 Giovanna Menduti
Giovanna Menduti Daniela Maria Rasà
Daniela Maria Rasà Serena Stanga
Serena Stanga Marina Boido
Marina Boido- Department of Neuroscience Rita Levi Montalcini, Neuroscience Institute Cavalieri Ottolenghi, University of Turin, Turin, Italy
Spinal muscular atrophy (SMA) is the most common genetic disease affecting infants and young adults. Due to mutation/deletion of the survival motor neuron (SMN) gene, SMA is characterized by the SMN protein lack, resulting in motor neuron impairment, skeletal muscle atrophy and premature death. Even if the genetic causes of SMA are well known, many aspects of its pathogenesis remain unclear and only three drugs have been recently approved by the Food and Drug Administration (Nusinersen—Spinraza; Onasemnogene abeparvovec or AVXS-101—Zolgensma; Risdiplam—Evrysdi): although assuring remarkable results, the therapies show some important limits including high costs, still unknown long-term effects, side effects and disregarding of SMN-independent targets. Therefore, the research of new therapeutic strategies is still a hot topic in the SMA field and many efforts are spent in drug discovery. In this review, we describe two promising strategies to select effective molecules: drug screening (DS) and drug repositioning (DR). By using compounds libraries of chemical/natural compounds and/or Food and Drug Administration-approved substances, DS aims at identifying new potentially effective compounds, whereas DR at testing drugs originally designed for the treatment of other pathologies. The drastic reduction in risks, costs and time expenditure assured by these strategies make them particularly interesting, especially for those diseases for which the canonical drug discovery process would be long and expensive. Interestingly, among the identified molecules by DS/DR in the context of SMA, besides the modulators of SMN2 transcription, we highlighted a convergence of some targeted molecular cascades contributing to SMA pathology, including cell death related-pathways, mitochondria and cytoskeleton dynamics, neurotransmitter and hormone modulation.
Introduction to Spinal Muscular Atrophy and Available Therapies
Spinal Muscular Atrophy Pathogenesis
Spinal muscular atrophy (SMA) is a severe neuromuscular disorder affecting children and young adults with an incidence of one in 3,900–16,000 live births. In Europe, 4,653 patients were genetically diagnosed between 2011 and 2015, with 992 diagnosed in 2015 alone (Verhaart et al., 2017). SMA is characterized by brainstem and spinal motor neuron (MN) degeneration, due to mutation/deletion of survival motor neuron 1 (SMN1) gene. In physiological conditions, the encoded SMN protein has many important roles such as in the assembly of the spliceosome, biogenesis of ribonucleoproteins, mRNA trafficking and local translation, cytoskeletal dynamics, cellular bioenergetics, endocytosis and autophagy (for an extensive review on SMN functions, see Chaytow et al., 2018). In SMA, its lack determines motor impairment, muscle atrophy and premature death. However, the ubiquitous deficiency of SMN protein leads to consider SMA a multisystemic disorder, since its depletion can dramatically affect many other organs/systems (including heart, pancreas and immune system) (Bottai and Adami, 2013).
More in details, in humans there are two SMN genes i) the telomeric form SMN1, which translates for a ubiquitous protein (full-length SMN or FL-SMN), and ii) its centromeric homologous SMN2 which mostly generates a truncated and rapidly degraded protein delta7-SMN (SMNΔ7) and only about 10% of FL-SMN (Lorson et al., 1999). Therefore, in SMA, the production of functional SMN protein depends only on SMN2 gene and the degree of the disease severity is based on SMN2 copy number. Indeed, there are four types of SMA (reviewed by D’Amico et al., 2011; Boido and Vercelli, 2016; Vaidya and Boes, 2018). SMA 1, also known as Werdnig-Hoffmann disease, is diagnosed within 6 months of age: it is the most severe and the most common type (60% of all SMA cases) and it is generally fatal early on in life. SMA one babies show severe muscle weakness and trouble breathing due to spared diaphragm and feeble intercostal muscles; they also have difficulties in coughing, swallowing and feeding. SMA two is usually diagnosed between 6 months and 2 years of age, and the life expectancy is reduced. SMA type 2 babies show significant delay in reaching motor milestones or fail to meet milestones entirely: they can sit up without help, though they may need assistance getting into a seated position, but they are unable to walk and require a wheelchair. SMA 3, also called Kugelberg-Welander disease or juvenile SMA, is usually diagnosed after 18 months of age. Individuals affected by SMA three can be divided in two subgroups depending on the disease onset: i) patients with onset before 3 years of age are initially able to walk, but have increasingly limited mobility as they grow due to scoliosis and many need to use a wheelchair; and ii) patients with onset after 3 years of age might continue to walk and show slight muscular weakness. Finally, SMA type 4 is very rare and usually appears in adulthood (after 18 years of age), leading to mild motor impairment and no respiratory and nutritional problems.
Spinal Muscular Atrophy Approved Drugs
Despite the disease severity and its well-known genetic causes, until 2017 no treatment was available for SMA. Indeed, the efforts of the scientific, pharmaceutical, academic and clinical communities led to the discovery of effective drugs able to restore SMN1 or to increase the expression of SMN2 gene, in order to compensate the lack of FL-SMN protein.
Nusinersen (Spinraza) from Biogen is the first drug approved by the Food and Drug Administration (FDA) (in December 2016) and by the European Medicines Agency (EMA) (in June 2017) for both infants and adults with SMA. It is a modified 2′-O-methoxyethyl antisense oligonucleotide (ASO) designed to increase the expression of the SMN protein (Chiriboga et al., 2016). Nusinersen increments the capability of SMN2 to produce FL-SMN by binding to the intron-splicing silencer region N1 in the SMN2 pre-messenger RNA (pre-mRNA) promoting exon seven inclusion (Singh et al., 2006). Since the drug cannot pass through the blood brain barrier (BBB), it must be intrathecally administered. On May 2019, another drug, AVXS-101 (Zolgensma) from AveXis, a Novartis company, has been approved by the FDA, after the publication of the positive results of the phase one study called START (Identifier: NCT01547871) on its safety and efficacy after a one-time infusion in SMA one patients with symptoms before 6 months of age. In March 2020, it also received a conditional marketing authorization and it has been approved in May 2020 from EMA (Zolgensma, 2020a; Novartis, 2020).
AVXS-101 is the non-replicating recombinant AAV9 containing the complimentary DNA of the human SMN gene under the control of the cytomegalovirus enhancer/chicken-β-actin-hybrid promoter. The phase 3, open-label, single-arm and single-dose study delivering AVXS-101 by intravenous infusion called STR1VE (Identifier: NCT03306277) has been concluded in November 2019. On March 2020, the company showed the results of the concluded study STR1VE-US: “nine of 22 patients in the completed pivotal study demonstrated the ability to thrive, a stringent composite endpoint remarkable compared to untreated children with SMA type 1; the study showed that patients achieved rapid and sustained improvement in motor function” (for the complete press-release see Zolgensma, 2020b). Up to now, different studies are still ongoing: START Long Term Follow Up (Identifier: NCT03421977) aims to estimate the long-term safety on patients who completed the study START; whereas SPR1NT (Identifier: NCT03505099), a phase 3 open-label, single-arm, multi-center trial has been designed to evaluate the safety and efficacy of a one-time intravenous infusion in pre-symptomatic patients with SMA 1. The overall expected advantage of the AAV9 is that by a single administration patients could have a systemic and long-term lasting expression of SMN1(Al-Zaidy et al., 2018). A recent review, which compared all the published data and clinical trials on AVXS-101, confirmed that it represents an effective therapy for younger pediatric patients with SMA 1 (Stevens et al., 2020).
Risdiplam (Evrysdi), from Genentech, a member of the Roche Group, has been approved by the FDA on August 7th, 2020 for the treatment of children from 2 months of age on of adult (FDA Approves Genentech’s Evrysdi (risdiplam) for Treatment of Spinal Muscular Atrophy (SMA) in Adults and Children 2 Months and Older, 2020). Risdiplam is a mRNA splicing modifier which increases SMN protein expression. It is a liquid medicine, which can be administered orally without the need for hospitalization. Risdiplam is currently under study in four open-label trials: i) FIREFISH (Identifier: NCT02913482) and ii) SUNFISH (Identifier: NCT02908685) to investigate safety, tolerability, pharmacodynamics and kinetics and Efficacy in SMA 1 or SMA2 and 3, respectively; iii) JEWELFISH (Identifier: NCT03032172) and iv) RAINBOWFISH (Identifier: NCT03779334) to reveal the long-term efficacy of Risdiplam in patients who previously received another SMA treatment and infants from birth till 6 weeks of age, respectively.
Besides the ascertained data reporting the safety profile of these treatments and their significant benefits for some cohorts of SMA patients, it is necessary to take into account also their limitations, not only from the medical point of view but also from the socio-economical side.
Limits of Current Therapies and the Need of New Targets
One important limit of the current available treatments for SMA is that such approaches are merely SMN-dependent strategies and overlook other molecular pathways contributing to SMA pathogenesis (see beyond). To overcome this problem, combinatorial therapies should be considered, in order to redefine the timing and parameters of administration of the SMN-enhancing therapies currently in use, and to consider the synergistic effects with other drugs. Overall, combinatorial treatment strategies are required to face the SMN-independent features of SMA pathology. Moreover, the efficacy of the available treatments strictly depends on the age/type of patients: indeed, the effects induced by SMN-enhancing therapies are most consistent in the early-treated patients, whereas delayed interventions lead to less efficient or none rescue of motor neuron defects (Hoolachan et al., 2019; Hensel et al., 2020; Poletti and Fischbeck, 2020). In fact, SMN-restoring approaches seem particularly effective when the MNs are still alive and muscle functions not irreversibly compromised, as in the early phase of SMA disease. Depending on the age at the beginning of the therapy, Nusinersen and AVXS-101 can significantly extend the survival of babies with SMA 1, allowing motor milestone achievement; similarly, young patients with SMA two also show progresses on different motor scales after treatment. However, when Nusinersen is administered in adults patients with SMA type 2 and 3 (20–68 years old), improvements did not reach similar significant levels and could just support the stabilization of motor functions and the reduction in the symptom worsening (Jochmann et al., 2020; Mercuri and Sansone, 2020). While on one hand this piece of evidence strongly encourages the improvement of newborn screening methods, on the other hand it explains the growing pressure from late-symptomatic patients and caregivers for accessing to additional treatments (Richelme, 2019; Ramdas and Servais, 2020). Furthermore, prospective studies with larger patient numbers as well longer follow-up durations are required to better define the safety and efficacy of the treatments (Lee et al., 2019; Malone et al., 2019; Zuluaga-Sanchez et al., 2019).
To note, the current therapies present important limits such as i) the invasiveness of the administration route (in the case of Nusinersen), ii) more or less severe side effects, and iii) their cost and commercial accessibility. Indeed, Nusinersen administration requires hospitalization since it is administered intrathecally at least three times per year, for the entire life of the patient. Moreover, SMA patients generally develop severe scoliosis and spinal deformities, which in turn complicate or hinder this way of administration. These limitations can be circumvented by the development of systemically (as AVXS-101) or orally administered drugs able to cross the BBB (as Risdiplam) (Poletti and Fischbeck, 2020). To date, other orally delivered compounds are in final phases of clinical development and trials (Ramdas and Servais, 2020): these include a mRNA splicing corrector, branaplam (LMI070, Novartis), and a fast-skeletal muscle troponin activator, reldesemtiv (CK-2127107, Cytokinetics) (Rao et al., 2018). These alternative administration routes can also assure peripheral SMN-restoration, complementing the SMN central effects.
Additionally, the available treatments can also cause important side effects such as headache, back pain (Nusinersen), acute liver damage, bleeding, and heart damage (AVXS-101) (Gidaro and Servais, 2019). Moreover, about 5% of AVXS-101-treated patients can develop anti-AAV9 antibodies (viral titer greater than 1:50) this can increase the risk for immune response to gene therapy and reduce its therapeutic benefit (Al-Zaidy and Mendell, 2019). Finally, Nusinersen and AVXS-101 are listed among the most expensive drugs in the world: the relative cost-effectiveness ratio data reports, constantly updated, must be considered (Rao et al., 2018; Hoot, 2019; Malone et al., 2019).
Considering the limitations of the approved drugs for SMA, it is evident that the search for other potential therapies is necessary.
Below, we firstly describe two common methods to find new drugs: drug screening (DS) and drug repositioning (DR). Secondly, we make a comprehensive review of DS and DR studies conducted specifically in the SMA field, describing procedures, models, drug-targeted signaling pathways and results.
Drug Screening and Drug Repositioning for SMA
DS is a process by which a huge amount of compounds can be relatively quickly tested and selected as effective, by means of appropriate experimental models. On the contrary, DR (also currently referred as drug repurposing, reprofiling, retasking, or therapeutic switching) consists in a strategy to attribute new uses to drugs (generally already FDA approved) that are outside the scope of the original medical indications reducing risks and costs associated with time consuming new drug development programs. Briefly, the two approaches, eventually in combination, exploit the availability of large compound libraries, which include thousands of chemical and natural compounds and/or FDA-approved substances: Prestwick Chemical Library, MicroSource Discovery Systems, ComGenex, National Institute for Neurological Disorders and Stroke, TimTec, IBS, and ChemBridge are just few libraries that have been used to find new SMA therapies during the last years (Kelley et al., 2004; Sleigh et al., 2011; Konieczny and Artero, 2020). Afterward, once screened in search of specific cellular/molecular readouts, the substances can be firstly tested on “simplified” SMA models (cell cultures and/or invertebrates models), and then (or directly, in some cases) on SMA mice and/or patient-derived iPSCs (Figure 1).
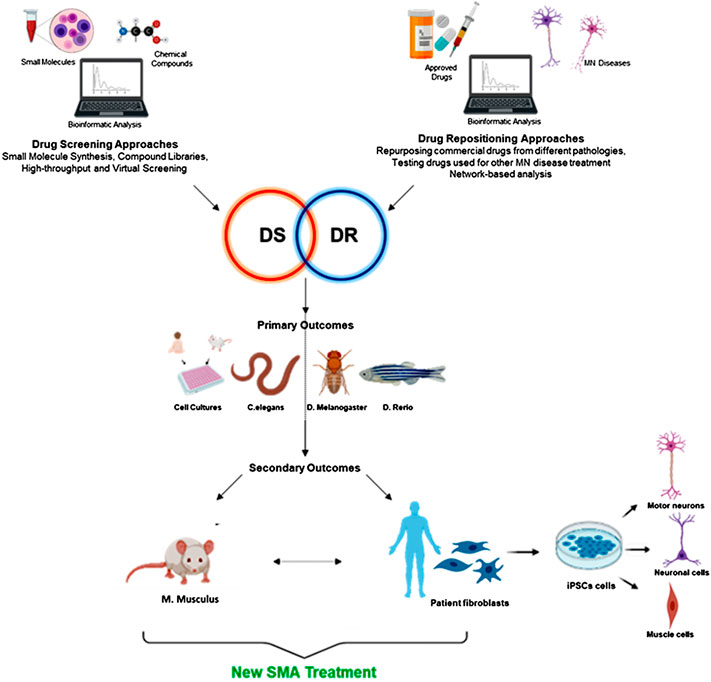
FIGURE 1. DS and DR methods as powerful approaches in SMA therapeutic research. The two approaches, eventually in combination, can pave the way for rapid identification of drugs for novel SMA treatments. The compounds (present in large screening libraries) can be tested for primary outcomes (continuous arrow, in the middle) firstly on “simplified” SMA models (cell cultures and/or invertebrates models), and then on SMA mice and/or patient-derived iPSCs (differentiated in neurons, MNs and muscle cells, eventually cocultured) to achieve secondary outcomes. In some cases (dotted arrow, in the middle) the hit compounds can be directly tested on murine models and iPSCs. Created with BioRender software. DR, drug repositioning; DS, drug screening; MN, motor neuron; SMA, spinal muscular atrophy.
Many kinds of DS approaches exist since a long time, including High-throughput, Focused, Fragment, Structural aided drug design, Virtual, Physiological, and Nuclear Magnetic Resonance screens (for an extensive review, see Hughes et al., 2011). These methods can be also extended to DR research, when FDA-approved drugs (already employed for the treatment of other pathologies) are investigated. Notably, High-throughput, Virtual and Physiological screenings are those ones mainly used in the SMA field.
High-throughput and Virtual screening are two rapid methods that allow wide-scale assays (by omics studies), in particular during the first screening phase to identify hit-compounds. As reviewed in Fox et al., (2006), to date, the High-throughput approaches, based on phenotypic screening of entire compound libraries, are commonly used in drug discovery processes, including different phases: target validation, assay development, secondary screening, ADME/Tox, and lead compound optimization (Fox et al., 2006). As an example, in SMA drug discovery, the 3,6-disubstituted pyridazine was identified through High-throughput screening in the NSC-34 cell line containing a SMN2 minigene reporter, and was then chemically modified leading to the synthesis of Branaplam (nowadays in clinical trial phase 2 for SMA treatment) (see section Direct and Indirect Modulation of Survival Motor Neuron 2 Transcription) (Cheung et al., 2018). On the other hand, virtual screening methods (such as ligand-based/structure-based virtual screening; as reviewed by McInnes, 2007), which exploit docking of new compounds that could bind known targets, are widely and successfully used to identify novel drugs for the treatment of neurodegenerative diseases (Makhouri and Ghasemi, 2017). Although these virtual screening paradigms have been poorly carried out in the SMA field, some successful applications of these methods have been reported: in particular, molecular docking studies highlighted both the binding mode of the C5-substituted quinazolines against a RNA metabolism regulator (the scavenger decapping enzyme, DcpS), and the binding mode of E-resveratrol, suberoylanilide hydroxamic acid and valproic acid against HDAC8 (see section Direct and Indirect Modulation of Survival Motor Neuron 2 Transcription) (Dayangaç-Erden et al., 2008; Singh et al., 2008; Makhouri and Ghasemi, 2017). Some of these compounds identified by DS (e.g., RG3039 and valproic acid) underwent clinical trial studies for SMA treatments (Kissel et al., 2011; Gogliotti et al., 2013). Moreover, molecular docking studies represent a rapid and cheap strategy to test FDA-approved drugs, paving the way to DR (Hughes et al., 2011; Pushpakom et al., 2018). Finally, as reviewed by Hughes et al., (2011), in the last stage of DS process, the Physiological screening is generally exploited to assay only hit-compounds (Hughes et al., 2011): indeed, this screening, based on tissue specific analyses and readouts (as muscular tissues in case of SMA), can require complex experimental models, such as transgenic mice (Le et al., 2005).
