 Junqi Bai1
Junqi Bai1 Qiyu Zou1
Qiyu Zou1 He Su1
He Su1 Baosheng Liao1
Baosheng Liao1 Ping Wang2
Ping Wang2 Juan Huang1
Juan Huang1 Danchun Zhang1
Danchun Zhang1 Lu Gong1
Lu Gong1 Wen Xu1
Wen Xu1 Jing Zhang1
Jing Zhang1 Zhihai Huang1*
Zhihai Huang1* Xiaohui Qiu1*
Xiaohui Qiu1*- 1Guangdong Provincial Hospital of Traditional Chinese Medicine, The Second Clinical Medical College of Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Shanghai Dehua Traditional Chinese Medicine Co., Ltd., Shanghai, China
Introduction: The root of Reynoutria multiflora (Thunb.) Moldenke (RM) has been used widely in formulations of herbal medicines in China for centuries. Raw R. multiflora (RRM) should be processed before use to reduce toxicity and increase efficacy. However, detailed regulation of the processing endpoint is lacking, and the duration of processing can vary considerably. We conducted in-depth research on stilbene glycosides in RM at different processing times. Previously, we discovered that 219 stilbene glycosides changed markedly in quantity and content. Therefore, we proposed that processing causes changes in various chemical groups.
Methods: To better explain the mechanism of RM processing for toxicity reduction and efficacy enhancement, we used a method of tandem mass spectrometry described previously to research gallic acid based and catechin based metabolites.
Results: A total of 259 metabolites based on gallic acid and 112 metabolites based on catechins were identified. Among these, the peak areas of 157 gallic acid and 81 catechins gradually decreased, those of another 71 gallic acid and 30 catechins first increased and then decreased, those of 14 gallic acid and 1 catechin gradually increased. However, 17 of the gallic acids showed no significant changes. We speculate that many gallic acid metabolites hydrolyze to produce gallic acid; moreover, the dimers/trimers of catechins, after being cleaved into catechins, epicatechin, gallic acid catechins, and epicatechin monomers, are cleaved into gallic acid and protocatechualdehyde under high temperature and high humidity, subsequently participating in the Maillard reaction and browning reactions.
Discussion: We showed that processing led to changes in chemical groups, clarification of the groups of secondary metabolites could provide a basis for research on the pharmacological and toxic mechanisms of RM, as well as the screening of related markers.
1 Introduction
The root of Reynoutria multiflora Thunb. (syn: Polygonum multiflorum Thunb.) is known as “He-shou-wu” in China. It has been used widely for centuries in traditional Chinese medicine (TCM) formulations (Li, et al., 2017). Several studies have shown that raw R. multiflora and its processed products have different pharmacological effects. RRM has the effects of detoxification, carbuncle elimination, and bowel relaxation. Processed R. multiflora (PRM) shows the effects of tonifying the liver and kidneys, and hair-blackening, and can be used in tonic medicines (Cheung, et al., 2014; Lin, et al., 2015; Chinese Pharmacopoeias commission, 2020). The difference between RRM and PRM is processing: steaming or stewing. However, detailed regulation of the processing endpoint is lacking, and the duration of processing can vary considerably. Previously (Bai et al., 2021; Bai et al., 2022), we showed that if RM was processed for 24 h then there were attributable differences between PRM and RRM. It has been shown that stilbene glycosides undergo marked changes during processing; 219 stilbene glycosides have been shown to undergo dramatic changes in number and content (not just a few chromatographic peaks on spectra). For instance, the content of 2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside has decreased, or the content of several other metabolites has increased or decreased, during processing (Liang, et al., 2010; Liu, et al., 2011; Chen, et al., 2016; Wang, et al., 2016).
Some researchers have reported that, under high temperatures, catechins are prone to degradation and isomerization (Yoshihiro, et al., 1993; Zulema, et al., 2004; Wang, et al., 2006). Catechins can be cracked into gallic acid. Isomerization, oxidative polymerization, and hydrolysis are important reactions that cause the chemical transformation of catechins. Isomerization of one molecule of a catechin can generate one molecule of the corresponding isomer, whereas hydrolysis of one molecule of a catechin can generate one molecule of gallic acid (Li, et al., 2018). Previously, we showed that the content of catechins and epicatechins decreased sharply (or even disappeared) during processing. What catechins are transformed into during the processing of RM and the manner of this transformation are not clear, but we believe that the answers to these questions are important for the processing of RM.
It has been reported that condensed tannins can undergo acid-catalyzed cleavage in the presence (or excess amount) of a nucleophile (Nonaka, et al., 1982; Torres et al., 2002). That is, condensed polymers are depolymerized into oligomers and monomers under thermal and acidic conditions. Oligomers and monomers (except gallic acid) are also unstable in thermal and acidic conditions. Hence, they are subject to structural transformation after additional processing, which causes catechin to be transformed into its isomeric compound: epicatechin (Ross, et al., 2011). Besides isomerization, polymers, and monomers are depolymerized further. Hence, the monomer protocatechuic aldehyde could finally be obtained after processing, and gallic acid (another derived monomer) could accumulate after each processing cycle. Studies have shown that the contents of other bioactive compounds (e.g., catechins, gallic acid, procyanidin B2) change after processing, which may lead to a change in their therapeutic effects (Yao, et al., 2006; Chen, et al., 2012; Han, et al., 2013; Li, et al., 2018).
PRM at 24 h was clearly different from RRM, we conducted more in-depth analyses of the important chemical metabolites of RM (stilbene glycosides) and studied changes in the peak area with the duration of processing (Bai et al., 2021). A total of 219 stilbene glycosides underwent marked changes in number and content (not only a few chromatographic peaks), so we continued to conduct in-depth research on metabolites related to gallic acid and catechins. First, we identified the changes in the number of these two types of metabolites. Second, we evaluated the change process and mechanism of the gradual deepening of color during the processing of RM.
2 Materials and methods
2.1 Materials and sample processing
RRM materials were collected from Deqing (Guangzhou, China), Miyi (Sichuan, China), Kaili (Guizhou, China), the geo-authentic producing area of RM. There were three batches (30 kg each) from each producing area. The corresponding batch numbers, producer, and image information were documented (Supplementary Table S1). Samples were authenticated by Professor Zhihai Huang, and voucher specimens were deposited in the Materials Medica Preparation Laboratory of the Second Affiliated Hospital of the Guangzhou University of Chinese Medicine (Guangzhou, China).
Galic acid, catechin and epicatechin were purchased from China institute for food and drug control (No. 110831-201906, 110877-202005, 110878-201703, beijing, China). Acetonitrile and methanol (HPLC grade), were supplied by E. Merck (Darmstadt, Germany), formic acid (HPLC grade) was purchased from Fisher Scientific (Massachusetts, United States), ultra-pure water was prepared by a Mili-Q water purification system (Millipore, MA, United States).
2.2 Preparation of samples
2.2.1 Processing
According to the processing technology we have researched earlier (Bai et al., 2021), Each batch of RRM was first moistened with black bean juice, and then steamed for 32 h. At the time of steaming for 4 h, 8 h, 12 h, 18 h, and 24 h, 2 kg of RRM were taken out and dried to obtain PRM of different times. Samples were processed by Shanghai Dehua Traditional Chinese Medicine Co., Ltd.
2.2.2 Extraction
All the samples were prepared using following method: 1 g sample powder was ultrasonicated for 30 min with 25 mL of 70% ethanol, followed by filtration and then evaporated the filtrate. 5 mL of ultrapure water were added to dissolve the residue and then extracted twice with 15 mL of ethyl acetate. The resulting mixture was combined with an ethyl acetate solution and evaporated over a water bath; after that, 1 mL of methanol was added to dissolve the residue and centrifugation (15,000 rpm, 4°C) for 10 min by a 1.5 mL centrifuge tube. Finally, the supernatant of the treatment samples was injected into the UPLC-Q-Exactive plus orbitrap MS/MS system.
2.3 UPLC-tandem Q-exactive plus orbitrap mass spectrometry analysis
All samples were analysed using an Ultimate 3000 UPLC system (Dionex, United States) that was controlled with Thermo Xcalibur software (Thermo Fisher Scientific, United States). The samples were separated using a Kinetex UPLC C18 column (100*2.1 mm, 1.7 µm) (Phenomenex, United States). The mobile phase consisted of solvent A (0.1% formic acid) and solvent B (acetonitrile). A gradient elution was applied using the following optimized gradient program: 8%–8% B at 0–3 min, 8%–28% B at 3–25 min, 28%–40% B at 25–26 min, 40%–50% B at 26–28 min, 50%–70% B at 28–30 min, 70%–90% B at 30–32 min, and 90%–90% B at 32–35 min. The flow rate was kept at 0.4 mL/min, the sample injection volume was 1 μL, and the column temperature was maintained at 25°C. Mass spectrometry (MS) was undertaken on a Q-Exactive Plus™ Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, United States) in negative ion mode. The scan mass range was set at m/z 100–1,200. The parameter settings were: a full scan and fragment spectral resolution of 70,000 full width at half maximum (FWHM) and 17,500 FWHM, respectively; capillary temperature was 350°C; temperature of the auxiliary gas heater was 350°C; spray voltage was −3.2 KV; sheath-gas flow rate was 40 Arb; auxiliary-gas flow rate was 15 Arb; S-lens RF level was set at 50. The acquisition mode of stepped normalized collision energy was 30, 50, and 70 eV. The accumulated resultant fragment ions were injected into the mass spectrometer for single-scan detection.
3 Results
3.1 Base peak chromatograms
The chemical profiles of RRM and PRM were analyzed by tandem mass spectrometry (MS2) using an UHPLC-Q-Exactive Plus Orbitrap mass spectrometer. The representative base peak chromatograms of RRM and PRM (24 h) are shown in Figure 1.
