 Katelin M. Allan
Katelin M. Allan Nigel Farrow3,4,5
Nigel Farrow3,4,5 Martin Donnelley
Martin Donnelley Shafagh A. Waters
Shafagh A. Waters- 1School of Women’s and Children’s Health, Faculty of Medicine, University of New South Wales, Sydney, Australia
- 2Molecular and Integrative Cystic Fibrosis Research Centre (miCF_RC), University of New South Wales and Sydney Children’s Hospital, Sydney, Australia
- 3Respiratory and Sleep Medicine, Women’s and Children’s Health Network, Adelaide, Australia
- 4Robinson Research Institute, The University of Adelaide, Adelaide, Australia
- 5Adelaide Medical School, The University of Adelaide, Adelaide, Australia
- 6Department of Respiratory Medicine, Sydney Children’s Hospital, Sydney, Australia
Prognosis of patients with cystic fibrosis (CF) varies extensively despite recent advances in targeted therapies that improve CF transmembrane conductance regulator (CFTR) function. Despite being a multi-organ disease, extensive lung tissue destruction remains the major cause of morbidity and mortality. Progress towards a curative treatment strategy that implements a CFTR gene addition-technology to the patients’ lungs has been slow and not yet developed beyond clinical trials. Improved delivery vectors are needed to overcome the body’s defense system and ensure an efficient and consistent clinical response before gene therapy is suitable for clinical care. Cell-based therapy–which relies on functional modification of allogenic or autologous cells ex vivo, prior to transplantation into the patient–is now a therapeutic reality for various diseases. For CF, pioneering research has demonstrated proof-of-principle for allogenic transplantation of cultured human airway stem cells into mouse airways. However, applying a cell-based therapy to the human airways has distinct challenges. We review CF gene therapies using viral and non-viral delivery strategies and discuss current advances towards autologous cell-based therapies. Progress towards identification, correction, and expansion of a suitable regenerative cell, as well as refinement of pre-cell transplant lung conditioning protocols is discussed.
CFTR Correction Strategies
Cystic fibrosis (CF) is an inherited, multi-organ disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene (Rowe et al., 2005). CF is progressive, with its major pathology impacting the lung, liver, pancreas and intestine. Mortality in CF patients is mostly due to respiratory failure (Elborn, 2016). The CFTR protein, an anion channel, is expressed in a diverse range of epithelial tissues (Riordan, 2008). CFTR dysfunction disrupts ion transport equilibrium, deregulating fluid absorption and secretion processes in epithelial tissue such as the airways, resulting in mucus accumulation and recurrent bacterial infections (Ratjen et al., 2015). Symptomatic therapies such as airway clearance by physiotherapy, mucus thinning agents, antibiotics and anti-inflammatories remain crucial for the management of CF airways (Flume et al., 2007).
Recently, targeted therapies have been approved for CF treatment. These small molecule compounds modulate CFTR protein abundance and/or function at the apical epithelial cell membrane (Davies, 2015). A combination of three CFTR modulating small molecules–elexacaftor/tezacaftor/ivacaftor–is the most advanced targeted therapeutic approved for patients with one or two Phe508del-CFTR alleles–the most common CFTR mutation in the population. CFTR modulator therapies result in improved lung function and better quality of life for patients with CF (Davies et al., 2018). Yet, variable therapeutic response, inadequate long-term efficacy data, adverse effects and unavailability of modulators for the 10% of CF patients with mutations that produce little or no CFTR protein has rekindled great interest in the development of CFTR mutation-independent corrective strategies.
In vivo transfer of a functional copy of CFTR has been envisioned as a CF airway treatment since 1989 when the CFTR gene was identified as the cause of this multisystemic disease (Tsui et al., 1985; Wainwright et al., 1985). Gene therapy has received FDA approval for treatment of monogenic disorders (U.S. Food and Drug Administration. 2020) such as spinal muscular atrophy (Kariyawasam et al., 2018), coagulative disorders (Batty and Lillicrap 2019), and immunodeficiency diseases (Booth et al., 2019), but not yet for CF. Numerous research programs and clinical trials have been undertaken to explicate the most effective vector (viral or non-viral) to deliver CFTR to airway cells (Griesenbach et al., 2015). However, clinical efficacy of these vectors in vivo in humans has been insignificant and inconsistent in improving lung function (Alton et al., 2015a). The greatest barrier to enabling clinical translation of gene therapy for CF remains the lack of an effective delivery system to the lungs. A successful gene therapy system for restoration of CFTR function needs to navigate the complexities of the lung clearance and innate immunity defense functions that are further complicated in the CF airways due to increased mucus volume and viscosity (reviewed in (Donnelley and Parsons 2018)). Even if these obstacles are circumvented, heterogeneous and highly regulated CFTR expression in various cell types of the lung raises the question of the most appropriate cellular target.
One proposed strategy to deal with the challenges associated with in vivo delivery of CFTR to the airway cells is to correct the airway cells ex vivo followed by transplanting the corrected cells to repopulate the patient’s lung with CFTR-corrected cells (Figure 1). This approach is the basis of the first ex vivo hematopoietic stem cell gene therapy, Strimvelis, which was approved for treatment of adenosine deaminase-severe combined immunodeficiency (Stirnadel-Farrant et al., 2018). In this review, we will first describe alternative strategies to CFTR DNA therapy, and discuss the advances in the main groups of viral and non-viral vectors that have shown promise in CF therapy. The second part of this review will focus on recent progress in cell-based therapies, including the gene editing technologies that facilitate CFTR correction in ex vivo cells.
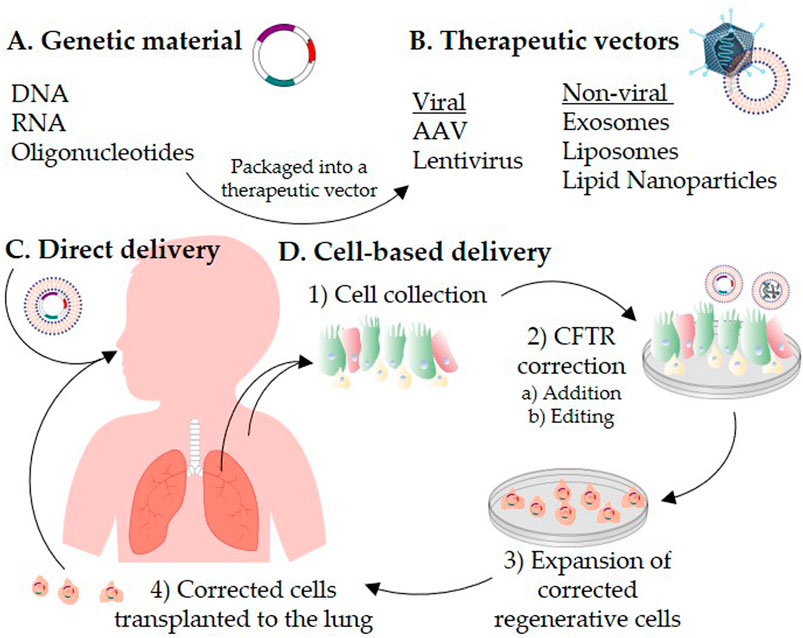
FIGURE 1. Schematic representation of CFTR correction strategies for the treatment of cystic fibrosis. Genetic materials (A) are packaged into a therapeutic vector (B). The therapeutic vector is delivered directly to the patient’s lungs (C) or introduced into cells ex vivo(D). For autologous cell-based therapy, 1) airway cells are isolated from the patient’s respiratory tract or induced pluripotent stem cells are generated from the patient’s skin fibroblasts or blood. 2) CFTR is corrected in vitro in the collected cells by a) addition or b) editing strategies. 3) The CFTR-corrected regenerative cells are expanded to reach a therapeutic dose, and then 4) transplanted back to repopulate the patient lung epithelium.
Therapeutic Genetic Material Other Than DNA: RNA Addition and Repair
The earliest efforts to deliver genetic material into diseased cells focused on directly introducing therapeutic CFTR DNA as an addition strategy to subsequently produce functional CFTR protein (reviewed in (Cooney et al., 2018)). A novel alternative to DNA therapeutics is based on addition of RNA. Since the functional site of messenger RNA (mRNA) is the cell cytoplasm, the challenge of nuclear translocation is eliminated (Hajj and Whitehead 2017).
Exogenous nucleic acids are susceptible to degradation by nucleases and can trigger an immune response upon cellular entry (Alexopoulou et al., 2001; Kariko et al., 2004). Therefore, current strategies utilize chemical modification of the nucleic acid bases to reduce immunogenicity and increase stability (Sahin et al., 2014; Pardi et al., 2015). Manufacturing and addition of modifications to RNA is easier than DNA, extending the usefulness of RNA therapy (Kuhn et al., 2012). Yet, repeat RNA administration remains necessary to sustain therapeutic levels of protein (Patel et al., 2019b). Successful delivery of chemically modified CFTR mRNA to patient-derived bronchial epithelial cells has demonstrated increased CFTR expression at the plasma membrane and rescue of chloride transport (Robinson et al., 2018). Aerosolized delivery of in vitro transcribed CFTR mRNA complexed with lipids to the lungs (Orphan drug MRT5005) is a therapeutic forerunner currently being tested in a phase 1/2 trial (NCT03375047) which has highly anticipated results for in vivo correction of CFTR.