In the last years, DS/DR methods allowed both an in depth understanding of SMA pathological mechanisms and the identification of therapeutic approaches, including enhancing SMN function and regulating the SMN exon seven splicing. Furthermore, new targets mainly related to the rescue of the downstream effects of SMN protein depletion have been also highlighted. With this purpose, DR strategies have been applied in SMA field, allowing either the usage of established approaches on new targets, or the development of novel approaches on established targets (Ashburn and Thor, 2004). Indeed, the advantages of this approach can be summarized in three main features: i) it provides a lower risk of drug failure; ii) it reduces time frame for drug development, by skipping several clinical trial steps; and iii) it needs lower investment in clinical trials. This is due to pre-existing proofs of safety and efficacy for the repurposed drugs. In fact, the success rates for a repurposed drug to the market are significantly higher compared to specific de novo clinical trials (more than 15% in phases II–III clinical trial) (Hoolachan et al., 2019). For this reason, DR became a helpful approach in orphan diseases (designation granted by FDA) such as SMA (Hoolachan et al., 2019). In addition, the development costs related to DR are estimated to be thousands folds lower than those of a new chemical compound (Lotfi Shahreza et al., 2018; Pushpakom et al., 2018).
To date, albeit successfully, the process of DR has been largely opportunistic and serendipitous, often lacking a systematic approach aimed at the identification of a drug off-target or new target effects, suggesting its repositioning (Lotfi Shahreza et al., 2018; Lipinski and Reaume, 2020). During the early stages of the DR process (hypothesis generation phase), a potential repurposable drug can be identified with a high level of confidence thanks to a systematic approach and by exploiting different types of data coming from modern DS methods (i.e., computational and experimental approaches).
The computational approaches, as reviewed in detail by Lotfi Shahreza et al. (2018), involve the main in silico methods, based on docking simulation and machine learning (drug-based/disease-based). These methods allow to evaluate new indications on chemical ligands and protein targets or to discover repositioning opportunities (Lotfi Shahreza et al., 2018). Furthermore, as a natural evolution of these studies, promising results have been achieved in network-based computational biology, which attempts to identify pharmacological targets by reconstructing the biological network in different pathologies, simulating its interactions and highlighting correlations between drug targets (Lotfi Shahreza et al., 2018; Sam and Athri, 2019; Singh et al., 2020). In this way, the integration of “big data” generated by high-performance DNA and RNA sequencing, mass spectrometry, metabolomics and transcriptomic studies leads to the generation of different types of network-based association studies (Lotfi Shahreza et al., 2018; Pushpakom et al., 2018; Álvarez-Machancoses et al., 2020).
DR computational methods have already been exploited in the SMA field. In particular, Artero’s group using the Prestwick Chemical Library drug database (constituted by almost 1,280 compounds) (Prestwick Chemical, 2013) discovered a repurposed FDA-approved small molecule, the antibiotic Moxifloxacin, with the potential to become a new therapy for SMA (see section Direct and Indirect Modulation of Survival Motor Neuron 2 Transcription) (Konieczny and Artero, 2020). However, DR is a complicated process in which the computational approach per se is not sufficient to achieve satisfactory results. Indeed (as reviewed by Pushpakom et al., 2018; Lipinski and Reaume, 2020), the intertwining of computational and experimental methods achieves the reliability of DR approach. Data from large-scale drug screens, combined with genomic data, binding assays and High-throughput phenotypic screening, can be used to identify novel targets of known drugs (Pushpakom et al., 2018; Lipinski and Reaume, 2020). As an example, in vivo phenotypic screenings have been also exploited in the SMA field, allowing the discovery of small molecules able to target the RNA splicing specifically enhancing the exon seven inclusion of SMN2 transcript and the SMN protein levels. Indeed, in 2014, Naryshkin and coll. published the first report of a phenotypic screen yielding selective SMN2 splice modulators, leading to Risdiplam (RG7916), a recently FDA-approved drug for SMA treatment (Naryshkin et al., 2014; FDA Approves Genentech’s Evrysdi (risdiplam) for Treatment of Spinal Muscular Atrophy (SMA) in Adults and Children 2 Months and Older, 2020), while in 2015, Palacino’s group identified the splicing modulator Branaplam (LMI070) (see section Direct and Indirect Modulation of Survival Motor Neuron 2 Transcription) (Palacino et al., 2015). Most importantly, DR phenotypic screening allowed uncovering novel SMN-independent targets and drug paradigms, such as Olesoxime (see section Mitochondria-Related Pathways). This compound has been identified as potential therapeutic agent for both SMA and ALS (Calder et al., 2016; Swalley, 2020), since it is able to preserve mitochondrial function and protect MNs from degeneration (Calder et al., 2016; Blasco et al., 2018; Rovini et al., 2019). Indeed, a successful repositioning strategy in SMA treatment may be to identify drugs that are currently used to treat other neuromuscular diseases [such as ALS, hereditary spastic paraplegia (HSP) and Duchenne dystrophy (DD) (Patten et al., 2014)]: FDA-approved drugs modulating the pathogenetic pathways shared by these diseases have been proposed for SMA patients, as riluzole (Rilutek™ or Teglutik™) (see section Neurotransmitters’ Modulation), commonly administered for ALS (Calder et al., 2016; Hoolachan et al., 2019; Hensel et al., 2020). Also other drugs originally developed for different neurodegenerative diseases (such as rasalgiline and masitinib) could potentially represent promising SMA treatments (Hoolachan et al., 2019; Hensel et al., 2020).
Overall, we will focus here on the different DR and DS approaches employed to screen and identify candidate molecules for SMA treatment. Typically, in vitro phenotypic screenings exploit a wide range of cell-based assays in a 96-well format, including cellular disease models, highly engineered immortalized cell lines or different kind of induced Pluripotent Stem Cells (iPSCs); moreover, whole-organism phenotypic assays (using C. elegans, Drosophila, zebrafish, and mouse models) are also very important for their physiological relevance.
Experimental Models for Drug Screening/Drug Repositioning Studies in the Spinal Muscular Atrophy Field
Over the years, several experimental models have been developed to identify and improve SMA therapies. In vitro models are needed for High-throughput screenings where hundreds or thousands of compounds must be tested, while animal models are more indicated for the final study phases to assess phenotypical effects of hit compounds (Figure 1).
To perform large High-throughput screenings for SMA therapy, different cell lines can be used, including NSC-34 (murine MNs), HEK-293 (human embryonic kidney cells) and C33A (human cervical squamous cell carcinoma) (Zhang et al., 2001; Sumner et al., 2003; Jarecki et al., 2005). A more recent approach consider the use of patient fibroblasts and iPSC models, allowing the possibility to tailor the medical treatments as personalized medicine (Lai et al., 2017; Wang et al., 2019).
These experimental models, to apply High-throughput screening systems, are pivotal for the rapid validation of compound libraries. For example, Jarecki and coll. used a SMN2 promoter β-lactamase reporter gene test in NSC-34 cell line (Jarecki et al., 2005), while Zhang and collaborators developed a wide-scale screening approach based on the insertion of SMN2-luciferase or SMN2-GFP mini-gene reporters into HEK-293 and C33A (Zhang et al., 2001). These cellular models allowed to easily identify compounds that interfere with SMN2 gene expression and, to date, SMN2-luciferases mini-gene reporter is the most used one (Son et al., 2019). In 2013, Letso’s group developed a High-throughput screening test, to evaluate through ELISA assays the endogenous SMN protein levels in SMA patient fibroblasts (GM03813, GM09677, and GM00232) (Letso et al., 2013). More recently, Wang and collaborators tested a small library of 980 compounds, using the HEK293 cell line in which human SMN2-GFP gene reporter was targeted by CRISPR/Cas9–mediated homologous recombination (Wang et al., 2019).
Cellular models can also be used to evaluate hit-compounds during the final screening phases; the most used are SMA patient fibroblast lines (GM03813, GM03814, and GM22592) (Cherry et al., 2013; Wang et al., 2019), human iPSCs-derived neurons (Lai et al., 2017) or MNs (Wang et al., 2019), and co-culture of human MNs and skeletal muscle cells (Santhanam et al., 2018). Very recently, a promising (but not yet tested for DS/DR) tool has been developed: it consists in an on-chip 3D neuromuscular junction (NMJ) model, with optogenetically controllable human iPSCs-derived MNs and skeletal muscle cells (Osaki et al., 2020).
However, in the majority of cases, the final screening phase to assess effectiveness of selected hit-compounds is carried out using animal models. The percentage of identity and evolutionary divergence among different species can be evaluated through the amino acid differences of SMN protein. Homo sapiens, Mus musculus, Danio rerio, Xenopus laevis, Caenorhabditis elegans, Drosophila melanogaster, and Schizosaccharomyces pombe have wide evolutionary distances, with range of SMN protein conservation from 83 to 18.9% identity. However, the N-terminal Gemin2 binding domain, the central Tudor domain, and the C-terminal YG box are highly homologous among all species (Osman et al., 2019). Among the invertebrate models, C. elegans, together with D. melanogaster, represents the most exploited animal model for High-throughput screening studies for in vivo, allowing to study the mechanisms of MN degeneration underlying SMA and to test compounds. Its small size, short life cycle, body transparency, ease to generate transgenic animals, and low maintenance costs contribute to its large use in experimental studies. Moreover its nervous and locomotor systems are well known, since its 302 neurons and 95 body wall muscles are all identified (C. elegans Sequencing Consortium, 1998; Dimitriadi and Hart, 2010; Wolozin et al., 2011; Cáceres et al., 2012; Lee et al., 2013; de Carlos Cáceres et al., 2018). To date, several C. elegans Smn mutants have been developed, such as smn-1(ok355) (null-mutant form) and smn-1(cb131). The first one bears a wide deletion of most Smn coding region, leading to growth and fertility defects, MN loss and to early death (Briese et al., 2009; Dimitriadi and Hart, 2010; Grice et al., 2011). On the other hand, smn-1(cb131) model shows a point mutation in N-terminal domain and, while displaying a similar MN degeneration to smn-1(ok355), it survives longer allowing screening progression (Grice et al., 2011; Sleigh et al., 2011). Neuromuscular and motor defects can be analyzed in the worm by the “thrashing assay,” a test to measure the number of lateral swimming movements (Buckingham and Sattelle, 2009). These evaluations can be also correlated to MN degeneration analysis. To this aim, the most recent automated system has been developed by de Carlos Cáceres group: it employs microfluidic and image analysis that assess worm phenotypes analyzing D-type ventral MN degeneration through a quick genetic screening technique (De Carlos Cáceres et al., 2018).
Another invertebrate animal, used for both DS and DR, is Drosophila melanogaster: it shares some advantages of C. elegans, as a rapid life cycle, an easy husbandry and a simple genetic manipulation (Cauchi and Van Den Heuvel, 2007; Lessing and Bonini, 2009; McGurk et al., 2015; Aquilina and Cauchi, 2018). Different D. melanogaster Smn mutants have been set up: smn73Ao (smnA) and smnB mutants bear point mutations in C-terminal YG box (Chan et al., 2003; Chang et al., 2008; Grice et al., 2011); smnf05960 (smnC) and smnf01109 (smnD) mutants carry a transposon insertion respectively downstream and inside the Tudor domain. All these mutant models display impaired motor behavior, neuronal transmission and NMJ defects, leading to early death at larval moult stage (Grice et al., 2011). Finally, the EY14384 (smnE33) mutant fly carries a transposon insertion upstream of putative transcription start site and, while showing similar motor defects to the other mutants, it has a higher survival (Rajendra et al., 2007; Chang et al., 2008; Grice et al., 2011). However, the most recent fly SMA model has been developed by inserting the human SMN2 minigene reporter, fused to luciferase, into the Drosophila genome, in order to easily obtain the exon 7-inclusion during SMN2 splicing process. This cheap and feasible model allows to rapidly screen thousands of chemicals, possibly increasing FL-SMN protein levels (Konieczny and Artero, 2020).
Danio rerio, also known as zebrafish, is another useful invertebrate model to screen many compounds by DS/DR approaches. It is commonly used because of its high gene homology with human species and low maintenance costs (Howe et al., 2013; Patten et al., 2014). However, it has been mainly employed for ALS and HSP studies, whereas few SMA zebrafish models exist. One SMA model was developed by injecting an antisense morpholino oligonucleotide against Smn gene. Other zebrafish models bear the following gene mutations: smnG264D, smnY262X and smnL265X and are all similarly characterized by muscle weakness and atrophy, with halved lifespan compared to WT controls (Boon et al., 2009; Patten et al., 2014). To further increase the fish survival, the human SMN2 gene has been inserted in the genome of smnY262X fishes: such insertion increased SMN protein levels and slightly improved the animal survival (Hao et al., 2011).
Among the vertebrate models, Mus musculus is the most valuable one, especially for the Phenotypic screening. Murine and human genes display 90% of homology, even though mouse has just 20 chromosome pairs, while humans 23. Mouse genome has been widely manipulated to develop several neuromuscular disease models (transgenic, knockin, chimeras, and knockout), including SMA (Mouse Genome Sequencing Consortium et al., 2002; Fisher and Bannerman, 2019), taking into account that mice possess only one Smn gene and none SMN2 gene copies (Monani, 1999). Some of the SMA murine models recapitulate the human severe disease form (type I), such as SMN− and FVB.SMN2;Smn− (referred also as Burghes’ severe model incipient congenic); other ones (such as FVB.SMNΔ7 model) mimic the intermediate disease form (type II); milder forms (type III and IV) have been also developed [respectively, Smn A2G (also referred as Burghes type III model incipient congenic) and Smn1c, and FVB.Cg-Smn1<tm1Msd> Tg (SMN2)566Ahmb/J] (Osborne et al., 2012; Edens et al., 2015; Eshraghi et al., 2016). Moreover there is also the “SMA-like” murine model, mainly referred as Taiwanese model: these animals, carrying the Smn−/−SMN2 genotype, are classified into the three pathological SMA form groups (type 1, 2 or 3) based on their phenotypes (Hsieh-Li et al., 2000).
The most used SMA murine model for DS/DR is certainly the SMNΔ7 mouse: it is a triple homozygote, characterized by selective spinal MN degeneration, progressive muscle atrophy, reduced body weight, early (from postnatal day 5, P5) motor performance impairment, and premature death (around P14) compared to WT littermates (Edens et al., 2015; Simon et al., 2017; Kannan et al., 2018; Rimer et al., 2019; Tejero et al., 2020). The other available SMA models are generally excluded from DS/DR studies, since they die too early (by P5, or even embryonic, respectively in the case of FVB.SMN2;Smn− and SMN−) or too late [Smn A2G and Smn1c, and FVB. Cg-Smn1<tm1Msd> Tg (SMN2)566Ahmb/J], hence making difficult to evaluate the tested compound efficacy or requiring excessively extended observational studies.
Pathways Targeted by Drug Screening and Drug Repositioning Approaches in Spinal Muscular Atrophy
As stated, only three drugs are currently authorized for administration to patients. To further increase the available therapeutic options, DS/DR approaches can be pivotal since they can identify new or repurposed effective drugs.
Obviously, the attention is primarily focused on targeting pathways involved in SMN-specific transcription regulation. However, current expert opinions suggest not neglecting SMN-independent cascades and different cell targets, such as pathways involving degradation processes, cytoskeletal modulation, cell signaling and oxidative mechanisms (Figure 2). Below we describe natural, chemical and/or FDA-approved compounds, which have been discovered and tested through DS/DR studies, classifying them on the basis of their molecular mechanisms of action. The entirety of the described compounds are also resumed in Table 1.
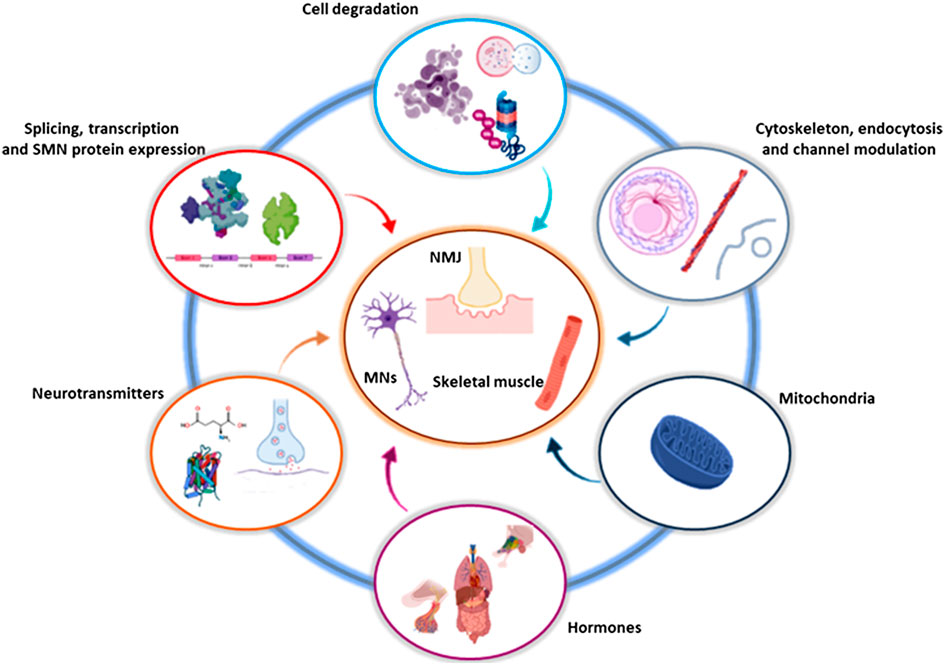
FIGURE 2. Involved and converging pathways targeted by DS and DR. The drugs identified by DS and DR influence and converge on a limited number of cellular and molecular pathways, that in turn act on specific districts, in particular involving MNs, NMJs and skeletal muscles. Created with BioRender software. DR, drug repositioning; DS, drug screening; MN, motor neuron; NMJ, neuromuscular junction; SMN, survival motor neuron.

TABLE 1. List of compounds discovered by DS or DR approaches for SMA treatment.
Direct and Indirect Modulation of Survival Motor Neuron 2 Transcription
During the last years, many studies are aiming to identify new SMN-dependent approaches, more effective and less invasive than the available ones.
