 Liliya N. Kirpotina1†
Liliya N. Kirpotina1† Igor A. Schepetkin1†
Igor A. Schepetkin1† Deepa Hammaker2Amanda Kuhs2
Deepa Hammaker2Amanda Kuhs2 Andrei I. Khlebnikov3,4
Andrei I. Khlebnikov3,4 Mark T. Quinn1*
Mark T. Quinn1*- 1Department of Microbiology and Immunology, Montana State University, Bozeman, MT, United States
- 2Division of Rheumatology, Allergy, and Immunology, School of Medicine, University of California, San Diego, La Jolla, CA, United States
- 3Kizhner Research Center, Tomsk Polytechnic University, Tomsk, Russia
- 4Research Institute of Biological Medicine, Altai State University, Barnaul, Russia
Rheumatoid arthritis (RA) is a chronic autoimmune disease involving joint and bone damage that is mediated in part by proteases and cytokines produced by synovial macrophages and fibroblast-like synoviocytes (FLS). Although current biological therapeutic strategies for RA have been effective in many cases, new classes of therapeutics are needed. We investigated anti-inflammatory properties of the natural alkaloid tryptanthrin (TRYP) and its synthetic derivative tryptanthrin-6-oxime (TRYP-Ox). Both TRYP and TRYP-Ox inhibited matrix metalloproteinase (MMP)-3 gene expression in interleukin (IL)-1β-stimulated primary human FLS, as well as IL-1β–induced secretion of MMP-1/3 by FLS and synovial SW982 cells and IL-6 by FLS, SW982 cells, human umbilical vein endothelial cells (HUVECs), and monocytic THP-1 cells, although TRYP-Ox was generally more effective and had no cytotoxicity in vitro. Evaluation of the therapeutic potential of TRYP and TRYP-Ox in vivo in murine arthritis models showed that both compounds significantly attenuated the development of collagen-induced arthritis (CIA) and collagen-antibody–induced arthritis (CAIA), with comparable efficacy. Collagen II (CII)-specific antibody levels were similarly reduced in TRYP- and TRYP-Ox-treated CIA mice. TRYP and TRYP-Ox also suppressed proinflammatory cytokine production by lymph node cells from CIA mice, with TRYP-Ox being more effective in inhibiting IL-17A, granulocyte-macrophage colony-stimulating factor (GM-CSF), and receptor activator of nuclear factor-κB ligand (RANKL). Thus, even though TRYP-Ox generally had a better in vitro profile, possibly due to its ability to inhibit c-Jun N-terminal kinase (JNK), both TRYP and TRYP-Ox were equally effective in inhibiting the clinical symptoms and damage associated with RA. Overall, TRYP and/or TRYP-Ox may represent potential new directions for the pursuit of novel treatments for RA.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease that involves inflammation and progressive damage to distal joints, as well as inflammation and injury to other organs of the body (Firestein, 2003; Sharif et al., 2018). Thus, optimal therapeutic approaches should be developed to prevent inflammation, immune system dysregulation, and bone destruction associated with this disease, while still exhibiting enhanced safety and efficacy. Natural compounds have been considered as potential alternative or complementary treatments, as these compounds have been shown to possess a broader diversity in chemical space and, as a result, have significantly impacted drug development for many diseases (Hong, 2011). Indeed, many natural products have been shown to exhibit potential for treatment of inflammatory diseases (Lu et al., 2015) and have been evaluated in pre-clinical and clinical trials. For example, triptolide and its derivatives have been evaluated for their therapeutic effects in RA (Han et al., 2012; Tang and Zuo, 2012). Likewise, the plant-derived drug paclitaxel has been shown to inhibit collagen-induced arthritis (CIA) in mice (Xu et al., 2019).
Tryptanthrin (TRYP) (indolo[2,1-b]quinazolin-6,12-dione) is a well-known alkaloid and antibiotic that can be isolated from Candida lypolica (Brufani et al., 1971), higher plants (Bergman et al., 1985), and several species of marine micro- and macroorganisms [for review (Agafonova and Moskovkina, 2018)]. This compound has various pharmacological properties, including anti-inflammatory (Recio et al., 2006; Iwaki et al., 2011; Pathania et al., 2014), antimicrobial (Honda et al., 1979), antiviral (Tsai et al., 2020), and anti-tumor activities (Kimoto et al., 2001; Liao and Leung, 2013). For example, TRYP has been reported to reduce leukotriene-formation in human neutrophils and rat pleural exudates (Pergola et al., 2012). Likewise, TRYP was found to be effective in protecting mice against experimentally-induced colitis via regulation of the tumor necrosis factor (TNF)/nuclear factor (NF)-κB and interleukin (IL)-6/signal transducer and activator of transcription 3 (STAT3) signaling pathways (Wang et al., 2018). Although there are no reported studies regarding the effects of TRYP on RA, the signaling pathways impacted by TRYP clearly play roles in RA pathogenesis [e.g., see (Lubberts, 2015; Mitchell and Carmody, 2018)]. Thus, we hypothesized that TRYP or its structural analogs might be effective treatments for RA.
Structural modification of natural compounds can increase compound potency and selectivity, enhance their pharmacological properties, and significantly diminish their detrimental effects (Guo, 2017). Several TRYP derivatives with various tetracyclic scaffold modifications have been developed, including compounds with anti-plasmodium and anti-toxoplasma properties (Krivogorsky et al., 2008; Onambele et al., 2015), indoleamine 2,3-dioxygenase inhibitors (Yang et al., 2013), and DNA triplex stabilizing agents (Chen et al., 2007). Recently, we found that tryptanthrin-6-oxime (TRYP-Ox) had high affinity for JNK1-3 and also blocked activation of NF-κB/AP-1 and the production of IL-6 by lipopolysaccharide-treated monocytic cells (Schepetkin et al., 2019). Since JNK inhibition has potential for reducing inflammation associated with RA, it is reasonable that JNK inhibitors could be developed as RA therapeutics (Han et al., 2001; Bogoyevitch et al., 2010; Koch et al., 2015). Indeed, we found that 11H-indeno[1,2-b]quinoxalin-11-one oxime salt (IQ-1S) was an effective JNK inhibitor that blocked proinflammatory cytokine production and that IQ-1S treatment had a beneficial therapeutic effect in a mouse model of RA (Schepetkin et al., 2012; Schepetkin et al., 2015). Moreover, we also found that the indolo[2,1-b]quinazo-lin-6,12-dione (TRYP) structure was analogous to the 11H-indeno[1,2-b]quinoxalin-11-one scaffold (Schepetkin et al., 2019).
In the present study, we expanded our search for potential biological targets of TRYP and TRYP-Ox using a reverse pharmacophore mapping approach and again found that JNK1 and JNK3 were potential therapeutic targets of TRYP-Ox but not for TRYP. Furthermore, we evaluated anti-inflammatory effects of TRYP and TRYP-Ox in vitro and found that TRYP-Ox effectively inhibited IL-1β-induced IL-6 secretion by FLS, SW-982 synovial cells, and THP-1 monocytic cells, whereas TRYP was generally less effective. We also investigated the effect of these compounds in vivo using collagen-induced arthritis (CIA) and collagen-antibody-induced arthritis (CAIA) models of RA and found that TRYP-Ox significantly reduced the clinical symptoms and cartilage damage in CIA and CAIA. Surprisingly, TRYP was also effective in treating CIA and CAIA, although inhibition of cartilage destruction was more effective with TRYP-Ox. The therapeutic effects of TRYP and TRYP-Ox in CIA were associated with reduced levels of CII-specific antibodies and inhibition of proinflammatory cytokine production by lymph node (LN) cells. Overall, TRYP-Ox has a relatively greater therapeutic potential for treatment of RA compared to TRYP and represents a potential new direction for pursuit of novel treatments for RA.
Materials and Methods
Compounds
TRYP was purchased from Combi-Blocks (San Diego, CA, USA). TRYP-Ox and IQ-1S were synthesized, as described previously (Schepetkin et al., 2019). The JNK inhibitor SP600125 was from Tocris Bioscience (Ellisville, MO, USA). For in vitro studies, the compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted into the desired buffer or culture media. For in vivo treatments, the compounds were suspended in sterile phosphate buffer saline (PBS).
CIA Induction, Treatment, and Clinical Evaluation
DBA1/J male mice (6–8 weeks) were obtained from The Jackson Laboratories (Bar Harbor, ME, USA). All animal experiments were performed in accordance with National Institutes of Health guidelines and approved by the Montana State University Institutional Animal Care and Use Committee.
To induce CIA in DBA1/J mice, immunization-grade bovine Type II collagen (CII) (Chondrex, Redmond, WA, USA) was solubilized in 0.05 M acetic acid (2 mg/ml), and 100 μg of CII emulsified in complete Freund’s adjuvant containing 4 mg/ml Mycobacterium tuberculosis (Chondrex) were injected subcutaneously (s.c.) in the tail (Kochetkova et al., 2010). Using this method, nearly 100% of mice consistently showed clinical symptoms by day 25.
Suspensions of TRYP and TRYP-Ox or sterile saline were injected intraperitoneally (i.p.) at 30 mg/kg daily beginning on day 8 after the CII challenge, as indicated, and continued until day 42 post-CII challenge. Mice were scored using a scale of 0 to 3 for each limb, with a maximal total score of 12, as previously described (Schepetkin et al., 2015): 0, no signs of inflammation; 1, mild redness or swelling of single digits; 2, significant swelling of ankle or wrist with erythema; and 3, severe swelling and erythema of multiple joints.
CAIA Induction, Treatment, and Clinical Evaluation
CAIA was induced in 7- to 8-week-old BALB/c mice from Charles River Laboratories (Wilmington, MA, USA) by i.p. injection of a cocktail of monoclonal anti-CII antibodies (Chondrex; 1.5 mg/mouse) on day 0, followed by i.p. lipopolysaccharide (LPS) injection (30 μg LPS from Escherichia coli strain 0111:B4 in PBS) on day 3. Control animals received an equal volume of PBS. Suspensions of TRYP, TRYP-Ox, and IQ-1S (all compounds in dose 30 mg/kg) or sterile saline were injected i.p. daily beginning at day 1 after injection of anti-CII Abs and continued until day 9.
Arthritis development was evaluated daily for 10 days post-injection of the monoclonal antibody cocktail. Arthritis symptoms were scored on scale of 0 to 4 in accordance with the Chondrex Mouse Arthritis Scoring System in a blinded manner, as follows: 0, no signs of inflammation; 1, mild redness or swelling of single joint (one of the interphalangeal joints, metacarpophalangeal joints, carpal joint for the front paw, and tarsal joint for the hind); 2, two joints have redness and swelling; 3, all three joint types have redness and swelling; 4, maximal redness and swelling of entire paw leads to the disappearance of anatomical definition.
Histopathology
At 44 days after CII injection (CIA) or 14 days after anti-CII antibody injection (CAIA), the mice were euthanized, and their hind limbs were collected and fixed in 10% neutral buffered formalin. The limbs were decalcified in 5% formic acid for 3 to 6 days, and the joints were embedded in paraffin. Eight 8-μm sections were cut using a sagittal projection with 40 µm spacing of the cuts. Hematoxylin and eosin (H&E) and toluidine blue (TB) staining were performed for each sample.
H&E histopathological scores for the joints were determined by using a graded scale (Schramm et al., 2004), separately for cartilage destruction, pannus formation, and synovial cells changes. For cartilage destruction, the scale was: 0—normal, superficial zone is smooth; 1—superficial fibrillations, some loss of surface lamina; 2—vertical clefts/erosion to the calcified cartilage up to 25% of the surface; 3—vertical clefts/erosion to the calcified cartilage more than 25% of the surface, severe chondrocytes, and cartilage matrix loss with new bone tissue substitution, bone destruction. For pannus formation, the scale was: 0—normal; 1—exudate in the joint; 2—mild infiltration of the inflammatory cells; 3—heavy inflammatory infiltration and debris in the joint). For synovial changes, the scale was: 0—normal; 1—initial hyperplasia of the cells, with occasional infiltration of inflammatory cells; 2—focal infiltration of inflammatory cells into the synovial layers; 3—diffused infiltration of inflammatory cells into synovium. All sections of the hind paws and knee joints were examined, the highest score was recorded, with a total maximum score of 36 possible per mouse. The loss of the proteoglycans and cartilage degeneration was also scored in TB-stained sections on a graded scale of 0–3, as previously described (Bernotiene et al., 2004): 0—no cartilage loss; 1 – minimal chondrocytes and proteoglycan loss in superficial zone; 2—moderate chondrocytes and proteoglycan loss into middle zone but above tidemark; and 3—severe cartilage degeneration through tidemark, with the maximum score of 12 possible per mouse.
In CAIA, animals were sacrificed on day 14, and their forward and hind limbs were processed as described above. Three sections in the sagittal projection of joints, 40 µm apart, were examined by the same system with the grades ranging 0 to 4. Total H&E histopathological score for three sections included cartilage destruction, pannus formation, and synovial cells changes. For cartilage destruction, the scale was: 0—normal, superficial zone is smooth; 1—superficial fibrillations, some loss of surface lamina; 2—vertical clefts/erosion to the calcified cartilage up to 25% of the surface; 3—vertical clefts/erosion to the calcified cartilage 25% to 50% of the surface, 4—vertical clefts/erosion to the calcified cartilage more than 50% of the surface, severe chondrocytes, and cartilage matrix loss with new bone tissue substitution. For pannus formation the scale was: 0—normal; 1—exudate in the joint; 2—single inflammatory cells in the exudate; 3—mild infiltration of the inflammatory cells; 4—heavy inflammatory infiltration and debris in the joint. For synovial membrane changes the scale was: 0 – normal, 1 to 2 layer of synovial lining cells; 1—initial hyperplasia of the cells and increased number of lining cells layers; 2—increased number of lining cell layers, more than 3 to 4 layers and/or proliferation of sub-synovial tissue; 3—more than 4 layers of lining cells, focal infiltration of inflammatory cells, proliferation of subsynovial tissue; 4—more than four layers of lining cells, diffused infiltration of inflammatory cells, proliferation of sub-synovial tissue.
Anti-CII Antibody Enzyme-Linked Immunosorbent Assay (ELISA)
Serum samples from mice with CIA were collected on day 44 after CII injection. Samples of the diluted sera were added to microtiter plates (Greiner Bio-One, Monroe, NC, USA) coated with 2 μg/ml ELISA grade mouse CII (Chondrex). Horseradish peroxidase-conjugated goat anti-bovine total IgG, IgG1, IgG2a, IgG2b, and IgG3 (Southern Biotechnology Associates, Birmingham, AL, USA) were used for detection. The samples were developed with 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (Moss Inc., Pasadena, MD, USA), and absorbance was monitored at 415 nm using a SpectraMax Plus microplate reader. Endpoint titer represents reciprocal log 2 for the serum dilution with absorbance ≥ 0.1 above negative control.
