Abstract
Colchicine binds to tubulin and destabilizes microtubules, stopping cell division and causing apoptosis. Its anti-cancer property affects microtubule integrity despite its reported toxicity. A series of novel colchicine derivatives were synthesized using the multi-component reaction method and evaluated for their antiproliferative properties, aiming to enhance their efficacy as anti-cancer agents compared to the parent compound, colchicine. With the SRB assay, we tested these derivatives for their anti-cancer efficacy against lung, breast, and melanoma using several human cancer cell lines, including A549, MCF-7, MDAMB-231, and A375. The study identified a derivative, 3g, as notably more effective against melanoma cells, with a selectivity index about two times higher than colchicine. We further investigated the anti-cancer efficacy of compound 3g on human melanoma cells using additional in vitro models, including the wound healing assay and colony formation assay. Compound 3g inhibited colony formation by up to 62.5% and reduced the migration potential of melanoma cells by 69%. The in silico studies reveal the probable interactions with the colchicine binding sites that have a comparable pose to colchicine. 3g formed a hydrogen bond with Cys241, Asn258 and a salt bridge with Lys352, which is important for its tubulin polymerization inhibitory activity. These findings suggest that 3g could selectively target melanoma cells, minimizing toxicity to healthy cells and potentially providing a safer and more effective treatment option with improved therapeutic outcomes.
1 Introduction
Colchicine is a tropolone alkaloid natural product and an FDA-approved drug to treat gout and familial Mediterranean fever (Schlesinger et al., 2020). It is an antimitotic compound that binds to β-tubulin, destabilizes microtubules, and promotes depolymerization, leading to cell cycle arrest, apoptosis, and cell death (Ghawanmeh et al., 2018). Despite its ability to induce apoptosis, colchicine is not widely used as an anticancer drug, due to relatively high toxicity in the gastrointestinal tract, which is one of the three phases of toxicity that limit the use of certain compounds in cancer chemotherapy (Banerjee et al., 1997; Imazio et al., 2015; Tardif et al., 2022). Chemical modifications, derivatizations, and structure optimizations of colchicine at rings A, B, and C in colchicine result in functionality conversion and affect biological activity, which improved its therapeutic potential and safety profile (Singh et al., 2015; Dubey et al., 2017; Gracheva et al., 2020; Iqbal Lone et al., 2024). In recent years, a low-dose colchicine tablet branded as Lodoco was the first anti-inflammatory atheroprotective therapy for cardiovascular disease to reduce the risk of myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adults (Banach and Penson, 2021; Halsey, 2023). Therefore, colchicine and its derivatives promise the potential of developing novel anti-cancer agents and therapeutic approaches (Kumar et al., 2017).
Cancer is a global public health challenge, marked by high prevalence, diverse types, and increasing incidence (Arnold et al., 2020). Advancing cancer drug discovery is imperative for improving treatments and reducing its impact on individuals and societies (Huang et al., 2021). Melanoma is an aggressive cancer with a high potential for metastasis. When it undergoes malignant transformation, it develops into one of the most lethal variants of skin cancer. While surgical excision is a frequently used successful treatment for early-stage melanomas, advanced cases have a grim prognosis, with an average survival duration of three to 11 months. Recent high-throughput genomic screening has shown the diverse character of melanoma by identifying several mutations driving its progression, and the cytotoxic effects and multidrug resistance linked to existing chemotherapeutic drugs restrict the treatment options for melanoma. Even though melanoma accounts for only 5% of all skin cancer cases, it is the cause of over 75% of skin cancer-related deaths, and the rate of incidence of melanoma is still on the rise (Smalley, 2010; Khan et al., 2023).
In this study, we synthesized novel colchicine derivatives using the Biginelli Multi-component Reaction (MCR) method, which has been vastly valuable for drug discovery (Kaur et al., 2017; Grazia Martina et al., 2023; Shahi et al., 2024) and screened them for antiproliferative activity against a panel of human cancer cell lines.
2 Experimental procedure
2.1 Extraction and isolation of colchicine and synthesis of colchicine aldehyde
A dried tuber (50.0 gm) of Gloriosa superba L. was extracted with MeOH: H2O (1:1) and colchicine was isolated by column chromatography using neutral alumina loaded with hexane. The eluting solvents chloroform and toluene in a ratio of 97:3. The fractions containing colchicine were collected, separated, and compared with standard colchicine, identified by thin layer chromatography (TLC); the fraction was evaporated in rotavaporatory and crystallized using chloroform as described in this procedure (Kannan et al., 2007). Purity of colchicine was determined by HPLC, as shown in Figure 1. Then, colchicine (1.0 mmol) was dissolved in dichloromethane (DCM, 25 mL) in a round-bottom flask, and dichloromethyl methyl ether (1.0 mmol) was added to the solution. The reaction mixture was stirred at 0°C for 30 min. After that, tin chloride (1.5 mmol) was added drop by drop to the reaction mixture, and stirring was continued at room temperature for 3.0 h. Once the reaction was complete (monitored by TLC), it was slowly quenched with ice water. The product was extracted with ethyl acetate, and the combined organic layer was dried with anhydrous sodium sulfate. The solution was concentrated using a rotary evaporator, and the residue was purified by silica gel column chromatography using hexane and ethyl acetate to obtain the pure product (Iqbal Lone et al., 2024).
FIGURE 1
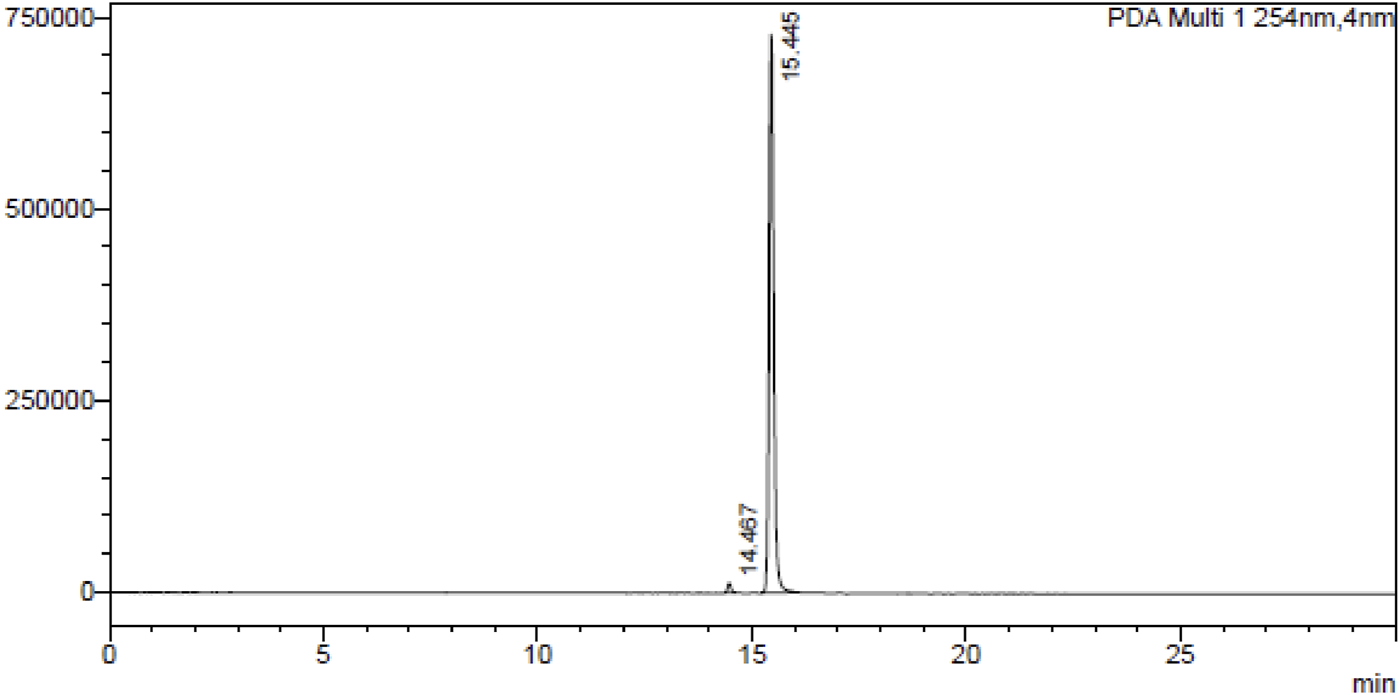
HPLC chromatogram of colchicine at λ254 nm.
2.2 Synthesis of colchicine derivatives
For the preparation of colchicine derivatives 3a to 3j, colchicine aldehyde (2) (1.0 mmol) and β-keto ester (2.0 mmol) were dissolved in ethanol (25 mL), taken in a round bottom flask after ammonia/urea/thiourea was added (2.0 mmol) dropwise. Then zinc chloride was added in a catalytic amount (0.3 mmol) and refluxed the mixture at 80°C for 5 h. After the reaction (product monitored by TLC) was completed, it was quenched slowly with sodium bicarbonate dissolved in water (for neutralizing acid). The pure product was obtained after column chromatography.
Compound 3a: 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.29–7.20 (m, 2H), 7.10 (d, J = 10.6 Hz, 1H), 4.38 (s, 1H), 3.93 (d, J = 4.4 Hz, 1H), 3.89 (d, J = 7.5 Hz, 3H), 3.81 (dd, J = 16.6, 8.2 Hz, 6H), 3.45 (d, J = 3.9 Hz, 3H), 2.16 (d, J = 16.6 Hz, 3H), 2.02 (dd, J = 17.5, 12.3 Hz, 1H), 1.90 (s, 3H), 1.72–1.62 (m, 1H), 1.19 (d, J = 7.4 Hz, 3H), 0.91–0.79 (m, 3H). 13C NMR (101 MHz, MeOD) δ 179.56, 171.32, 166.14, 164.18, 152.70, 151.02, 136.99, 136.41, 132.93, 129.33, 113.53, 60.23, 60.17, 60.07, 59.53, 55.65, 52.42, 34.87, 31.69, 30.76, 29.38, 29.10, 28.68, 24.63, 22.36, 21.05, 16.74, 13.19, 13.08. LC-MS (ESI) calcd for Chemical Formula: C30H35N3O8S 598.2223 and, found 598.2226.
