 You Gao
You Gao Yunfeng Li
Yunfeng Li Xiaomei Zhuang
Xiaomei Zhuang- State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing, China
YL-IPA08, exerting rapid antidepressant-like and anxiolytic-like effects on behaviors by translocator protein (TSPO) mediation, is a novel compound that has been discovered and developed at our institute. Fit-for-purpose pharmacokinetic properties is urgently needed to be discovered as early as possible for a new compound. YL-IPA08 exhibited low bioavailability (∼6%) during the preliminary pharmacokinetics study in rats after oral administration. Our aim was to determine how metabolic disposition by microsomal P450 enzymes in liver and intestine limited YL-IPA08’s bioavailability and further affected brain penetration to the target. Studies of in vitro metabolic stability and permeability combined with in vivo oral bioavailability, panel CYP inhibitor co-administration via different routes, and double cannulation rats were conducted to elucidate the intestinal and hepatic first-pass effect of YL-IPA08 on bioavailability. Unbound brain-to-plasma ratio (Kp,uu) in rats was determined at steady state. Results indicated that P450-mediated elimination appeared to be important for its extensive first-pass effect with comparative contribution of gut (35%) and liver (17%), and no significant species difference was observed. The unbound concentration of YL-IPA08 in rat brain (6.5 pg/ml) was estimated based on Kp,uu (0.18) and was slightly higher than in vitro TSPO-binding activity (4.9 pg/ml). Based on the onset efficacy of YL-IPA08 toward TPSO in brain and Kp,uu, therapeutic human plasma concentration was predicted to be ∼27.2 ng/ml would easily be reached even with unfavorable bioavailability.
Introduction
Major depressive disorder (MDD) is a chronic and debilitating disorder with high rates of medical and psychiatric co-morbidity. Translocator protein (TSPO, 18 kDa) has drawn growing attention in the pathophysiology of stress-response and stress-related disorders (Rupprecht et al., 2009; Pinna, 2010). YL-IPA08, upon binding to TSPO, stimulates the de novo synthesis of neurosteroids, which potentiates the GABAA receptor function and, consequently, conducts its antidepressant- and anxiolytic-like effects (Zhang et al., 2017).
Oral bioavailability and target exposure related to the pharmacological efficacy of orally administered drugs is a key aspect of new drug development (Obach, 2001; Li et al., 2019). However, YL-IPA08 exhibited low bioavailability (∼6%) during the preliminary pharmacokinetics (PK) study in rats after oral intake. It is well recognized that a drug with low oral bioavailability can be impacted by multiple factors including absorption barrier, intestinal and hepatic metabolism before going into systemic circulation (Fan et al., 2019). Both CYP and UGT enzymes are major drug metabolizing enzymes in gut and liver that facilitate the elimination of xenobiotics. Although the liver contains higher amount of CYPs and UGTs, the small intestine is usually exposed to higher concentration of xenobiotics. Thus, both liver and intestine play important roles in bioavailability (Ohno and Nakajin, 2009; Court et al., 2012; Karlsson et al., 2013). Several studies have demonstrated the relative importance of intestinal metabolism to low bioavailability, and the situations were substrate-dependent (Cubitt et al., 2009; Fan et al., 2019).
To address the roles of the liver and intestine in YL-IPA08 first-pass metabolism, gastrointestinal absorption and hepatic and gut first-pass metabolism of YL-IPA08 were evaluated to understand of the causes of low bioavailability. Then, the antidepressant-like efficacy achieved in rat models were taken to explore the effective brain exposure of YL-IPA08 under the same oral dose regiment combined with in vitro TSPO-binding assessment. Liver microsomes and intestinal microsomes of rat and human were firstly used to conduct in vitro stability of YL-IPA08 in the presence of NADPH and UDPGA as cofactors to identify the major enzymes and metabolic organs responsible for the first-pass elimination. In the view of rat model is commonly used in pharmacokinetic studies (Zeng et al., 2019), rat pharmacokinetic studies were conducted using ABT as CYP inhibitor to knock out the function of hepatic CYPs or hepatic and intestinal CYPs via different routes (Strelevitz et al., 2006). In addition, a rat model with double cannulation of portal and jugular veins (Murakam et al., 2003) was used to testify the contribution of hepatic metabolism. As a central nervous system (CNS) drug, CNS penetration assessment was performed in rat under steady-state to obtain the brain/plasma partition. Kp,uu was calculated by correction of unbound fractions of YL-IPA08 in plasma and brain. Brain exposure of YL-IPA08 was estimated in the case of limited systemic exposure. Based on the current results, we attempt to elucidate the disposition characters of YL-IPA08 associated with its efficacy and explore the potential of clinical application based on our mechanistic understanding.
Materials and Methods
Chemical and Reagents
YL-IPA08 and AC-5216 internal standard (IS) (Figure 1) were supplied by chemical synthesis laboratory of our Institute (Beijing, China) with purity greater than 99%. Midazolam (MDZ), 1′-OH-MDZ, phenacetin, acetaminophen, diclofenac, S-mephenytoin, 4-OH-diclofenac, 4-OH-mephenytoin, bupropion, OH-bupropion, amodiaquine, N-desethylamodiaquine, dextromethorphan, dextrorphan, atenolol, propranolol, and digoxin were all purchased from Sigma-Aldrich (St. Louis, MO). Human liver microsomes (pool of 50, mixed gender), male rat liver microsomes (pool of 495), human intestinal microsomes (pool of 15, mixed gender) and male rat intestinal microsomes (pool of 100) were purchased from XENOTECH (Lenexa, KS). NADPH was purchased from Roche Life Science (Basel-Stadt, Switzerland). Other reagents were of HPLC grade or better.
FIGURE 1. Chemical structural of YL-IPA08 (A) and AC-5216 (IS) (B).