Determination of Cytokines in LN Supernatants
At the end of the study (day 44), we isolated the axillary, inguinal, and popliteal LN and purified mononuclear cells from the LN. These cells were cultured in 6-well plates (5 × 106 cells/ml) without or with 50 μg/ml CII for 3 days in RPMI-1640 medium supplemented with 10% of fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids (Kochetkova et al., 2008). The levels of cytokines, including tumor necrosis factor (TNF), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1β, IL-5, IL-6, IL-10, IL-17A, and receptor activator of nuclear factor-κB ligand (RANKL) were measured in LN culture supernatants using ELISA kits (BD Biosciences, San Jose, CA, USA) for mouse cytokines.
Cell Lines
The SW982 human synovial cell line was obtained from the America Type Culture Collection (ATCC, Rockville, MD, USA). SW982 cells were maintained in ATCC-formulated Leibovitz’s L-15 Medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Human monocytic THP-1 cells (ATCC) were cultured in RPMI-1640 medium (Mediatech Inc., Herndon, VA, USA) supplemented with 10% (v/v) FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Human umbilical vein endothelial cells (HUVECs) (ATCC, Rockville, MD, USA) were cultured in vascular cell basal medium (ATCC) supplemented with components from the endothelial cell growth kit-VEGF (ATCC): 2% FBS, 10 mM L-glutamine, 5 ng/ml recombinant human epidermal growth factor (rh EGF), 5 ng/ml recombinant human vascular endothelial growth factor (rh VEGF), 15 ng/ml recombinant human insulin-like growth factor (rh IGF-1), 1 μg/ml hydrocortisone hemisuccinate, 0.75 U/ml heparin sulfate, 50 μg/ml ascorbic acid, 100 U/ml penicillin, and 100 μg/ml streptomycin according to the manufacturer’s specifications. All cell cultures were maintained in polystyrene tissue culture flasks at 37°C and 5% CO2.
Isolation and Culture of Primary Fibroblast-Like Synoviocytes
This study was approved by the Institutional Review Board of University of California, San Diego School of Medicine (La Jolla, CA, USA). Informed consent was obtained from all participants. Anonymous synovial tissue samples were obtained from six patients with RA at the time of total joint replacement (5 females, 1 male; age range, 44–72 years), as previously described (Alvaro-Gracia et al., 1990). The diagnosis of RA conformed to American College of Rheumatology 1987 revised criteria (Arnett et al., 1988). The synovium was minced and incubated with collagenase type VIII (0.5 mg/ml) (Sigma-Aldrich) in serum-free RPMI-1640 (Life Technologies, Grand Island, NY, USA) for 2 h at 37°C, filtered, extensively washed, and cultured in DMEM (Life Technologies) supplemented with 10% FBS (Gemini Bio Products, Calabasas, CA, USA), penicillin, streptomycin, gentamicin, and glutamine in a humidified 5% CO2 atmosphere. Cells were allowed to adhere overnight, nonadherent cells were removed, and adherent fibroblast-like synoviocytes (FLS) were split at 1:3 when 70%–80% confluent. FLS were used from passages 3 through 9, during which time they are a homogeneous population of cells (<1% CD11b positive, <1% phagocytic, and <1% FcγRII and FcγRIII receptor positive).
Analysis of Matrix Metalloproteinase (MMP) Expression in FLS
For MMP mRNA analysis, FLS were plated in six-well plates, cultured until 80% confluence, and subsequently serum starved (1% FBS/DMEM) for 24 h. The cells were treated with TRYP or TRYP-Ox (4, 10, and 25 μM) or vehicle (DMSO) for 1 h before IL-1β stimulation (2 ng/ml) for 6 h. The mRNA was isolated and reverse transcribed to obtain cDNA. Quantitative real-time polymerase chain reaction was performed using primer probe sets for MMP3 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Life Technologies). Threshold cycle values were obtained and normalized to GAPDH expression.
Analysis of c-Jun Phosphorylation in FLS
Cultured FLS (passage 4) were plated at 2 × 105 cells in six-well plates overnight in DMEM containing 10% FBS and synchronized for 24 h in DMEM/0.1% FBS. The cells were pretreated for 1 h with different concentrations of TRYP, TRYP-Ox, or DMSO (vehicle control) and then stimulated with 2 ng/ml IL-1β or medium for 15 min at 37°C. The cells were then washed once with ice-cold PBS and lysed with modified radioimmunoprecipitation assay buffer [50 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 2.5 mM MgCl2, 1.0 mM EDTA, 20 mM β-glycerophosphate, 10 mM NaF, 1 mM Na3VO4, and Protease inhibitor cocktail (Roche, Indianapolis, IN)]. Protein concentration of the lysates was measured using a MicroBCA Assay kit (Pierce, Rockford, IL), and 40 μg lysate was subjected to 10% SDS-PAGE and Western blot analysis. Anti–phospho-c-Jun (Ser63) was purchased from Cell Signaling Technology (Danvers, MA), and anti-mouse GAPDH was from Santa Cruz Biotechnology (Santa Cruz, CA).
Determination of MMP-1/3 and IL-6 in Cell Culture Supernatants
FLS, HUVEC, SW982, and THP-1 cells were plated in 96 well plates (104 to 105 cells/well) for 48 h before treatment. At 2 days post-confluency, the cells were serum-starved and washed twice (with the exception of THP-1 cell, which were not washed) with FBS-free medium prior to the addition of test compounds. The cells were incubated in the presence or absence of test compounds or DMSO (vehicle control) for 30 min at 37°C in a 5% CO2 atmosphere, IL-1β (5 ng/ml) was added, and the cells were incubated for an additional 24 h. Culture supernatants were collected and stored at –80°C. The levels of MMP-1 and MMP-3 were determined in culture supernatants using commercially available ELISA kits according to the manufacturer’s instructions (R&D System, Minneapolis, MN, USA). IL-6 levels were also determined in the supernatants using a human IL-6 ELISA kit (BD Biosciences, San Jose, CA, USA).
AChE Inhibition Assay
The inhibitory effect of test compounds on AChE activity was performed using an AChE inhibitor screening kit from Sigma-Aldrich. The kit is based on an improved Ellman method, whereby thiocholine produced from AChE activity forms a yellow color with 5,5’-dithiobis(2-nitrobenzoic acid), and the intensity the color (412 nm) is proportional to enzyme activity. The concentration of compound required to cause 30% inhibition (IC30) was used. IC30 was obtained by graphing the % inhibition of enzyme activity versus the logarithm of concentration of test compound using 5–7 tested concentrations.
Cytotoxicity Assay
Cytotoxicity was analyzed using a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega), according to the manufacturer’s protocol. Briefly, cells were cultured at a density of 104 to 105 cells/well with different concentrations of test compounds (final concentration of DMSO was 1%) for the indicated periods of time (2, 12, or 24 h) at 37°C and 5% CO2. Following treatment, substrate was added to the cells, and the samples were analyzed with a Fluoroscan Ascent FL microplate reader.
PharmMapper Target Identification
The protein targets of TRYP and TRYP-Ox were analyzed using PharmMapper (Liu et al., 2010). PharmMapper is a tool that identifies potential targets for a given small molecule using an “invert” pharmacophore mapping procedure. PharmMapper utilizes reference databases of protein drug targets encoded by sets of pharmacophore points for faster mapping. Initial structures of the test compounds were prepared using ChemDraw 16.0 software and saved in Tripos MOL2 format. The MOL2 files of TRYP and Z- and E-isomers of TRYP-Ox were uploaded into the PharmMapper server with automatic generation of up to 300 conformers for each compound switched on. We selected the “Human Protein Targets Only” database containing 2241 targets for pharmacophore mapping. The top 250 potential targets were retrieved and sorted by normalized fit score value.
The physicochemical properties of selected compounds were computed using SwissADME (http://www.swissadme.ch) (Daina et al., 2017).
Statistical Analysis
A nonparametric Mann–Whitney U test was used for statistical analysis of CIA and CAIA clinical scores, histology scores, and cartilage destruction. All other data were analyzed by ANOVA with no correction, and differences were considered statistically significant if p<0.05.
Results
Identification of Potential Therapeutic Targets for TRYP and TRYP-Ox
Previously we used kinase profiling to demonstrate that TRYP-Ox had high binding affinity for JNK1-3, whereas TRYP had very low affinity for JNK1 and had no binding affinity for JNK2 and JNK3 (Schepetkin et al., 2019). To expand and enhance our understanding of the potential biological targets of TRYP/TRYP-Ox, we performed reverse pharmacophore mapping on TRYP and the Z- and E-isomers of TRYP-Ox using PharmMapper. This program compared a database of pharmacophore patterns with our test compounds and generated target information, including normalized fitness scores and pharmacophoric characteristics. As shown in Table 1, PharmMapper analysis indicated that the top three ranked potential targets for the Z- and E-isomers of TRYP-Ox were JNK1, JNK3, and complement factor B (CFB), confirming the high binding affinity for JNK1 and JNK3 that we found using kinase scanning and verifying that these are indeed relevant therapeutic targets for TRYP-Ox. In contrast, PharmMapper analysis of the parent alkaloid TRYP showed relatively low fitness scores for JNK1 and JNK3, with the top three ranked targets for TRYP being acetylcholinesterase (AChE), carboxylesterase 1 (CES-1), and transthyretin (TTR) (Table 1). The low fitness for TRYP binding to JNK is consistent with our previous kinase scanning studies (Schepetkin et al., 2019).
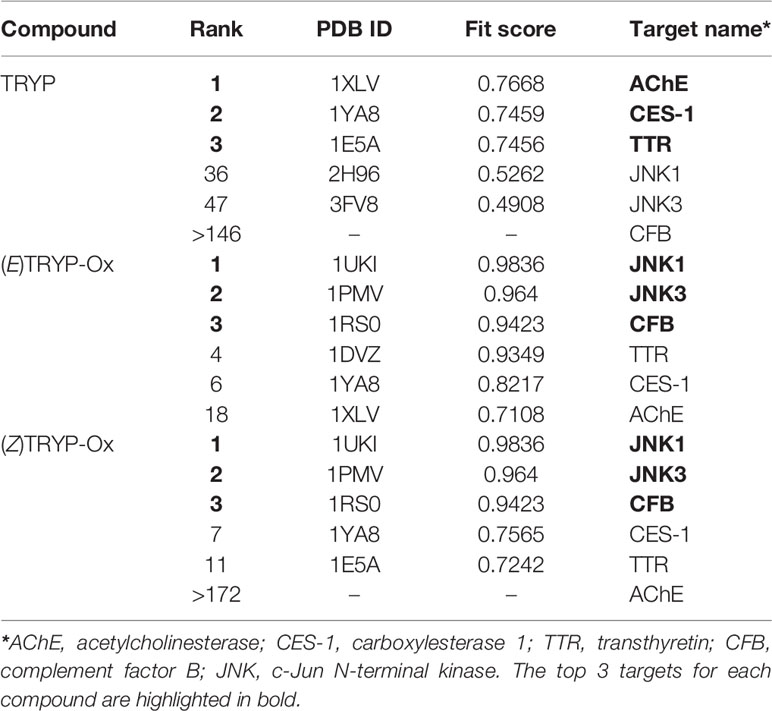
Table 1 Potential protein targets of TRYP and E/Z isomers of TRYP-Ox.
Since AChE was found to be the top target for TRYP and was also on the target list for TRYP-Ox, although not close to the top targets, we evaluated the direct AChE inhibitory effects of both TRYP and TRYP-Ox using an enzymatic test-system but found that both compounds had a relatively low anti-AChE activity, with TRYP being somewhat more active (Table 2). Thus, PharmMapper analysis supported the role of JNK1 and JNK3 as targets for TRYP-Ox, which is relevant to therapeutic intervention in RA, whereas no obvious targets related to RA intervention were found for TRYP.

Table 2 Effect of TRYP and TRYP-Ox on the top ranked molecular targets.
In Silico ADME Predictions
The ADME properties determining either the access of a potential drug to the target or its elimination by the organism are necessary during initial stages of drug discovery (Doogue and Polasek, 2013). TRYP and TRYP-Ox were analyzed for the most important physicochemical properties in comparison with indenoquinoxaline analogs IQ-1 (an oxime formed after spontaneous hydrolysis of the JNK inhibitor IQ-1S) and IQ-18 (the inactive ketone precursor of IQ-1) using the SwissADME tool (Daina et al., 2017). We found that these compounds were similar regarding many ADME properties (Table 3). Nevertheless, they differed markedly in the number of H-bond acceptors and donors, which we showed previously affected JNK binding affinities and locations within the JNK binding site for (Schepetkin et al., 2012; Schepetkin et al., 2015). We also created bioavailability radar plots that display an assessment of the drug-likeness of each compound. Six physicochemical properties are considered: lipophilicity, size, polarity, solubility, flexibility, and saturation. The physicochemical range on each axis is depicted as a pink area in which the radar plot of the molecule has to fall entirely to be considered drug-like (Daina et al., 2017). We found that all four compounds had similar bioavailability radar plots (Figure 1). Moreover, the charge distributions for IQ-1 and TRYP-Ox were very similar, which is not surprising based on their similar structures (Schepetkin et al., 2019).