Compound 3b: Ethyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-2-imino-6-methyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.28–7.21 (m, 2H), 7.09 (dd, J = 11.0, 4.4 Hz, 1H), 4.37 (dd, J = 11.8, 6.0 Hz, 1H), 3.89 (d, J = 8.3 Hz, 3H), 3.81 (d, J = 4.4 Hz, 5H), 3.44 (d, J = 7.4 Hz, 3H), 2.18 (s, 3H), 2.07–1.96 (m, 1H), 1.90 (d, J = 3.0 Hz, 3H), 1.69 (dd, J = 11.7, 5.8 Hz, 1H), 1.19 (s, 1H), 0.90 (dt, J = 31.2, 7.0 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 179.57, 171.32, 164.16, 152.67, 150.69, 129.70, 129.31, 129.18, 113.54, 60.19, 60.02, 59.34, 59.31, 55.63, 52.41, 52.34, 34.95, 29.39, 24.56, 21.01, 20.98, 17.24, 13.21. LC-MS (ESI) calcd for Chemical Formula: C30H36N4O8 Exact Mass: 581.25, found 582.22.
Compound 3c: phenyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.25–7.19 (m, 1H), 7.10 (d, J = 2.5 Hz, 2H), 7.02 (s, 2H), 6.91–6.81 (m, 2H), 5.00 (dd, J = 26.7, 12.5 Hz, 1H), 4.68 (d, J = 12.2 Hz, 1H), 4.31 (dd, J = 11.7, 6.3 Hz, 1H), 3.92 (s, 3H), 3.75 (d, J = 12.0 Hz, 5H), 3.44 (s, 3H), 2.88 (dd, J = 14.0, 4.8 Hz, 1H), 2.19 (d, J = 18.6 Hz, 3H), 2.09 (s, 1H), 1.84 (d, J = 3.8 Hz, 3H), 1.55–1.39 (m, 1H). 13C NMR (101 MHz, MeOD) δ 179.54, 171.28, 166.05, 164.10, 150.63, 136.50, 136.29, 129.18, 128.04, 127.51, 113.51, 65.23, 60.22, 60.02, 55.70, 52.39, 34.98, 24.12, 21.04, 20.97, 17.37. LC-MS (ESI) calcd for Chemical Formula: C34H35N3O9 Exact Mass: 629.24 and, found 644.30 [M+Na]+.
Compound 3d: Ethyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-2-oxo-6-(trichloromethyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.23 (ddd, J = 23.2, 13.9, 7.6 Hz, 2H), 7.10 (d, J = 10.9 Hz, 1H), 4.40–4.27 (m, 1H), 4.12–3.99 (m, 1H), 3.96–3.92 (m, 2H), 3.91 (s, 3H), 3.89–3.79 (m, 6H), 3.51 (s, 1H), 3.47 (q, J = 4.5 Hz, 3H), 2.23–2.00 (m, 2H), 1.92 (d, J = 2.1 Hz, 3H), 1.81–1.70 (m, 1H), 1.22–1.11 (m, 3H), 1.01–0.67 (m, 2H). 13C NMR (101 MHz, MeOD) δ 184.25, 179.57, 171.37, 164.40, 164.30, 164.26, 153.73, 152.29, 151.82, 136.61, 136.55, 136.49, 136.37, 129.38, 129.31, 113.43, 88.61, 60.40, 60.33, 60.30, 60.22, 60.16, 55.68, 52.16, 51.73, 35.04, 29.37, 21.01, 12.97, 12.61. LC-MS (ESI) calcd for Chemical Formula: C30H32Cl3N3O9 Exact Mass: 683.12 and, found 704.20 [M+Na]+.
Compound 3e: Ethyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-6-(2,6-dichloro-5-fluoropyridin-3-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.71 (ddd, J = 20.5, 10.9, 6.7 Hz, 1H), 7.29–7.23 (m, 2H), 7.10 (d, J = 11.0 Hz, 1H), 4.38 (dd, J = 11.6, 6.0 Hz, 1H), 3.96 (dd, J = 17.5, 8.2 Hz, 3H), 3.91 (s, 3H), 3.86 (d, J = 6.4 Hz, 2H), 3.81 (s, 1H), 3.78–3.70 (m, 1H), 3.62 (ddd, J = 13.9, 7.0, 4.0 Hz, 1H), 3.47 (t, J = 4.1 Hz, 3H), 2.09 (dd, J = 15.1, 5.2 Hz, 1H), 1.90 (s, 3H), 1.74 (dd, J = 17.0, 8.0 Hz, 1H), 1.19 (s, 1H), 0.72–0.60 (m, 3H). 13C NMR (101 MHz, MeOD) δ 179.58, 175.01, 171.33, 164.20, 129.28, 119.17, 116.70, 113.49, 110.59, 60.51, 60.21, 60.16, 60.12, 60.07, 59.92, 58.84, 55.63, 52.43, 52.36, 52.32, 31.15, 29.37, 20.99, 20.65, 12.70. LC-MS (ESI) calcd for Chemical Formula: C34H33Cl2FN4O9 Exact Mass: 731.1687 and, found 731.1685.
Compound 3f: Ethyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-2-oxo-6-(perfluoroethyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.26 (s, 2H), 7.21 (d, J = 10.7 Hz, 2H), 7.10 (d, J = 11.1 Hz, 3H), 5.17 (d, J = 11.2 Hz, 2H), 4.31–4.22 (m, 4H), 3.92–3.89 (m, 15H), 3.87 (d, J = 3.7 Hz, 7H), 3.83–3.75 (m, 10H), 3.47 (s, 6H), 1.91 (s, 8H), 1.19 (s, 4H), 0.83 (t, J = 7.1 Hz, 7H). 13C NMR (101 MHz, MeOD) δ 179.57, 171.18, 169.77, 164.31, 154.65, 152.23, 151.92, 145.88, 136.48, 134.05, 129.44, 129.38, 121.54, 113.43, 61.06, 60.38, 60.26, 60.24, 55.67, 53.42, 52.09, 48.88, 34.71, 25.80, 24.97, 20.95, 13.03. LC-MS (ESI) calcd for Chemical Formula: C31H32F5N3O9 Exact Mass: 685.21 and, found 704.20 [M+Na]+.
Compound 3g: Ethyl 4-((S)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-2-oxo-6-(perfluoroethyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.26 (s, 1H), 7.22 (d, J = 10.8 Hz, 1H), 7.10 (d, J = 11.0 Hz, 1H), 4.37 (dd, J = 11.9, 6.3 Hz, 1H), 4.03 (d, J = 11.2 Hz, 1H), 3.93 (s, 2H), 3.91 (s, 2H), 3.86 (s, 1H), 3.83 (s, 2H), 3.82 (s, 1H), 3.46 (d, J = 1.7 Hz, 3H), 1.94 (s, 1H), 1.91 (s, 2H), 1.84 (s, 1H), 1.19 (s, 1H), 0.77 (d, J = 7.1 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 179.57, 171.38, 169.11, 164.26, 154.53, 152.31, 151.79, 146.18, 136.53, 136.31, 133.99, 129.47, 129.42, 121.91, 113.42, 60.80, 60.20, 60.13, 55.68, 55.66, 52.18, 35.06, 20.98, 12.67. LC-MS (ESI) calcd for Chemical Formula: C31H32F5N3O9 Exact Mass: 685.21 and, found 704.20 [M+Na]+.
Compound 3h: ethyl 4-((R)-7-acetamido-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-4-yl)-6-ethoxy-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate.
1H NMR (400 MHz, MeOD) δ 7.26 (s, 1H), 7.18 (d, J = 10.8 Hz, 1H), 7.09 (d, J = 11.1 Hz, 1H), 5.84 (d, J = 11.3 Hz, 1H), 4.27 (dd, J = 12.0, 6.5 Hz, 1H), 4.17–4.08 (m, 2H), 4.04 (s, 3H), 4.01 (d, J = 11.3 Hz, 1H), 3.90 (s, 3H), 3.76 (ddd, J = 17.9, 12.5, 7.1 Hz, 2H), 3.45 (s, 3H), 2.44–2.32 (m, 1H), 1.97 (dd, J = 13.9, 6.2 Hz, 1H), 1.90 (s, 3H), 1.70 (td, J = 12.1, 5.5 Hz, 1H), 1.18 (t, J = 7.1 Hz, 3H), 0.81 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 179.59, 171.42, 167.37, 166.94, 164.17, 159.21, 153.03, 152.61, 151.09, 144.85, 136.97, 136.10, 132.80, 129.41, 128.89, 124.62, 113.46, 78.08, 61.45, 61.04, 60.22, 60.17, 56.98, 55.62, 52.34, 35.01, 25.11, 21.01, 13.02, 12.76. LC-MS (ESI) calcd for Chemical Formula: C31H37N3O10 Exact Mass: 611.25 and, found 630 [M+NH4]+.
Compound 3i: N-((7R)-4-(5-(cyclopropanecarbonyl)-6-methoxy-2-oxo-1,2,3,4-tetrahydro pyrimidin-4-yl)-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-7-yl) acetamide.
1H NMR (400 MHz, MeOD) δ 7.29–7.22 (m, 2H), 7.09 (dd, J = 10.7, 5.7 Hz, 1H), 5.75 (s, 1H), 4.38 (dd, J = 9.0, 4.9 Hz, 1H), 3.89 (d, J = 11.4 Hz, 3H), 3.82 (d, J = 10.6 Hz, 6H), 3.45 (d, J = 9.3 Hz, 3H), 3.40 (d, J = 17.8 Hz, 3H), 2.83–2.61 (m, 1H), 2.32–2.16 (m, 1H), 2.02 (td, J = 14.1, 6.0 Hz, 1H), 1.90 (d, J = 10.2 Hz, 3H), 1.77–1.63 (m, 1H), 1.19 (s, 1H), 0.84 (d, J = 8.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 179.58, 171.34, 171.30, 167.08, 164.17, 164.15, 152.75, 152.59, 150.72, 150.65, 145.43, 136.37, 132.72, 129.39, 129.33, 129.22, 113.55, 60.18, 60.02, 59.94, 55.63, 52.41, 52.22, 49.93, 49.91, 35.01, 24.17, 21.03, 21.01, 11.36 LC-MS (ESI) calcd for Chemical Formula: C31H35N3O9 Exact Mass: 594.2452 and, found 594.2453.