SMN2 pre-mRNA splicing modulators (as Nusinersen) can significantly improve child health status. In this context, Branaplam is the first small compound orally administered and it is currently in phase II clinical trial (involving SMA type 1 patients; ClinicalTrials.gov identifier NCT02268552). It was developed by improving effectiveness, bioavailability and safety of pyridinazine, discovered as hit compound by High-throughput screening in NSC34 cell line with SMN2 mini-gene reporter inclusion (Palacino et al., 2015; Cheung et al., 2018). In detail, it stabilizes U1 snRNP binding with 5′ splice site, leading to exon seven inclusion in SMN2 mRNA, thus increasing SMN2-derived FL-SMN level (Palacino et al., 2015; Shorrock et al., 2018). Another small molecule, known as RG7800, was discovered using a similar screening. PTC Therapeutics, in collaboration with Roche, identified RG7800, then tested in the MOONFISH clinical trial. However, the trial was preventively stopped between phase I and II because of a RG7800-dependent eye toxicity observed in a long-term concomitant treatment study performed in monkeys (Identifier: NCT02240355) (Naryshkin et al., 2014; Kletzl et al., 2019). Therefore, RG7800 was optimized developing a new oral drug, RG7916, known as Risdiplam, which was tested in different cell lines and animal models (mice, rats, and monkeys) (Poirier et al., 2018; Ratni et al., 2018). Risdiplam is currently under evaluation in the four clinical studies mentioned above in Spinal Muscular Atrophy Approved Drugs (Kletzl et al., 2019; Haniff et al., 2020).
From the beginning of 21st century until now, other synthetic, inorganic and natural compounds were discovered via DS approaches, acting as splicing modifiers and up-regulators of SMN2-derived FL-SMN. Among them, the 2,4-diaminoquinazoline class was found by Jarecki and coll., after the remarkable screening of 550,000 synthetic compounds using NSC-34 cell line containing the SMN2 promoter β-lactamase reporter gene (Jarecki et al., 2005). The quinazoline analog compound RG3039 is a DcpS inhibitor that assured an overall improvement of disease phenotype when tested on Taiwanese and 2B/− and on SMA2B/− mice (Gogliotti et al., 2013). Based on this experimental evidence, Repligen conducted a clinical trial which initially provided successful results, but, following the take-over by Pfizer, the phase I trial was suspended on June 2014, because of a limited SMN increase in SMA children blood (Pfizer Pulls Plug on Repligen SMA Collaboration, 2015).
In addition, SMN2 modulators such as Sodium vanadate, small molecules (LDN-76070 and LDN-75654) and Brucea Javanica extract were originally identified via cell-signaling assays, but they gave insufficient results to be tested in SMA mice (Zhang et al., 2001; Cherry et al., 2013; Baek et al., 2019). In particular, Sodium vanadate showed to be effective in Taiwanese type III SMA mice when administered in combination with the detoxification agent L-ascorbic acid: however, their effects remain to be confirmed in more severe mouse models of SMA (Zhang et al., 2001; Liu et al., 2013; Seo et al., 2013). While Brucea Javanica has been recently tested on SMAΔ7 mice and the available preliminary results still require more detailed studies, LDN-76070 and LDN-75654 administration in the same mouse model increased lifespan, motor functions and SMN protein levels (Cherry et al., 2013; Baek et al., 2019).
In addition, histone deacetylase (HDAC) inhibitors, revealed by DR studies, could be promising also in case of SMA. Several FDA-approved HDAC inhibitors (sodium phenylbutyrate, valproic acid, Vorinostat, trichostatin A, and Panobinostat) or not yet approved (sodium butyrate) have been investigated for SMA treatment (Chang et al., 2001; Sumner et al., 2003; Andreassi et al., 2004; Hahnen et al., 2006; Avila et al., 2007; Garbes et al., 2009). In addition to promoting SMN2 transcription, they can affect the expression of many other genes (Calder et al., 2016; Poletti and Fischbeck, 2020). In combination with Nusinersen, HDAC inhibitors exerted synergistic effects, further enhancing the expression of SMN2 in human SMA fibroblasts (Pagliarini et al., 2020). Importantly, this combinatorial strategy could lead patient benefits, hypothesizing lower or less frequent Nusinersen doses, and consequently reducing repeated intrathecal administrations and high costs (Poletti and Fischbeck, 2020; Ramdas and Servais, 2020).
Other FDA-approved drugs arisen from DR studies and able to modulate SMN2 are celecoxib and Hydroxyurea; they are both important enzymatic inhibitors. Celecoxib is a non-steroidal anti-inflammatory cyclo-oxygenase two inhibitor, mainly used to treat rheumatoid arthritis and osteoarthritis (Seibert et al., 1994; Lipsky and Isakson, 1997). Its potential role in SMA therapy was deepened by Farooq’s lab, which focused on p38 MAPK pathway and showed that it upregulates cytoplasmic HuR protein, in turn able to stabilize mRNA-binding, also involving SMN (Farooq et al., 2013). Celecoxib, tested on Human neuron-committed teratocarcinoma (NT2) and mouse motor neuron-derived (MN-1) cell lines and on SMAΔ7 mice, induced the SMN2-derived FL-SMN mRNA stabilization. In addition, since it crosses the BBB, celecoxib is an optimal candidate for SMA therapy; indeed, it is currently in phase II clinical trial (Identifier: NCT02876094 (Farooq et al., 2013; Wadman et al., 2020). Instead, Hydroxyurea, a ribonucleoside diphosphate reductase inhibitor, prevents the exit from cell cycle G1/S phase and promotes fetal hemoglobin production. For these reasons, it is used to treat many neoplasias such as melanoma, chronic myelogenous leukemia, polycythemia vera, cervical and ovarian cancers, head and neck tumors, and sickle cell anemia (Rodgers et al., 1990; Madaan et al., 2012). Due to its gene interaction ability, Hydroxyurea was evaluated as therapeutic candidate for SMA, showing excellent results in lymphoblastoid cell lines isolated from type I, II, and III SMA patients (Grzeschik et al., 2005). However, three clinical trials (Identifiers: NCT00568698, NCT00568802, and NCT00485511) did not confirm its efficacy in improving motor functions of SMA types II or III (Wadman et al., 2020).
Although DS and DR are often carried out separately, their combination allowed to discover unusual drugs for neurodegenerative disease treatment. For example, Aclarubicin, an oligosaccharide anthracycline antineoplastic antibiotic, used in case of Acute Myeloid Leukemia treatment (Mitrou, 1990), was identified in 2001 via High-throughput screening on SMA type I fibroblast cell line and NSC-34 cell line containing SMN2 minigene reporter: Aclarubicin increases SMN2 exon seven inclusion, upregulating FL-SMN expression. Indeed, Aclarubicin seems to act as a transcriptional activator, inducing by indirect pathways SMN2 exon seven inclusion (Morceau et al., 1996; Andreassi et al., 2001). Likewise, Moxifloxacin, a synthetic fluoroquinolone antibiotic used to treat several infections, was identified by performing High-throughput screening study in a Drosophila SMN2 minigene reporter model, as described above (Konieczny and Artero, 2020). Moxifloxacin modulates SMN2 splicing by promoting exon seven inclusion and crosses the Drosophila BBB; moreover, it increases FL-SMN expression in HeLa cell lines. The authors showed that Moxifloxacin exerts a dose-dependent increase of Serine/arginine Rich Splicing Factor 1 (SRSF1) levels promoting the SMN2 exon seven inclusion (Konieczny and Artero, 2020).
Finally, Rigosertib and Indoprofene have been also proposed for SMA therapy, after DS identification. Rigosertib is a synthetic benzyl styryl sulfone analogue currently in phase III clinical trial for chronic myelomonocytic leukemia care (Garcia-Manero et al., 2016); it acts as a SMN2 splicing modifier, as suggested by a screening of small molecules carried out on C33A cell lines containing a SMN2-luciferase minigene reporter and SMA type I fibroblast cell lines (Son et al., 2019). Similarly, Indoprofene is a cyclooxygenase (COX) inhibitor, used as analgesic and anti-inflammatory drug (Paeile et al., 1989): it can increase SMN levels both in SMN2-luciferase cells and in type I SMA patient fibroblasts, and enhance the viability of a transgenic Type I SMA mouse model (Monani, 2000). Moreover, while showing also positive effects mediated by PDK1/AKT pathway on muscle wasting as demonstrated in aged mice (Kim et al., 2020), up to now it was never tested in clinical trials for SMA.
Cell Death and Degradation Pathways
The above-mentioned approaches are merely SMN-dependent strategies, but there are numerous studies suggesting that further cellular mechanisms can affect the severity of SMA. Indeed, DS and DR approaches represent a valid strategy to identify molecules also acting on SMN-independent pathways.
Recent studies suggest that the lack of SMN increases the cleavage of caspase-3, and triggers the apoptotic pathway and MN degeneration (Piras et al., 2017; Maretina et al., 2018; Schellino et al., 2018). In this context, the repositioning of the anti-epileptic drug Levetiracetam (trade name Keppra) decreased the cleaved-caspase three expression in SMA-iPSCs-MNs (Ando et al., 2019). Likewise, Bax and Bcl-xL proteins, respectively mediating anti- and pro-apoptotic pathways, seem to be influenced by SMN lack, with consequent downstream effects. The expression levels of Bax are significantly increased in the spinal cord of SMA mice, and the overexpression of Bcl-xL increases SMN-reduced MN survival (Garcera et al., 2011) and can extend SMA mice lifespan (Tsai et al., 2006; Tsai et al., 2008). Moreover, a decrease in the levels of Bcl2 in postmortem spinal cord from SMA I fetuses and SMAΔ7 mice was reported (Soler-Botija et al., 2003; Tsai et al., 2008; Piras et al., 2017). Based on this evidence, some authors proposed the repositioning of the water extract of Liuwei dihuang (LWDH-WE): despite not being FDA approved, it is a herbal formula widely used in the traditional Chinese medicine as treatment for kidney and liver disorders, asthma and geriatric diseases (Sangha et al., 2012; Tseng et al., 2014). When tested on Parkinson’s disease (PD) mouse models, it improved their motor activity, suggesting its use also for SMA treatment (Tseng et al., 2014; Tseng et al., 2017). Indeed, LWDH-WE was able to attenuate SMN deficiency-induced down-regulation of Bcl-2 and decreased cytosolic cytochrome c and cleaved caspase-3; the drug also counteracted cell death in NSC34 cells thanks to its SMN-promoting and antiapoptotic activities (Tseng et al., 2017).
Moreover, the ubiquitin/proteasome system is also altered in SMA, due to SMN lack (Powis et al., 2016). DS-based comparative proteomics revealed a significantly decrease of ubiquitin-like modifier activating enzyme 1 (UBA1) level in SMA mice. Since SMN directly influences UBA1 level, and its overexpression in SMA mice improves motor functions and increases their survival (Wishart et al., 2014; Powis et al., 2016), several works suggest to pharmacologically modulate its levels to influence SMA progression (Patten et al., 2014; Maretina et al., 2018). On the other hand, by using repositioning strategy involving the use of proteasome inhibitors, it was showed that the chemotherapy drug Bortezomib blocks SMN degradation in peripheral tissues and improves motor functions in SMAΔ7 mice (Kwon et al., 2011; Foran et al., 2016).
Other degradation pathways have been recently investigated. Wang and collaborators tested different drugs from a small library and demonstrated the involvement of both non-lysosomal (calpain 1/2) and lysosomal cysteine proteases (CTSL/CTSB) in degrading SMN proteins (Wang et al., 2019). In particular, establishing a versatile SMN2-GFP reporter cell line, they identified a novel role of the cysteine protease inhibitor Z-FA-FMK: this compound increased the level of functional SMN by inhibiting the protease-mediated degradation of both FL-SMN and delta7 SMN. Z-FA-FMK and the analogous compound E64days, previously used as Alzheimer’s disease (AD) and brain injury treatments, rescued MN degeneration in SMA, suggesting that inhibiting protease-mediated degeneration could be a potential therapeutic for SMA (Wang et al., 2019).
Finally, the repositioning of Edaravone and the Acetyl-L-carnitine (both drugs effective in oxidative stress modulation) has been also evaluated. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a free radical scavenger, showed efficacy in acute brain infarction and in ALS, and worldwide it is now approved for the treatment of both these pathologies in several countries (Jackson et al., 2019; Sun et al., 2019). Edaravone reversed oxidative stress-induced apoptosis and inhibited mitochondrial reactive oxygen species upregulation in SMA-iPSCs-derived spinal MNs (Ando et al., 2017). Instead, the L-carnitine is an anti-oxidant natural compound involved in cellular lipid peroxidation and known to inhibit mitochondrial damage and mitochondrial-mediated apoptosis both in vitro and in vivo (Bigini et al., 2002). Its exogenous administration in the acetylated form (Acetyl-L-carnitine, ALC), alone (Merlini et al., 2002, 2010; Wadman et al., 2020) or in combination with the valproic acid (Swoboda et al., 2010; Kissel et al., 2011; Wadman et al., 2020), was tested in two different SMA trials. However, on one hand, although the administration of ALC alone seemed effective, it did not allow to draw a final conclusion due to a too small cohort of SMA patients (Merlini et al., 2010); on the other hand, the combinatorial treatment of ALC with valproic acid compared to placebo did not reach significant improvements of motor function and muscle strength (Swoboda et al., 2010; Kissel et al., 2011; Wadman et al., 2020).
Mitochondria-Related Pathways
Mitochondria are organelles highly impacted at the very early stage of many neurodegenerative diseases, to the point to be considered as a possible unifying trait in the pathogenesis of these disorders (for an extended review see Stanga et al., 2020). Mitochondrial dysfunctions are reported as important pathological mechanisms in MN disorders (Patten et al., 2014). In SMA, mitochondrial impairment occurs in many tissues, both at the level of central and peripheral nervous system. This is probably due to the fact that i) mitochondria are particularly present in axons of neuronal cells, heart cells and skeletal muscles, and ii) SMN is ubiquitously expressed in the body. Indeed, SMN deficiency has been correlated to oxidative stress, mitochondrial dysfunction and deregulation of bioenergetic pathways. Therefore, treatments targeting mitochondria could represent a new promising solution not only for SMA, but also for many other disorders (Panuzzo et al., 2020).
Also in the case of mitochondria-related pathways, DR and DS approaches helped in the identification of promising drugs targeting mitochondria in SMA. Drugs targeting mitochondrial proteins and channels, such as the Na+/Ca2+exchanger (Annunziato et al., 2020), could be promising in the SMA treatment: the modulation of NCX2 (sodium calcium exchanger isoform 2) expression, by microRNA-206 administration in SMAΔ7 mice, delayed the disease progression and improved behavioral performance in mice (Valsecchi et al., 2020).
Another example is Olesoxime, originally evaluated for diabetes since able to promote the survival of the pancreatic β-cells, which are particularly rich in mitochondria. Interestingly, DR studies revealed important positive effects also for SMA. Indeed, Olesoxime is able to bind proteins of the outer mitochondrial membrane: there, it reduces its permeability when exposed to stress (Pruss et al., 2010) preventing, in turn, apoptosis by reducing release of pro-apoptotic factors and maintaining energy production (Kariyawasam et al., 2018). In this way, Olesoxime can preserve the integrity of MNs (Bordet et al., 2007). Moreover, Olesoxime showed neuroprotective and neuroregenerative effects in several animal models of motor nerve degeneration and in a transgenic mouse model of severe SMA (SmnF7/F7; NSE-Cre mice; mutant mice carrying a deletion of Smn exon seven directed to neurons): daily administrations of Olesoxime extended the survival of the treated mice (Bordet et al., 2010). Taken together, these data suggest that Olesoxime might maintain MN function and might be a therapeutic drug in the treatment of SMA. However, unfortunately, oral administration of Olesoxime in phase II trial for SMA (and phase III for ALS) failed, because it did not show sufficient efficacy (Bordet et al., 2007; Swalley, 2020).
Cytoskeleton Dynamics, Endocytic Pathway and Channel Modulators
The exploitation of modern DS and DR approaches also allowed to identify new treatments aimed at improving or maintaining integrity and functionality of SMA axons/NMJs. In this context, perturbations of cytoskeleton dynamics, known to impair SMA MN neurogenesis, have been studied (Bowerman et al., 2007); therefore, the overexpression or inhibition of cytoskeletal remodeling modulators represent intriguing therapeutic strategies.
One of the main actin dynamics regulator is the RhoA-ROCK pathway. ROCK is a serine-threonine kinases and a downstream effector of the RhoA small GTPase. The RhoA/ROCK pathway is mainly involved in shape regulation and neuronal cells movement (extension and branching), by acting on the cytoskeleton dynamics (growth, differentiation, and retraction) and critically influencing MN synapse functions. An aberrant upregulation of RhoA/ROCK pathway was observed in SMA neuronal cell models and in SMA patients fibroblasts (Bowerman et al., 2007; Bowerman et al., 2010; Nölle et al., 2011; Koch et al., 2018). Moreover, the profilin IIa upregulation (due to SMN-deficiency) causes an upstream dysregulation of RhoA/ROCK pathway (Maretina et al., 2018). This evidence suggests that ROCK inhibition can induce beneficial effects in SMA, as already demonstrated for several neurodegenerative diseases (Hensel et al., 2015), including SMA: indeed, Y-27632 and Fasudil are two RhoA/ROCK inhibitors, able to extend lifespan and improve motor functions in Taiwanese and SMN2B/− SMA mice (Bowerman et al., 2010; Bowerman et al., 2012; Bowerman, 2014; Hensel et al., 2017).
Hensel tested the combination of Y-27632 with an ERK pathway inhibitor, using an automated MN cell-bodies High-throughput detection screening on primary spinal cord cultures. The simultaneous inhibition of both pathways induced synergistic beneficial effects, significantly increasing MN viability, with respect to the single inhibition of one of them (Hensel et al., 2017).
Fasudil is a vasodilator drug, used for the treatment of cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. In Japan its use has been approved by the Pharmaceuticals and Medical Devices Agency since 1995 (Shibuya and Suzuki, 1993; Zhao et al., 2006) and is currently tested in clinical studies for disorders such as the Raynaud phenomenon, atherosclerosis and ALS (ROCK-ALS trial, NCT03792490, Eudra-CT-Nr.: 2017-003676-31). Interestingly, Fasudil improved survival and promoted skeletal muscle development in SMA Smn2B/− mice, restoring the correct function of actin in MNs and supporting the formation of functional NMJs (Bowerman et al., 2012; Koch et al., 2018). Therefore, while Y-27632 is mainly used as an experimental biochemical tool in the study of ROCK signaling pathways, the ongoing clinical study of Fasudil for ALS patients could pave the way for the therapeutic evaluation of ROCK inhibitors in MN diseases by strengthening its DR in SMA field (Bowerman et al., 2017).