In addition to direct RNA supplementation, an alternative therapeutic option is to alter the defective RNA to restore proper CFTR protein function. This can be achieved using antisense oligonucleotides (ASOs). ASOs are short synthetic oligonucleotides that are chemically modified to bind to target RNA, offering a tool for direct mRNA restoration (Oren et al., 2017). In vivo delivery of ASOs to the airway epithelium of mice initially resulted in no significant uptake (Griesenbach et al., 2006). It was postulated that this inefficiency was due to physical barriers to airway epithelial cell delivery (Griesenbach et al., 2006). Advances in RNA chemical modifications have been key to the development of a successful ASO-mediated therapy, with the accelerated FDA approval of eteplirsen (Exondys 51) being granted in 2016 for patients with Duchenne muscular dystrophy that have a mutation in the dystrophin gene which can be treated by exon 51 skipping (reviewed in (Bennett et al., 2017)). An ASO candidate for ASO-directed mRNA restoration, Eluforsen, binds to and restores the mRNA region encoding the Phe508 deletion (Beumer et al., 2019). In a phase 1 study, repeated administration of Eluforsen improved CFTR activity in patients homozygous for the Phe508 mutation (Sermet-Gaudelus et al., 2019) (NCT02564354). In the following phase 1b safety study with 70 enrolled participants Eluforsen was well-tolerated (NCT02532764), supporting the therapeutic translation of novel mRNA-based therapies for patients with CF. ASOs have also been tested for their ability to correct CFTR splicing mutations, which constitute 13% of disease-causing CFTR mutations, and result in no functional protein being produced (Igreja et al., 2016). Single-stranded DNA oligonucleotides designed to hybridize to pre-mRNA and modify aberrant splicing restored splicing in immortalized cells expressing the CFTR splicing mutation, c.2657 + 5G > A-CFTR (Igreja et al., 2016).
Although clinical translation of DNA or RNA therapy has been proven feasible, the efficient targeting and delivery of these genetic materials into the lung epithelium for treatment of CF remains exceedingly challenging.
Therapeutic Delivery Vectors
Nucleic acids are unable to cross the cell membrane because of their high negative charge, and therefore require assistance for intracellular delivery (reviewed in (Ni et al., 2019)). Numerous viral and non-viral delivery systems have been investigated for their ability to transfer genetic material–RNA (Therapeutic Genetic Material Other Than DNA: RNA Addition and Repair), DNA (this section) and gene editing system components (Gene Editing Technologies)–to the lung. The main groups of viral and non-viral vectors used in CF therapy and advances toward their clinical translation will be summarized below.
Viral Vectors for Airway Gene Therapy
Viral vector-mediated gene delivery systems take advantage of a virus’s natural ability to evade the lung mucus barrier and transduce the airway by inserting their DNA into epithelial cells. The natural tropism of viral vectors enables preferential targeting of the airway progenitor cells–the basal cells–so that multi-lineage transgene expression can be achieved (Havenga et al., 2002). Other advantages and disadvantages of available vectors have been discussed in depth elsewhere (Miah et al., 2019). Here we briefly discuss the most commonly used vectors for CF.
Adeno-Associated Virus
In the early 2000s, human CF clinical trials tested aerosolized viral vectors, including AAV serotype 2 (AAV2). Based on delivery of CFTR complementary DNA (cDNA) to the human nasal cavity and lung, AAV was determined safe, but failed to demonstrate significant clinical benefit in restoration of lung function (Wagner et al., 2002; Flotte et al., 2003; Moss et al., 2004; Moss et al., 2007). These findings have been attributed to issues regarding the packaging capacity of the AAV2 genome to encompass a CFTR expression cassette (Wu et al., 2010) and also low transduction efficiency of the airway epithelial cells (AECs) due to the limited binding of AAV2 to the apical surface of human airway epithelia (Zabner et al., 2000).
To overcome the packaging capacity limitation of AAV, a split AAV gene delivery approach can be implemented, wherein a large transgene is split across multiple separate AAV vectors (Patel et al., 2019a). As a means of screening for AAV serotypes that display airway tropism and permit efficient gene delivery to the human lung parenchyma, lung bud organoids–a model of lung parenchyma derived from human embryonic stem cells–have been utilized (Meyer-Berg et al., 2020). A 2020 study identified AAV2 and AAV serotype 6 (AAV6) as having the highly efficient transduction of the human lung parenchyma. Additionally, it was recently discovered that unlike other AAV serotypes, AAV6 is capable of efficiently diffusing through mucus in primary differentiated CF airway cultures (Duncan et al., 2018). Inhaled administration of AAV6 provided high-level transgene expression (mediating roughly 30% airway coverage) in a mouse model with airway mucus obstruction (Duncan et al., 2018). Yet, the high immunogenicity of AAV draws concern to the potential for participants of viral-based gene therapy clinical trials to acquire AAV immunity, and for potential preexisting immunity in some patients (reviewed in (Ronzitti et al., 2020). These are major concerns because immunity to AAV is likely to prevent repeat administration of AAV gene therapy to patients (George et al., 2020), therefore risking their eligibility for future AAV-based therapies. In 2020, the imlifidase (IdeS) enzyme was tested in rodents and non-human primates and successfully demonstrated inhibition of the host immune response to AAV resulting from preexisting immunity and following AAV gene therapy (Leborgne et al., 2020). If IdeS treatment in human patients were to enable repeat administration of AAV-based therapies, this could have big implications for the future course of AAV gene therapy. To improve the gradual decrease in CFTR expression due to the transient episomal expression nature of AAV, and avoid the need for repeat dosing of the AAV vector, Cooney and colleagues designed a novel integrating AAV-based CFTR-expressing vector (termed piggyBac/AAV), which demonstrated efficient transduction and persistent expression in primary human CF airway cells in vitro and in mouse airways in vivo (Cooney et al., 2015). A follow-up large animal study delivered aerosolized piggyBac/AAV-CFTR to CF pig airways and demonstrated phenotypic restoration of CFTR function (Cooney et al., 2019).
Lentivirus
In comparison to AAVs, lentivirus vectors (LV) have a larger packaging capacity (approximately 8 kb) which makes them more compatible for full length CFTR packaging (Castellani and Conese, 2010). Additionally, LVs can transduce and integrate into the genome of both dividing and non-dividing cells (Naldini, Blomer et al., 1996; Wang et al., 1999). Proof-of-principle that a lentiviral vector could correct the CFTR defect in vivo and provide persistent CFTR expression was first demonstrated in mouse airways (Limberis et al., 2002). This was followed by a demonstration of partially restored in vivo CFTR channel activity following aerosol delivery of Feline immunodeficiency virus (IV)-CFTR to CF pig airways (Cooney et al., 2016). More recently, insertion of a LacZ marker gene into airway basal cells via a LV vector produced persistent transgene expression and importantly, the basal cells successfully passed on the introduced gene to their daughter cells in a mouse airway (Farrow et al., 2018b). In support of a first-in-man CF trial, a simian IV-based lentiviral vector, pseudotyped with Sendai virus fusion protein and Hemagglutinin/Neuraminidase envelope proteins exhibited efficient transduction of human airway cells in vitro and murine lung epithelium in vivo (Alton et al., 2017).
Non-viral Vectors
Progression of viral vectors as a clinical therapy for CF remains contingent on demonstrating successful long-term transduction efficacy, the safety of delivery to the CF lung and the ability for repeat dosing (Donnelley and Parsons, 2018). Some of these barriers have necessitated the development of more cost-effective non-viral vectors to provide an increased safety profile and limitless genetic material packaging size, while obtaining more reproducible delivery outcomes. Non-viral vectors are naturally produced by the body and can also be engineered in the laboratory. Despite reduced immunogenicity, the transfection efficiency of non-viral vectors is still low compared to viral vectors. Specifically, unmodified non-viral vectors are rapidly cleared and have low accumulation in target tissues and cells (Takahashi et al., 2013; Smyth et al., 2015), thus new compounds are constantly engineered and investigated (Murphy et al., 2019).
Exosomes
Exosomes are naturally occurring nanoscale extracellular vesicles which, following their release from cells, facilitate intercellular communication by transporting material to neighboring or distant recipient cells (Doyle and Wang, 2019). Due to this ability to function as an endogenous intercellular cargo transfer system, they have been exploited as vehicles for the delivery of genetic material, and as such, are a promising vector for in vivo gene therapy. Exosomes have been demonstrated to deliver human CFTR mature glycoprotein, as well as CFTR mRNA, in both Chinese hamster ovarian cells (Gonzalez et al., 2012) and CFTR-deficient cells derived from CF patients (Vituret et al., 2016; Villamizar et al., 2019). In both models, CFTR channel function was shown to be corrected in exosome-recipient cells. Exosomes have similarly shown utility in delivering small interfering RNA in well-differentiated human AECs (Singh et al., 2020), suggesting value beyond gene transfer for the delivery of gene editing materials. These studies demonstrated the potential application of exosomes as vectors for CFTR transfer and functional correction of the genetic defect in human CF cells.
