 Robert W. Malone1,17*
Robert W. Malone1,17* Philip Tisdall2
Philip Tisdall2 Philip Fremont-Smith3
Philip Fremont-Smith3 Yongfeng Liu4Xi-Ping Huang4Kris M. White5,6
Yongfeng Liu4Xi-Ping Huang4Kris M. White5,6 Lisa Miorin5,6Elena Moreno5,6Assaf Alon7Elise Delaforge8Christopher D. Hennecker8
Lisa Miorin5,6Elena Moreno5,6Assaf Alon7Elise Delaforge8Christopher D. Hennecker8 Guanyu Wang8
Guanyu Wang8 Joshua Pottel9Robert V. Blair10,11
Joshua Pottel9Robert V. Blair10,11 Chad J. Roy10,12Nora Smith3Julie M. Hall13Kevin M Tomera14Gideon Shapiro15
Chad J. Roy10,12Nora Smith3Julie M. Hall13Kevin M Tomera14Gideon Shapiro15 Anthony Mittermaier8Andrew C. Kruse7
Anthony Mittermaier8Andrew C. Kruse7 Adolfo García-Sastre5,6,16,17
Adolfo García-Sastre5,6,16,17 Bryan L. Roth4Jill Glasspool-Malone1
Bryan L. Roth4Jill Glasspool-Malone1 Darrell O. Ricke3
Darrell O. Ricke3- 1RW Malone MD LLC, Madison, VA, United States
- 2Medical School Companion LLC, Marco Island, FL, United States
- 3MIT Lincoln Laboratory, Lexington, MA, United States
- 4Department of Pharmacology, University of North Carolina, Chapel Hill, Chapel Hill, NC, United States
- 5Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 6Global Health and Emerging Pathogens Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 7Department of Biological Chemistry and Molecular Pharmacology, Blavatnik Institute, Harvard Medical School, Boston, MA, United States
- 8Department of Chemistry, McGill University, Montreal, QC, Canada
- 9Molecular Forecaster Inc, Montreal, QC, Canada
- 10Tulane National Primate Research Center, Covington, LA, United Sates
- 11Department of Pathology and Laboratory Animal Medicine, Tulane University School of Medicine, New Orleans, LA, United States
- 12Department of Microbiology and Immunology, Tulane University School of Medicine, New Orleans, LA, United States
- 13Frank H. Netter MD School of Medicine – Quinnipiac University, Hamden, CT, United States
- 14Department of Urology, Beloit Memorial Hospital, Beloit, WI, United States
- 15Pharmorx LLC, Gainesville, FL, United States
- 16Division of Infectious Diseases, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 17Icahn School of Medicine at Mount Sinai, The Tisch Cancer Institute, New York, NY, United States
SARS-CoV-2 infection is required for COVID-19, but many signs and symptoms of COVID-19 differ from common acute viral diseases. SARS-CoV-2 infection is necessary but not sufficient for development of clinical COVID-19 disease. Currently, there are no approved pre- or post-exposure prophylactic COVID-19 medical countermeasures. Clinical data suggest that famotidine may mitigate COVID-19 disease, but both mechanism of action and rationale for dose selection remain obscure. We have investigated several plausible hypotheses for famotidine activity including antiviral and host-mediated mechanisms of action. We propose that the principal mechanism of action of famotidine for relieving COVID-19 symptoms involves on-target histamine receptor H2 activity, and that development of clinical COVID-19 involves dysfunctional mast cell activation and histamine release. Based on these findings and associated hypothesis, new COVID-19 multi-drug treatment strategies based on repurposing well-characterized drugs are being developed and clinically tested, and many of these drugs are available worldwide in inexpensive generic oral forms suitable for both outpatient and inpatient treatment of COVID-19 disease.
Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is a highly infectious and pathogenic betacoronavirus first detected in human infections during December 2019 (Wu D. et al., 2020; Wu and McGoogan, 2020; Zhu et al., 2020). Coronavirus infectious disease—2019 (COVID-19) is a disease spectrum causally associated with infection by SARS-CoV-2. Definitive COVID-19 diagnosis requires the presence of the virus, which can be isolated, grown, or otherwise detected as unique SARS-CoV-2 viral nucleic acid sequences. There are SARS-CoV-2 virus shedding or nucleic acid positive patients that do not manifest clinical COVID-19 (Danis et al., 2020; Furukawa et al., 2020; Ki and Task Force for -nCo, 2020; Lai et al., 2020; Pan et al., 2020; Zou et al., 2020). 13–20% of patients with symptoms develop severe respiratory compromise requiring oxygenation, with radiological findings of ground glass opacities and consolidation (Huang et al., 2020; Shi et al., 2020; Wang et al., 2020). Up to 80% of SARS-CoV-2 infected patients are asymptomatic and do not appear to progress to COVID-19 (Tian et al., 2020a; BMJ, 2020; Day, 2020; He et al., 2020; Hu et al., 2020; Mizumoto et al., 2020; WHO, 2020). Therefore, SARS-CoV-2 infection is necessary but not sufficient for development of clinical COVID-19 disease.
Patients with COVID-19 disease can present with a range of mild to severe non-specific clinical signs and symptoms which develop 2–14 days after exposure to SARS-CoV-2. These symptoms include cough or shortness of breath, and at least two of the following; fever, chills, repeated rigor, myalgia, headache, oropharyngitis, anosmia, and ageusia (Giacomelli et al., 2020; Lechien et al., 2020). More severe symptoms warranting hospital admission include difficulty breathing, a persistent sense of chest pain or pressure, confusion or difficulty to arouse, and central cyanosis. Of hospitalized patients, 20–42% develop Acute Respiratory Disease Syndrome (ARDS), the most common cause for admission to hospital intensive care units (ICU). 39–72% of patients admitted to the ICU will die (CDC, 2020).
Clinical data from a variety of sources indicate that famotidine treatment may reduce morbidity and mortality associated with COVID-19, but other studies suggest no clinical benefits from famotidine treatment. An early retrospective cohort study of 1,620 hospitalized COVID-19 patients indicates that 84 propensity score matched patients receiving famotidine during hospitalization (oral or IV, 20 mg or 40 mg daily) had a statistically significant reduced risk for death or intubation (adjusted hazard ratio (aHR) 0.42, 95% CI 0.21–0.85) and also a reduced risk for death alone (aHR 0.30, 95% CI 0.11–0.80) (Freedberg et al., 2020). More recent retrospective studies concerning famotidine and COVID-19 have been more variable and generally less sanguine, ranging from no effect (Cheung et al., 2020; Shoaibi et al., 2020; Yeramaneni et al., 2020) to effects similar to those originally reported (Odds Ratio—OR-death 0.37, OR-death or intubation 0.47) (Mather et al., 2020). The difference in measured outcomes between the two most recent studies reporting no effect (Shoaibi et al., 2020; Yeramaneni et al., 2020) and the study from Hartford hospital (Mather et al., 2020) may have multiple explanations, including famotidine dosage. An early limited case series suggested that it is necessary to administer famotidine at higher than standard over-the-counter doses to relieve COVID-19 symptoms (ergo, high dose [HD] famotidine treatment) (Janowitz et al., 2020). Together, these data have been interpreted as indicating that the increased survival pattern initially reported (Freedberg et al., 2020) is due to an off-target, non-histamine receptor-mediated property of famotidine not shared with cimetidine (Borrell, 2020; Freedberg et al., 2020; Janowitz et al., 2020). HD famotidine was initially being tested in the United States (US) under an Investigational New Drug (IND) waiver for treating COVID-19. This early double blind randomized clinical trial involved high intravenous famotidine doses in combination with either hydroxychloroquine or remdesivir (ClinicalTrials.gov Identifier: NCT04370262), but that trial has been delayed and is yet to complete enrollment or report results. HD famotidine for treatment of COVID-19 is now also being clinically tested in Bangladesh (ClinicalTrials.gov Identifier: NCT04504240), and Iran (Samimagham et al., 2020). Recent encouraging clinical reports include simultaneous administration of famotidine and cetirizine at standard OTC doses (Hogan et al., 2020) and HD famotidine with celecoxib (Tomera and Kittah, 2020a; Tomera and Kittah, 2020b).
Herein we aim to investigate how famotidine may act to relieve early phase COVID-19 clinical symptoms. The most likely mechanisms of actions include: via antiviral activity, via novel human targets, or via the on-target mechanism described in the current FDA market authorization—famotidine is a histamine receptor H2 antagonist (and inverse agonist).
Methods
Experimental Analysis of the Mechanism of Action of Famotidine
Famotidine was originally selected by the authors for advancement as a potential repurposed drug candidate therapeutic for COVID-19 based on molecular docking data to the SARS-CoV-2 papain-like protease (PLpro). Based on this analysis the US Food and Drug Administration (FDA) granted an IND waiver for the subsequent double blinded randomized clinical trial currently in progress (ClinicalTrials.gov Identifier: NCT04370262). Briefly, a ranked list of licensed compounds with predicted binding activity in the PLpro catalytic site was computationally generated, and the PLpro catalytic site binding pose of each of the top compounds was examined and ranked by a team of pharmaceutical chemists. Package inserts or product monographs for the licensed compounds which generated high computational binding scores and passed inspection were then reviewed and used to rank compounds based on adverse events, FDA warnings, drug interactions on-target mechanisms, pharmacokinetic and absorption, metabolism, excretion and toxicity (ADMET), protein binding and available therapeutic window considerations. Famotidine (“PEPCID®”), a histamine H2 antagonist (inverse agonist) widely available in tablet form over-the-counter, as well as in solution form for intravenous administration, was repeatedly computationally ranked as among the most promising of the compounds tested and was associated with the most favorable pharmacokinetic and safety profile.
Recognizing that computational docking predictions are typically associated with about a 20% success rate, we applied the method of multiple working hypotheses (1890) to assess the mechanism of action of famotidine as a potential treatment for COVID-19. Relevant hypotheses tested included;
• Direct binding and action as an inhibitor of SARS-CoV-2 PLpro.
• Action as a direct acting inhibitor of SARS-CoV-2 infection or replication.
• Off-target binding and inhibition of either sigma receptors or a non-histamine H2 G-coupled protein receptor.
• Histamine H2 receptor inhibition.
Initial lay press journalistic reports based on unverified hearsay from the People’s Republic of China (PRC) (Borrell, 2020) suggested that famotidine provided clinical benefit for treatment of COVID-19, but other H2 receptor antagonists (cimetidine) did not. This resulted in an inference that famotidine did not act via its known mechanism of action as an H2 receptor inhibitor. Pharmacokinetic analyses were performed to model systemic circulating levels of famotidine and cimetidine at various doses.
If famotidine relieves clinical symptoms of COVID-19, and acts via known inhibitory and inverse agonist interactions with H2 receptors, then there must be a histopathological source of histamine release in damaged tissues including peripheral lung. One of the most common cellular sources of histamine are mast cells, so SARS-CoV-2 was used to experimentally infect African green monkeys (AGM). At necropsy, AGM lung sections from diseased and control lung parenchyma were sampled and stained for presence and density of mast cells.
Does Famotidine Directly Bind and Act as an Inhibitor of SARS-CoV-2 PLpro?
Production of Recombinant SARS-CoV-2 Plpro
An expression plasmid containing the sequence for (His)6-TEVsite-SARS-CoV-2 PLpro (nsp3 from Wuhan-Hu-1 isolate, polyprotein 1ab 1564-1878) was obtained commercially from ATUM. The plasmid was transformed into E. coli BL21 (DE3) pLysS. The expression and purification protocols were adapted from prior work (Lindner et al., 2005).
Production of Recombinant ISG15
The expression plasmid for proISG15 (2-165) was a gift from David Komander (Addgene plasmid #110762; http://n2t.net/addgene:110762 ; RRID:Addgene_110762) (Swatek et al., 2018). Expression and purification protocols were adapted from (Swatek et al., 2018). A size exclusion chromatography step on a Superdex 75 column (GE Healthcare) was added as a final step.
PLpro Activity Assays
Cleavage of ISG15 by SARS-CoV-2 PLpro was tested by incubating 4 nM of PLpro in 50 mM Tris-HCl (pH 7.3), 150 mM NaCl, 2 mM DTT, 0.1 mg ml−1 BSA, with 10 µM of ISG15 in a final volume of 20 µL for 1 h at room temperature. Control was incubated without enzyme. Samples were subjected to SDS-PAGE.
Does Famotidine Directly Inhibit SARS-CoV-2 Infection or Replication in Vero Cells?
Viral Growth and Cytotoxicity Assays in the Presence of Inhibitors
2,000 Vero E6 cells were seeded into 96-well plates in Dulbecco’s Modified Eagles Medium (DMEM, 10% FBS) and incubated for 24 h at 37°C, 5% CO2. Two hours before infection, the medium was replaced with 100°ul of DMEM (2% FBS) containing the compound of interest at concentrations 50% greater than those indicated, including a DMSO control. Plates were then transferred into the Biosafety Level 3 (BSL3) facility and 100 PFU (MOI 0.025) was added in 50°ul of DMEM (2% FBS), bringing the final compound concentration to those indicated. Plates were then incubated for 48 h at 37°C. After infection, supernatants were removed and cells were fixed with 4% formaldehyde for 24 h prior to being removed from the BSL3 facility. The cells were then immunostained for the viral NP protein with a DAPI counterstain. Infected cells (488 nM) and total cells (DAPI) were quantified using the Celigo (Nexcelcom) imaging cytometer. Percent infection was quantified as ((Infected cells/Total cells) − Background) *100 and the DMSO control was then set to 100% infection for analysis. The IC50 and IC90 for each experiment were determined using the Prism (GraphPad Software) software. For select inhibitors, infected supernatants were assayed for infectious viral titer using the TCID50 method. Cytotoxicity was also performed using the MTT assay (Roche), according to the manufacturer’s instructions. Cytotoxicity was performed in uninfected Vero E6 cells with same compound dilutions and concurrent with viral replication assay.
TCID50 Assay
Infectious supernatants were collected at 48 h post infection and frozen at −80°C until later use. Infectious titers were quantified by limiting dilution titration on Vero E6 cells. Briefly, Vero E6 cells were seeded in 96-well plates at 20,000 cells/well. The next day, SARS-CoV2-containing supernatant was applied at serial 10-fold dilutions ranging from 10−1 to 10−6 and, after 5 days, viral CPE was detected by staining cell monolayers with crystal violet. Median tissue culture infectious doses (TCID50)/ml were calculated using the method of Reed and Muench.
Does Famotidine Bind and Interact with the Sigma 1 or 2 Receptors?