In addition, several SMA modifiers have been proposed as SMA therapy in combination with SMN-enhancing treatments. In particular, the contribution to SMA pathogenesis of Plastin 3 (PLS3) and Neurocalcin Delta (NCALD) proteins has been deeply evaluated. PLS3 is a Ca2+-dependent actin-binding protein, important for axonogenesis by increasing F-actin levels, and acting as positive regulator of endocytosis process. PLS3 is a powerful modifier of SMA: high levels of PLS3 have been reported in unaffected subjects carrying SMN1 mutations; moreover PLS3 overexpression increased cell survival, supported neurite overgrowth and restored impaired endocytosis in vitro and in vivo SMA models (Oprea et al., 2008; Hosseinibarkooie et al., 2016a; Alrafiah et al., 2018; Maretina et al., 2018). PLS3 overexpression, combined with the subcutaneous injection of ASOs, has been recently confirmed to improve the survival of SMA mice, motor functions and NMJ size. Since their beneficial synergistic effects were greater than those obtained with ASO administration alone, further DS studies are recommended to better define effective therapeutic combinatorial strategies (Hosseinibarkooie et al., 2016b; Kaifer et al., 2017).
Analogous positive results have been obtained by combining NCALD and ASO treatment (Riessland et al., 2017). Riessland’ group identified NCALD as a potential SMA modifier by Genome-Wide Linkage and Transcriptome-Wide differential expression analysis performed on samples of SMA type 1 patients and fully asymptomatic people, both carrying homozygous SMN1 deletions (Riessland et al., 2017). Furthermore, this work demonstrated the role of NCALD as negative regulator of endocytosis, since its knockdown effectively ameliorated these dysfunctions, supporting motor axon development and improving MN circuitry and NMJ presynaptic function in SMA models (worm, zebrafish, and mice) (Riessland et al., 2017). Given this evidence, DS and DR approaches confirmed their important contribution in revealing potential SMA disease modifiers involved in the modulation of different signaling pathways.
Another important example comes from Sleigh’s group, that in 2011, using a SMA type III C. elegans model, screened 1040 FDA-approved compounds of the NINDS library, to identify effective drugs targeting nerve/muscle activity (Sleigh et al., 2011). The results of this analysis suggested 4-aminopyridine (4-AP) as a candidate drug for SMA treatment: it is a dose-dependent potassium channel blocker, able to restore demyelinated neuron conductance and enhance synaptic transmission (by increasing pre-synaptic calcium influx into neurons). Administration of 4-AP rescued mutant C. elegans motility (Sleigh et al., 2011). Interestingly, 4-AP is the active ingredient of Fampiridine drug, also named Ampyra or Fampyra respectively in United States and Europe, and approved by FDA and EMA in 2011. It is already administered to multiple sclerosis and Lambert-Eaton myasthenic syndrome patients; but it has been also tested for spinal cord injury and PD (Jensen et al., 2011; Pérez Luque et al., 2015; Acorda Therapeutics Inc., 2017).
Therapeutic strategy of drug targeting signaling pathways involving Ca2+ influx modulation has been developed specifically to enhance muscle functions, as suggested by Citokinetics-Astellas ongoing trial of Reseldemtiv. Reldesemtiv, previously known as CY 5021 and CK-2127107, is a selective small-molecule fast skeletal muscle troponin activator, to date in trials for chronic obstructive pulmonary disease and ALS treatment (Shefner et al., 2018; Rossiter et al., 2019). It improves muscle contractility by increasing the affinity between troponin C and Ca2+, providing a therapeutic target for several skeletal muscle-related diseases, including SMA. Indeed, FDA attributed to Reldesemtiv the orphan drug designation as potential SMA therapeutic (Orphan designation: Reldesemtiv, Treatment of SMA, 2019). Its efficacy and tolerability in increasing muscle contraction force was demonstrated respectively in preclinical studies and phase one SMA trials (Calder et al., 2016). The following phase 2 trial (Identifier: NCT02644668) confirmed Reldesemtiv efficacy, when orally administered to patients with SMA types II, III, and IV, without observing dose-limiting safety or tolerability issues. A confirmatory phase 3 study is planned in the next years (Rudnicki et al., 2018; Ramdas and Servais, 2020).
Hormone Signaling Pathways
The negative effects of SMN-deficiency, although mainly involved in MN impairment, also extend, as multisystem pathologies, to other components of the motor circuits and modulators of skeletal muscle development and function (Zhou et al., 2016). Among the others, the pharmacological modulation of various hormones and their signaling pathways is being studied in order to minimize muscle atrophy and injury by acting on neuronal regeneration, peripheral reinnervation and muscle growth (Tuffaha et al., 2016; Lopez et al., 2019). In addition, these drugs, mostly already approved by the FDA, could quickly be suitable for clinical translation.
For example, the administration of hypothalamic and pituitary hormones [growth hormone (GH) and thyrotropin releasing hormone (TRH)] or synthetic glucocorticoids (e.g., prednisolone), administered in case of several neuromuscular diseases can exert beneficial effects on muscle functions (Wadman et al., 2020). Indeed, GH induces insulin-like growth factor-1 (IGF-1) secretion at muscle and liver level: then, IGF-1 stimulates the physiological growth of long bones and soft tissues and muscle development, whereas in case of trauma, it supports muscle regeneration by promoting myogenic differentiation and myocyte hypertrophy (Tritos and Klibanski, 2016). GH administration is already clinically employed in chronic renal failure, Turner and Prader-Willi syndromes, growth disorders; furthermore, it has been studied for its beneficial effects in enhancing nerve regeneration and muscle reinnervation, following peripheral nerve injuries (Tuffaha et al., 2016; Lopez et al., 2019). Therefore, a DR for these natural or synthetic hormones can be suggested for SMA treatment.
Indeed, several studies have shown that intracerebral injections of IGF-1 in SMA mice supported survival and improved motor functions, preventing muscle atrophy and preserving NMJs (Bosch-Marcé et al., 2011; Shababi et al., 2011; Tsai, 2012; Tsai et al., 2014; Wadman et al., 2020); however, similar results have not yet been obtained in SMA patients (Kirschner et al., 2014).
Beneficial effects of TRH tripeptide, Glu-His-Pro-NH2, have been also observed in SMA skeletal muscles (Wadman et al., 2020). The TRH, in addition to stimulate the release of thyroid-stimulating hormone, seems involved in neuronal activity by its association with serotonin (Tzeng et al., 2000). Its synthetic analogs, i.e., Protirelin and Taltirelin hydrate, have been used to treat epilepsy, spinal cord injury, spinocerebellar ataxia, and neonatal respiratory distress (Shimizu et al., 1989; Tzeng et al., 2000; Urayama et al., 2002). Since TRH has been found in the spinal MNs, its role in the pathogenesis of MN diseases has been suggested, even if a TRH-based trial in ALS was unsuccessful (Brooks et al., 1987). However, its beneficial effects on motor functions and electromyographic results of SMA type II/III patients have been reported in different studies (small groups of infants with SMA type I in children with types II and III) and in a clinical trial (Takeuchi et al., 1994; Tzeng et al., 2000; Wadman et al., 2020).
Also the repositioning of a synthetic glucocorticoid drug prednisolone, currently used for Duchenne muscular dystrophy treatment, has been recently suggested for SMA therapy (Hoolachan et al., 2019). Prednisolone is also employed for the treatment of a wide spectrum of inflammatory conditions, involving allergies, autoimmune disorders and cancers (Gambertoglio et al., 1980; Frey, 1987). The beneficial effects of intermittent dosage of prednisolone in recovering skeletal muscles from injury, promoting sarcolemmal repair and limiting atrophic remodeling have been showed in the treatment of Duchenne muscular dystrophy patients and also in other neuromuscular diseases models (such as acute muscle injuries and muscular dystrophy mouse models) (Beenakker et al., 2005; Matthews et al., 2016; Quattrocelli et al., 2017). Moreover, prednisolone promotes the expression of Klf15, a transcription factor involved in muscle homeostasis and deregulated in pre-symptomatic SMA mice: this can further justify the positive effects, observed in SMA mice, including the improvement of muscle trophism and functioning and lifespan extension. Thus, this suggests that further investigations on the possibility of prednisolone repositioning as SMA therapy (Walter et al., 2018; Hoolachan et al., 2019).
Nevertheless, the studies were not completely free of bias, and further evaluations are required, in particular regarding TRH (Wadman et al., 2020). However, SMA-specific DS and DR studies concerning such hormone-based therapies still seem to be promising, as suggested by different works on SMA models reporting the benefits of their administration and recommending further related screenings (Kato et al., 2009; Ohuchi et al., 2016; Wadman et al., 2020).
Neurotransmitters’ Modulation
Impairment of synaptic transmission has been also widely reported in SMA, suggesting that the pharmacological modulation of synaptic plasticity mechanisms could represent another therapeutic target and sustain the survival of MNs.
Different studies aimed at characterizing alterations of neurotransmission and abnormalities in SMA spinal circuitries both in vitro and in vivo models revealed hyperexcitability and loss of afferent proprioceptive synapses on MNs (Mentis et al., 2011; Gogliotti et al., 2012; Zhou et al., 2016; Bowerman et al., 2018), suggesting an impairment of glutamatergic synaptic transmission (Bowerman et al., 2018; Sun and Harrington, 2019). Impaired glutamate transport and excitotoxicity are involved in the pathogenesis of many MN diseases, as already well known for ALS (Rothstein et al., 1995). Such perturbations can also contribute to SMA disease, by enhancing MN death: thus, Riluzole and ceftriaxone, two different kind of drugs able to influence glutamatergic signaling and both FDA-approved for ALS (albeit with modest efficacy), have been proposed as repurposed compounds in clinical SMA trials. Riluzole exerts its anti-glutamatergic action, by enhancing the uptake of glutamate into astrocytes and by inhibiting its release blocking voltage-gated Na+ currents, thus preventing the neurotransmitter accumulation in the extracellular space and degeneration of MNs by excitotoxicity. When administered to SmnF7/F7; NSE-Cre mice SMA mice, Riluzole significantly attenuated disease progression (Haddad et al., 2003). Similarly, in a small phase I clinical study with enrolled SMA type I infants, Riluzole was proved to be safe and able to mitigate the natural course of the disease (Russman et al., 2003; Wadman et al., 2020). Although some aspects of Riluzole mechanism of action still need to be clarified in SMA, Dimitriadi and coll. proved that the drug acts on Ca2+-activated K channels, thus determining the improvement of MN functionality in several SMA models (Dimitriadi et al., 2013).
Ceftriaxone (a β-lactam antibiotic, also known as Rocephin) is used for the treatment of a number of bacterial infections (Koster, 1986; Schito and Keenan, 2005). Given its potential to reduce glutamate toxicity by modulating glial glutamate transporters (GLT1, EAAT2) (Calder et al., 2016; Ramalho et al., 2018), ceftriaxone efficacy has been tested in several MN disease models. Its administration in SMNΔ7 mice provided neuroprotective effects by modulating the glutamate transporter GLT1, the transcription factor Nrf2 and SMN protein levels, improving neuromuscular phenotype and increasing animal survival (Nizzardo et al., 2011). Therefore, also considering the safety and tolerability of ceftriaxone administration in ALS patients, the potential repositioning of β-lactam antibiotics as a treatment for SMA has been suggested (Nizzardo et al., 2011; Cudkowicz et al., 2014; Calder et al., 2016).
Another successful DR concerns the glutamate inhibitor Lamotrigine, commonly used for the treatment of various neuropsychiatric disorders and epilepsy. Its prolonged administration to adult type II and III SMA patients (Ng et al., 2008; Naguy and Al-Enezi, 2019) prevented the deterioration of motor functions for almost 5 years of treatment (Nascimento et al., 2010; Wadman et al., 2020).
Moreover, the imbalance in excitatory/inhibitory signaling have been also speculated in SMA. In particular, the enhancement of GABAergic neurotransmission in C. elegans SMA models was able to correct the locomotor dysfunctions (Wu et al., 2018). Moreover, in the work of Sleigh et al., a chemical library DS highlighted the rescuing of phenotypic dysfunction by Gaboxadol, a potent agonist of a specific extrasynaptic GABAA receptor subtype (Sleigh et al., 2011). Gaboxadol has been proposed for the treatment of insomnia (Mathias et al., 2005; Roth et al., 2010) and two neurological development disorders, Fragile X syndrome and Angelman syndrome (Identifier: NCT04106557), in which it improves behavioral and motor dysfunctions by enhancing GABAergic transmission (Cogram et al., 2019; Keary et al., 2020). Its repositioning for SMA treatment could be also suggested. Likewise, Gabapentin (Neurontin), a FDA-approved drug whit a molecular structure similar to GABA, acts by inhibiting calcium channels. Gabapentin is used for the treatment of different forms of neuropathic pain, for anxiety disorders and alcoholism (Field et al., 1997; Caraceni et al., 2004; Pfizer Inc., 2009; Moore et al., 2011; Levine et al., 2019). Considering the neuroprotective effect of gabapentin in nerve damage-induced chronic neuropathic pain (Wiffen et al., 2017), two different clinical trials were conducted enrolling SMA type II and III patients (Miller et al., 2001; Merlini et al., 2003). These trials confirmed a significant gabapentin-dependent improvements in the so-called “leg megascore” (calculated by summing knee flexion, knee extension and foot extension scores) and muscle strength, recommending further studies to evaluate prolonged administration of the drug in SMA patients (Merlini et al., 2003; Wadman et al., 2020).
Neuromuscular Junction Stabilization
The NMJ represents the interaction core between the motor nerve terminal and the skeletal muscle fiber. SMN, neurotrophic factors (Stanga et al., 2016), “auxiliary proteins” (as neuregulins, dystrophin-glycoprotein complex, ErbB receptors, Wnts), miRNAs (as miRNA-9 and miRNA-206) and agrin (a heparan sulfate proteoglycan) seem to contribute to maturation and/or stabilization of NMJs (Valsecchi et al., 2015; Boido and Vercelli, 2016; Boido et al., 2018; Guarino et al., 2020). Given this evidence, DS and DR approaches have been proposed to improve NMJ development/stabilization in SMA patients.
To this purpose, Amifampridine, a FDA-approved drug used to treat Lambert-Eaton myasthenic syndrome, is a promising compound. Its efficacy has been also tested in Myasthenia Gravis (MG) patients: indeed, although Amifampridine acts on presynaptic terminal blocking K+ channels regulating ACh release, its positive effects were also observed in postsynaptic disorder such as MG (Bonanno et al., 2018). Since NMJs show both presynaptic and postsynaptic defects in SMA, Amifampridine can be considered a possible candidate to improve overall conditions of SMA patients (Hoolachan et al., 2019): to this aim, the recruiting-phase II clinical trial NCT03781479 is currently ongoing (Maddison et al., 1998; Hoolachan et al., 2019).
Similar effects are expected from Tideglusib, a glycogen synthase kinase 3 beta (GSK-3β) inhibitor, not yet FDA-approved, mainly investigated for AD and progressive supranuclear palsy treatments (Lovestone et al., 2015; Stamelou et al., 2016). However, Tideglusib was also proposed for a clinical trial on congenital myotonic dystrophy (Identifier NCT02858908, completed in January 2018) in whom excellent results were obtained with improvement in muscle growth (Horrigan et al., 2018); moreover, another trial is ongoing (Identifier NCT03692312). In addition, an image-based screening of chemical libraries showed that GSK-3β chemical inhibitors and short hairpin RNAs increase SMN protein levels and block cell death (Makhortova et al., 2011). Altogether, these data suggest that Tideglusib, likewise Amifampridine, could be a valuable drug candidate for SMA treatment (Hoolachan et al., 2019).
DS also allowed the development and identification of myostatin-follistatin modulators. Myostatin, a member of the transforming growth factor β (TGFβ) superfamily of growth factors, is primarily expressed by skeletal muscle cells, where acts as a negative regulator of muscle mass. Follistatin binds and inhibits myostatin and other members of the TGFβ family, contributing to the correct balance of muscle protein metabolism (Lee and McPherron, 2001). Dysregulation of myostatin-follistatin signaling pathway has been studied in several neuromuscular diseases, including SMA (Mariot et al., 2017; Shorrock et al., 2018). Myostatin expression is impaired in serum and muscle biopsies of SMA patients (Mariot et al., 2017), and its inhibition with intramuscular injection of AAV1-follistatin or muSRK-015P monoclonal antibody ameliorated muscle mass functions in different models of SMA mice (Feng et al., 2016; Long et al., 2019). Therefore, preventing myostatin activation has been widely suggested as therapeutic approach in SMA. To this aim, Scholar Rock developed several laboratory-made monoclonal antibodies against myostatin: by carrying out a phenotypic screening on a dexamethasone-induced muscle atrophy murine model, the SRK-015 antibody was proved as able to fully prevent muscle function loss (Pirruccello-Straub et al., 2018). Given this evidence, it has been tested in two groups of SMNΔ7 mice (which received different pharmacologic restoration of SMN, to reflect early or late therapeutic intervention) leading to positive effects by increasing muscle mass and function (Long et al., 2019). The efficacy of SRK-105 is currently evaluated in a phase 2 trial (TOPAZ, Identifier: NCT03921528) involving type II and III SMA patients (see Scholar Rock Reports First Quarter, 2020). Finally, another FDA-approved drug, successfully repositioned for SMA therapy, is Salbutamol (also known as albuterol; brand name Ventolin), widely used to reduce bronchospasm in some respiratory pathological conditions (i.e., asthma and chronic obstructive pulmonary disease). As a short-acting compound with selective agonist activity on β2-adrenergic receptors, Salbutamol has been evaluated for its possible beneficial effects on impaired SMA neuromuscular synaptic transmission and NMJ functions. It has been shown that Salbutamol determines a rapid and significant increase of FL-SMN mRNA and protein in SMA fibroblasts, predominantly by promoting exon seven inclusion in SMN2 transcripts (Angelozzi et al., 2008): these results were further confirmed in peripheral blood leukocytes of SMA type II–III patients (Tiziano et al., 2010). Moreover, when administered to SMA type II and III patients, Salbutamol induced an overall improvement of motor performances and lung/inspiratory functions and a reduction of the perceived fatigue, suggesting further studies on the molecular mechanisms underlying these effects and its influence on muscles and NMJs (Pane et al., 2008; McCullagh et al., 2011; Giovannetti et al., 2016; Khirani et al., 2017; Frongia et al., 2019; Wadman et al., 2020).