Liposomes
Liposomes are artificially created vesicles with a lipid bilayer membrane and an aqueous core (Akbarzadeh et al., 2013). In a single dose, dose-escalation phase 1/2a safety trial assessment of a liposomal mixture that included the cationic lipid mixture (GL67A) complexed with CFTR complementary DNA, patients with CF showed improved lung function and no adverse effects (Alton et al., 2015b). This was followed by a double-blind, placebo-controlled multi-dose phase 2b trial (NCT01621867) that found that repeated administration of GL67A liposome significantly, yet modestly, stabilized lung function in the treatment group (n = 62) vs. placebo (n = 54) in CF adults or children aged 12 years or older enrolled in this trial (Alton et al., 2015a). Although this trial reached its primary efficacy endpoint (lung function improvement), the magnitude and variability in effect did not support progression to phase 3 trials. These results established for the first time a proof of principle that gene therapy was capable of favorably modulating CF lung function, with no safety concerns with repeat dosing. However, they also highlighted the need for more efficacious methods of gene delivery transport, such as viral vectors, before large-scale clinical translation can be achieved. Gene delivery systems likely face greater barriers in adults with established lung disease and irreversible fibrotic scarring in comparison to children, with less progressed lung disease. Strong reasoning to include children in gene therapy clinical trials has been given previously (Jaffe et al., 2006; Delhove et al., 2020) and may present the most rational avenue forward as a clinical target age.
Lipid Nanoparticles
Like liposomes, lipid nanoparticles (LNPs) are used to encapsulate and deliver therapeutic RNA formulations to the lung via intranasal or systemic routes. However, LNPs differ in their composition, often carrying cargo within a non-aqueous core (Kraft et al., 2014). Recent advances have focused on exploiting the dynamic structure of LNPs to engineer specialized formulations tailored to increase mucus penetrance (Nafee et al., 2018; Wan et al., 2018) and evade host immune detection (Vencken et al., 2019). This work climaxed in the successful LNP-mediated delivery of chemically modified CFTR mRNA to CF airway cells (Robinson et al., 2018). However, it has since been shown that LNPs only modestly increase CFTR expression in patient-derived human nasal epithelial cells (Villamizar et al., 2019). It is probable that the inefficiency of LNP delivery is due to endosome retention (Sahay et al., 2013), and as such, further work to engineer particles capable of escaping endosome retention is needed. Since CFTR expression in the epithelial airway is cell type specific (Plasschaert et al., 2018), further testing in vitro and in vivo models will be required before LNPs can be established as efficient and clinically relevant delivery systems.
Cell-Based Therapies
Cell-based therapies offer an alternative therapeutic opportunity to in vivo gene therapy. This approach integrates advances in stem cell biology with the well-established platform of in vitro gene delivery and repair strategies. The exemplar for cell-based therapy is hematopoietic stem cell transplantation, which is successfully performed in over 50,0000 patients per year world-wide (Gratwohl and Niederwieser 2012). The safety of correcting a patient’s cells ex vivo via a gene editing technology, before reintroducing them into the same patient (autologous transplantation), has been demonstrated in a clinical trial for hematological malignancies (Stadtmauer et al., 2020). Autologous cell therapy has also been used for treatment of epidermolysis bullosa (Eichstadt et al., 2019; Lwin et al., 2019; Marinkovich and Tang 2019), whilst treatment of insulin-dependent diabetes has shown success with transplant of allogenic islet cells (Kumagai et al., 2002; Yamada et al., 2011).
The efficacy of these regenerative cellular therapies has propagated investigation of cell-based therapy for CF. Yet, in the CF airways, the volume, viscosity and composition of the mucus that protects the airway from foreign particles and irritants is altered in such way that penetrating this barrier poses a great challenge. Successful clinical translation of a cell therapy approach will require regenerative cell 1) identification, 2) CFTR correction (addition or editing), 3) expansion, and 4) transplantation back into the patient’s lungs (Figure 1). The remainder of this review will discuss the progress and challenges toward these milestones.
Identification of Suitable Regenerative Cells With Differentiation Capacity
One of the obstacles to cell-based therapy for CF is the identification of an optimal self-renewing cell that can also differentiate into the cells of the airway epithelium. In this review we discuss therapeutic perspectives of using adult tissue-resident basal stem cells, yet first we will briefly describe advances in the field using mesenchymal and induced pluripotent stem cells (iPSCs).
Mesenchymal stem cells (MSCs) are an allogenic source of cells that have been investigated for their application in CF cell-based therapy due to their immunomodulatory and anti-inflammatory properties (reviewed in (Caretti et al., 2019)). In favor of their potential for CF cell therapy, MSCs co-cultured with CF immortalized airway epithelial cells at air-liquid interface have demonstrated acquisition of an epithelial phenotype, and subsequent restoration of functional CFTR protein (Carbone et al., 2014; Carbone et al., 2018). However, there is concern over low engraftment levels of administered MSCs in the lung (Ortiz et al., 2003; Xu et al., 2007). Furthermore, recent studies suggest that MSCs function transiently to reduce inflammation via the secretion of extra-cellular vesicles such as exosomes (Zhu et al., 2014; Zulueta et al., 2018) that can be used for delivery of drugs or gene editing material (reviewed in (Almeida-Porada et al., 2020)). Exosomes derived from MSCs genetically engineered to carry a transcription activator protein have demonstrated success in targeting and activating CFTR transcription in primary human bronchial epithelial cells from patients with CF (Villamizar et al., 2021). Further in vitro and in vivo studies are needed before this approach is considered viable. Until then, there remains uncertainty surrounding the capacity of MSCs to effectively restore CFTR function in the respiratory epithelium.
iPSCs are routinely generated from skin or blood cells that are reprogrammed back into an embryonic-like pluripotent state. In addition, they are amenable to gene editing and expansion, thereby providing a large potential source of gene-corrected cells which upon differentiation into lung basal stem cells can be engrafted into the lung for therapeutic use. Two iPSC-derived cell populations have been isolated for the purpose of CF cell therapy: one- airway epithelial cells (AECs) or 2-basal stem cells. The strengths and limitations of both these will be discussed here briefly. Firstly, several reports have demonstrated successful directed differentiation of human iPSCs into AECs in vitro (Firth et al., 2014; Huang et al., 2014; Dye et al., 2015; Konishi et al., 2016; McCauley et al., 2017). The iPSC-derived AEC cultures are a heterogenous cell population containing limited numbers of basal cells with self-renewing capacity (McCauley et al., 2017). As such, transplanting AECs will only provide a short-term solution, even if engraftment of cells was successful. Therefore, although this method can generate ample AECs, transplantation of terminally differentiated AECs would not be an effective CF cell therapy.
More recently, iPSCs have been differentiated into basal cells, termed induced basal cells (iBCs) (Hawkins et al., 2020). These iBCs are functionally indistinct from native basal cells and have been shown in vitro to differentiate into a pseudostratified airway epithelium exhibiting CFTR activity (Hawkins et al., 2020). However, the rarer cell types of the epithelium such as the CFTR-expressing ionocytes, and brush cells, were not observed in vitro cultures of iBCs differentiated at air-liquid interface (Hawkins et al., 2020). Furthermore, the long-term effects of these manipulations are not well understood, and several questions regarding the in vivo competence of iBCs remain unanswered. For these reasons, development of additional screening assays similar to the dual fluorescent transporter assay used by Hawkins and colleagues will be necessary to verify the cellular phenotype and differentiation capacity of iBCs in comparison to their endogenous counterparts both in vitro and in vivo before clinical translation of iPSC-based CF cell therapy can be considered viable.
To improve the chances of successful cell transplantation to the lung, it is possible that tissue-resident adult stem cell from the lung itself would be a better suited candidate. For some time, there had been a lack of clarity as to the identity of airway stem cells. However, increased knowledge of stem cell biology together with the characterization of lung progenitor lineage has brought emerging consensus that basal cells are a stem cell or progenitor cell of the airway epithelium (Rock et al., 2009; Bertoncello, 2016). Importantly, basal cells have the capacity to differentiate into all the cell types of the pseudostratified airway epithelium, including the rare population of CFTR high-expressing ionocytes (Montoro et al., 2018; Plasschaert et al., 2018). This means that upon CFTR correction of basal cells, multi-lineage expression of the corrected CFTR could be achieved in the airway epithelium.
The existence of multiple progenitors with various differentiation capacities has been detected in human bronchial xenografts (Engelhardt et al., 1995). This is in concordance with reports that mouse tracheal basal cells comprise two molecularly distinct subpopulations of multipotent stem cells and committed secretory precursors (Hong et al., 2004; Watson et al., 2015). More recently, a single-cell RNA-seq study on human airway epithelial cells identified a heterogenous population of basal cells that include multipotent and secretory-primed subsets (Carraro et al., 2020). However, it remains unknown whether secretory-primed basal cells represent a transitory state of basal cells or a phenotypically stable state. The implication is that cell therapy approaches will have to account for basal cell subtypes when selecting a suitable cell to target.
Correcting CFTR in Regenerative Cells
Correction of CFTR in vitro in airway epithelial cells can be achieved by direct addition of genetic material as discussed in the sections above (Therapeutic Genetic Material Other Than DNA: RNA Addition and Repair and Therapeutic Delivery Vectors). An alternative to introducing new material is to correct the defective CFTR via gene editing.
Gene Editing Technologies
The advent of gene editing technologies has refueled excitement over gene therapy and is an area of vast development. The three major gene editing technologies are clustered regularly interspaced short palindromic repeat (CRISPR)–Cas-associated nucleases, programmable nucleases, such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). The first in vivo application of CRISPR/Cas9 was employed for a person with a mutation in the CEP290 gene which causes retinal degeneration (Ledford, 2020). Clinical trials are also underway to evaluate these therapeutic approaches for treatment of cancer and sickle cell disease (Ernst et al., 2020). In the context of CF, emerging gene editing technologies hold the potential to repair specific CFTR gene mutations and restore their function, offering the ultimate opportunity for precision medicine. However, to date, gene editing technologies for CF remain in the preclinical realm.