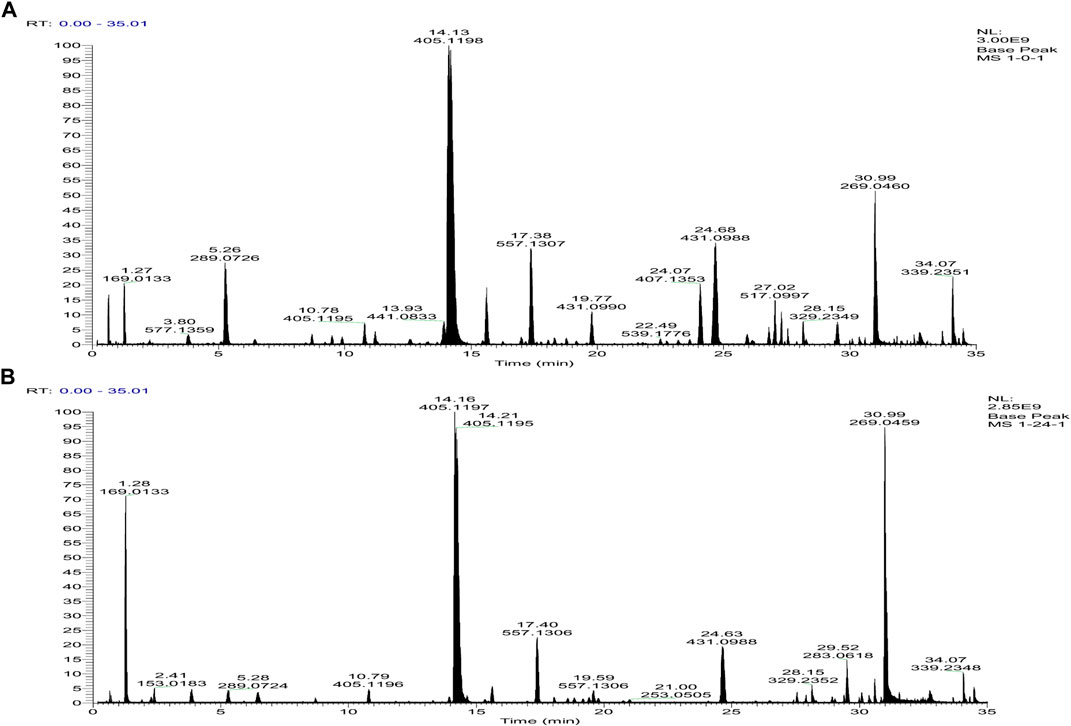
FIGURE 1. Base peak intensity chromatograms of samples of raw R. multiflora Thunb. [RRM, (A)] and processed Reynoutria multiflora Thunb. [PRM for 24 h, (B)] derived from MS2 using the UHPLC-Q-Exactive Plus Orbitrap mass spectrometer.
3.2 Fragmentation pathway of gallic acid and derivatives
To identify the derivatives of gallic acid in the processing of RM, a standard of gallic acid was first analyzed by MS2 using the UPLCQ-Exactive Plus Orbitrap mass spectrometer under the condition mentioned above. Gallic acid (A1-1, retention time (tR) = 1.28 min) had a [M-H]− ion at m/z 169.0133 (C7H5O5) with only a dominant ion at m/z 125.0231 (C6H5O3) in MS2 spectrum, but a difference of 44 Da between the mass of the precursor ion and product ion, which involved a neutral loss of CO2. These two ions could be used as diagnostic ions to identify gallic acid. Metabolite A1-2 (tR = 2.03 min) also had an [M-H]− ion at m/z 169.0133 (C7H5O5), and showed a fragment ion at m/z 125.0231 in MS2 spectrum, indicated that it was an isomer of gallic acid. In the present study, most of the metabolites were formed by dehydration of gallic acid glycosides and other small molecules, gallic acid and different substituents are linked to different hydroxyl groups of glucose. Under electron bombardment, substituents and glucose are cleaved, and the oxygen linked to glucose and substituents breaks off from glucose, forming glucose residues (161.04) and glycosides formed by dehydration of gallic acid (152.01). Therefore, the ion formed after the substituent is 313.0564 instead of 331.0667. As shown in Figures 2–5.
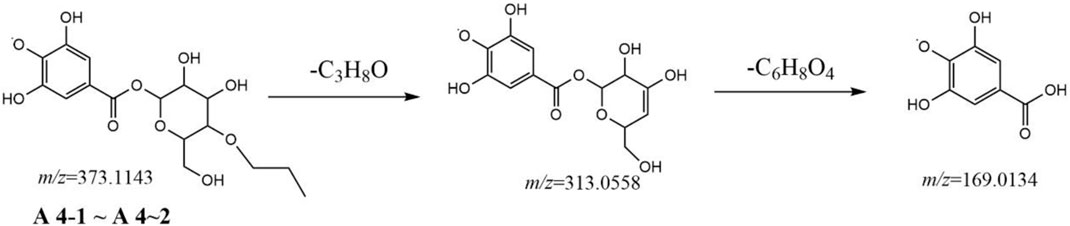
FIGURE 2. Postulated fragmentation pathways of metabolites A4-1 to A4-2.
Metabolites A2-1, A2-2, and A2-3 showed the same [M-H]− ion at m/z 183.0292 (C8H7O5) and MS2 spectra gave ions at m/z 183.0292 (C8H7O5), 168.0053(C7H4O5), and 124.0147 (C6H4O3) from a continuous loss of CH3 and CO2. Thus, metabolites A2-1, A2-2, and A2-3 were identified as methyl gallate.
Metabolites A3-1, A3-2, A3.3, and A3-4 showed the same [M-H]− ion at m/z 331.0673 (C13H15O10) and MS2 spectra gave identical ions at m/z 331.0667 (C13H15O10), 271.0461 (C11H11O8), 211.0241 (C9H7O6), and 169.0132 (C7H5O5), respectively. Comparison with the literature revealed metabolites A3-1 to A3-4 to be gallic acid-O-glycoside.
Metabolites A4-1 and A4-2 showed the same [M-H]− ion at m/z 373.1143 (C16H21O10) and MS2 spectra gave identical ions at m/z 313.0558 (C13H13O9) and 169.0134 (C7H5O5), respectively. Comparison with the literature revealed metabolites A4-1 and A4-2 to be gallic acid-O-glycoside-O-propanoyl. A fragmentation pathway was postulated (Figure 2). Similarly, metabolites A5-1, A5-2, and A5-3 were characterized tentatively to be gallic acid-O-glycoside-O-hydroxyphenyl because of the [M-H]− ion at m/z 423.0924 (C19H19O12) and because MS2 spectra gave ions at m/z 313.0558 (C13H13O9), 169.0134 (C7H5O5), and 125.0231 (C6H5O3), respectively.
Metabolites A9-1 to A9-8 showed the same [M-H]− ion at m/z 453.1048 (C20H21O12) and MS2 spectra gave identical ions at m/z 313.0563 (C13H13O9) and 169.0134 (C7H5O5). The molecular formula of the substituent should be C7H8O3, the most likely was methoxycatechol. Therefore, metabolites A9-1 to A9-8 were determined to be gallic acid-O-glycoside-O-methoxycatechol acyl. Similarly, metabolites A10-1 to A10-5 were characterized tentatively to be gallic acid-O-glycoside-O-dimethoxycatechol acyl because of the [M-H]− ion at m/z 467.1196 (C21H23O12) and because MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 169.0134 (C7H5O5), and 153.0545 (C8H9O3). The molecular formula of the substituent could be C8H10O3, which was 14 Da (CH2) higher than that of metabolites A9, so it was determined to be dimethoxyphenol.
Metabolites A11-1 to A11-4 showed the same [M-H]− ion at m/z 477.1038 (C22H21O12) and MS2 spectra gave identical ions at m/z 313.0563 (C13H13O9), 169.0134 (C7H5O5), 163.0390 (C9H7O3), and 125.0231 (C6H5O3). The molecular formula of substituent should be C9H8O3, It may be hydroxycinnamic acid. Therefore, metabolites A11-1 to A11-4 were determined as gallic acid-O-glycoside-O-hydroxycinnamoyl. Similarly, metabolites A12-1 to A12-9 tentatively characterized to be gallic acid-O-glycoside-O-hydroxyphenyl propionyl. Since the [M-H]− ion at m/z 479.1198 (C22H23O12) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 169.0134 (C7H5O5), 165.0547 (C9H9O3), and 153.0545 (C7H5O3). The molecular formula of the substituent may be C9H10O3, which was 2 Da (H2) higher than that of metabolites A11, the substituent may be hydroxyphenylpropionic acid.
Metabolites A13-1 to A13-6 gave an [M-H]− ion at m/z 481.0990 (C21H21O13) and product ions at m/z 313.0563 (C13H13O9) and 169.0134 (C7H5O5). The molecular formula of the substituent may be C8H8O4, the substituent may be hydroxymethoxybenzoic acid. Therefore, metabolites A13-1 to A13-6 were determined as gallic acid-O-glycoside-O-hydroxymethoxybenzoyl.
Metabolites A14-1 and A14-2 were tentatively characterized as gallic acid-O-glycoside-O- hydroxymethoxyphenylethanol acyl because of the [M-H]− ion at m/z 481.1353 (C22H25O12) and because MS2 spectra gave ions at m/z 313.0563 (C13H13O9) and 169.0134 (C7H5O5). The molecular formula of the substituent may be C9H12O3, the substituent may be hydroxymethoxyphenylethanol.
Metabolites A15-1 to A15-9 displayed a high resolution [M-H]− ion at m/z 483.0785 and gave an element composition of C20H19O14, product ions at m/z 331.0667 (C13H15O10), 313.0564 (C13H13O9), 271.0461 (C11H11O8), 211.0241 (C9H7O6), and 169.0134 (C7H5O5). The fragment ion m/z 331.0667 was obtained from 152 Da (C7H4O4) loss of parent ion, suggesting that the substituent was galloyl. Other fragment ions were similar to metabolites A4. Therefore, metabolites A15-1 to A15-9 were identified to be gallic acid-O-glycoside-O-galloyl (Figure 3).