In Vitro Study
NADPH- and UDPGA-Dependent Hepatic Clearance in Liver and Intestinal Microsomes of Rat and Human
Pilot in vitro metabolic stability experiments were conducted to identify the incubation conditions to capture the linear elimination of YL-IPA08 including concentration of YL-IPA08, protein concentration of microsomes, and stop times. The CYP-mediated hepatic and intestinal metabolic stability test was performed in incubations containing YL-IPA08 (1 μM, dissolved in saline) in pooled rat or human liver and intestinal microsomes (0.2 mg/ml protein) in 100 mM of potassium phosphate buffer with 3 mM of MgCl2, pH 7.4. The mixture was pre-incubated at 37°C for 5 min. The reaction was started with the addition of NADPH (with final concentration of 1 mM). For UGT-mediated metabolic clearance assays, alamethicin at final concentration of 50 μg/mg protein, 1 μM of YL-IPA08 and rat or human liver and intestinal microsomes in 100 mM of potassium phosphate buffer (pH 7.4) were mixed on ice for 15 min. The mixture was then pre-incubated at 37°C for 5 min. The reaction was started with the addition of UDPGA (2.5 mM). Aliquots of the incubation were removed at different time points in the duration of 60 min after dosing of cofactors (NADPH or UDPGA) and diluted with 6× volume of chilled acetonitrile containing internal standard to stop the reactions. After centrifugation at 13,000 g for 10 min, the supernatant was collected and stored at −20°C until LC-MS/MS analysis. Negative control without NADPH and positive control with cocktailed probe compounds (phenacetin, diclofenac, S-mephenytoin, bupropion, amodiaquine, dextromethorphan, and midazolam) in liver microsomes and midazolam in intestinal microsomes were conducted simultaneously.
Transcellular Transport Experiment With Caco-2 Cells
Caco-2 cell lines were cultured (ATCC, Manassas, VA, United States) as described previously (Liu et al., 2014). Briefly, the cells were cultured at 37°C in 5% CO2 at 90% humidity in DMEM high glucose medium containing 20% fetal bovine serum, 1% nonessential amino acids, 100 U/ml penicillin and 1% streptomycin. For the efflux studies, the cells were seeded onto polyethylene terephthalate Millicell® cell culture inserts (0.4 μm pore size, 6.5 mm diameter, Millipore Corporation, Billerica, MA, United States) at a density of 1.7 × 105 cells/ml. The culture medium was refreshed on the day after seeding, after which it was refreshed every other day and on the day before the transport experiment. The cells were cultured for 20–22 days after seeding and then evaluated by measuring the transepithelial electrical resistance (TEER) (Millicell ERS®, Millipore Corporation) before experiments. Batches of Caco-2 cells were certified by measuring the TEER values and the apparent permeability coefficient (Papp) of control compounds: atenolol (10 μM, low permeability), propranolol (10 μM, high permeability), and P-gp substrate digoxin (10 μM). Transepithelial permeability studies for YL-IPA08 (10 μM) were conducted from apical to basolateral side (A-B) and basolateral to apical side (B-A) for 120 min 0.2% BSA was included in the receiver solution to avoid nonspecific binding. At 60, 90, and 120 min, half volume of solution from receiver side was withdrawn, and same volume of HBSS was immediately added. Obtained samples were precipitated by adding 4× volumes of acetonitrile containing IS and centrifuged. The supernatants were stored in −20°C until further analysis by LC-MS/MS.
Blood/Plasma Partitional Ratio
The blood/plasma partitional ratio (Rb/p) in rat and human blood was measured in vitro using fresh pooled blood including heparin. Whole blood was preincubated at 37°C in a water bath, and spiked with YL-IPA08 at 1 µM. The blood samples were incubated at 37°C for 1 h. After incubation, aliquots of 25 µl spiked whole blood were removed, and the remaining blood was centrifuged at 2,000 × g for 10 min, after which 25 µl aliquots of plasma were removed. Obtained blood and plasma samples were spiked with same volume of blank plasma or blood, respectively, and then immediately precipitated by adding 4× volumes of acetonitrile containing IS to get the same matrix. The concentrations of YL-IPA08 in blood and plasma were determined by LC-MS/MS.
Protein Binding in Different Biomatrices
The fraction of unbound YL-IPA08 in plasma and liver microsomes of rat and human, and brain homogenates of rat were determined utilizing Rapid Equilibrium Dialysis method (Waters et al., 2008). Rat brain homogenate was harvested in 4-fold volume (w/v) of PBS (pH 7.4). YL-IPA08 were spiked into fresh plasma, rat brain homogenate, liver microsomes (0.2 mg/ml protein) at concentration of 1 µM, and then dialyzed against biomatrices on a shaker at 37°C for 4 h. To avoid the high non-specific binding of YL-IPA08, 0.01% Tween 80 was added in the incubation mixture. At the end of the incubation, protein was precipitated with acetonitrile (containing IS) and analyzed by LC-MS/MS. Phenacetin, quinidine, and warfarin were selected as positive control for the protein binding test.
In Vivo Study
Male SD rats (200–240 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. Animals were housed in a temperature- and humidity-controlled room with a 12 h light/dark cycle. They were fasted 12 h before the experiments and had ad libitum access to water. The animal experiments were conducted in the Beijing Center for Drug Safety Evaluation and according to a protocol (IACUC-DWZX-2020-691) approved by the Institutional Animal Care and Use Committee of the Center, which followed the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Bioavailability Study
Male rats (n = 6) were administered YL-IPA08 by oral gavage at 1, 3 and 10 mg/kg or i.v. injection at 0.3 mg/kg. Blood samples were collected before and after dosing at 0.033 (i.v. dosing only), 0.083, 0.25, 0.5, 1, 2, 4, 6, 8,12 and 24 h. Blood sample (no more than 100 µl) was collected from the jugular vein into tubes containing heparin on ice and centrifuged within 1 h of collection. Plasma was harvested and stored at −20°C for bioanalysis.
Surgical Procedure for Portal and Jugular Vein Double Cannulation and the Pharmacokinetic Study With the Same Parameters as Normal Rats After p.o.