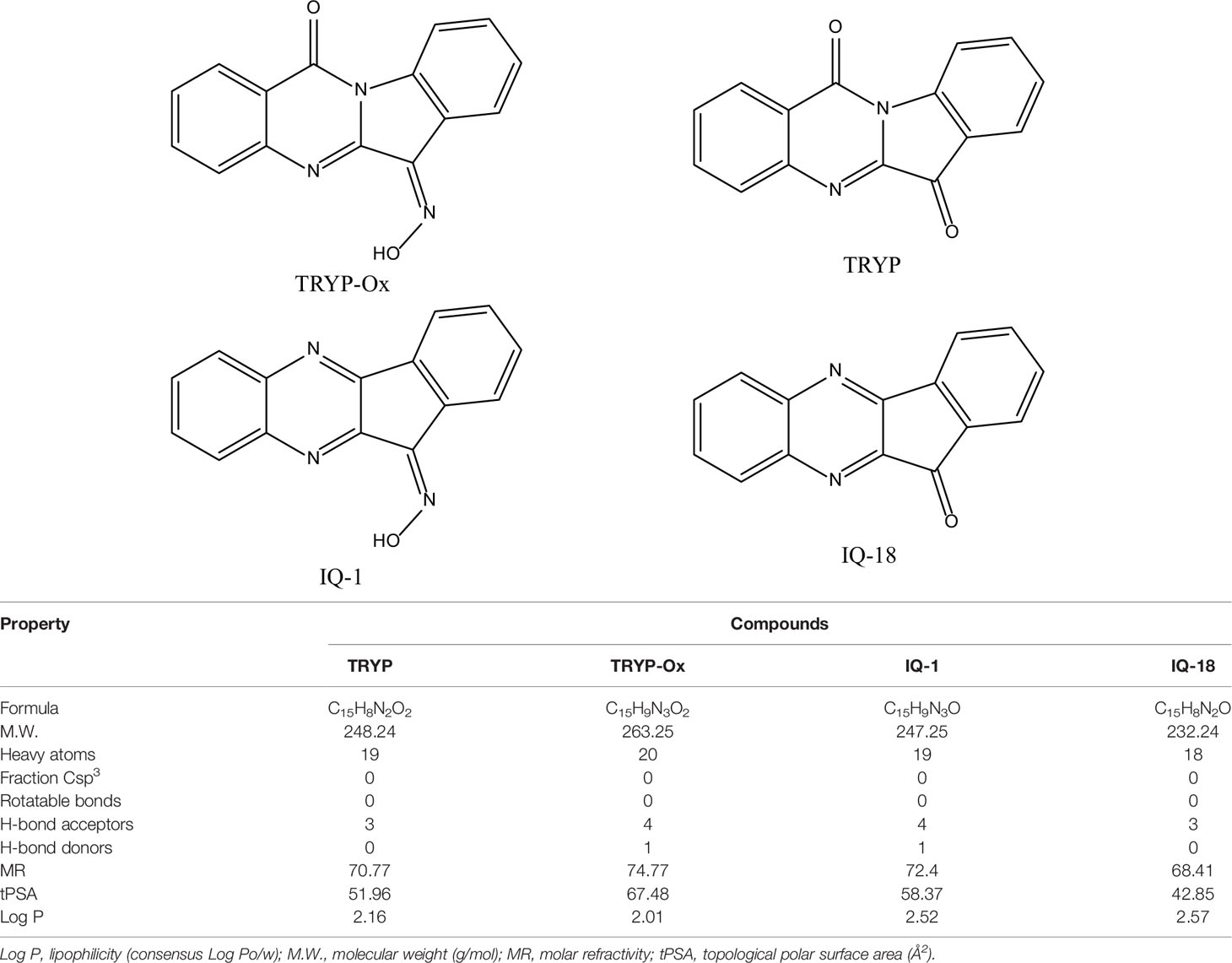
Table 3 Chemical structures and physicochemical properties of JNK inhibitors TRYP-Ox and IQ-1 and their ketone precursors TRYP and IQ-18.
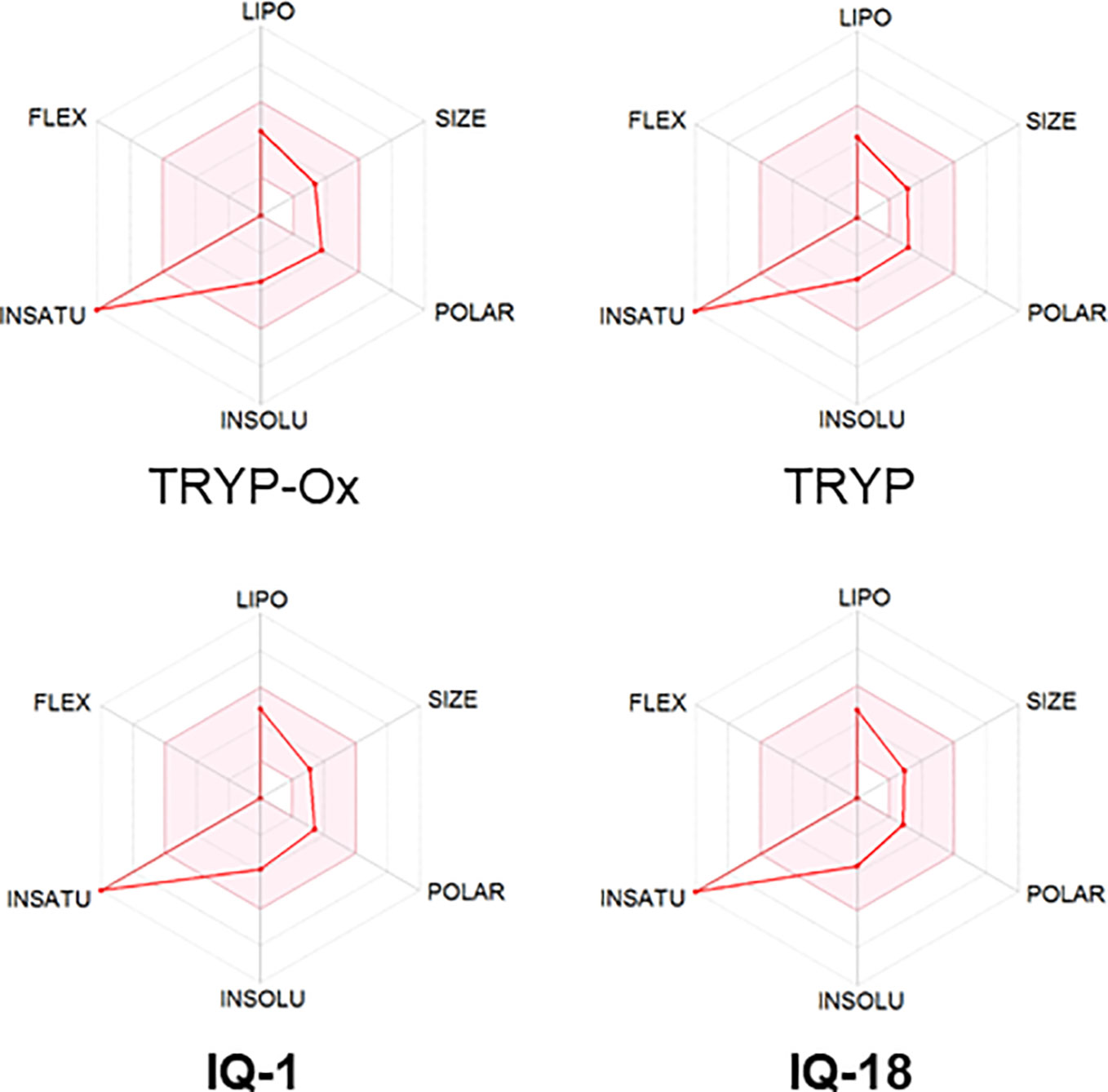
Figure 1 Bioavailability radar plots of JNK inhibitors TRYP-Ox and IQ-1 and their ketone precursors TRYP and IQ-18. The plots depict the LIPO (lipophilicity), SIZE (molecular weight), POLAR (polarity), INSOLU (insolubility), INSATU (unsaturation), and FLEX (rotatable bond flexibility) parameters.
TRYP and TRYP-Ox Inhibit MMP3 Gene Expression and MMP1/3 and IL-6 Secretion
FLS obtained by arthroplasty from tissue of patients with RA are an excellent in vitro model for screening novel compounds with antiarthritic potential (Bartok and Firestein, 2010). We evaluated whether TRYP and TRYP-Ox altered MMP3 expression by IL-1β–stimulated human FLS and found that cells pretreated with either of these compounds exhibited a dose-dependent reduction in MMP3 mRNA levels compared with the vehicle-treated group, but that TRYP-Ox was significantly more active than TRYP (Figure 2). Numerous studies suggest that human SW982 synovial sarcoma cells share similar physiological and immunological properties with primary FLS and that IL-1β stimulation of SW982 cells can mimic the inflammatory status of synovial cells typically seen in RA patients (Tsuji et al., 1999; Kim et al., 2008; Chang et al., 2014). Thus, SW982 cells can be useful in screening of potential antiarthritic compounds, as FLS are in limited supply. Indeed, we found that TRYP and TRYP-Ox inhibited IL-1β–induced IL-6 secretion by FLS and SW982 cells, as well as by HUVECs and THP-1 monocytic cells, in a dose-dependent manner, with the most potent being TRYP-Ox (Table 4A). These compounds also inhibited IL-1β–induced MMP-1/3 secretion by FLS, SW982 cells, and HUVECs, with the most potent being TRYP-Ox in FLS and SW982 cells (Table 4A). As examples, the dose-dependent effects of TRYP and TRYP-Ox on IL-1β–induced secretion of IL-6 by FLS, SW982 cells, and HUVECs are shown in Figure 3. Note that TRYP-Ox was significantly more effective than TRYP in inhibiting IL-6 production in all three of these cell types.
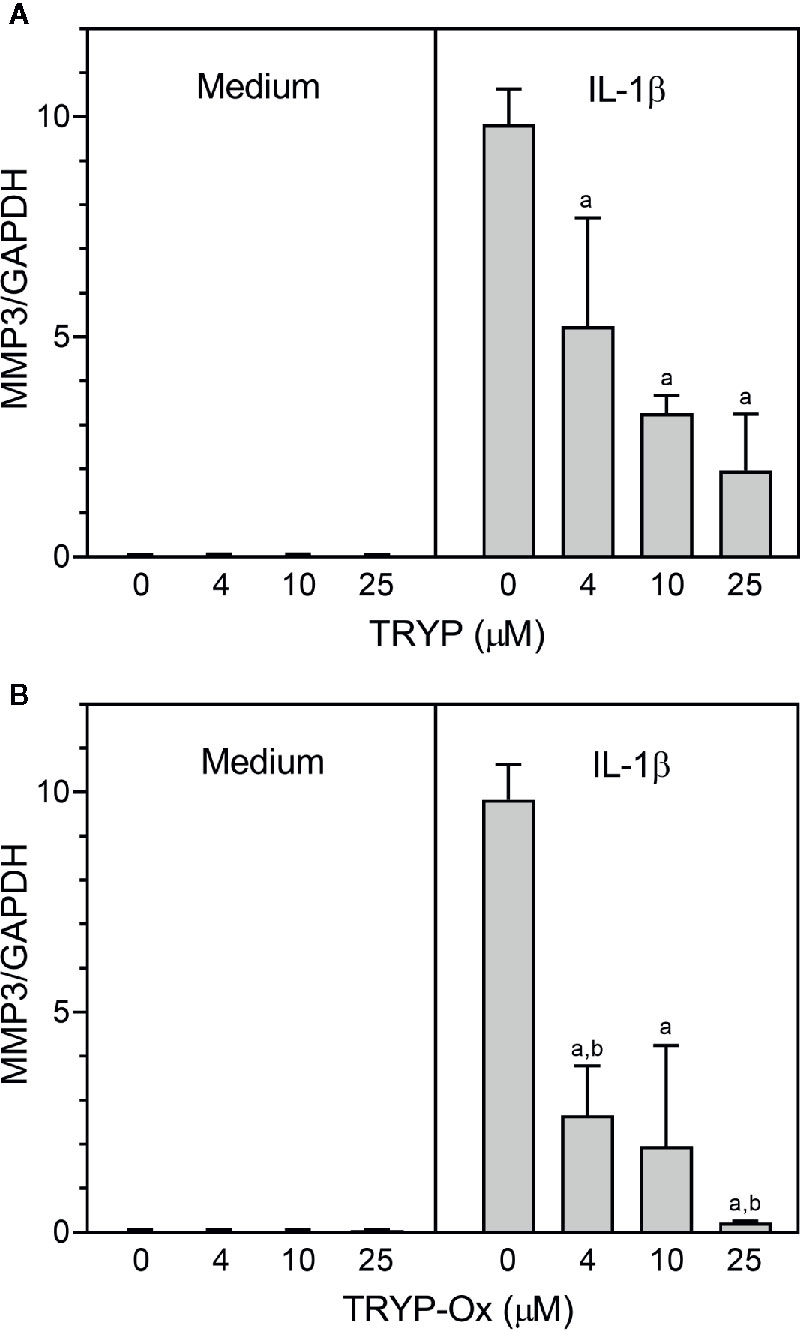
Figure 2 TRYP and TRYP-Ox decrease MMP3 gene expression in FLS from RA patients. FLS were treated with the indicated concentrations of TRYP (Panel A) or TRYP-Ox (Panel B) and stimulated with control medium or IL-1β (2 ng/ml) for 6 h. MMP3 gene expression was evaluated by quantitative polymerase chain reaction and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as described under Materials and Methods. The results are shown as the mean ± S.D. of three different RA lines. ap < 0.05 compared with the DMSO control; bp < 0.05 compared with TRYP.
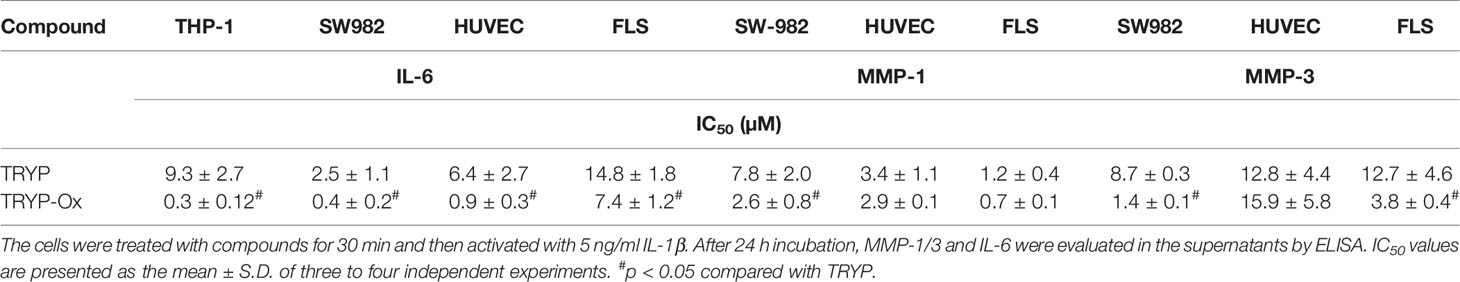
Table 4A Inhibitory effect of TRYP and TRYP-Ox on IL-1β-induced IL-6 and MMP-1/3 secretion by monocytic THP-1 cells, synovial SW982 cells, and HUVEC.