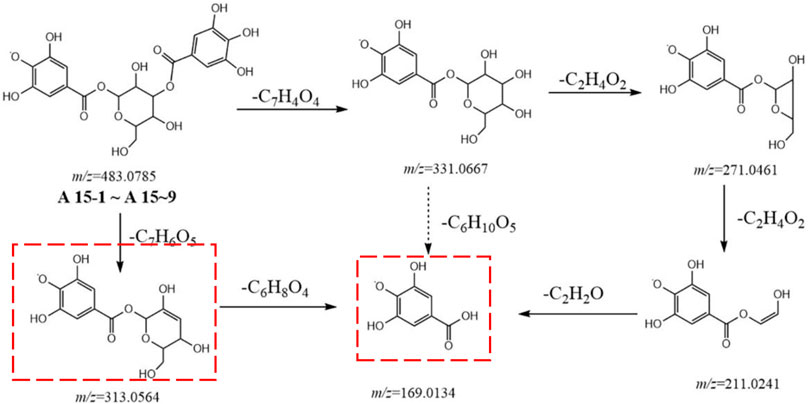
FIGURE 3. Postulated fragmentation pathways of metabolites A15-1 to A15-9. The ion fragments in the red box are characteristic ion fragments of gallic-acid metabolites.
Metabolites A16-1 to A16-6 displayed a high resolution [M-H]− ion at m/z 493.1352 and gave an element composition of C23H25O12, product ions at m/z 313.0563 (C13H13O9), 179.0704 (C10H11O3), 169.0134 (C7H5O5), and 151.0022 (C7H3O4). The molecular formula of the substituent was C8H10O3, which was 14 Da (CH2) higher than that of metabolites A12, and it was determined to be methoxyphenylpropionic acid. Therefore, metabolites A16-1 to A16-6 were determined to be gallic acid-O-glycoside-O-methoxyphenylpropionyl. Similarly, metabolites A17-1 and A17-2 were tentatively characterized as gallic acid-O-glycoside-O-hydroxy-methoxy-phenylpropanol acyl, since the [M-H]− ion at m/z 495.1512 (C23H27O12) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9) and 169.0134 (C7H5O5).
Metabolite A18 gave an [M-H]− ion at m/z 497.0941 (C21H21O14) and product ions at m/z 345.0823 (C14H17O10), 313.0558 (C13H13O9), 183.0291 (C8H7O5), and 169.0134 (C7H5O5). Fragment ions m/z 313.0558 and 169.0134 followed to the above rules, fragment ion m/z 345.0823 was obtained by loss of a galloyl, and fragment ion m/z 183.0291 was the substituent. Therefore, metabolite A18 was tentatively identified to be gallic acid-O-glycoside-O-methoxyphenoyl. Similarly, metabolites A19-1 and A19-2 were tentatively characterized to be gallic acid-O-glycoside-O-trimethoxyphenol acyl because of the [M-H]− ion at m/z 497.1305 (C22H25O12) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 169.0134 (C7H5O5), and 151.0024 (C7H3O4).
Metabolites A20-1 to A20-6 showed a high resolution [M-H]− ion at m/z 503.1201 and gave an element composition of C24H23O12, product ions at m/z 313.0563 (C13H13O9), 189.0547 (C11H9O3), 169.0134 (C7H5O5), and 151.0024 (C7H3O4). Therefore, metabolites A20-1 to A20-6 were determined to be gallic acid-O-glycoside-O-dimethyl-hydroxycoumarin acyl. Similarly, metabolites A21-1 to A21-4 were tentatively characterized to be gallic acid-O-glycoside-O-carboxy-cinnamoyl because of the [M-H]− ion at m/z 505.0994 (C23H21O13) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 191.0341 (C10H7O4), and 169.0134 (C7H5O5). Metabolites A22-1 and A22-2 were characterized to be gallic acid-O-glycoside-O-hydroxy-methoxy-cinnamoyl, since the [M-H]−ion at m/z 507.1145 (C23H23O13) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 193.0499 (C10H9O4), and 169.0134 (C7H5O5).Then, metabolites A23-1 and A23-2 were tentatively identified to be gallic acid-O-glycoside-O-dimethoxy-phenylacetate acyl, because of the [M-H]− ion at m/z 509.1302 (C23H25O13) and because MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 195.0659 (C10H11O4), and 169.0134 (C7H5O5).
Metabolite A24 displayed a high resolution [M-H]− ion at m/z 511.1095 and gave an element composition of C22H23O14, product ions at m/z 467.1206 (C19H13O6), 313.0563 (C13H13O9), and 169.0134 (C7H5O5). Therefore, metabolite A24 was tentatively identified to be gallic acid-O-glycoside-O-hydroxy-dimethoxybenzoyl.
Metabolites A25-1 to A25-6 were eluted at 16.40 min, 16.99 min, 17.77 min, 18.40 min, 18.78 min, and 19.69 min. They both showed an accurate [M-H]− ion at m/z 541.1359 (C26H27O12). In their MS2 spectra, the [M-H]− ion showed fragment ions at m/z 313.0563 (C13H13O9), 243.0661 (C14H11O4), 227.0707 (C14H11O3), and 169.0134 (C7H5O5). Metabolites A25-1 to A25-6 were identified to be gallic acid-O-glycoside-O-trihydroxystilbene. Similarly, metabolites A26-1 and A26-2 were characterized to be gallic acid-O-glycoside-O-dihydrotrihydroxystilbene. Since the [M-H]− ion at m/z 543.1513 (C27H27O12) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 229.0865 (C14H13O3), and 169.0134 (C7H5O5).
Metabolites A27-1 to A27-8 displayed a high resolution [M-H]− ion at m/z 545.1306 and gave an element composition of C26H25O13. MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 231.0658 (C13H11O4), and 169.0134 (C7H5O5). Comparison with the literature revealed metabolites A27-1 to A27-8 were tentatively identified to be gallic acid-O-glycoside-O-hydroxymusizin.
Metabolites A29-1 to A29-5 showed a high resolution [M-H]− ion at m/z 555.1508 and gave an element composition of C28H27O12. MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 241.0865 (C15H13O3), and 169.0134 (C7H5O5). Comparison with the literature revealed metabolites A29-1 to A29-5 were tentatively identified as gallic acid-O-glycoside-O-tetrahydroxyphenanthryl. Similarly, metabolites A30-1 to A30-7 were characterized to be gallic acid-O-glycoside-O-tetrahydroxystilbene-acyl (Figure 4), because of the [M-H]− ion at m/z 557.1307 (C27H25O13) and because MS2 spectra gave ions at m/z 405.1187 (C20H21O9), 313.0563 (C13H13O9), 243.0658 (C14H11O4), and 169.0134 (C7H5O5).
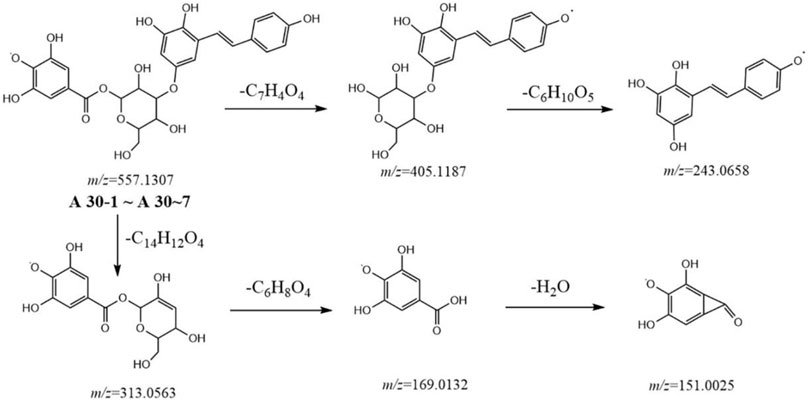
FIGURE 4. Postulated fragmentation pathways of metabolites A30-1 to A30-7.
Metabolites A 31-1 to A 31-4 gave precursor ion [M-H]− at m/z 559.1460 (C27H27O13) and eluted at 16.02 min, 25.60 min, 26.70 min, 27.66 min. In MS2 spectra, the [M-H]− ions showed at m/z 559.1460 (C27H27O13), 407.1342 (C20H23O9), 313.0563 (C13H13O9), 245.0817 (C14H13O4), 169.0134 (C7H5O5), and 139.0386 (C7H7O3). By comparison with the literature, metabolites A 31-1 to A 31-4 were tentatively characterized to be gallic acid-O-glycoside-O-dihydrotetrahydroxystilbeneacyl.
Metabolite A32 displayed a high resolution [M-H]− ion at m/z 563.1170 and gave an element composition of C29H23O12, product ions at m/z 313.0563 (C13H13O9), 249.0533 (C15H10O3), and 169.0134 (C7H5O5). The substituent was suggested to be hydroxy-methyl-anthraquinone by comparison with literature. Metabolite A32 was tentatively identified to be gallic acid-O-glycoside-O-hydroxy-methyl-anthraquinoyl. Similarly, metabolite A33 was characterized to be gallic acid-O-glycoside-O-emodin anthrone acyl, since the [M-H]− ion at m/z 569.1309 (C28H25O13) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 255.0663(C15H11O4), and 169.0134 (C7H5O5). By comparison with literature, the substituent was suggested to be emodin anthrone. Metabolites A34-1, A34-2, and A34-3 were characterized to be gallic acid-O-glycoside-O-salicylate diester, because of the [M-H]− ion at m/z 571.1094 (C27H23O14) and MS2 spectra gave ions at m/z 419.0987 (C20H19O10), 313.0563 (C13H13O9), 257.0453 (C15H13O4), and 169.0134 (C7H5O5). The substituent was suggested to be a salicylate diester.