Sigma-1 and Sigma-2 Competition Binding Assays
Sigma-1 receptor [3H](+)-pentazocine competition curves testing the binding of Famotidine, Cimetidine, and PB-28 (as positive control), were performed with Expi293 cells (Thermo Fisher) overexpressing the human sigma-1 receptor. Membranes were incubated in a 100 μL reaction buffered with 50 mM Tris (pH 8.0), with 10 nM [3H](+)-pentazocine, 0.1% BSA, and seven concentrations (ranging from 10 μM to 0.1 nM) of the competing cold ligand. Reactions were incubated for 2 h at 37°C and then were terminated by filtration through a glass fiber filter using a Brandel cell harvester. Glass fiber filters were soaked in 0.3% polyethylenimine for at least 30 min at room temperature before harvesting. All reactions were performed in triplicate using a 96-well block. After the membranes were transferred to the filters and washed, the filters were soaked in 5 ml Cytoscint scintillation fluid overnight, and radioactivity was measured using a Beckman Coulter LS 6500 scintillation counter. Data were analyzed using GraphPad Prism software. Ki values were computed by directly fitting the data and using the experimentally determined probe Kd to calculate a Ki value, using the GraphPad Prism software. This process implicitly uses a Cheng–Prusoff correction, so no secondary correction was applied.
Sigma-2 competition curves were performed in a similar manner, using Expi293 cells overexpressing the human sigma-2 (TMEM97) and using [3H] DTG as the radioactive probe.
Does Famotidine Act via an Off-Target Activity Involving a G-Coupled Protein Receptor (GPCR) Other than the Histamine H2 GPCR?
Human G-coupled Protein Receptor genome (GPCRome) screening was carried out according to published procedure (Kroeze et al., 2015) with minor modifications. In brief, HTLA cells were subcultured into poly-L-lysine coated clear bottom 384-well white plates at a density of 6,000 cells/well in 40 ul of DMEM supplemented with 1% dialyzed FBS for overnight, transfected with 20 ng/well Tango constructs for 24 h, received drug stimulation (10 uM final) for another 24 h. Medium and drug solutions were removed and Bright-Glo Reagents (Promega) were added for luminescence counting. For concentration response assays, HTLA cells were transfected with Tango constructs for 24 h, plated in poly-L-Lysine coated clear bottom 384-well white plates at a density of 10,000 cells/well in 40 ul of DMEM supplemented with 1% dialyzed FBS for 6 h before receiving drugs for overnight stimulation. Plates were then counted as above.
Does Famotidine Act by Blocking Histamine H2 receptor(s)?
The known on-target activity of famotidine considered the known primary mechanism of action is as an antagonist of the histamine H2 receptor. This hypothesis was originally rejected due to unverified reports that clinical researchers in PRC (Wuhan) had observed that famotidine use was associated with protection from COVID-19 mortality, while the histamine H2 receptor antagonist cimetidine was not. Positing that this difference in clinical effectiveness for the two different H2 receptor antagonists may reflect absorption, pharmacokinetic and pharmacodistribution differences between famotidine and cimetidine, steady state concentrations were calculated for both drugs when administered at standard oral doses as well as the elevated doses of famotidine which are being prescribed off-label for outpatient clinical use to treat COVID-19 or are being used in the ongoing inpatient clinical trial (NCT04370262), and these were compared to the published H2 receptor IC50 for each drug.
GloSensor cAMP Assays
cAMP production was determined in transiently transfected HEK293T cells (Stauch et al., 2019) with minor modifications. In brief, HEK293 T cells were co-transfected with H2 and GloSensor cAMP reporter DNA (Promega) overnight and plated in poly-l-lysine coated clear bottom 384-well white plate at a density of 15,000 cells/well in 40 ul of DMEM supplemented with 1% dialyzed FBS for 6 h. Medium was removed and cells were loaded with 3 mM luciferin in drug buffer (20 ul/well, 1x HBSS, 20 mM HEPES, pH 7.40) for 30 min. Test compounds were prepared in the same drug buffer supplemented with 1 mg/ml BSA and added (20 ul/well at 2x) to cells. Luminescence was counted after 20 min and results were analyzed in Prism 8.4.
Radioligand binding assays with human histamine receptors. Radioligand binding assays with human H1, H2, H3, and H4 receptors were conducted according to the NIMH PDSP assay protocol (https://pdsp.unc.edu/pdspweb/?site=assays) and published procedures (Besnard et al., 2012).
Famotidine and Cimetidine Pharmacokinetic Analyses
Steady-state values of famotidine and cimetidine at various doses were calculated as follows using standard pharmacokinetic calculations. Bioavailability, and clearance values were obtained from data reported by Lin (1991) for tablet and capsule dosing of famotidine and cimetidine; for 60 mg famotidine bid, kinetic data were obtained from a report by Yeh et al. (1987). For intravenous administration of famotidine (IV 120 mg q8 hours), kinetic values were obtained from the famotidine New Drug Applications (NDAs 19-510/S-029, 20-249/S-012).
Calculations: Area under the curve (AUC) determinations were made as follows: AUC (mg h/L) = (F x Dose)/Cl; where F = bioavailability, Cl = clearance. Steady (Css) state values were calculated as follows: Css (μg/L) = AUC/T; where T = dosing interval (h). Css levels were converted to μM using the molecular weights of famotidine (337.45) and cimetidine (252.34), respectively.
Are Increased Numbers Mast Cells Present in Lung Parenchyma of SARS-CoV-2 Infected AGM?
African green monkeys were exposed by aerosol to SARS-CoV-2 and euthanized after progression of clinical signs or at the end of the study period (four weeks post-infection) (Blair et al., 2020). Sections of lung were stained with toluidine blue and digitally scanned with a Zeiss Axio Scan.Z1 slide scanner (Carl Zeiss Microscopy, White Plains, NY). Whole slide images were analyzed using the digital image analysis software, HALO Multiplex IHC v2.1.1 (Indica Labs, Albuquerque, NM). Mast cells were quantified by the detection of metachromasia within the cytoplasm. The positive threshold for the detection of metachromasia was set by a veterinary pathologist using a slide of a mast cell tumor as a positive control. Annotation regions were drawn around the entire lung section and the analysis was run on the annotation region. The proportion of mast cells over the total number of cells detected were reported.
Results
Analysis of the Mechanism of Action of Famotidine
Famotidine Does Not Bind to SARS-CoV-2 Proteases
Initial testing of famotidine as a medical countermeasure for COVID-19 emerged from a computational molecular docking effort aimed at identifying inhibitors of the papain-like protease (PLpro) of SARS-CoV-2 (Baez-Santos et al., 2015; Daczkowski et al., 2017). In addition to processing the viral polyprotein, the papain-like protease from coronaviruses (PLpro) is known to remove the cellular substrates ubiquitin and the interferon stimulated gene 15 (ISG15) from host cell proteins by cleaving the C-terminal end of the consensus sequence LXGG, a process termed deISGylation (Han et al., 2005; Mielech et al., 2014). Here, we used the enzymatic reaction of SARS-CoV-2 PLpro on ISG15 to assess the potential inhibition of PLpro by famotidine. The cleavage of the 8 C-terminal amino acids of ISG15 by PLpro is clearly detected by SDS-PAGE (Figure 1, lanes 2 and 3). However, the addition of 1–100 µM famotidine to the reaction does not significantly reduce the amount of ISG15 cleaved during the assay (Figure 1, lanes 4–6), thus suggesting that famotidine does not inhibit SARS-CoV-2 PLpro. A previous virtual screening report (Wu C. et al., 2020) suggested that famotidine might bind to the 3 chymotrypsin-like protease (3CLpro), more commonly referred to as the main protease (Mpro), however this mechanism was recently discounted (Anson et al., 2020).
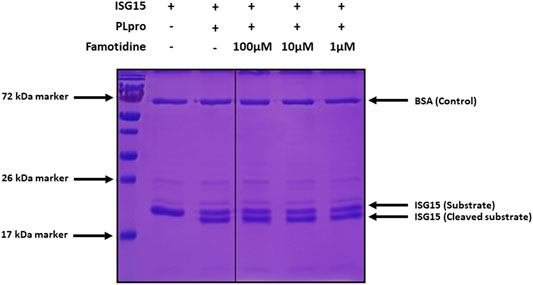
FIGURE 1. Cleavage of ISG15 C-terminal 8 amino acids by SARS-CoV-2 PLpro purified from E. coli. ISG15 was incubated with SARS-CoV-2 PLpro (lanes 3–6). SARS-CoV-2 PLpro was present at 4 nM, ISG15 was present at 10 µM. For lane 4 to 6, famotidine was present at 100 µM, 10 µM and 1 µM respectively. Control was without enzyme (lane 2). Proteins were resolved by 15% SDS-PAGE and revealed by Coomassie blue staining. The molecular weights of the marker proteins are indicated on the left of the gel.
Famotidine Does Not Directly Inhibit SARS-CoV-2 Infection
To assess the possibility that famotidine may inhibit SARS-CoV-2 infection by other routes, a Vero E6 cell-based assay was performed to compare median tissue culture infectious doses (TCID50/mL) of famotidine, remdesivir, and hydroxychloroquine (Figure 2). While both remdesivir and hydroxychloroquine demonstrated antiviral activity, no inhibition of SARS-CoV-2 infection was observed with famotidine.
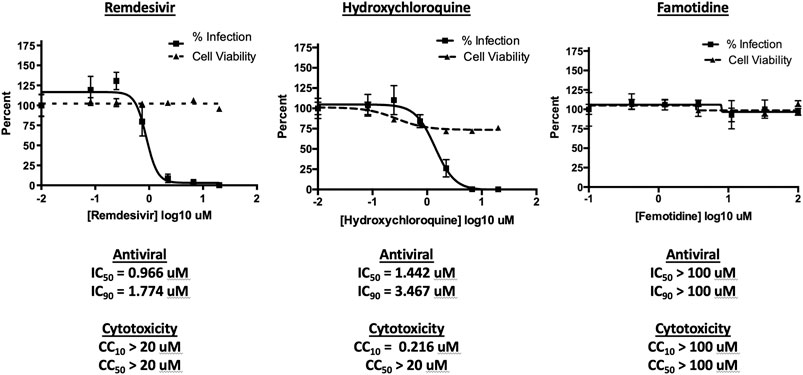
FIGURE 2. Famotidine does not directly inhibit SARS-CoV-2 infection. To assess the possibility that famotidine may inhibit SARS-CoV-2 infection by other routes, a Vero E6 cell-based assay was performed to compare median tissue culture infectious doses (TCID50/mL) of famotidine, remdesivir, and hydroxychloroquine. Vero E6 cells were cultured and infected as described in methods and scored for presence or absence of infection using the surrogate of an immunohistochemical stain for cell-associated SARS-CoV-2 NP protein as scored by imaging cytometer. Non-specific cytotoxicity (inverse of viability) was assessed using an MTT assay. As appropriate, infected supernatants were assayed for infectious viral titer using the TCID50 method. Results are displayed as % inhibition of the viral infection Vero E6 cells as a function of tested pharmaceutical, % infected cell viability, pharmaceutical agent concentration necessary to achieve 50% or 90% replication inhibition (IC50, IC90 respectively), and pharmaceutical agent concentration required to yield 10% or 50% reduction in cell viability (CC10, CC50).
Human Receptors
Famotidine Does Not Act via Sigma-1 or -2 Receptor Binding
A wide-ranging study recently presented a map of interactions between viral and host proteins (Gordon et al., 2020). It was shown that regulation of the sigma-1 and sigma-2 receptors had antiviral effects. Sigma and histamine receptors share several ligands in common, like the antipsychotic haloperidol, the antihistamines astemizole and clemastine, the antidepressive clomipramine, and many more. As such, we tested for possible interaction between famotidine and sigma-1 or sigma-2 receptors (Figure 3). We performed radioligand competition binding experiments using cloned sigma receptors, following established procedures (Schmidt et al., 2016;Alon et al., 2017). In these assays, famotidine showed no detectable displacement of radioligand probes for either sigma-1 or sigma-2 receptors at famotidine concentrations up to 10 μM. Hence, famotidine’s binding to sigma-1 and sigma-2 receptors is likely negligible at physiologically relevant concentrations.
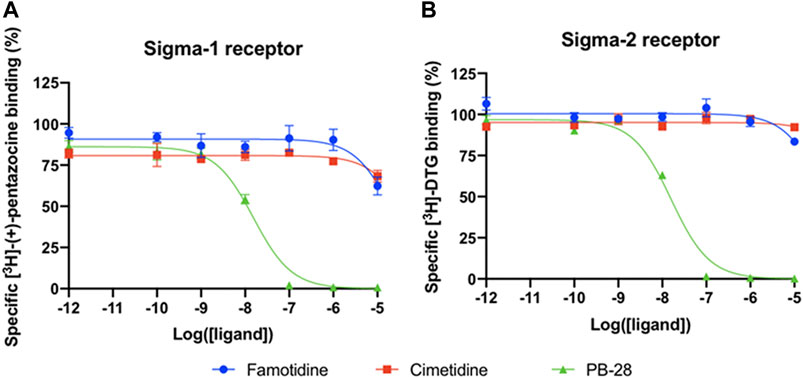
FIGURE 3. Competition binding curves of Famotidine (blue circles), Cimetidine (red squares), and PB-28 (green triangle), a potent sigma receptor ligand as positive control. (A) [3H](+)-pentazocine competition curves in Expi293 membranes expressing sigma-1. (B) [3H]DTG competition curves in Expi293 membranes expressing sigma-2 (TMEM97).
Famotidine is Selective for Receptor H2
As is well-known (Bertaccini et al., 1986), famotidine is a selective blocker of the histamine H2 receptor with a measured affinity of approximately 14 nM, substantially more active than the affinity (590 nM) of cimetidine for histamine H2 receptors (Figure 4A). As shown below, data generated demonstrate that famotidine has highly efficacious inverse agonist activity (reducing basal activity by 75%) with a potency of 33 nM (Figure 4C). Intriguingly, and unlike cimetidine, while famotidine acts to block Gs protein signaling, it actually acts as a partial agonist of arrestin recruitment, with an efficacy of about 15% that of histamine, and an EC50 of 105 nM (Figure 4D), suggesting that the molecule promotes arrestin-scaffolded signaling--such as through the ERK pathway (Alonso et al., 2015), and promotes internalization of the receptor and further non-canonical signaling once internalized (Irannejad and von Zastrow, 2014; Jean-Charles et al., 2017) through an arrestin-biased mechanism. These features distinguish famotidine certainly from cimetidine, and potentially from other H2 blockers, as such biased activation of arrestin recruitment for GPCR antagonists, while not unprecedented, is not common.