The surgical procedure for portal vein catheter insertion was performed according to reference (Matsuda et al., 2012). Male SD rats (250–280 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Rats were anesthetized with 2% pentobarbital sodium (0.2–0.3 mg/100 g) intraperitoneally dosed. Cannulation into the jugular vein was performed by the method of Murakami reported (Murakami et al., 2003) with minor modification. Briefly, an incision of ∼0.5 cm near right clavicle was made and the jugular vein was exposed. Next, this vein was cannulated with ∼12.5 cm of silicone gel tubing (0.6 mm I.D. × 0.9 mm O.D., Skillsmodel Inc., Beijing, China). The other side of the tubing was passed through the back skin. 2.5 cm midline incision was made in the abdominal cavity close to ensiform process and the portal vein was detached. To prevent bleeding, the portal vein was ligated temporarily as the catheter was inserted. The silicone tubing (22 cm, 0.6 mm I.D. × 0.9 mm O.D., Skillsmodel Inc., Beijing, China) was inserted immediately and fixed by a purse-string suture on the portal vein. The time to reperfusion was about 1 min after intercepted blood flow. In addition, a catheter with trumpet-shaped opening was used to prevent the catheter from slipping out of the vessel and minimize the effect on blood flow. Another end of the catheter was passed subcutaneously to the skin close to the scapula.
Male rats (n = 5), after recovery from successful double cannulation, were administered YL-IPA08 by oral gavage at 1 mg/kg. Blood samples were collected at 0, 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 12 h. Blood (approximately 100 µl) was collected from the jugular and portal veins into tubes containing heparin on ice and centrifuged within 1 h of collection. Plasma was harvested and stored at −20°C for bioanalysis.
Intestinal and Hepatic Availability of YL-IPA08 Using ABT Block the CYP Metabolism
ABT was used to differentiate the gastrointestinal and hepatic first-pass elimination of YL-IPA08 by conducting four groups of rat in vivo experiments (n = 6 per group). YL-IPA08 was formulated in saline and dosed p.o. (1 mg/kg) or i.v. (0.1 mg/kg or 0.5 mg/kg) in rats pretreated for 1 h with ABT i.v. (50 mg/kg in saline, 5 ml/kg) or 15 h with ABT p.o. (100 mg/kg in saline, 10 ml/kg). Blood was collected from the jugular vein at 0.033 (i.v. dosing only), 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h postdosing. Blood samples were immediately transferred into tubes containing heparin, and plasma was obtained following centrifugation at 2,000 g for 10 min at 4°C. The plasma samples were stored at −20°C until analysis.
Brain Distribution Under Steady State
Rats (n = 3) was i.v. injected YL-IPA08 at loading dose of 0.28 mg/kg, then continuous i.v. infusion at the speed of 0.22 mg/h lasted for 1 h via tail vein. Jugular vein blood samples were collected at 30, 40, 50, and 60 min of i.v. infusion to verify attainment of steady state. Immediately after collecting the last blood sample (60 min), rats were terminally anesthetized and brain tissues were harvested, weighted and frozen at −80°C until analysis. Brain samples were homogenized with 4× fold volume of cold buffer. The homogenates were precipitated with acetonitrile (containing IS) and analyzed.
Bioanalysis Methods
On the day of bioanalysis, all in vitro and in vivo samples were precipitated with acetonitrile (containing IS) and analyzed by LC-MS/MS. Separation was performed using a C18 column (3.0 mm × 50 mm, 2.6 µm, Phenomenex). The mobile phase consisted of water containing 5 mM ammonium acetate (A) and acetonitrile containing 5 mM ammonium acetate (B). Separation was achieved with a 3.5-min run time with the following gradient program: initial conditions of 40% B held for 0.3 min followed by an increase to 95% B over 2.0 min, hold at 95% B for 0.5 min, and return to 40% B over 1 min. The flow rate is 0.7 ml/min. Analyte detection was achieved with an AB Sciex API 5000 Triple quadrupole mass spectrometer operated in positive ion mode with multiple reaction monitoring (MRM). The precursor and product ions transition for YL-IPA08 and internal standard were 453.195/317.100 and 402.389/227.5.
Data Analysis
The in vitro t1/2 in liver microsomal incubation was calculated from the semi-log plot of percentage remaining vs. incubation time and intrinsic clearance (Clint, ml/min/mg protein) was calculated as Eq. 1:
(Suzuki et al., 2003).Apparent permeability was obtained according to the equation Papp = (dQ/dt)/(A × C0), where dQ/dt is the mass transport rate (determined from the slope of the amount transported vs. time plot), A is the surface area of the monolayer, and C0 is the initial concentration of YL-IPA08 in the donor chamber (Suzuki et al., 2003). The efflux ratio was calculated for each study using the following equation: endoplasmic reticulum (ER) = Papp(B–A)/Papp(A–B), where Papp(B–A) and Papp(A–B) represent apparent drug permeability in the B to A and A to B direction, respectively. An efflux ratio greater than 2 indicates net efflux.
The unbound fraction of YL-IPA08 in plasma or liver microsomes (ƒu,x) was calculated as shown in Eq. 2, and unbound fraction in the tissue homogenate (ƒu,b) was calculated according to Eq. 3.
The measured unbound fractions would be higher when a tissue is homogenized and diluted in buffer, the unbound fractions in an undiluted tissues (ƒu,brain) were calculated using Eq. 4 (Kalvass and Maurer, 2002).
Where D represents the fold of dilution factor in brain homogenates. The free fraction of plasma and undiluted tissue is used in tissue-to-plasma exposure ratio calculation.
Pharmacokinetic parameters were calculated by the noncompartmental method using WinNonlin 7.0 (Pharsight, CA). The area under the plasma concentration time curve (AUC) from time 0 to the last time point with a measurable concentration (AUC0–t) was calculated by trapezoidal method. Bioavailability (%F) was calculated as the ratio of the mean dose-normalized AUC values for oral and intravenous dosed groups [Fp.o. = AUCp.o. × Dosei.v. × 100/(AUCi.v. × Dosep.o.)]. In vivo plasma clearance and volume of distribution were archived from the pharmacokinetic study via i.v. injection.
FaFg was calculated using mass balance method and rat portal blood flow (Qpv) of 32 ml/min/kg, where Rb/p is used to convert plasma concentrations to blood concentrations.
Liver and gut extraction ratios in ABT treatment were calculated using the following formula:
Results
LC-MS/MS Methodology
The quantification of YL-IPA08 in rat plasma was fully validated, and in vitro samples were particularly validated by selectivity, precision, and stability. The calibration curves of YL-IPA08 in rat plasma and in vitro incubates ranged in the concentrations of 0.5–500 ng/ml and 1–1,000 nM, respectively. The intra-day precision and inter-day precision (error from the true value) were less than 15% at QC concentrations (full methodology validation results were presented in Supplementary Tables S1–S3).