Compound 3j: N-((7S)-4-(5-(cyclopropanecarbonyl)-6-methoxy-2-oxo-1,2,3,4-tetrahydro pyrimidin-4-yl)-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo [a]heptalen-7-yl) acetamide.
1H NMR (400 MHz, MeOD) δ 7.29–7.23 (m, 2H), 7.10 (dd, J = 11.1, 4.5 Hz, 1H), 4.38 (dd, J = 11.9, 6.1 Hz, 1H), 3.90 (d, J = 9.4 Hz, 3H), 3.84 (s, 3H), 3.80 (d, J = 5.3 Hz, 3H), 3.55 (dd, J = 12.4, 5.5 Hz, 2H), 3.45 (d, J = 3.4 Hz, 3H), 3.39 (d, J = 16.3 Hz, 3H), 2.74–2.65 (m, 2H), 1.99 (dd, J = 14.5, 7.3 Hz, 3H), 1.91 (d, J = 4.0 Hz, 3H), 1.69 (td, J = 11.9, 6.0 Hz, 1H), 1.19 (s, 1H). 13C NMR (101 MHz, MeOD) δ 179.57, 171.35, 166.40, 164.14, 152.59, 150.78, 150.64, 136.48, 132.81, 129.48, 129.25, 129.23, 113.57, 60.23, 60.16, 60.05, 60.02, 55.63, 52.41, 52.24, 49.92, 43.76, 43.68, 31.16, 28.92, 24.20, 21.03. LC-MS (ESI) calcd for Chemical Formula: C31H35N3O9 Exact Mass: 594.2452 and, found 594.2453.
2.3 Antiproliferative study
2.3.1 Cell culture and cell lines
Roswell Park Memorial Institute (RPMI)-1640 medium, Dulbecco’s modified eagle medium high glucose (DMEM; Gibco) supplemented with 10% Fetal Bovine Serum (FBS; Gibco, and 1% antibiotic-antimycotic (Gibco; Anti-Anti 100x), phosphate-buffered saline (PBS; Gibco), 0.25% trypsin (Gibco), sodium pyruvate (Sigma-Aldrich), HEPES (Gibco). Human cancer cell lines A549 (lung carcinoma), MCF-7 (breast carcinoma), MDAMB-231 (breast carcinoma), A375 (melanoma), and normal human embryonic kidney cells (HEK-293) were taken from Dr. Sheikh Tasduq Abdullah and Dr. Boobalan Gopu (CSIR-IIIM).
2.3.2 In vitro cytotoxicity
2.3.2.1 Sulforhodamine B (SRB) assay
Sulforhodamine B (SRB), a semi-automated colorimetric assay, was used to evaluate the anti-cancerous properties of indicated colchicine derivatives. Their activity was tested against human cancer cell lines A549 (lung carcinoma), MCF-7 (breast carcinoma), MDAMB-231 (breast carcinoma), and A375 (melanoma). The selectivity index (SI) was calculated against normal human embryonic kidney cells (HEK-293) to determine cytotoxic selectivity. The cells (7 × 103 cells/well) were seeded on 96 well plates, followed by overnight incubation. Multiple concentrations (50 µM, 20 µM, 10 µM, 5 µM, and 1 µM) of compounds were used to treat the cells, except for control wells that contained only cells with media. After 48 h of incubation, the cells attached to wells were fixed with 50 µL of chilled 50% TCA, and the plate was kept at 4°C for 1 h, as described previously (Malik et al., 2023; Kumar et al., 2024). The plate was washed (3–4 times) with distilled water (250 µL/well) and air-dried for 24 h. The next day, 100 µL of 0.4% SRB dye was added to each well, and the cells were incubated at room temperature for 1 h. The plate contents were discarded and washed 3 times with 1% glacial acetic acid (250 µL/well), followed by one last wash with distilled water and air drying for another 24 h. Subsequently, 100 µL of Tris base (10 mM, pH 10.5) was added to each well to dissolve the dye bound to fixed cells. The dissolved dye indicating growth inhibition was quantified by calculating optical density at 540 nm via microplate reader (Tecan infinite M200PRO, Switzerland). The IC50 values were calculated using GraphPad Prism Software Version 5.0. A favorable Selectivity Index (SI) is the most relevant parameter to detect anti-cancer potential in vitro as it indicates drug safety against cancer cells versus normal cells, efficacy of the drug, and selectivity. The selectivity index is calculated by using the formula.
2.3.2.2 MTT assay
Cell viability was assessed using the MTT assay, as outlined in previous studies (Majeed et al., 2014; Amin et al., 2025). Briefly, A549, A375, MCF-7 and MDA-MB231 cells (10 × 103 cells/well) were seeded in 96-well plates and incubated for 24 h at 37°C in a 5% CO2 atmosphere. After this incubation, the cells were treated with test compounds for another 48 h. At 44th hour of treatment, the cells were incubated with MTT (2.5 mg/mL) for additional 4 h. At 48th hour, DMSO was added (100 µL/well), to dissolve formazan crystals. Absorbance was measured at 570 nm. Cell viability was calculated relative to untreated controls. IC50 values were determined from independent triplicate experiments using GraphPad™ Prism 8.0 software.
2.3.3 Colony formation assay
A colony formation assay was performed to assess the ability of 3g to inhibit the reproductive and colony forming ability of A375 cells. The cells were seeded into a 6-well plate with a 1 × 103 cells/well density. Next day, the cells were treated with multiple concentrations of 3g, such as 30 µM, 20 µM, 10 µM and 1 µM, in fresh media for 48 h. The media was changed on alternate days till day 10, till untreated (negative control) became confluent. On day 10, the cells were washed with PBS, and colonies were observed under a microscope. The cells/colonies were fixed with 100% methanol for 20 min at 4°C and stained with 0.5% solution of crystal violet for 30 min at room temperature, as described previously (Kumar et al., 2024). Subsequently, the dye was removed from plates, and excess dye was rinsed by washing it with distilled water (3–5 times), followed by air drying. The colonies were counted using ImageJ software, and photographs were taken with a digital camera. The percentage of colonies formed was calculated using the formula mentioned below.
2.3.4 Wound healing assay
Cell scratch assay, also known as wound healing assay, was performed to evaluate the impact of 3g on migration of A375 cells. The cells were seeded into the 6-well plate with 4 × 103 cells/well density. The next day, in fresh media, a horizontal scratch was administered in the 90% confluent monolayer using a sterile 200 µL tip, as described previously, with slight modification (Kumar et al., 2024). The cells were incubated for 48 h with multiple concentrations of 3g such as 30 µM, 20 µM, 10 µM and 1 µM. Wound healing was observed using a Digital Inverted Fluorescence Microscope (Life Technologies EVOS FLc). The percentage of wound healing was calculated as.
2.3.5 Molecular docking experiment
Before molecular docking studies, colchicine and its derivative, 3g, underwent preparation using the OpenBabel module within PyRx software (O’Boyle et al., 2011). This process involved structural minimization via the universal force field (UFF) and subsequent conversion to PDBQT format, rendering them suitable for docking studies. The 3D structure of the tubulin target protein co-crystalized with the drug colchicine (PDB ID: 1SA0) was downloaded from the protein data bank. The structure was subsequently prepared using the Chimera DockPrep module by removing co-crystallized water, adding hydrogen atoms, and optimizing the pKa states (Pettersen et al., 2004). Chain B of the protein was used for molecular docking. The grid was generated around the drug colchicine at coordinates X = 117.219, Y = 90.179, and Z = 6.289 Å as described (Moussaoui et al., 2024). Molecular docking was performed using the AutoDock Vina module. The docking protocol was verified by redocking the co-crystalized colchicine to its binding site and comparing the root mean square deviation with the crystallized pose. After docking, the binding poses were visualized with the free Maestro visualizer.
2.3.6 Pharmacokinetic property prediction
Pharmacokinetic (PK) properties, including absorption, distribution, metabolism, excretion, and toxicity (ADMET), are critical for drug development. In this study, pkCSM web-based platform (https://biosig.lab.uq.edu.au/pkcsm/) was used. For the prediction of ADMET properties. pkCSM uses graph-based signatures and machine learning algorithms to predict pharmacokinetic properties. It provides predictions for key ADMET parameters, such as water solubility, intestinal absorption, blood-brain barrier permeability, CYP450 interactions, and toxicity endpoints. For prediction of human liver microsomal stability PredMS (https://predms.netlify.app.), a random forest model based tool was used.
3 Results and discussion
3.1 Chemistry
Colchicine is a tricyclic system containing one six-membered and two seven-membered rings with an (R)-stereogenic center at the C-7 position. First, colchicine (1) was converted into colchicine aldehyde (2) at the C-4 position by using the dichloromethylmethyl ether, which acts as a formylating agent to formylate aromatic ring in the presence of Lewis acid tin tetrachloride at 0°C to obtained colchicine aldehyde (Scheme 1a). Further, we have synthesized colchicine derivatives using the Biginelli multi-component chemical reaction method to synthesize dihydropyrimidones. This multi-component reaction requires the colchicine aldehyde, β-keto ester, and urea/thiourea/guanidine (Scheme 1b). For the synthesis 3a, we used thiourea with ethyl acetoacetate as a β-keto ester. Guanidine and ethyl acetoacetate as β-keto ester were used to synthesize 3b. The rest of the compounds (3c, 3d, 3e, 3f, 3g, 3h, 3i, and 3j) were synthesized using urea and different β-keto ester in the presence of ZnCl2 catalytic amounts. We observed diastereomers in the case of compounds 3f and 3g and 3i and 3j, which were separated by HPLC, as shown in 3f, 3g, 3i, and 3j. In other compounds, we have only found single stereoisomers and other stereoisomers in a trace, so we only isolated the single compounds 3a, 3b, 3c, 3d, 3e, and 3h.