Metabolites A35-1 to A35-5 showed a high resolution [M-H]− ion at m/z 573.1259 and gave an element composition of C27H25O14. MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 259.0610 (C14H11O5), and 169.0134 (C7H5O5). Comparison with the literature revealed metabolites A35-1 to A35-5 were tentatively identified as gallic acid-O-glycoside-O-tetrahydroxyphenanthryl.
Metabolites A36-1 to A36-9 displayed a high resolution [M-H]− ion at m/z 583.1102 and gave an element composition of C28H23O14, product ions at m/z 583.1102 (C28H23O14), 431.0983 (C21H19O10), 313.0564 (C13H13O9), 269.0458 (C15H9O5), and 169.0134 (C7H5O5). Among these, fragment ion 431.0983 was obtained from the loss of 152 Da (C7H4O4) of the parent ion, suggesting that the substituent was gallic acid. The ion of m/z 269.0458 suggested that it was emodin. Thus, metabolites A36-1 to A36-9 were identified to be gallic acid-O-glycoside-O-emodin acyl.
Metabolites A37-1 to A37-6 gave a [M-H]− ion at m/z 585.1248 (C28H25O14) and product ions at m/z 585.1248 (C28H25O14), 541.1355(C27H25O12), 313.0563 (C13H13O9), and 169.0134 (C7H5O5). The lion of 585.1248 was 44 Da (CO2) higher than that of metabolites A25, and other fragment ions were consistent with metabolites A25. Therefore, metabolites A37-1 to A37-6 were determined to be gallic acid-O-glycoside-O-tetrahydroxystilbene-COOH.
Metabolites A38-1 to A38-7 were characterized as gallic acid-O-glycoside-O-afzelechin acyl, as the [M-H]− ion m/z 587.1415 (C28H27O14) and MS2 ions at m/z 313.0563 (C13H13O9), 273.0769 (C15H13O5), 169.0134 (C7H5O5), and 149.0232 (C8H5O3).
Metabolites A39-1 to A39-4 showed the same [M-H]− ion at m/z 589.1206 (C27H25O15) and MS2 spectra gave ions at m/z 465.0675 (C20H17O13), 437.1085 (C20H21O11), 313.0564 (C13H13O9), and 169.0136 (C7H5O5). MS2 showed that the ions m/z 465.0675 and 437.1085 of C7H8O2 and C7H14O4 were lost due to the hydroxybenzene hexanol and gallic acid moieties. By literature comparison, metabolites 39-1 to 39-4 were identified to be gallic acid-O-glycoside-O- hexahydroxystilbene.
Metabolites A40-1 to A40-4 showed the same [M-H]− ion at m/z 597.1258 (C29H25O14) and MS2 spectra gave ions at m/z 313.0564 (C13H13O9), 283.0612 (C16H11O5), 269.0457 (C15H9O5), and 169.0132 (C7H5O5). Therefore, metabolites A40-1 to A40-4 were determined to be gallic acid-O-glycoside-O-physcion. Similarly, metabolites A41-1 and A41-2 were characterized to be gallic acid-O-glycoside-O-hydroxyemodin. The [M-H]− ion at m/z 599.1049 (C28H23O15) and MS2 spectra gave ions at m/z 313.0563 (C13H13O9), 255.0663(C15H11O4), and 169.0134 (C7H5O5).
Metabolites A42-1 and A42-2 showed the same [M-H]− ion at m/z 589.1206 (C27H25O15) and MS2 spectra gave ions at m/z 465.0675 (C20H17O13), 437.1085 (C20H21O11), 313.0564 (C13H13O9), and 169.0136 (C7H5O5). MS2 showed that the ions m/z 465.0675 and 437.1085 were losses of C7H8O2 and C7H14O4 due to hydroxybenzene hexanol and gallic acid moiety. Fragment ions m/z 465.0675, 313.0564, 169.0136 were the determination ion of di-gallic acid glucoside. By literature comparison, metabolites A42-1 and A42-2 were identified to be eriodictyol 7-O-(6″-O-galloyl)-β-D-glucopyranoside.
Metabolites A43-1 to A43-6 gave a [M-H]− ion at m/z 603.1364 (C28H27O15) and product ions at m/z 313.0563 (C13H13O9), 289.0718 (C15H13O6), and 169.0134 (C7H5O5). The substituent can be catechin or epicatechin. Their relative retention time were 5.45 min, 8.82 min, 9.33 min, 10.43 min, 10.76 min, and 11.32 min. According to the retention time of catechin and epicatechin, the substituents of A43-1, A43-2, and A43-3 should be catechin, and the substituents of A43-4, A43-4 and A43-5 should be epicatechin. Therefore, metabolites A43-1 ∼ A43-3 were determined to be gallic acid-O-glycoside-O-catechin, and metabolites A43-4 ∼ A43-6 were determined to be gallic acid-O-glycoside-O-epicatechin.
Metabolite A45 gave a [M-H]− ion at m/z 605.1156 (C27H25O16) and product ions at m/z 465.0681(C20H17O13), 313.0558(C13H13O9), 169.0134 (C7H5O5), and 125.0225 (C6H5O3). Therefore, metabolite A45 was tentatively identified as di-gallic acid-O-glucoside-O-benzyl alcohol. Metabolites A47-1 to A47-5 displayed a high resolution [M-H]− ion at m/z 613.1207 and gave an element composition of C29H25O15. MS2 spectra gave ions at 613.1201 (C29H25O15), 569.1323 (C28H25O13), 313.0563 (C13H13O9), 299.0562 (C16H11O6), 255.0665 (C15H11O4), and 169.0134 (C7H5O5). By literature comparison, metabolites A47-1 to A47-5 were tentatively identified as gallic acid-O-glycoside-O-questinol.
Metabolite A48 gave an [M-H]− ion at m/z 617.1157 (C28H25O16) and product ions at m/z 599.1043 (C28H23O15), 313.0558 (C13H13O9), 303.0513 (C15H11O7), 285.0410 (C15H9O6), 169.0134 (C7H5O5), and 125.0225 (C6H5O3). By comparison with the literature, metabolite A48 was tentatively characterized as taxillusin. Similarly, metabolites A49-1 and A49-2 showed a [M-H]−ion at m/z 619.1075 (C31H23O14) and MS2 spectra gave ions at m/z 313.0558 (C13H13O9), 305.0427 (C18H9O5), 271.0462 (C15H11O7), and 169.0134 (C7H5O5). By comparison with the literature, metabolites A49-1 and A49-2 were tentatively characterized to be gallic acid-O-glycoside-O-trihydroxynaphthacenequinone.
Metabolite A50 gave an [M-H]− ion at m/z 629.1158 (C29H25O16) and product ions at m/z 477.1046 (C22H21O12), 465.0681(C20H17O13), 313.0558 (C13H13O9), 303.0513 (C15H11O7), 285.0410 (C15H9O6), 169.0134 (C7H5O5), and 125.0225 (C6H5O3). Fragment ions m/z 465.0681, 313.0558, and 169.0134 indicated that there were two gallic acid substitutions and fragment ions m/z 477.1046, 313.0558, and 169.0134 indicated that there was also a p-coumaric acid substituent. Therefore, metabolite A50 was tentatively characterized as di-gallic acid-O-glycoside-O-coumaric acid.
Metabolites A52-1 and A52-2 showed the same [M-H]− ion at m/z 631.1311 (C29H27O16), which was 44 Da (CO2) higher than that of metabolites A38. MS2 spectra gave ions at m/z 313.0564 (C13H13O9), 273.0771 (C15H13O5), 255.0666 (C15H11O4), 193.0132 (C15H11O4), 169.0136 (C7H5O5), and 149.0232 (C8H5O3), and all fragment ions were consistent with metabolites A38. Therefore, the metabolites A52-1 and A52-2 were tentatively characterized to be gallic acid-O-glycoside-O-afzelechin acyl-cooh.
Metabolites A53-1, A53-2, and A53-3 showed same [M-H]− ion at m/z 635.0899 (C27H23O18) and MS2 spectra gave ions at m/z 465.0681(C20H17O13), 313.0558 (C13H13O9), and 169.0136 (C7H5O5). Fragment ion m/z 465.0681 was obtained by removing one molecule of gallic acid from the parent ion. Metabolites A53-1, A53-2, and A53-3 were identified to be trigalloyl glucose.
Metabolites A54-1 to A54-4 showed the same [M-H]− ion at m/z 709.1422 (C34H29O17) and MS2 spectra gave ions at m/z 557.1300 (C27H25O13), 465.0681(C20H17O13), 405.1185 (C20H21O9), 313.0563 (C13H13O9), and 169.0134 (C7H5O5). The ion m/z 557.1300 of C7H4O4 was lost due to gallic acid moiety. Therefore, metabolites A54-1 to A54-4 were determined as di-gallic acid-O-glycoside-O-tetrahydroxystilbene acyl and metabolites A55-1 to A55-7 were tentatively characterized as gallic acid-O-di-glycoside-O-tetrahydroxystilbene-acyl. The [M-H]− ion at m/z 719.1839 (C33H35O18) and MS2 spectra gave ions at m/z 557.1300 (C27H25O13), 465.0681(C20H17O13), 405.1185 (C20H21O9), 313.0563 (C13H13O9), and 169.0134 (C7H5O5).