In vitro Metabolic Elimination of YL-IPA08
YL-IPA08 was discovered to be eliminated in liver microsomes and intestinal microsomes of rat and human to different extents in the presence of CYPs (NADPH) and/or UGTs (UDPGA) co-factors. The disappearance of YL-IPA08 at various time points are presented in Figure 2. The intrinsic clearances (Table 1) were calculated based on data presented in Figure 2. The amounts of YL-IPA08 remained stable during the 60-mininute incubation in the presence of UDPGA in RLM, RIM, HLM and HIM, which indicated that glucuronidation of YL-IPA08 cannot take place both in liver and in gut. The depletions of YL-IPA08 were very rapid in the presence of NADPH in RLM and HLM, with the CYP-mediated intrinsic clearances of 1.13 ± 0.02 and 2.36 ± 0.03 ml/min/mg protein, respectively. If those intrinsic clearance values were normalized by respective CYP contents in rat and human, the results would be 2.76 ± 0.05 and 3.47 ± 0.04 ml/min/nmol CYP protein, respectively. However, in the intestine, the CYP-mediated intrinsic clearances in HIM and RIM were 0.20 ± 0.03 and 0.053 ± 0.02 ml/min/mg protein, respectively. Although statistical significance was observed in the intrinsic clearances between rat and human liver microsomes, the major contributions of hepatic metabolizing enzymes were almost identical. As for the contributions of intestine, the differences between rat and human were remarkable. The main reason for the difference may be the metabolic activity of intestinal microsomes. The eliminations of cocktailed probe compounds in HLM and RLM were well accepted by the literature to verify the model. Midazolam clearances in HIM and RIM were identical to the indicators provided by the vendor.
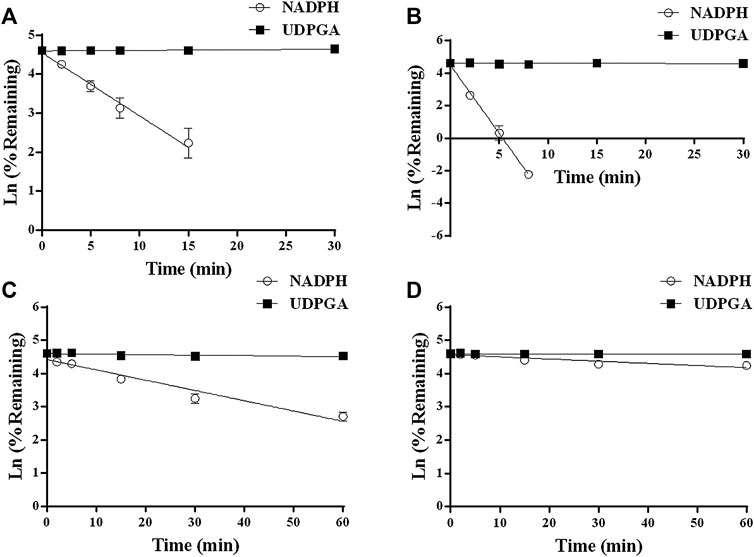
FIGURE 2. Comparison of P450 and UGT depletion profiles of YL-IPA08 in HLM (A), RLM (B), HIM (C), and RIM (D). Open circle represents P450 metabolism, closed square represents glucuronidation (n = 3).
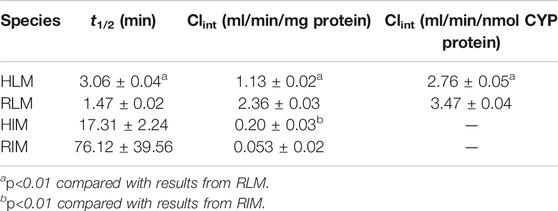
TABLE 1. In vitro metabolic clearance YL-IPA08 in liver and intestinal microsomes of rats and humans in the presence of NADPH (n = 3).
Bidirectional Transport of YL-IPA08 Across Caco-2 Cells
As shown in Table 2, transcellular transport of YL-IPA08 across Caco-2 cells showed high permeability similar to positive control propranolol. P-gp activity was confirmed by the transcellular transport of digoxin with a flux ratio of 6.25. The basal-to-apical permeability of YL-IPA08 was comparable to apical-to-basal permeability, with a flux ratio of 0.66, suggesting the transportation through gut wall is mainly passive penetration. Furthermore, combined with the high aqueous solubility and high permeability, YL-IPA08 belongs to BCS class I compound.
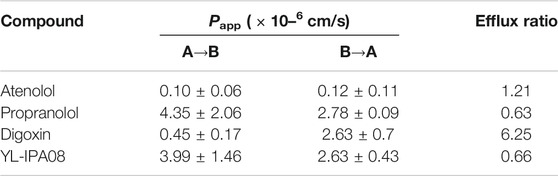
TABLE 2. Transcellular transport of YL-IPA08 in Caco-2 cell line (n = 3).
Blood/Plasma Partitioning and Unbound Fractions in Biomatrices
The mean blood/plasma partitioning (Rb/P) of YL-IPA08 in human and rat whole blood were 1.58 ± 0.06 and 1.12 ± 0.08, respectively. The individual protein binding in rat plasma is 58.48 ± 2.26, 78.08 ± 4.49, and 99.67 ± 0.08% for phenacetin, quinidine, and warfarin, respectively, which is well accepted by the literature to verify the model. Protein bindings of YL-IPA08 in biomatrices were presented in Table 3. Plasma Protein binding is almost 99% bound. Unbound fractions of YL-IPA08 were generated according to the protein bindings. Rb/p and unbound fractions are important parameters to perform theoretical translations between in vitro and in vivo systems.
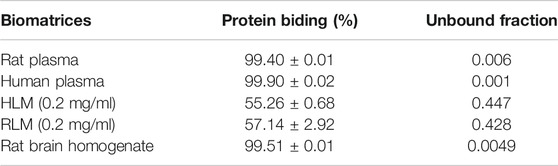
TABLE 3. Protein bindings (%) and unbound fractions of YL-IPA08 in different sorts of biomatrices (n = 3).