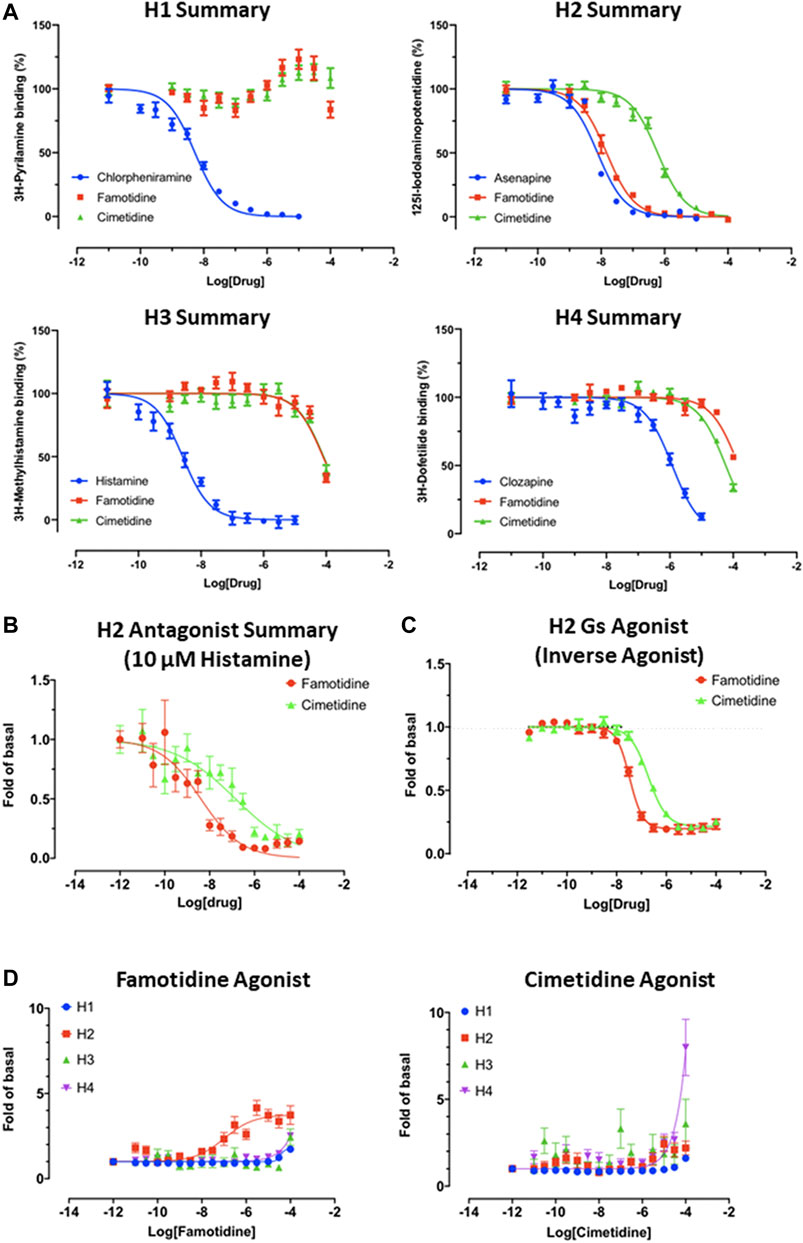
FIGURE 4. Famotidine and cimetidine activity on histamine receptors. Experiments performed in duplicate. (A) Competitive binding dose-response curves for famotidine and cimetidine on four histamine receptors with reference compounds. (B) The partial agonist, famotidine, shows antagonist activity of H2 in the presence of potent endogenous agonist, histamine. (C) Inverse agonism of famotidine and cimetidine on H2, whereas histamine stimulated cAMP production by 20-fold of basal (N = 2). (D) Arrestin recruitment by famotidine (left) and cimetidine (right) upon interaction with histamine receptors.
Famotidine May Activate Other GPCRs
A screen for famotidine activation of 318 receptors of the GPCR-ome was performed and revealed only seven receptors with an average fold of basal increase above 3.0, including H2 (Figure 5). In all cases, the quadruplicate replicates were not in agreement. Excluding H2, none of the activations were reproducible in follow-up studies.
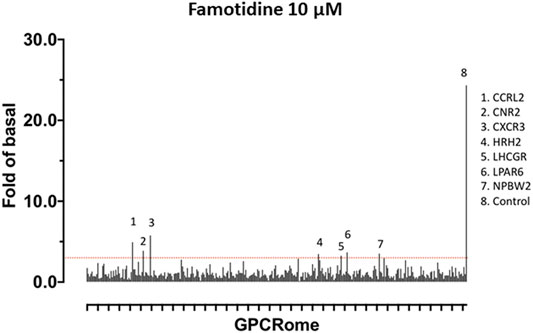
FIGURE 5. Screen for activation of 318 receptors of the GPCR-ome. To test whether famotidine may act via other G-coupled protein receptors (GPCRs) in addition to its activity as an inverse agonist for the histamine H2 receptor, a screening assay method was applied to detect potential agonist activity of famotidine (10 microM final) when interacting with each of the 318 known human GPCRs. Prior surveys with other pharmaceuticals have defined the baseline for non-specific signal in this assay at 3x greater than the corresponding basal signal. GPCRs meeting the screening criteria of >3x baseline are listed. None of these screening signals were verified in follow up studies, yielding the conclusion that famotidine has no agonist activity for other human GPCRs.
Famotidine Reaches Functionally Relevant Systemic Concentrations, Whereas Cimetidine Does Not
We calculated predicted steady state concentrations of famotidine and cimetidine at different doses based on published pharmacokinetic and biodistribution data (Lin et al., 1987; Yeh et al., 1987; Lin, 1991). This modeling demonstrated that the different clinical outcomes exhibited by COVID-19 patients taking famotidine vs. cimetidine could be readily explained by the distinctive pharmacokinetic and pharmacodistribution properties of the two agents.
Therapeutic efficacy of a pharmacological antagonist requires that it achieves a steady-state concentration that substantially exceeds the half maximal inhibitory concentration (IC50) for its target. Thus, to evaluate the relative systemic effects of famotidine and cimetidine, the IC50 values of each agent for the H2 receptor were compared to the steady-state plasma concentrations (Css) predicted at standard clinical doses. As demonstrated above, famotidine binds to H2 with a concentration of inhibitor at which (under saturating substrate conditions) the reaction rate is half of the maximum reaction rate Vmax (ergo Ki) of 14 nM, whereas cimetidine binds to H2 with Ki 586 nM. Previous reports suggest functional IC50s are approximately 3x higher, and these data were used for the current analyses (Lin et al., 1987; Lin, 1991). In these reports, the total concentration of inhibitor required to reach 50% inhibition (IC50) for the H2 receptor were reported as 13 μg/L (0.039 μM) for famotidine and 400–780 μg/L (1.59–3.09 μM) for cimetidine. Steady state concentration (Css) values were calculated using pharmacokinetic data for dosing, clearance, bioavailability, and volume of distribution as summarized previously (Lin, 1991). Table 1 lists the Css values for both famotidine and cimetidine.
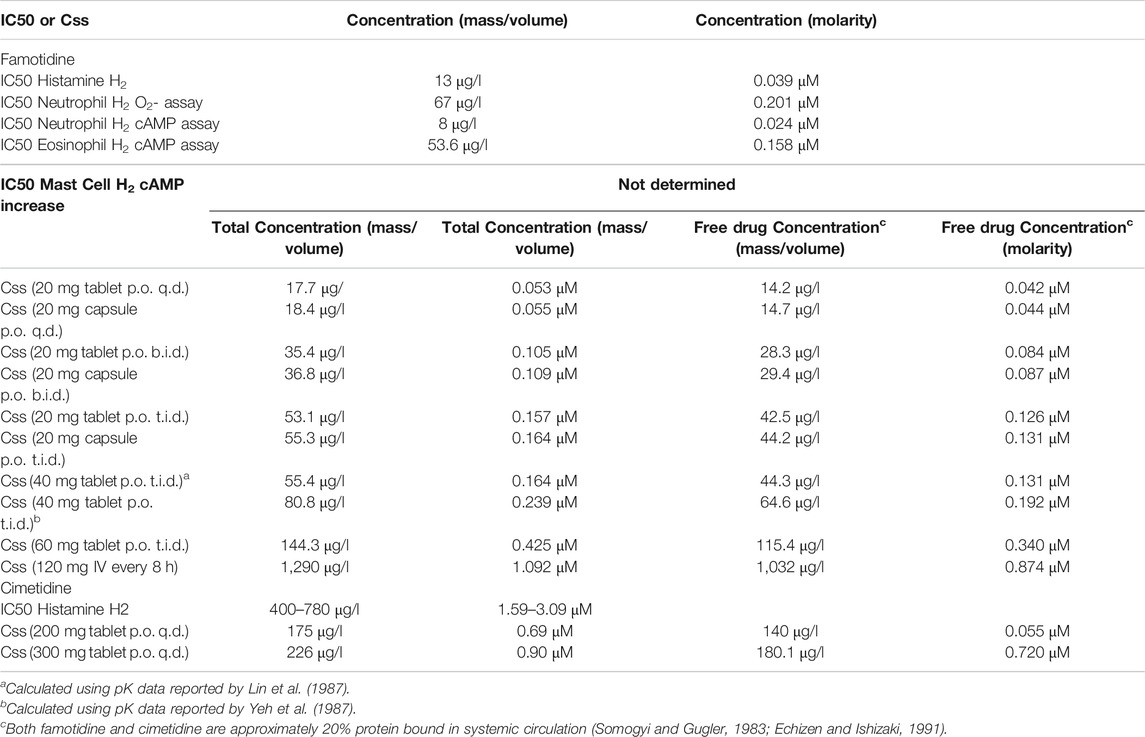
TABLE 1. Steady-state concentrations (Css) of Famotidine and Cimetidine at standard doses compared to the half maximal inhibitory concentration (IC50) value of Famotidine or Cimetidine for histamine H2 receptor antagonism.
In primary human neutrophils and eosinophils, H2 activation by histamine inhibits neutrophil effector functions including O2- release (Gespach and Abita, 1982; Burde et al., 1989), platelet-activating-factor induced chemotaxis (Rabier et al., 1989) and leukotriene biosynthesis (Flamand et al., 2004). Eosinophil functions are also inhibited by H2 activation; histamine binding diminishes eosinophil peroxidase release (Ezeamuzie and Philips, 2000) and, at high concentrations, inhibits eosinophil chemotaxis (Clark et al., 1975; Wadee et al., 1980). Famotidine is one of the most effective antagonists of these H2-mediated histamine effects on neutrophils and eosinophils (Reher et al., 2012). IC50 for two measures that relate to these phenotypes are also listed in Table 1. Mast cells express histamine H2 and H4 receptors, and histamine-induced increase of cAMP in mast cells is inhibited by famotidine (Lippert et al., 2004). 10 μM famotidine pre-incubation blocks histamine-induced cAMP increase in human skin mast cells, however, the IC50 for this effect has not been determined (Lippert et al., 2004).
At all dosing regimens, the Css for famotidine exceeds the general IC50 value for the H2 receptor, and at the twice daily (b.i.d.) and thrice daily (t.i.d.) dosing of 20 and 40 mg, the Css for unbound famotidine is 2-5 fold greater than the H2 IC50. Also calculated and summarized is the Css for the intravenous dosage currently being administered in clinical trial NCT04370262 and that dose exceeds famotidine IC50 by greater than 20-fold. In contrast, unbound cimetidine levels at standard doses of 200 or 300 mg daily (q.d.), achieve a Css that is a fraction of the reported IC50 range of 400–780 μg/l.
Comparative Analysis of Mast Cell Density in Lung Parenchyma of SARS-CoV-2 Infected African Green Macaques
Images and data summarizing studies of infiltration of mast cells into the pulmonary parenchyma of SARS-CoV-2 African Green monkeys (AGMs) are summarized in Figure 6. As shown in Panel A (20x magnification), in naïve AGM lung parenchyma, mast cells (arrow) are rarely observed within alveolar interstitium. Panels B and C (20x), increased numbers of mast cells (arrows) are seen within the interstitium of SARS-CoV-2 infected AGMs that both developed mild (B) and severe pneumonia (C). Panel D summarizes the proportion of total cells that are mast cells is elevated in all lobes of SARS-CoV-2 infected AGMs in comparison to naïve control tissue.
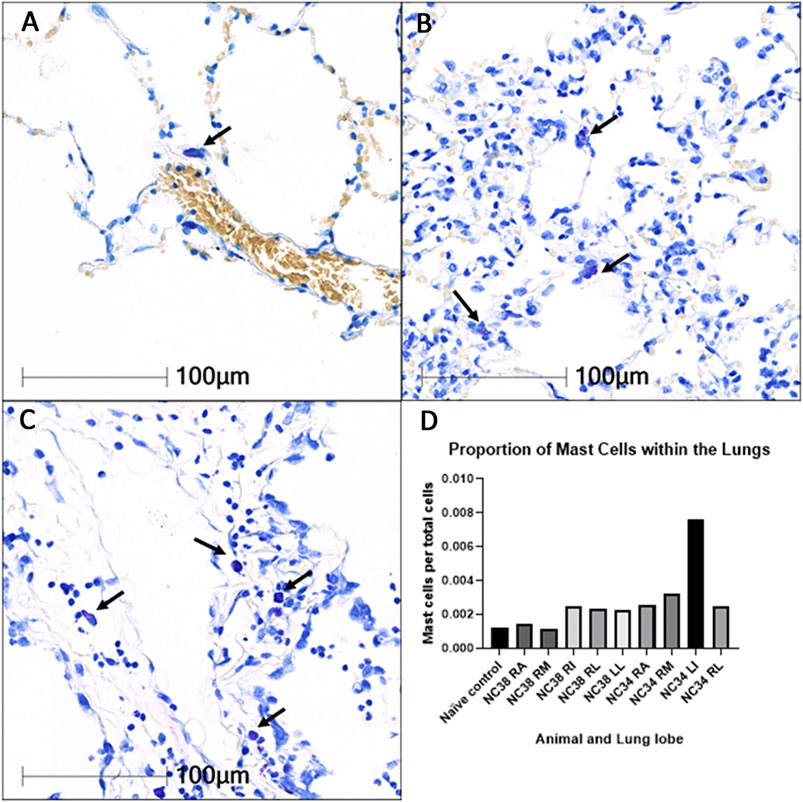
FIGURE 6. Infiltration of mast cells into the pulmonary parenchyma of SARS-CoV-2 infected African Green monkeys (AGMs). 20× magnification. Toluidine blue stain. RA: right anterior; RM: right middle; RI: right intermediate; RL: right lower; LL: left lower.