Conclusion
The traditional process for drug discovery (starting from preclinical testing to several clinical trial phases) is a time and money consuming procedure that, possibly, after 10–15 years could bring to a new potential molecule for the targeted disease (Kraljevic et al., 2004). In the last years, DS and DR became powerful strategies for finding new molecules and/or for giving a new birth to drugs already in use for other purposes. For sure, the development of potent computational approaches, the collection of omics data (genomics, proteomics or metabolomics) and the availability of databases, in combination with new biological experimental approaches, represent the key for DS and DR success and exploitation worldwide. Altogether, DS and DR represent a challenging field, which requires constant technological update but is extremely promising.
In this work, we reviewed both the models used for the screenings and the molecules identified by DS/DR in the context of SMA; exploring these approaches could represent a potential interesting market, especially for rare disorders such as SMA.
Moreover, interestingly, we highlighted that the molecules until now identified by DS/DR are involved and act on a limited number of molecular pathways (Figure 2). This indirectly contributes to shed light on the pathogenesis of SMA, clarifying which molecular cascades, organelles and cellular structures (including cell degradation, cytoskeletal dynamics, neurotransmitter and channel modulation) are particularly affected by SMN lack, and may represent therapeutic targets in combination or alternative to the SMN-dependent approaches.
Unexpectedly, pathways involving the transcription factor nuclear factor-κB (NF-κB), calpains and autophagy are not still be directly targeted by DR/DS studies, although they are known to be strongly related to SMA pathogenesis and their direct modulation already demonstrated to be effective. Indeed, NF-κB, it is known to play a fundamental role in the survival of neuronal cells (Bhakar et al., 2002) and is able to induce the release of different neurotrophic factors which are decisive for the survival of cultured MNs (Mincheva et al., 2011). In severe SMA mice, it has been observed that NF-κB is less expressed and its inhibition, by lentiviral delivery, causes the decrease of SMN protein levels (Arumugam et al., 2018). Calpains, a family of calcium-dependent proteases, are able to regulate the expression of SMN protein by direct cleavage. This has been observed both in muscle cells (Walker et al., 2008; Fuentes et al., 2010) and in MNs cells (de la Fuente et al., 2020): indeed, calpains’ activation induces SMN cleavage in MNs, while its knockdown or inhibition increases SMN level and prevents neurite degeneration. In vivo, the treatment with calpeptin, a calpain inhibitor, improves both lifespan and motor function of SMA mice (de la Fuente et al., 2019). Interestingly, calpain levels and activity are linked to autophagy, a finely tuned process which is fundamental for the maintenance of cellular homeostasis and which has already been observed as altered in SMA (Piras et al., 2017). Calpain reduction by lentiviruses in SMA cultured MNs can induce the expression of LC3-II, a well-known marker of autophagy (Periyakaruppiah et al., 2016). The activation of autophagy has also been observed after SMN reduction, underlining its important role in SMA pathophysiology (Garcera et al., 2013; Custer and Androphy, 2014). Although, current DS/DR studies did not yet identify molecules or drugs directly acting on NF-kB, calpain and autophagy.
Furthermore, the research field of biomarkers, besides supporting diagnosis it is important i) for evaluating the efficacy of new molecules, and ii) for revealing both specific and cross-disease pathological mechanisms: for this reason, it is largely exploited for neurodegenerative disorders such as SMA, ALS, and AD (Lanni et al., 2010; Stanga et al., 2012; Kariyawasam et al., 2018; Stanga et al., 2018a; Stanga et al., 2018b).
However, besides the positive aspects deepened in this review, it is evident that DS/DR methodologies still need to be further improved. Indeed, sometimes, the promising molecules identified in preclinical studies then fail to assure an equivalent efficacy in human patients or, if effective, can encounter difficulties in the patenting phase (Cagan, 2016; Mohs and Greig, 2017; Pushpakom et al., 2018): a higher methodological standardization together with more stringent parameters could further implement the validity and the success of these powerful screening approaches.
Author Contributions
Conceptualization MB; writing original draft GM, DR, and SS; supervision SS and MB; review editing MB; funding acquisition MB. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the AFM Telethon (project number 22346) to MB, and by Girotondo/ONLUS and SMArathon-ONLUS foundations.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Prof. Alessandro Vercelli (University of Turin) for the critical reading of the review.
References
Acorda Therapeutics Inc (2017). Ampyra highlights of prescribing information. New York, NY: Acorda Ther. Inc.
Alrafiah, A., Karyka, E., Coldicott, I., Iremonger, K., Lewis, K. E., Ning, K., et al. (2018). Plastin 3 promotes motor neuron axonal growth and extends survival in a mouse model of spinal muscular atrophy. Mol. Ther. Methods Clin. Dev. 9, 81–89. doi:10.1016/j.omtm.2018.01.007
Azzouz, Ó., DeAndrés Galiana, E. J., Cernea, A., Fernández Sánchez de la Viña, J., and Fernández-Martínez, J. L. (2020). On the role of artificial intelligence in genomics to enhance precision medicine. Pharm. Pers. Med. 13, 105–119. doi:10.2147/PGPM.S205082
Al‐Zaidy, S., Pickard, A. S., Kotha, K., Alfano, L. N., Lowes, L., Paul, G., et al. (2018). Health outcomes in spinal muscular atrophy type 1 following AVXS‐101 gene replacement therapy. Pediatr. Pulmonol. 54, 179–185. doi:10.1002/ppul.24203.
Al-Zaidy, S. A., and Mendell, J. R. (2019). From clinical trials to clinical practice: practical considerations for gene replacement therapy in SMA type 1. Pediatr. Neurol. 100, 3–11. doi:10.1016/j.pediatrneurol.2019.06.007
Ando, S., Funato, M., Ohuchi, K., Inagaki, S., Sato, A., Seki, J., et al. (2019). The protective effects of levetiracetam on a human iPSCs-derived spinal muscular atrophy model. Neurochem. Res. 44, 1773–1779. doi:10.1007/s11064-019-02814-4
Ando, S., Funato, M., Ohuchi, K., Kameyama, T., Inagaki, S., Seki, J., et al. (2017). Edaravone is a candidate agent for spinal muscular atrophy: in vitro analysis using a human induced pluripotent stem cells-derived disease model. Eur. J. Pharmacol. 814, 161–168. doi:10.1016/j.ejphar.2017.08.005
Andreassi, C., Angelozzi, C., Tiziano, F. D., Vitali, T., De Vincenzi, E., Boninsegna, A., et al. (2004). Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur. J. Hum. Genet. 12, 59–65. doi:10.1038/sj.ejhg.5201102
Andreassi, C., Jarecki, J., Zhou, J., Coovert, D. D., Monani, U. R., Chen, X., et al. (2001). Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum. Mol. Genet. 10, 2841–2849. doi:10.1093/hmg/10.24.2841
Angelozzi, C., Borgo, F., Tiziano, F. D., Martella, A., Neri, G., and Brahe, C. (2008). Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J. Med. Genet. 45, 29–31. doi:10.1136/jmg.2007.051177
Annunziato, L., Secondo, A., Pignataro, G., Scorziello, A., and Molinaro, P. (2020). New perspectives for selective NCX activators in neurodegenerative diseases. Cell Calcium 87, 102170. doi:10.1016/j.ceca.2020.102170
Aquilina, B., and Cauchi, R. J. (2018). Modelling motor neuron disease in fruit flies: lessons from spinal muscular atrophy. J. Neurosci. Methods 310, 3–11. doi:10.1016/j.jneumeth.2018.04.003
Arumugam, S., Mincheva-Tasheva, S., Periyakaruppiah, A., de la Fuente, S., Soler, R. M., and Garcera, A. (2018). Regulation of survival motor neuron protein by the nuclear factor-kappa B pathway in mouse spinal cord motoneurons. Mol. Neurobiol. 55, 5019–5030. doi:10.1007/s12035-017-0710-4
Ashburn, T. T., and Thor, K. B. (2004). Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 3, 673–683. doi:10.1038/nrd1468
Avila, A. M., Burnett, B. G., Taye, A. A., Gabanella, F., Knight, M. A., Hartenstein, P., et al. (2007). Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117, 659–671. doi:10.1172/JCI29562
Baek, J., Jeong, H., Ham, Y., Jo, Y. H., Choi, M., Kang, M., et al. (2019). Improvement of spinal muscular atrophy via correction of the SMN2 splicing defect by Brucea javanica (L.) Merr. extract and Bruceine D. Phytomedicine 65, 153089. doi:10.1016/j.phymed.2019.153089
Beenakker, E. A. C., Fock, J. M., Van Tol, M. J., Maurits, N. M., Koopman, H. M., Brouwer, O. F., et al. (2005). Intermittent Prednisone Therapy in Duchenne Muscular Dystrophy. Arch. Neurol. 62, 128. doi:10.1001/archneur.62.1.128
Bhakar, A. L., Tannis, L.-L., Zeindler, C., Russo, M. P., Jobin, C., Park, D. S., et al. (2002). Constitutive nuclear factor-κB activity is required for central neuron survival. J. Neurosci.22,8466–8475. doi:10.1523/jneurosci.22-19-08466.2002
Bigini, P., Larini, S., Pasquali, C., Muzio, V., and Mennini, T. (2002). Acetyl-l-carnitine shows neuroprotective and neurotrophic activity in primary culture of rat embryo motoneurons. Neurosci. Lett. 329, 334–338. doi:10.1016/S0304-3940(02)00667-5
Blasco, H., Patin, F., Descat, A., Garçon, G., Corcia, P., Gelé, P., et al. (2018). A pharmaco-metabolomics approach in a clinical trial of ALS: identification of predictive markers of progression. PLoS One 13, e0198116-14. doi:10.1371/journal.pone.0198116
Boido, M., de Amicis, E., Valsecchi, V., Trevisan, M., Ala, U., Ruegg, M. A., et al. (2018). Increasing agrin function antagonizes muscle atrophy and motor impairment in spinal muscular atrophy. Front. Cell. Neurosci. 12, 1–14. doi:10.3389/fncel.2018.00017
Boido, M., and Vercelli, A. (2016). Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front. Neuroanat. 10, 6. doi:10.3389/fnana.2016.00006
Bordet, T., Berna, P., Abitbol, J.-L., and Pruss, R. M. (2010). Olesoxime (TRO19622): a novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals 3, 345–368. doi:10.3390/ph3020345
Bonanno, S., Pasanisi, M. B., Frangiamore, R., Maggi, L., Antozzi, C., Andreetta, F., et al. (2018). Amifampridine phosphate in the treatment of muscle-specific kinase myasthenia gravis: a phase IIb, randomized, double-blind, placebo-controlled, double crossover study, SAGE Open Med. 6, 205031211881901. doi:10.1177/2050312118819013
Boon, K.-L., Xiao, S., McWhorter, M. L., Donn, T., Wolf-Saxon, E., Bohnsack, M. T., et al. (2009). Zebrafish survval motor neuron mutants exhibit presynaptic neuromuscular junction defects. Hum. Mol. Genet. 18, 3615–3625. doi:10.1093/hmg/ddp310
Bordet, T., Buisson, B., Michaud, M., Drouot, C., Galéa, P., Delaage, P., et al. (2007). Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Therapeut. 322, 709. doi:10.1124/jpet.107.123000
Bottai, D., and Adami, R. (2013). Spinal muscular atrophy: new findings for an old pathology. Brain Pathol. 23, 613–622. doi:10.1111/bpa.12071
Bosch-Marcé, M., Wee, C. D., Martinez, T. L., Lipkes, C. E., Choe, D. W., Kong, L., et al. (2011). Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum. Mol. Genet. 20, 1844–1853. doi:10.1093/hmg/ddr067
Bowerman, M. (2014). ROCK inhibition as a therapy for spinal muscular atrophy : understanding the repercussions on multiple cellular targets. Front. Neurosci. 8, 1–12. doi:10.3389/fnins.2014.00271
Bowerman, M., Beauvais, A., Anderson, C. L., and Kothary, R. (2010). Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 19, 1468–1478. doi:10.1093/hmg/ddq021
Bowerman, M., Becker, C. G., Ya, R. J., Ning, K., Wood, M. J. A., Gillingwater, T. H., et al. (2017). Therapeutic strategies for spinal muscular atrophy : SMN and beyond. Dis. Model Mech. 10, 943–954. doi:10.1242/dmm.030148
Bowerman, M., Murray, L. M., Boyer, J. G., Anderson, C. L., and Kothary, R. (2012). Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 10, 24. doi:10.1186/1741-7015-10-24
Bowerman, M., Murray, L. M., Scamps, F., Schneider, B. L., Kothary, R., and Raoul, C. (2018). Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: converging roads to therapeutic development. Eur. J. Med. Genet. 61, 685–698. doi:10.1016/j.ejmg.2017.12.001
Bowerman, M., Shafey, D., and Kothary, R. (2007). Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J. Mol. Neurosci. 32, 120–131. doi:10.1007/s12031-007-0024-5
Briese, M., Esmaeili, B., Fraboulet, S., Burt, E. C., Christodoulou, S., Towers, P. R., et al. (2009). Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum. Mol. Genet. 18, 97–104. doi:10.1093/hmg/ddn320
Brooks, B. R., Sufit, R. L., Montgomery, G. K., Beaulieu, D. A., and Erickson, L. M. (1987). Intravenous thyrotropin-releasing hormone in patients with amyotrophic lateral sclerosis: dose-response and randomized concurrent placebo-controlled pilot studies. Neurol. Clin. 5, 143–158. doi:10.1016/S0733-8619(18)30939-3
Buckingham, S. D., and Sattelle, D. B. (2009). Fast, automated measurement of nematode swimming (thrashing) without morphometry. BMC Neurosci. 10, 1–6. doi:10.1186/1471-2202-10-84
C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology [published correction appears in Science 283 (5398), 35. doi:10.1126/science.282.5396.2012
Cáceres, I. d. C., Valmas, N., Hilliard, M. A., and Lu, H. (2012). Laterally Orienting C. elegans using geometry at microscale for high-throughput visual screens in neurodegeneration and neuronal development studies. PLoS One 7, e35037. doi:10.1371/journal.pone.0035037
Cagan, R. (2016). Drug screening using model systems: some basics. Dis. Model. Mech. 9, 1241–1244. doi:10.1242/dmm.028159
Calder, A. N., Androphy, E. J., and Hodgetts, K. J. (2016). Small molecules in development for the treatment of spinal muscular atrophy. J. Med. Chem. 59, 10067. doi:10.1021/acs.jmedchem.6b00670
Caraceni, A., Zecca, E., Bonezzi, C., Arcuri, E., Tur, R. Y., Maltoni, M., et al. (2004). Gabapentin for neuropathic cancer pain: a randomized controlled trial from the gabapentin cancer pain study group. J. Clin. Oncol. 22, 2909–2917. doi:10.1200/JCO.2004.08.141
Cauchi, R. J., and Van Den Heuvel, M. (2007). The fly as a model for neurodegenerative diseases: is it worth the jump? Neurodegener. Dis. 3, 338–356. doi:10.1159/000097303
Chan, Y. B., Miguel-Aliaga, I., Franks, C., Thomas, N., Trülzsch, B., Sattelle, D. B., et al. (2003). Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum. Mol. Genet. 12, 1367–1376. doi:10.1093/hmg/ddg157
Chang, H. C.-H., Dimlich, D. N., Yokokura, T., Mukherjee, A., Kankel, M. W., Sen, A., et al. (2008). Modeling spinal muscular atrophy in Drosophila. PLoS One 3, e3209–e3218. doi:10.1371/journal.pone.0003209
Chang, J.-G., Hsieh-Li, H.-M., Jong, Y.-J., Wang, N. M., Tsai, C.-H., and Li, H. (2001). Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. U.S.A. 98, 9808–9813. doi:10.1073/pnas.171105098
Chaytow, H., Huang, Y.-T., Gillingwater, T. H., and Faller, K. M. E. (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci., 75, 3877 doi:10.1007/s00018-018-2849-1
Cherry, J. J., Osman, E. Y., Evans, M. C., Choi, S., Xing, X., Cuny, G. D., et al. (2013). Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug‐like compounds. EMBO Mol. Med. 5, 1103–1118. doi:10.1002/emmm.201202305
Cheung, A. K., Hurley, B., Kerrigan, R., Shu, L., Chin, D. N., Shen, Y., et al. (2018). Discovery of small molecule splicing modulators of survival motor neuron-2 (SMN2) for the treatment of spinal muscular atrophy (SM). J. Med. Chem. 61, 11021–11036. doi:10.1021/acs.jmedchem.8b01291
Chiriboga, C. A., Swoboda, K. J., Darras, B. T., Iannaccone, S. T., Montes, J., De Vivo, D. C., et al. (2016). Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology, 86, 890. doi:10.1212/WNL.0000000000002445
Cogram, P., Deacon, R. M. J., Warner-Schmidt, J. L., von Schimmelmann, M. J., Abrahams, B. S., and During, M. J. (2019). Gaboxadol normalizes behavioral abnormalities in a mouse model of fragile x syndrome. Front. Behav. Neurosci. 13, 1–9. doi:10.3389/fnbeh.2019.00141
Cudkowicz, M. E., Titus, S., Kearney, M., Yu, H., Sherman, A., Schoenfeld, D., et al. (2014). Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 13, 1083–1091. doi:10.1016/S1474-4422(14)70222-4
Custer, S. K., and Androphy, E. J. (2014). Autophagy dysregulation in cell culture and animals models of spinal muscular atrophy. Mol. Cell. Neurosci.61,133–140. doi:10.1016/j.mcn.2014.06.006