Metabolites A56-1 to A56-5 showed the same [M-H]− ion at m/z 727.1313 (C37H27O16) and MS2 spectra gave ions at m/z 575.1183 (C30H23O12), 539.0986 (C30H19O10), 449.0883 (C24H17O9), 407.0781 (C22H15O8), 289.0721 (C15H13O6), 285.0407 (C15H9O6), 269.0457 (C15H9O5), 241.0504 (C14H9O4), 169.0134 (C7H5O5), and 125.0231 (C6H5O3). Fragment ions m/z 407.0781 and 289.0721 indicated that the metabolites were procyanidins, and fragment ion m/z 169.0134 indicated that the metabolites were procyanidins and gallic acid. By comparison with the literature, metabolites A56-1 to A56-5 were identified to be procyanidin A-O-galloyl. Similarly, metabolites A57-1 to A57-12 were tentatively characterized to be procyanidin B2-O-galloyl. Since the [M-H]− ion at m/z 729.1470 (C37H29O16) and MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). Similarly, metabolites A59-1 to A59-5 were tentatively characterized to be procyanidin-O-galloyl, because of the [M-H]− ion at m/z 745.1421 (C37H29O17) and because MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 169.0134 (C7H5O5), 177.0183 (C9H5O4), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
Metabolites A60-1 to A60-7 showed the same [M-H]− ion at m/z 881.1581 (C44H33O20) and MS2 spectra gave ions at m/z 577.1356 (C30H25O12), 407.0781 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). Fragment ion m/z 577.1356 was obtained from the loss of 304 Da (C14H8O8) of the parent ion, which should be two gallic acid substituents. By comparison with the literature, metabolites A60-1 to A60-7 were identified to be procyanidin B2-O-di-galloyl. Similarly, metabolites A61-1, A61-2, and A61-3 were tentatively characterized to be epicatechin-O-gallate (4β-8)-(−)-epigallocatechin-O-gallate. Since the [M-H]− ion at m/z 897.1533 (C44H33O21) and MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 177.0183 (C9H5O4),169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
Metabolites A62-1 to A62-10 showed the same [M-H]− ion at m/z 1,017.2110 (C52H41O22) and MS2 spectra gave ions at m/z 677.1306 (C37H25O13), 525.0829 (C29H17O10), 451.1047 (C24H19O9), 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 177.0183 (C9H5O4), 169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). Comparison with the literature revealed metabolites A62-1 to A62-10 to be [Epicatechin-(4β->8)]2-epicatechin 3‴-gallate. Similarly, metabolites A63-1 to A63-5 were characterized tentatively to be procyanidin C-1 3′, 3″-di-O-gallate. The [M-H]− ion at m/z 1,169.2220 (C59H45O26) was 152 Da (C7H4O4) higher than that of metabolites A62, MS2 spectra gave ions at m/z 677.1306 (C37H25O13), 559.1238 (C30H23O11), 525.0829 (C29H17O10), 451.1047 (C24H19O9), 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 177.0183 (C9H5O4), 169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
In addition, metabolites A28-1 to A28-4 displayed a high-resolution [M-H]− ion at m/z 547.1464, gave an element composition of C26H27O13, and product ions at m/z 529.1357 (C26H25O12), 503.1205 (C24H23O12), 313.0563 (C13H13O9), 233.0815 (C13H13O4), 169.0134 (C7H5O5), and 151.0024 (C7H3O4). Metabolites A44-1 to A44-4 gave a [M-H]− ion at m/z 605.0936 (C30H21O14) and product ions at m/z 605.0936 (C30H21O14), 431.0986 (C20H19O10), 413.0874 (C21H17O9), 313.0564 (C13H13O9), 291.0276 (C17H7O5), 269.0457 (C15H9O5), and 169.0134 (C7H5O5), \. Among them, ions at m/z 431.0986 and 269.0457 indicated that they should be emodin glucoside, ions at m/z 605.0936, 313.0564, and 169.0134 indicated that gallic-acid glucoside was linked to the ion at m/z 291.0276. Similarly, metabolites A46-1 to A46-4 gave a [M-H]− ion at m/z 609.1233 (C30H25O14) and product ions at m/z 609.1221 (C30H21O14), 461.0709 (C21H17O12), 417.1190 (C21H21O9), 313.0564 (C13H13O9), 295.0587 (C17H11O5), 273.0768 (C15H13O5), and 169.0134 (C7H5O5), which were 4-Da higher than metabolites A44, so they were attributed to be homologous metabolites.
Metabolites A51-1 and A51-2 showed the same [M-H]− ion at m/z 629.1522 (C30H29O15) and MS2 spectra gave identical ions at m/z 315.0872 (C17H15O6), 313.0564 (C13H13O9), 297.0771 (C17H13O5), 271.0977 (C16H15O4), and 169.0136 (C7H5O5). The group of the fragment ion at m/z 315.0872 was the substituent of the metabolites, but the structure was not known temporarily. Similarly, metabolites A58-1 and A58-2 gave a [M-H]− ion at m/z 733.1744 (C37H33O16) and MS2 spectra gave ions at m/z 419.1135 (C24H19O7), 313.0563 (C13H13O9), and 169.0134 (C7H5O5). These metabolites could not be identified accurately based on fragment ions or the literature, and were classified temporarily as “unknown,” all of which were potential new metabolites.
3.3 Fragmentation pathway of catechins and derivatives
To identify the derivatives of catechins in the processing of RM, standards of catechin and epicatechin were first analyzed by MS2 using the UPLC-Q-Exactive Plus mass spectrometer under the condition mentioned above.
Catechin (B2-1, tR = 5.26 min) and epicatechin (B2-2, tR = 8.70 min) had an [M-H]− ion at m/z 289.0726 (C15H13O6) with MS2 ions at m/z 289.0721 (C15H13O6), 271.0620 (C15H11O5), 245.0810 (C14H13O4), 227.0705 (C14H11O3), 205.0498 (C11H9O4), 203.0706 (C12H11O3), 187.0391 (C11H7O3), 179.0340 (C9H7O4), 165.0183 (C8H5O4), 161.0698 (C10H9O2), 151.0390 (C8H7O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). The fragmentation pathway we postulated is shown in Figure 5. Among them, ions at m/z 289.0726 and 205.0498 were the identification ions of the monomer of catechin metabolites.
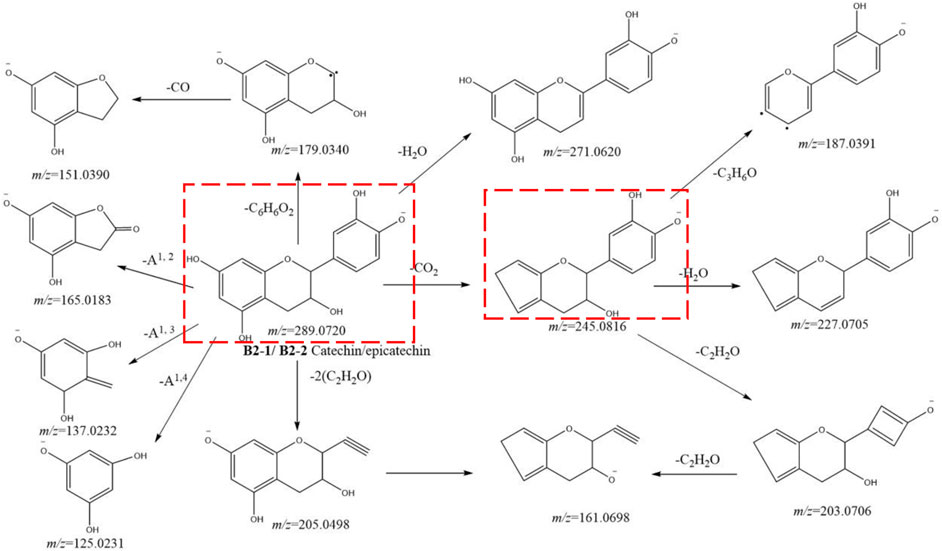
FIGURE 5. Postulated fragmentation pathways of metabolites B2-1 to B2-2. The ion fragments in the red box are characteristic ion fragments of monomer catechin metabolites.
Metabolites B1-1 and B1-2 showed the same [M-H]− ion at m/z 137.0233 (C7H5O3) and MS2 spectra gave ions at m/z 137.0233 (C7H5O3). Comparison with the literature suggested metabolites B1-1 and B1-2 were protocatechualdehyde.
Metabolites B3-1, B3-2, and B3-3 showed the same [M-H]− ion at m/z 305.0668 (C15H13O7), 16 Da higher than that of metabolites B2. MS2 spectra gave ions at m/z 305.0665 (C15H13O7), 261.0771 (C14H13O5), 243.0663 (C14H11O4), 221.0450 (C11H9O4), 219.0665 (C12H11O4), 179.0340 (C9H7O4), 165.0183 (C8H5O4), 137.0232 (C7H5O3), and 125.0231 (C6H5O3), which were 16 Da higher than those of metabolites B2. Comparison with the literature and the relative tR suggested metabolite B3-1 to be gallic catechin and metabolites B3-2 and B3-3 to be epigallocatechin.
Metabolites B4-1 and B4-2 showed the same [M-H]− ion at m/z 441.0833 (C7H5O3) and MS2 spectra gave ions at m/z 289.0721 (C15H13O6), 245.0810 (C14H13O4), 227.0705 (C14H11O3), 205.0498 (C11H9O4), and 169.0134 (C7H5O5). Comparison with the literature suggested that metabolites B4-1 and B4-2 were catechin-O-galloyl. Similarly, metabolites B6-1 and B6-2 were characterized as gallic catechin-O-galloyl because the [M-H]− ions at m/z 457.0781 (C22H17O11) and MS2 spectra gave identical ions at m/z 305.0668 (C15H13O7) and 169.0134 (C7H5O5).
Metabolites B5-1 and B5-2 showed the same [M-H]− ion at m/z 451.1249 (C21H23O11) and MS2 spectra gave ions at m/z 289.0721 (C15H13O6), 245.0810 (C14H13O4), 205.0498 (C11H9O4), and 179.0335 (C9H7O4). The ion m/z 289.0721 was derived from the loss of C6H10O5 (hexoside). Thus, metabolites B5-1 and B5-2 were identified as catechin-O-glycoside.