In Vivo Pharmacokinetic Behaviors
Pharmacokinetic profiles of YL-IPA08 were archived in rats via i.v. and p.o. dosing (Figure 3). As shown in Table 4, YL-IPA08 exhibited short half-life and high clearance in vivo, which agreed with in vitro results. After oral dosing, the plasma exposures of YL-IPA08 were proportionate within the dose range of 1–10 mg/kg, possessing a low bioavailability of ∼6%.
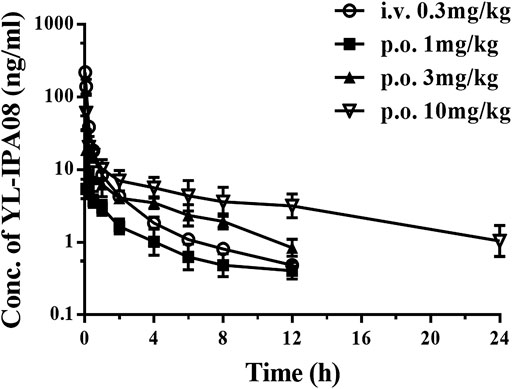
FIGURE 3. Plasma concentration-time profiles of YL-IPA08 after intravenous (0.3 mg/kg) and oral administration of different doses (1, 3, 10 mg/kg) of YL-IPA08 in rats. Error bars represent the standard deviation of the mean concentration (n = 6).
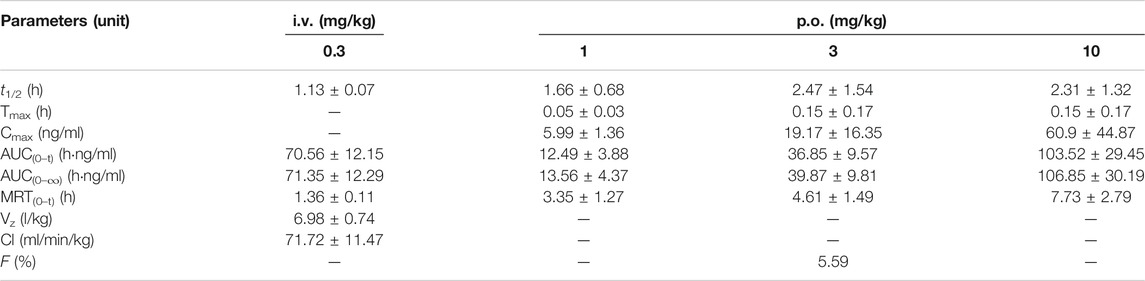
TABLE 4. Pharmacokinetic parameters of YL-IPA08 in rats after intravenous and oral administration (n = 6).
The Effect of ABT on YL-IPA08 Pharmacokinetics
As shown in Figure 4, combination administration with ABT significantly elevated the YL-IPA08 plasma concentrations. The pharmacokinetic parameters are presented in Table 5. After pretreated with ABT through i.v. injection and oral gavage, systemic clearance of YL-IPA08 reduced by about 75 and 80%, with dramatic increased oral bio-availabilities of ∼20 and ∼70%, respectively. Hepatic and gut extraction ratios in different route of ABT treatment groups were elucidated and shown in Table 6. ABT pretreated via i.v. injection or oral gavage, resulted in either hepatic metabolism inhibition or both gut and hepatic metabolism inhibition. Under such conditions, gut and liver have similar contributions to the low bioavailability of YL-IPA08. The extraction ratios of gut and liver were 65 and 83%, respectively, when gut and hepatic metabolism worked normally, while the corresponding extraction ratios were 16 and 17%, respectively, after the majority of the CYP enzymes were inactivated, excepted for the weak inactivation toward CYP2C9 (Linder, et al., 2009).
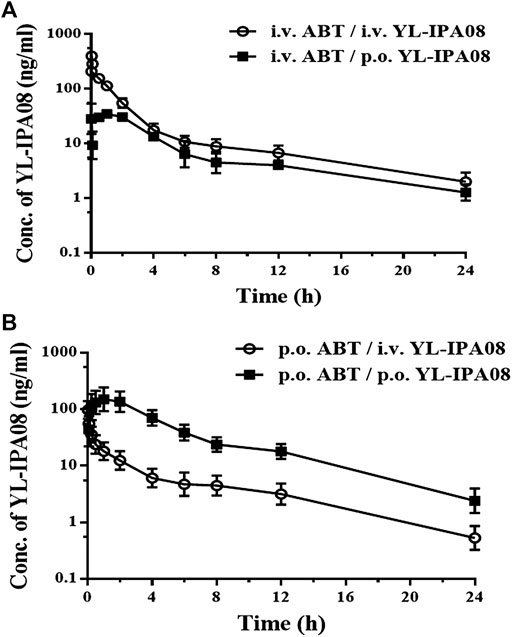
FIGURE 4. Plasma concentration-time profiles of YL-IPA08 after intravenous (0.1 mg/kg or 0.5 mg/kg) and oral administration (1 mg/kg) of YL-IPA08 combined with ABT pretreatment via intravenous (50 mg/kg) and oral dosing (100 mg/kg) in rats. Error bars represent the standard deviation of the mean concentration (n = 6). (A) i.v. 50 mg/kg ABT/i.v. 0.5 mg/kg YL-IPA08 & i.v. 50 mg/kg ABT/p.o. 1 mg/kg YL-IPA08; B: p.o. 100 mg/kg ABT/i.v. 0.1 mg/kg YL-IPA08 & p.o. 100 mg/kg ABT/p.o. 1 mg/kg YL-IPA08.
