed in an open-label clinical trial of tafamidis in patients with non-Val30Met and non-Val122Ile hereditary transthyretin amyloidosis. J. Cardiovasc. Transl. Res.8 (2), 117–127. doi: 10.1007/s12265-015-9613-9
36
De StrooperB.IwatsuboT.WolfeM. S. (2012). Presenilins and gamma-secretase: structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med.2 (1), a006304. doi: 10.1101/cshperspect.a006304
37
DemberL. M.SanchorawalaV.SeldinD. C.WrightD. G.LaValleyM.BerkJ. L.et al. (2001). Effect of dose-intensive intravenous melphalan and autologous blood stem-cell transplantation on al amyloidosis-associated renal disease. Ann. Intern. Med.134 (9 Pt 1), 746–753. doi: 10.7326/0003-4819-134-9_part_1-200105010-00011
38
DiasA.SantosD.CoelhoT.Alves-FerreiraM.SequeirosJ.AlonsoI.et al. (2019). C1QA and C1QC modify age-at-onset in familial amyloid polyneuropathy patients. Ann. Clin. Transl. Neurol.6 (4), 748–754. doi: 10.1002/acn3.748
39
DispenzieriA.LacyM. Q.KatzmannJ. A.RajkumarS. V.AbrahamR. S.HaymanS. R.et al. (2006). Absolute values of immunoglobulin free light chains are prognostic in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood107 (8), 3378–3383. doi: 10.1182/blood-2005-07-2922
40
DittrichT.BochtlerT.KimmichC.BeckerN.JauchA.GoldschmidtH.et al. (2017). AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis. Blood130 (5), 632–642. doi: 10.1182/blood-2017-02-767475
41
EdwardsC. V.BhutaniD.MaparaM.RadhakrishnanJ.ShamesS.MaurerM. S.et al. (2019). One year follow up analysis of the phase 1a/b study of chimeric fibril-reactive monoclonal antibody 11-1F4 in patients with AL amyloidosis. Amyloid26 (sup1), 115–116. doi: 10.1080/13506129.2019.1584892
42
GamezJ.SalvadoM.ReigN.SuneP.CasasnovasC.Rojas-GarciaR.et al. (2019). Transthyretin stabilization activity of the catechol-O-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: proof-of-concept study(). Amyloid26 (2), 74–84. doi: 10.1080/13506129.2019.1597702
43
GertzM. A.BensonM. D.DyckP. J.GroganM.CoelhoT.CruzM.et al. (2015). Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J. Am. Coll. Cardiol.66 (21), 2451–2466. doi: 10.1016/j.jacc.2015.09.075
44
GertzM. A.LandauH.ComenzoR. L.SeldinD.WeissB.ZonderJ.et al. (2016). First-in-Human Phase I/II Study of NEOD001 in Patients With Light Chain Amyloidosis and Persistent Organ Dysfunction. J. Clin. Oncol.34 (10), 1097–1103. doi: 10.1200/JCO.2015.63.6530
45
GhilardiG.StussiG.MazzucchelliL.RockenC.RossiD.GerberB. (2019). Venetoclax plus daratumumab induce hematological CR and organ response in an AL amyloidosis patient with t(11;14). Amyloid26 (3), 173–174. doi: 10.1080/13506129.2019.1615428
46
GillmoreJ. D.MaurerM. S.FalkR. H.MerliniG.DamyT.DispenzieriA.et al. (2016). Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation133 (24), 2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612
47
Gonzalez-LopezE.GagliardiC.DominguezF.QuartaC. C.de Haro-Del MoralF. J.MilandriA.et al. (2017). Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur. Heart J.38 (24), 1895–1904. doi: 10.1093/eurheartj/ehx043
48
HammarstromP.SchneiderF.KellyJ. W. (2001). Trans-suppression of misfolding in an amyloid disease. Science293 (5539), 2459–2462. doi: 10.1126/science.1062245
49
HammarstromP.WisemanR. L.PowersE. T.KellyJ. W. (2003). Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science299 (5607), 713–716. doi: 10.1126/science.1079589
50
HartleyD. M.WalshD. M.YeC. P.DiehlT.VasquezS.VassilevP. M.et al. (1999). Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci.19 (20), 8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999
51
HawkinsP. N.PepysM. B. (1990). A primed state exists in vivo following histological regression of amyloidosis. Clin. Exp. Immunol.81 (2), 325–328. doi: 10.1111/j.1365-2249.1990.tb03339.x
52