The first study of CRISPR/Cas9 as a potential therapy for CF used site-specific knock-in of the correct CFTR sequence to robustly restore CFTR function in human intestinal stem cell organoids derived from patients homozygous for the Phe508del mutation (Schwank et al., 2013). This approach has since been implemented in cell culture by Ruan and colleagues to achieve greater than 20% repair of patient-derived induced pluripotent stem cells (iPSCs) (Ruan et al., 2019). Furthermore, in recent advancements, an AAV-delivered Cas9 gene editing platform facilitated correction of >30% Phe508del-CFTR in patient-derived airway basal cells, prior to transplantation to an ex vivo engraftment scaffold and near-normal levels of CFTR function were restored (Vaidyanathan et al., 2020). Considering reports that suggest as little as 4.7% of WT CFTR expression can lead to a milder CF phenotype (Ramalho et al., 2002), prospects for clinical translation are promising. However, a shortcoming of these gene editing technologies is that they are mutation specific. As such, different mutations must be individually corrected, with some being more amenable to this strategy than others.
CRISPR/Cas9 has successfully corrected CFTR splicing mutations (Sanz et al., 2017) and mutations leading to the formation of a premature termination codon that produce no functional CFTR protein (Erwood et al., 2020). As such, an approach for allele-specific editing has been outlined. Gene editing using Zinc finger nucleases (ZFNs) has also facilitated the correction of iPSC-derived CF AECs (Crane et al., 2015). Crane and colleagues showed that CFTR-corrected iPSCs, following induced differentiation in vitro, expressed functional CFTR protein (Crane et al., 2015). A similar study used ZFNs to correct Phe508del and demonstrated restored CFTR protein expression and function in air-liquid interface cultures established from the edited basal cells (Suzuki et al., 2020). TALEN technologies have also been used to restore normal CFTR expression and activity in organoids derived from Phe508del patient-derived iPSCs (Fleischer et al., 2020). The ability to achieve CFTR correction in additional stem/progenitor cells such as airway basal cells, either as primary airway basal cells or those derived from iPSCs, has specific relevance for progress toward cell-based therapies for CF.
Expansion of CFTR-Corrected Regenerative Cells That Retain Differentiation Capacity
Irrespective of which regenerative cell type or in vitro correction method is to be used, cell-based therapy will require large numbers of viable cells to repopulate the lung by replacing the CFTR defective endogenous AECs. Hayes and colleagues have estimated that 60 million regenerative cells will be required to treat a human patient with cystic fibrosis cell therapy (Hayes et al., 2019). However, procurement of basal lung epithelial cells via bronchial lavage, sputum collection or endobronchial biopsy provides only low cell numbers (Mou et al., 2012; Pollock et al., 2013; Butler et al., 2016). To overcome the issue of low initial cell numbers, extensive cell expansion will be required prior to implantation.
Bronchial basal cells have a limited lifespan (Ghosh et al., 2011) with differentiation capacity that decreases over time in vitro (Gentzsch et al., 2017). Various culture protocols for cell expansion have been adapted to overcome these limitations (reviewed in (Awatade et al., 2018)). The conditionally reprogrammed cell (CRC) methodology (Liu et al., 2012) is one approach to increase bronchial cell yield (Martinovich et al., 2017) and maintain differentiation potential for multiple passages (Lee et al., 2020). CF and non-CF bronchial basal cells have been expanded to a therapeutic dose of 60 million cells via a modified CRC method, albeit the frequency of regenerative cells having decreased to 20% by the time the therapeutic dose was reached (Hayes et al., 2019). Importantly, the bronchial basal cells were demonstrated to retain their differentiation potential from passages 2 to 15 using this approach (Hayes et al., 2019). Yet, further concern is raised by reports of in vitro basal cell expansion altering CFTR functional activity (Awatade et al., 2021) and differentiation cell-fate composition, leading to a reduced number of ciliated cells in expanded cells compared to freshly isolated cells (Eenjes et al., 2018). If expanded basal cells differentiate to pseudostratified epithelium with an altered cell type composition, this would have implications for their suitability for cell therapy, therefore necessitating further investigation.
Another challenge faced by cell expansion for the purpose of CF cell-based therapy is reports that differentiated cultures established from extended in vitro expansion of basal cells often have decreased CFTR ion transport function (Peters-Hall et al., 2018). Lee and colleagues report that although CRC expanded cells have increased CFTR ion transport compared to conventionally expanded cells, short-circuit current (a proxy for CFTR function) still decreased by passage three in CRC expanded cells (Lee et al., 2020). A better understanding of the impact of extended cell expansion on CFTR function is required.
Transplantation of CFTR-Corrected Regenerative Cells
Basal cells are located within the surface epithelium, adjacent to the basement membrane, and their differentiation leads to the cellular diversity of the airway epithelium. As such, if CFTR-corrected basal cells are to be the basis of a cell therapy, then they would need to be engrafted onto the basement membrane of the airway epithelium. This should replenish the airways with CFTR-corrected ciliated and secretory cells, and ionocytes. Meanwhile, the engrafted basal cells, corrected by integrating CFTR addition or mutation-specific gene editing, would retain their capacity for self-renewal to establish a long-term corrected cell population.
Studies of hematopoietic stem cell transplantation show that transplanted cells compete with endogenous bone marrow stroma cells (Abbuehl et al., 2017). Similar concerns have been raised for competition between endogenous and transplanted cells used to repair the lung epithelium. In a competitive repopulation assay, a mixed culture of CF and healthy (non-CF) basal cells were differentiated at air-liquid interface to generate a pseudostratified airway epithelium in vitro (Lee et al., 2020). Lee and colleagues found that non-CF cells outcompeted CF donor cells, suggesting that endogenous airway epithelial repair and regeneration is likely to hinder cell engraftment. However, more work is required to fully comprehend and address factors such as these which may impact stable integration of transplanted cells.
The innate barrier properties of the CF airway epithelium–the thick mucus layer and the ciliated pseudostratified multi-layered nature of the epithelial tissue–will likely make the delivery of the corrected basal cells to the basement membrane difficult. An effective cell delivery method will need to first overcome the mucus layer covering the epithelial surface. Whilst aerosolization of stem cell MSCs has demonstrated feasibility as a new method of cell delivery in vivo in rabbit airways (Kardia et al., 2014; Kardia et al., 2018; Halim et al., 2019), this technique has not been trialed using basal stem cells. Most studies of basal cell engraftment delivered via intratracheal instillation (Ghosh et al., 2017) or injection (Millerl et al., 2018) use rodent models (Ghosh et al., 2017; Miller et al., 2018). However, these animal models are limited in their capacity to accurately recreate human CF airway pathogenesis. Evidently further testing in appropriate in vivo animal models is required to determine an effective approach for cell delivery capable of overcoming the altered mucus barrier in CF. Yet, even if an optimal method is achieved, it is anticipated that disruption of the epithelial cell layer via conditioning or transient injury will be necessary to facilitate effective cell transplantation. Similar strategies have been successful for conditioning of the bone marrow prior to haemopoietic stem cell transplantation (Gyurkocza and Sandmaier, 2014).
Experimental evidence for successful donor cell engraftment in the lungs of animal models has been shown with various injury protocols. Following conditioning with naphthalene, both iPSCs (Miller et al., 2018) and airway basal cells (Ghosh et al., 2017) successfully transplant into mouse airways and persist for up to six weeks. Comparatively, partially stripping the AECs by treatment with polidocanol, an agent that creates a larger site for donor cell engraftment than naphthalene, has enabled basal cells to engraft into live mouse airways and the transplanted cells remained viable for at least three weeks (Farrow et al., 2018a). The long-term effect of these conditioning protocols upon cell engraftment is yet to be examined. In the context of CF, no ideal strategy has yet been developed. To injure the already inflamed and infected lungs of CF recipients might appear counterintuitive. The question remains: what is the optimal conditioning scenario that will cause minimal injury, whilst facilitating effective engraftment of corrected cells in sufficient numbers to restore lung function?
Conclusion
There are clear challenges to the successful translation of gene and cell therapies designed to correct the CFTR defect in the airways. Various novel viral and non-viral therapeutic vectors have advanced to clinical trials; however, no significant clinical benefit has been achieved. The greatest barrier to the success of these gene therapeutics is their delivery to the human airways. Overcoming this barrier will be central to ongoing research and paramount to the achievement of an efficacious in vivo CF gene therapy.
Cell-based therapies represent a promising alternative strategy wherein CFTR is corrected ex vivo. Yet, questions remain unanswered. Even if regenerative basal cells that are corrected via integrating CFTR addition or mutation-specific gene editing can efficaciously transplant at the basement membrane of the airway epithelium, how safe are these ex vivo-corrected cells? We know that reprogramming, expansion, and editing increases the probability of tumorigenicity (reviewed in (Berical et al., 2019)). Can basal cells only be expanded to the necessary therapeutic dose at the cost of preserving the transcriptome, epigenome, and differentiation capacity of these basal cells? Long-term investigations are needed to confirm transplanted basal cells are free from mutations, that they are stably engrafted and that CFTR function is retained. Future studies to investigate and improve the findings discussed in this review are required to validate the feasibility of cell-based therapy for treatment of CF.
Moreover, challenges will likely be met in transitioning gene- and cell-based therapies to clinical care. Discussions of the difficulties in developing and sustaining a successful business model for cell-based therapies, and the changes in clinical care necessary to make this potentially transformative therapeutic approach accessible to patients, are ongoing (Elverum and Whitman, 2019). As we continue to forge ahead in this era of personalized medicine, improvements to current CFTR modulator drugs will likely herald increased patient benefit. However, clinical translation of gene or cell-based therapies, though still an ambitious goal, offers future promise of a mutation-agnostic cure for CF.