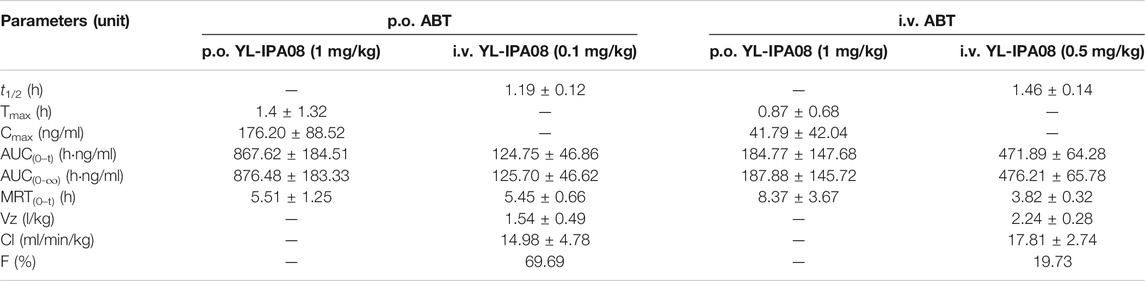
TABLE 5. Pharmacokinetic parameters of YL-IPA08 in rats after intravenous and oral combination with ABT (n = 6).
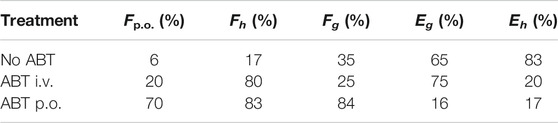
TABLE 6. Oral bioavailability and estimated hepatic and gastrointestinal extraction values for YL-IPA08 using ABT blocking metabolism (n = 6).
Pharmacokinetics of YL-IPA08 in Double Cannulated Rats
Figure 5 showed the concentration-time profiles of YL-IPA08 in portal and jugular venous plasma after oral administration of YL-IPA08 in double cannulated rats at a dose of 1 mg/kg. The plasma concentrations in portal veins is slightly higher than those in the jugular vein. No observed difference in clearance kinetics indicated the metabolic clearance in gut and liver is almost equal. The AUC values of YL-IPA08 were 441 ± 93 and 339 ± 93 h・ng/ml in portal and jugular venous plasma, respectively. FaFg value was 22% after calculating with Eq. 5 using the plasma AUC corrected by Rb/p.
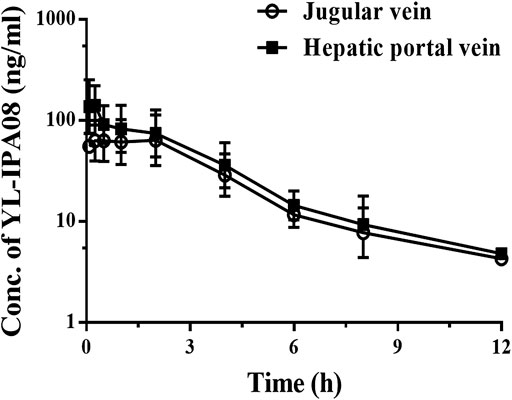
FIGURE 5. Jugular vein and portal vein plasma concentrations of YL-IPA08 after oral administration (1 mg/kg) in double cannulated rats. Error bars represent the standard deviation of the mean concentration (n = 5).
Unbound Concentration of YL-IPA08 in Brain Connects With Its Pharmacological Effect
Unbound fractions of YL-IPA08 in rat plasma and brain homogenate were 0.6 and 0.49%, respectively. Kp value of brain to plasma in rats was obtained under steady state to reach brain equilibrium in a transient dosing regimen. Then, Kp,uu achieved 0.18. The unbound concentration of YL-IPA08 in brain can be estimated based on the plasma concentrations after rats receiving different dosages of YL-IPA08 via oral dosing (Table 4). For example, the unbound concentration of 6.5 pg/g in brain can be achieved after 1 mg/kg oral dosing. According to in vitro TPSO-binding activity reported by Zhang et al. (2017), YL-IPA08 showed a high affinity for TSPO (IC50 of 0.23 nM) in the crude mitochondrial fraction prepared from rat cerebellum, homogenized in 10 volumes of ice-cold HS buffer. The unbound concentration in the incubates was obtained with 0.0108 nM (e.g., 4.9 pg/ml) by correcting the binding fraction of 10-volume brain homogenate (0.047). Thus, it is rational that marked acute antidepressant-like effects were observed in forced swim rats at the dosage of 1, 3, and 10 mg/kg (Zhang et al., 2017).
Discussion
YL-IPA08, eliciting rapid anti-PTSD-like effects upon binding to TSPO, is a promising new drug with a novel CNS target and mechanism. The pharmacokinetic behavior, especially the CNS exposure, is closely associated with its pharmacology. In a series of rat behavior models, YL-IPA08 (1–10 mg/kg) produced significant antidepressant-like and anxiolytic-like effects (Zhang et al., 2017) following oral dosing. Thus, the pharmacokinetic study was performed in rats at the same dose regiment first. The liner PK behaviors of YL-IPA08 were observed after orally administrated with 1, 3, 10 mg/kg (Table 4; Figure 3). YL-IPA08 was absorbed rapidly and exposures were dose dependent with proportionate increasing of Cmax and AUC. However, the bioavailability is pretty low (∼6%) by comparing the AUC obtained following i.v. dosing. It is clear that the fraction of dose absorbed (Fa), fraction of absorbed dose escaping first-pass clearance in the gut wall (Fg) and fraction escaping liver first-pass clearance (Fh) are three major determinants toward oral bioavailability (F) (Darwich, et al., 2010; Nakanishi and Tamai, 2015).
Since identification of Fa, Fg and Fh is of importance to mechanistic understanding the systemic exposure, the related efficacy, and the potent drug-drug interactions, a number of in vivo and in vitro models have been developed to elucidate Fa, Fg and Fh. As for in vitro methods, liver microsomes, S9 or hepatocytes are all well accepted for the generation of hepatic clearance and Fh via in vitro-in vivo extrapolation (IVIVE) (Houston and Carlile, 1998). Furthermore, functionally mature enterocytes mainly express CYPs and UGTs. Among UGT isoforms, UGT1A8 and UGT1A10 show gut-specific expression patterns (Kaminsky and Zhang, 2003; Court et al., 2012). However, the achievement of intestinal clearance and (Fg) from intestinal microsomes is limited by a lack of consensus on the appropriate scaling factors and a low availability of high-quality tissue. In the present study, in vitro metabolic stability of YL-IPA08 was performed in liver microsomes and intestinal microsomes of rat and human in the presence of NADPH and UDPGA to identify the metabolizing enzyme, organ, and species difference. 1 µM of YL-IPA08 was incubated in liver microsomes due to the Km values (1–3 µM) had been obtained in pilot enzyme kinetic study. The results indicated that CYP-mediated intrinsic clearances in HLM and RLM were very rapid. UGT is not involved in the depletion of YL-IPA08. Although the CYP-mediated elimination in HIM and RIM were also observed, with great difference of enzyme expression level between liver and gut, the contribution of intestinal metabolism to the oral bioavailability is hard to define. Especially, as for the RIM stability study, the significantly minimal turnover rate was mainly due to lack of normal activity. In vitro results from rat and human regents provided an evidence that metabolic characters of YL-IPA08 had no obvious species difference between rat and human. The contributions of hepatic and intestinal metabolism of YL-IPA08 were further investigated in in vivo models.