HoshinoJ.YamagataK.NishiS.NakaiS.MasakaneI.IsekiK.et al. (2016). Significance of the decreased risk of dialysis-related amyloidosis now proven by results from Japanese nationwide surveys in 1998 and 2010. Nephrol. Dial. Transplant.31 (4), 595–602. doi: 10.1093/ndt/gfv276
53
HwaY. L.KumarS. K.GertzM. A.LacyM. Q.BuadiF. K.KourelisT. V.et al. (2016). Induction therapy pre-autologous stem cell transplantation in immunoglobulin light chain amyloidosis: a retrospective evaluation. Am. J. Hematol.91 (10), 984–988. doi: 10.1002/ajh.24453
54
IshiiT.SekijimaY.AndoY. (2019). Patient profile with ATTR-FAP and evaluation of the safety and efficacy of tafamidis meglumine in Japan - interim analysis in post-marketing surveillance. Amyloid26 (sup1), 45–46. doi: 10.1080/13506129.2019.1582508
55
JadoulM.GarbarC.NoelH.SennesaelJ.VanholderR.BernaertP.et al. (1997). Histological prevalence of beta 2-microglobulin amyloidosis in hemodialysis: a prospective post-mortem study. Kidney Int.51 (6), 1928–1932. doi: 10.1038/ki.1997.262
56
JudgeD. P.HeitnerS. B.FalkR. H.MaurerM. S.ShahS. J.WittelesR. M.et al. (2019). Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol.74 (3), 285–295. doi: 10.1016/j.jacc.2019.03.012
57
KanekoS.YamagataK. (2018). Hemodialysis-related amyloidosis: Is it still relevant? Semin. Dial.31 (6), 612–618. doi: 10.1111/sdi.12720
58
KarlstedtE.Jimenez-ZepedaV.HowlettJ. G.WhiteJ. A.FineN. M. (2019). Clinical Experience With the Use of Doxycycline and Ursodeoxycholic Acid for the Treatment of Transthyretin Cardiac Amyloidosis. J. Card. Fail25 (3), 147–153. doi: 10.1016/j.cardfail.2019.01.006
59
KastritisE.KostopoulosI. V.TerposE.PaivaB.FotiouD.GavriatopoulouM.et al. (2018). Evaluation of minimal residual disease using next-generation flow cytometry in patients with AL amyloidosis. Blood Cancer J.8 (5), 46. doi: 10.1038/s41408-018-0086-3
60
Kluve-BeckermanB.HardwickJ.DuL.BensonM. D.MoniaB. P.WattA.et al. (2011). Antisense oligonucleotide suppression of serum amyloid A reduces amyloid deposition in mice with AA amyloidosis. Amyloid18 (3), 136–146. doi: 10.3109/13506129.2011.597464
61
KollmerJ.HundE.HornungB.HegenbartU.SchonlandS. O.KimmichC.et al. (2015). In vivo detection of nerve injury in familial amyloid polyneuropathy by magnetic resonance neurography. Brain138 (Pt 3), 549–562. doi: 10.1093/brain/awu344
62
KollmerJ.SahmF.HegenbartU.PurruckerJ. C.KimmichC.SchonlandS. O.et al. (2017). Sural nerve injury in familial amyloid polyneuropathy: MR neurography vs clinicopathologic tools. Neurology89 (5), 475–484. doi: 10.1212/WNL.0000000000004178
63
KolstoeS. E.MangioneP. P.BellottiV.TaylorG. W.TennentG. A.DerooS.et al. (2010). Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc. Natl. Acad. Sci. U.S.A.107 (47), 20483–20488. doi: 10.1073/pnas.1008255107
64
KourelisT. V.KumarS. K.GoR. S.KapoorP.KyleR. A.BuadiF. K.et al. (2014). Immunoglobulin light chain amyloidosis is diagnosed late in patients with preexisting plasma cell dyscrasias. Am. J. Hematol.89 (11), 1051–1054. doi: 10.1002/ajh.23827
65
KumarS.DispenzieriA.LacyM. Q.HaymanS. R.BuadiF. K.ColbyC.et al. (2012). Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol.30 (9), 989–995. doi: 10.1200/JCO.2011.38.5724
66
KumarS.KaufmanJ. L.GasparettoC.MikhaelJ.VijR.PegourieB.et al. (2017). Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood130 (22), 2401–2409. doi: 10.1182/blood-2017-06-788786
67
KumarS.MurrayD.DasariS.MilaniP.BarnidgeD.MaddenB.et al. (2019). Assay to rapidly screen for immunoglobulin light chain glycosylation: a potential path to earlier AL diagnosis for a subset of patients. Leukemia33 (1), 254–257. doi: 10.1038/s41375-018-0194-x
68
LachmannH. J.GilbertsonJ. A.GillmoreJ. D.HawkinsP. N.PepysM. B. (2002). Unicentric Castleman’s disease complicated by systemic AA amyloidosis: a curable disease. QJM95 (4), 211–218. doi: 10.1093/qjmed/95.4.211
69
LachmannH. J.GoodmanH. J.GilbertsonJ. A.GallimoreJ. R.SabinC. A.GillmoreJ. D.et al. (2007). Natural history and outcome in systemic AA amyloidosis. N. Engl. J. Med.356 (23), 2361–2371. doi: 10.1056/NEJMoa070265