Author Contributions
SW was responsible for conception and design. KA wrote the initial draft, in consultation with and edited by SW, NF, and MD, with critical input from AJ. All authors approved the final manuscript.
Funding
KA was supported by an Australian Government Research Training Program Scholarship. SW gratefully acknowledges support from National Health and Medical Research Council (NHMRC) Australia (GNT1188987), CF Foundation Australia and Sydney Children’s Hospital Foundation. NF was supported by a MS McLeod Postdoctoral Fellowship. MD was supported in part by the NHMRC (GNT1160011), and Cystic Fibrosis Foundation (PARSON18GO).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AAV, adeno-associated virus; AAV2, adeno-associated virus serotype 2; AAV6, adeno-associated virus serotype 6; AECs, airway epithelial cells; ASO, antisense oligonucleotide; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; CRISPR, clustered regularly interspaced short palindromic repeat; CRC, conditionally reprogrammed cell; iPSCs, induced pluripotent stem cells; IV, immunodeficiency virus; LNPs, lipid nanoparticles; LV, lentivirus vector; mRNA, messenger RNA; MSCs, mesenchymal stem cells; NMD, nonsense mediated decay; TALENs, transcription activator-like effector nucleases; ZFNs, zinc-finger nucleases
References
Abbuehl, J.-P., Tatarova, Z., Held, W., and Huelsken, J. (2017). Long-term engraftment of primary bone marrow stromal cells repairs niche damage and improves hematopoietic stem cell transplantation. Cell Stem Cell 21 (2), 241–255. doi:10.1016/j.stem.2017.07.004 |
Akbarzadeh, A., Rezaei-Sadabady, R., Davaran, S., Joo, S. W., Zarghami, N., Hanifehpour, Y., et al. (2013). Liposome: classification, preparation, and applications. Nanoscale Res. Lett. 8 (1), 102. doi:10.1186/1556-276x-8-102 |
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413 (6857), 732–738. doi:10.1038/35099560 |
Almeida-Porada, G., Atala, A. J., and Porada, C. D. (2020). Therapeutic mesenchymal stromal cells for immunotherapy and for gene and drug delivery. Mol. Ther.—Methods Clin. Develop. 16, 204–224. doi:10.1016/j.omtm.2020.01.005
Alton, E. W. F. W., Armstrong, D. K., Ashby, D., Bayfield, K. J., Bilton, D., Bloomfield, E. V., et al. (2015a). Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 3 (9), 684–691. doi:10.1016/s2213-2600(15)00245-3 |
Alton, E. W. F. W., Beekman, J. M., Boyd, A. C., Brand, J., Carlon, M. S., Connolly, M. M., et al. (2017). Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 72 (2), 137–147. doi:10.1136/thoraxjnl-2016-208406 |
Alton, E. W. F. W., Boyd, A. C., Porteous, D. J., Davies, G., Davies, J. C., Griesenbach, U., et al. (2015b). A phase I/IIa safety and efficacy study of nebulized liposome-mediated gene therapy for cystic fibrosis supports a multidose trial. Am. J. Respir. Crit. Care Med. 192 (11), 1389–1392. doi:10.1164/rccm.201506-1193le |
Awatade, N. T., Wong, S. L., Capraro, A., Pandzic, E., Slapetova, I., Zhong, L., et al. (2021). Significant functional differences in differentiated Conditionally Reprogrammed (CRC)- and Feeder-free Dual SMAD inhibited-expanded human nasal epithelial cells. J. Cyst. Fibros. S1569-1993(20), 30951–30956. doi:10.1016/j.jcf.2020.12.019
Awatade, N. T., Wong, S. L., Hewson, C. K., Fawcett, L. K., Kicic, A., Jaffe, A., et al. (2018). Human primary epithelial cell models: promising tools in the era of cystic fibrosis personalized medicine. Front. Pharmacol. 9, 1429. doi:10.3389/fphar.2018.01429 |
Batty, P., and Lillicrap, D. (2019). Advances and challenges for hemophilia gene therapy. Hum. Mol. Genet. 28 (R1), R95–R101. doi:10.1093/hmg/ddz157 |
Bennett, C. F., Baker, B. F., Pham, N., Swayze, E., and Geary, R. S. (2017). Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 57, 81–105. doi:10.1146/annurev-pharmtox-010716-104846 |
Berical, A., Lee, R. E., Randell, S. H., and Hawkins, F. (2019). Challenges facing airway epithelial cell-based therapy for cystic fibrosis. Front. Pharmacol. 10, 74. doi:10.3389/fphar.2019.00074 |
Bertoncello, I. (2016). Properties of adult lung stem and progenitor cells. J. Cel. Physiol. 231 (12), 2582–2589. doi:10.1002/jcp.25404
Beumer, W., Swildens, J., Leal, T., Noel, S., Anthonijsz, H., van der Horst, G., et al. (2019). Evaluation of eluforsen, a novel RNA oligonucleotide for restoration of CFTR function in in vitro and murine models of p.Phe508del cystic fibrosis. PLoS One 14 (6), e0219182. doi:10.1371/journal.pone.0219182 |
Booth, C., Romano, R., Roncarolo, M. G., and Thrasher, A. J. (2019). Gene therapy for primary immunodeficiency. Hum. Mol. Genet. 28 (R1), R15–R23. doi:10.1093/hmg/ddz170 |
Butler, C. R., Hynds, R. E., Gowers, K. H. C., Lee, D. D. H., Brown, J. M., Crowley, C., et al. (2016). Rapid expansion of human epithelial stem cells suitable for airway tissue engineering. Am. J. Respir. Crit. Care Med. 194 (2), 156–168. doi:10.1164/rccm.201507-1414oc |
Carbone, A., Castellani, S., Favia, M., Diana, A., Paracchini, V., Di Gioia, S., et al. (2014). Correction of defective CFTR/EN aC function and tightness of cystic fibrosis airway epithelium by amniotic mesenchymal stromal (stem) cells. J. Cel. Mol. Med. 18 (8), 1631–1643. doi:10.1111/jcmm.12303
Carbone, A., Zefferino, R., Beccia, E., Casavola, V., Castellani, S., Di Gioia, S., et al. (2018). Gap junctions are involved in the rescue of CFTR-dependent chloride efflux by amniotic mesenchymal stem cells in coculture with cystic fibrosis CFBE41o- cells. Stem Cell Int 2018, 1203717. doi:10.1155/2018/1203717 |
Caretti, A., Peli, V., Colombo, M., and Zulueta, A. (2019). Lights and shadows in the use of mesenchymal stem cells in lung inflammation, a poorly investigated topic in cystic fibrosis. Cells 9 (1), 20. doi:10.3390/cells9010020
Carraro, G., Mulay, A., Yao, C., Mizuno, T., Konda, B., Petrov, M., et al. (2020). Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am. J. Respir. Crit. Care Med. 202 (11), 1540–1550. doi:10.1164/rccm.201904-0792oc |
Castellani, S., and Conese, M. (2010). Lentiviral vectors and cystic fibrosis gene therapy. Viruses 2 (2), 395–412. doi:10.3390/v2020395 |
Cooney, A. L., Abou Alaiwa, M. H., Shah, V. S., Bouzek, D. C., Stroik, M. R., Powers, L. S., et al. (2016). Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 1 (14), e88730. doi:10.1172/jci.insight.88730
Cooney, A. L., McCray, P. B., and Sinn, P. L. (2018). Cystic fibrosis gene therapy: looking back, looking forward. Genes (Basel) 9 (11), 538. doi:10.3390/genes9110538
Cooney, A. L., Singh, B. K., and Sinn, P. L. (2015). Hybrid nonviral/viral vector systems for improved piggyBac DNA transposon in vivo delivery. Mol. Ther. 23 (4), 667–674. doi:10.1038/mt.2014.254 |
Cooney, A. L., Thornell, I. M., Singh, B. K., Shah, V. S., Stoltz, D. A., McCray, P. B., et al. (2019). A novel AAV-mediated gene delivery system corrects CFTR function in pigs. Am. J. Respir. Cel. Mol. Biol. 61 (6), 747–754. doi:10.1165/rcmb.2019-0006oc
Crane, A. M., Kramer, P., Bui, J. H., Chung, W. J., Li, X. S., Gonzalez-Garay, M. L., et al. (2015). Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cel. Rep. 4 (4), 569–577. doi:10.1016/j.stemcr.2015.02.005
Davies, J. C., Moskowitz, S. M., Brown, C., Horsley, A., Mall, M. A., McKone, E. F., et al. (2018). VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 379 (17), 1599–1611. doi:10.1056/nejmoa1807119 |
Davies, J. C. (2015). The future of CFTR modulating therapies for cystic fibrosis. Curr. Opin. Pulm. Med. 21 (6), 579–584. doi:10.1097/mcp.0000000000000211 |
Delhove, J., Osenk, I., Prichard, I., and Donnelley, M. (2020). Public acceptability of gene therapy and gene editing for human use: a systematic review. Hum. Gene Ther. 31 (1-2), 20–46. doi:10.1089/hum.2019.197 |
Donnelley, M., and Parsons, D. W. (2018). Gene therapy for cystic fibrosis lung disease: overcoming the barriers to translation to the clinic. Front. Pharmacol. 9, 1381. doi:10.3389/fphar.2018.01381 |
Doyle, L. M., and Wang, M. Z. (2019). Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 8 (7), 727. doi:10.3390/cells8070727
Duncan, G. A., Kim, N., Colon-Cortes, Y., Rodriguez, J., Mazur, M., Birket, S. E., et al. (2018). An adeno-associated viral vector capable of penetrating the mucus barrier to inhaled gene therapy. Mol. Ther.—Methods Clin. Develop. 9, 296–304. doi:10.1016/j.omtm.2018.03.006
Dye, B. R., Hill, D. R., Ferguson, M. A., Tsai, Y. H., Nagy, M. S., Dyal, R., et al. (2015). In vitro generation of human pluripotent stem cell derived lung organoids. Elife 4, e05098. doi:10.7554/elife.05098
Eenjes, E., Mertens, T. C. J., Buscop-van Kempen, M. J., van Wijck, Y., Taube, C., Rottier, R. J., et al. (2018). A novel method for expansion and differentiation of mouse tracheal epithelial cells in culture. Sci. Rep. 8 (1), 7349. doi:10.1038/s41598-018-25799-6 |
Eichstadt, S., Barriga, M., Ponakala, A., Teng, C., Nguyen, N. T., Siprashvili, Z., et al. (2019). Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 4 (19), e130554. doi:10.1172/jci.insight.130554
Elborn, J. S. (2016). Cystic fibrosis. The Lancet 388 (10059), 2519–2531. doi:10.1016/s0140-6736(16)00576-6
Elverum, K., and Whitman, M. (2019). Delivering cellular and gene therapies to patients: solutions for realizing the potential of the next generation of medicine. Gene Ther. 27, 537–544. doi:10.1038/s41434-019-0074-7 |
Engelhardt, J. F., Schlossberg, H., Yankaskas, J. R., and Dudus, L. (1995). Progenitor cells of the adult human airway involved in submucosal gland development. Development 121 (7), 2031–2046.
