 Vanessa Petrucci1
Vanessa Petrucci1 Patrizia Dragone
Patrizia Dragone- 1Angelini Pharma S.p.A., Rome, Italy
- 2Angelini Pharma France SASU, Rueil-Malmaison, France
Background: Trazodone, an antidepressant indicated for the treatment of major depressive disorder (MDD), undergoes complex metabolism involving multiple cytochrome P450 (CYP) isoforms. Despite its widespread clinical use, there is limited contemporary research on trazodone biotransformation, particularly regarding interspecies differences mediated by CYP enzymes. This study investigates the hepatic metabolic stability, pathways, and inhibitory effects of trazodone and its metabolites.
Methods: The hepatic metabolic stability of trazodone was evaluated in cryopreserved hepatocytes from human, mouse, and rat sources. Metabolic profiling was conducted using human and rat liver microsomes, hepatocytes, and rat plasma to identify primary and secondary metabolites. CYP isoforms involved in trazodone metabolism were identified through selective CYP inhibitors and recombinant enzymes. Additionally, the inhibitory potential of trazodone and m-chlorophenylpiperazine (mCPP) —the only pharmacologically active metabolite—on CYP enzymes was assessed in both human and mouse hepatocytes.
Results: Trazodone demonstrated significant interspecies differences in intrinsic clearance rates, with human hepatocytes exhibiting the slowest conversion, suggesting prolonged exposure and potential for stable plasma levels. CYP3A4 was identified as the primary enzyme mediating trazodone metabolism, particularly in the formation of mCPP while CYP2D6, CYP2C19, and FMOs contributed to the formation of other major, inactive metabolites such as M9 and M2.
Conclusion: CYP3A enzymes were identified as the primary mediators of trazodone metabolism, particularly for the formation of its only active metabolite, mCPP, with additional contributions from CYP2D6, CYP2C19, and FMOs to other pathways. Interspecies differences were most pronounced for CYP3A-mediated metabolism and hepatic clearance, reinforcing the importance of human-specific models to characterize trazodone’s pharmacokinetics. These findings advance the understanding of its biotransformation and support its optimized clinical use in major depressive disorder—particularly in complex therapeutic regimens and genetically diverse populations.
1 Introduction
Trazodone, first synthesized and introduced in the 1970s, is a widely prescribed antidepressant classified as a serotonin receptor antagonist and reuptake inhibitor (SARI). It is approved by several European regulatory agencies for the treatment of major depressive episodes, including those with co-occurring anxiety symptoms (Stahl, 2009; Fagiolini et al., 2012; Khouzam, 2017). Its pharmacological profile combines serotonin reuptake inhibition with potent antagonism at 5-HT2A receptors, distinguishing it from selective serotonin reuptake inhibitors (SSRIs), serotonin–norepinephrine reuptake inhibitors (SNRIs), and tricyclic antidepressants (TCAs) (Stahl, 2009; Fagiolini et al., 2012). This dual mechanism also confers sedative properties via antagonism at α1-adrenergic and H1-histaminergic receptors, supporting its use in patients with anxiety, sleep disturbances, or sexual disfunction (Fagiolini et al., 2012). Trazodone has shown efficacy comparable to TCAs and several second-generation antidepressants in treating symptoms of major depressive disorder (MDD) (Gartlehner et al., 2011; Cuomo et al., 2019). Its favorable safety profile and its great availability of formulations allow to personalize trazodone treatment according to patient profile characteristics (Cuomo et al., 2021).
Pharmacokinetically, trazodone is rapidly absorbed after oral administration, with immediate-release formulations achieving peak plasma levels within 0.5–2 h (DESYREL, 2024, OLEPTRO, 2024, Trittico, 2024). Extended-release formulations were developed to improve plasma concentration stability and minimize peak-related adverse effects (Karhu et al., 2011). Trazodone undergoes extensive hepatic metabolism, predominantly via cytochrome P450 (CYP) 3A4 (CYP3A4), which converts it into several metabolites, including m-chlorophenylpiperazine (mCPP)—its only known active metabolite (Rotzinger et al., 1998a). mCPP has high affinity for various serotonin receptors, including 5-HT2C, 5-HT3, 5-HT2A, 5-HT1B, and 5-HT1A (Caccia et al., 1981; Rotzinger et al., 1998a; Zalma et al., 2000; Kalgutkar et al., 2005). In humans, systemic levels of mCPP are typically below 10% of those of trazodone (Otani et al., 1997). Unlike trazodone, which primarily antagonizes 5-HT2A and 5-HT2C receptors, mCPP acts as an agonist, potentially modulating the net pharmacodynamic profile (Fiorella et al., 1995; Mihara et al., 2002). Trazodone’s metabolism involves not only CYP3A4 but also other hepatic enzymes such as CYP2D6, CYP2C19, and flavin-containing monooxygenases (FMOs), which contribute to secondary pathways like hydroxylation and N-oxidation (Rotzinger et al., 1998a). Despite its clinical relevance, detailed studies on the specific metabolic routes, enzyme contributions, and interspecies differences remain limited.