Case History, Severe COVID-19 Outpatient Treatment With Famotidine
Patient JM is a 47 year old male who received PCR diagnosis of COVID-19 after 8 days of complaints of diarrhea, abdominal cramping, eructation, low energy, dry cough, arthralgia, myalgia, anosmia and ageusia. The patient has a history of hypertension (10 years), Type II diabetes (4 years), hypercholesterolemia (3 years) and gout (10 years). Current medications included Metformin, Allopurinol, Lisinopril, and Atorvastatin. He is employed as a hospital maintenance worker in the hospital to which he presented.
Contact tracing revealed that his son (same household) had developed COVID-19 symptoms 12 days prior. Receipt on day 8 of positive PCR diagnosis (from a prior outpatient intranasal swab sample) coincided with onset of fever (102oF), night sweats, shortness of breath and a feeling of chest pressure. Famotidine (“PEPCID AC ®” 60 mg p.o. t.i.d. = 2.24 mg/ft2 t.i.d) was initiated upon receiving the PCR diagnosis due to symptoms meeting FDA criteria for severe COVID-19, combined with high-risk pre-existing conditions. The famotidine drug regime was continued for 30 days. After initiating famotidine in the evening, the patient was able to sleep through the night and reported complete relief from the chest pressure sensation, reduction in cough, but continued to be febrile (101.6°F).
On day 10, he presented to the emergency room (ER) with continuing complaints of diarrhea, abdominal cramping, eructation, low energy, dry cough, arthralgia, myalgia, anosmia and ageusia and shortness of breath on exertion. Day 10ER physical examination, including the chest, was unremarkable and vital signs were normal. The patient body mass index (BMI) was 36 (Du Bois BSA 26.78 ft2). SpO2 was 93%, rising to 97 and 99% on 3 L/min by nasal cannula over the next 30 min. An intranasal sample was obtained for SARS-CoV-2 rtPCR diagnostic analysis. Comprehensive metabolic panel showed a mild decrease in serum sodium and chloride with hyperglycemia (260 mg/dl). Complete blood count (CBC) was normal, specifically including the lymphocyte count. Urinalysis showed a specific gravity of 1.025 but was otherwise normal. A portable chest X-ray had poor inspiration but was interpreted as showing “bibasilar areas of airspace disease” consistent with COVID-19 (Figure 7, chest x-ray [CXR] day 10). The patient was diagnosed as dehydrated, given ondansetron IV, 1 L IV of normal saline and discharged home with a hospital pulse oximeter. At the time of departure, he had an SpO2 of 94% on room air that did not drop with ambulation.
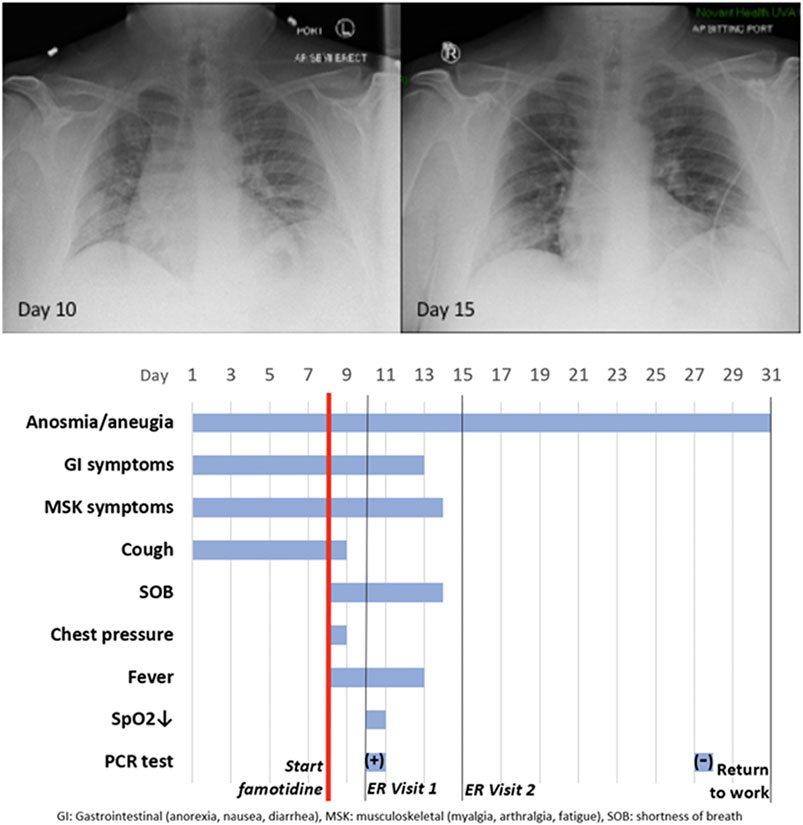
FIGURE 7. ase Study JM: CXR and Timeline. Famotidine (60 mg PO tid) was started on Day 8 from start of symptoms. It was continued for 30 days. The anosmia and ageusia are still present at Day 50.
The patient again presented to the emergency room on day 15 after experiencing near-syncope during showering. Physical examination was unremarkable. Vital signs were normal. SpO2 showed values of 98, 93, and 97% on room air over the 2 h period. Basic metabolic panel showed only hyperglycemia (266 mg/dl). CBC was normal except for a mild lymphopenia (0.96; reference range 1.00–3.00 × 103/μL) and mild monocytosis (0.87; 0.20–0.80 × 103/μL). Chest X-ray was interpreted as showing “Faint patchy consolidation of lung bases bilaterally, similar to perhaps minimally improved at the lower left lung base compared to prior” (Figure 6 CXR day 15). The patient was placed on azithromycin and discharged to home.
On days 27 and 28 after initial symptoms, he tested negative (2x, successive days) for SARS-CoV-2 nucleic acid by PCR (intranasal swab) and returned to his work at the local hospital 31 days after initial symptoms. 47 days after first developing COVID-19 symptoms he continues to note a lack of ability to taste or smell, but otherwise considers himself largely recovered from COVID-19 (Figure 7 timeline).
Use of famotidine in this patient was recommended due to meeting FDA criteria for severe COVID-19 and his COVID-19 risk factors: male, 47yo, hypertension, obesity (Divoux et al., 2012) and diabetes mellitus Type 2. Although this is an anecdotal example, the patient experienced relief of symptoms overnight upon initiating use of famotidine. While not sufficient to demonstrate proof of cause and effect, this case does provide context for typical COVID-19 presentation and symptoms, as well as support for additional well-controlled famotidine therapeutic clinical trials in an outpatient setting.
Discussion
Famotidine is an off-patent drug available as either branded (“PEPCID®”) or generic medicines in tablet, capsule or intravenous forms. The general pharmacology of famotidine is well-characterized, with an excellent absorption, distribution, metabolism, excretion and toxicology profile (FDA, 1986). Famotidine is unique among the drugs currently being tested for treatment of COVID-19, in that it is an H2 receptor antagonist (and inverse agonist). Famotidine is currently being tested for treating COVID-19 in a double blind randomized clinical trial at high intravenous doses in combination with either hydroxychloroquine or remdesivir (ClinicalTrials.gov Identifier: NCT04370262). A retrospective cohort study of 1,620 hospitalized COVID-19 patients indicates that 84 propensity score matched patients receiving famotidine during hospitalization (oral or IV, 20 mg or 40 mg daily) had a statistically significant reduced risk for death or intubation (adjusted hazard ratio (aHR) 0.42, 95% CI 0.21–0.85) and also a reduced risk for death alone (aHR 0.30, 95% CI 0.11–0.80) (Freedberg et al., 2020). Subsequent famotidine/COVID-19 retrospective data analysis reports have been less clear, ranging from reporting no effect (Cheung et al., 2020; Shoaibi et al., 2020; Yeramaneni et al., 2020) to various levels of protective effect (Mather et al., 2020). To the extent that these retrospective studies report famotidine dosage levels, in all cases the dosages are either unreported (Cheung et al., 2020) or predominantly standard dosing for relief of gastro-esophageal reflux disease (GERD). No retrospective studies involving high dose famotidine (60–80 mg oral 3–4 times per day) have been reported to date. Anecdotal reports and undisclosed data indicating that famotidine provided protection from COVID-19 mortality while neither cimetidine nor proton pump inhibitors were similarly protective lead to an initial inference that the beneficial effects of famotidine were not related to the known on-target activity of the drug (Borrell, 2020). Studies detailed in this report and others, however, indicate that famotidine does not act by directly inhibiting either of the principal SARS-CoV-2 proteases (PLpro or Mpro) (Anson et al., 2020). Vero E6-based cell assays also indicate that famotidine has no direct antiviral activity in this cell line, although antiviral activity in cells that express H2 has not been tested. Additional hypotheses that famotidine may act via binding either the sigma-1 or -2 receptors have not been supported by the studies summarized herein.
The most straightforward explanation of the apparent famotidine activity as a COVID-19 therapy is that the drug acts via its antagonism or inverse-agonism of histamine signaling and via its arrestin biased activation—all a result of famotidine binding to histamine receptor H2. If true, then it is reasonable to infer that a SARS-CoV-2 infection that results in COVID-19 is at least partially mediated by pathologic histamine release. The anecdotal lack of protection provided by oral administration of the H2 antagonist cimetidine can be accounted for by insufficient systemic drug levels after oral administration and does not contradict potential benefit provided by famotidine H2 binding. Intravenous cimetidine at sufficient doses may achieve levels high enough for clinical benefit and would further support this hypothesis. Failure to achieve clinical COVID-19 responses with cimetidine may indicate that inverse agonism or other GPCR-mediated effects of famotidine may play an important role in the (preliminary) observed clinical benefits. Analysis of famotidine activity in histamine receptor competition assays indicate that, over the range of clinical steady state famotidine drug levels being tested, famotidine is specific for H2. Therefore, off-target antagonism of histamine H1 receptor, H3 receptor, or H4 receptor is unlikely to contribute to famotidine-mediated effects.
Steady state famotidine concentrations sufficient to elicit H2 antagonism (and inverse agonism) are readily achieved using inexpensive oral tablets and safe dosage levels. As summarized above, the famotidine dosage employed in the retrospective hospital studies currently available which examine famotidine effects on COVID-19 outcomes appears to be levels (20–40 mg daily) which are unlikely to fully inhibit histamine-mediated systemic effects at the H2 receptor (Freedberg et al., 2020). In contrast, study NCT04370262 administers intravascular famotidine doses that are more than 20-fold greater than the IC50 for antagonism of H2. Other prospective randomized clinical studies involving high dose famotidine for treating COVID-19 are in progress [Samimagham et al. (2020), NCT NCT04504240]. The data presented herein provides a rationale for famotidine dose selection to maintain a steady state concentration at a reasonable multiple of the IC50 for systemic antagonism of H2 and indicate that oral tablet dosages of between 40 mg every 8 h to 80 mg every 8 h should be sufficient to insure maximal H2 target effects. As famotidine is primarily cleared by the kidney, adequate renal function is required for higher dosages (FDA, 1986).
In addition to H2 antagonism, famotidine may also act as an inverse agonist thereby lowering the concentration of cyclic-Adenosine Monophosphate (c-AMP) (Alonso et al., 2015). Endothelial cell permeability has been attributed to histamine H2 activation and is blunted by famotidine pretreatment (Luo et al., 2013). Histamine, bradykinin and des-arg-bradykinin receptor engagements can lead to increased endothelial permeability through a common pathway that results in AKT-1 activation (Di Lorenzo et al., 2009). The H2 receptor also signals through Gq/11 proteins, resulting in inositol phosphate formation and increases in cytosolic Ca2+ concentrations which may account for the increased endothelial cell fluid permeability (Panula et al., 2015).
One alternative hypothesis is that famotidine may not only inhibit signaling through the H2 receptor but may also engage in cross talk with the kinin B1 receptor, which moderates the response of endothelial cells to DABK and DAKD ligands. Data provided here in are not consistent with this hypothesis; no activation of bradykinin receptor B1 or B2 were observed in quadruplicate replicate TANGO assay.
Another alternative hypothesis is that famotidine is active in mitigating the effects of neutrophil sensitivity to activation yielding extracellular traps (Radermecker et al., 2020). At autopsy, the microvascular thrombi of COVID-19 demonstrate large number of neutrophils, and it has now been shown that overshooting, global neutrophil activation is present in severe and critical.
COVID-19 (Nicolai et al., 2020; Radermecker et al., 2020). Hyperactivated neutrophils and enhanced neutrophil extracellular traps (NETS) function in the NET-like structures subsequently identified in pulmonary, kidney, and heart specimens. Reactive Oxygen species are important for NETosis, and these effects are at least partially mitigated by histamine H2 blockers including famotidine and cimetidine (Mikawa et al., 1999). Previous pharmacological studies of famotidine have shown a dose dependent attenuation of intracellular calcium concentrations in neutrophils and at high doses that reactive oxygen species are attenuated [see Tomera and Kittah (2020b) for further details]. This would explain the clinical observations that elevated D-dimers and pulmonary emboli become responsive to standard anticoagulation therapy when given HD famotidine (Tomera and Kittah, 2020a; Radermecker et al., 2020).
While COVID-19 symptoms affect multiple organ systems, respiratory failure due to acute respiratory distress syndrome (ARDS) is the most common cause of death. Examination of RNA expression profiles of the cells which contribute to lung anatomy and function demonstrate the presence of multiple ACE2/TMPRSS2 positive cell types susceptible to SARS-CoV-2 infection in the lung. In addition, these and other associated lung cells that are positive for histamine receptors H1 and H2 could respond to local histamine release following mast cell degranulation (Krystel-Whittemore et al., 2015), and therefore those cells positive for H2 may be responsive to famotidine effects.
To understand how famotidine may act to reduce pulmonary COVID-19 symptoms requires an understanding of COVID-19 lung pathophysiology, which appears to have two principal disease phases. In turn, this requires an appreciation of pulmonary tissue and cell types. Pulmonary edema results from loss of a regulation of fluid transfer that occurs at several levels in the alveolus, as diagrammed in Figure 8. In the capillary wall, there are the glycocalyx, the endothelial cell with associated tight junctions, and the basement membrane. In the epithelium there is a surfactant layer on the alveolar lining fluid, manufactured and secreted by the Type II pneumocyte, and the Type I pneumocyte itself with its tight junctions and negatively charged basement membrane which restricts albumin. The pulmonary pericytes located in the terminal conducting airway region play a critical role in synthesizing the endothelial basement membrane and regulating blood flow in the precapillary arteriole, the capillary and the postcapillary venule. Disruption of any of these cells or layers can lead to edema. This edema fluid may be a transudate in milder dysfunctions or an exudate when inflammation or necrosis develop. Two possible pathologies that could result in edema of the alveolar wall and space include infection of cells by SARS-CoV-2 and mast cell degranulation with release of hundreds of compounds that can impact on cellular and basement membrane functions, glycocalyx and tight junction integrity. These compounds include histamine, bradykinin, heparin, tryptase and cytokines.