Ernst, M. P. T., Broeders, M., Herrero-Hernandez, P., Oussoren, E., van der Ploeg, A. T., and Pijnappel, W. W. M. P. (2020). Ready for repair? Gene editing enters the clinic for the treatment of human disease. Mol. Ther.—Methods Clin. Develop. 18, 532–557. doi:10.1016/j.omtm.2020.06.022
Erwood, S., Laselva, O., Bily, T. M. I., Brewer, R. A., Rutherford, A. H., Bear, C. E., et al. (2020). Allele-specific prevention of nonsense-mediated decay in cystic fibrosis using homology-independent genome editing. Mol. Ther.—Methods Clin. Develop. 17, 1118–1128. doi:10.1016/j.omtm.2020.05.002
Farrow, N., Cmielewski, P., Donnelley, M., Rout-Pitt, N., Moodley, Y., Bertoncello, I., et al. (2018a). Epithelial disruption: a new paradigm enabling human airway stem cell transplantation. Stem Cell Res Ther. 9 (1), 153. doi:10.1186/s13287-018-0911-4 |
Farrow, N., Donnelley, M., Cmielewski, P., Roscioli, E., Rout-Pitt, N., McIntyre, C., et al. (2018b). Role of basal cells in producing persistent lentivirus-mediated airway gene expression. Hum. Gene Ther. 29 (6), 653–662. doi:10.1089/hum.2017.059 |
Firth, A. L., Dargitz, C. T., Qualls, S. J., Menon, T., Wright, R., Singer, O., et al. (2014). Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. 111 (17), E1723–E1730. doi:10.1073/pnas.1403470111 |
Fleischer, A., Vallejo-Díez, S., Martín-Fernández, J. M., Sánchez-Gilabert, A., Castresana, M., Del Pozo, A., et al. (2020). iPSC-Derived Intestinal Organoids from Cystic Fibrosis Patients Acquire CFTR Activity upon TALEN-Mediated Repair of the p.F508del mutation. Mol. Ther.—Methods Clin. Develop. 17, 858–870. doi:10.1016/j.omtm.2020.04.005
Flotte, T. R., Zeitlin, P. L., Reynolds, T. C., Heald, A. E., Pedersen, P., Beck, S., et al. (2003). Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a two-part clinical study. Hum. Gene Ther. 14 (11), 1079–1088. doi:10.1089/104303403322124792 |
Flume, P. A., O’Sullivan, B. P., Robinson, K. A., Goss, C. H., Mogayzel, P. J., Willey-Courand, D. B., et al. (2007). Cystic fibrosis pulmonary guidelines. Am. J. Respir. Crit. Care Med. 176 (10), 957–969. doi:10.1164/rccm.200705-664oc |
Gentzsch, M., Boyles, S. E., Cheluvaraju, C., Chaudhry, I. G., Quinney, N. L., Cho, C., et al. (2017). Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 56 (5), 568–574. doi:10.1165/rcmb.2016-0276ma |
George, L. A., Ragni, M. V., Rasko, J. E. J., Raffini, L. J., Samelson-Jones, B. J., Ozelo, M., et al. (2020). Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol. Ther. 28 (9), 2073–2082. doi:10.1016/j.ymthe.2020.06.001 |
Ghosh, M., Ahmad, S., White, C. W., and Reynolds, S. D. (2017). Transplantation of airway epithelial stem/progenitor cells: a future for cell-based therapy. Am. J. Respir. Cell. Mol. Biol. 56 (1), 1–10. doi:10.1165/rcmb.2016-0181ma |
Ghosh, M., Helm, K. M., Smith, R. W., Giordanengo, M. S., Li, B., Shen, H., et al. (2011). A single cell functions as a tissue-specific stem cell and theIn VitroNiche-forming cell. Am. J. Respir. Cell Mol. Biol. 45 (3), 459–469. doi:10.1165/rcmb.2010-0314oc |
Gonzalez, G., Vituret, C., Di Pietro, A., Chanson, M., Boulanger, P., and Hong, S. S. (2012). Microparticle-mediated transfer of the viral receptors CAR and CD46, and the CFTR channel in a CHO cell model confers new functions to target cells. PLoS One 7 (12), e52326. doi:10.1371/journal.pone.0052326 |
Gratwohl, A., and Niederwieser, D. (2012). History of hematopoietic stem cell transplantation: evolution and perspectives. Curr. Probl. Dermatol. 43, 81–90. doi:10.1159/000335266 |
Griesenbach, U., Kitson, C., Escudero Garcia, S., Farley, R., Singh, C., Somerton, L., et al. (2006). Inefficient cationic lipid-mediated siRNA and antisense oligonucleotide transfer to airway epithelial cells in vivo. Respir. Res. 7, 26. doi:10.1186/1465-9921-7-26 |
Griesenbach, U., Pytel, K. M., and Alton, E. W. F. W. (2015). Cystic fibrosis gene therapy in the UK and elsewhere. Hum. Gene Ther. 26 (5), 266–275. doi:10.1089/hum.2015.027 |
Gyurkocza, B., and Sandmaier, B. M. (2014). Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood 124 (3), 344–353. doi:10.1182/blood-2014-02-514778 |
Hajj, K. A., and Whitehead, K. A. (2017). Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2 (10), 17056. doi:10.1038/natrevmats.2017.56
Halim, N. S. S., Ch’ng, E. S., Kardia, E., Ali, S. A., Radzi, R., and Yahaya, B. H. (2019). Aerosolised mesenchymal stem cells expressing angiopoietin-1 enhances airway repair. Stem Cel. Rev. Rep. 15 (1), 112–125. doi:10.1007/s12015-018-9844-7
Havenga, M. J. E., Lemckert, A. A. C., Ophorst, O. J. A. E., van Meijer, M., Germeraad, W. T. V., Grimbergen, J., et al. (2002). Exploiting the natural diversity in adenovirus tropism for therapy and prevention of disease. J. virol. 76 (9), 4612–4620. doi:10.1128/jvi.76.9.4612-4620.2002 |
Hawkins, F. J., Suzuki, S., Beermann, M. L., Barilla, C., Wang, R., Villacorta-Martin, C., et al. (2020). Derivation of airway basal stem cells from human pluripotent stem cells. Cell Stem Cell 28, 79–95. doi:10.1016/j.stem.2020.09.017 |
Hayes, D., Kopp, B. T., Hill, C. L., Lallier, S. W., Schwartz, C. M., Tadesse, M., et al. (2019). Cell therapy for cystic fibrosis lung disease: regenerative basal cell amplification. Stem Cells Translational Med. 8 (3), 225–235. doi:10.1002/sctm.18-0098 |
Hong, K. U., Reynolds, S. D., Watkins, S., Fuchs, E., and Stripp, B. R. (2004). In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am. J. Physiology-Lung Cell Mol. Physiol. 286 (4), L643–L649. doi:10.1152/ajplung.00155.2003
Huang, S. X. L., Islam, M. N., O’Neill, J., Hu, Z., Yang, Y.-G., Chen, Y.-W., et al. (2014). Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 32 (1), 84–91. doi:10.1038/nbt.2754 |
Igreja, S., Clarke, L. A., Botelho, H. M., Marques, L., and Amaral, M. D. (2016). Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat. 37 (2), 209–215. doi:10.1002/humu.22931 |
Jaffe, A., Prasad, S. A., Larcher, V., and Hart, S. (2006). Gene therapy for children with cystic fibrosis--who has the right to choose?. J. Med. Ethics 32 (6), 361–364. doi:10.1136/jme.2005.012740 |
Kardia, E., Ch’ng, E. S., and Yahaya, B. H. (2018). Aerosol‐based airway epithelial cell delivery improves airway regeneration and repair. J. Tissue Eng. Regen. Med. 12 (2), e995–e1007. doi:10.1002/term.2421 |
Kardia, E., Yusoff, N. M., Zakaria, Z., and Yahaya, B. (2014). Aerosol-based delivery of fibroblast cells for treatment of lung diseases. J. Aerosol. Med. Pulm. Drug Deliv. 27 (1), 30–34. doi:10.1089/jamp.2012.1020 |
Karikó, K., Ni, H., Capodici, J., Lamphier, M., and Weissman, D. (2004). mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279 (13), 12542–12550. doi:10.1074/jbc.m310175200 |
Kariyawasam, D., Carey, K. A., Jones, K. J., and Farrar, M. A. (2018). New and developing therapies in spinal muscular atrophy. Paediatric Respir. Rev. 28, 3–10. doi:10.1016/j.prrv.2018.03.003
Konishi, S., Gotoh, S., Tateishi, K., Yamamoto, Y., Korogi, Y., Nagasaki, T., et al. (2016). Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 6 (1), 18–25. doi:10.1016/j.stemcr.2015.11.010
Kraft, J. C., Freeling, J. P., Wang, Z., and Ho, R. J. Y. (2014). Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 103 (1), 29–52. doi:10.1002/jps.23773 |
Kuhn, A. N., Beiβert, T., Simon, P., Vallazza, B., Buck, J., Davies, B. P., et al. (2012). mRNA as a versatile tool for exogenous protein expression. Curr. Gene Ther. 12 (5), 347–361. doi:10.2174/156652312802762536 |