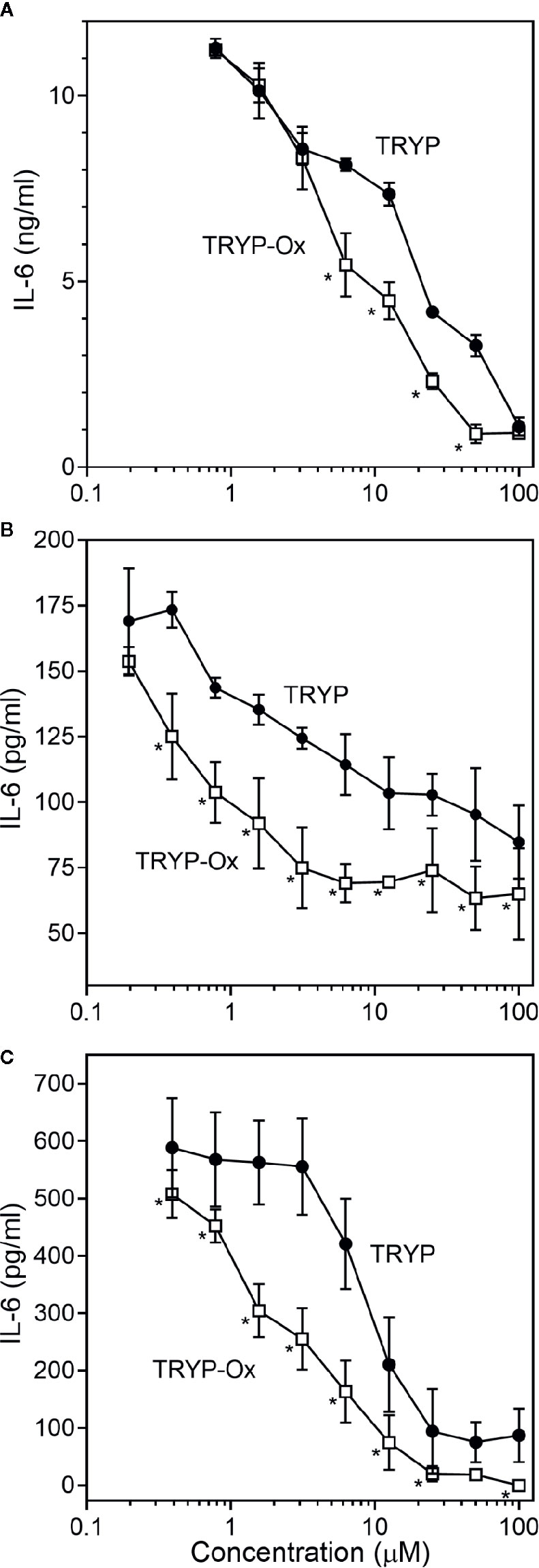
Figure 3 Effects of TRYP and TRYP-Ox on IL-1β-induced IL-6 secretion by FLS, SW982 cells, and HUVECs. FLS (A), SW982 cells (B), and HUVECs (C) were treated with the indicated compounds or DMSO for 30 min, followed by stimulation with 5 ng/ml IL-1β for 24 h. IL-6 levels were measured by ELISA. Values are expressed as mean ± S.D. from one experiment that is representative of 3 independent experiments. *p<0.05 compared with TRYP.
To ensure that the effect of our compounds on functional activity in vitro were not affected by possible toxicity, we evaluated cytotoxicity of TRYP and TRYP-Ox at various concentrations (up to 50 µM) in all cultured cells (Table 4B). While TRYP-Ox exhibited no cytotoxicity in all of the cells tested, TRYP exhibited some cytotoxic effects, albeit at higher concentrations that those required to inhibit IL-6 and MMP-1/3 production. Thus, TRYP cytotoxicity may have some contribution to the inhibition of pro-inflammatory cytokine/enzyme production by some cell lines.

Table 4B Cytotoxic effect of TRYP and TRYP-Ox on THP-1 cells, SW982 cells, HUVECs, and FLS.
We also evaluated the effects of SP600125, a commonly used JNK inhibitor (Bennett et al., 2001), on IL-6 secretion in SW982 synovial cells. Since SP600125 increased cytotoxicity of TRYP during a 24 h incubation, we used a shorter incubation period (12 h) that did not affect cell viability. We found that SP600125 dose-dependently inhibited IL-1β–induced IL-6 secretion by SW982 cells (IC50 = 1.3 ± 0.5 µM). In addition, combined treatment of SP600125 plus TRYP was evaluated. In the presence of 5 µM TRYP, the IC50 value for SP600125 was the similar to that observed when TRYP was absent (1.2 ± 0.2 µM), although TRYP acted synergistically with SP00125 to significantly increase the overall inhibition compared to SP00125 alone (Figure 4). Thus, TRYP and SP600125 may inhibit IL-6 production through different pathways.
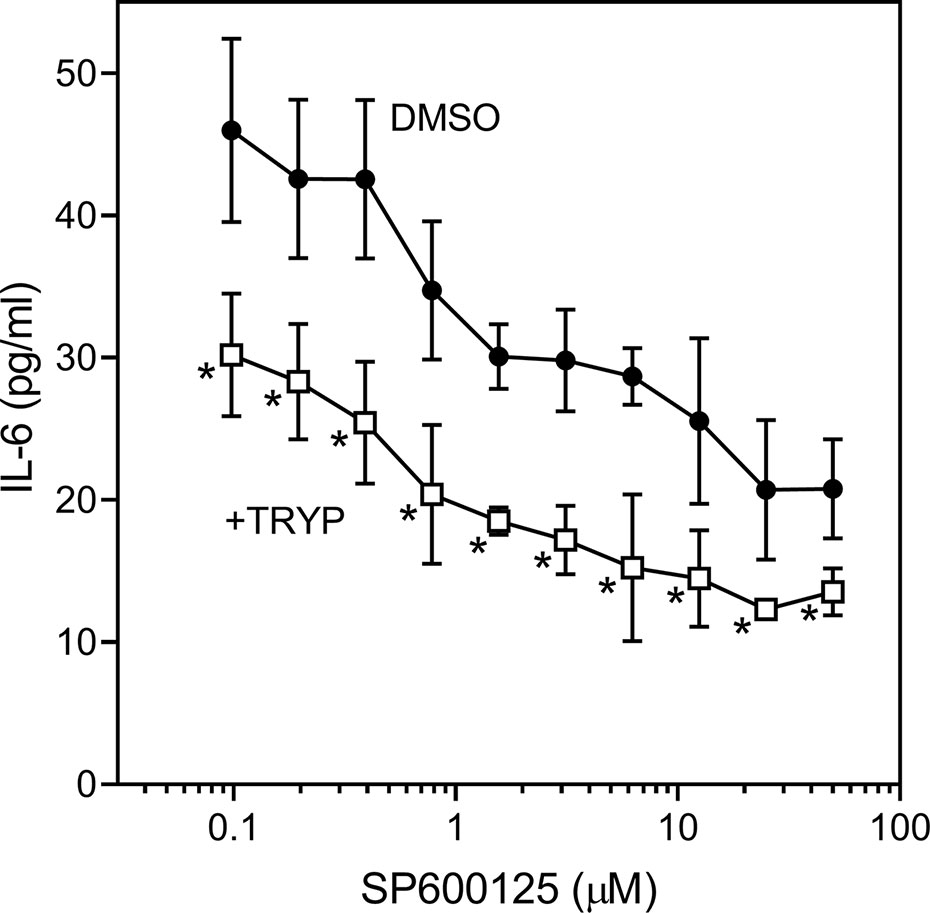
Figure 4 Effects of JNK inhibitor SP600125 and TRYP on IL-1β-induced IL-6 secretion by SW982 cells. Cells were treated with 5 µM TRYP or DMSO combined with the indicated concentrations of SP600125 for 30 min, followed by stimulation with 5 ng/ml IL-1β for 10 h. IL-6 levels were measured by ELISA. Values are expressed as mean ± S.D. of one experiment that is representative of 2 independent experiments. *p < 0.05 compared with SP600125 treatment alone.
TRYP-Ox Inhibits c-Jun Phosphorylation in Rheumatoid FLS
Previously, we showed that the JNK inhibitor IQ-1S, which is a close chemical analog of TRYP-Ox (see Table 3), inhibited c-Jun phosphorylation in FLS (Schepetkin et al., 2015). Analysis of the effects of TRYP and TRYP-Ox on c-Jun phosphorylation in IL-1β–stimulated FLS showed that TRYP-Ox also inhibited c-Jun phosphorylation, whereas TRYP had no effect (Figure 5), confirming our previous conclusion that TRYP-Ox, but not TRYP, is a JNK inhibitor (Schepetkin et al., 2019). Note that neither TRYP nor TRYP-Ox affected FLS viability during the 90-min period.
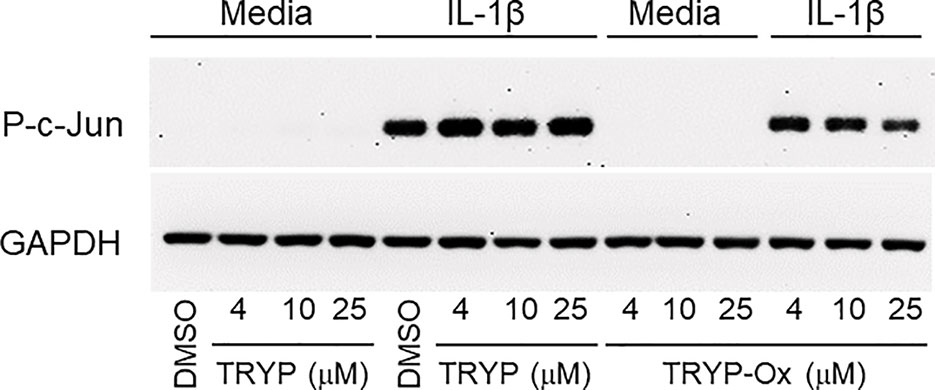
Figure 5 TRYP-Ox inhibits c-Jun phosphorylation induced by IL-1β in FLS from RA patients. FLS were pretreated for 1 h with the indicated concentrations of TRYP-Ox, TRYP, or DMSO and then stimulated with 2 ng/ml IL-1β or medium for 15 min at 37°C. Equal concentrations of cell lysates were separated by SDS-PAGE, followed by Western blotting for phospho-c-Jun (P-c-Jun) and GAPDH, as described. The blot shown is from one experiment that is representative of 2 independent experiments.
TRYP and TRYP-Ox Reduce CIA Severity
Previously, we evaluated a range of IQ-1S doses (5, 20, 30, and 50 mg/kg i.p.) and found that 5 mg/kg was not very effective, whereas 30 and 50 mg/kg were equally effective in reducing arthritis in the CIA model (Schepetkin et al., 2015). Preclinical pharmacokinetic studies in rats demonstrated that after i.p. administration of IQ-1S, a rapid rise in the serum concentration was observed, peaking at 5 min, and the half-life in circulation was 12 h (Plotnikov et al., 2020). When mice were dosed with 30 mg/kg IQ-1S i.p., the serum exposure of the compound was also good, with AUC 0–12 h values of 7.4 µM/h (Schepetkin et al., 2012). Thus, based on the similarity in physicochemical properties of TRYP-Ox and IQ-1S, their high JNK binding affinity, the effectiveness of 30 mg/kg IQ-1S in previous CIA experiments, and the known preclinical pharmacokinetic studies for IQ-1S, we selected a 30 mg/kg dose for TRYP-Ox and TRYP treatment in our in vivo studies.
CIA was induced in arthritis-susceptible DBA1/J mice by immunization with bovine CII emulsified in complete Freund’s adjuvant. To test the therapeutic potential of TRYP and TRYP-Ox on CIA, treatment was initiated at day 8 after CII challenge. Although disease symptoms appear usually at day 21 post-induction, immune pathologic processes begin soon after CII injection (Brand et al., 2003). Thus, we consider administration of compounds between days 0 and 21 following CIA initiation to be a combined prevention/treatment model. We found that mice treated i.p. daily with 30 mg/kg of TRYP or TRYP-Ox displayed significant reductions in CIA symptoms compared with vehicle-treated mice (Figure 6). Both TRYP and TRYP-Ox seem to be equally effective treatments for reducing clinical scores in the CIA model.
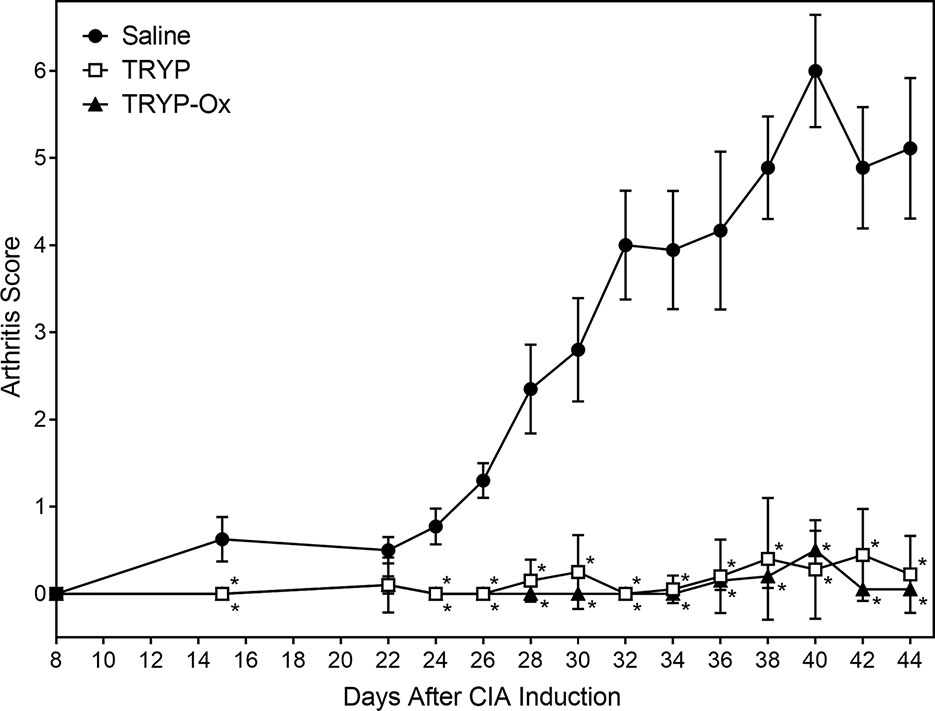
Figure 6 Therapeutic effects of TRYP and TRYP-Ox in CIA mice. Starting at day 8 after CII challenge, mice were treated i.p. daily with 30 mg/kg TRYP ( ) or 30 mg/kg (
) or 30 mg/kg ( ) TRYP-Ox until day 42, and clinical scores were determined, as described. Control mice were treated i.p. daily with saline (
) TRYP-Ox until day 42, and clinical scores were determined, as described. Control mice were treated i.p. daily with saline ( ). Results are shown as the mean of 10 mice per group ± S.D. *p < 0.05 compared with saline control.
). Results are shown as the mean of 10 mice per group ± S.D. *p < 0.05 compared with saline control.