70
LambertM. P.BarlowA. K.ChromyB. A.EdwardsC.FreedR.LiosatosM.et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A.95 (11), 6448–6453. doi: 10.1073/pnas.95.11.6448
71
LaneT.LoefflerJ. M.RowczenioD. M.GilbertsonJ. A.BybeeA.RussellT. L.et al. (2013). AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum.65 (4), 1116–1121. doi: 10.1002/art.37827
72
LaneT.PinneyJ. H.GilbertsonJ. A.HuttD. F.RowczenioD. M.MahmoodS.et al. (2017a). Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral centre. Amyloid24 (3), 162–166. doi: 10.1080/13506129.2017.1342235
73
LaneT.WechalekarA. D.GillmoreJ. D.HawkinsP. N.LachmannH. J. (2017b). Safety and efficacy of empirical interleukin-1 inhibition using anakinra in AA amyloidosis of uncertain aetiology. Amyloid24 (3), 189–193. doi: 10.1080/13506129.2017.1352503
74
LaneT.FontanaM.Martinez-NaharroA.QuartaC. C.WhelanC. J.PetrieA.et al. (2019). Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation140 (1), 16–26. doi: 10.1161/CIRCULATIONAHA.118.038169
75
LeungN.ThomeS. D.DispenzieriA. (2018). Venetoclax induced a complete response in a patient with immunoglobulin light chain amyloidosis plateaued on cyclophosphamide, bortezomib and dexamethasone. Haematologica103 (3), e135–e137. doi: 10.3324/haematol.2017.183749
76
LongJ. M.HoltzmanD. M. (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell179 (2), 312–339. doi: 10.1016/j.cell.2019.09.001
77
LundmarkK.Vahdat ShariatpanahiA.WestermarkG. T. (2013). Depletion of spleen macrophages delays AA amyloid development: a study performed in the rapid mouse model of AA amyloidosis. PloS One8 (11), e79104. doi: 10.1371/journal.pone.0079104
78
MallucciG.DickinsonA.LinehanJ.KlohnP. C.BrandnerS.CollingeJ. (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science302 (5646), 871–874. doi: 10.1126/science.1090187
79
MallucciG. R.WhiteM. D.FarmerM.DickinsonA.KhatunH.PowellA. D.et al. (2007). Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron53 (3), 325–335. doi: 10.1016/j.neuron.2007.01.005
80
MandreoliM.CasanovaS.VianelliN.PasqualiS.ZucchelliP. (2002). Remission of nephrotic syndrome due to AA amyloidosis and initiation of glomerular repair after surgical resection of localized Castleman’s disease. Nephron90 (3), 336–340. doi: 10.1159/000049070
81
ManwaniR.HegenbartU.MahmoodS.SachchithananthamS.KyriakouC.YongK.et al. (2018). Deferred autologous stem cell transplantation in systemic AL amyloidosis. Blood Cancer J.8 (11), 101. doi: 10.1038/s41408-018-0137-9
82
ManwaniR.CohenO.SharpleyF.MahmoodS.SachchithananthamS.FoardD.et al. (2019). A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood134 (25), 2271–2280. doi: 10.1182/blood.2019000834
83
MarcouxJ.MangioneP. P.PorcariR.DegiacomiM. T.VeronaG.TaylorG. W.et al. (2015). A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med.7 (10), 1337–1349. doi: 10.15252/emmm.201505357
84
MartinE. B.WilliamsA.WooliverC.HeidelR. E.AdamsS.DunlapJ.et al. (2017). Differential recruitment efficacy of patient-derived amyloidogenic and myeloma light chain proteins by synthetic fibrils-A metric for predicting amyloid propensity. PloS One12 (3), e0174152. doi: 10.1371/journal.pone.0174152
85
MartinE. B.WilliamsA. D.HeidelR. E.FosterJ. S.LandsR. H.KennelS. J.et al. (2018). A functional assay to identify amyloidogenic light chains. Amyloid25 (2), 93–100. doi: 10.1080/13506129.2018.1456425
86
MatsuzonoK.HondaH.SatoK.MoriharaR.DeguchiK.HishikawaN.et al. (2016). ‘PrP systemic deposition disease’: clinical and pathological characteristics of novel familial prion disease with 2-bp deletion in codon 178. Eur. J. Neurol.23 (1), 196–200. doi: 10.1111/ene.12905
87
MaurerM. S.SchwartzJ. H.GundapaneniB.ElliottP. M.MerliniG.Waddington-CruzM.et al. (2018). Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med.379 (11), 1007–1016. doi: 10.1056/NEJMoa1805689