Kumagai, N., LaMattina, J. C., Kamano, C., Vagefi, P. A., Barth, R. N., O’Neil, J. J., et al. (2002). Vascularized islet cell transplantation in miniature Swine: islet-kidney allografts correct the diabetic hyperglycemia induced by total pancreatectomy. Diabetes 51 (11), 3220–3228. doi:10.2337/diabetes.51.11.3220 |
Leborgne, C., Barbon, E., Alexander, J. M., Hanby, H., Delignat, S., Cohen, D. M., et al. (2020). IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 26 (7), 1096–1101. doi:10.1038/s41591-020-0911-7 |
Ledford, H. (2020). CRISPR treatment inserted directly into the body for first time. Nature 579 (7798), 185. doi:10.1038/d41586-020-00655-8 |
Lee, R. E., Miller, S. M., Mascenik, T. M., Lewis, C. A., Dang, H., Boggs, Z. H., et al. (2020). Assessing human airway epithelial progenitor cells for cystic fibrosis cell therapy. Am. J. Respir. Cell Mol. Biol. 63 (3), 374–385. doi:10.1165/rcmb.2019-0384oc |
Limberis, M., Anson, D. S., Fuller, M., and Parsons, D. W. (2002). Recovery of airway cystic fibrosis transmembrane conductance regulator function in mice with cystic fibrosis after single-dose lentivirus-mediated gene transfer. Hum. Gene Ther. 13 (16), 1961–1970. doi:10.1089/10430340260355365 |
Liu, X., Ory, V., Chapman, S., Yuan, H., Albanese, C., Kallakury, B., et al. (2012). ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 180 (2), 599–607. doi:10.1016/j.ajpath.2011.10.036 |
Lwin, S. M., Syed, F., Di, W. L., Kadiyirire, T., Liu, L., Guy, A., et al. (2019). Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight 4 (11), e126243. doi:10.1172/jci.insight.126243
Martinovich, K. M., Iosifidis, T., Buckley, A. G., Looi, K., Ling, K. M., Sutanto, E. N., et al. (2017). Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci. Rep. 7 (1), 17971. doi:10.1038/s41598-017-17952-4 |
Marinkovich, M. P., and Tang, J. Y. (2019). Gene therapy for epidermolysis bullosa. J. Invest. Dermatol. 139 (6), 1221–1226. doi:10.1016/j.jid.2018.11.036 |
McCauley, K. B., Hawkins, F., Serra, M., Thomas, D. C., Jacob, A., and Kotton, D. N. (2017). Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of wnt signaling. Cell Stem Cell 20 (6), 844–857. doi:10.1016/j.stem.2017.03.001 |
Meyer-Berg, H., Zhou Yang, L., Pilar de Lucas, M., Zambrano, A., Hyde, S. C., and Gill, D. R. (2020). Identification of AAV serotypes for lung gene therapy in human embryonic stem cell-derived lung organoids. Stem Cell Res. Ther. 11 (1), 448. doi:10.1186/s13287-020-01950-x |
Miah, K. M., Hyde, S. C., and Gill, D. R. (2019). Emerging gene therapies for cystic fibrosis. Expert Rev. Respir. Med. 13 (8), 709–725. doi:10.1080/17476348.2019.1634547 |
Miller, A. J., Hill, D. R., Nagy, M. S., Aoki, Y., Dye, B. R., Chin, A. M., et al. (2018). In Vitro induction and in vivo engraftment of lung bud tip progenitor cells derived from human pluripotent stem cells. Stem Cell Rep. 10 (1), 101–119. doi:10.1016/j.stemcr.2017.11.012
Montoro, D. T., Haber, A. L., Biton, M., Vinarsky, V., Lin, B., Birket, S. E., et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560 (7718), 319–324. doi:10.1038/s41586-018-0393-7 |
Moss, R. B., Rodman, D., Spencer, L. T., Aitken, M. L., Zeitlin, P. L., Waltz, D., et al. (2004). Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis. Chest 125 (2), 509–521. doi:10.1378/chest.125.2.509 |
Moss, R. B., Milla, C., Colombo, J., Accurso, F., Zeitlin, P. L., Clancy, J. P., et al. (2007). Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 18 (8), 726–732. doi:10.1089/hum.2007.022 |
Mou, H., Zhao, R., Sherwood, R., Ahfeldt, T., Lapey, A., Wain, J., et al. (2012). Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell 10 (4), 385–397. doi:10.1016/j.stem.2012.01.018 |
Murphy, D. E., de Jong, O. G., Brouwer, M., Wood, M. J., Lavieu, G., Schiffelers, R. M., et al. (2019). Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Exp. Mol. Med. 51 (3), 1–12. doi:10.1038/s12276-019-0223-5
Nafee, N., Forier, K., Braeckmans, K., and Schneider, M. (2018). Mucus-penetrating solid lipid nanoparticles for the treatment of cystic fibrosis: proof of concept, challenges and pitfalls. Eur. J. Pharmaceutics Biopharmaceutics 124, 125–137. doi:10.1016/j.ejpb.2017.12.017
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272 (5259), 263–267. doi:10.1126/science.272.5259.263 |
Ni, R., Feng, R., and Chau, Y. (2019). Synthetic approaches for nucleic acid delivery: choosing the right carriers. Life (Basel) 9 (3), 59. doi:10.3390/life9030059
Oren, Y. S., Pranke, I. M., Kerem, B., and Sermet-Gaudelus, I. (2017). The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr. Opin. Pharmacol. 34, 125–131. doi:10.1016/j.coph.2017.09.017 |
Ortiz, L. A., Gambelli, F., McBride, C., Gaupp, D., Baddoo, M., Kaminski, N., et al. (2003). Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Pnas 100 (14), 8407–8411. doi:10.1073/pnas.1432929100 |
Pardi, N., Tuyishime, S., Muramatsu, H., Kariko, K., Mui, B. L., Tam, Y. K., et al. (2015). Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Release 217, 345–351. doi:10.1016/j.jconrel.2015.08.007
Patel, A., Zhao, J., Duan, D., and Lai, Y. (2019a). Design of AAV vectors for delivery of large or multiple transgenes. Methods Mol. Biol. 1950, 19–33. doi:10.1007/978-1-4939-9139-6_2
Patel, A. K., Kaczmarek, J. C., Bose, S., Kauffman, K. J., Mir, F., Heartlein, M. W., et al. (2019b). Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv. Mater. 31 (8), e1805116. doi:10.1002/adma.201970053 |
Peters-Hall, J. R., Coquelin, M. L., Torres, M. J., LaRanger, R., Alabi, B. R., Sho, S., et al. (2018). Long-term culture and cloning of primary human bronchial basal cells that maintain multipotent differentiation capacity and CFTR channel function. Am. J. Physiology-Lung Cell Mol. Physiol. 315 (2), L313–L327. doi:10.1152/ajplung.00355.2017
Plasschaert, L. W., Žilionis, R., Choo-Wing, R., Savova, V., Knehr, J., Roma, G., et al. (2018). A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560 (7718), 377–381. doi:10.1038/s41586-018-0394-6 |
Pollock, K., Albares, L., Wendt, C., and Hubel, A. (2013). Isolation of fibroblasts and epithelial cells in bronchoalveolar lavage (BAL). Exp. Lung Res. 39 (3), 146–154. doi:10.3109/01902148.2013.781720 |
Ramalho, A. S., Beck, S., Meyer, M., Penque, D., Cutting, G. R., and Amaral, M. D. (2002). Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cel. Mol. Biol. 27 (5), 619–627. doi:10.1165/rcmb.2001-0004oc
Ratjen, F., Bell, S. C., Rowe, S. M., Goss, C. H., Quittner, A. L., and Bush, A. (2015). Cystic fibrosis. Nat. Rev. Dis. Primers 1, 15010. doi:10.1038/nrdp.2015.10 |
Riordan, J. R. (2008). CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726. doi:10.1146/annurev.biochem.75.103004.142532 |
Robinson, E., MacDonald, K. D., Slaughter, K., McKinney, M., Patel, S., Sun, C., et al. (2018). Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 26 (8), 2034–2046. doi:10.1016/j.ymthe.2018.05.014 |
Rock, J. R., Onaitis, M. W., Rawlins, E. L., Lu, Y., Clark, C. P., Xue, Y., et al. (2009). Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. 106 (31), 12771–12775. doi:10.1073/pnas.0906850106 |
Ronzitti, G., Gross, D. A., and Mingozzi, F. (2020). Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 11, 670. doi:10.3389/fimmu.2020.00670 |
Rowe, S. M., Miller, S., and Sorscher, E. J. (2005). Cystic fibrosis. N. Engl. J. Med. 352 (19), 1992–2001. doi:10.1056/nejmra043184 |