SCHEME 1

Synthesis of colchicine derivatives. Reagents and condition: (a) Dichloromethyl methyl ether (1.0 mmol), SnCl2 (1.5 mmol), DCM, 0°C, 70%; (b) β-keto ester (2.0 mmol), guanidine/urea/thiourea (2.0 mmol), ZnCl2 (cat), ethanol, 80°C, 10 h, 50%–60%.
3.2 Biology
We screened a series of colchicine derivatives for their in vitro cytotoxicity in different cancer cell lines. We used SRB assay, a semi-automated colorimetric assay to evaluate the antiproliferative activity on different human cancer cell lines such as A549 (lung carcinoma), MCF-7 (breast carcinoma), MDAMB-231 (breast carcinoma) and A375 (melanoma) (Table 1). To access the cytotoxic activity levels, the effect of individual derivatives was assessed on a healthy non-cancerous cell line (HEK-293) to confirm their selectivity towards cancerous cells. Among all the derivatives, 3g resulted in a marked reduction in cell viability with a comparatively improved selectivity index compared to colchicine. Detailed information about half-maximal inhibitory concentration, IC50 ± SD, and SI can be found in Table 1, demonstrating 3g as a more selective cytotoxic agent against human melanoma cells than colchicine. It has improved selectivity (SI = 1.52) with IC50 (10.35 ± 0.56) (Table 1) and exhibits 60% growth inhibition at 20 µM in A375 cells (Figure 2). Additionally, 3g exhibited potent cytotoxicity against MCF-7 cells with IC50 = 15.69 ± 0.39 and a selectivity index of 1.005 (Table 1). The IC50 and Selectivity index (SI) of 3g against A375, a human melanoma cell line, as shown in Figure 3. The IC50 of Colchicine against mentioned human cancer cell lines has also been shown in Supplementary Table S1 along with Paclitaxel via MTT assay.
TABLE 1
| A375 | MCF-7 | MDA-MB 231 | A549 | HEK-293 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Compounds | IC50 (µM) | SI | IC50 (µM) | SI | IC50 (µM) | SI | IC50 (µM) | SI | IC50 (µM) |
| Colchicine | 0.076 ± 0.01 | 0.802 | 4.44 ± 0.28 | 0.013 | 4.83 ± 0.31 | 0.012 | 0.174 ± 0.028 | 0.35 | 0.061 ± 0.002 |
| 3a | >50 | 40.55 ± 0.91 | 0.73 | >50 | >50 | 29.66 ± 0.07 | |||
| 3b | >50 | 44.72 ± 0.64 | 0.85 | >50 | >50 | 38.33 ± 1.36 | |||
| 3c | >50 | 44.75 ± 0.68 | 0.79 | >50 | >50 | 35.61 ± 2.97 | |||
| 3d | >50 | 43.79 ± 0.33 | 0.82 | >50 | >50 | 36.09 ± 1.79 | |||
| 3e | >50 | 45.073 ± 0.73 | 0.89 | >50 | >50 | 40.29 ± 1.71 | |||
| 3f | 28.27 ± 2.19 | 0.62 | 26.39 ± 2.79 | 0.67 | 46.59 ± 0.86 | 0.38 | >50 | 17.81 ± 1.30 | |
| 3g | 10.35 ± 0.56 | 1.52 | 15.69 ± 0.39 | 1.005 | 24.92 ± 2.70 | 0.63 | >50 | 15.78 ± 0.81 | |
| 3h | 35.16 ± 2.79 | 0.75 | 49.92 ± 2.72 | 0.53 | >50 | >50 | 26.55 ± 2.65 | ||
| 3i | >50 | >50 | >50 | 39.52 ± 1.36 | |||||
| 3j | >50 | >50 | >50 | >50 | 38.97 ± 0.79 |
Half-maximal inhibitory concentration (IC50, µmol/L) and selectivity index (SI) values of synthesized colchicine derivatives against various human cancer cell lines and a normal cell line using sulphorhodamine B assay.
The IC50 data are expressed as mean ± SD (n = 3) and were determined using GraphPad™. Prism 5.0 software. The selectivity index (SI) was calculated HEK-293 cell line (human embryonic kidney cells).
FIGURE 2
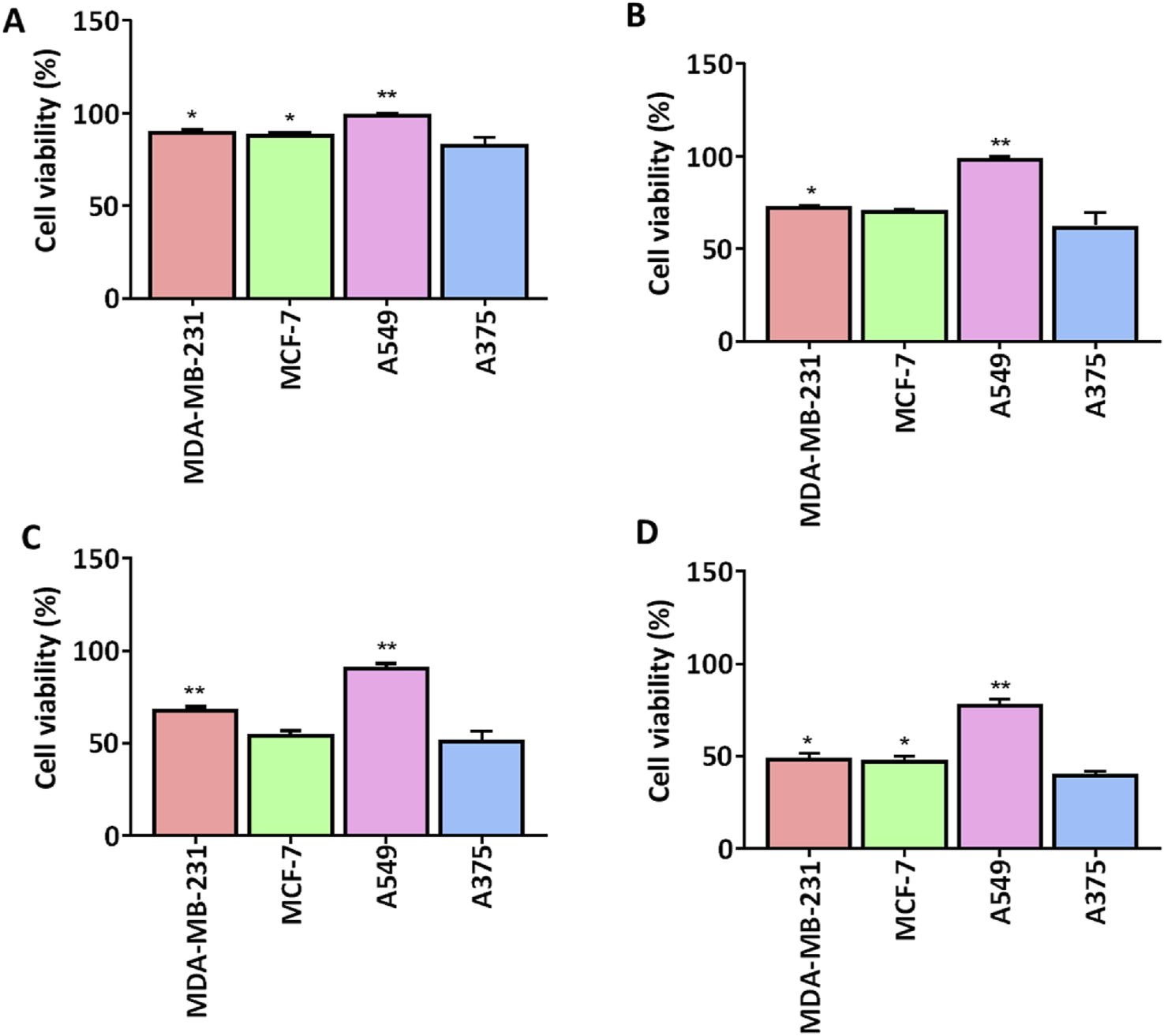
Comparison of cell viability (%) of 3g at different concentrations in different human cell lines. 3g was tested for cell viability (n = 3) at four different concentrations (A) 1 µM, (B) 5 µM, (C) 10 µM, (D) 20 µM. Cell viability graphs are expressed as mean ± SD (n = 3) and plotted using GraphPadTM Prism 8.0 software. A375 cells were compared against other cancer cells for significance by applying One-way ANOVA with Dunnet’s multiple comparison test. *p < 0.05, **p < 0.001.
FIGURE 3
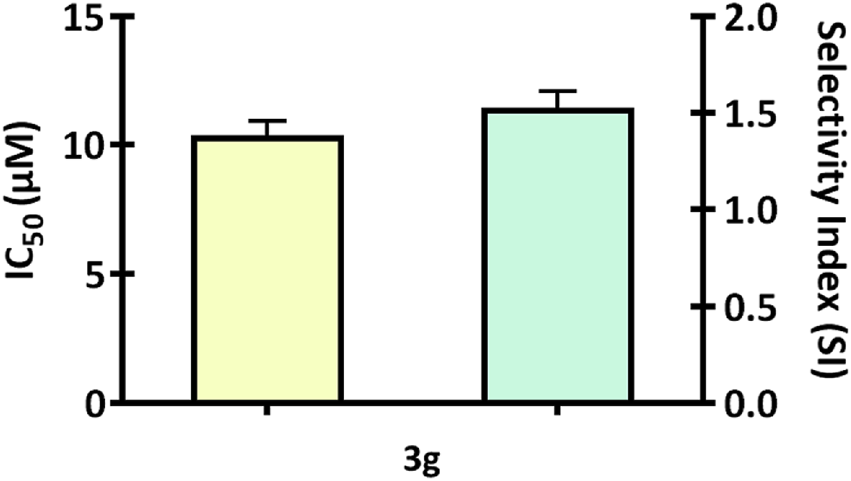
The bar graph illustrates the IC50 and Selectivity index (SI) of 3g against A375, a human melanoma cell line. The graphs are expressed as mean ± SD (n = 3) and plotted using GraphPad™ Prism 8.0 software.