88
McCauslandK. L.WhiteM. K.GuthrieS. D.QuockT.FinkelM.LousadaI.et al. (2018). Light Chain (AL) Amyloidosis: The Journey to Diagnosis. Patient11 (2), 207–216. doi: 10.1007/s40271-017-0273-5
89
MeadS.GandhiS.BeckJ.CaineD.GallujipaliD.CarswellC.et al. (2013). A novel prion disease associated with diarrhea and autonomic neuropathy. N. Engl. J. Med.369 (20), 1904–1914. doi: 10.1056/NEJMoa1214747
90
MerliniG.BellottiV. (2003). Molecular mechanisms of amyloidosis. N. Engl. J. Med.349 (6), 583–596. doi: 10.1056/NEJMra023144
91
MerliniG.PalladiniG. (2012). Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematol. Am. Soc. Hematol. Educ. Program2012, 595–603. doi: 10.1182/asheducation-2012.1.595
92
MerliniG.AscariE.AmboldiN.BellottiV.ArbustiniE.PerfettiV.et al. (1995). Interaction of the anthracycline 4’-iodo-4’-deoxydoxorubicin with amyloid fibrils: inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. U.S.A.92 (7), 2959–2963. doi: 10.1073/pnas.92.7.2959
93
MerliniG.Plante-BordeneuveV.JudgeD. P.SchmidtH.ObiciL.PerliniS.et al. (2013). Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J. Cardiovasc. Transl. Res.6 (6), 1011–1020. doi: 10.1007/s12265-013-9512-x
94
MerliniG.DispenzieriA.SanchorawalaV.SchonlandS. O.PalladiniG.HawkinsP. N.et al. (2018). Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers4 (1), 38. doi: 10.1038/s41572-018-0034-3
95
MikhaelJ. (2018). Turn off the Tap! The Need for Induction Therapy for AL Amyloidosis Before Transplantation. Biol. Blood Marrow Transplant.24 (11), e1–e2. doi: 10.1016/j.bbmt.2018.09.019
96
MilandriA.FarioliA.GagliardiC.LonghiS.SalviF.CurtiS.et al. (2020). Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur. J. Heart Fail.22 (3), 507–515. doi: 10.1002/ejhf.1742
97
MilaniP.BassetM.RussoF.FoliA.MerliniG.PalladiniG. (2017). Patients with light-chain amyloidosis and low free light-chain burden have distinct clinical features and outcome. Blood130 (5), 625–631. doi: 10.1182/blood-2017-02-767467
98
MillerM.PalA.AlbusairiW.JooH.PappasB.Haque TuhinM. T.et al. (2018). Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis. J. Med. Chem.61 (17), 7862–7876. doi: 10.1021/acs.jmedchem.8b00817
99
MinamisawaM.ClaggettB.AdamsD.KristenA. V.MerliniG.SlamaM. S.et al. (2019). Association of Patisiran, an RNA Interference Therapeutic, With Regional Left Ventricular Myocardial Strain in Hereditary Transthyretin Amyloidosis: The APOLLO Study. JAMA Cardiol.4 (5), 466–472. doi: 10.1001/jamacardio.2019.0849
100
MontagnaG.CazzulaniB.ObiciL.UggettiC.GiorgettiS.PorcariR.et al. (2013). Benefit of doxycycline treatment on articular disability caused by dialysis related amyloidosis. Amyloid20 (3), 173–178. doi: 10.3109/13506129.2013.803463
101
MonteiroC.MesgazardehJ. S.AnselmoJ.FernandesJ.NovaisM.RodriguesC.et al. (2019). Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight4 (12), e126526. doi: 10.1172/jci.insight.126526
102
MoreauP.Chanan-KhanA.RobertsA. W.AgarwalA. B.FaconT.KumarS.et al. (2017). Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood130 (22), 2392–2400. doi: 10.1182/blood-2017-06-788323
103
MorillaM. E.KocherJ.HarmatyM. (2009). Localized amyloidosis at the site of enfuvirtide injection. Ann. Intern. Med.151 (7), 515–516. doi: 10.7326/0003-4819-151-7-200910060-00017
104
MorrisA. D.SmithR. N.StoneJ. R. (2019). The pathology and changing epidemiology of dialysis-related cardiac beta-2 microglobulin amyloidosis. Cardiovasc. Pathol.42, 30–35. doi: 10.1016/j.carpath.2019.05.002
105
MuchtarE.DispenzieriA.KumarS. K.KetterlingR. P.DingliD.LacyM. Q.et al. (2017). Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia31 (7), 1562–1569. doi: 10.1038/leu.2016.369
106
MuchtarE.DispenzieriA.LeungN.LacyM. Q.BuadiF. K.DingliD.et al. (2018). Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia32 (10), 2240–2249. doi: 10.1038/s41375-018-0060-x