Histopathological analysis was performed on ankle joints collected from CIA mice treated with TRYP or TRYP-Ox and compared with control, saline-treated mice. Representative images of stained joint tissue from mice treated with 30 mg/kg compounds are shown in Figure 7A. In contrast to the severe cartilage erosion, synovial hyperplasia, and infiltration of inflammatory cells observed in the joints of saline-treated CIA mice, there was little cartilage erosion, synovial hyperplasia, or cellular infiltration in the joints of TRYP and TRYP-Ox-treated CIA mice. Furthermore, histological scoring showed significant differences between treated and control groups (Figures 7B, C), whereas the differences between TRYP- and TRYP-Ox-treated mice were not significant. Thus, consistent with the reductions in CIA clinical score, TRYP and TRYP-Ox treatment clearly protected these mice from cartilage destruction.
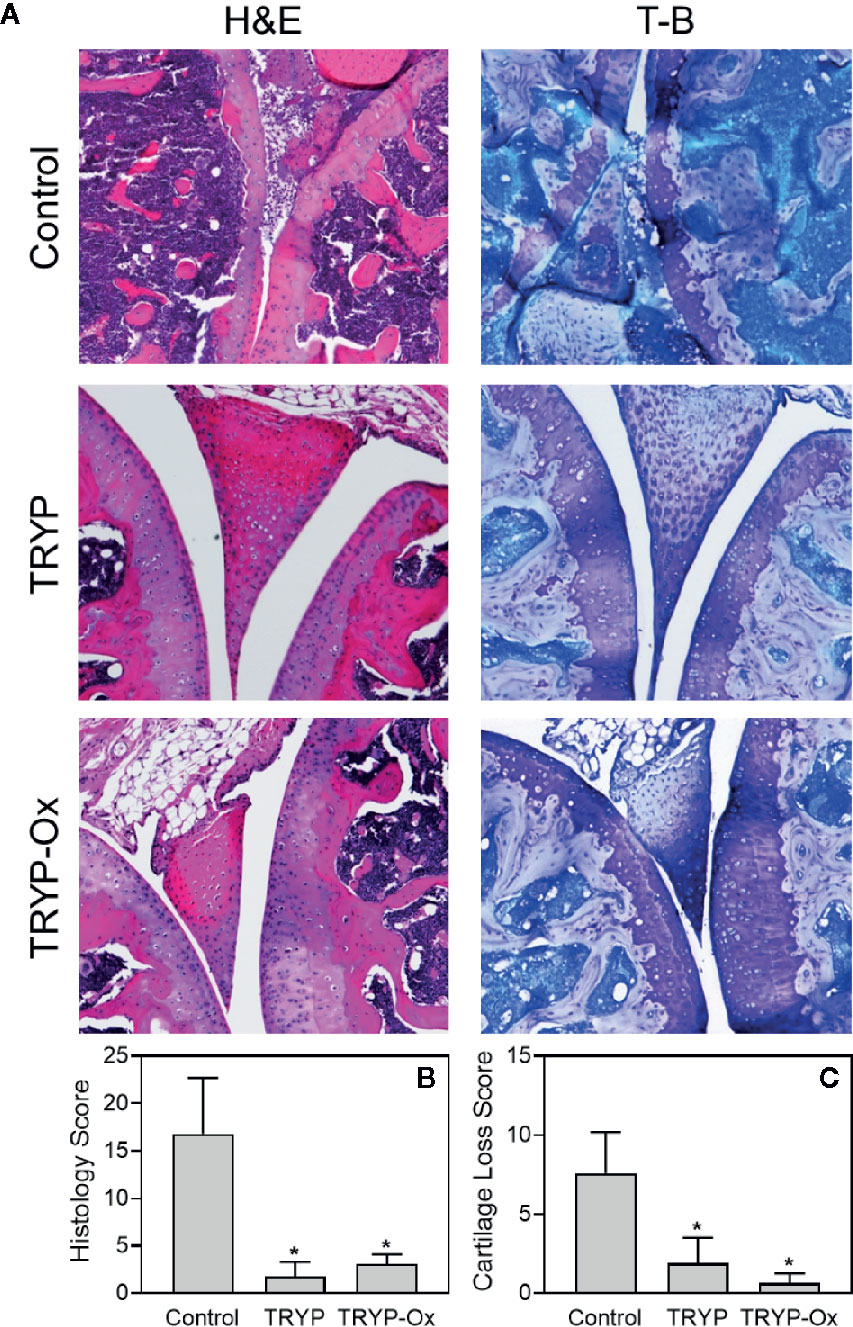
Figure 7 Effects of TRYP and TRYP-Ox on inflammation and cartilage loss in CIA mice. Starting at day 8 after CII challenge, mice were treated i.p. daily with 30 mg/kg TRYP or 30 mg/kg TRYP-Ox in sterile saline until day 42, when mice were euthanized. Control mice received saline alone. (A) Hematoxylin and eosin (H&E)- and toluidine blue (T-B)-stained knee sections. (B, C) Histology scores (0–3 per section) and cartilage loss scores (0–3 per section) were graded for each limb and knee section. The results show the mean of five mice per group ± S.D. *p < 0.05 compared with saline control.
TRYP and TRYP-Ox Decrease Serum Titer of CII-Specific Abs
Injected CII-specific Abs can induce transient arthritis, indicating that Ab responses are important in CIA pathogenesis (Myers et al., 1997; Nandakumar et al., 2003), and it has been shown that the anti-CII IgG antibody titer correlates with arthritis severity in CIA mice (Williams et al., 1998). We measured the levels of CII-specific Abs in TRYP-, TRYP-Ox-, and vehicle-treated CIA mice and found that the titers of the IgG, IgG1, IgG2a, IgG2b, and IgG3 were significantly lower in the compound-treated groups compared to those in the saline-treated group (Figure 8).
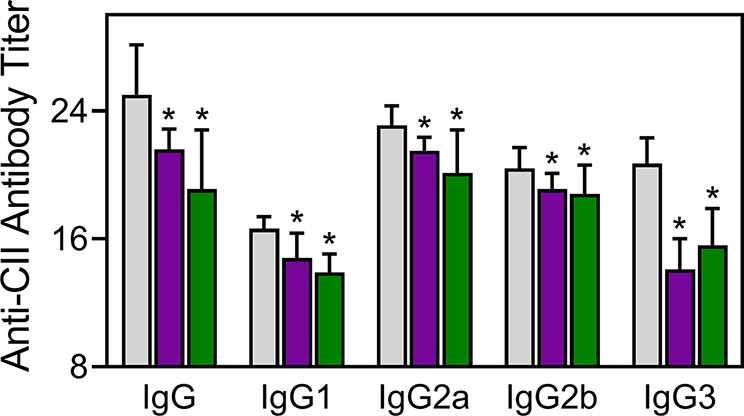
Figure 8 Effect of TRYP and TRYP-Ox on the antibody response to CII. Starting at day 8 after CII challenge, mice were treated i.p. daily with control saline ( ), 30 mg/kg TRYP (
), 30 mg/kg TRYP ( ), or 30 mg/kg TRYP-Ox (
), or 30 mg/kg TRYP-Ox ( ) until day 42. Mice were euthanized, and sera were collected for measurement of anti-CII IgG, IgG1, IgG2a, IgG2b, and IgG3. The results show the mean of 10 mice per group ± S.D. *p < 0.05 compared with saline control.
) until day 42. Mice were euthanized, and sera were collected for measurement of anti-CII IgG, IgG1, IgG2a, IgG2b, and IgG3. The results show the mean of 10 mice per group ± S.D. *p < 0.05 compared with saline control.
TRYP and TRYP-Ox Decrease Proinflammatory Cytokine Levels in LN Cells
Proinflammatory cytokines produced by myeloid cells can facilitate inflammation, cartilage damage, and joint destruction in RA (Schurgers et al., 2011; Zhou et al., 2011; Azizi et al., 2013). To test whether TRYP and TRYP-Ox affected the production of inflammatory cytokines by CII-specific T cells, we evaluated CII-induced cytokine production by LN cells isolated from control and treated mice. We cultured the same numbers of cells from compound-treated and saline-treated CIA mice for 72 h in the presence of CII and found that the generation of proinflammatory cytokines (IL-1β, IL-5, IL-6, IL-17A, TNF, GM-CSF, and RANKL) was significantly reduced in compound-treated mice (30 mg/kg) compared to saline-treated CIA mice. In contrast, the production of the anti-inflammatory cytokine IL-10 was higher in compound-treated mice (Figure 9). Note that the levels of IL-17A, GM-CSF, and RANKL production by LN cells isolated from TRYP-Ox-treated mice were significantly lower compared to mice treated with TRYP.
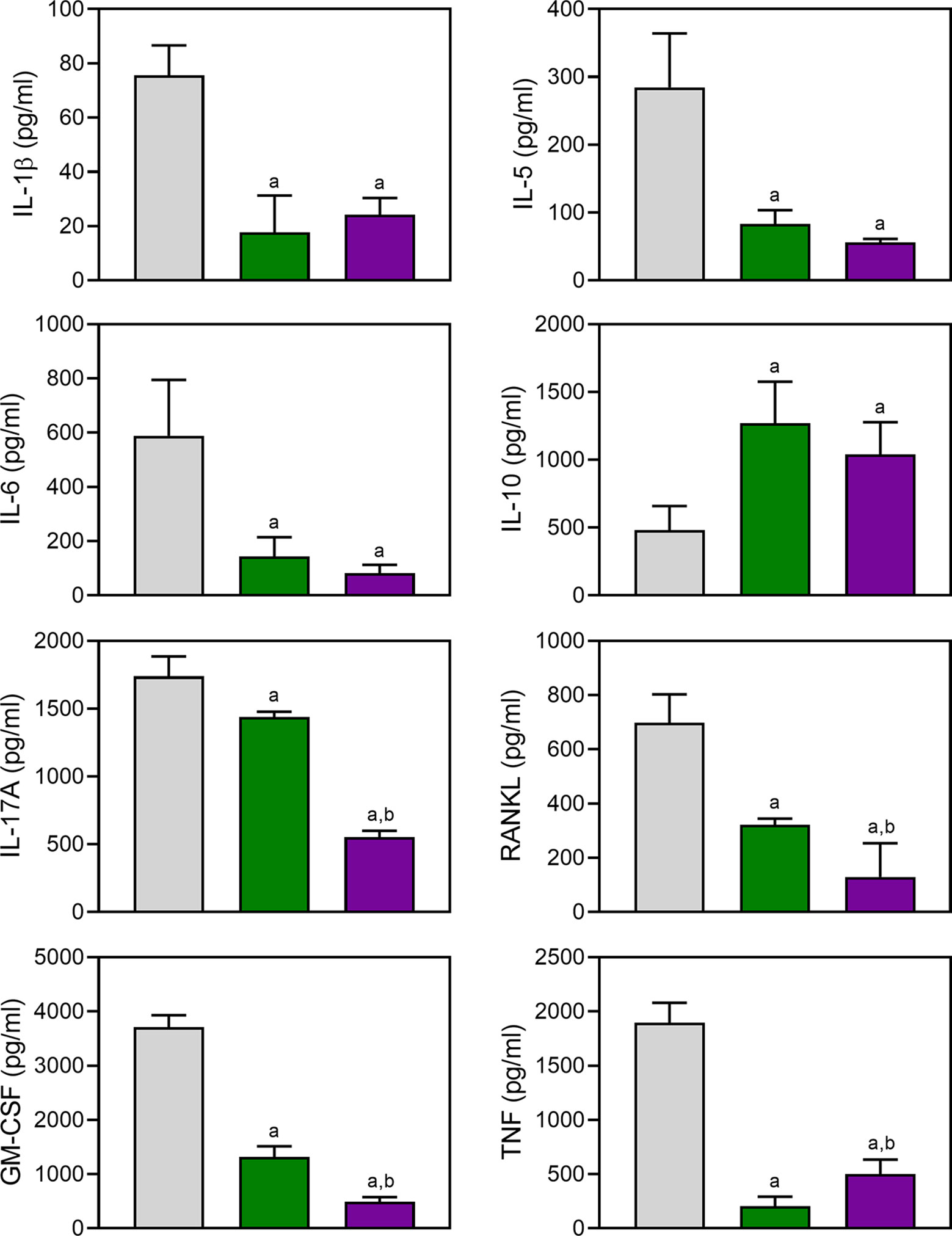
Figure 9 Effect of TRYP and TRYP-Ox on cytokine production by LN cells. Starting at day 8 after CII challenge, mice were treated i.p. daily with saline (0), 30 mg/kg TRYP, or 30 mg/kg TRYP-Ox until day 42. Mice were euthanized, and their LN mononuclear cells were purified and cultured for 3 days with CII. Cytokine levels were measured in supernatants by ELISA. The results show the mean of one experiment (three to four replicates) ± S.D., which is representative of three independent experiments. ap < 0.05 compared with the DMSO controls; bp < 0.05 compared with TRYP.
TRYP and TRYP-Ox Suppress CAIA Severity
The CAIA mouse model is another widely used animal model to screen for potential anti-RA compounds [for example (Chang et al., 2011)]. To evaluate the therapeutic potential of TRYP and TRYP-Ox in CAIA, treatment was initiated at day 1 after anti-CII antibody injection. We found that mice treated i.p. daily with 30 mg/kg TRYP or TRYP-Ox displayed significant reductions in the severity of CAIA compared with vehicle-treated mice, as assessed by the mean arthritis score (Figure 10). Although the differences in therapeutic effects of TRYP and TRYP-Ox were not significant, the therapeutic effects of TRYP and TRYP-Ox were significantly greater at days 6 and 7 compared to mice treated with IQ-1S, a JNK inhibitor that we previously showed to inhibit arthritis in the CIA model (Schepetkin et al., 2015).