Portal vein and jugular vein double cannulation rat and ABT pretreated via different routes to inhibit CYP functions of rats were available models to differentiate liver and gut contribution to first-pass effect. In rats, administration of the panel CYP inhibitor ABT via the intravenous route (which inhibits only hepatic CYP enzymes) and the oral route (which inhibits both intestinal and hepatic CYP enzymes) can be used to assess the relative roles of the intestine and liver in the first-pass metabolism (Strelevitz et al., 2006). Double cannulation rats are also useful for separately assessing intestinal and hepatic first-pass effects (Murakam et al., 2003). This method, in which the animal is not restricted or under anesthesia, allows us to obtain reliable measurement of individual first pass effects in the intestine and liver. In the current study, we compared the two models simultaneously. After pretreated with ABT through i.v. injection and oral gavage according to recommended dosages, oral bio-availabilities elevated from 6 to ∼20% and then to ∼70%. Under the assumption of 100% absorption, hepatic and gut extraction ratios of YL-IPA08 were calculated to be 65 and 83%, respectively, indicating the contributions of liver and intestine to the first-pass effect are similar. In double cannulation rat model, FaFg value was 22%, which also indicated that if Fa is 100%, extraction of gut metabolism is 78%. This outcome is very close to the ABT inhibition study. However, at the same dose (1 mg/kg) via oral administration, the plasma concentrations from jugular vein in double cannulation rats were higher than the normal rats (Figures 3, 5). The difference may be explained that the surgery altered the physiological and hematological conditions to some extent.
Apart from metabolism of intestine and liver, absorption fraction (Fa) can also affect oral bioavailability. Aqueous solubility, permeability and efflux potential of YL-IPA08 are determine factors. YL-IPA08 employed high solubility. In the present study, Caco-2 cell model was utilized to evaluate the permeability across gut wall. PappA-B and PappB-A values of YL-IPA08 was 3.99 × 10–6 and 2.63 × 10–6 cm/s along with reasonable quality controls, suggesting that concentration gradient-driven passive transport cannot limit the oral bioavailability of YL-IPA08. Additionally, minimal parent recovery in feces with well less than 1% (0.004%, data not shown) in mass balance study in rats via oral administration also reflected complete absorption of YL-IPA08.
As a CNS targeted drug, BBB penetration assessment is an extra consideration to connect pharmacology. Definitive Kp,uu is a well-recognized parameter to estimate brain espouse. Kp,uu consists of two concepts, a) free (unbound) drug concentration at the site of action leads to pharmacological activity, and b) the free drug concentration at steady state is the same across any bio-membrane. Typically, Kp,uu is obtained from the unbound AUC of brain and of plasma. Owing to the rapid metabolism of YL-IPA08 in gut and liver, steady state is hard to be maintained following a single dosing. In the current study, efforts were made to rapidly obtain steady-state via i.v. loading dosage combined with i.v. infusion. By using this model, dosing and sample collection is completed only in three rats after steady-state was verified by continuous measuring plasma concentrations (data not shown).
Kp,uu in the current investigation is critical to link the PD of in vitro and in vivo results (see the result part). What is more important is that Kp,uu is often preserved across species (Di et al., 2013). Human Kp,uu can be estimated using that of rat (
Finally, with gut and liver confirmed as the equal contribution clearance pathway of YL-IPA08, CYP3A is the major metabolizing enzyme involved. The Med ID and pharmacokinetic drug interactions of YL-IPA08 deserve further investigation.
Conclusion
Although YL-IPA08 has shown potential antidepressant effect in in vitro binding assessment and in vivo animal model, great challenge still exists in its clinical therapy in patients, due to uncertain target exposure of the CNS agent. In this study, our result indicated P450-mediated elimination appeared to be important for its extensive first-pass effect with comparative contribution of gut and liver and no species difference was observed. Then, efforts were made to achieve the brain distribution at steady-state. Therapeutic human plasma concentration was predicted in advance with confidence.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by Beijing Center for Drug Safety Evaluation. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
YG, CY, CW, and YX conducted the experiments. YG analyzed the data. YL provided YL-IPA08. XZ and WZ conceived the research. XZ contributed reagents, wrote the manuscript, is responsible for the corresponding. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by Chinese Major Scientific and Technological Special Project of China (2018ZX09304017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.588127/full#supplementary-material
References
Court, M. H., Zhang, X., Ding, X., Yee, K. K., Hesse, L. M., and Finel, M. (2012). Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica 42, 266–277. doi:10.3109/00498254.2011.618954
Cubitt, H. E., Houston, J. B., and Galetin, A. (2009). Relative importance of intestinal and hepatic glucuronidation-impact on the prediction of drug clearance. Pharm. Res. (N. Y.) 26, 1073–1083. doi:10.1007/s11095-008-9823-9
Darwich, A. S., Neuhoff, S., Jamei, M., and Rostami-Hodjegan, A. (2010). Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the “advanced dissolution, absorption, metabolism (ADAM)” model. Curr. Drug Metabol. 11 (9), 716–729. doi:10.2174/138920010794328913