107
NakagawaM.SekijimaY.YazakiM.TojoK.YoshinagaT.DodenT.et al. (2016). Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid23 (1), 58–63. doi: 10.3109/13506129.2015.1135792
108
NuvoloneM.MerliniG. (2017a). Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis. Expert Opin. Ther. Targets21 (12), 1095–1110. doi: 10.1080/14728222.2017.1398235
109
NuvoloneM.MerliniG. (2017b). Systemic amyloidosis: novel therapies and role of biomarkers. Nephrol. Dial. Transplant.32 (5), 770–780. doi: 10.1093/ndt/gfw305
110
NuvoloneM.AguzziA.HeikenwalderM. (2009). Cells and prions: a license to replicate. FEBS Lett.583 (16), 2674–2684. doi: 10.1016/j.febslet.2009.06.014
111
NystromS. N.WestermarkG. T. (2012). AA-Amyloid is cleared by endogenous immunological mechanisms. Amyloid19 (3), 138–145. doi: 10.3109/13506129.2012.711391
112
ObiciL.MerliniG. (2012). Amyloidosis in autoinflammatory syndromes. Autoimmun. Rev.12 (1), 14–17. doi: 10.1016/j.autrev.2012.07.016
113
ObiciL.CorteseA.LozzaA.LucchettiJ.GobbiM.PalladiniG.et al. (2012). Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid19 Suppl 1, 34–36. doi: 10.3109/13506129.2012.678508
114
PalladiniG.LavatelliF.RussoP.PerliniS.PerfettiV.BosoniT.et al. (2006). Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood107 (10), 3854–3858. doi: 10.1182/blood-2005-11-4385
115
PalladiniG.DispenzieriA.GertzM. A.KumarS.WechalekarA.HawkinsP. N.et al. (2012). New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J. Clin. Oncol.30 (36), 4541–4549. doi: 10.1200/JCO.2011.37.7614
116
PalladiniG.HegenbartU.MilaniP.KimmichC.FoliA.HoA. D.et al. (2014). A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood124 (15), 2325–2332. doi: 10.1182/blood-2014-04-570010
117
PalladiniG.BassetM.MilaniP.VarettoniM.MangiacavalliS.RussoF.et al. (2017a). Biomarker-Based Screening of Organ Dysfunction in Patients with MGUS Allows Early Diagnosis of AL Amyloidosis. Blood130 (Suppl 1), 1760–1760. doi: 10.1182/blood.V130.Suppl_1.1760.1760
118
PalladiniG.RivaE.BassetM.RussoF.MilaniP.PasquinucciE.et al. (2017b). Prognostication of survival and progression to dialysis in AA amyloidosis. Amyloid24 (sup1), 136–137. doi: 10.1080/13506129.2017.1289917
119
PalladiniG.KastritisE.MaurerM. S.ZonderJ. A.MinnemaM. C.WechalekarA. D.et al. (2020). Daratumumab Plus CyBorD for Patients With Newly Diagnosed AL Amyloidosis: Safety Run-in Results of ANDROMEDA. Blood. doi: 10.1182/blood.2019004460
120
PapaR.LachmannH. J. (2018). Secondary, AA, Amyloidosis. Rheum. Dis. Clin. North Am.44 (4), 585–603. doi: 10.1016/j.rdc.2018.06.004
121
PenchalaS. C.ConnellyS.WangY.ParkM. S.ZhaoL.BaranczakA.et al. (2013). AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. U.S.A.110 (24), 9992–9997. doi: 10.1073/pnas.1300761110
122
PerfettiV.BellottiV.MaggiA.ArbustiniE.De BenedettiF.PaulliM.et al. (1994). Reversal of nephrotic syndrome due to reactive amyloidosis (AA-type) after excision of localized Castleman’s disease. Am. J. Hematol.46 (3), 189–193. doi: 10.1002/ajh.2830460306
123
PiccoliG. B.HachemiM.MolfinoI.CoindreJ. P.BoursotC. (2017). Doxycycline treatment in dialysis related amyloidosis: discrepancy between antalgic effect and inflammation, studied with FDG-positron emission tomography: a case report. BMC Nephrol.18 (1), 285. doi: 10.1186/s12882-017-0698-z
124
PinneyJ. H.LachmannH. J.SattianayagamP. T.GibbsS. D.WechalekarA. D.VennerC. P.et al. (2013). Renal transplantation in systemic amyloidosis-importance of amyloid fibril type and precursor protein abundance. Am. J. Transplant.13 (2), 433–441. doi: 10.1111/j.1600-6143.2012.04326.x
125
Plante-BordeneuveV.GorramF.SalhiH.NordineT.AyacheS. S.Le CorvoisierP.et al. (2017). Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J. Neurol.264 (2), 268–276. doi: 10.1007/s00415-016-8337-3
126