Metabolites B7-1 and B7-2 gave a [M-H]− ion at m/z 561.1405 (C30H25O11) and product ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 205.0497 (C11H9O4), 179.0337 (C9H7O4), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). As described for metabolites A56, the characteristic fragment ion was a procyanidin. By comparison with the literature, metabolites B7-1 and B7-2 were tentatively characterized to be fisetinidol-(4α,8)-catechin. Similarly, metabolites B8-1 to B8-4 were characterized as procyanidin A, because the [M-H]− ion at m/z 575.1201 (C30H23O12) and MS2 spectra gave ions at m/z 575.1201 (C30H23O12), 539.0986 (C30H19O10), 449.0883 (C24H17O9), 407.0770 (C22H15O8), 289.0721 (C15H13O6), 285.0407 (C15H9O6), 241.0504 (C14H9O4), and 125.0231 (C6H5O3). And metabolites B10-1 to B10-5 showed the same [M-H]− ion at m/z 591.1150 (C30H23O13), 16 Da higher than that of metabolites B8. MS2 spectra gave ions at m/z 555.0932 (C30H19O11), 465.0829 (C24H17O10), 407.0770 (C22H15O8), 327.0509 (C17H11O7), 301.0354 (C15H9O7), 289.0721 (C15H13O6), and 165.0182 (C8H5O4). The ions of 555.0932 and 465.0829 were 16 Da higher than those of metabolites B2. Therefore, metabolites B10-1 to B10-5 were characterized as procyanidin A-OH.
Metabolites B9-1 to B9-10 showed the same [M-H]− ion at m/z 557.1359 (C30H25O12) and MS2 spectra gave ions at m/z 451.1046 (C24H19O9), 425.0879 (C22H17O9), 407.0770 (C22H15O8), 381.0972 (C21H17O7), 339.0870 (C19H15O6), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 225.0500 (C11H9O3), 179.0335 (C9H7O4), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). By comparison with the literature, metabolites B9-1 to B9-10 were identified as procyanidin B2 (Figure 6) (Qiu, et al., 2013). Similarly, metabolites B11-1 to B11-4 were characterized as procyanidin, because of the [M-H]− ion at m/z 593.1309 (C30H25O13) and MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 285.0407 (C15H9O6), 245.0810 (C14H13O4), 177.0183 (C9H5O4), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
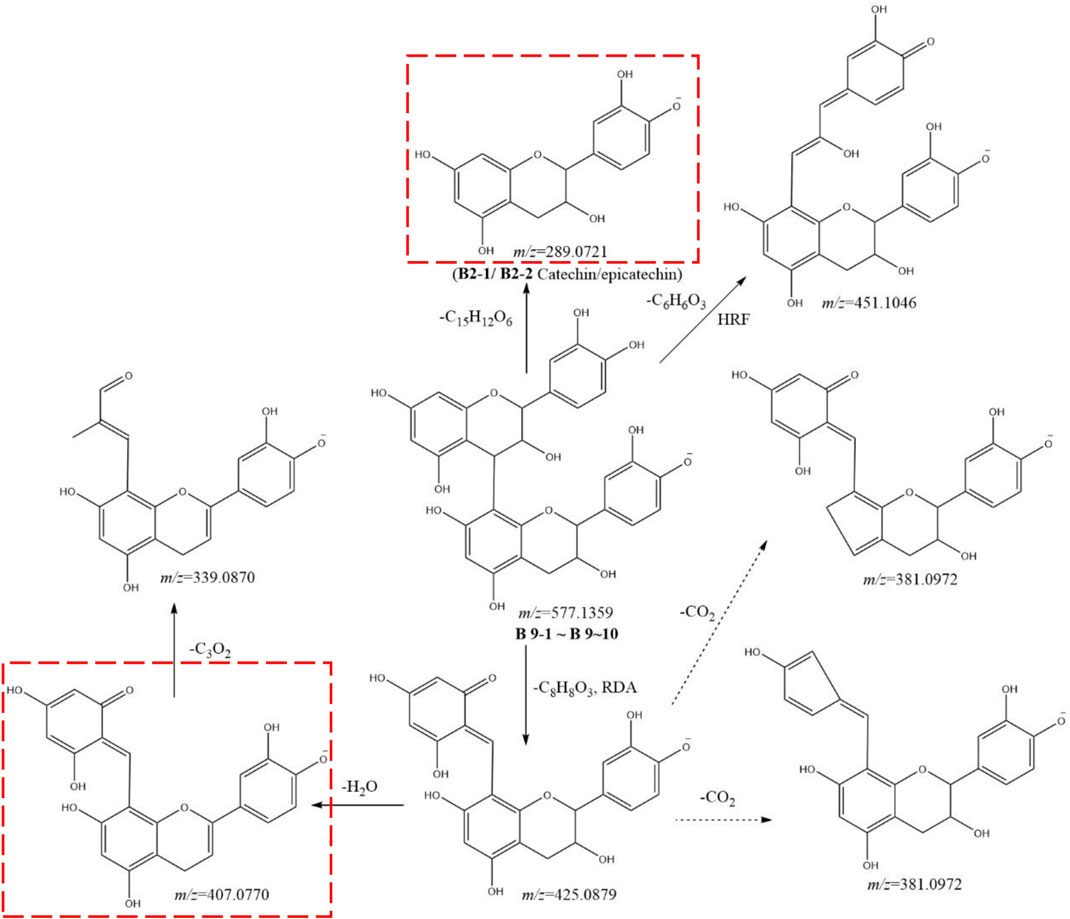
FIGURE 6. Postulated fragmentation pathways of metabolites B9-1 to B9-10. The ion fragments in the red box are characteristic ion fragments of oligomer catechin metabolites. RDA: retro Diels–Alder reaction; HRF: heterocyclic ring fission.
Metabolites B13-1 to B13-5 showed the same [M-H]− ion at m/z 695.1990 (C35H35O15) and MS2 spectra gave ions at m/z 601.1569 (C29H29O14), 439.1029 (C23H19O9), 393.0973 (C22H17O7), 289.0721 (C15H13O6), and 245.0810 (C14H13O4). By comparison with literature, metabolites B13-1 to B13-5 were identified as unknown. Similarly, metabolites B15-1, B15-2, and B15-3 were tentatively characterized as metabolites B13-OH. Since the [M-H]− ions at m/z 711.1940(C35H35O16) and MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0810 (C14H13O4), 177.0183 (C9H5O4),169.0134 (C7H5O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
Metabolite B14 gave a [M-H]-ion at m/z 697.1573 (C37H29O14) and MS2 spectra gave ions at m/z 407.0772 (C22H15O8), 315.0879 (C17H15O6), 289.0721 (C15H13O6), 271.0610 (C15H11O5), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3). By comparison with literature, metabolite B14 was tentatively characterized to be procyanidin B2-O-hydroxybenzoyl. Similarly, metabolites B16-1 and B16-2 were characterized to be procyanidin B2-O-dihydroxybenzoyl. Since the [M-H]− ion at m/z 713.1523 (C37H29O15) was 16 Da (O) higher than that of metabolite B14, and MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 271.0610 (C15H11O5), 245.0810 (C14H13O4), 229.0491 (C13H9O4), 137.0232 (C7H5O3), and 125.0231 (C6H5O3).
Metabolites B20-1 to B20-7 showed same [M-H]− ion at m/z 865.1994 (C45H37O18) and MS2 spectra gave identical ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 245.0825 (C14H13O4), 179.0335 (C9H7O4), 161.0233 (C9H5O3), 137.0232 (C7H5O3), and 125.0231 (C6H5O3), respectively. Comparison with the literature suggested metabolites B20-1 to B20-7 to be procyanidin C. Similarly, metabolites B24-1 to B24-3 were characterized as cinnamtannin A2 because of the [M-H]− ion at m/z 1,153.2628 (C60H49O24) and because MS2 spectra gave ions at m/z 407.0770 (C22H15O8), 289.0721 (C15H13O6), 287.0568 (C15H11O6), 243.0298 (C13H7O5), and 125.0231 (C6H5O3).
Some metabolites had both catechin and gallic-acid substituents and were identified and analyzed in gallic acid metabolites. These included metabolites B12-1 to B12-6 and A43-1 to A43-6, B17-1 to B17-5 and A56-1 to A56∼5, B18-1 to B18-12 and A57-1 to A57-12, B19-1 to B19-5 and A59-1 to A59-5; B21-1 to B21-7 and A60-1 to 60-7, B22-1 to B22-3 and A61-1 to A61-3, B23-1 to B23-10 and A62-1 to A62-10, B25-1 to B25-6 and A63-1 to A63-6.
3.4 Mechanisms of chemical transformation
Several studies have demonstrated that the contents of catechin and epicatechin decrease sharply (and even disappear) after processing, whereas the content of gallic acid increases. Liliang et al. showed that procyanidin was depolymerized into monomeric metabolites, catechin, epicatechin, and epigallocatechin. Then, these monomeric metabolites at high temperatures and high humidity produced protocatechualdehyde and gallic acid. The general metabolic profile was such that our more nuanced resolution identified catechins and gallic acid-based metabolites. Of these, 259 metabolites were based on gallic acid, and 112 metabolites were based on catechins. Many metabolites were dehydrated, hydrolyzed, or subjected to more complex reactions during processing. We speculated that procyanidins (including dimers, trimers, and tetramers) generated monomeric metabolites of catechin, epicatechin, gallate catechin, epigallocatechin, and epicatechin due to isomerization during processing at a high temperature and high humidity (Li, et al., 2018). The C-ring, C3-site acyl group of gallate-type metabolites was prone to deglutamylation and hydrolysis reactions.