3.3 3g reduces colony-forming ability of A375 cells
The ability of individual cells to endure, multiply, and form colonies is determined by a colony formation assay used to assess how pharmaceuticals or radiation therapy affect the viability and proliferation of cells (Stone et al., 2016; Pouget et al., 2022). The antiproliferative effect of 3g was examined using colony formation assay. The A375 cells were treated with various concentrations of 3g (1 µM, 10 µM, 20 µM, 30 µM) for 48 h. It was observed that treatment with 3g induced the irreversible growth arrest ability and significantly attenuated the size, number of colonies, and colony growth of A375 cells compared to untreated control cells (Figure 4). The percentage of colony growth formation was 100% in untreated control cells wheras it was 37.5% in case of 3g at 20 µM in A375 cells.
FIGURE 4
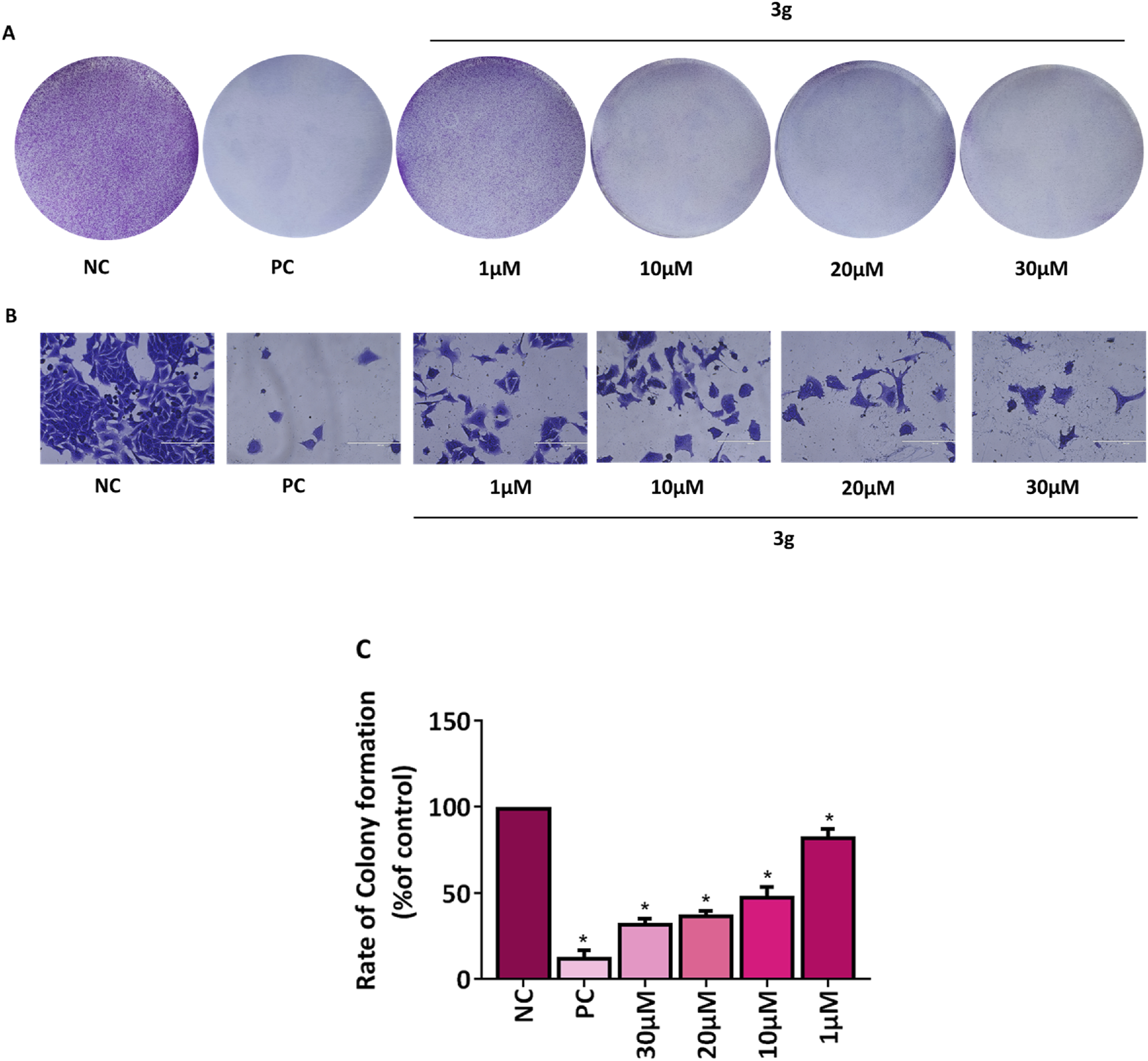
3g inhibits colony formation in A375 cells. (A,B) Representative images of colony growth inhibition of A375 cells treated with an indicated concentration of 3g for 48 h. Camptothecin (0.1 µM) was used as a positive control (PC) (Rodríguez-Berna et al., 2014). (A) No magnification, (B) 20x, Sacle Bar 200 µm. (C) Colonies were quantified using ImageJ software (ver. 1.49 V), and graphs were plotted as mean ± SD (n = 3) using GraphPad™ Prism 8.0 software. The statistical significance was determined by comparing the treated samples with an untreated negative control (NC) using One-way ANOVA with Dunnet’s multiple comparison test. *p < 0.001 in comparison to NC.
3.4 3g reduces migration potential of A375 cells
Tumor cell invasion and metastases are significantly influenced by cell migration (Shi et al., 2024). A fundamental characteristic of a chemotherapeutic agent is its ability to inhibit cell migration and invasion and its capacity to induce targeted cell death in cancerous tissues. Agents for anti-metastatic therapy may include molecules that block the migration of cancer cells (Anderson et al., 2019). Wound healing assay was performed to analyze the effect of compound 3g on the wound closure rate and migration potential of cells. The A375 cells were treated with various concentrations of 3g (30 µM, 20 μM, 10 µM, and 1 µM) for 48 h. 3g was found to decrease the migration of A375 cells toward the margin of the wound, resulting in poor closure of the gap and healing of the wound compared to untreated control cells (Figure 5). The percentage of wound closure was 84% in untreated control cells whereas 69% in case of 3g at 20 µM in A375 cells (Figure 5).
FIGURE 5
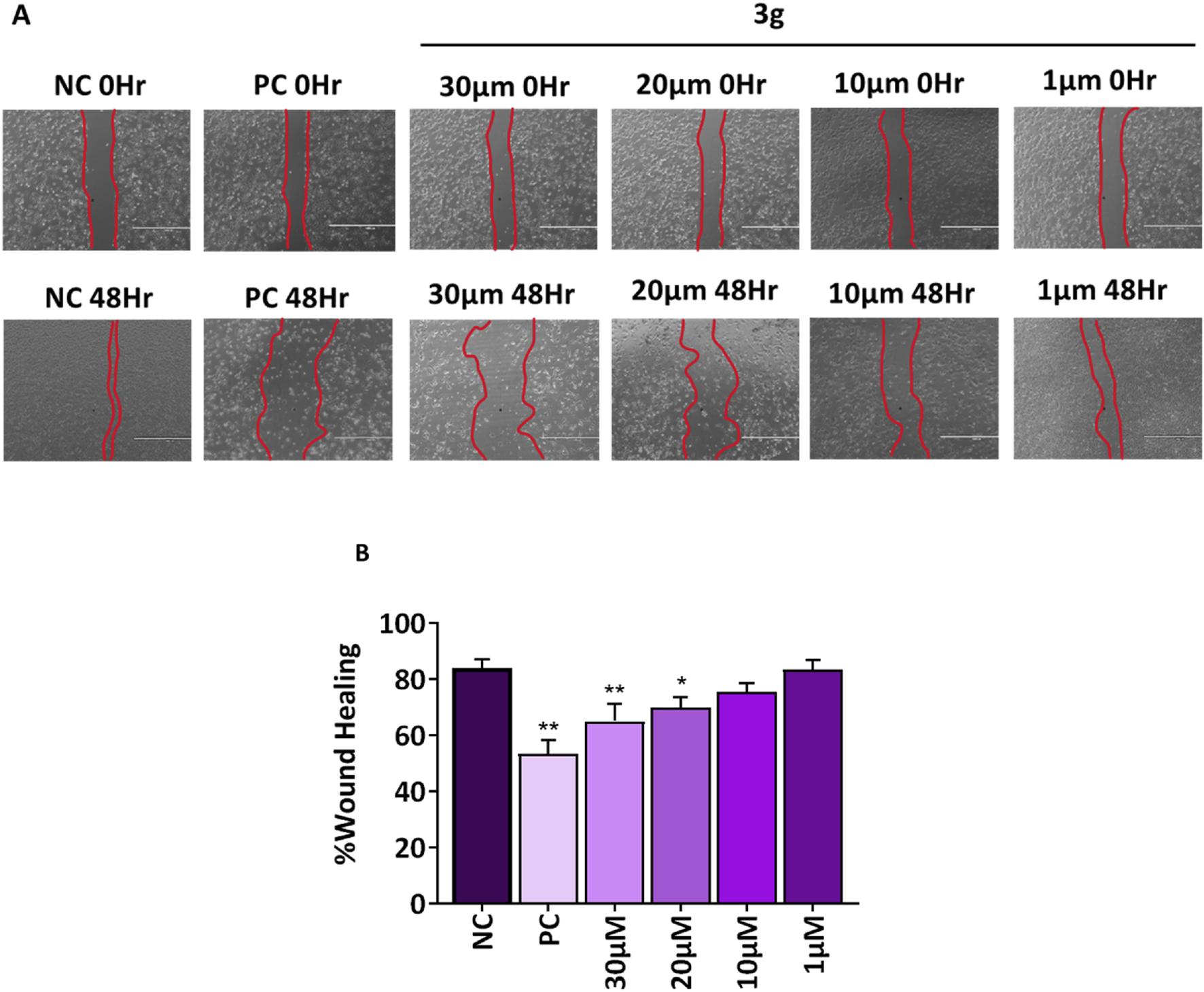
3g inhibits the migration of A375 cells into the wound region after 48h treatment with indicated concentrations. (A) Representative images from a bright field microscope (4x, Scale bar 1000 µm) showing inhibition of A375 cell invasion in vitro scratch/wound healing assay with indicated concentrations. Camptothecin (0.1 µM) was used as a positive control (PC). (B) The decrease in wound closure or gap filling with an increase in concentration of 3g was quantified using ImageJ software (ver 1.49 V) and plotted as mean ± SD (n = 3) using GraphPad™ Prism 8.0 software. The statistical significance was determined by comparing the treated samples with an untreated negative control (NC) using One-way ANOVA with Dunnet’s multiple comparison test. *p < 0.05, **p < 0.001 in comparision to NC.