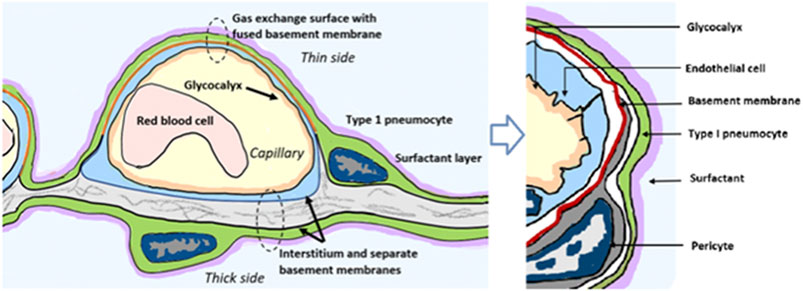
FIGURE 8. Lung alveolus cell interactions and gas exchange. Schematic diagram illustrating relevant cellular and tissue microanatomy of the pulmonary alveolus. Pulmonary edema results from loss of a regulation of fluid transfer that occurs at several levels in the alveolus, including disrupted capillary wall components, surfactant, Type I and II pneumocytes, as well as the pulmonary pericytes which are a histamine-responsive contractile cell which both synthesize the endothelial basement membrane and regulate blood flow in the precapillary arteriole, the capillary and the postcapillary venule via contraction and relaxation response to histamine and other signaling molecules.
Gene expression patterns of these pulmonary cells provide insight into which cells are likely to be infected, and which express the H2 receptor that could be directly impacted by famotidine treatment and resulting H2 antagonism or inverse agonism (Figure 9). These patterns suggest that epithelial cells and endothelial cells are more likely to be infected based on ACE2 and TMPRSS2 expression patterns in those cell types. The cells most likely to show a famotidine effect include Type 2 pneumocytes, smooth muscle cells, pericytes, and myeloid granulocytes (which includes mast cells, neutrophils and eosinophils).
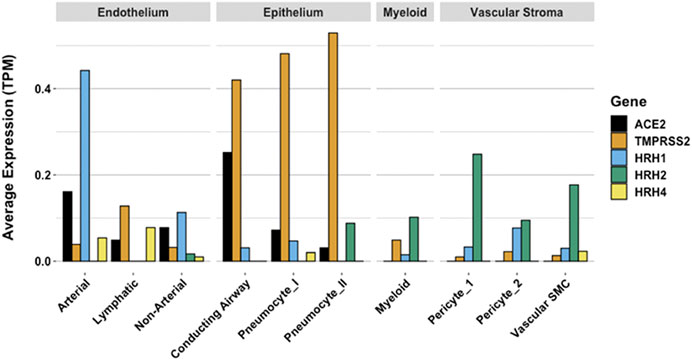
FIGURE 9. Human single cell lung gene expression normalized to transcripts per million (TPM) from LunGENS web portal (Du et al., 2015). Single cell lung gene expression patterns from the Dropseq PND1 experiment for angiotensin-converting enzyme 2 (ACE2: black), transmembrane protease, serine 2 (TMPRSS2; orange), and histamine receptors H1 (blue), H2 (green), and H4 (yellow).
The limited tissue pathology available from early COVID-19 cases seems to support both viral infection as well as histamine effects in the lung. In a singular study of early COVID-19, Sufang Tian et al. (2020b) describe the viral lung pathology of early COVID-19 in tissue resected for cancer. Their photomicrographs show two different patterns of disease. As shown in Figure 9 panel B, some samples of this lung tissue demonstrate the usual mononuclear inflammatory pattern of interstitial pneumonitis and fibrinous exudate that one would associate with a viral infection. It is striking that no neutrophils or eosinophils are observed in the inflammatory infiltrate. One explanation is that H2 activation of neutrophils inhibits neutrophil effector functions including O2- release (Gespach and Abita, 1982; Burde et al., 1989), platelet-activating-factor induced chemotaxis (Rabier et al., 1989) and leukotriene biosynthesis (Flamand et al., 2004). Eosinophil functions are also inhibited by H2 activation; histamine binding diminishes eosinophil peroxidase release (Ezeamuzie and Philips, 2000) and, at high concentrations, inhibits eosinophil chemotaxis (Clark et al., 1975; Wadee et al., 1980).
The reports of Tian et al. (2020b) and Zeng et al. (2020) also include images in which there is interstitial and alveolar edema while the alveolar septae retain normal architecture (Figure 10 panel A). This is not a pattern typically observed in viral infection, as there is no inflammation, and the fluid appears to be a transudate. It is consistent with dysregulation of the fluid barrier due to the effect of histamine or other mast cell products on endothelial cells, pericytes or Type II pneumocytes. Increased endothelial permeability due to histamine is driven by H1 receptor activation, and so if any potential famotidine treatment effect on these cells occurs it would most likely be indirect by inhibition of mast cell degranulation. Forskolin activates the enzyme adenylyl cyclase and increases intracellular levels of cAMP, and can be used to inhibit the release of histamine from human basophils and mast cells (Marone et al., 1986). Histamine may act as an autocrine regulator of mast cell cytokine and TNF-a release in a PGE2-dependent fashion. Based on in vitro studies, this autocrine feedback appears to be mediated by H2 and H3. Endothelial cells are also susceptible to infection by SARS-CoV-2. Mast cell degranulation-related pulmonary edema could correlate with the early phase silent hypoxia and the high compliance non-ARDS ventilation pattern associated with shortness of breath (Couzin-Frankel, 2020). The image in Figure 10 panel B does not permit evaluation for microvascular thrombi.
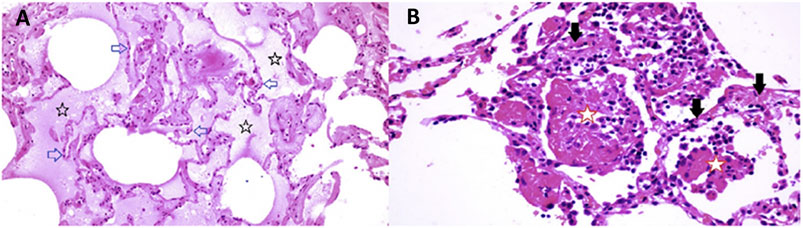
FIGURE 10. Lung pathology of early COVID-19. Early COVID-19 pulmonary histopathology, illustrating an atypical viral pathology pattern of interstitial and alveolar edema together with alveolar septae which retain normal architecture. Atypical for viral pneumonia, this resection from early in the course of COVID-19 disease lacks inflammation, and the accumulated fluid appears to be a transudate.
Eighty four year old female undergoing right middle lobe (RML) resection for adenocarcinoma. On Day 6 of hospitalization a CT scan showed a ground glass opacity (GGO) in the RML in addition to the tumor mass. Lobectomy was performed on Day 12. On Day 13 (Day 1 post-operation), CT scan showed bilateral bibasilar GGO. On Day 16, she developed typical COVID-19 symptoms with cough, dyspnea and chest tightness. Capillary O2 saturation ranged from 77–88%. Death ensued on Day 29. SARS-CoV-2 was confirmed by nasal swab (Tian et al., 2020b)
Panel A (RML): There is extensive pulmonary edema consistent with a transudate (open black stars). Alveolar septae appear normal and there is no inflammation (open blue arrows). Features are not suggestive of an infection.
Panel B (RML): There is fibrinous exudate in the alveolar spaces (open red stars). Alveolar septae show edema and a mononuclear infiltrate (solid black arrows). No neutrophils are identified. There is no significant diffuse alveolar damage of ARDS. Features are typical of an interstitial viral pneumonia.
These findings are supported in a separate autopsy case report of a patient dying 5 days after onset of COVID-19 symptoms. In this case, photomicrographs also show a non-inflammatory transudative-type edema (Schweitzer et al., 2020). In both of these studies, the observed non-inflammatory edema in early stage COVID-19 pulmonary disease is consistent with histamine release by mast cells.
Mast cell degranulation correlates with the COVID-19 natural history that progresses through functionally and clinically different early and later phases. Most SARS-CoV-2 infections follow the typical early phase pattern of any lower respiratory virus, in which a majority of patients have asymptomatic or minimal disease, while a minority go on to later phase acute respiratory distress syndrome (ARDS). Within this spectrum typical of any severe viral disease, COVID-19 has a number of distinctive features. In the out-patient setting, early COVID-19 is usually indistinguishable from other “influenza-like illnesses”, presenting with various non-specific symptoms ranging from sore throat, headache and diarrhea to fever, cough, and myalgias. In these first few days however, COVID-2 may also be associated with anosmia, a unique feature (Eliezer et al., 2020). It is toward the end of the first week of symptoms that COVID-19 patients develop shortness of breath (SOB). This follows cough and fever by several days, a feature not typical of other viruses (Cohen et al., 2020). On physical examination of COVID-19 patients with SOB, the oxygen saturation drops dramatically on exertion. CT scan will usually show bilateral bibasilar ground glass opacifications consistent with pulmonary edema. Nasopharyngeal swabs test positive for SARS-CoV-19. This SOB correlates with a distinctive clinical phenotype of hypoxia with near normal compliance (i.e., >50 mLcmH2O). Some authors attribute this to a loss of pulmonary vasoconstriction, one cause of which could be histamine effect on the H2 receptors of pericytes and/or vascular smooth muscle. H1-related edema and microthrombosis of lung vessels could also be causes. These are the patients that PEEP ventilation will not help, as there are no recruitable alveoli. These patients are helped by lying prone (Gattinoni et al., 2020). It is at this stage that the patient is at greatest risk to progress onto the serious complications of later disease, especially ARDS with its 60–80% mortality if ventilation is required. Patients may also present with additional neurological symptoms and complications including ischemic stroke (Filatov et al., 2020; Mao et al., 2020; Qureshi et al., 2020). Cardiac complications of later COVID-19 include myocarditis, acute myocardial infarction, heart failure, dysrhythmias, and venous thromboembolic events (Long et al., 2020; Mahmud et al., 2020).
Multiple studies have demonstrated a hypercoagulable state in COVID-19 patients requiring hospitalization. Results from a recent large autopsy study suggests that there is also a novel lung-centric coagulopathy that manifests as a small vessel microthrombosis. Based on this study, there are indications that over 50% of patients dying of COVID-19 have pulmonary microthrombosis (Carsana et al., 2020). This thrombosis is not only in arterial vessels, but also can be found in alveolar capillaries in the absence of inflammation and ARDS, as seen in Figure 11 (Magro et al., 2020).
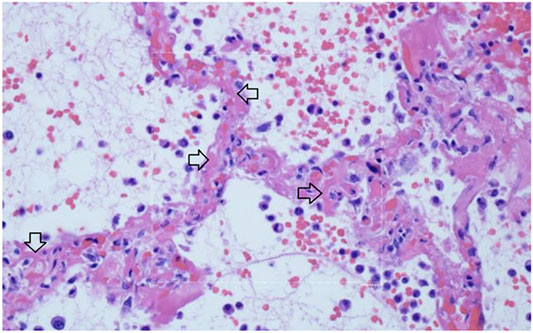
FIGURE 11. Micro-thrombosis in the pulmonary microvasculature in COVID-19 at autopsy (Magro et al., 2020). There is widening of the alveolar septae by extensive fibrinous occlusion of capillaries (open black arrows). There is alveolar space edema with red blood cell extravasation. Septae show a mild mononuclear infiltrate. Alveolar edema shows neutrophils in proportion to the blood.
Capillary wall disruption accompanied by fibrin deposition and red cell extravasation, with neutrophils in the septa and within the alveolar spaces (Hematoxylin and eosin, 1000×). For further discussion of microvascular coagulation associated with COVID-19, see (Magro et al., 2020).
Because small microthrombi are difficult to identify on CT scan even with iodinated contrast (Oudkerk et al., 2020), pre-mortem diagnosis is difficult. Laboratory coagulation tests have typically shown normal or mildly prolonged Prothrombin time (PT) and activated partial thromboplastin time (aPTT), normal to increased or slightly decreased platelet counts, elevated fibrinogen levels and very elevated D-dimers (Panigada et al., 2020). Although referred to by some authors as a DIC-like state, this pulmonary microthrombosis does not appear as a typical coagulation factor consumptive bleeding condition typical of overt DIC, but instead more closely resembles hypercoagulable thrombosis. This coagulopathy appears to be a core pathophysiology of COVID-19 as rising D-dimer levels, correlate with a poor prognosis, as do rising levels of IL-6 and CRP. IL-6 levels have been correlated to fibrinogen levels in one study, possibly through the acute phase reactant response (Ranucci et al., 2020). The pathogenesis of microthrombosis of the lung in COVID-19 is not known. There are multiple working hypotheses concerning this finding currently being assessed (Ackermann et al., 2020). Damage to the vascular endothelial glycocalyx can be caused by TNF-α, ischemia and bacterial lipopolysaccharide. As well, activated mast cells release cytokines, proteases, histamine, and heparinase, which degrade the glycocalyx (Alphonsus and Rodseth, 2014) and may thereby contribute to microthrombosis. Disruption of the glycocalyx exposes endothelial cell adhesion molecules, triggering further inflammation, rolling and adhesion of white blood cells and platelets (Becker et al., 2010). Glycocalyx components measured in serum positively correlate with increased mortality in septic patients (Nelson et al., 2008). Other causes of hypercoagulability include direct damage to ACE2 positive endothelial cells by viral invasion or secondary damage from the inflammatory response to the infection. Mast cells release heparin which activates the contact system, producing plasmin and bradykinin. Plasmin activation could account for the singular rise in D-dimer levels. Activation of platelets also seems likely as part of the thrombo-inflammatory response but their precise role in thrombus formation remains to be elucidated (Jackson et al., 2019). A more complete understanding awaits further study.