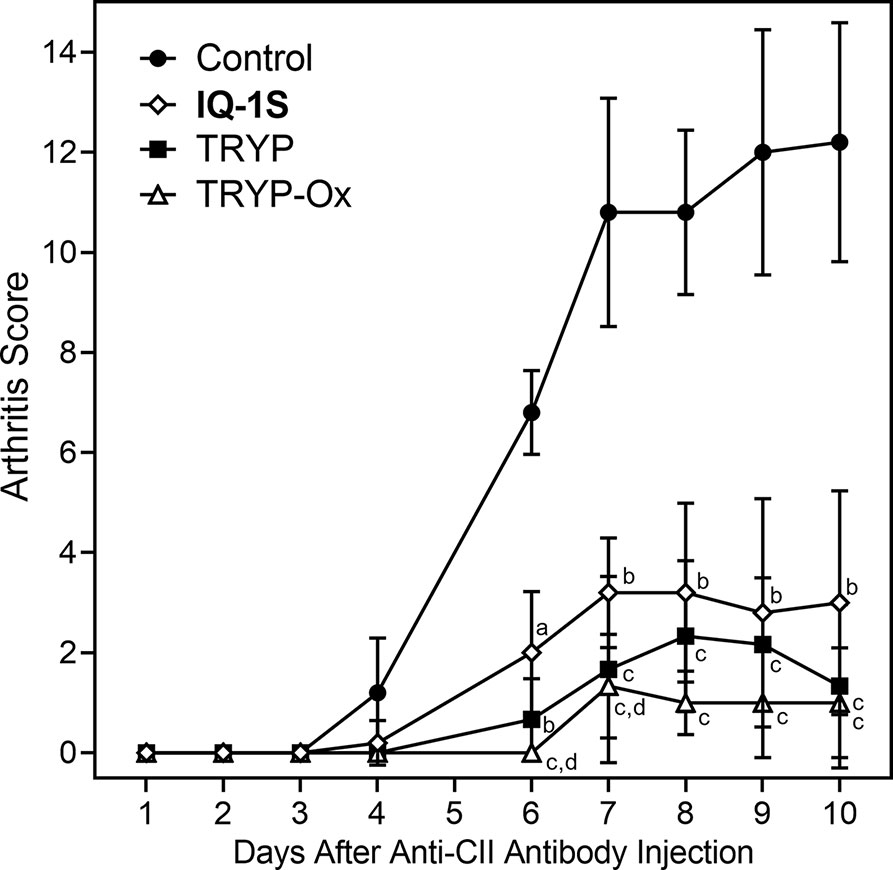
Figure 10 Therapeutic effects of TRYP and TRYP-Ox in CAIA mice. Starting at day 1 after the anti-CII antibody injection, mice were treated i.p. daily with control saline ( ), TRYP (
), TRYP ( ), TRYP-Ox (
), TRYP-Ox ( ), or IQ-1S (
), or IQ-1S ( ) (30 mg/kg of each compound) until day 9, and clinical scores were determined. Results are shown as the mean of 5 mice per group ± S.D. ap < 0.05; bp < 0.01; cp < 0.001 compared with saline control, and dp < 0.05 compared with the IQ-1S–treated group.
) (30 mg/kg of each compound) until day 9, and clinical scores were determined. Results are shown as the mean of 5 mice per group ± S.D. ap < 0.05; bp < 0.01; cp < 0.001 compared with saline control, and dp < 0.05 compared with the IQ-1S–treated group.
Histopathological analysis was also performed on ankle joints collected from CAIA mice treated with 30 mg/kg TRYP or TRYP-Ox and compared to that of control mice (Figure 11). Severe cartilage erosion, synovial hyperplasia, and infiltration of inflammatory cells was seen in the joints of saline-treated CAIA mice. In contrast, there was little cartilage erosion, synovial hyperplasia, or cellular infiltration in TRYP and TRYP-Ox-treated CAIA mice. Furthermore, histological scoring showed significant differences between treated and control groups (Figures 11B, C). Thus, consistent with the reduced CAIA clinical score and the results using the CIA model, TRYP and TRYP-Ox treatment clearly protected CAIA mice from cartilage damage, as well as from severe joint inflammation.
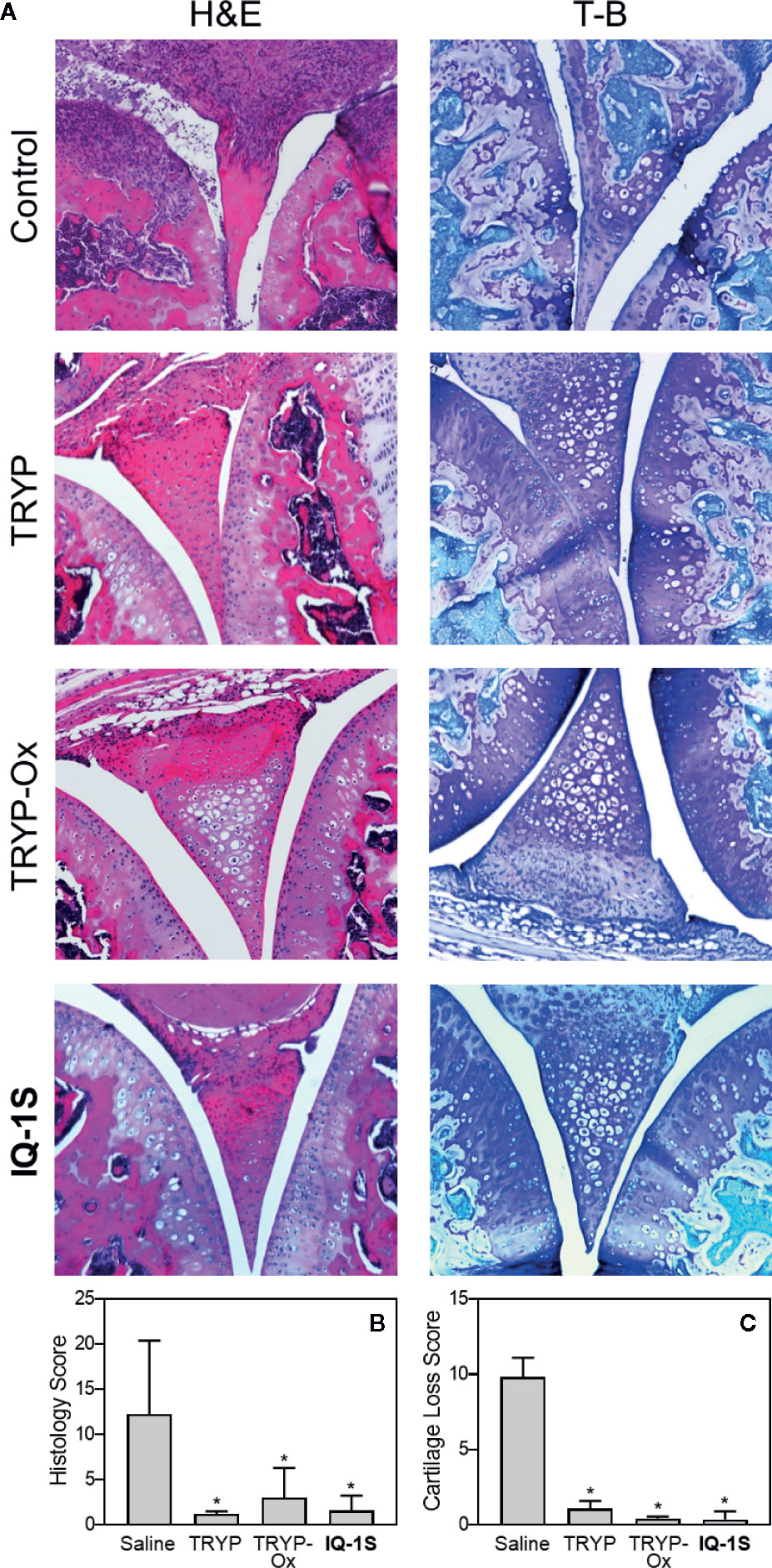
Figure 11 Effect of TRYP and TRYP-Ox on inflammation and cartilage loss in CAIA mice. Starting at day 1 after injection of anti-CII antibodies, mice were treated i.p. daily with 30 mg/kg of TRYP, TRYP-Ox, or IQ-1S in sterile saline until day 9. The mice were euthanized at day 14. Control mice received saline alone. (A) Hematoxylin and eosin (H&E)- and toluidine blue (T-B)-stained knee sections. (B, C) Histology scores (0–3 per section) and cartilage loss scores (0–3 per section) were graded for each limb and knee section. The results show the mean of five mice per group ± S.D. *p < 0.01 compared with saline controls.
Discussion
RA is a chronic destructive autoimmune disorder that commonly leads to significant joint destruction that is mediated in part by proteases and cytokines produced by macrophages and FLS. Although current biological therapeutic strategies for RA have been effective in many cases, there are still issues of cost, loss of efficacy over time, toxicity, and protein degradation. Thus, new classes of therapeutics are needed that can be used alone or in combination with the current biologic RA treatments. In the present study, we identify TRYP-Ox and possibly TRYP as potentially novel RA therapeutics that could be pursued in drug development.
Previously, we demonstrated that TRYP-Ox had high affinity for JNKs and also inhibited pro-inflammatory cytokine production in vitro (Schepetkin et al., 2019), suggesting that it might be have potential in the development of treatments for inflammatory diseases, such as RA. To further evaluate potential targets of TRYP and TRYP-Ox, we performed a broad pharmacophore mapping screen using PharmMapper, which screens a large database of human protein targets. Screening of TRYP-Ox showed that JNK1, JNK3, and CFB were the top potential protein targets. These mapping results are supported by our previous kinase screening, which demonstrated that TRYP-Ox had affinity for all three JNKs, with the highest affinity for JNK1 (150 nM) and JNK3 (275 nM) (Schepetkin et al., 2019). Indeed, a number of studies have shown that JNKs play an important role in RA (Han et al., 2001; Bogoyevitch et al., 2010; Koch et al., 2015), as well as many other inflammatory diseases (Thalhamer et al., 2008; Nikoloudaki et al., 2020). CFB may also represent a therapeutic target in RA, as CFB deficient mice (CFB-/-) are highly resistant to CIA and CAIA and have a decreased CII-specific IgG antibody response in CIA (Hietala et al., 2002; Banda et al., 2006). Furthermore, local complement production, including CFB, may play a role in RA pathogenesis. Thus, the reduction local complement generation may represent a reasonable therapeutic approach for RA (Neumann et al., 2002).
In contrast to TRYP-Ox, the three top ranked targets for TRYP were AChE, CES-1, and TTR, which do not seem to be obvious targets associated with RA. Thus, it is interesting that TRYP was still fairly effective in vivo in treating CIA/CAIA. Although TTR has been reported to be significantly upregulated in the plasma of RA patients (Sharma et al., 2014), this protein has not yet been established as a pharmacological target for RA treatment. CES-1 may play important role in pharmacokinetics and pharmacodynamics of some anti-inflammatory drugs (Mishima et al., 1991), but CES-1 also has not been reported as a therapeutic target for RA. Recently rivastigmine, a known AChE inhibitor, was reported to have therapeutic effects in a complete Freund’s adjuvant-induced model of arthritis (Shafiey et al., 2018). Furthermore, it was found that AChE activity was increased with age and may increase RA risk and severity in elderly people (Sato et al., 2015). Therefore, AChE could be an additional co-target for agents with anti-arthritic activity. However, we found here that both TRYP and TRYP-Ox have relatively low anti-AChE effects, suggesting that AChE is probably not a therapeutic target for these compounds in CIA or CAIA. Note that TRYP has been reported to act on multiple signaling cascades associated with inflammation, including TNF/NF-κB and IL-6/STAT3 (Wang et al., 2018), leukotriene secretion (Pergola et al., 2012), and several cytokines required for TH17 polarization (Cheng et al., 2020). TRYP has also been reported to inhibit macrophage nitric oxide and prostaglandin E production upon exposure to oxidative stress (Ishihara et al., 2000), as well as inhibit cyclooxygenase-2 and indoleamine 2,3-dioxygenase activities (Danz et al., 2001; Pergola et al., 2012; Yang et al., 2013). Overall, the putative TRYP anti-inflammatory target associated with RA is still not clear and will require further investigation.
We demonstrate here for the first time that TRYP and TRYP-Ox may be promising therapeutics for treating RA, as shown by their ability to ameliorate clinical outcomes in vivo in CIA and CAIA mice, as well as reduce disease-associated anti-CII antibody production in CIA mice. CIA progression requires both adaptive and innate immune responses through the production of anti-CII-specific antibodies and the development of CII-specific T cells (Brand et al., 2003). We found that both TRYP and TRYP-Ox inhibited the production of anti-CII antibodies by B cells, especially IgG1 and IgG3, which represent T cell-dependent and T cell-independent responses, respectively (Stevens et al., 1988). Thus, therapeutic effects of TRYP and TRYP-Ox in CIA may be associated with direct inhibition of B cell activity or indirect effects on innate immune cells. Indeed, in both CIA and CAIA models, anti-CII antibodies have been reported to form immune complexes that activate myloid cell FcγR, leading to infiltration of monocyte/macrophages, neutrophils, and mast cells into the joints, where they release inflammatory mediators that initiate synovitis and bone destruction (Delire, 1994; Firestein and Corr, 2005).
Macrophages and FLS invade the synovium in RA and secrete proinflammatory cytokines and MMPs that enhance and sustain the inflammatory response (Kinne et al., 2000; Davignon et al., 2013). The primary cytokines required for inducing arthritis in autoimmune models are IL-1β, IL-6, IL-17A, TNF, GM-CSF, and RANKL (Nakae et al., 2003; Guma et al., 2010; Li et al., 2012; Tanaka, 2019). These cytokines play a key role in RA pathogenesis through their ability to recruit and stimulate macrophages, neutrophils, and mast cells (Drexler et al., 2008; Guma et al., 2010; Tanaka, 2019). Here, we found that TRYP and TRYP-Ox down-regulated IL-1β, IL-5, IL-6, IL17A, TNF, GM-CSF, and RANKL production by LN cells, while IL-10 production was increased. Levels of IL-17A, GM-CSF, and RANKL production by LN cells isolated from TRYP-Ox-treated mice were much lower compared to mice treated with TRYP. This finding may suggest JNK-dependent regulation of these cytokines. Indeed, RANKL and GM-CSF expression and(or) secretion in other cells, such as macrophages, osteocytes, and FLS, can be blocked by SP600125, another JNK inhibitor (Xu et al., 2015; Liu et al., 2016; Takeno et al., 2018). Among the different T cell subsets, a population of IL17-producing T helper cells (TH17) has been shown to be involved in model systems of autoimmunity (Weaver et al., 2007). Recently Cheng et al. (Cheng et al., 2020) reported that TRYP exhibits anti-TH17 activity through its ability to repress the expression of several cytokines required for TH17 polarization.
IL-1β plays important roles in inflammation and destruction of synovial tissue, cartilage, and bone in patients with RA (Tak and Bresnihan, 2000). IL-1β induces synoviolin, an E3 ubiquitin ligase, in synovial fibroblasts, which is involved in the overgrowth of synovial cells during RA (Gao et al., 2006). Therapeutic effects of some kinase inhibitors is associated with decreased IL-1β levels in the joints of CIA mice (McIntyre et al., 2003; Nishikawa et al., 2003). Here, we show that TRYP and TRYP-Ox significantly inhibited IL-1β–induced IL-6 secretion by SW982 synovial sarcoma cells and THP-1 monocytic cells, with TRYP-Ox being far more effective than TRYP.