Colchicine blocks cell division by disrupting microtubules, and the spindle microtubules are more sensitive to colchicine than the interphase microtubules. It penetrates the cells and equilibrates with the external colchicine rapidly; however, a more extended period is required to attain saturation. Differential sensitivity at different stages of cell division as it binds to microtubules dissociates to tubulin dimers. For instance, colchicine at a concentration of 50 nM blocks almost all the prophase, metaphase, and anaphase cells at the mitosis cell cycle. This abnormal mitotic cycle is sometimes called C-mitosis or Colchicine-mitosis (Taylor, 1965). Moreover, it can inhibit the function of several ion channels and alter the membrane potential of the mitochondria, resulting in the release of proapoptotic factors like caspases, cytochrome-C, and apoptotic features, leading to cell cycle arrest and death (Volbracht et al., 2001). Microtubule targeting agents (MTA) are also named antimitotic agents that bind to the tubulin in the microtubules and prohibit the proliferation of the cells by inhibiting or destabilizing agents or agents that bind to the colchicine (Jordan and Wilson, 2004; Arnst, 2018; Mirzaei et al., 2020). Tubulin-targeting agents and microtubule-associated proteins as targets have gained much attention since vincristine was approved by the US FDA in 1963 in cancer treatment and anti-cancer drug development (Lu et al., 2012; Khwaja et al., 2021). Many colchicine-based derivatives have been synthesized and generated to develop novel and increasing pharmacological profiles to improve their efficacies, toxicity, and therapeutic importance (Zhang et al., 2015; Majcher et al., 2018; Pyta et al., 2022; Iqbal Lone et al., 2024).
The study found that a derivative of colchicine, identified as 3g, demonstrated enhanced effectiveness against human melanoma cells. Specifically, 3g exhibited a selectivity index approximately two times higher than colchicine. We sought to further evaluate the efficacy of 3g against human melanoma in A375 cells using other in vitro assays such as colony formation assay and wound healing assay. We found that 3g reduced the number and growth of colonies of these cells. The percentage of colony growth formation was 100% in untreated control cells wheras it was 37.5% in case of 3g at 20 µM in A375 cells. Moreover, 3g decreased the migration of A375 cells toward the margin of the wound, resulting in poor wound healing. The percentage of wound closure was 84% in untreated control cells whereas 69% in case of 3g at 20 µM in A375 cells. These findings suggest we may have identified a more selective colchicine derivative targeting melanoma cells. This increased selectivity could reduce toxicity to healthy cells, thereby enhancing its therapeutic potential. With its higher selectivity and effectiveness, compound 3g could present a substantial advantage over colchicine as a treatment for melanoma, potentially resulting in improved outcomes and reduced side effects.
3.5 Molecular docking
The molecular docking study investigated the binding interactions between colchicine, its derivative, and chain B of the tubulin protein. The docking protocol successfully reproduced the binding pose of the co-crystallized ligand with a low RMSD value of 1.33 Å, demonstrating the accuracy of the docking approach (Figure 6). Additionally, molecular docking of colchicine derivative (binding energy −6.498 kcal/mol) showed a comparable pose to colchicine (binding energy −7.963 kcal/mol). The colchicine derivative formed a hydrogen bond with Cys241, Asn258 and a salt bridge with Lys352 (Figure 7). Notably, the interaction with Cys241 is also shown by co-crystalized colchicine in PDB ID 1SA0 (Figure 7). Therefore, these interactions might be important for binding the colchicine derivative with tubulin.
FIGURE 6
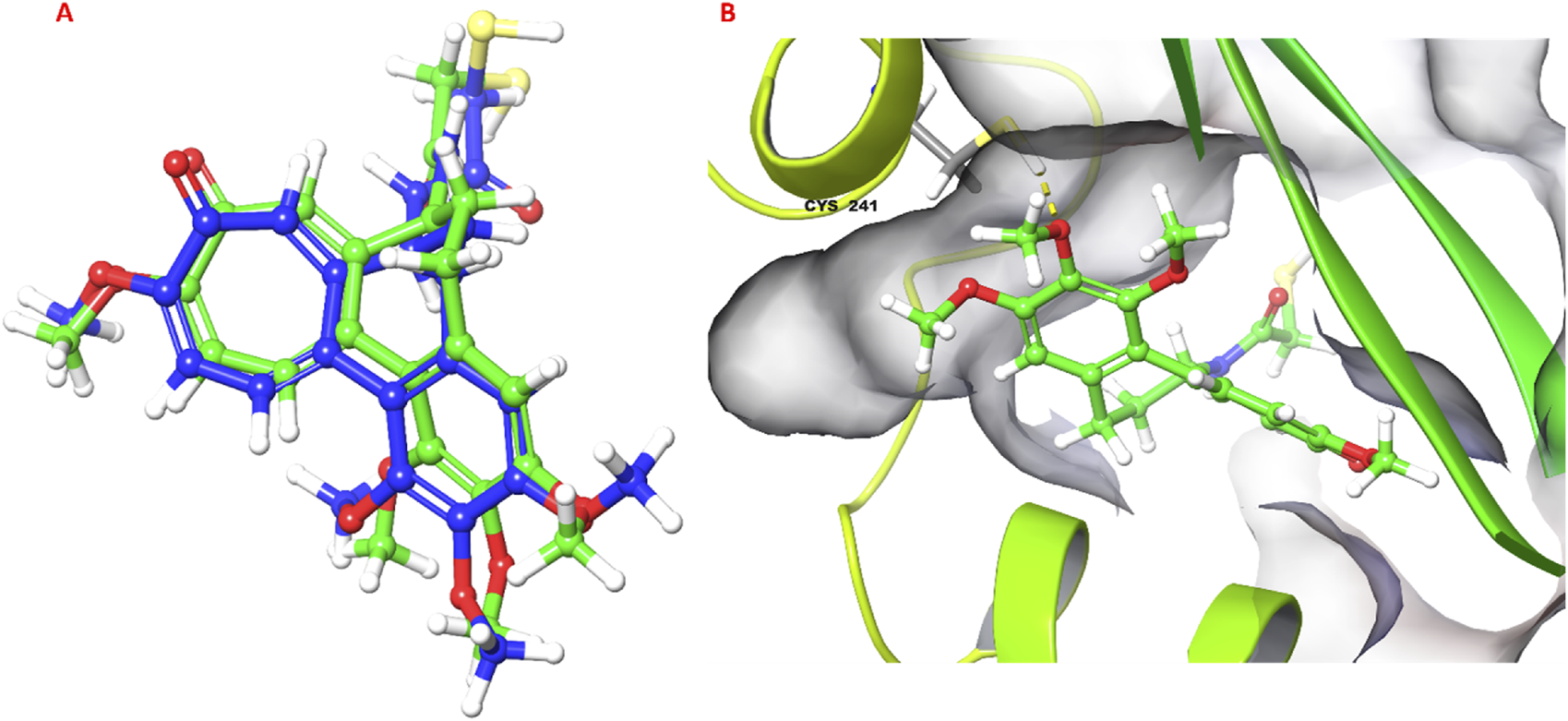
Validation of docking protocol. (A) Superimposed pose of colchicine co-crystalized with tubulin (PDB ID: 1SA0) with respect to molecular docking pose; (B) 3D pose of colchicine obtained from molecular docking, hydrogen bond is shown as yellow dashed line.
FIGURE 7
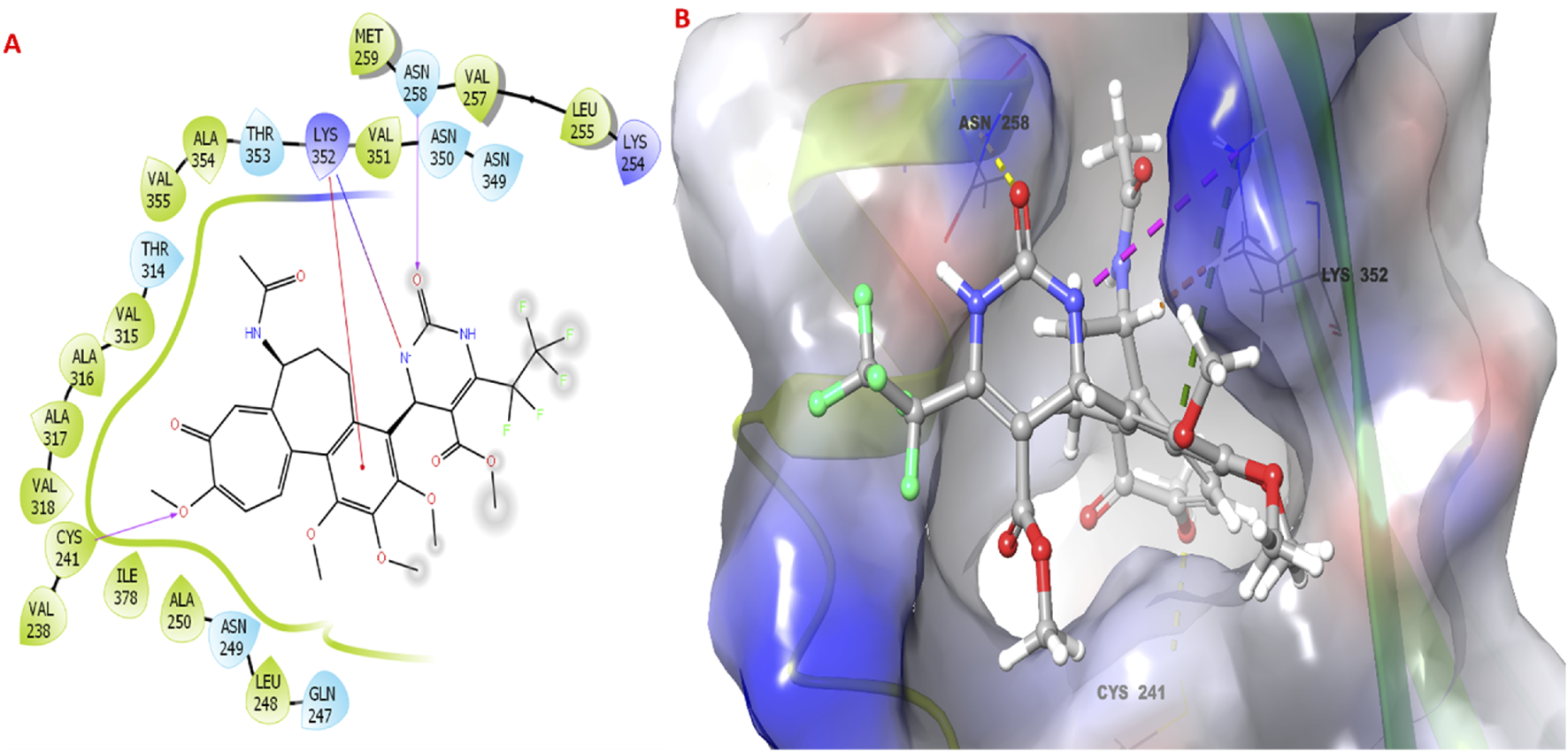
Molecular docking studies of colchicine derivative 3g with the targeted protein. (A,B) 2D & 3D poses of colchicine derivative.