Ruan, J., Hirai, H., Yang, D., Ma, L., Hou, X., Jiang, H., et al. (2019). Efficient gene editing at major CFTR mutation loci. Mol. Ther.—Nucleic Acids 16, 73–81. doi:10.1016/j.omtn.2019.02.006
Sahay, G., Querbes, W., Alabi, C., Eltoukhy, A., Sarkar, S., Zurenko, C., et al. (2013). Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 31 (7), 653–658. doi:10.1038/nbt.2614 |
Sahin, U., Türeci, K. Ö., and Tureci, O. (2014). mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13 (10), 759–780. doi:10.1038/nrd4278 |
Sanz, D. J., Hollywood, J. A., Scallan, M. F., and Harrison, P. T. (2017). Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One 12 (9), e0184009. doi:10.1371/journal.pone.0184009 |
Schwank, G., Koo, B.-K., Sasselli, V., Dekkers, J. F., Heo, I., Demircan, T., et al. (2013). Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13 (6), 653–658. doi:10.1016/j.stem.2013.11.002 |
Sermet-Gaudelus, I., Clancy, J. P., Nichols, D. P., Nick, J. A., De Boeck, K., Solomon, G. M., et al. (2019). Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J. Cystic Fibrosis 18 (4), 536–542. doi:10.1016/j.jcf.2018.10.015
Singh, B. K., Cooney, A. L., Krishnamurthy, S., and Sinn, P. L. (2020). Extracellular vesicle-mediated siRNA delivery, protein delivery, and CFTR complementation in well-differentiated human airway epithelial cells. Genes (Basel) 11 (4), 351. doi:10.3390/genes11040351
Smyth, T., Kullberg, M., Malik, N., Smith-Jones, P., Graner, M. W., and Anchordoquy, T. J. (2015). Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Controlled Release 199, 145–155. doi:10.1016/j.jconrel.2014.12.013
Stadtmauer, E. A., Fraietta, J. A., Davis, M. M., Cohen, A. D., Weber, K. L., Lancaster, E., et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367 (6481), eaba7365. doi:10.1126/science.aba7365 |
Stirnadel-Farrant, H., Kudari, M., Garman, N., Imrie, J., Chopra, B., Giannelli, S., et al. (2018). Gene therapy in rare diseases: the benefits and challenges of developing a patient-centric registry for Strimvelis in ADA-SCID. Orphanet. J. Rare Dis. 13 (1), 49. doi:10.1186/s13023-018-0791-9 |
Suzuki, S., Crane, A. M., Anirudhan, V., Barillà, C., Matthias, N., Randell, S. H., et al. (2020). Highly efficient gene editing of cystic fibrosis patient-derived airway basal cells results in functional CFTR correction. Mol. Ther. 28 (7), 1684–1695. doi:10.1016/j.ymthe.2020.04.021 |
Takahashi, Y., Nishikawa, M., Shinotsuka, H., Matsui, Y., Ohara, S., Imai, T., et al. (2013). Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 165 (2), 77–84. doi:10.1016/j.jbiotec.2013.03.013 |
Tsui, L., Buchwald, M., Barker, D., Braman, J., Knowlton, R., Schumm, J., et al. (1985). Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 230 (4729), 1054–1057. doi:10.1126/science.2997931 |
U.S. Food and Drug Administration (2020). Approved cellular and gene therapy products Retrieved from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products ( (October 21, 2020)
Vaidyanathan, S., Salahudeen, A. A., Sellers, Z. M., Bravo, D. T., Choi, S. S., Batish, A., et al. (2020). High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell 26 (2), 161–171. doi:10.1016/j.stem.2019.11.002 |
Vencken, S., Foged, C., Ramsey, J. M., Sweeney, L., Cryan, S. A., MacLoughlin, R. J., et al. (2019). Nebulised lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells. ERJ Open Res. 5 (2), 00161. doi:10.1183/23120541.00161-2018 |
Villamizar, O., Waters, S. A., Scott, T., Saayman, S., Grepo, N., Urak, R., et al. (2019). Targeted activation of cystic fibrosis transmembrane conductance regulator. Mol. Ther. 27 (10), 1737–1748. doi:10.1016/j.ymthe.2019.07.002 |
Villamizar, O., Waters, S. A., Scott, T., Grepo, N., Jaffe, A., and Morris, K. V. (2021). Mesenchymal Stem Cell exosome delivered Zinc Finger Protein activation of cystic fibrosis transmembrane conductance regulator. J. Extracell. Vesicles 10 (3), e12053. doi:10.1002/jev2.12053 |
Vituret, C., Gallay, K., Confort, M.-P., Ftaich, N., Matei, C. I., Archer, F., et al. (2016). Transfer of the cystic fibrosis transmembrane conductance regulator to human cystic fibrosis cells mediated by extracellular vesicles. Hum. Gene Ther. 27 (2), 166–183. doi:10.1089/hum.2015.144 |
Wagner, J. A., Nepomuceno, I. B., Messner, A. H., Moran, M. L., Batson, E. P., Dimiceli, S., et al. (2002). A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 13 (11), 1349–1359. doi:10.1089/104303402760128577 |
Wainwright, B. J., Scambler, P. J., Schmidtke, J., Watson, E. A., Law, H.-Y., Farrall, M., et al. (1985). Localization of cystic fibrosis locus to human chromosome 7cen-q22. Nature 318 (6044), 384–385. doi:10.1038/318384a0 |
Wan, F., Nylander, T., Klodzinska, S. N., Foged, C., Yang, M., Baldursdottir, S. G., et al. (2018). Lipid shell-enveloped polymeric nanoparticles with high integrity of lipid shells improve mucus penetration and interaction with cystic fibrosis-related bacterial biofilms. ACS Appl. Mater. Inter. 10 (13), 10678–10687. doi:10.1021/acsami.7b19762
Wang, G., Slepushkin, V., Zabner, J., Keshavjee, S., Johnston, J. C., Sauter, S. L., et al. (1999). Feline immunodeficiency virus vectors persistently transduce nondividing airway epithelia and correct the cystic fibrosis defect. J. Clin. Invest. 104 (11), R55–R62. doi:10.1172/jci8390 |
Watson, J. K., Rulands, S., Wilkinson, A. C., Wuidart, A., Ousset, M., Van Keymeulen, A., et al. (2015). Clonal dynamics reveal two distinct populations of basal cells in slow-turnover airway epithelium. Cell Rep. 12 (1), 90–101. doi:10.1016/j.celrep.2015.06.011 |
Wu, Z., Yang, H., and Colosi, P. (2010). Effect of genome size on AAV vector packaging. Mol. Ther. 18 (1), 80–86. doi:10.1038/mt.2009.255 |
Xu, J., Woods, C. R., Mora, A. L., Joodi, R., Brigham, K. L., Iyer, S., et al. (2007). Prevention of endotoxin-induced systemic response by bone marrow-derived mesenchymal stem cells in mice. Am. J. Physiology-Lung Cell Mol. Physiol. 293 (1), L131–L141. doi:10.1152/ajplung.00431.2006
Yamada, K., Hirakata, A., Tchipashvili, V., Shimizu, A., Iwaki, H., Griesemer, A., et al. (2011). Composite islet-kidneys from single baboon donors cure diabetes across fully allogenic barriers. Am. J. Transpl. 11 (12), 2603–2612. doi:10.1111/j.1600-6143.2011.03733.x
Zabner, J., Seiler, M., Walters, R., Kotin, R. M., Fulgeras, W., Davidson, B. L., et al. (2000). Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J. Virol. 74 (8), 3852–3858. doi:10.1128/jvi.74.8.3852-3858.2000 |
Zhu, Y.-g., Feng, X.-m., Abbott, J., Fang, X.-h., Hao, Q., Monsel, A., et al. (2014). Human mesenchymal stem cell microvesicles for treatment ofEscherichia coliEndotoxin-induced acute lung injury in mice. Stem Cells 32 (1), 116–125. doi:10.1002/stem.1504 |
Keywords: cystic fibrosis, CFTR, gene therapy, cell-based therapy, therapeutic vectors, stem cells
Citation: Allan KM, Farrow N, Donnelley M, Jaffe A and Waters SA (2021) Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front. Pharmacol. 12:639475. doi: 10.3389/fphar.2021.639475
Received: 09 December 2020; Accepted: 27 January 2021;
Published: 16 March 2021.
Edited by:
Miquéias Lopes-Pacheco, University of Lisbon, PortugalReviewed by:
Susan Reynolds, Nationwide Children’s Hospital, Columbus, United StatesMassimo Conese, University of Foggia, Italy
John Sheridan, Cystic Fibrosis Foundation, Bethesda, United States
Copyright © 2021 Allan, Farrow, Donnelley, Jaffe and Waters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shafagh A. Waters, c2hhZmFnaC53YXRlcnNAdW5zdy5lZHUuYXU=