D’Amico, A., Mercuri, E., Tiziano, F. D., and Bertini, E. (2011). Spinal muscular atrophy. Orphanet J. Rare Dis. 6, 71. doi:10.1186/1750-1172-6-71
Dayangaç-Erden, D., Topaloğlu, H., and Erdem-Yurter, H. (2008). A preliminary report on spinal muscular atrophy lymphoblastoid cell lines: are they an appropriate tool for drug screening? Adv. Ther. 25, 274–279. doi:10.1007/s12325-008-0030-1
de Carlos Cáceres, I., Porto, D. A., Gallotta, I., Santonicola, P., Rodríguez-Cordero, J., Di Schiavi, E., et al. (2018). Automated screening of C. elegans neurodegeneration mutants enabled by microfluidics and image analysis algorithms. Integr. Biol. 10, 539–548. doi:10.1039/C8IB00091C
de la Fuente, S., Sansa, A., Hidalgo, I., Vivancos, N., Romero-Guevara, R., Garcera, A., et al. (2020). Calpain system is altered in survival motor neuron-reduced cells from in vitro and in vivo spinal muscular atrophy models. Cell Death Dis.11, 487. doi:10.1038/s41419-020-2688-5
de la Fuente, S., Sansa, A., Periyakaruppiah, A., Garcera, A., and Soler, R. M. (2019). Calpain inhibition increases SMN protein in spinal cord motoneurons and ameliorates the spinal muscular atrophy phenotype in mice. Mol. Neurobiol. 56, 4414–4427. doi:10.1007/s12035-018-1379-z
Dimitriadi, M., and Hart, A. C. (2010). Neurodegenerative disorders: insights from the nematode Caenorhabditis elegans. Neurobiol. Dis. 40, 4–11. doi:10.1016/j.nbd.2010.05.012
Dimitriadi, M., Kye, M. J., Kalloo, G., Yersak, J. M., Sahin, M., and Hart, A. C. (2013). The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci. 33, 6557–6562. doi:10.1523/JNEUROSCI.1536-12.2013
Edens, B. M., Ajroud-Driss, S., Ma, L., and Ma, Y.-C. (2015). Molecular mechanisms and animal models of spinal muscular atrophy. Biochim. Biophys. Acta 1852, 685–692. doi:10.1016/j.bbadis.2014.07.024
Eshraghi, M., McFall, E., Gibeault, S., and Kothary, R. (2016). Effect of genetic background on the phenotype of theSmn2B/-mouse model of spinal muscular atrophy. Hum. Mol. Genet. 25, ddw278. doi:10.1093/hmg/ddw278
Farooq, F., Abadía-Molina, F., MacKenzie, D., Hadwen, J., Shamim, F., O'Reilly, S., et al. (2013). Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum. Mol. Genet. 22, 3415–3424. doi:10.1093/hmg/ddt191
Feng, Z., Ling, K. K. Y., Zhao, X., Zhou, C., Karp, G., Welch, E. M., et al. (2016). Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 25, 964–975. doi:10.1093/hmg/ddv629
Field, M. J., Oles, R. J., Lewis, A. S., McCleary, S., Hughes, J., and Singh, L. (1997). Gabapentin (neurontin) and S -(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Br. J. Pharmacol. 121, 1513–1522. doi:10.1038/sj.bjp.0701320
Fisher, E. M. C., and Bannerman, D. M. (2019). Mouse models of neurodegeneration: know your question, know your mouse. Sci. Transl. Med. 11, eaaq1818-16. doi:10.1126/scitranslmed.aaq1818
Foran, E., Kwon, D. Y., Nofziger, J. H., Arnold, E. S., Hall, M. D., Fischbeck, K. H., et al. (2016). CNS uptake of bortezomib is enhanced by P-glycoprotein inhibition: implications for spinal muscular atrophy. Neurobiol. Dis. 88, 118–124. doi:10.1016/j.nbd.2016.01.008
Fox, S., Farr-Jones, S., Sopchak, L., Boggs, A., Nicely, H. W., Khoury, R., et al. (2006). High-throughput screening: update on practices and success. J. Biomol. Screen 11, 864–869. doi:10.1177/1087057106292473
Frey, F. J. (1987). Kinetics and dynamics of prednisolone*. Endocr. Rev. 8, 453–473. doi:10.1210/edrv-8-4-453
Frongia, A. L., Natera-de Benito, D., Ortez, C., Alarcón, M., Borrás, A., Medina, J., et al. (2019). Salbutamol tolerability and efficacy in patients with spinal muscular atrophy type II. Neuromuscul. Disord. 29, 517–524. doi:10.1016/j.nmd.2019.04.003
Fuentes, J. L., Strayer, M. S., and Matera, A. G. (2010). Molecular determinants of survival motor neuron (SMN) protein cleavage by the calcium-activated protease, calpain. PLoS One. 5, e15769. doi:10.1371/journal.pone.0015769
Gambertoglio, J. G., Amend, W. J. C., and Benet, L. Z. (1980). Pharmacokinetics and bioavailability of prednisone and prednisolone in healthy volunteers and patients: a review. J. Pharmacokinet. Biopharm. 8, 1–52. doi:10.1007/BF01059447
Garbes, L., Riessland, M., Hölker, I., Heller, R., Hauke, J., Tränkle, C., et al. (2009). LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 18, 3645–3658. doi:10.1093/hmg/ddp313
Garcera, A., Bahi, N., Periyakaruppiah, A., Arumugam, S., and Soler, R. M. (2013). Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell Death Dis. 4:e686. doi:10.1038/cddis.2013.209
Garcera, A., Mincheva, S., Gou-Fabregas, M., Caraballo-Miralles, V., Lladó, J., Comella, J. X., et al. (2011). A new model to study spinal muscular atrophy: neurite degeneration and cell death is counteracted by BCL-XL Overexpression in motoneurons. Neurobiol. Dis. 42, 415–426. doi:10.1016/j.nbd.2011.02.003
Garcia-Manero, G., Fenaux, P., Al-Kali, A., Baer, M. R., Sekeres, M. A., Roboz, G. J., et al. (2016). Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol. 17, 496–508. doi:10.1016/S1470-2045(16)00009-7
Gaidano, T., and Servais, L. (2019). Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps. Dev. Med. Child Neurol. 61, 19–24. doi:10.1111/dmcn.14027
Genentech (2020). FDA approves Genentech’s Evrysdi (risdiplam) for treatment of spinal muscular atrophy (SMA) in adults and children 2 months and older. Available at: https://www.gene.com/media/press-releases/14866/2020-08-07/fda-approves-genentechs-evrysdi-risdipla (Accessed August 7, 2020)
Giovannetti, A. M., Pasanisi, M. B., Černiauskaitė, M., Bussolino, C., Leonardi, M., and Morandi, L. (2016). Perceived efficacy of salbutamol by persons with spinal muscular atrophy: a mixed methods study. Muscle Nerve 54, 843–849. doi:10.1002/mus.25102
Gogliotti, R. G., Cardona, H., Singh, J., Bail, S., Emery, C., Kuntz, N., et al. (2013). The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 22, 4084–4101. doi:10.1093/hmg/ddt258
Gogliotti, R. G., Quinlan, K. A., Barlow, C. B., Heier, C. R., Heckman, C. J., and DiDonato, C. J. (2012). Motor Neuron Rescue in Spinal Muscular Atrophy Mice Demonstrates That Sensory-Motor Defects Are a Consequence, Not a Cause, of Motor Neuron Dysfunction. J. Neurosci. 32, 3818–3829. doi:10.1523/JNEUROSCI.5775-11.2012
Grice, S. J., Sleigh, J. N., Liu, J.-L., and Sattelle, D. B. (2011). Invertebrate models of spinal muscular atrophy: insights into mechanisms and potential therapeutics. Bioessays 33, 956–965. doi:10.1002/bies.201100082
Grzeschik, S. M., Ganta, M., Prior, T. W., Heavlin, W. D., and Wang, C. H. (2005). Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann. Neurol. 58, 194–202. doi:10.1002/ana.20548
Guarino, S. R., Canciani, A., and Forneris, F. (2020). Dissecting the extracellular complexity of neuromuscular junction organizers. Front. Mol. Biosci. 6. doi:10.3389/fmolb.2019.00156
Haddad, H., Cifuentes-Diaz, C., Miroglio, A., Roblot, N., Joshi, V., and Melki, J. (2003). Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 28, 432–437. doi:10.1002/mus.10455
Hahnen, E., Eyüpoglu, I. Y., Brichta, L., Haastert, K., Tränkle, C., Siebzehnrübl, F. A., et al. (2006). In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem. 98, 193–202. doi:10.1111/j.1471-4159.2006.03868.x
Haniff, H. S., Knerr, L., Chen, J. L., Disney, M. D., and Lightfoot, H. L. (2020). Target-directed approaches for screening small molecules against RNA targets. SLAS Discov. 25, 869. doi:10.1177/2472555220922802
Hao, L. T., Burghes, A. H., and Beattie, C. E. (2011). Generation and Characterization of a genetic zebrafish model of SMA carrying the human SMN2 gene. Mol. Neurodegener. 6, 24–29. doi:10.1186/1750-1326-6-24
Hensel, N., Baskal, S., Walter, L. M., Brinkmann, H., Gernert, M., and Claus, P. (2017). ERK and ROCK functionally interact in a signaling network that is compensationally upregulated in spinal muscular atrophy. Neurobiol. Dis. 108, 352–361. doi:10.1016/j.nbd.2017.09.005
Hensel, N., Kubinski, S., and Claus, P. (2020). The need for SMN-independent treatments of spinal muscular atrophy (SMA) to complement SMN-enhancing drugs. Front. Neurol. 11, 1–10. doi:10.3389/fneur.2020.00045
Hensel, N., Rademacher, S., and Claus, P. (2015). Chatting with the neighbors: crosstalk between Rho-kinase (ROCK) and other signaling pathways for treatment of neurological disorders. Front. Neurosci. 9, 198. doi:10.3389/fnins.2015.00198
Hoolachan, J. M., Sutton, E. R., and Bowerman, M. (2019). Teaching an old drug new tricks: repositioning strategies for spinal muscular atrophy. Future Neurol. 14, FNL25. doi:10.2217/fnl-2019-0006
Hoot, N. R. (2019). Nusinersen for type 1 spinal muscular atrophy: a father’s perspective. Pediatrics 144, e20190226, doi:10.1542/peds.2019-0226
Horrigan, J., McMorn, A., Snape, M., Nikolenko, N., Gomes, T., and Lochmuller, H. (2018). AMO-02 (tideglusib) for the treatment of congenital and childhood onset myotonic dystrophy type 1. Neuromuscul. Disord. 28, S14. doi:10.1016/S0960-8966(18)30330-4
Hosseinibarkooie, S., Peters, M., Torres-Benito, L., Rastetter, R. H., Hupperich, K., Hoffmann, A., et al. (2016). The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 99, 647–665. doi:10.1016/j.ajhg.2016.07.014
Hosseinibarkooie, S. M., Peters, M., Torres-Benito, L., Heesen, L., Peitz, M., Markus, S., et al. (2016). Plastin 3, a human protective modifier is highly up regulated in iPSC-derived motoneurons in asymptomatic SMN1-deteted individuals and rescues spinal muscular atrophy in mice by restored endocytosis. Med. Genet. 28, 84–232. doi:10.1007/s11825-016-0083-5
Howe, K., Clark, M. D., Torroja, C. F., Torrance, J., Berthelot, C., Muffato, M., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. doi:10.1038/nature12111
Hsieh-Li, H. M., Chang, J.-G., Jong, Y.-J., Wu, M.-H., Wang, N. M., Tsai, C. H., et al. (2000). A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66–70. doi:10.1038/71709
Hughes, J., Rees, S., Kalindjian, S., and Philpott, K. (2011). Principles of early drug discovery. Br. J. Pharmacol. 162, 1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
Jackson, C., Heiman-Patterson, T., Kittrell, P., Baranovsky, T., McAnanama, G., Bower, L., et al. (2019). Radicava (edaravone) for amyotrophic lateral sclerosis: US experience at 1 year after launch. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 605–610. doi:10.1080/21678421.2019.1645858
Jarecki, J., Chen, X., Bernardino, A., Coovert, D. D., Whitney, M., Burghes, A., et al. (2005). Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 14, 2003–2018. doi:10.1093/hmg/ddi205
Jensen, H. B., Stenager, E., and Ravnborg, M. H. (2011). Aminopyridines for symptomatic treatment of multiple sclerosis. Ugeskr. Laeger 173, 3259–3263. doi:10.1002/14651858.CD001330
Jochmann, E., Steinbach, R., Jochmann, T., Chung, H.-Y., Rödiger, A., Neumann, R., et al. (2020). Experiences from treating seven adult 5q spinal muscular atrophy patients with Nusinersen. Ther. Adv. Neurol. Disord. 13, 175628642090780. doi:10.1177/1756286420907803
Kaifer, K. A., Villalón, E., Osman, E. Y., Glascock, J. J., Arnold, L. L., Cornelison, D. D. W., et al. (2017). Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2, e89970. doi:10.1172/jci.insight.89970
Kaneko, C., Burdine, R., Saulnier, C., Chen, S., Pudussery, G., Rakhit, A., et al. (2020). The pivotal Phase 3 NEPTUNE trial investigating gaboxadol in Angelman syndrome: study design (1327). Am. Acad. Neurol.
Kannan, A., Bhatia, K., Branzei, D., and Gangwani, L. (2018). Combined deficiency of senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 46, 8326–8346. doi:10.1093/nar/gky641
Kariyawasam, D., Carey, K. A., Jones, K. J., and Farrar, M. A. (2018). New and developing therapies in spinal muscular atrophy. Paediatr. Respir. Rev. 28, 3–10. doi:10.1016/j.prrv.2018.03.003
Kato, Z., Okuda, M., Okumura, Y., Arai, T., Teramoto, T., Nishimura, M., et al. (2009). Oral administration of the thyrotropin-releasing hormone (TRH) analogue, taltireline hydrate, in spinal muscular atrophy. J. Child Neurol. 24, 1010–1012. doi:10.1177/0883073809333535
Kelley, B. P., Lunn, M. R., Root, D. E., Flaherty, S. P., Martino, A. M., and Stockwell, B. R. (2004). A flexible data analysis tool for chemical genetic screens. Chem. Biol. 11, 1495–1503. doi:10.1016/j.chembiol.2004.08.026
Khirani, S., Dabaj, I., Amaddeo, A., Olmo Arroyo, J., Ropers, J., Tirolien, S., et al. (2017). Effect of salbutamol on respiratory muscle strength in spinal muscular atrophy. Pediatr. Neurol. 73, 78–87.e1. doi:10.1016/j.pediatrneurol.2017.04.013
Kim, H., Cho, S. C., Jeong, H. J., Lee, H. Y., Jeong, M. H., Pyun, J. H., et al. (2020). Indoprofen prevents muscle wasting in aged mice through activation of PDK1/AKT pathway. J. Cachex., Sarco. Muscle 11, 1070. doi:10.1002/jcsm.12558
Kirschner, J., Schorling, D., Hauschke, D., Rensing-Zimmermann, C., Wein, U., Grieben, U., et al. (2014). Somatropin treatment of spinal muscular atrophy: a placebo-controlled, double-blind crossover pilot study. Neuromuscul. Disord., 24, 134 doi:10.1016/j.nmd.2013.10.011
Kissel, J. T., Scott, C. B., Reyna, S. P., Crawford, T. O., Simard, L. R., Krosschell, K. J., et al. (2011). Sma carni-val trial part II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One 6, e21296-11. doi:10.1371/journal.pone.0021296
Kletzl, H., Marquet, A., Günther, A., Tang, W., Heuberger, J., Groeneveld, G. J., et al. (2019). The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 29, 21–29. doi:10.1016/j.nmd.2018.10.001
Koch, J. C., Tatenhorst, L., Roser, A.-E., Saal, K.-A., Tönges, L., and Lingor, P. (2018). ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 189, 1–21. doi:10.1016/j.pharmthera.2018.03.008
Konieczny, P., and Artero, R. (2020). Drosophila SMN2 minigene reporter model identifies moxifloxacin as a candidate therapy for SMA. FASEB J. 34, 3021–3036. doi:10.1096/fj.201802554RRR
Kraljevic, S., Stambrook, P. J., and Pavelic, K. (2004). Accelerating drug discovery. EMBO Rep. 5, 837–842. doi:10.1038/sj.embor.7400236
Kwon, D. Y., Motley, W. W., Fischbeck, K. H., and Burnett, B. G. (2011). Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum. Mol. Genet. 20, 3667–3677. doi:10.1093/hmg/ddr288
Lai, J.-I., Leman, L. J., Ku, S., Vickers, C. J., Olsen, C. A., Montero, A., et al. (2017). Cyclic tetrapeptide HDAC inhibitors as potential therapeutics for spinal muscular atrophy: screening with iPSC-derived neuronal cells. Bioorg. Med. Chem. Lett 27, 3289–3293. doi:10.1016/j.bmcl.2017.06.027
Lanni, C., Racchi, M., Stanga, S., Mazzini, G., Ranzenigo, A., Polotti, R., et al. (2010). Unfolded p53 in blood as a predictive signature signature of the transition from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. 20, 97–104. doi:10.3233/JAD-2010-1347
Le, T. T., Pham, L. T., Butchbach, M. E. R., Zhang, H. L., Monani, U. R., Coovert, D. D., et al. (2005). SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857. doi:10.1093/hmg/ddi078
Lee, H., Crane, M. M., Zhang, Y., and Lu, H. (2013). Quantitative screening of genes regulating tryptophan hydroxylase transcription in Caenorhabditis elegans using microfluidics and an adaptive algorithm. Integr. Biol. 5, 372–380. doi:10.1039/C2IB20078C
Lee, M., França, U. L., Graham, R. J., and McManus, M. L. (2019). Pre-Nusinersen hospitalization costs of children with spinal muscular atrophy. Pediatr. Neurol. 92, 3–5. doi:10.1016/j.pediatrneurol.2018.11.002
Lee, S.-J., and McPherron, A. C. (2001). Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U.S.A. 98, 9306–9311. doi:10.1073/pnas.151270098
Lessing, D., and Bonini, N. M. (2009). Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat. Rev. Genet. 10, 359–370. doi:10.1038/nrg2563
Letso, R. R., Bauer, A. J., Lunn, M. R., Yang, W. S., and Stockwell, B. R. (2013). Small molecule screen reveals regulation of survival motor neuron protein abundance by Ras proteins. ACS Chem. Biol. 8, 914–922. doi:10.1021/cb300374h
Levine, A. R., Carrasquillo, L., Mueller, J., Nounou, M. I., Naut, E. R., and Ibrahim, D. (2019). High‐dose gabapentin for the treatment of severe alcohol withdrawal syndrome: a retrospective cohort analysis. Pharmacotherapy 39, 881–888. doi:10.1002/phar.2309
Lipinski, C. A., and Reaume, A. G. (2020). High throughput in vivo phenotypic screening for drug repurposing: discovery of MLR-1023 a novel insulin sensitizer and novel Lyn kinase activator with clinical proof of concept. Bioorg. Med. Chem. 28, 115425. doi:10.1016/j.bmc.2020.115425
Lipsky, P. E., and Isakson, P. C. (1997). Outcome of specific COX-2 inhibition in rheumatoid arthritis. J. Rheumatol. 49, 9–14.