3.6 Pharmacokinetic properties of compound 3g
The pkCSM results provide a comprehensive overview of the pharmacokinetic and toxicity properties of the compound (Table 2). In terms of absorption, the compound exhibits moderate intestinal absorption in humans (75.376%) but has low water solubility (−5.195) and poor Caco2 permeability (−0.229). The compound is a substrate for P-glycoprotein and acts as an inhibitor for both P-glycoprotein I and II, which may influence its absorption and efflux. The distribution volume (VDss) is low (−1.367), and the fraction unbound in plasma is 0.148, indicating significant protein binding. The compound has poor blood-brain barrier (BBB) permeability (−2.15) and very low CNS permeability (−3.483), suggesting it is unlikely to cross into the central nervous system.
TABLE 2
| S. No. | Property | Model name | Predicted value | Unit |
|---|---|---|---|---|
| 1. | Absorption | Water solubility | −5.195 | Numeric (log mol/L) |
| 2. | Absorption | Caco2 permeability | −0.229 | Numeric (log Papp in 10−6 cm/s) |
| 3. | Absorption | Intestinal absorption (human) | 75.376 | Numeric (% Absorbed) |
| 4. | Absorption | Skin Permeability | −2.768 | Numeric (log Kp) |
| 5. | Absorption | P-glycoprotein substrate | Yes | Categorical (Yes/No) |
| 6. | Absorption | P-glycoprotein I inhibitor | Yes | Categorical (Yes/No) |
| 7. | Absorption | P-glycoprotein II inhibitor | Yes | Categorical (Yes/No) |
| 8. | Distribution | VDss (human) | −1.367 | Numeric (log L/kg) |
| 9 | Distribution | Fraction unbound (human) | 0.148 | Numeric (Fu) |
| 10. | Distribution | BBB permeability | −2.15 | Numeric (log BB) |
| 11. | Distribution | CNS permeability | −3.483 | Numeric (log PS) |
| 12. | Metabolism | CYP2D6 substrate | No | Categorical (Yes/No) |
| 13. | Metabolism | CYP3A4 substrate | Yes | Categorical (Yes/No) |
| 14. | Metabolism | CYP1A2 inhibitior | No | Categorical (Yes/No) |
| 15. | Metabolism | CYP2C19 inhibitior | No | Categorical (Yes/No) |
| 16. | Metabolism | CYP2C9 inhibitior | No | Categorical (Yes/No) |
| 17. | Metabolism | CYP2D6 inhibitior | No | Categorical (Yes/No) |
| 18. | Metabolism | CYP3A4 inhibitior | Yes | Categorical (Yes/No) |
| 19. | Excretion | Total Clearance | −0.214 | Numeric (log mL/min/kg) |
| 20. | Excretion | Renal OCT2 substrate | No | Categorical (Yes/No) |
| 21. | Toxicity | AMES toxicity | No | Categorical (Yes/No) |
| 22. | Toxicity | Max. tolerated dose (human) | −0.139 | Numeric (log mg/kg/day) |
| 23. | Toxicity | hERG I inhibitor | No | Categorical (Yes/No) |
| 24. | Toxicity | hERG II inhibitor | Yes | Categorical (Yes/No) |
| 25. | Toxicity | Oral Rat Acute Toxicity (LD50) | 2.358 | Numeric (mol/kg) |
| 26. | Toxicity | Oral Rat Chronic Toxicity (LOAEL) | 1.285 | Numeric (log mg/kg_bw/day) |
| 27. | Toxicity | Hepatotoxicity | No | Categorical (Yes/No) |
| 28. | Toxicity | Skin Sensitisation | No | Categorical (Yes/No) |
| 29. | Toxicity | T.Pyriformis toxicity | 0.291 | Numeric (log ug/L) |
| 30. | Toxicity | Minnow toxicity | 1.622 | Numeric (log mM) |
Pharmacokinetic properties of compound 3g predicted by pkCSM tool.
Regarding metabolism, the compound is a substrate for CYP3A4 but not CYP2D6. It inhibits CYP3A4 but does not inhibit other major CYP enzymes, which may reduce the risk of drug-drug interactions. Excretion data indicate low total clearance (−0.214), and the compound is not a substrate for renal OCT2, suggesting renal excretion may not be a major elimination pathway. Toxicity profiling shows the compound is non-toxic in AMES tests and non-hepatotoxic. It does not inhibit hERG I but inhibits hERG II. The compound has moderate acute toxicity (LD50 = 2.358) and low chronic toxicity (LOAEL = 1.285) in rats. It is non-sensitizing to the skin and shows low toxicity in T. Pyriformis (0.291) and moderate toxicity in minnows (1.622). PredMS predicts the compound to be metabolically stable in human liver micosomes.
4 Conclusion
In summary, a series of novel colchicine analogs derivatized at the C-4 position was utilized to obtain via multi-component reaction methods and screen for in vitro cytotoxicity against human cancer cell lines and normal human embryonic kidney cells (HEK-293) to evaluate for their anti-cancer potency. The MCR method has high utility in drug discovery, especially for structurally optimizing complex natural products for their biological properties (Kaur et al., 2017). Derivative 3g reduced cell viability and showed a significantly improved selectivity index against melanoma compared to colchicine. It has a selectivity index of 1.52 with 60% growth inhibition at 20 µM in melanoma cancer cells with IC50 of 10.35 ± 0.56. Additionally, 3g exhibited significant cytotoxicity against MCF-7 cells with IC50 of 15.69 ± 0.39 and a selectivity index of 1.005. Among the tested derivatives, compound 3g is the most promising candidate, as elucidated by the molecular docking studies, demonstrating significant potential as a colchicine-based anti-cancer agent that can overcome drug resistance and improve efficacy and pharmacokinetic properties for cancer therapy treatment strategies.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
PK: Formal Analysis, Investigation, Writing – original draft. TK: Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. WL: Investigation, Writing – review & editing. YA: Formal Analysis, Investigation, Writing – review & editing. HT: Software, Visualization, Writing – review & editing. AS: Software, Visualization, Writing – review & editing. ZA: Supervision, Resources, Funding acquisition, Writing – review and editing. SR: Conceptualization, Project administration, Supervision, Writing – review & editing. JA: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors are grateful for the financial support from the Director, CSIR-IIIM, Jammu. SR acknowledges the financial support from Ramalingaswami Fellowship (Ref. No. BT/RLF/Re-entry/13/2019) received from the Department of Biotechnology, Government of India, and Ministry of Earth Sciences, Grant No. F. No. MoES/PAMC/DOM/55/2023 (E-12933).
Acknowledgments
We also acknowledge the technical support from the central instrumentation facility of CSIR-IIIM, Jammu. The institutional manuscript communication number is CSIR-IIIM/IPR/00823.
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1528235/full#supplementary-material
References
1
Amin H. Kaur G. Goswami A. Bhat K. A. (2025). Facile synthesis of ureidic derivatives of betulinic acid with antiproliferative effects on colorectal and prostate cancer cells. Chem. Biodiversitye202402669.
2
Anderson R. L. Balasas T. Callaghan J. Coombes R. C. Evans J. Hall J. A. et al (2019). A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol.16, 185–204. 10.1038/s41571-018-0134-8
3
Arnold M. Abnet C. C. Neale R. E. Vignat J. Giovannucci E. L. McGlynn K. A. et al (2020). Global burden of 5 major types of gastrointestinal cancer. Gastroenterology159, 335–349.e15. 10.1053/j.gastro.2020.02.068
4
Arnst K. E. (2018). Targeting the colchicine binding site on tubulin to overcome multidrug resistance and anti-cancer efficacy of selective survivin inhibitors. ProQuest Diss. Theses209. 10.21007/etd.cghs.2018.0469
5
Banach M. Penson P. E. (2021). Colchicine and cardiovascular outcomes: a critical appraisal of recent studies. Curr. Atheroscler. Rep.23, 32. 10.1007/s11883-021-00932-5
6
Banerjee S. Chakrabarti G. Bhattacharyya B. (1997). Colchicine binding to tubulin monomers: a mechanistic study. Biochemistry36, 5600–5606. 10.1021/bi962648n
7
Dubey K. K. Kumar P. Labrou N. E. Shukla P. (2017). Biotherapeutic potential and mechanisms of action of colchicine. Crit. Rev. Biotechnol.37, 1038–1047. 10.1080/07388551.2017.1303804
8
Ghawanmeh A. A. Chong K. F. Sarkar S. M. Bakar M. A. Othaman R. Khalid R. M. (2018). Colchicine prodrugs and codrugs: chemistry and bioactivities. Eur. J. Med. Chem.144, 229–242. 10.1016/j.ejmech.2017.12.029
9
Gracheva I. A. Shchegravina E. S. Schmalz H. G. Beletskaya I. P. Fedorov A. Y. (2020). Colchicine alkaloids and synthetic analogues: current progress and perspectives. J. Med. Chem.63, 10618–10651. 10.1021/acs.jmedchem.0c00222
10
Grazia Martina M. Giannessi L. Radi M. (2023). Multi-component synthesis of purines and pyrimidines: from the origin of life to new sustainable approaches for drug-discovery applications. Eur. J. Org. Chem.26, e202201288. 10.1002/ejoc.202201288
11
Halsey G. (2023). FDA approves low-dose colchicine as first anti-inflammatory therapy to reduce cardiovascular events. Patient Care.