In addition to the usual features of a viral infection, early COVID-19 often presents with anosmia, ageusia, skin rashes including pruritis and urticaria, neuropsychiatric symptoms (including altered dream states), and silent hypoxia. These symptoms are all consistent with histamine signaling. Anosmia, ageusia, and other symptoms relating to cachexia are often reported in both COVID-19 and mast cell degranulation syndrome, and the potential role of histamine signaling in driving the pathophysiology of cachexia has been reviewed (Becker et al., 2012; Zwickl et al., 2019). As summarized in Figure 12, the distinctive later findings of abnormal coagulation, involvement of other organ systems and ARDS occur in the second week after the appearance of symptoms. This is coincidental with a rising immunoglobulin response to SARS-CoV-2 antigens. For a subset of patients, disease progress may suddenly worsen at days 7–10, and this correlates with the onset of SARS-CoV-2 spike protein neutralizing antibody titers (Suthar et al., 2020). In this study, it was shown that IgG starts to rise within 4 days post-symptoms, inconsistent with a first antigenic exposure (Suthar et al., 2020). Rapid onset of specific neutralizing antibody responses beginning less than seven days after exposure to SARS-CoV-2 implies a recall rather than primary B cell response, and therefore the response is being driven by a pre-existing memory cell population. SARS-CoV-2-reactive CD4+ T cells have been detected in ∼40–60% of unexposed individuals, suggesting cross-reactive T cell recognition between circulating ‘common cold’ coronaviruses and SARS-CoV-2 (Grifoni et al., 2020). These memory cells may have been educated by prior exposure to another coronavirus (e.g. circulating alphanumeric coronaviruses), raising concerns that this second phase of COVID-19 disease progression could share an immunologic basis with Dengue hemorrhagic fever (Mongkolsapaya et al., 2003). If pre-existing cross-reactive IgE “common cold” antibodies and/or associated memory B cell populations are also present, this may help account for or otherwise exacerbate dysfunctional mast cell degranulation. Antibodies produced from this early rapid humoral response may also drive further mast cell degranulation. During this phase rising D-dimer levels correlate with poor prognosis, as do measured levels of CRP and IL-6.
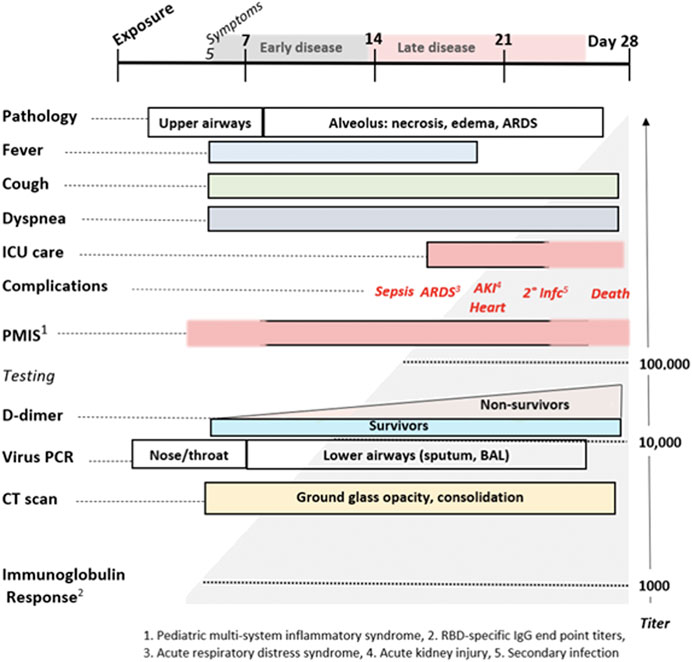
FIGURE 12. The Natural History of COVID-19. Modified from Oudkerk et al. (2020).
Current reviews seek to explain COVID-19 clinical and pathologic findings based on standard models of antiviral innate and adaptive immune responses which do not consider the potential role of mast cell activation and degranulation. Reviews emphasize the inflammatory cell response cascade associated with monocytes, macrophages (Merad and Martin, 2020), and adaptive T and B cell helper and effector responses (Vabret et al., 2020). These types of immune responses are also invoked to explain the novel microvascular pulmonary intravascular coagulopathy associated with COVID-19 (McGonagle et al., 2020).
We propose an alternative paradigm; SARS-CoV-2 infection-induced mast cell activation could account for some of the core pathologic cascade and much of the unusual symptomatology associated with COVID-19 (Kritas et al., 2019). Many of the unique clinical symptoms observed during the early phase of COVID-19 are consistent with known effects of histamine release (Conti et al., 2020). Histamine may act as an autocrine regulator of mast cell cytokine and TNF-a release in a PGE2-dependent fashion and based on in vitro studies the autocrine feedback appears to be mediated by H2 and H3 (Bissonnette, 1996). This model is consistent with the histopathologic findings seen at surgery, autopsies, and is supported by clinical pharmacologic findings suggesting potential benefits of histamine H2 receptor blockade using famotidine. This model is also supported by the significant overlap in the clinical signs and symptoms of the initial phase of COVID-19 disease and those of mast cell activation syndrome (MCAS) (Afrin et al., 2020; Butterfield, 2020; Weiler, 2020; Weinstock et al., 2020) as well similarities to Dengue hemorrhagic fever and shock syndrome (including T cell depletion) during the later phase of COVID-19 (Mongkolsapaya et al., 2003; Guzman and Harris, 2015; Redoni et al., 2020). The cardiac events, stroke, and related outcomes associated with COVID-19 also appear consistent with the Kounis syndrome (Gonzalez-de-Olano et al., 2011; Kounis, 2016; Kounis et al., 2020). This hypothesis is supported by the findings herein of increased numbers of mast cells in the alveolar septal walls and pulmonary parenchyma of SARS-CoV-2 infected AGM, as well as in the alveolar septa of COVID-19 patients (Motta Junior et al., 2020).
If COVID-19 is partially driven by dysfunctional mast cell degranulation, then a variety of medical interventions employing marketed drugs useful for treating mast cell-related disorders may help to reduce death and disease associated with SARS-CoV-2 infection. Examples include drugs with mast cell stabilizing activity such as beta-2 adrenoceptor antagonists (Scola et al., 2004) or cromolyn sodium (Zhang et al., 2016; Han et al., 2016), other histamine antagonists (for example H1 and H4 types) (Okayama et al., 1994; Marone et al., 2003; Hogan et al., 2020), leukotriene antagonists and leukotriene receptor antagonists (Fidan and Aydogdu, 2020), anti-inflammatory agents such as those developed for inflammatory bowel diseases, and mast cell activation inhibitors (Castells and Butterfield, 2019; Theoharides et al., 2019). If such repurposed drugs are used in combination with pharmaceuticals that directly inhibit SARS-CoV-2 infection or replication, it may be possible to rapidly develop potent, safe and effective outpatient treatments for preventing or treating COVID-19 until such time as a safe and effective SARS-CoV-2 vaccine becomes available.
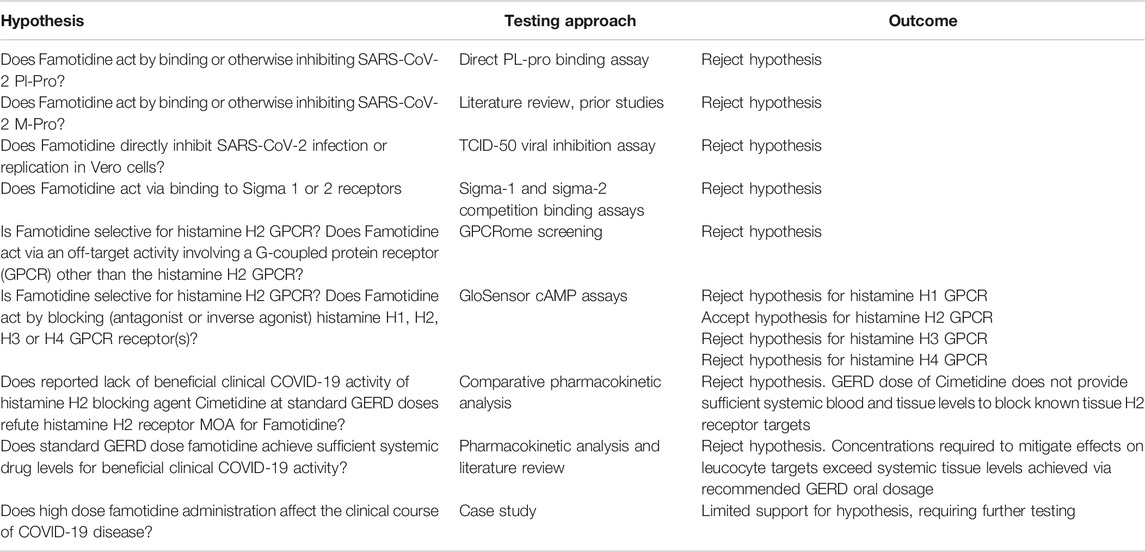
TABLE 2. Summary of multiple working hypotheses tested
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and the institutional requirements. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Tulane University.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Primary research design and data were generated by YL, X-PH, KW, LM, EM, AA, ED, GW., RB, CR, Modeling, data interpretation, analysis/theory development, experimental design guidance, figure and manuscript authorship were primarily performed by RM, PT, CH, PF-S, JP, RB, CR, NS, JH, KT, GS, AM, AK, AS, BR, JG-M, and DR.
Funding
This material is based upon work supported under Air Force Contract No. FA8702-15-D-0001. Any opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the U.S. Air Force. Funding was also provided by grants from the Defense Advanced Research Projects Agency HR0011-19-2-0020 (to A. G-S.); by CRIP (Center for Research for Influenza Pathogenesis), a NIAID supported Center of Excellence for Influenza Research and Surveillance (CEIRS, contract # HHSN272201400008C) and by supplements to NIAID grant U19AI135972 and DoD grant W81XWH-20-1-0270 to A.G.-S.
Conflict of Interest
RM, PT, and GS were employed by the companies RW Malone MD LLC, Medical School Companion LLC, and Pharmorx LLC, respectively. In all three cases, their contributions to the work described were voluntary and uncompensated. By joint agreement, no patent rights relating to these findings have been asserted by any of the authors.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the Department of Defense (DoD), Defense Threat Reduction Agency (DTRA), and the Joint Science and Technology Office (JSTO) of the Chemical and Biological Defense Program (CBDP) for funding under the Discovery of Medical countermeasures Against Novel Entities (DOMANE) initiative. This work has also benefitted from advice, guidance, information and comments provided by Drs. Revell Phillips, Howard Haimes, David Hone, and Roland Seifert. We appreciate the valuable input provided by Dr Frank Weichold of the Office of Regulatory Science and Innovation (ORSI), Office of the Chief Scientist (OCS), Office of the Commissioner (OC), FDA/HHS, and Dr. Lawrence Callahan of the Office of Health Informatics, Office of the Chief Scientist (OCS), Office of the Commissioner (OC), FDA/HHS. We also thank Dr Anton Yuryev from Elsevier for his assistance in literature mining and reconstruction of the histamine signaling model. D.R. gratefully acknowledges the support of the MIT SuperCloud team. In the interests of expediting COVID-19 related scientific communications, the initial version of this manuscript was made available via the Research Square preprint server 23 May 2020 as DOI 10.21203/rs.3.rs-30934/v1.
References
Ackermann, M., Verleden, S. E., Kuehnel, M., Haverich, A., Welte, T., Laenger, F., et al. (2020). Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128. doi:10.1056/NEJMoa2015432
Afrin, L. B., Ackerley, M. B., Bluestein, L. S., Brewer, J. H., Brook, J. B., Buchanan, A. D., et al. (2020). Diagnosis of mast cell activation syndrome: a global "consensus-2”. Diagnosis (Berl). doi:10.1515/dx-2020-0005
Alon, A., Schmidt, H. R., Wood, M. D., Sahn, J. J., Martin, S. F., and Kruse, A. C. (2017). Identification of the gene that codes for the σ2 receptor. Proc. Natl. Acad. Sci. USA 114 (27), 7160–7165. doi:10.1073/pnas.1705154114
Alonso, N., Zappia, C. D., Cabrera, M., Davio, C. A., Shayo, C., Monczor, F., et al. (2015). Physiological implications of biased signaling at histamine H2 receptors. Front. Pharmacol. 6, 45. doi:10.3389/fphar.2015.00045
Alphonsus, C. S., and Rodseth, R. N. (2014). The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia 69 (7), 777–784. doi:10.1111/anae.12661
Anson, B. J., Chapman, M. E., Lendy, E. K., Pshenychnyi, S., D’Aquila, R. T., Satchell, K. J. F., et al. (2020). Broad-spectrum inhibition of coronavirus main and papain-like proteases by HCV drugs. PREPRINT (Version 1) Available at Research Square. (Accessed May, 2020). doi:10.21203/rs.3.rs-26344/v1
Baez-Santos, Y. M., St John, S. E., and Mesecar, A. D. (2015). The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 115, 21–38. doi:10.1016/j.antiviral.2014.12.015
Becker, B. F., Chappell, D., Bruegger, D., Annecke, T., and Jacob, M. (2010). Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc. Res. 87 (2), 300–310. doi:10.1093/cvr/cvq137
Becker, S., Pflugbeil, C., Groger, M., Canis, M., Ledderose, G. J., and Kramer, M. F. (2012). Olfactory dysfunction in seasonal and perennial allergic rhinitis. Acta Otolaryngol. 132 (7), 763–768. doi:10.3109/00016489.2012.656764
Bertaccini, G., Coruzzi, G., Poli, E., and Adami, M. (1986). Pharmacology of the novel H2 antagonist famotidine: in vitro studies. Agents Actions 19 (3, 4), 180–187. doi:10.1007/BF01966204
Besnard, J., Ruda, G. F., Setola, V., Abecassis, K., Rodriguiz, R. M., Huang, X. P., et al. (2012). Automated design of ligands to polypharmacological profiles. Nature 492 (7428), 215–220. doi:10.1038/nature11691
Bissonnette, E. Y. (1996). Histamine inhibits tumor necrosis factor alpha release by mast cells through H2 and H3 receptors. Am. J. Respir. Cell Mol Biol. 14 (6), 620–626. doi:10.1165/ajrcmb.14.6.8652190
Blair, R. V., Vaccari, M., Doyle-Meyers, L. A., Roy, C. J., Russell-Lodrigue, K., Fahlberg, M., et al. (2021). Acute respiratory distress in aged, SARS-CoV-2 infected african green monkeys but not rhesus Macaques. Am. J. Pathol. 191, 274. doi:10.1016/j.ajpath.2020.10.016
BMJ (2020). Covid-19: four fifths of cases are asymptomatic, China figures indicate. Br. Med. J. 369, 375. doi:10.1136/bmj.m1375
Borrell, B. (2020). New York clinical trial quietly tests heartburn remedy against coronavirus [Online]. Science Magazine. Available at: https://www.sciencemag.org/news/2020/04/new-york-clinical-trial-quietly-tests-heartburn-remedy-against-coronavirus (Accessed May 17, 2020). doi:10.1126/science.abc4739
Burde, R., Seifert, R., Buschauer, A., and Schultz, G. (1989). Histamine inhibits activation of human neutrophils and HL-60 leukemic cells via H2-receptors. Arch. Pharmacol. 340 (6), 671–678. doi:10.1007/BF00717743
Butterfield, J. H. (2020). Survey of mast cell mediator levels from patients presenting with symptoms of mast cell activation. Int. Arch. Allergy. Immunol. 181 (1), 43–50. doi:10.1159/000503964
Carsana, L., Sonzogni, A., Nasr, A., Rossi, R., Pellegrinelli, A., Zerbi, P., et al. (2020). Pulmonary post-mortem findings in a large series of COVID-19 cases from Northern Italy, medRxiv 20, 1132–1140. doi:10.1101/2020.04.19.20054262
Castells, M., and Butterfield, J. (2019). Mast cell activation syndrome and mastocytosis: initial treatment options and long-term management. J. Allergy Clin. Immunol. Pract. 7 (4), 1097–1106. doi:10.1016/j.jaip.2019.02.002
CDC (2020). Nterim clinical guidance for management of patients with confirmed coronavirus diseaseCOVID-19) [Online]. Available at: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html (Accessed May 17, 2020).