Liu, H.-C., Ting, C.-H., Wen, H.-L., Tsai, L.-K., Hsieh-Li, H.-M., Li, H., et al. (2013). Sodium vanadate combined with l-ascorbic acid delays disease progression, enhances motor performance, and ameliorates muscle atrophy and weakness in mice with spinal muscular atrophy. BMC Med. 11, 38. doi:10.1186/1741-7015-11-38
Long, K. K., O’Shea, K. M., Khairallah, R. J., Howell, K., Paushkin, S., Chen, K. S., et al. (2019). Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 28, 1076–1089. doi:10.1093/hmg/ddy382
Lopez, J., Quan, A., Budihardjo, J., Xiang, S., Wang, H., Koshy, K., et al. (2019). Growth hormone improves nerve regeneration, muscle re-innervation, and functional outcomes after chronic denervation injury. Sci. Rep. 9, 3117. doi:10.1038/s41598-019-39738-6
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 6307, doi:10.1073/pnas.96.11.6307
Lotfi Shahreza, M., Ghadiri, N., Mousavi, S. R., Varshosaz, J., and Green, J. R. (2018). A review of network-based approaches to drug repositioning. Briefings Bioinf. 19, 878–892. doi:10.1093/bib/bbx017
Lovestone, S., Boada, M., Dubois, B., Hüll, M., Rinne, J. O., Huppertz, H.-J., et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. doi:10.3233/JAD-141959
Madaan, K., Kaushik, D., and Verma, T. (2012). Hydroxyurea: a key player in cancer chemotherapy. Expet Rev. Anticancer Ther. 12, 19–29. doi:10.1586/era.11.175
Maddison, P., Newsom-Davis, J., and Mills, K. R. (1998). Effect of 3,4-diaminopyridine on the time course of decay of compound muscle action potential augmentation in the Lambert-Eaton myasthenic syndrome. Muscle Nerve 21, 1196–1198. doi:10.1002/(SICI)1097-4598(199809)21:9<1196::AID-MUS11>3.0.CO;2-Q
Makhortova, N. R., Hayhurst, M., Cerqueira, A., Sinor-Anderson, A. D., Zhao, W.-N., Heiser, P. W., et al. (2011). A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 7, 544–552. doi:10.1038/nchembio.595
Makhouri, F. R., and Ghasemi, J. B. (2017). In silico studies in drug research against neurodegenerative diseases. Curr. Neuropharmacol. 16, 664–725. doi:10.2174/1570159x15666170823095628
Malone, D. C., Dean, R., Arjunji, R., Jensen, I., Cyr, P., Miller, B., et al. (2019). Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy 7, 1601484. doi:10.1080/20016689.2019.1601484
Mariot, V., Joubert, R., Hourdé, C., Féasson, L., Hanna, M., Muntoni, F., et al. (2017). Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 8, 1859. doi:10.1038/s41467-017-01486-4
Maru, M. A., Zheleznyakova, G. Y., Lanko, K. M., Egorova, A. A., Baranov, V. S., and Kiselev, A. V. (2018). Molecular factors involved in spinal muscular atrophy pathways as possible disease-modifying candidates. Curr. Genomics 19, 339–355. doi:10.2174/1389202919666180101154916
Mathias, S., Zihl, J., Steiger, A., and Lancel, M. (2005). Effect of repeated gaboxadol administration on night sleep and next-day performance in healthy elderly subjects. Neuropsychopharmacology 30, 833–841. doi:10.1038/sj.npp.1300641
Matthews, E., Brassington, R., Kuntzer, T., Jichi, F., and Manzur, A. Y. (2016). Corticosteroids for the treatment of duchenne muscular dystrophy. Cochrane Database Syst. Rev. 5, CD003725. doi:10.1002/14651858.CD003725.pub4
Mercuri, E., and Sansone, V. (2020). Nusinersen in adults with spinal muscular atrophy: new challenges. Lancet Neurol. 19, 283–284. doi:10.1016/S1474-4422(20)30068-5
Mentis, G. Z., Blivis, D., Liu, W., Drobac, E., Crowder, M. E., Kong, L., et al. (2011). Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69, 453–467. doi:10.1016/j.neuron.2010.12.032
Merlini, L., Basoglu, B., Dahna-Schwake, C., Febrer, A., Hausmanova-Petrusewicz, I., and Jedrzejonska, M. (2010). Eurosmart: european spinal muscular atrophy randomised trial of acetyl-L-carnitine in spinal muscular atrophy. Rev. Neurol. (Paris). 166, 353–362. doi:10.1016/j.neurol.2009.07.016.
Merlini, L., Estournet-Mathiaud, B., Iannaccone, S., Melki, J., Muntoni, F., Rudnik-Schöneborn, S., et al. (2002). 90th ENMC international workshop: European spinal muscular atrophy randomised trial (EuroSMART) 9-10 February 2001, Naarden, The Netherlands. Neuromuscul. Disord. 12, 201–210. doi:10.1016/s0960-8966(01)00272-3
Merlini, L., Solari, A., Vita, G., Bertini, E., Minetti, C., Mongini, T., et al. (2003). Role of gabapentin in spinal muscular atrophy. J. Child Neurol. 18, 537–541. doi:10.1177/08830738030180080501
McCullagh, G., Main, M., Muntoni, F., Manzur, A. Y., Scoto, M.-C., and Robb, S. (2011). Response to daily salbutamol in spinal muscular atrophy type III. Eur. J. Paediatr. Neurol. 15, S96. doi:10.1016/S1090-3798(11)70332-4
McGurk, L., Berson, A., and Bonini, N. M. (2015). Drosophilaas anIn VivoModel for human neurodegenerative disease. Genetics 201, 377–402. doi:10.1534/genetics.115.179457
McInnes, C. (2007). Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 11, 494–502. doi:10.1016/j.cbpa.2007.08.033
Miller, R. G., Moore, D. H., Dronsky, V., Bradley, W., Barohn, R., Bryan, W., et al. (2001). A placebo-controlled trial of gabapentin in spinal muscular atrophy. J. Neurol. Sci. 191, 127–131. doi:10.1016/S0022-510X(01)00632-3
Mincheva, S., Garcera, A., Gou-Fabregas, M., Encinas, M., Dolcet, X., and Soler, R. M. (2011). The canonical nuclear factor- B pathway regulates cell survival in a developmental model of spinal cord motoneurons. J. Neurosci. 31, 6493–6503. doi:10.1523/JNEUROSCI.0206-11.2011
Mitrou, P. S. (1990). “Aclarubicin in single agent and combined chemotherapy of adult acute myeloid leukemia,” inNew findings on aclarubicin in the treatment of acute myeloid leukemia (Berlin, Heidelberg: Springer Berlin Heidelberg), 1–7. doi:10.1007/978-3-642-75720-4_1
Mohs, R. C., and Greig, N. H. (2017). Drug discovery and development: role of basic biological research. Alzheimer's Dement. 3, 651–657. doi:10.1016/j.trci.2017.10.005
Monani, U. R. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi:10.1093/hmg/8.7.1177
Monani, U. R. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn-/- mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339. doi:10.1093/hmg/9.3.333
Moore, R. A., Wiffen, P. J., Derry, S., and McQuay, H. J. (2011). “Gabapentin for chronic neuropathic pain and fibromyalgia in adults,” in Cochrane database of systematic reviews. Editor R. A. Moore (Chichester, UK: John Wiley & Sons, Ltd), CD007938. doi:10.1002/14651858.CD007938.pub2.
Morceau, F., Chénais, B., Gillet, R., Jardillier, J. C., Jeannesson, P., and Trentesaux, C. (1996). Transcriptional and posttranscriptional regulation of erythroid gene expression in anthracycline-induced differentiation of human erythroleukemic cells. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 7, 1023–1029.
Mouse Genome Sequencing Consortium Waterston, R. H., Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. doi:10.1038/nature01262
Naguy, A., and Al-Enezi, N. (2019). Lamotrigine uses in psychiatric practice. Am. J. Therapeut. 26, e96–e102. doi:10.1097/MJT.0000000000000535
Naryshkin, N. A., Dakka, A., Narasimhan, J., Zhao, X., Feng, Z., et al. (2014). SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 345 (6197), 688–693. doi:10.1126/science.1250127
Nascimento, O. J. M., Orsini Neves, M. A., and Quintanilha, G. (2010). Lamotrigina en los síntomas motores de las atrofias musculares espinales. RevNeurol 50, 127–128. doi:10.33588/rn.5002.2009267
Ng, F., Berk, M., Dean, O., and Bush, A. I. (2008). Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. 11, 851–876. doi:10.1017/s1461145707008401
Nizzardo, M., Nardini, M., Ronchi, D., Salani, S., Donadoni, C., Fortunato, F., et al. (2011). Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms. Exp. Neurol. 229, 214–225. doi:10.1016/j.expneurol.2011.01.017
Nölle, A., Zeug, A., van Bergeijk, J., Tönges, L., Gerhard, R., Brinkmann, H., et al. (2011). The spinal muscular atrophy disease protein SMN is linked to the rho-kinase pathway via profilin. Hum. Mol. Genet. 20, 4865–4878. doi:10.1093/hmg/ddr425
Novartis (2020). Media release media release media release Zolgensma ® data shows rapid, significant, clinically meaningful benefit in SMA including prolonged event- free survival, motor milestone achievement and durability now up to 5 years post-dosing. EMA
Ohuchi, K., Funato, M., Kato, Z., Seki, J., Kawase, C., Tamai, Y., et al. (2016). Established stem cell model of spinal muscular atrophy is applicable in the evaluation of the efficacy of thyrotropin-releasing hormone analog. Stem Cells Transl. Med. 5, 152 doi:10.5966/sctm.2015-0059
Oprea, G. E., Kröber, S., McWhorter, M. L., Rossoll, W., Müller, S., Krawczak, M., et al. (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320 (5875), 524–527. doi:10.1126/science.1155085
Orphan. Bassell: reldesemtiv, treatment of spinal muscular atrophy. (2019). Case Med. Res. doi:10.31525/cmr-1e36741
Osaki, T., Uzel, S. G. M., and Kamm, R. D. (2020). On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat. Protoc. 15, 421–449. doi:10.1038/s41596-019-0248-1
Osborne, M., Gomez, D., Feng, Z., Mcewen, C., Beltran, J., Cirillo, K., et al. (2012). Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum. Mol. Genet. 21, 4431–4447. doi:10.1093/hmg/dds285
Osman, E. Y., Bolding, M. R., Villalón, E., Kaifer, K. A., Lorson, Z. C., Tisdale, S., et al. (2019). Functional characterization of SMN evolution in mouse models of SMA. Sci. Rep. 9, 1–12. doi:10.1038/s41598-019-45822-8
Paeile, C., Lombard, M. C., Saavedra, H., Bustos, E., and Pelissier, T. (1989). Evidence of a central component in the analgesic effect of indoprofene. Gen. Pharmacol. Vasc. Syst. 20, 519–524. doi:10.1016/0306-3623(89)90206-1
Pagliarini, V., Guerra, M., Di Rosa, V., Compagnucci, C., and Sette, C. (2020). Combined treatment with the histone deacetylase inhibitor LBH589 and a splice‐switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in spinal muscular atrophy cells. J. Neurochem. 153, 264–275. doi:10.1111/jnc.14935
Palacino, J., Swalley, S. E., Song, C., Cheung, A. K., Shu, L., Zhang, X., et al. (2015). SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 11, 511–517. doi:10.1038/nchembio.1837
Pane, M., Staccioli, S., Messina, S., D’Amico, A., Pelliccioni, M., Mazzone, E. S., et al. (2008). Daily salbutamol in young patients with SMA type II. Neuromuscul. Disord. 18, 536–540. doi:10.1016/j.nmd.2008.05.004
Panuzzo, C., Jovanovski, A., Pergolizzi, B., Pironi, L., Stanga, S., Fava, C., et al. (2020). Mitochondria: a galaxy in the hematopoietic and leukemic stem cell universe. Int. J. Mol. Sci. 21, 3928. doi:10.3390/ijms21113928
Patten, S. A., Armstrong, G. A. B., Lissouba, A., Kabashi, E., Parker, J. A., and Drapeau, P. (2014). Fishing for causes and cures of motor neuron disorders. Dis. Models Mech. 7, 799–809. doi:10.1242/dmm.015719
Pérez Luque, A., Rus Hidalgo, M., Arellano Velázquez, E., Becerril Ríos, N., García Alonso, J. A., and Escudero Uribe, S. (2015). Uso de fampyra para la mejoría de la marcha en el paciente con esclerosis múltiple y sus repercusiones en la calidad de vida del paciente. Rev. Cientí. Soc. Esp. Enfer. Neuroló 42, 25–28. doi:10.1016/j.sedene.2015.05.002
Periyakaruppiah, A., de la Fuente, S., Arumugam, S., Bahí, N., Garcera, A., and Soler, R. M. (2016). Autophagy modulators regulate survival motor neuron protein stability in motoneurons. Exp. Neurol. 283, 287–297. doi:10.1016/j.expneurol.2016.06.032
Pfizer Inc (2009). Neurontin® (gabapentin) capsules Neurontin® (gabapentin) tablets neurontin ® (gabapentin) oral solution. Neurontin Package Insert.
Pfizer Pulls Plug on Repligen SMA Collaboration (2015). Available at: https://www.genengnews.com/topics/omics/update-pfizer-pulls-plug-on-repligen-sma-collaboration (Accessed January 30, 2015)
Piras, A., Schiaffino, L., Boido, M., Valsecchi, V., Guglielmotto, M., De Amicis, E., et al. (2017). Inhibition of autophagy delays motoneuron degeneration and extends lifespan in a mouse model of spinal muscular atrophy. Cell Death Dis. 8, 3223. doi:10.1038/s41419-017-0086-4
Poletti, A., and Fischbeck, K. H. (2020). Combinatorial treatment for spinal muscular atrophy. J. Neurochem., 153, 264. doi:10.1111/jnc.14974
Poirier, A., Heinig, K., Bucheli, F., Schoenlein, K., Alsenz, J., et al. (2018). Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 6, e00447-12. doi:10.1002/prp2.447
Powis, R. A., Karyka, E., Boyd, P., Côme, J., Jones, R. A., Zheng, Y., et al. (2016). Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 1, e87908. doi:10.1172/jci.insight.87908
Prestwick Chemical Library (2013). Available at: http://www.prestwickchemical.com/index.php?pa=26
Pruss, R. M., Giraudon-Paoli, M., Morozova, S., Berna, P., Abitbol, J.-L., and Bordet, T. (2010). Drug discovery and development for spinal muscular atrophy: lessons from screening approaches and future challenges for clinical development. Future Med. Chem. 2, 1429–1440. doi:10.4155/fmc.10.228
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2018). Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58. doi:10.1038/nrd.2018.168
Puyal, M., Jackson, J., Wawersik, S., Webster, M. T., Salta, L., Long, K., et al. (2018). Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci. Rep. 8, 2292. doi:10.1038/s41598-018-20524-9
Quattrocelli, M., Barefield, D. Y., Warner, J. L., Vo, A. H., Hadhazy, M., Earley, J. U., et al. (2017). Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J. Clin. Invest. 127, 2418–2432. doi:10.1172/JCI91445
Rajendra, T. K., Gonsalvez, G. B., Walker, M. P., Shpargel, K. B., Salz, H. K., and Matera, A. G. (2007). A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J. Cell Biol. 176, 831–841. doi:10.1083/jcb.200610053
Ramalho, T. C., de Castro, A. A., Tavares, T. S., Silva, M. C., Silva, D. R., Cesar, P. H., et al. (2018). Insights into the pharmaceuticals and mechanisms of neurological orphan diseases: current status and future expectations. Prog. Neurobiol. 169, 135–157. doi:10.1016/j.pneurobio.2018.06.011
Ramdas, S., and Servais, L. (2020). New treatments in spinal muscular atrophy: an overview of currently available data. Expet Opin. Pharmacother. 21, 307–315. doi:10.1080/14656566.2019.1704732
Rao, V. K., Kapp, D., and Schroth, M. (2018). Gene therapy for spinal muscular atrophy: an emerging treatment option for a devastating disease. J. Manag. Care Spec. Pharm. 24, S3–S16. doi:10.18553/jmcp.2018.24.12-a.s3
Ratni, H., Ebeling, M., Baird, J., Bendels, S., Bylund, J., Chen, K. S., et al. (2018). Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517. doi:10.1021/acs.jmedchem.8b00741
Richelme, C. (2019). Nusinersen chez l’enfant : évolution ou révolution ? Med. Sci. 35, 22–25. doi:10.1051/medsci/2019049
Riessland, M., Kaczmarek, A., Schneider, S., Swoboda, K. J., Löhr, H., Bradler, C., et al. (2017). Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am. J. Hum. Genet. 100, 297–315. doi:10.1016/j.ajhg.2017.01.005
Rimer, M., Seaberg, B. L., Yen, P.-F., Lam, S., Hastings, R. L., Lee, Y. i., et al. (2019). Nerve sprouting capacity in a pharmacologically induced mouse model of spinal muscular atrophy. Sci. Rep. 9, 1–9. doi:10.1038/s41598-019-44222-2
Rodgers, G. P., Dover, G. J., Noguchi, C. T., Schechter, A. N., and Nienhuis, A. W. (1990). Hematologic responses of patients with sickle cell disease to treatment with hydroxyurea. N. Engl. J. Med. 322, 1037–1045. doi:10.1056/NEJM199004123221504
Rossiter, H. B., Casaburi, R., Sciurba, F. C., Porszasz, J., Stringer, W. W., Valluri, U., et al. (2019). “A randomized, double-blind, placebo-controlled, crossover study to assess the effect of reldesemtiv on exercise tolerance in subjects with chronic obstructive pulmonary disease,” in C17. Pulmonary rehabilitation 2019 (American Thoracic Society), A4272. doi:10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A4272.