The phenolic hydroxyl group of the B-ring passes through the “catechin-O-quinone-macromolecular polymer” pathway readily, so oxidation, polymerization, and condensation reactions occur and a yellow-brown oxidation product is generated (Andrew et al., 2008). It is presumed that catechin monomeric metabolites in RM form brown (and even black-brown) products via this pathway at a high temperature and high humidity. Also, there is partial decomposition of monomeric metabolites into protocatechualdehyde and gallic acid (Li, et al., 2018). This action also leads to a dramatic decrease in the number of catechins with increasing processing time, which may even be undetectable on equipment with suboptimal detection.
A hydrolysis reaction occurs on the acyl group of gallic acid metabolites. Several metabolites of gallic acid will undergo a hydrolysis reaction. Hence, the content of gallic acid should increase obviously. In reality, the content of gallic acid increases but not very obviously or in a regular fashion. Hence, gallic acid may be consumed by other pathways. Gallic acid may be involved in the Maillard reaction during processing, but so may monosaccharides and amino acids, to generate melanoid metabolites.
The color change during the processing of RM should result from the two pathways: Maillard reaction and browning reaction (the appearance and cross-sectional color are shown in Supplementary Table S2), in which many small-molecule metabolites (e.g., monosaccharides, amino acids, gallic acid, catechins) are consumed.
3.5 Trend in content change of gallic acid- and catechin based metabolites
The peak areas of all metabolites were based on the extracted ion chromatographic peaks. Mean values were calculated and the column diagram of each metabolite was drawn, as shown in Supplementary Table S1. In the latter: the black image indicates that the peak area of the metabolites decreases with processing time; the red image indicates that the peak area first increases and then decreases with processing time; the blue image indicates that the peak area increases with processing time; the green image indicates that the change is not significant with processing time. A total of 259 gallic acid metabolites were identified. Of these; 157 gallic acid metabolites had peak areas that decreased with processing time; 71 metabolites had peak areas that increased first and then decreased; 14 metabolites had peak areas that increased with processing time; 17 metabolites had peak areas that did not change significantly during processing. We summed the peak areas of each part. The proportion that decreased with processing time was 48.24%, the proportion that increased with processing time was 36.73%, the proportion that increased first and then decreased was 13.57%, and the proportion that did not change significantly was 1.46%. The content of gallic acid increased with processing time, and the total area of a single metabolite accounted for 35.64%. However, according to the histogram, the increase in the peak area was not very high. In addition to gallic acid, the total area of the other 13 metabolites accounted for 1.09%. This observation bolstered our inference that many gallic acid metabolites lose the galloyl group and form gallic acid during processing, while some gallic acid participates in the Maillard reaction.
We identified 112 catechin metabolites. Of these: 81 catechin metabolites had peak areas that decreased with processing time; 30 metabolites had peak areas that increased first and then decreased; one metabolite had a peak area that increased with processing time. We summed the peak areas of each part. The proportion that decreased with processing time was 92.57%, the proportion that increased with processing time was 0.02%, and the proportion that increased first and then decreased was 7.41%. The content of many procyanidins decreased (or even disappeared) during processing, and the content of most of them decreased sharply after 4 h of processing. This process produced catechin, epicatechin, and other metabolites, and the content of these metabolites also decreased gradually. The content of catechin and epicatechin decreased sharply after 4 h of processing and nearly disappeared after 18–24 h of processing. Some studies [14, 17] have shown that catechin and epicatechin break down into protocatechuic aldehyde and gallic acid at a high temperature, but the peak area of protocatechuic aldehyde decreases with processing time. In summary, metabolites such as procyanidins, catechin, and epicatechin are thermally unstable. They break down into many small-molecule metabolites in the early stage of processing, and small-molecule metabolites participate rapidly in the Maillard reaction or other darkening reactions (Figure 7). The literature suggests that procyanidins have antioxidant, anti-inflammatory, immunoregulatory, hepatoprotective, and other effects, but the present study showed that the number of procyanidins decreased (or even disappeared) with processing. The antioxidant effect of RRM is much higher than that of PRM (Gu, et al., 2005; Pu, 2021). Hence, the effects of RM on tonifying the liver and kidneys, nourishing blood essence, and anti-aging are not likely to be related to procyanidins.
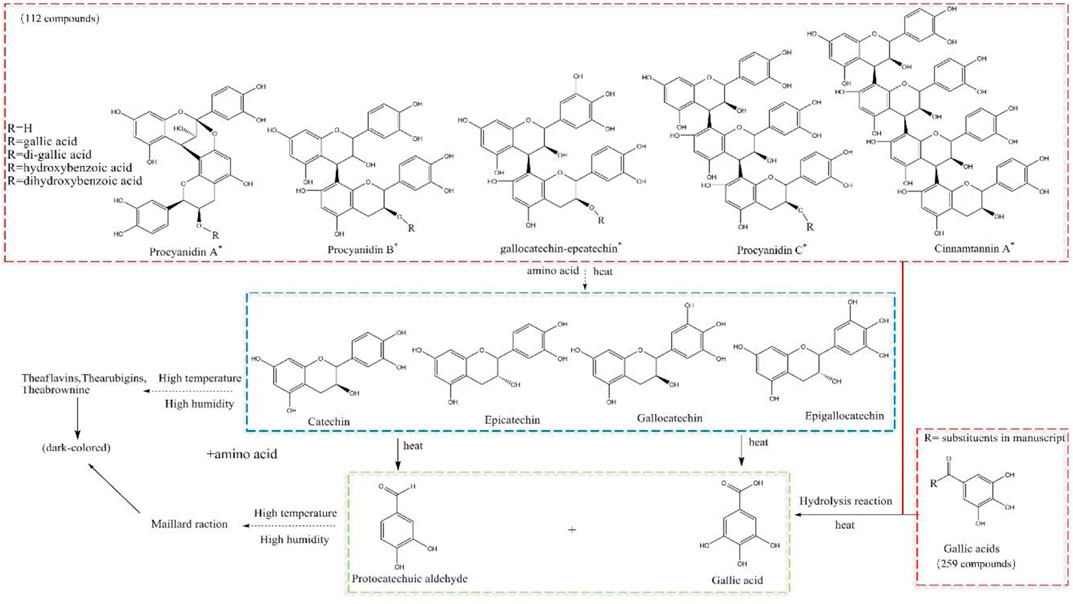
FIGURE 7. Postulated transformation mechanism of gallic-acid and catechin metabolites during processing, and speculation of surface and section browning.
4 Discussion
In this study, gallic acid and catechin metabolites were identified, and metabolic pathways were analyzed by MS2. In the latter, gallicacid metabolites were distinguished by fragment ions, accurate measurement of mass, and fragment pathways of m/z 313.0564 and 169.0133. The identification of catechins was relatively complicated. Catechin monomers were identified as diagnostic fragment ions at m/z 289.0720 and 245.0816, or diagnostic fragment ions involving oxygenation. Dimers, and trimers at m/z 407.0770, 289.0720, and 245.0816. Linked galloyl would also have diagnostic fragment ions from gallic acid. Next, we extracted the ion chromatogram at m/z 313.0564 and 289.0720. Then, we set the extraction-ion atlas at ppm <5. Next, we extracted their secondary cracking ions and parent ions one by one to ensure the complete identification of gallic acids and catechin metabolites. Finally, 259 gallic acid and 112 catechin metabolites were identified. The peak areas of 157 gallic acids (accounting for 48.24% of the total area) and 81 catechins (accounting for 92.47% of the total area) decreased gradually. The peak areas of 71 gallic acids (13.57%) and 30 catechins (7.41%) increased first and then decreased. The peak areas of 14 gallic acids (36.73%, of which gallic acid accounted for 35.64%) and one catechin (0.02%) increased gradually, and 17 gallic acids (1.46%) showed no significant change. These data indicated that the processing of RM involved the participation of chemical groups and that the change process was extremely complicated.
We speculate that many gallic-acid metabolites are hydrolyzed to produce gallic acid and that the dimers/trimers of catechins are cleaved into catechins, epicatechin, gallic-acid catechins, and epicatechin monomers, followed by being cleaved into gallic acid and protocatechualdehyde under a high temperature and high humidity and, subsequently, participating in the Maillard reaction and browning reactions, which two reactions deepen the color of RM. Many studies support our speculation (Nonaka et al., 1982; Torres et al., 2002; Wang et al., 2006; li et al., 20,122; Nie, 2017), and similar speculations have also been made in studies related to Polygonum multiflorum (Li et al., 2018). We are conducting a comprehensive identification and analysis of the changes in the binding content of metabolites for speculation.
5 Conclusion
This article provides an in-depth identification of the metabolites of gallic acid and catechins, and investigates their transformation during processing. We found that the content of catechins decreased (or even disappeared) after processing, and although the content of gallic acid increased, the amount of gallic acid metabolites also decreased significantly, as reported in literature (Liu et al., 2009; Li et al., 2022). Anthocyanins have antioxidant, anti-inflammatory, immunomodulatory, and hepatoprotective effects. There are also studies on the stronger antioxidant activity of RRM (Liu et al., 2009; Yu et al., 2012), indicating that processing can significantly reduce anthocyanins. Therefore, the role of RM in tonifying the liver and kidney, nourishing blood essence, and anti-aging is not related to anthocyanins. The regularity study of gallic acid and catechins at different processing times in this article can guide the research of processing technology and endpoint. For the study of RM efficacy and toxicity, attention should be paid to the degree of processing of RM and the significant reduction of original catechins, in order to obtain true efficacy and toxicity results.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: http://www.gpgenome.com/species/378.
Author contributions
JB: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing–original draft, Writing–review and editing. QZ: Data curation, Investigation, Software, Writing–original draft. HS: Formal Analysis, Software, Validation, Writing–original draft. BL: Conceptualization, Formal Analysis, Resources, Software, Supervision, Validation, Writing–original draft. PW: Resources, Writing–original draft. JH: Data curation, Formal Analysis, Funding acquisition, Validation, Writing–original draft. DZ: Investigation, Project administration, Software, Validation, Writing–original draft. LG: Methodology, Resources, Validation, Writing–original draft. WX: Methodology, Project administration, Software, Writing–original draft. JZ: Data curation, Formal Analysis, Methodology, Project administration, Software, Writing–original draft. ZH: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Writing–original draft. XQ: Funding acquisition, Project administration, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (81373967, 82204622), Quality standard system construction for the whole industry chain of Chinese medicinal detection pieces from Guangdong Provincial Drug Administration of China (2019KT1261/2020ZDB25).