12
Huang M. Lu J. J. Ding J. (2021). Natural products in cancer therapy: past, present and future. Nat. Prod. Bioprospecting11, 5–13. 10.1007/s13659-020-00293-7
13
Imazio M. Gaita F. LeWinter M. (2015). Evaluation and treatment of pericarditis: a systematic review. JAMA - J. Am. Med. Assoc.314, 1498–1506. 10.1001/jama.2015.12763
14
Iqbal Lone W. Chand J. Kumar P. Garg Y. Ahmed Z. Mukherjee D. et al (2024). Discovery of colchicine aryne cycloadduct as a potent molecule for the abrogation of epithelial to mesenchymal transition via modulating cell cycle regulatory CDK-2 and CDK-4 kinases in breast cancer cells. Bioorg. Chem.150, 107581. 10.1016/j.bioorg.2024.107581
15
Jordan M. A. Wilson L. (2004). Microtubules as a target for anti-cancer drugs. Nat. Rev. Cancer4, 253–265. 10.1038/nrc1317
16
Kannan S. Wesley S. D. Ruba A. Rajalakshmi A. R. Kumaragurubaran K. (2007). Optimization of solvents for effective isolation of colchicines from Gloriosa superba L. seeds. Nat. Prod. Res.21, 469–472. 10.1080/14786410601129507
17
Kaur R. Chaudhary S. Kumar K. Gupta M. K. Rawal R. K. (2017). Recent synthetic and medicinal perspectives of dihydropyrimidinones: a review. Eur. J. Med. Chem.132, 108–134. 10.1016/j.ejmech.2017.03.025
18
Khwaja S. Kumar K. Das R. Negi A. S. (2021). Microtubule associated proteins as targets for anti-cancer drug development. Bioorg. Chem.116, 105320. 10.1016/j.bioorg.2021.105320
19
Kumar A. Sharma P. R. Mondhe D. M. (2017). Potential anti-cancer role of colchicine-based derivatives: an overview. Anti-cancer drugs28, 250–262. 10.1097/CAD.0000000000000464
20
Kumar P. Singh R. Sharma D. Hassan Q. P. Gopu B. Anal J. M. H. (2024). Design, synthesis, and biological evaluation of chalcone acetamide derivatives against triple negative breast cancer. Bioorg. & Med. Chem. Lett.107, 129795. 10.1016/j.bmcl.2024.129795
21
Lu Y. Chen J. Xiao M. Li W. Miller D. D. (2012). An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res.29, 2943–2971. 10.1007/s11095-012-0828-z
22
Majcher U. Klejborowska G. Kaik M. Maj E. Wietrzyk J. Moshari M. et al (2018). Synthesis and biological evaluation of novel triple-modified colchicine derivatives as potent tubulin-targeting anti-cancer agents. Cells7, 216. 10.3390/cells7110216
23
Majeed R. Hamid A. Sangwan P. L. Chinthakindi P. K. Koul S. Rayees S. et al (2014). Inhibition of phosphotidylinositol-3 kinase pathway by a novel naphthol derivative of betulinic acid induces cell cycle arrest and apoptosis in cancer cells of different origin. Cell Death Dis.5 (10), e1459–e1459.
24
Malik S. Mintoo M. J. Reddy C. N. Kumar R. Kotwal P. Bharate S. B. et al (2023). In vitro and in vivo anti-cancer potential and molecular targets of the new colchicine analog IIIM-067. J. Integr. Med.21, 62–76. 10.1016/j.joim.2022.09.006
25
Mirzaei S. Hadizadeh F. Eisvand F. Mosaffa F. Ghodsi R. (2020). Synthesis, structure-activity relationship and molecular docking studies of novel quinoline-chalcone hybrids as potential anti-cancer agents and tubulin inhibitors. J. Mol. Struct.1202, 127310. 10.1016/j.molstruc.2019.127310
26
Moussaoui M. Baammi S. Soufi H. Baassi M. El Allali A. Belghiti M. E. et al (2024). QSAR, ADMET, molecular docking, and dynamics studies of 1,2,4-triazine-3(2H)-one derivatives as tubulin inhibitors for breast cancer therapy. Sci. Rep.14, 16418. 10.1038/s41598-024-66877-2
27
O’Boyle N. M. Banck M. James C. A. Morley C. Vandermeersch T. Hutchison G. R. (2011). Open Babel: an open chemical toolbox. J. cheminformatics3, 33. 10.1186/1758-2946-3-33
28
Pettersen E. F. Goddard T. D. Huang C. C. Couch G. S. Greenblatt D. M. Meng E. C. et al (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem.25, 1605–1612. 10.1002/jcc.20084
29
Pouget J.-P. Santoro L. Piron B. Paillas S. Ladjohounlou R. Pichard A. et al (2022). From the target cell theory to a more integrated view of radiobiology in Targeted radionuclide therapy: the Montpellier group’s experience. Nucl. Med. Biol.104, 53–64. 10.1016/j.nucmedbio.2021.11.005
30
Pyta K. Skrzypczak N. Ruszkowski P. Bartl F. Przybylski P. (2022). Regioselective approach to colchiceine tropolone ring functionalization at C(9) and C(10) yielding new anti-cancer hybrid derivatives containing heterocyclic structural motifs. J. Enzyme Inhibition Med. Chem.37, 597–605. 10.1080/14756366.2022.2028782
31
Rodríguez-Berna G. Mangas-Sanjuán V. Gonzalez-Alvarez M. Gonzalez-Alvarez I. García-Giménez J. L. Cabañas M. J. D. et al (2014). A promising camptothecin derivative: semisynthesis, antitumor activity and intestinal permeability. Eur. J. Med. Chem.83, 366–373. 10.1016/j.ejmech.2014.06.050
32
Schlesinger N. Firestein B. L. Brunetti L. (2020). Colchicine in COVID-19: an old drug, new use. Curr. Pharmacol. Rep.6, 137–145. 10.1007/s40495-020-00225-6
33
Shahi A. Manhas R. Bhattacharya S. Rathore A. Kumar P. Samanta J. et al (2024). Synthesis and antibacterial potential of novel thymol derivatives against methicillin-resistant Staphylococcus aureus and P. aeruginosa pathogenic bacteria. Front. Chem.12, 1482852. 10.3389/fchem.2024.1482852
34
Shi X. Wang X. Yao W. Shi D. Shao X. Lu Z. et al (2024). Mechanism insights and therapeutic intervention of tumor metastasis: latest developments and perspectives. Signal Transduct. Target. Ther.9, 192. 10.1038/s41392-024-01885-2
35
Singh B. Kumar A. Joshi P. Guru S. K. Kumar S. Wani Z. A. et al (2015). Colchicine derivatives with potent anti-cancer activity and reduced P-glycoprotein induction liability. Org. Biomol. Chem.13, 5674–5689. 10.1039/c5ob00406c
36
Smalley K. S. M. (2010). Understanding melanoma signaling networks as the basis for molecular targeted therapy. J. Investigative Dermatology130, 28–37. 10.1038/jid.2009.177
37
Stone H. B. Bernhard E. J. Coleman C. N. Deye J. Capala J. Mitchell J. B. et al (2016). Preclinical data on efficacy of 10 drug-radiation combinations: evaluations, concerns, and recommendations. Transl. Oncol.9, 46–56. 10.1016/j.tranon.2016.01.002
38
Tardif J.-C. Cossette M. Guertin M.-C. Bouabdallaoui N. Dubé M.-P. Boivin G. et al (2022). Predictive risk factors for hospitalization and response to colchicine in patients with COVID-19. Int. J. Infect. Dis.116, 387–390. 10.1016/j.ijid.2022.01.020
39
Taylor E. W. (1965). The mechanism of colchicine inhibition of mitosis: I. Kinetics of inhibition and the binding of h3-colchicine. J. cell Biol.25, 145–160. 10.1083/jcb.25.1.145
40
Volbracht C. Leist M. Kolb S. A. Nicotera P. (2001). Apoptosis in caspase-inhibited neurons. Mol. Med.7, 36–48. 10.1007/bf03401837
41
Zhang X. Kong Y. Zhang J. Su M. Zhou Y. Zang Y. et al (2015). Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem.95, 127–135. 10.1016/j.ejmech.2015.03.035
Summary
Keywords
Colchicum autumnale , colchicine, cancer, melanoma, proliferation, SRB assay
Citation
Kumar P, Kotra T, Lone WI, Arfath Y, Tiwari H, Shukla AK, Ahmed Z, Rayees S and Anal JMH (2025) Design, synthesis and antiproliferative activity of novel colchicine derivatives: selective inhibition of melanoma cell proliferation. Front. Pharmacol. 16:1528235. doi: 10.3389/fphar.2025.1528235
Received
14 November 2024
Accepted
28 May 2025
Published
27 June 2025
Volume
16 - 2025
Edited by
José Carlos Menéndez, Complutense University of Madrid, Spain
Reviewed by
Debasish Bandyopadhyay, The University of Texas Rio Grande Valley, United States
Souvik Banerjee, Middle Tennessee State University, United States
Updates
Copyright
© 2025 Kumar, Kotra, Lone, Arfath, Tiwari, Shukla, Ahmed, Rayees and Anal.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jasha Momo H. Anal, hmunshel.jasha@iiim.res.in; Sheikh Rayees, rayees.h@iiim.res.in
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.