Roth, T., Lines, C., Vandormael, K., Ceesay, P., Anderson, D., and Snavely, D. (2010). Effect of gaboxadol on patient-reported measures of sleep and waking function in patients with primary insomnia: results from two randomized, controlled, 3-month studies. J. Clin. Sleep Med 6, 30–39. doi:10.5664/jcsm.27707
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi:10.1002/ana.410380114
Rovini, A., Gurnev, P. A., Beilina, A., Queralt-Martín, M., Rosencrans, W., Cookson, M. R., et al. (2019). Molecular mechanism of olesoxime-mediated neuroprotection through targeting α-synuclein interaction with mitochondrial VDAC. Cell. Mol. Life Sci. 77, 3611. doi:10.1007/s00018-019-03386-w
Rudnicki, S. A., Andrews, J. A., and Malik, F. I. (2018). “CY 5021 A phase 2, double-blind, randomized, placebo-controlled, multiple-dose study of reldesemtiv 2 ascending-dose cohorts of patients with spinal muscular atrophy (SMA),”in Communication presented at the Cure SMA 2018, Dallas, TX, June, 16, 2018.
Russman, B. S., Iannaccone, S. T., and Samaha, F. J. (2003). A phase 1 trial of riluzole in spinal muscular atrophy. Arch. Neurol. 60, 1601. doi:10.1001/archneur.60.11.1601
Sangha, J. S., Sun, X., Wally, O. S. D., Zhang, K., Ji, X., Wang, Z., et al. (2012). Liuwei dihuang (LWDH), a traditional Chinese medicinal formula, protects against β-Amyloid toxicity in transgenic Caenorhabditis elegans. PLoS One 7, e43990. doi:10.1371/journal.pone.0043990
Santhanam, N., Kumanchik, L., Guo, X., Sommerhage, F., Cai, Y., Jackson, M., et al. (2018). Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials 166, 64–78. doi:10.1016/j.biomaterials.2018.02.047
Sam, E., and Athri, P. (2019). Web-based drug repurposing tools: a survey. Briefings Bioinf. 20, 299–316. doi:10.1093/bib/bbx125
Schellino, R., Boido, M., Borsello, T., and Vercelli, A. (2018). Pharmacological c-Jun NH2-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 11, 308. doi:10.3389/fnmol.2018.00308
Schito, G. C., and Keenan, M. H. J. (2005). Predicting the clinical efficacy of generic formulations of ceftriaxone. J. Chemother. 17, 33–40. doi:10.1179/joc.2005.17.Supplement-2.33
Scholar Rock Reports First Quarter (2020). Financial results and highlights business progress. Available at: https://www.businesswire.com/news/home/20200507006017/en/Scholar-Rock-Reports-Quarter-2020-Financial-Results.
Seibert, K., Zhang, Y., Leahy, K., Hauser, S., Masferrer, J., Perkins, W., et al. (1994). Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. U.S.A. 91, 12013–12017. doi:10.1073/pnas.91.25.12013
Seo, J., Howell, M. D., Singh, N. N., and Singh, R. N. (2013). Spinal muscular atrophy: an update on therapeutic progress. Biochim. Biophys. Acta 1832, 2180 doi:10.1016/j.bbadis.2013.08.005
Shababi, M., Glascock, J., and Lorson, C. L. (2011). Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 22, 135–144. doi:10.1089/hum.2010.114
Shefner, J. M., Andrews, J. A., Genge, A., Jackson, C., Lechtzin, N., Miller, T. M., et al. (2018). Theme 9 clinical trials and trial design. Amyotroph. Lateral Scler. Front. Degener. 19, 264–281. doi:10.1080/21678421.2018.1510576.
Shibuya, M., and Suzuki, Y. (1993). Treatment of cerebral vasospasm by a protein kinase inhibitor AT 877. No To Shinkei 45, 819–824 [in Japanese].
Shimizu, M., Yamamoto, M., and Kobayashi, M. (1989). Effects of a new TRH analogue, YM-14673, on spontaneous motor activity in rats. Arch. Int. Pharmacodyn. Ther. 301, 100–111.
Shorrock, H. K., Gillingwater, T. H., and Groen, E. J. N. (2018). Overview of current drugs and molecules in development for spinal muscular atrophy therapy. Drugs 78, 293–305. doi:10.1007/s40265-018-0868-8
Singh, N., Chaput, L., and Villoutreix, B. O. (2020). Virtual screening web servers: designing chemical probes and drug candidates in the cyberspace. Briefings Bioinf., bbaa034. doi:10.1093/bib/bbaa034
Singh, J., Salcius, M., Liu, S.-W., Staker, B. L., Mishra, R., Thurmond, J., et al. (2008). DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem. Biol. 3, 711–722. doi:10.1021/cb800120t
Singh, N. K., Singh, N. N., Androphy, E. J., and Singh, R. N. (2006). Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 26, 1333 doi:10.1128/mcb.26.4.1333-1346.2006
Simon, C. M., Dai, Y., Van Alstyne, M., Koutsioumpa, C., Pagiazitis, J. G., Chalif, J. I., et al. (2017). Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 21, 3767–3780. doi:10.1016/j.celrep.2017.12.003
Sleigh, J. N., Buckingham, S. D., Esmaeili, B., Viswanathan, M., Cuppen, E., Westlund, B. M., et al. (2011). A novel Caenorhabditis elegans allele, smn-1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre-approved compounds. Hum. Mol. Genet. 20, 245–260. doi:10.1093/hmg/ddq459
Soler-Botija, C., Ferrer, I., Alvarez, J. L., Baiget, M., and Tizzano, E. F. (2003). Downregulation of Bcl-2 proteins in type I spinal muscular atrophy motor neurons during fetal development. J. Neuropathol. Exp. Neurol. 62, 420–426. doi:10.1093/jnen/62.4.420
Son, Y. S., Choi, K., Lee, H., Kwon, O., Jung, K. B., Cho, S., et al. (2019). A SMN2 splicing modifier rescues the disease phenotypes in an in vitro human spinal muscular atrophy model. Stem Cell. Dev. 28, 438–453. doi:10.1089/scd.2018.0181
Stanga, S., Brambilla, L., Tasiaux, B., Dang, A. H., Ivanoiu, A., Octave, J.-N., et al. (2018a). A role for GDNF and soluble APP as biomarkers of amyotrophic lateral sclerosis pathophysiology. Front. Neurol. 9, 384. doi:10.3389/fneur.2018.00384
Stanga, S., Caretto, A., Boido, M., and Vercelli, A. (2020). Mitochondrial dysfunctions: a red thread across neurodegenerative diseases. Int. J. Mol. Sci. 21, 3719. doi:10.3390/ijms21103719
Stanga, S., Lanni, C., Sinforiani, E., Mazzini, G., and Racchi, M. (2012). Searching for predictive blood biomarkers: misfolded p53 in mild cognitive impairment. Curr. Alzheimer Res. 9, 1191–1197. doi:10.2174/156720512804142886
Stanga, S., Vrancx, C., Tasiaux, B., Marinangeli, C., Karlström, H., and Kienlen-Campard, P. (2018b). Specificity of presenilin-1- and presenilin-2-dependent γ-secretases towards substrate processing. J. Cell Mol. Med. 22, 823–833. doi:10.1111/jcmm.13364
Stanga, S., Zanou, N., Audouard, E., Tasiaux, B., Contino, S., Vandermeulen, G., et al. (2016). APP‐dependent glial cell line‐derived neurotrophic factor gene expression drives neuromuscular junction formation. FASEB J. 30, 1696–1711. doi:10.1096/fj.15-278739
Stamelou, M., Schöpe, J., Wagenpfeil, S., Del Ser, T., Bang, J., Lobach, I. Y., et al. (2016). Power calculations and placebo effect for future clinical trials in progressive supranuclear palsy. Mov. Disord. 31, 742–747. doi:10.1002/mds.26580
Stevens, D., Claborn, M. K., Gildon, B. L., Kessler, T. L., and Walker, C. (2020). Onasemnogene abeparvovec-xioi: gene therapy for spinal muscular atrophy. Ann. Pharmacother. 54, 1001. doi:10.1177/1060028020914274
Sun, J., and Harrington, M. A. (2019). The alteration of intrinsic excitability and synaptic transmission in lumbar spinal motor neurons and interneurons of severe spinal muscular atrophy mice. Front. Cell. Neurosci. 13, 15. doi:10.3389/fncel.2019.00015
Sun, Z., Xu, Q., Gao, G., Zhao, M., and Sun, C. (2019). Clinical observation in edaravone treatment for acute cerebral infarction. Niger. J. Clin. Pract. 22, 1324. doi:10.4103/njcp.njcp_367_18
Sumner, C. J., Huynh, T. N., Markowitz, J. A., Perhac, J. S., Hill, B., Coovert, D. D., et al. (2003). Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 54, 647–654. doi:10.1002/ana.10743
Swalley, S. E. (2020). Expanding therapeutic opportunities for neurodegenerative diseases: A perspective on the important role of phenotypic screening. Bioorg. Med. Chem. 28, 115239. doi:10.1016/j.bmc.2019.115239
Swoboda, K. J., Scott, C. B., Crawford, T. O., Simard, L. R., Reyna, S. P., Krosschell, K. J., et al. (2010). SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 5, e12140. doi:10.1371/journal.pone.0012140
Takeuchi, Y., Miyanomae, Y., Komatsu, H., Oomizono, Y., Nishimura, A., Okano, S., et al. (1994). Efficacy of thyrotropin-releasing hormone in the treatment of spinal muscular atrophy. J. Child Neurol. 9, 287–289. doi:10.1177/088307389400900313
Tejero, R., Balk, S., Franco-Espin, J., Ojeda, J., Hennlein, L., Drexl, H., et al. (2020). R-roscovitine improves motoneuron function in mouse models for spinal muscular atrophy. iScience 23, 100826. doi:10.1016/j.isci.2020.100826
Tiziano, F. D., Lomastro, R., Pinto, A. M., Messina, S., D'Amico, A., Fiori, S., et al. (2010). Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: relevance for clinical trial design. J. Med. Genet. 47, 856–858. doi:10.1136/jmg.2010.080366
Tritos, N. A., and Klibanski, A. (2016). Effects of growth hormone on bone. Prog. Mol. Biol. Transl. Sci. 138, 193–211. doi:10.1016/bs.pmbts.2015.10.008
Tsai, L.-K. (2012). Therapy development for spinal muscular atrophy in SMN independent targets. Neural Plast. 2012 1–13. doi:10.1155/2012/456478
Tsai, L.-K., Chen, C.-L., Ting, C.-H., Lin-Chao, S., Hwu, W.-L., Dodge, J. C., et al. (2014). Systemic administration of a recombinant AAV1 vector encoding IGF-1 improves disease manifestations in SMA mice. Mol. Ther. 22, 1450–1459. doi:10.1038/mt.2014.84
Tsai, L.-K., Tsai, M.-S., Ting, C.-H., Wang, S.-H., and Li, H. (2008). Restoring Bcl-xL levels benefits a mouse model of spinal muscular atrophy. Neurobiol. Dis. 31, 361–367. doi:10.1016/j.nbd.2008.05.014
Tsai, M. S., Chiu, Y. T., Wang, S. H., Hsieh-Li, H. M., Lian, W. C., and Li, H. (2006). Abolishing bax-dependent apoptosis shows beneficial effects on spinal muscular atrophy model mice. Mol. Ther. 13, 1149–1155. doi:10.1016/j.ymthe.2006.02.008
Tseng, Y.-T., Chang, F.-R., and Lo, Y.-C. (2014). The Chinese herbal formula Liuwei dihuang protects dopaminergic neurons against Parkinson's toxin through enhancing antioxidative defense and preventing apoptotic death. Phytomedicine 21, 724–733. doi:10.1016/j.phymed.2013.11.001
Tseng, Y.-T., Jong, Y.-J., Liang, W.-F., Chang, F.-R., and Lo, Y.-C. (2017). The water extract of Liuwei dihuang possesses multi-protective properties on neurons and muscle tissue against deficiency of survival motor neuron protein. Phytomedicine 34, 97–105. doi:10.1016/j.phymed.2017.08.018
Tuffaha, S. H., Singh, P., Budihardjo, J. D., Means, K. R., Higgins, J. P., Shores, J. T., et al. (2016). Therapeutic augmentation of the growth hormone axis to improve outcomes following peripheral nerve injury. Expert Opin. Ther. Targets 20, 1259–1265. doi:10.1080/14728222.2016.1188079
Tzeng, A. C., Cheng, J., Fryczynski, H., Niranjan, V., Stitik, T., Sial, A., et al. (2000). A study of thyrotropin-releasing hormone for the treatment of spinal muscular atrophy. Am. J. Phys. Med. Rehabil. 79, 435–440. doi:10.1097/00002060-200009000-00005
Urayama, A., Yamada, S., Kimura, R., Zhang, J., and Watanabe, Y. (2002). Neuroprotective effect and brain receptor binding of taltirelin, a novel thyrotropin-releasing hormone (TRH) analogue, in transient forebrain ischemia of C57BL/6J mice. Life Sci. 72, 601–607. doi:10.1016/S0024-3205(02)02268-3
Vaidya, S., and Boes, S. (2018). Measuring quality of life in children with spinal muscular atrophy: a systematic literature review. Qual. Life Res. 27 (12), 3087–3094, doi:10.1007/s11136-018-1945-x
Valsecchi, V., Anzilotti, S., Serani, A., Laudati, G., Brancaccio, P., Guida, N., et al. (2020). miR-206 reduces the severity of motor neuron degeneration in the facial nuclei of the brainstem in a mouse model of SMA. Mol. Ther. 28, 1154–1166. doi:10.1016/j.ymthe.2020.01.013
Valsecchi, V., Boido, M., De Amicis, E., Piras, A., and Vercelli, A. (2015). Expression of muscle-specific MiRNA 206 in the progression of disease in a murine SMA model. PLoS One 10, e0128560-17. doi:10.1371/journal.pone.0128560
Verhaart, I. E. C., Robertson, A., Leary, R., McMacken, G., König, K., Kirschner, J., et al. (2017). A multi-source approach to determine SMA incidence and research ready population. J. Neurol. 264, 1465–1473. doi:10.1007/s00415-017-8549-1
Wadman, R. I., van der Pol, W. L., Bosboom, W. M., Asselman, F.-L., van den Berg, L. H., Iannaccone, S. T., et al. (2020). Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 1, CD006282. doi:10.1002/14651858.CD006282.pub5
Wang, Y., Xu, C., Ma, L., Mou, Y., Zhang, B., Zhou, S., et al. (2019). Drug screening with human SMN2 reporter identifies SMN protein stabilizers to correct SMA pathology. Life Sci. Alliance 2, e201800268-19. doi:10.26508/lsa.201800268
Walter, L. M., Deguise, M.-O., Meijboom, K. E., Betts, C. A., Ahlskog, N., van Westering, T. L. E., et al. (2018). Interventions targeting glucocorticoid-krüppel-like factor 15-branched-chain amino acid signaling improve disease phenotypes in spinal muscular atrophy mice. EBioMedicine 31, 226–242. doi:10.1016/j.ebiom.2018.04.024
Walker, M. P., Rajendra, T. K., Saieva, L., Fuentes, J. L., Pellizzoni, L., and Matera, A. G. (2008). SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet. 17,3399–3410. doi:10.1093/hmg/ddn234
Wiffen, P. J., Derry, S., Bell, R. F., Rice, A. S., Tölle, T. R., Phillips, T., et al. (2017). Gabapentin for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 6, CD007938. doi:10.1002/14651858.CD007938.pub4
Wishart, T. M., Mutsaers, C. A., Riessland, M., Reimer, M. M., Hunter, G., Hannam, M. L., et al. (2014). Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J. Clin. Invest. 124, 1821–1834. doi:10.1172/JCI71318
Wolozin, B., Gabel, C., Ferree, A., Guillily, M., and Ebata, A. (2011). Watching worms whither. Prog. Mol. Biol. Transl. Sci. 100, 499–514. doi:10.1016/B978-0-12-384878-9.00015-7
Wu, C.-Y., Gagnon, D. A., Sardin, J. S., Barot, U., Telenson, A., Arratia, P. E., et al. (2018). Enhancing GABAergic transmission improves locomotion in a caenorhabditis elegans model of spinal muscular atrophy. eNeuro. 5 (6), ENEURO.0289-18.2018. doi:10.1523/ENEURO.0289-18.2018
Zhang, M., Lorson, C., Androphy, E., and Zhou, J. (2001). An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA. Gene Ther. 8, 1532–1538. doi:10.1038/sj.gt.3301550
Zhao, J., Zhou, D., Guo, J., Ren, Z., Zhou, L., Wang, S., et al. (2006). Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol. Med.-Chir. 46, 421–428. doi:10.2176/nmc.46.421
Zhou, C., Feng, Z., and Ko, C.-P. (2016). Defects in motoneuron-astrocyte interactions in spinal muscular atrophy. J. Neurosci. 36, 2543–2553. doi:10.1523/JNEUROSCI.3534-15.2016
Zolgensma (2020a). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma#product-information-section (Accessed May 18, 2020).
Zolgensma (2020b). Available at: https://www.novartis.com/news/media-releases/zolgensma-data-shows-rapid-significant-clinically-meaningful-benefit-sma-including-prolonged-event-free-survival-motor-milestone-achievement-and-durability-now (Accessed March 24, 2020).
Keywords: survival motor neuron, motor neuron disease, therapy, cell death and degradation, mitochondria, cytoskeleton dynamics, neurotransmitter modulation, neuromuscular junction stabilization
Citation: Menduti G, Rasà DM, Stanga S and Boido M (2020) Drug Screening and Drug Repositioning as Promising Therapeutic Approaches for Spinal Muscular Atrophy Treatment. Front. Pharmacol. 11:592234. doi: 10.3389/fphar.2020.592234
Received: 06 August 2020; Accepted: 29 September 2020;
Published: 12 November 2020.
Edited by:
Anna R. Carta, University of Cagliari, ItalyReviewed by:
Rosa M. Soler, Universitat de Lleida, SpainFederico Forneris, University of Pavia, Italy
Copyright © 2020 Menduti, Rasà, Stanga and Boido. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Boido, bWFyaW5hLmJvaWRvQHVuaXRvLml0
† These authors have contributed equally to this work