The joint tissue damage associated with RA is due primarily to the aggressiveness of FLS in the synovial intima, and it is known that rheumatoid FLS produce MMPs that participate in extracellular matrix destruction in articular cartilage and cytokines that prolong inflammation (Burrage et al., 2006; Bartok and Firestein, 2010). Furthermore, JNK plays a key role in FLS MMP gene expression (Han et al., 1999; Liacini et al., 2002; Inoue et al., 2005; Kanai et al., 2020). In our studies, TRYP and TRYP-Ox inhibited IL-1β–induced MMP-1/3 secretion by HUVECs and SW982 cells and MMP3 gene expression in FLS, with the most potent being TRYP-Ox in FLS and SW982 cells. Thus, part of the therapeutic effects of TRYP and TRYP-Ox may be due to their ability to target FLS and potentially reduce their aggressiveness in RA [e.g., (McIntyre et al., 2003; Nishikawa et al., 2003; Hegen et al., 2008)]. These results are consistent with our observations in CIA mouse model, where TRYP and TRYP-Ox decreased joint edema, cell migration, and cartilage erosion.
JNKs play important roles in many pathological processes, including autoimmune inflammatory disorders, such as RA (Bogoyevitch et al., 2004; Bogoyevitch et al., 2010; Mehan et al., 2011). A number of JNK inhibitors with anti-inflammatory properties have been developed (Bhagwat, 2007), yet few have been evaluated for treatment of arthritis. Previous studies demonstrated that SP600125 and IQ-1S, which are both JNK inhibitors, could reduce the development and pathogenesis of arthritis when evaluated in animal models (Han et al., 2001; Han et al., 2012; Schepetkin et al., 2015). Interestingly, the IQ-1S structure is related to TRYP-Ox, and, similarly to IQ-1S, TRYP-Ox was specific for JNK as compared to a variety of other kinases screened (Schepetkin et al., 2019). Moreover, TRYP-Ox is likely a prodrug for TRYP, because microsomal metabolism of aryl-oximes leads to the formation of ketone derivatives (Andronik-Lion et al., 1992). Although IQ-18, the ketone precursor of IQ-1S, did not have any anti-inflammatory activity or therapeutic effects in CIA (Schepetkin et al., 2012; Schepetkin et al., 2015), we show here that TRYP, the ketone precursor of TRYP-Ox, has therapeutic effects in both CIA and CAIA, although with lower efficacy. Thus, the therapeutic effects observed for TRYP-Ox in vivo are likely due to the combined effects of both TRYP-Ox and its metabolite TRYP.
In conclusion, we demonstrated that TRYP and TRYP-Ox have anti-inflammatory properties and potentially represent a novel class of quinazoline-based therapeutic agents that could be used in the development of treatment for RA. TRYP-Ox has additive beneficial therapeutic properties as a JNK inhibitor.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board, University of California, San Diego School of Medicine, La Jolla, CA. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Animal Care and Use Committee, Montana State University, Bozeman, MT 59717.
Author Contributions
IS, LK, DH, AK, and MQ participated in research design. LK, IS, DH, AIK, and AK performed experiments. LK, IS, DH, AIK, AK, and MQ performed data analysis; IS, LK, DH, AIK, and MQ wrote or contributed to the writing of the manuscript.
Funding
This research was supported in part by National Institutes of Health IDeA Program Grants GM110732, GM115371, and GM103474; USDA National Institute of Food and Agriculture Hatch project 1009546; the Montana State University Agricultural Experiment Station, and the Tomsk Polytechnic University Competitiveness Enhancement Program. Molecular modeling studies were supported by the Russian Science Foundation grant 17-15-01111.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agafonova, I. G., Moskovkina, T. V. (2018). Low-dose action of tryptanthrin and its derivatives against developing embryos of the sea urchin Strongylocentrotus intermedius. Environ. Monit. Assess. 190 (9), 502. doi: 10.1007/s10661-018-6808-y
Alvaro-Gracia, J. M., Zvaifler, N. J., Firestein, G. S. (1990). Cytokines in chronic inflammatory arthritis. V. Mutual antagonism between interferon-γ and tumor necrosis factor-alpha on HLA-DR expression, proliferation, collagenase production, and granulocyte macrophage colony-stimulating factor production by rheumatoid arthritis synoviocytes. J. Clin. Invest. 86 (6), 1790–1798. doi: 10.1172/JCI114908
Andronik-Lion, V., Boucher, J. L., Delaforge, M., Henry, Y., Mansuy, D. (1992). Formation of nitric oxide by cytochrome P450-catalyzed oxidation of aromatic amidoximes. Biochem. Biophys. Res. Commun. 185 (1), 452–458. doi: 10.1016/s0006-291x(05)81006-x
Arnett, F. C., Edworthy, S. M., Bloch, D. A., McShane, D. J., Fries, J. F., Cooper, N. S., et al. (1988). The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 31 (3), 315–324. doi: 10.1002/art.1780310302
Azizi, G., Jadidi-Niaragh, F., Mirshafiey, A. (2013). Th17 Cells in Immunopathogenesis and treatment of rheumatoid arthritis. Int. J. Rheum. Dis. 16 (3), 243–253. doi: 10.1111/1756-185X.12132
Banda, N. K., Thurman, J. M., Kraus, D., Wood, A., Carroll, M. C., Arend, W. P., et al. (2006). Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J. Immunol. 177 (3), 1904–1912. doi: 10.4049/jimmunol.177.3.1904
Bartok, B., Firestein, G. S. (2010). Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 233 (1), 233–255. doi: 10.1111/j.0105-2896.2009.00859.x
Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., et al. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 98 (24), 13681–13686. doi: 10.1073/pnas.251194298
Bergman, J., Lindstrom, J. O., Tilstam, U. (1985). The structure and properties of some indolic constituents in Couroupita guianensis Aubl. Tetrahedron 41 (14), 2879–2881. doi: 10.1016/S0040-4020(01)96609-8
Bernotiene, E., Palmer, G., Talabot-Ayer, D., Szalay-Quinodoz, I., Aubert, M. L., Gabay, C. (2004). Delayed resolution of acute inflammation during zymosan-induced arthritis in leptin-deficient mice. Arthritis Res. Ther. 6 (3), R256–R263. doi: 10.1186/Ar1174
Bhagwat, S. S. (2007). MAP kinase inhibitors in inflammation and autoimmune disorders. Annu. Rep. Med. Chem. (42), 265–278. doi: 10.1016/S0065-7743(07)42017-6
Bogoyevitch, M. A., Boehm, I., Oakley, A., Ketterman, A. J., Barr, R. K. (2004). Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochim. Biophys. Acta 1697 (1-2), 89–101. doi: 10.1016/j.bbapap.2003.11.016
Bogoyevitch, M. A., Ngoei, K. R., Zhao, T. T., Yeap, Y. Y., Ng, D. C. (2010). c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim. Biophys. Acta 1804 (3), 463–475. doi: 10.1016/j.bbapap.2009.11.002
Brand, D. D., Kang, A. H., Rosloniec, E. F. (2003). Immunopathogenesis of collagen arthritis. Springer Semin. Immunopathol. 25 (1), 3–18. doi: 10.1007/s00281-003-0127-1
Brufani, M., Fedeli, W., Mazza, F., Gerhard, A., Kellersc, W. (1971). Structure of tryptanthrin. Experientia 27 (11), 1249–1250. doi: 10.1007/bf02136666
Burrage, P. S., Mix, K. S., Brinckerhoff, C. E. (2006). Matrix metalloproteinases: role in arthritis. Front. Biosci. 11, 529–543. doi: 10.2741/1817
Chang, B. Y., Huang, M. M., Francesco, M., Chen, J., Sokolove, J., Magadala, P., et al. (2011). The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 13 (4), R115. doi: 10.1186/ar3400
Chang, J. H., Lee, K. J., Kim, S. K., Yoo, D. H., Kang, T. Y. (2014). Validity of SW982 synovial cell line for studying the drugs against rheumatoid arthritis in fluvastatin-induced apoptosis signaling model. Indian J. Med. Res. 139 (1), 117–124.
Chen, G. S., Bhagwat, B. V., Liao, P. Y., Chen, H. T., Lin, S. B., Chern, J. W. (2007). Specific stabilization of DNA triple helices by indolo[2,1-b]quinazolin-6,12-dione derivatives. Bioorg. Med. Chem. Lett. 17 (6), 1769–1772. doi: 10.1016/j.bmcl.2006.12.079
Cheng, H. M., Kuo, Y. Z., Chang, C. Y., Chang, C. H., Fang, W. Y., Chang, C. N., et al. (2020). The anti-TH17 polarization effect of Indigo naturalis and tryptanthrin by differentially inhibiting cytokine expression. J. Ethnopharmacol. 112760. doi: 10.1016/j.jep.2020.112760
Daina, A., Michielin, O., Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. doi: 10.1038/srep42717
Danz, H., Stoyanova, S., Wippich, P., Brattstrom, A., Hamburger, M. (2001). Identification and isolation of the cyclooxygenase-2 inhibitory principle in Isatis tinctoria. Planta Med. 67 (5), 411–416. doi: 10.1055/s-2001-15805
Davignon, J. L., Hayder, M., Baron, M., Boyer, J. F., Constantin, A., Apparailly, F., et al. (2013). Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology 52 (4), 590–598. doi: 10.1093/rheumatology/kes304
Delire, M. (1994). Different modes of action of high-dose immunoglobulins in rheumatoid arthritis. Acta Univ. Carol Med. (Praha) 40 (1-4), 95–99.
Doogue, M. P., Polasek, T. M. (2013). The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 4 (1), 5–7. doi: 10.1177/2042098612469335
Drexler, S. K., Kong, P. L., Wales, J., Foxwell, B. M. (2008). Cell signalling in macrophages, the principal innate immune effector cells of rheumatoid arthritis. Arthritis Res. Ther. 10, 216. doi: 10.1186/Ar2481
Firestein, G. S., Corr, M. (2005). Common mechanisms in immune-mediated inflammatory disease. J. Rheumatol. Suppl. 73, 8–13.
Firestein, G. S. (2003). Evolving concepts of rheumatoid arthritis. Nature 423 (6937), 356–361. doi: 10.1038/nature01661
Gao, B., Calhoun, K., Fang, D. (2006). The proinflammatory cytokines IL-1β and TNF-α induce the expression of Synoviolin, an E3 ubiquitin ligase, in mouse synovial fibroblasts via the Erk1/2-ETS1 pathway. Arthritis Res. Ther. 8 (6), R172. doi: 10.1186/ar2081
Guma, M., Kashiwakura, J.-i., Crain, B., Kawakami, Y., Beutler, B., Firestein, G. S., et al. (2010). JNK1 controls mast cell degranulation and IL-1β production in inflammatory arthritis. Proc. Natl. Acad. Sci. U.S.A. 107 (51), 22122–22127. doi: 10.1073/pnas.1016401107
Guo, Z. R. (2017). The modification of natural products for medical use. Acta Pharm. Sin. B 7 (2), 119–136. doi: 10.1016/j.apsb.2016.06.003
Han, Z. N., Boyle, D. L., Aupperle, K. R., Bennett, B., Manning, A. M., Firestein, G. S. (1999). Jun N-terminal kinase in rheumatoid arthritis. J. Pharmacol. Exp. Therapeut. 291 (1), 124–130.
Han, Z., Boyle, D. L., Chang, L., Bennett, B., Karin, M., Yang, L., et al. (2001). c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest. 108 (1), 73–81. doi: 10.1172/JCI12466
Han, H., Lee, K. S., Rong, W., Zhang, G. (2012). Different roles of peripheral mitogen-activated protein kinases in carrageenan-induced arthritic pain and arthritis in rats. Anesth. Analg. 115 (5), 1221–1227. doi: 10.1213/ANE.0b013e318266c1ee
Han, R., Rostami-Yazdi, M., Gerdes, S., Mrowietz, U. (2012). Triptolide in the treatment of psoriasis and other immune-mediated inflammatory diseases. Br. J. Clin. Pharmacol. 74 (3), 424–436. doi: 10.1111/j.1365-2125.2012.04221.x
Hegen, M., Keith, J. C., Collins, M., Nickerson-Nutter, C. L. (2008). Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann. Rheum. Dis. 67 (11), 1505–1515. doi: 10.1136/ard.2007.076430
Hietala, M. A., Jonsson, I. M., Tarkowski, A., Kleinau, S., Pekna, M. (2002). Complement deficiency ameliorates collagen-induced arthritis in mice. J. Immunol. 169 (1), 454–459. doi: 10.4049/jimmunol.169.1.454
Honda, G., Tabata, M., Tsuda, M. (1979). The antimicrobial specificity of tryptanthrin. Planta Med. 37 (2), 172–174. doi: 10.1055/s-0028-1097320
Hong, J. Y. (2011). Role of natural product diversity in chemical biology. Curr. Opin. Chem. Biol. 15 (3), 350–354. doi: 10.1016/j.cbpa.2011.03.004
Inoue, T., Hammaker, D., Boyle, D. L., Firestein, G. S. (2005). Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast-like synoviocytes. J. Immunol. 174 (7), 4301–4306. doi: 10.4049/jimmunol.174.7.4301
Ishihara, T., Kohno, K., Ushio, S., Iwaki, K., Ikeda, M., Kurimoto, M. (2000). Tryptanthrin inhibits nitric oxide and prostaglandin E(2) synthesis by murine macrophages. Eur. J. Pharmacol. 407 (1-2), 197–204. doi: 10.1016/S0014-2999(00)00674-9
Iwaki, K., Ohashi, E., Arai, N., Kohno, K., Ushio, S., Taniguchi, M., et al. (2011). Tryptanthrin inhibits Th2 development, and IgE-mediated degranulation and IL-4 production by rat basophilic leukemia RBL-2H3 cells. J. Ethnopharmacol. 134 (2), 450–459. doi: 10.1016/j.jep.2010.12.041
Kanai, T., Kondo, N., Okada, M., Sano, H., Okumura, G., Kijima, Y., et al. (2020). The JNK pathway represents a novel target in the treatment of rheumatoid arthritis through the suppression of MMP-3. J. Orthop. Surg. Res. 15 (1), 87. doi: 10.1186/s13018-020-01595-9
Kim, K. O., Park, S. Y., Han, C. W., Chung, H. K., Yoo, D. H., Han, J. S. (2008). Effect of sildenafil citrate on interleukin-1beta-induced nitric oxide synthesis and iNOS expression in SW982 cells. Exp. Mol. Med. 40 (3), 286–293. doi: 10.3858/emm.2008.40.3.286
Kimoto, T., Hino, K., Koya-Miyata, S., Yamamoto, Y., Takeuchi, M., Nishizaki, Y., et al. (2001). Cell differentiation and apoptosis of monocytic and promyelocytic leukemia cells (U-937 and HL-60) by tryptanthrin, an active ingredient of Polygonum tinctorium Lour. Pathol. Int. 51 (5), 315–325. doi: 10.1046/j.1440-1827.2001.01204.x
Kinne, R. W., Brauer, R., Stuhlmuller, B., Palombo-Kinne, E., Burmester, G. R. (2000). Macrophages in rheumatoid arthritis. Arthritis Res. 2 (3), 189–202. doi: 10.1186/Ar86
Koch, P., Gehringer, M., Laufer, S. A. (2015). Inhibitors of c-Jun N-Terminal Kinases: An Update. J. Med. Chem. 58 (1), 72–95. doi: 10.1021/Jm501212r
Kochetkova, I., Trunkle, T., Callis, G., Pascual, D. W. (2008). Vaccination without autoantigen protects against collagen II-induced arthritis via immune deviation and regulatory T cells. J. Immunol. 181 (4), 2741–2752. doi: 10.4049/jimmunol.181.4.2741
Kochetkova, I., Golden, S., Holderness, K., Callis, G., Pascual, D. W. (2010). IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J. Immunol. 184 (12), 7144–7153. doi: 10.4049/jimmunol.0902739
Krivogorsky, B., Grundt, P., Yolken, R., Jones-Brando, L. (2008). Inhibition of toxoplasma gondii by indirubin and tryptanthrin analogs. Antimicrob. Agents Chemother. 52 (12), 4466–4469. doi: 10.1128/AAC.00903-08
Li, J., Hsu, H. C., Mountz, J. D. (2012). Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 14 (5), 445–454. doi: 10.1007/s11926-012-0272-4
Liacini, A., Sylvester, J., Li, W. Q., Zafarullah, M. (2002). Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor κB (NF-κB) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 21 (3), 251–262. doi: 10.1016/s0945-053x(02)00007-0
Liao, X., Leung, K. N. (2013). Tryptanthrin induces growth inhibition and neuronal differentiation in the human neuroblastoma LA-N-1 cells. Chem. Biol. Interact. 203 (2), 512–521. doi: 10.1016/j.cbi.2013.03.001
Liu, X., Ouyang, S., Yu, B., Liu, Y., Huang, K., Gong, J., et al. (2010). PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 38, W609–W614. doi: 10.1093/nar/gkq300
Liu, Q., Dong, L., Li, H., Yuan, J., Peng, Y., Dai, S. (2016). Lentinan mitigates therarubicin-induced myelosuppression by activating bone marrow-derived macrophages in an MAPK/NF-κB-dependent manner. Oncol. Rep. 36 (1), 315–323. doi: 10.3892/or.2016.4769
Lu, S. W., Wang, Q. S., Li, G. Y., Sun, S., Guo, Y. Y., Kuang, H. X. (2015). The treatment of rheumatoid arthritis using Chinese medicinal plants: From pharmacology to potential molecular mechanisms. J. Ethnopharmacol. 176, 177–206. doi: 10.1016/j.jep.2015.10.010
Lubberts, E. (2015). Role of T lymphocytes in the development of rheumatoid arthritis. implications for treatment. Curr. Pharmaceut. Des. 21 (2), 142–146. doi: 10.2174/1381612820666140825122247
McIntyre, K. W., Shuster, D. J., Gillooly, K. M., Dambach, D. M., Pattoli, M. A., Lu, P., et al. (2003). A highly selective inhibitor of I κB kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 48 (9), 2652–2659. doi: 10.1002/Art.11131
Mehan, S., Meena, H., Sharma, D., Sankhla, R. (2011). JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J. Mol. Neurosci. 43 (3), 376–390. doi: 10.1007/s12031-010-9454-6
Mishima, M., Kobayashi, S., Hashida, R., Yuzuriha, T., Sato, T., Satoh, T. (1991). Enzymatic hydrolysis of indometacin farnesil, a prodrug of indomethacin, by carboxylesterase in cultured synovial cells. Res. Commun. Chem. Pathol. Pharmacol. 72 (2), 183–190.