Chamberlin, T. (1890). The method of multiple working hypotheses. Sci. 15 (366), 92–6. doi:10.1126/science.ns-15.366.92
Cheung, K. S., Hung, I. F., and Leung, W. K. (2020). Association between famotidine use and COVID-19 severity in Hong Kong: a territory-wide study. Gastroenterology. doi:10.1053/j.gastro.2020.05.098
Clark, R. A., Gallin, J. I., and Kaplan, A. P. (1975). The selective eosinophil chemotactic activity of histamine. J. Exp. Med. 142 (6), 1462–1476. doi:10.1084/jem.142.6.1462
Cohen, P. A., Hall, L., Johns, J. N., and Rapoport, A. B. (2020). The early natural history of SARS-CoV-2 infection: clinical observations from an urban, ambulatory COVID-19 clinic. Mayo Clin. Proc. 95, 1124. doi:10.1016/j.mayocp.2020.04.010
Conti, P., Caraffa, A., Tete, G., Gallenga, C. E., Ross, R., Kritas, S. K., et al. (2020). Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J. Biol. Regul. Homeost. Agents 34 (5), 1629–1632. doi:10.23812/20-2EDIT
Couzin-Frankel, J. (2020). The mystery of the pandemic's 'happy hypoxia. Science 368 (6490), 455–456. doi:10.1126/science.368.6490.455
Daczkowski, C. M., Dzimianski, J. V., Clasman, J. R., Goodwin, O., Mesecar, A. D., and Pegan, S. D. (2017). Structural insights into the interaction of coronavirus papain-like proteases and interferon-stimulated gene product 15 from different species. J. Mol. Biol. 429 (11), 1661–1683. doi:10.1016/j.jmb.2017.04.011
Danis, K., Epaulard, O., Benet, T., Gaymard, A., Campoy, S., Bothelo-Nevers, E., et al. (2020). Cluster of coronavirus disease 2019 (Covid-19) in the French alps, 2020. Clin. Infect. Dis. 71 (15), 825–832. doi:10.1093/cid/ciaa424
Day, M. (2020). Covid-19: identifying and isolating asymptomatic people helped eliminate virus in Italian village. BMJ 368, m1165. doi:10.1136/bmj.m1165
Di Lorenzo, A., Fernandez-Hernando, C., Cirino, G., and Sessa, W. C. (2009). Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc. Natl. Acad. Sci. USA 106 (34), 14552–14557. doi:10.1073/pnas.0904073106
Divoux, A., Moutel, S., Poitou, C., Lacasa, D., Veyrie, N., Aissat, A., et al. (2012). Mast cells in human adipose tissue: link with morbid obesity, inflammatory status, and diabetes. J. Clin. Endocrinol. Metab. 97 (9), E1677–E1685. doi:10.1210/jc.2012-1532
Du, Y., Guo, M., Whitsett, J. A., and Xu, Y. (2015). LungGENS': a web-based tool for mapping single-cell gene expression in the developing lung. Thorax 70 (11), 1092–1094. doi:10.1136/thoraxjnl-2015-207035
Echizen, H., and Ishizaki, T. (1991). Clinical pharmacokinetics of famotidine. Clin. Pharmacokinet. 21 (3), 178–194. doi:10.2165/00003088-199121030-00003
Eliezer, M., Hautefort, C., Hamel, A.-L., Verillaud, B., Herman, P., Houdart, E., et al. (2020). Sudden and complete olfactory loss function as a possible symptom of COVID-19. JAMA Otolaryngol. Head Neck Surg. 146, 674–675. doi:10.1001/jamaoto.2020.0832
Ezeamuzie, C. I., and Philips, E. (2000). Histamine H(2) receptors mediate the inhibitory effect of histamine on human eosinophil degranulation. Br. J. Pharmacol. 131 (3), 482–488. doi:10.1038/sj.bjp.0703556
FDA (1986). EPCID® (famotidine) tablets, for oral use [Online]. US Food and Drug Administration, Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/019462s039lbl.pdf (Accessed May 17, 2020).
Fidan, C., and Aydogdu, A. (2020). As a potential treatment of COVID-19: Montelukast. Med. Hypotheses 142, 109828. doi:10.1016/j.mehy.2020.109828
Filatov, A., Sharma, P., Hindi, F., and Espinosa, P. S. (2020). Neurological complications of coronavirus disease (COVID-19): encephalopathy. Cureus 12 (3), e7352. doi:10.7759/cureus.7352
Flamand, N., Plante, H., Picard, S., Laviolette, M., and Borgeat, P. (2004). Histamine-induced inhibition of leukotriene biosynthesis in human neutrophils: involvement of the H2 receptor and cAMP. Br. J. Pharmacol. 141 (4), 552–561. doi:10.1038/sj.bjp.0705654
Freedberg, D. E., Conigliaro, J., Wang, T. C., Tracey, K. J., Callahan, M. V., Abrams, J. A., et al. (2020). Famotidine use is associated with improved clinical outcomes in hospitalized COVID-19 patients: a propensity score matched retrospective cohort study. Gastroenterology, 159, 1129–1131. doi:10.1053/j.gastro.2020.05.053
Furukawa, N. W., Brooks, J. T., and Sobel, J. (2020). Evidence supporting transmission of severe acute respiratory syndrome coronavirus 2 while presymptomatic or asymptomatic. Emerg. Infect. Dis. 26 (7). doi:10.3201/eid2607.201595
Gattinoni, L., Chiumello, D., and Rossi, S. (2020). COVID-19 pneumonia: ARDS or not? Crit. Care 24 (1), 154. doi:10.1186/s13054-020-02880-z
Gespach, C., and Abita, J. P. (1982). Human polymorphonuclear neutrophils. Pharmacological characterization of histamine receptors mediating the elevation of cyclic AMP. Mol. Pharmacol. 21 (1), 78–85.
Giacomelli, A., Pezzati, L., Conti, F., Bernacchia, D., Siano, M., Oreni, L., et al. (2020). Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study. Clin. Infect. Dis., 71, 889. doi:10.1093/cid/ciaa330
Gonzalez-de-Olano, D., Alvarez-Twose, I., Matito, A., Sanchez-Munoz, L., Kounis, N. G., and Escribano, L. (2011). Mast cell activation disorders presenting with cerebral vasospasm-related symptoms: a "Kounis-like" syndrome? Int. J. Cardiol. 150 (2), 210–211. doi:10.1016/j.ijcard.2011.05.007
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 583 (7816), 459–468. doi:10.1038/s41586-020-2286-9
Grifoni, A., Weiskopf, D., Ramirez, S. I., Mateus, J., Dan, J. M., Moderbacher, C. R., et al. (2020). Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 187 (7), 15. doi:10.1016/j.cell.2020.05.015
Guzman, M. G., and Harris, E. (2015). Dengue. Lancet 385 (9966), 453–465. doi:10.1016/S0140-6736(14)60572-9
Han, D., Wei, T., Zhang, S., Wang, M., Tian, H., Cheng, J., et al. (2016). The therapeutic effects of sodium cromoglycate against influenza A virus H5N1 in mice. Influenza Other Respir. Viruses 10 (1), 57–66. doi:10.1111/irv.12334
Han, Y. S., Chang, G. G., Juo, C. G., Lee, H. J., Yeh, S. H., Hsu, J. T., et al. (2005). Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry 44 (30), 10349–10359. doi:10.1021/bi0504761
He, G., Sun, W., Fang, P., Huang, J., Gamber, M., Cai, J., et al. (2020). The clinical feature of silent infections of novel coronavirus infection (COVID-19) in Wenzhou. J. Med. Virol., 92, 1761–1763. doi:10.1002/jmv.25861
Hogan, R. B., Hogan, R. B., Cannon, T., Rappai, M., Studdard, J., Paul, D., et al. (2020). Dual-histamine receptor blockade with cetirizine - famotidine reduces pulmonary symptoms in COVID-19 patients. Pulm. Pharmacol. Ther. 63, doi:10.1016/j.pupt.2020.101942
Hu, Z., Song, C., Xu, C., Jin, G., Chen, Y., Xu, X., et al. (2020). Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Sci. China Life Sci. 63 (5), 706–711. doi:10.1007/s11427-020-1661-4
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395 (10223), 497–506. doi:10.1016/S0140-6736(20)30183-5
Irannejad, R., and von Zastrow, M. (2014). GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol 27, 109–116. doi:10.1016/j.ceb.2013.10.003
Jackson, S. P., Darbousset, R., and Schoenwaelder, S. M. (2019). Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 133 (9), 906–918. doi:10.1182/blood-2018-11-882993
Janowitz, T., Gablenz, E., Pattinson, D., Wang, T. C., Conigliaro, J., Tracey, K., et al. (2020). Famotidine use and quantitative symptom tracking for COVID-19 in non-hospitalised patients: a case series. Gut, 69, 1592. doi:10.1136/gutjnl-2020-321852
Jean-Charles, P. Y., Kaur, S., and Shenoy, S. K. (2017). G protein-coupled receptor signaling through beta-arrestin-dependent mechanisms. J. Cardiovasc. Pharmacol. 70 (3), 142–158. doi:10.1097/FJC.0000000000000482
Ki, M., and Task Force for -nCo, V. (2020). Epidemiologic characteristics of early cases with 2019 novel coronavirus (2019-nCoV) disease in Korea. Epidemiol. Health 42, e2020007. doi:10.4178/epih.e2020007
Kounis, N. G. (2016). Kounis syndrome: an update on epidemiology, pathogenesis, diagnosis and therapeutic management. Clin. Chem. Lab. Med. 54 (10), 1545–1559. doi:10.1515/cclm-2016-0010
Kounis, N. G., Koniari, I., Tzanis, G., Soufras, G. D., Velissaris, D., and Hahalis, G. (2020). Anaphylaxis-induced atrial fibrillation and anesthesia: pathophysiologic and therapeutic considerations. Ann. Card. Anaesth. 23 (1), 1–6. doi:10.4103/aca.ACA_100_19
Kritas, S., Ronconi, G., Caraffa, A., Gallenga, C., Ross, R., and Conti, P. (2019). Mast cells contribute to coronavirus-induced inflammation: new anti-inflammatory strategy. J. Biol. Regul. Homeost. Agents 34. 9–14. doi:10.23812/20-Editorial-Kritas
Kroeze, W. K., Sassano, M. F., Huang, X. P., Lansu, K., McCorvy, J. D., Giguere, P. M., et al. (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22 (5), 362–369. doi:10.1038/nsmb.3014
Krystel-Whittemore, M., Dileepan, K. N., and Wood, J. G. (2015). Mast cell: a multi-functional master cell. Front Immunol. 6, 620. doi:10.3389/fimmu.2015.00620
Lai, C. C., Liu, Y. H., Wang, C. Y., Wang, Y. H., Hsueh, S. C., Yen, M. Y., et al. (2020). Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): facts and myths. J. Microbiol. Immunol. Infect., 53, 404–412. doi:10.1016/j.jmii.2020.02.012
Lechien, J. R., Chiesa-Estomba, C. M., De Siati, D. R., Horoi, M., Le Bon, S. D., Rodriguez, A., et al. (2020). Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur. Arch. Otorhinolaryngol., 277, 2251. doi:10.1007/s00405-020-05965-1
Lin, J. H., Chremos, A. N., Kanovsky, S. M., Schwartz, S., Yeh, K. C., and Kann, J. (1987). Effects of antacids and food on absorption of famotidine. Br. J. Clin. Pharmacol. 24 (4), 551–553. doi:10.1111/j.1365-2125.1987.tb03211.x
Lin, J. H. (1991). Pharmacokinetic and pharmacodynamic properties of histamine H2-receptor antagonists. Relationship between intrinsic potency and effective plasma concentrations. Clin. Pharmacokinet. 20 (3), 218–236. doi:10.2165/00003088-199120030-00004
Lindner, H. A., Fotouhi-Ardakani, N., Lytvyn, V., Lachance, P., Sulea, T., and Menard, R. (2005). The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79 (24), 15199–15208. doi:10.1128/JVI.79.24.15199-15208.2005
Lippert, U., Artuc, M., Grutzkau, A., Babina, M., Guhl, S., Haase, I., et al. (2004). Human skin mast cells express H2 and H4, but not H3 receptors. J. Invest. Dermatol. 123 (1), 116–123. doi:10.1111/j.0022-202X.2004.22721.x
Long, B., Brady, W. J., Koyfman, A., and Gottlieb, M. (2020). Cardiovascular complications in COVID-19. Am. J. Emerg. Med., 38, 1504. doi:10.1016/j.ajem.2020.04.048
Luo, T., Chen, B., Zhao, Z., He, N., Zeng, Z., Wu, B., et al. (2013). Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 108 (3), 342. doi:10.1007/s00395-013-0342-4
Magro, C., Mulvey, J. J., Berlin, D., Nuovo, G., Salvatore, S., Harp, J., et al. (2020). Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 220, 1–13. doi:10.1016/j.trsl.2020.04.007
Mahmud, E., Dauerman, H. L., Welt, F. G., Messenger, J. C., Rao, S. V., Grines, C., et al. (2020). Management of acute myocardial infarction during the COVID-19 pandemic. J. Am. Coll. Cardiol., 76, 1375. doi:10.1016/j.jacc.2020.04.039
Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., et al. (2020). Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in wuhan, China. JAMA Neurol. 77, 683–690. doi:10.1001/jamaneurol.2020.1127
Marone, G., Columbo, M., Triggiani, M., Vigorita, S., and Formisano, S. (1986). Forskolin inhibits the release of histamine from human basophils and mast cells. Agents Actions 18 (1-2), 96–99. doi:10.1007/BF01987993
Marone, G., Granata, F., Spadaro, G., Genovese, A., and Triggiani, M. (2003). The histamine-cytokine network in allergic inflammation. J. Allergy Clin. Immunol. 112 (4 Suppl), S83–88. doi:10.1016/s0091-6749(03)01881-5
Mather, J. J. F., Seip, R. L., and McKay, R. G. (2020). Impact of famotidine use on clinical outcomes of hospitalized patients with COVID-19. Am. J. Gastroenterol., 115, 1617. doi:10.14309/ajg.0000000000000832