PremkumarV.ComenzoR.LentzschS. (2019). Venetoclax in Immunoglobulin Light Chain Amyloidosis: Is This the Beginning or the End? Clin. Lymphoma Myeloma Leuk.19 (10), 686–688. doi: 10.1016/j.clml.2019.07.003
127
ProkaevaT.SpencerB.KautM.OzonoffA.DorosG.ConnorsL. H.et al. (2007). Soft tissue, joint, and bone manifestations of AL amyloidosis: clinical presentation, molecular features, and survival. Arthritis Rheum.56 (11), 3858–3868. doi: 10.1002/art.22959
128
PrusinerS. B. (2001). Shattuck lecture–neurodegenerative diseases and prions. N. Engl. J. Med.344 (20), 1516–1526. doi: 10.1056/NEJM200105173442006
129
QuartaC. C.BuxbaumJ. N.ShahA. M.FalkR. H.ClaggettB.KitzmanD. W.et al. (2015). The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med.372 (1), 21–29. doi: 10.1056/NEJMoa1404852
130
RapezziC.MerliniG.QuartaC. C.RivaL.LonghiS.LeoneO.et al. (2009). Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation120 (13), 1203–1212. doi: 10.1161/CIRCULATIONAHA.108.843334
131
RapezziC.QuartaC. C.RivaL.LonghiS.GallelliI.LorenziniM.et al. (2010). Transthyretin-related amyloidoses and the heart: a clinical overview. Nat. Rev. Cardiol.7 (7), 398–408. doi: 10.1038/nrcardio.2010.67
132
RavichandranS.LachmannH. J.WechalekarA. D. (2020). Epidemiologic and Survival Trends in Amyloidosis 1987-2019. N. Engl. J. Med.382 (16), 1567–1568. doi: 10.1056/NEJMc1917321
133
RazaviH.PalaninathanS. K.PowersE. T.WisemanR. L.PurkeyH. E.MohamedmohaideenN. N.et al. (2003). Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew. Chem. Int. Ed. Engl.42 (24), 2758–2761. doi: 10.1002/anie.200351179
134
RichardsD. B.CooksonL. M.BergesA. C.BartonS. V.LaneT.RitterJ. M.et al. (2015). Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N. Engl. J. Med.373 (12), 1106–1114. doi: 10.1056/NEJMoa1504942
135
RichardsD. B.CooksonL. M.BartonS. V.LiefaardL.LaneT.HuttD. F.et al. (2018). Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci. Transl. Med.10 (422), eaan3128. doi: 10.1126/scitranslmed.aan3128
136
RowczenioD.QuartaC. C.FontanaM.WhelanC. J.Martinez-NaharroA.TrojerH.et al. (2019). Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum. Mutat.40 (1), 90–96. doi: 10.1002/humu.23669
137
RubergF. L.GroganM.HannaM.KellyJ. W.MaurerM. S. (2019). Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol.73 (22), 2872–2891. doi: 10.1016/j.jacc.2019.04.003
138
RubinJ.AlvarezJ.TeruyaS.CastanoA.LehmanR. A.WeidenbaumM.et al. (2017). Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: can we identify affected patients earlier? Amyloid24 (4), 226–230. doi: 10.1080/13506129.2017.1375908
139
SanchorawalaV.SunF.QuillenK.SloanJ. M.BerkJ. L.SeldinD. C. (2015). Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood126 (20), 2345–2347. doi: 10.1182/blood-2015-08-662726
140
Sant’AnnaR.GallegoP.RobinsonL. Z.Pereira-HenriquesA.FerreiraN.PinheiroF.et al. (2016). Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun.7, 10787. doi: 10.1038/ncomms10787
141
SantosD.CoelhoT.Alves-FerreiraM.SequeirosJ.MendoncaD.AlonsoI.et al. (2016). Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). Eur. J. Hum. Genet.24 (5), 756–760. doi: 10.1038/ejhg.2015.180
142
SantosD.SantosM. J.Alves-FerreiraM.CoelhoT.SequeirosJ.AlonsoI.et al. (2018). mtDNA copy number associated with age of onset in familial amyloid polyneuropathy. J. Neurol. Neurosurg. Psychiatry89 (3), 300–304. doi: 10.1136/jnnp-2017-316657
143
SantosD.CoelhoT.Alves-FerreiraM.SequeirosJ.MendoncaD.AlonsoI.et al. (2019). Large normal alleles of ATXN2 decrease age at onset in transthyretin familial amyloid polyneuropathy Val30Met patients. Ann. Neurol.85 (2), 251–258. doi: 10.1002/ana.25409
144
SaraivaM. J. (2002). Hereditary transthyretin amyloidosis: molecular basis and therapeutical strategies. Expert Rev. Mol. Med.4 (12), 1–11. doi: 10.1017/S1462399402004647
145
ScarpioniR.RicardiM.AlbertazziV.De AmicisS.RastelliF.ZerbiniL. (2016). Dialysis-related amyloidosis: challenges and solutions. Int. J. Nephrol. Renovasc. Dis.9, 319–328. doi: 10.2147/IJNRD.S84784