Acknowledgments
We thank LetPub for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
Author PW was employed by Shanghai Dehua Traditional Chinese Medicine Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1356876/full#supplementary-material
References
Bai, J., Chen, W., Huang, J., Su, H., Zhang, D., Xu, W., et al. (2022). Transformation of stilbene glucosides from Reynoutria multiflora during processing. Front. Pharmacol. 13, 757490. doi:10.3389/fphar.2022.757490
Bai, J., He, S., Liang, Y., Shi, X., Huang, J., Xu, W., et al. (2021). Screening of quality markers during the processing of Reynoutria multiflora based on the UHPLC-Q-Exactive plus orbitrap MS/MS metabolomic method. Front. Pharmacol. 12, 695560. doi:10.3389/fphar.2021.695560
Chen, H., Chen, Y., Liu, C., Wang, L., Chen, X., Yu, B. Y., et al. (2016). Integrated chemometric fingerprints of antioxidant activities and HPLC–DAD–CL for assessing the quality of the processed roots of Polygonum multiflorum Thunb. (Heshouwu). Chin. Med. 11, 18. doi:10.1186/s13020-016-0087-8
Chen, Q. T., Zhuo, L. H., Xu, W., Huang, Z. H., and Qiu, X. H. (2012). Content changes of 5 components in Polygonum multiflorum during processing. Chin. J. Exp. Tradit. Med. Form. 18, 66–71.
Cheung, F. W., Leung, A. W., Liu, W. K., and Che, C. T. (2014). Tyrosinase inhibitory activity of a glucosylated hydroxystilbene in mouse melan-A melanocytes. J. Nat. Prod. 77, 1270–1274. doi:10.1021/np4008798
Chinese Pharmacopoeia Commission (2020). Pharmacopoeia of the people’s Republic of China. Beijing: China Medical Science and Technology Publishing House, 184.
Clark, A. C. (2008). The production of yellow pigments from(+)-catechin and dihydroxyfumaric acid in a model wine system. Eur. Food Res. Technol. 226 (4), 925–931. doi:10.1007/s00217-007-0615-y
Han, D. Q., Zhao, J., Xu, J., Peng, H. S., Chen, X. J., and Li, S. P. (2013). Quality evaluation of Polygonum multiflorum in China based on HPLC analysis of hydrophilic bioactive compounds and chemometrics. J. Pharm. Biomed. Anal. 72, 223–230. doi:10.1016/j.jpba.2012.08.026
Jin Gu, P. L., and Ma, F. (2005). Study on the antioxidation activity of different processed products of Polygonum multiflorum Thunb. China Pharm. 11, 875–876. doi:10.3969/j.issn.1001-0408.2005.11.033
Komatsu, Y., Shinichi, S., Yoshihiro, H., Saigo, H., Matsuda, R., and Hara, K. (1993). Effects of pH and temperature on reaction kinetics of catechins in green tea infusion. Biosci. Biotechnol. Biochem. 57 (6), 907–910. doi:10.1271/bbb.57.907
Li, C., Niu, M., Bai, Z., Zhang, C., Zhao, Y., Li, R., et al. (2017). Screening for main components associated with the idiosyncratic hepatotoxicity of a tonic herb, Polygonum multiflorum. Polyg. Multiflorum. Front. Med. 11, 253–265. doi:10.1007/s11684-017-0508-9
Li, Na, Taylor, L. S., Ferruzzi, M. G., and Mauer, L. J. (2018). Color and chemical stability of tea polyphenol (-)-epigallocatechin-3-gallate in solution and solid states. Food Res. Int. 53 (2), 909–921. doi:10.1016/j.foodres.2012.11.019
Li, Y., Wang, Y., Zhang, N., Yang, J., Liu, Y., Wang, Q., et al. (2022). Effects of duration of steaming on 26 chemical compositions of Polygoni Multiflori Radix based on UPLC-MS/MS detection technology. Chin. J. Pharmacovigil. 19 (12), 1295–1302. doi:10.19803/j.1672-8629.2022.07.24
Liang, L., Xu, J., Zhou, W., Brand, E., Chen, H. B., and Zhao, Z. Z. (2018). Integrating targeted and untargeted metabolomics to investigate the processing chemistry of polygoni multiflori radix. Front. Pharmacol. 9, 934. doi:10.3389/fphar.2018.00934
Liang, Z., Chen, H., Yu, Z., and Zhao, Z. (2010). Comparison of raw and processed Radix Polygoni Multiflori (Heshouwu) by high performance liquid chromatography and mass spectrometry. Chin. Med. 5, 29–9. doi:10.1186/1749-8546-5-29
Lin, L., Ni, B., Lin, H., Zhang, M., Li, X., Yin, X., et al. (2015). Traditional usages, botany, phytochemistry, Pharmacology and toxicology of Polygonum multiflorum Thunb.: a review. J. Ethnopharmacol. 159, 158–183. doi:10.1016/j.jep.2014.11.009
Liu, Z., Liu, Y., Wang, C., Guo, N., Song, Z., et al. (2011). Comparative analyses of chromatographic fingerprints of the roots of Polygonum multiflorum Thunb and their processed products using RRLC/DAD/ESI-MSn. Planta Med. 77, 1855–1860. doi:10.1055/s-0030-1271200
Liu, Z., Song, Z., Cao, Z., Wang, C., Lv, S., Wang, C., et al. (2009). HPLC determination of the changes in the content of antioxidant active ingredients gallic acid and catechins in Polygonum multiflorum before and after processing. Chin. Tradit. Pat. Med. 31 (9), 1392–1394.
Nie, Y. (2017). Study on heat- and photo-induced chemical conversions of catechins and relevant mechanisns. Zhejiang University. MA thesis.
Nonaka, G.-L., Miwa, N., and Nishioka, I. (1982). Stilbene glycoside gallates and proanthocyanidins from Polygonum multiflorum. Phytochemistry 21, 429–432. doi:10.1016/S0031-9422(00)95282-8
Pu., J. (2021). Study on antioxidant activity of different processed Polygonum multiflorum in vitro. Hunan University of Chinese Medicine. doi:10.27138/d.cnki.ghuzc.2021.000215
Qiu, X., Zhang, J., Huang, Z., Zhu, D., and Xu, W. (2013). Profiling of phenolic constituents in Polygonum multiflorum Thunb. by combination of ultra-high-pressure liquid chromatography with linear ion trap-Orbitrap mass spectrometry. J. Chroma A 1292, 121–131. doi:10.1016/j.chroma.2012.11.051
Ross, C. F., Hoye, C., and Fernandez-Plotka, V. C. (2011). Influence of heating on the polyphenolic content and antioxidant activity of grape seed flour. J. Food Sci. 76, 884–890. doi:10.1111/j.1750-3841.2011.02280.x
Torres, J. L., Lozano, C., Julià, L., Sánchez-Baeza, F. J., Anglada, J. M., Centelles, J. J., et al. (2002). Cysteinyl-flavan-3-ol conjugates from grape procyanidins. Antioxidant and antiproliferative properties. Bioorg. Med. Chem. 10, 2497–2509. doi:10.1016/S0968-0896(02)00127-X
Wang, R., Zhou, W., and Wen, R. H. (2006). Kinetic study of the thermal stability of tea catechins in aqueous systems using a microwave reactor. J. Agric. Food Chem. 64 (16), 5924–5932. doi:10.1021/jf0611419
Wang, T., Zhang, J., Qiu, X., Bai, J. Q., Gao, Y. H., and Xu, W. (2016). Application of ultra-high-performance liquid chromatography coupled with LTQ-orbitrap mass spectrometry for the qualitative and quantitative analysis of Polygonum multiflorum thumb. And its processed products. Molecules 21, E40. doi:10.3390/molecules21010040
Yao, S., Li, Y., and Kong, L. (2006). Preparative isolation and purification of chemical constituents from the root of Polygonum multiflorum by high-speed counter-current chromatography. J. Chromatogr. A 1115, 64–71. doi:10.1016/j.chroma.2006.02.071
Yu, J., Xie, J., Mao, X., Wei, H., Zhao, S. L., Ma, Y. G., et al. (2012). Comparison of laxative and antioxidant activities of raw, processed and fermented Polygoni Multiflori Radix. Chin. J. Nat. Med. 10 (1), 63–67. doi:10.1016/S1875-5364(12)60014-4
Keywords: Reynoutria multiflora (Thunb.) Moldenke, gallic acid, catechin, processing, tandem mass spectrometry
Citation: Bai J, Zou Q, Su H, Liao B, Wang P, Huang J, Zhang D, Gong L, Xu W, Zhang J, Huang Z and Qiu X (2024) Processing of Reynoutria multiflora: transformation of catechin and gallic acid derivatives and their identification. Front. Pharmacol. 15:1356876. doi: 10.3389/fphar.2024.1356876
Received: 16 December 2023; Accepted: 19 January 2024;
Published: 26 February 2024.
Edited by:
Rong-Rong He, Jinan University, ChinaReviewed by:
Tushar Dhanani, Florida Agricultural and Mechanical University, United StatesQing-Feng Zhang, Jiangxi Agricultural University, China
Copyright © 2024 Bai, Zou, Su, Liao, Wang, Huang, Zhang, Gong, Xu, Zhang, Huang and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihai Huang, emhodWFuZzczMDhAMTYzLmNvbQ==; Xiaohui Qiu, cWl1eGlhb2h1aUBnenVjbS5lZHUuY24=