McGonagle, D., O'Donnell, J. S., Sharif, K., Emery, P., and Bridgewood, C. (2020). Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2 (7), e437–e445. doi:10.1016/S2665-9913(20)30121-1
Merad, M., and Martin, J. J. C. (2020). Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 20, 355. doi:10.1038/s41577-020-0331-4
Mielech, A. M., Chen, Y., Mesecar, A. D., and Baker, S. C. (2014). Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus. Res. 194, 184–190. doi:10.1016/j.virusres.2014.01.025
Mikawa, K., Akamatsu, H., Nishina, K., Shiga, M., Maekawa, N., Obara, H., et al. (1999). The effects of cimetidine, ranitidine, and famotidine on human neutrophil functions. Anesth. Analg. 89 (1), 218–224. doi:10.1097/00000539-199907000-00040
Mizumoto, K., Kagaya, K., Zarebski, A., and Chowell, G. (2020). Estimating the asymptomatic proportion of coronavirus disease 2019 (COVID-19) cases on board the Diamond Princess cruise ship, Yokohama, Japan, 2020. Euro Surveill. 25 (10), 180. doi:10.2807/1560-7917.ES.2020.25.10.2000180
Mongkolsapaya, J., Dejnirattisai, W., Xu, X. N., Vasanawathana, S., Tangthawornchaikul, N., Chairunsri, A., et al. (2003). Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat. Med. 9 (7), 921–927. doi:10.1038/nm887
Motta Junior, J. D. S., Miggiolaro, A., Nagashima, S., de Paula, C. B. V., Baena, C. P., Scharfstein, J., et al. (2020). Mast cells in alveolar septa of COVID-19 patients: a pathogenic pathway that may link interstitial edema to immunothrombosis. Front Immunol. 11, 574862. doi:10.3389/fimmu.2020.574862
Nelson, A., Berkestedt, I., Schmidtchen, A., Ljunggren, L., and Bodelsson, M. (2008). Increased levels of glycosaminoglycans during septic shock: relation to mortality and the antibacterial actions of plasma. Shock 30 (6), 623–627. doi:10.1097/SHK.0b013e3181777da3
Nicolai, L., Leunig, A., Brambs, S., Kaiser, R., Weinberger, T., Weigand, M., et al. (2020). Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 142 (12), 1176–1189. doi:10.1161/CIRCULATIONAHA.120.048488
Okayama, Y., Benyon, R. C., Lowman, M. A., and Church, M. K. (1994). In vitro effects of H1-antihistamines on histamine and PGD2 release from mast cells of human lung, tonsil, and skin. Allergy 49 (4), 246–253. doi:10.1111/j.1398-9995.1994.tb02657.x
Oudkerk, M., Buller, H. R., Kuijpers, D., van Es, N., Oudkerk, S. F., McLoud, T. C., et al. (2020). Diagnosis, prevention, and treatment of thromboembolic complications in COVID-19 Report of the National Institute for Public Health of the Netherlands. Radiology 297 (1), E216–E222. doi:10.1148/radiol.2020201629
Pan, X., Chen, D., Xia, Y., Wu, X., Li, T., Ou, X., et al. (2020). Asymptomatic cases in a family cluster with SARS-CoV-2 infection. Lancet Infect. Dis. 20 (4), 410–411. doi:10.1016/S1473-3099(20)30114-6
Panigada, M., Bottino, N., Tagliabue, P., Grasselli, G., Novembrino, C., Chantarangkul, V., et al. (2020). Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 18, 1738–1740. doi:10.1111/jth.14850
Panula, P., Chazot, P. L., Cowart, M., Gutzmer, R., Leurs, R., Liu, W. L., et al. (2015). International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 67 (3), 601–655. doi:10.1124/pr.114.010249
Qureshi, A. I., Abd-Allah, F., Alsenani, F., Aytac, E., Borhani-Haghighi, A., Ciccone, A., et al. (2020). Management of acute ischemic stroke in patients with COVID-19 infection: report of an international panel. Int. J. Stroke 15, 540–554. doi:10.1177/1747493020923234
Rabier, M., Damon, M., Chanez, P., Mencia-Huerta, J. M., Braquet, P., Bousquet, J., et al. (1989). Inhibition by histamine of platelet-activating-factor-induced neutrophil chemotaxis in bronchial asthma. Int. Arch. Allergy Appl. Immunol. 89 (2, 3), 314–317. doi:10.1159/000234967
Radermecker, C., Detrembleur, N., Guiot, J., Cavalier, E., Henket, M., d'Emal, C., et al. (2020). Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 217 (12), 1012. doi:10.1084/jem.20201012
Ranucci, M., Ballotta, A., Di Dedda, U., Bayshnikova, E., Dei Poli, M., Resta, M., et al. (2020). The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost., 18, 1747–1751. doi:10.1111/jth.14854
Redoni, M., Yacoub, S., Rivino, L., Giacobbe, D. R., Luzzati, R., and Di Bella, S. (2020). Dengue: status of current and under-development vaccines. Rev. Med. Virol. 30, e2101. doi:10.1002/rmv.2101
Reher, T. M., Brunskole, I., Neumann, D., and Seifert, R. (2012). Evidence for ligand-specific conformations of the histamine H(2)-receptor in human eosinophils and neutrophils. Biochem. Pharmacol. 84 (9), 1174–1185. doi:10.1016/j.bcp.2012.08.014
Samimagham, H. R., Hassani Azad, M., Haddad, M., Arabi, M., Hooshyar, D., and KazemiJahromi, M. (2020). The Efficacy of Famotidine in improvement of outcomes in Hospitalized COVID-19 Patients: a structured summary of a study protocol for a randomised controlled trial. Trials 21 (1), 848. doi:10.1186/s13063-020-04773-6
Schmidt, H. R., Zheng, S., Gurpinar, E., Koehl, A., Manglik, A., and Kruse, A. C. (2016). Crystal structure of the human σ1 receptor. Nature 532 (7600), 527–530. doi:10.1038/nature17391
Schweitzer, W., Ruder, T., Baumeister, R., Bolliger, S., Thali, M., Meixner, E., et al. (2020). Implications for forensic death investigations from first Swiss post-mortem CT in a case of non-hospital treatment with COVID-19. Forensic Imaging 21, 200378. doi:10.1016/j.fri.2020.200378
Scola, A. M., Chong, L. K., Suvarna, S. K., Chess-Williams, R., and Peachell, P. T. (2004). Desensitisation of mast cell beta2-adrenoceptor-mediated responses by salmeterol and formoterol. Br. J. Pharmacol. 141 (1), 163–171. doi:10.1038/sj.bjp.0705599
Shi, H., Han, X., Jiang, N., Cao, Y., Alwalid, O., Gu, J., et al. (2020). Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect. Dis. 20 (4), 425–434. doi:10.1016/S1473-3099(20)30086-4
Shoaibi, A., Fortin, S., Weinstein, R., Berlin, J., and Ryan, P. (2020). Comparative effectiveness of famotidine in hospitalized COVID-19 patients. Am J Gastroenterol., 2023. doi:10.1101/2020.09.23.20199463
Somogyi, A., and Gugler, R. (1983). Clinical pharmacokinetics of cimetidine. Clin. Pharmacokinet. 8 (6), 463–495. doi:10.2165/00003088-198308060-00001
Stauch, B., Johansson, L. C., McCorvy, J. D., Patel, N., Han, G. W., Huang, X. P., et al. (2019). Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 569 (7755), 284–288. doi:10.1038/s41586-019-1141-3
Suthar, M. S., Zimmerman, M., Kauffman, R., Mantus, G., Linderman, S., Vanderheiden, A., et al. (2020). Rapid generation of neutralizing antibody responses in COVID-19 patients. medRxiv 1 (3), 100040. doi:10.1101/2020.05.03.20084442
Swatek, K. N., Aumayr, M., Pruneda, J. N., Visser, L. J., Berryman, S., Kueck, A. F., et al. (2018). Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc. Natl. Acad. Sci. USA 115 (10), 2371–2376. doi:10.1073/pnas.1710617115
Theoharides, T. C., Tsilioni, I., and Ren, H. (2019). Recent advances in our understanding of mast cell activation - or should it be mast cell mediator disorders? Expert Rev. Clin. Immunol. 15 (6), 639–656. doi:10.1080/1744666X.2019.1596800
Tian, S., Hu, N., Lou, J., Chen, K., Kang, X., Xiang, Z., et al. (2020a). Characteristics of COVID-19 infection in Beijing. J. Infect. 80 (4), 401–406. doi:10.1016/j.jinf.2020.02.018
Tian, S., Hu, W., Niu, L., Liu, H., Xu, H., and Xiao, S. Y. (2020b). Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J. Thorac. Oncol. 15 (5), 700–704. doi:10.1016/j.jtho.2020.02.010
Tomera KM, M. R., and Kittah, J. K. (2020a). Brief report: rapid clinical recovery from severe COVID-19 with high dose famotidine high dose celecoxib adjuvant therapy. Enliven. Pharmacovigil. Drug Saf. 6 (3).
Tomera KM, M. R., and Kittah, J. K. (2020b). Hospitalized COVID-19 patients treated with celecoxib and high dose famotidine adjuvant therapy SHOW significant clinical responses. SSRN.
Vabret, N., Britton, G. J., Gruber, C., Hegde, S., Kim, J., Kuksin, M., et al. (2020). Immunology of COVID-19: current state of the science. Immunity 52, 910–941. doi:10.1016/j.immuni.2020.05.002
Wadee, A. A., Anderson, R., and Sher, R. (1980). In vitro effects of histamine on eosinophil migration. Int. Arch. Allergy Appl. Immunol. 63 (3), 322–329. doi:10.1159/000232643
Wang, D., Hu, B., Hu, C., Zhu, F., Liu, X., Zhang, J., et al. (2020). Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA., 323, 1061–1069. doi:10.1001/jama.2020.1585
Weiler, C. R. (2020). Mast cell activation syndrome: tools for diagnosis and differential diagnosis. J. Allergy Clin. Immunol. Pract. 8 (2), 498–506. doi:10.1016/j.jaip.2019.08.022
Weinstock, L. B., Pace, L. A., Rezaie, A., Afrin, L. B., and Molderings, G. J. (2020). Mast cell activation syndrome: a primer for the gastroenterologist. Dig. Dis. Sci. doi:10.1007/s10620-020-06264-9
Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., et al. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin B 10, 766. doi:10.1016/j.apsb.2020.02.008
Wu, D., Wu, T., Liu, Q., and Yang, Z. (2020). The SARS-CoV-2 outbreak: what we know. Int. J. Infect. Dis. 94, 44–48. doi:10.1016/j.ijid.2020.03.004
Wu, Z., and McGoogan, J. M. (2020). Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA 323, 1239. doi:10.1001/jama.2020.2648
Yeh, K. C., Chremos, A. N., Lin, J. H., Constanzer, M. L., Kanovsky, S. M., Hucker, H. B., et al. (1987). Single-dose pharmacokinetics and bioavailability of famotidine in man. Results of multicenter collaborative studies. Biopharm. Drug Dispos. 8 (6), 549–560. doi:10.1002/bdd.2510080606
Yeramaneni, S., Doshi, P., Sands, K., Cooper, M., Kurbegov, D., and Fromell, G. (2020). Famotidine use is not associated with 30-day mortality: a coarsened exact match study in 7158 hospitalized COVID-19 patients from a large Healthcare system. Gastroenterology 160, 919–921. doi:10.1053/j.gastro.2020.10.011
Zeng, Z., Xu, L., Xie, X.-y., Yan, H.-l., Xie, B.-j., Xu, W.-z., et al. (2020). Pulmonary pathology of early phase COVID-19 pneumonia in a patient with a benign lung lesion. Histopathology.77 (5), 823–831. doi:10.1111/his.14138
Zhang, T., Finn, D. F., Barlow, J. W., and Walsh, J. J. (2016). Mast cell stabilisers. Eur. J. Pharmacol. 778, 158–168. doi:10.1016/j.ejphar.2015.05.071
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., et al. (2020). A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382 (8), 727–733. doi:10.1056/NEJMoa2001017
Zou, L., Ruan, F., Huang, M., Liang, L., Huang, H., Hong, Z., et al. (2020). SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382 (12), 1177–1179. doi:10.1056/NEJMc2001737
Keywords: famotidine (PubChem CID: 3325), mast cell activating disorder, COVID-19, hyperinflammation state, GPCR (G Protein Coupled Receptors), histamine (H2) receptor
Citation: Malone RW, Tisdall P, Fremont-Smith P, Liu Y, Huang X-P, White KM, Miorin L, Moreno E, Alon A, Delaforge E, Hennecker CD, Wang G, Pottel J, Blair RV, Roy CJ, Smith N, Hall JM, Tomera KM, Shapiro G, Mittermaier A, Kruse AC, García-Sastre A, Roth BL, Glasspool-Malone J and Ricke DO (2021) COVID-19: Famotidine, Histamine, Mast Cells, and Mechanisms. Front. Pharmacol. 12:633680. doi: 10.3389/fphar.2021.633680
Received: 25 November 2020; Accepted: 25 January 2021;
Published: 23 March 2021.
Edited by:
Narasaiah Kolliputi, University of South Florida, United StatesReviewed by:
Sudhir Putty Reddy, Moffitt Cancer Center, United StatesRiccardo Inchingolo, Catholic University of the Sacred Heart, Italy
Copyright © 2021 Malone, Tisdall, Fremont-Smith, Liu, Huang, White, Miorin, Moreno, Alon, Delaforge, Hennecker, Wang, Pottel, Blair, Roy, Smith, Hall, Tomera, Shapiro, Mittermaier, Kruse, García-Sastre, Roth, Glasspool-Malone and Ricke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert W. Malone, UldNYWxvbmVNREBnbWFpbC5jb20=