Di, L., Rong, H., and Feng, B. (2013). Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 56, 2–12. doi:10.1021/jm301297f
Fan, X., Li, H., Ding, X., and Zhang, Q. Y. (2019). Contributions of hepatic and intestinal metabolism to the disposition of niclosamide, a repurposed drug with poor bioavailability. Drug Metab. Dispos. 7, 756–763. doi:10.1124/dmd.119.086678
Houston, J. B., and Carlile, D. J. (1998). Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab. Dispos. 31, 140–144. doi:10.3109/03602539709002237
Kajbaf, M., Ricci, R., Zambon, S., and Fontana, S. (2013). Contribution of rat intestinal metabolism to the xenobiotics clearance. Eur. J. Drug Metab. Pharmacokinet. 38, 33–41. doi:10.1007/s13318-012-0098-5
Kalvass, J. C., and Maurer, T. S. (2002). Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Metab Dispos. 23 (8), 327–338. doi:10.1002/bdd.325
Kaminsky, L. S., and Zhang, Q. Y. (2003). The small intestine as a xenobiotic-metabolizing organ. Drug Metab. Dispos. 31, 1520–1525. doi:10.1124/dmd.31.12.1520
Karlsson, F. H., Bouchene, S., Hilgendorf, C., Dolgos, H., and Peters, S. A. (2013). Utility of in vitro systems and preclinical data for the prediction of human intestinal first-pass metabolism during drug discovery and preclinical development. Drug Metab. Dispos. 41, 2033–2046. doi:10.1124/dmd.113.051664
Li, Z., Zhang, Y., Gao, Y., Xiang, Y., Zhang, W., Lu, C., et al. (2019). Atipamezole is a promising non-discriminative inhibitor against pan-CYP450 including diclofenac 49-hydroxylation: a comparison with ABT for drug ADME optimization and mechanism study. Eur. J. Pharmaceut. Sci. 130, 156–165. doi:10.1016/j.ejps.2019.01.010
Linder, C., Renaud, N., and Hutzler, J. (2009). Is 1-aminobenzotriazole an appropriate in vitro tool as a nonspecific cytochrome P450 inactivator? Drug Metab. Dispos. 37, 10–13. doi:10.1124/dmd.108.024075
Liu, F., Zhuang, X., Yang, C., Li, Z., Xiong, S., Zhang, Z., et al. (2014). Characterization of preclinical in vitro and in vivo ADME properties and prediction of human PK using a physiologically based pharmacokinetic model for YQA-14, a new dopamine D3 receptor antagonist candidate for treatment of drug addiction. Biopharm. Drug Metab Dispos. 35, 296–307. doi:10.1002/bdd.1897
Matsuda, Y., Konno, Y., Satsukawa, M., Kobayashi, T., Takimoto, Y., Morisaki, K, et al. (2012). Assessment of intestinal availability of various drugs in the oral absorption process using portal vein-cannulated rats. Drug Metab. Dispos. 40, 2231–2238. doi:10.1124/dmd.112.048223
Murakami, T., Nakanishi, M., Yoshimori, T., Okamura, N., Norikura, R., and Mizojiri, K. (2003). Separate assessment of intestinal and hepatic first-pass effect using a rat model with double cannulation of the portal and jugular veins. Drug Metabol. Pharmacokinet. 18 (4), 252–260. doi:10.2133/dmpk.18.252
Nakanishi, T., and Tamai, I. (2015). Interaction of drug or food with drug transporters in intestine and liver. Curr. Drug Metabol. 16 (9), 753–764. doi:10.2174/138920021609151201113537
Obach, R. S. (2001). The prediction of human clearance from hepatic microsomal metabolism data. Curr Opin Drug Devel. 4 (1), 36–44.
Ohno, S., and Nakajin, S. (2009). Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab. Dispos. 37, 32–40. doi:10.1124/dmd.108.023598
Pinna, G. (2010). In a mouse model relevant for post-traumatic stress disorder, selective brain steroidogenic stimulants (SBSS) improve behavioral deficits by normalizing allopregnanolone biosynthesis. Behav. Pharmacol. 21, 438–450. doi:10.1097/FBP.0b013e32833d8ba0
Rupprecht, R., Rammes, G., Eser, D., Baghai, T. C., Schule, C., Kucher, K., et al. (2009). Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 325, 490–493. doi:10.1126/science.1175055
Strelevitz, T. J., Foti, R. S., and Fisher, M. B. (2006). In vivo use of the P450 inactivator 1-aminobenzotriazole in the rat: varied dosing route to elucidate gut and liver contributions to first-pass and systemic clearance. J. Pharmaceut. Sci. 95, 1334–1341. doi:10.1002/jps.20538
Suzuki, A., Iida, I., Hirota, M., Akimoto, M., Higuchi, S., Suwa, T., et al. (2003). CYP isoforms involved in the metabolism of clarithromycin in vitro: comparison between the identification from disappearance rate and that from formation rate of metabolites. Drug Metabol. Pharmacokinet. 18, 104–113. doi:10.2133/dmpk.18.104
Waters, N. J., Jones, R., Williams, G., and Sohal, B. (2008). Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharm. Sci. 97, 4586–4595. doi:10.1002/jps.21317
Zeng, X., Su, W., Zheng, Y., He, Y., He, Y., Rao, H., et al. (2019). Pharmacokinetics, tissue distribution, metabolism, and execration of naringin in aged rats. Front. Pharmacol. 10, 34. doi:10.3389/fphar.2019.00034
Keywords: YL-IPA08, pharmacokinetics, bioavailability, hepatic metabolism, intestinal metabolism, brain exposure, in vitro-in vivo extrapolation
Citation: Gao Y, Yang C, Wang L, Xiang Y, Zhang W, Li Y and Zhuang X (2020) Comparable Intestinal and Hepatic First-Pass Effect of YL-IPA08 on the Bioavailability and Effective Brain Exposure, a Rapid Anti-PTSD and Anti-Depression Compound. Front. Pharmacol. 11:588127. doi: 10.3389/fphar.2020.588127
Received: 28 July 2020; Accepted: 26 October 2020;
Published: 27 November 2020.
Edited by:
Victor A. Voicu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Stanislav Yanev, Bulgarian Academy of Sciences (BAS), BulgariaZhihao Liu, United States Department of Agriculture, United States
Copyright © 2020 Zhuang, Gao, Yang, Wang, Xiang, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaomei Zhuang, eGlhb21laXpodWFuZ0AxNjMuY29t