Mitchell, J. P., Carmody, R. J. (2018). NF-κB and the Transcriptional Control of Inflammation. Transcriptional Gene Regul. Health Dis. 335, 41–84.
Myers, L. K., Rosloniec, E. F., Cremer, M. A., Kang, A. H. (1997). Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 61 (19), 1861–1878. doi: 10.1016/s0024-3205(97)00480-3
Nakae, S., Nambu, A., Sudo, K., Iwakura, Y. (2003). Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171 (11), 6173–6177. doi: 10.4049/jimmunol.171.11.6173
Nandakumar, K. S., Andren, M., Martinsson, P., Bajtner, E., Hellstrom, S., Holmdahl, R., et al. (2003). Induction of arthritis by single monoclonal IgG anti-collagen type II antibodies and enhancement of arthritis in mice lacking inhibitory FcγRIIB. Eur. J. Immunol. 33 (8), 2269–2277. doi: 10.1002/eji.200323810
Neumann, E., Barnum, S. R., Tarner, I. H., Echols, J., Fleck, M., Judex, M., et al. (2002). Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum. 46 (4), 934–945. doi: 10.1002/art.10183
Nikoloudaki, G., Brooks, S., Peidl, A. P., Tinney, D., Hamilton, D. W. (2020). JNK signaling as a key modulator of soft connective tissue physiology, pathology, and healing. Int. J. Mol. Sci. 21 (3), 1015. doi: 10.3390/ijms21031015
Nishikawa, M., Myoui, A., Tomita, T., Takahi, K., Nampei, A., Yoshikawa, H. (2003). Prevention of the onset and progression of collagen-induced arthritis in rats by the potent p38 mitogen-activated protein kinase inhibitor FR167653. Arthritis Rheum. 48 (9), 2670–2681. doi: 10.1002/art.11227
Onambele, L. A., Riepl, H., Fischer, R., Pradel, G., Prokop, A., Aminake, M. N. (2015). Synthesis and evaluation of the antiplasmodial activity of tryptanthrin derivatives. Int. J. Parasitol. Drugs Drug Resist. 5 (2), 48–57. doi: 10.1016/j.ijpddr.2015.03.002
Pathania, A. S., Kumar, S., Guru, S. K., Bhushan, S., Sharma, P. R., Aithagani, S. K., et al. (2014). The synthetic tryptanthrin analogue suppresses STAT3 signaling and induces caspase dependent apoptosis via ERK up regulation in human leukemia HL-60 cells. PLoS One 9 (11), e110411. doi: 10.1371/journal.pone.0110411
Pergola, C., Jazzar, B., Rossi, A., Northoff, H., Hamburger, M., Sautebin, L., et al. (2012). On the inhibition of 5-lipoxygenase product formation by tryptanthrin: mechanistic studies and efficacy in vivo. Br. J. Pharmacol. 165 (3), 765–776. doi: 10.1111/j.1476-5381.2011.01605.x
Plotnikov, M. B., Aliev, O. I., Shamanaev, A. Y., Sidekhmenova, A. V., Anishchenko, A. M., Fomina, T. I., et al. (2020). Antihypertensive activity of a new c-Jun N-terminal kinase inhibitor in spontaneously hypertensive rats. Hypertens. Res. doi: 10.1038/s41440-020-0446-9
Recio, M. C., Cerda-Nicolas, M., Potterat, O., Hamburger, M., Rios, J. L. (2006). Anti-inflammatory and antiallergic activity in vivo of lipophilic Isatis tinctoria extracts and tryptanthrin. Planta Med. 72 (6), 539–546. doi: 10.1055/s-2006-931562
Sato, T., Enoki, Y., Sakamoto, Y., Yokota, K., Okubo, M., Matsumoto, M., et al. (2015). Donepezil prevents RANK-induced bone loss via inhibition of osteoclast differentiation by downregulating acetylcholinesterase. Heliyon 1 (1):e00013. doi: 10.1016/j.heliyon.2015.e00013
Schepetkin, I. A., Kirpotina, L. N., Khlebnikov, A. I., Hanks, T. S., Kochetkova, I., Pascual, D. W., et al. (2012). Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 81 (6), 832–845. doi: 10.1124/mol.111.077446
Schepetkin, I. A., Kirpotina, L. N., Hammaker, D., Kochetkova, I., Khlebnikov, A. I., Lyakhov, S. A., et al. (2015). Anti-inflammatory effects and joint protection in collagen-induced arthritis after treatment with IQ-1S, a selective c-Jun N-terminal kinase inhibitor. J. Pharmacol. Exp. Ther. 353 (3), 505–516. doi: 10.1124/jpet.114.220251
Schepetkin, I. A., Khlebnikov, A. I., Potapov, A. S., Kovrizhina, A. R., Matveevskaya, V. V., Belyanin, M. L., et al. (2019). Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 161, 179–191. doi: 10.1016/j.ejmech.2018.10.023
Schramm, C., Kriegsmann, J., Protschka, M., Huber, S., Hansen, T., Schmitt, E., et al. (2004). Susceptibility to collagen-induced arthritis is modulated by TGFβ responsiveness of T cells. Arthritis Res. Ther. 6 (2), R114–R119. doi: 10.1186/Ar1039
Schurgers, E., Billiau, A., Matthys, P. (2011). Collagen-induced arthritis as an animal model for rheumatoid arthritis: focus on interferon-γ. J. Interferon Cytokine Res. 31 (12), 917–926. doi: 10.1089/jir.2011.0056
Shafiey, S. I., Mohamed, W. R., Abo-Saif, A. A. (2018). Paroxetine and rivastigmine mitigates adjuvant-induced rheumatoid arthritis in rats: Impact on oxidative stress, apoptosis and RANKL/OPG signals. Life Sci. 212, 109–118. doi: 10.1016/j.lfs.2018.09.046
Sharif, K., Sharif, A., Jumah, F., Oskouian, R., Tubbs, R. S. (2018). Rheumatoid arthritis in review: Clinical, anatomical, cellular and molecular points of view. Clin. Anat. 31 (2), 216–223. doi: 10.1002/ca.22980
Sharma, S., Ghosh, S., Singh, L. K., Sarkar, A., Malhotra, R., Garg, O. P., et al. (2014). Identification of autoantibodies against transthyretin for the screening and diagnosis of rheumatoid arthritis. PLoS One 9 (4), e93905. doi: 10.1371/journal.pone.0093905
Stevens, T. L., Bossie, A., Sanders, V. M., Fernandezbotran, R., Coffman, R. L., Mosmann, T. R., et al. (1988). Regulation of antibody isotype secretion by subsets of antigen-specific helper T-cells. Nature 334 (6179), 255–258. doi: 10.1038/334255a0
Tak, P. P., Bresnihan, B. (2000). The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum. 43 (12), 2619–2633. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V
Takeno, A., Kanazawa, I., Notsu, M., Tanaka, K. I., Sugimoto, T. (2018). Glucose uptake inhibition decreases expressions of receptor activator of nuclear factor-κB ligand (RANKL) and osteocalcin in osteocytic MLO-Y4-A2 cells. Am. J. Physiol. Endocrinol. Metab. 314 (2), E115–E123. doi: 10.1152/ajpendo.00159.2017
Tanaka, S. (2019). RANKL is a therapeutic target of bone destruction in rheumatoid arthritis. F1000Res 8, 533. doi: 10.12688/f1000research.17296.1
Tang, W., Zuo, J. P. (2012). Immunosuppressant discovery from Tripterygium wilfordii Hook f: the novel triptolide analog (5R)-5-hydroxytriptolide (LLDT-8). Acta Pharmacol. Sin. 33 (9), 1112–1118. doi: 10.1038/aps.2012.108
Thalhamer, T., McGrath, M. A., Harnett, M. M. (2008). MAPKs and their relevance to arthritis and inflammation. Rheumatology 47 (4), 409–414. doi: 10.1093/rheumatology/kem297
Tsai, Y. C., Lee, C. L., Yen, H. R., Chang, Y. S., Lin, Y. P., Huang, S. H., et al. (2020). Antiviral action of tryptanthrin isolated from Strobilanthes cusia leaf against human coronavirus NL63. Biomolecules 10 (3), 366. doi: 10.3390/biom10030366
Tsuji, F., Oki, K., Senda, T., Horiuchi, M., Mita, S. (1999). Effects of mitogen-activated protein kinase inhibitors or phosphodiesterase inhibitors on interleukin-1-induced cytokines production in synovium-derived cells. Immunol. Lett. 68 (2-3), 275–279. doi: 10.1016/s0165-2478(99)00051-6
Wang, Z., Wu, X., Wang, C. L., Wang, L., Sun, C., Zhang, D. B., et al. (2018). Tryptanthrin protects mice against dextran sulfate sodium-induced colitis through inhibition of TNF-α/NF-κB and IL-6/STAT3 pathways. Molecules 23 (5), 1062. doi: 10.3390/molecules23051062
Weaver, C. T., Hatton, R. D., Mangan, P. R., Harrington, L. E. (2007). IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25, 821–852. doi: 10.1146/annurev.immunol.25.022106.141557
Williams, P. J., Jones, R. H., Rademacher, T. W. (1998). Correlation between IgG anti-type II collagen levels and arthritic severity in murine arthritis. Autoimmunity 27 (4), 201–207. doi: 10.3109/08916939808993831
Xu, L., Feng, X., Shi, Y., Wang, X., Kong, X., Zhang, M., et al. (2015). Interleukin-29 induces receptor activator of NF-κB ligand expression in fibroblast-like synoviocytes via MAPK signaling pathways. Int. J. Rheum. Dis. 18 (8), 842–849. doi: 10.1111/1756-185X.12747
Xu, J., Feng, Z., Chen, S., Zhu, J., Wu, X., Chen, X., et al. (2019). Taxol alleviates collagen-induced arthritis in mice by inhibiting the formation of microvessels. Clin. Rheumatol. 38 (1), 19–27. doi: 10.1007/s10067-017-3646-1
Yang, S., Li, X., Hu, F., Li, Y., Yang, Y., Yan, J., et al. (2013). Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in Lewis lung cancer (LLC) tumor-bearing mice. J. Med. Chem. 56 (21), 8321–8331. doi: 10.1021/jm401195n
Keywords: c-Jun N-terminal kinase, tryptanthrin, tryptanthrin-6-oxime, kinase inhibitor, arthritis, inflammation, collagen-induced arthritis, collagen antibody-induced arthritis
Citation: Kirpotina LN, Schepetkin IA, Hammaker D, Kuhs A, Khlebnikov AI and Quinn MT (2020) Therapeutic Effects of Tryptanthrin and Tryptanthrin-6-Oxime in Models of Rheumatoid Arthritis. Front. Pharmacol. 11:1145. doi: 10.3389/fphar.2020.01145
Received: 07 April 2020; Accepted: 13 July 2020;
Published: 24 July 2020.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Aleksander M. Grabiec, Jagiellonian University, PolandThomas Karonitsch, Medical University of Vienna, Austria
Gabriel Criado, Research Institute Hospital October 12, Spain
Copyright © 2020 Kirpotina, Schepetkin, Hammaker, Kuhs, Khlebnikov and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark T. Quinn, bXF1aW5uQG1vbnRhbmEuZWR1
†These authors have contributed equally to this work