146
ScharmanC. D.ShatzelJ. J.MaziarzR. T.MedvedovaE.StentzA.RhyneG.et al. (2017). Autologous Stem Cell Transplant in the Era of Bortezomib-Based Induction for AL Amyloidosis Results in Improved Hematologic Response Rates: a Single Institution 11 Year Experience. Blood130 (Suppl 1), 4552–4552. doi: 10.1182/blood.V130.Suppl_1.4552.4552
147
SchifflH. (2014). Impact of advanced dialysis technology on the prevalence of dialysis-related amyloidosis in long-term maintenance dialysis patients. Hemodial. Int.18 (1), 136–141. doi: 10.1111/hdi.12057
148
ScullyP. R.TreibelT. A.FontanaM.LloydG.MullenM.PuglieseF.et al. (2018). Prevalence of Cardiac Amyloidosis in Patients Referred for Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol.71 (4), 463–464. doi: 10.1016/j.jacc.2017.11.037
149
ScullyP. R.PatelK. P.TreibelT. A.ThorntonG. D.HughesR. K.ChadalavadaS.et al. (2020). Prevalence and outcome of dual aortic stenosis and cardiac amyloid pathology in patients referred for transcatheter aortic valve implantation. Eur. Heart J. doi: 10.1093/eurheartj/ehaa170
150
SekijimaY.DendleM. A.KellyJ. W. (2006). Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid13 (4), 236–249. doi: 10.1080/13506120600960882
151
SelkoeD. J.HardyJ. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med.8 (6), 595–608. doi: 10.15252/emmm.201606210
152
SidanaS.TandonN.DispenzieriA.GertzM. A.BuadiF. K.LacyM. Q.et al. (2018). Clinical presentation and outcomes in light chain amyloidosis patients with non-evaluable serum free light chains. Leukemia32 (3), 729–735. doi: 10.1038/leu.2017.286
153
SidanaS.SidiqiM. H.DispenzieriA.BuadiF. K.LacyM. Q.MuchtarE.et al. (2019). Fifteen year overall survival rates after autologous stem cell transplantation for AL amyloidosis. Am. J. Hematol.94 (9), 1020–1026. doi: 10.1002/ajh.25566
154
SidanaS.MuchtarE.SidiqiM. H.JevremovicD.DispenzieriA.GonsalvesW.et al. (2020). Impact of minimal residual negativity using next generation flow cytometry on outcomes in light chain amyloidosis. Am. J. Hematol.95 (5), 497–502. doi: 10.1002/ajh.25746
155
SidiqiM. H.AljamaM. A.BuadiF. K.WarsameR. M.LacyM. Q.DispenzieriA.et al. (2018). Stem Cell Transplantation for Light Chain Amyloidosis: Decreased Early Mortality Over Time. J. Clin. Oncol.36 (13), 1323–1329. doi: 10.1200/JCO.2017.76.9554
156
SimonsJ. P.Al-ShawiR.EllmerichS.SpeckI.AslamS.HutchinsonW. L.et al. (2013). Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. U.S.A.110 (40), 16115–16120. doi: 10.1073/pnas.1306621110
157
SolomonS. D.AdamsD.KristenA.GroganM.Gonzalez-DuarteA.MaurerM. S.et al. (2019). Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation139 (4), 431–443. doi: 10.1161/CIRCULATIONAHA.118.035831
158
SousaM. M.CardosoI.FernandesR.GuimaraesA.SaraivaM. J. (2001). Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol.159 (6), 1993–2000. doi: 10.1016/s0002-9440(10)63050-7
159
SperryB. W.ReyesB. A.IkramA.DonnellyJ. P.PhelanD.JaberW. A.et al. (2018). Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J. Am. Coll. Cardiol.72 (17), 2040–2050. doi: 10.1016/j.jacc.2018.07.092
160
SponarovaJ.NuvoloneM.WhicherC.FreiN.KanaV.SchwarzP.et al. (2013). Efficient amyloid A clearance in the absence of immunoglobulins and complement factors. Am. J. Pathol.182 (4), 1297–1307. doi: 10.1016/j.ajpath.2012.12.035
161
StaronA.BurksE. J.LeeJ. C.SarosiekS.SloanJ. M.SanchorawalaV. (2020). Assessment of minimal residual disease using multiparametric flow cytometry in patients with AL amyloidosis. Blood Adv.4 (5), 880–884. doi: 10.1182/bloodadvances.2019001331
162
StevensF. J. (2000). Four structural risk factors identify most fibril-forming kappa light chains. Amyloid7 (3), 200–211. doi: 10.3109/13506120009146835
163
StorkelS.SchneiderH. M.MunteferingH.KashiwagiS. (1983). Iatrogenic, insulin-dependent, local amyloidosis. Lab. Invest.48 (1), 108–111.
164
TanskanenM.PeuralinnaT.PolvikoskiT.NotkolaI. L.SulkavaR.HardyJ.et al. (2008). Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann. Med.40 (3), 232–239. doi: 10.1080/07853890701842988
165
UedaM.HoribataY.ShonoM.MisumiY.OshimaT.SuY.et al. (2011). Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod. Pathol.24 (12), 1533–1544. doi: 10.1038/modpathol.2011.117
166
VallabhS. M.NobuharaC. K.LlorensF.ZerrI.ParchiP.CapellariS.et al. (2019). Prion protein quantification in human cerebrospinal fluid as a tool for prion disease drug development. Proc. Natl. Acad. Sci. U.S.A.116 (16), 7793–7798. doi: 10.1073/pnas.1901947116
167
VerbruggheW.MaesB. D.KnockaertD. C. (2005). Localised plasma-cell type Castleman’s disease and AA-amyloidosis cured by resection. A case report and review of the literature. Acta Clin. Belg.60 (1), 22–27. doi: 10.1179/acb.2005.006
168
WaldenstromH. (1928). On the formation and disappearance of amyloid in man. Acta Chir. Scand.63, 479–530.
169
WalshD. M.KlyubinI.FadeevaJ. V.CullenW. K.AnwylR.WolfeM. S.et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature416 (6880), 535–539. doi: 10.1038/416535a
170
WechalekarA. D.WhelanC. (2017). Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J.7 (3), e546. doi: 10.1038/bcj.2017.26
171
WechalekarA. D.SchonlandS. O.KastritisE.GillmoreJ. D.DimopoulosM. A.LaneT.et al. (2013). A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood121 (17), 3420–3427. doi: 10.1182/blood-2012-12-473066
172
WestermarkG. T.WestermarkP. (2013). Islet amyloid polypeptide and diabetes. Curr. Protein Pept. Sci.14 (4), 330–337. doi: 10.2174/13892037113149990050
173
WestermarkG. T.FandrichM.WestermarkP. (2015). AA amyloidosis: pathogenesis and targeted therapy. Annu. Rev. Pathol.10, 321–344. doi: 10.1146/annurev-pathol-020712-163913
174
WillR. G.IronsideJ. W.ZeidlerM.CousensS. N.EstibeiroK.AlperovitchA.et al. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet347 (9006), 921–925. doi: 10.1016/s0140-6736(96)91412-9
175
Zegri-ReirizI.de Haro-Del MoralF. J.DominguezF.SalasC.de la CuadraP.PlazaA.et al. (2019). Prevalence of Cardiac Amyloidosis in Patients with Carpal Tunnel Syndrome. J. Cardiovasc. Transl. Res.12 (6), 507–513. doi: 10.1007/s12265-019-09895-0
176
ZhouP.KugelmassA.ToskicD.GodaraA.FogarenT.VargaC.et al. (2019). Seeking light-chain amyloidosis very early: The SAVE trial—identifying clonal lambda light chain genes in patients with MGUS or smoldering multiple myeloma. J. Clin. Oncol.37 (15_suppl), 8010–8010. doi: 10.1200/JCO.2019.37.15_suppl.8010
177
ZingraffJ. J.NoelL. H.BardinT.AtienzaC.ZinsB.DruekeT. B.et al. (1990). Beta 2-microglobulin amyloidosis in chronic renal failure. N. Engl. J. Med.323 (15), 1070–1071. doi: 10.1056/NEJM199010113231514
Summary
Keywords
protein misfolding (conformational) diseases, AL amyloidosis, ATTR amyloidosis, AA amyloidosis, amyloidosis, early intervention
Citation
Nevone A, Merlini G and Nuvolone M (2020) Treating Protein Misfolding Diseases: Therapeutic Successes Against Systemic Amyloidoses. Front. Pharmacol. 11:1024. doi: 10.3389/fphar.2020.01024
Received
29 April 2020
Accepted
24 June 2020
Published
10 July 2020
Volume
11 - 2020
Edited by
Cesare Mancuso, Catholic University of the Sacred Heart, Italy
Reviewed by
Stefania Schiavone, University of Foggia, Italy; Ines Moreno-Gonzalez, University of Malaga, Spain
Updates
Copyright
© 2020 Nevone, Merlini and Nuvolone.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Nuvolone, mario.nuvolone@unipv.it
This article was submitted to Experimental Pharmacology and Drug Discovery, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.