Research on central nervous system (CNS)-expressed CYP enzymes has underscored their relevance in modulating neurotransmitter dynamics, with significant implications for depressive disorders (Sheng et al., 2021). For instance, CYP2D6 in the brain is critical for the synthesis of serotonin from 5-methoxytryptamine and dopamine from tyramine, which influences serotonergic tone and dopaminergic signalling, both of which are fundamental to mood regulation and cognitive functions (Bromek et al., 2010; Bromek et al., 2011; Haduch et al., 2013; Haduch et al., 2015). Variability in CYP2D6 activity, whether due to genetic polymorphisms or drug-drug interactions, can influence serotonin and dopamine levels, potentially impacting antidepressant response and susceptibility to MDD (Ingelman-Sundberg et al., 2014; Penas-Lledo et al., 2015). In the substantia nigra, CYP2E1 participates in dopamine metabolism, and its role in producing reactive oxygen species (ROS) links it to neurodegenerative diseases like Parkinson’s, where oxidative stress exacerbates neuronal loss (Nissbrandt et al., 2001; Niazi Shahabi et al., 2003). CYP3A4, while predominantly hepatic, also plays a role in brain metabolism of psychotropic medications, affecting their local concentrations and possibly influencing both therapeutic and adverse effects (Ghosh et al., 2011; Sheng et al., 2021).
Despite its widespread use, current research on trazodone metabolic pathways, particularly CYP-mediated transformations, is limited and dated (Wen et al., 2008; Cuomo et al., 2019). While trazodone’s human metabolic pathways—principally CYP3A4-mediated N-dealkylation to mCPP and CYP2D6-mediated hydroxylation—are well characterized (Rotzinger et al., 1998a), early microsomal studies in rodents and rabbits demonstrate qualitatively different metabolic rates across species (Yamato et al., 1976). However, no peer-reviewed work has systematically quantified intrinsic clearance and mCPP formation in human versus rat hepatocytes. Given the significant role of CYP enzymes in both hepatic and CNS metabolism, filling this gap is essential to enhance the predictive power of preclinical in vitro models for human pharmacokinetics and drug–drug interaction risk (Sheng et al., 2021).
This study aims to further characterize the involvement of specific CYP isoforms in trazodone hepatic metabolism and to examine the inhibitory potential of trazodone and mCPP on major CYP enzymes. Comparative analyses across species and systems (microsomes, hepatocytes, and recombinant enzymes) were performed to clarify interspecies differences, with implications for translational pharmacokinetics and in vitro-to-in vivo extrapolation.
2 Materials and methods
2.1 Hepatic metabolic stability
The metabolic stability of trazodone was assessed in cryopreserved hepatocytes from mouse, rat, and human sources to evaluate hepatic intrinsic clearance (CLint) and to characterize metabolic pathways. Trazodone hydrochloride was prepared at a final concentration of 10 μM, using William’s medium E supplemented with 8.9 mM glutamine and 89 mM Hepes as the incubation medium. Cryopreserved hepatocytes were pooled from mixed-gender human (H634/DCT, XenoTech/Bioreclamation IVT), pooled male mouse (CD-1, XenoTech), and pooled male Sprague-Dawley rat (XenoTech) sources, with a final concentration of approximately 0.5 million cells/mL. Incubations were conducted in duplicate at 37 °C for 180 min in a reaction volume of 200 μL. Standard deviations were not calculated due to small replicate size. To validate metabolic activity, testosterone (10 μM) and 7-hydroxycoumarin (5 μM) were used as phase I and phase II positive controls, respectively. An internal standard, trazodone D6 hydrochloride, was included at 500 ng/mL in the acetonitrile solution used for termination. Samples were collected at 12 designated time points (0, 5, 10, 15, 20, 30, 45, 60, 90, 120, 150, and 180 min) to capture detailed metabolic data, and reactions were terminated by adding acetonitrile at a 1:2 ratio to precipitate proteins. Following termination, samples were centrifuged. Supernatants were diluted with water to adjust the final organic solvent concentration (acetonitrile) to approximately 37%. Processed samples were analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to monitor the disappearance of the parent compound over time (Bihan et al., 2015). Items were analyzed by LC-MS/MS on an Agilent HP1100 HPLC with a Synergi MAX RP (30 × 2 mm, 4 µm) column, using water + 0.1% formic acid (A) and acetonitrile + 0.1% formic acid (B) at 1.5 mL/min with a gradient from 5% B (0 min) to 95% B (1.5 min), held to 1.7 min, then returned to 5% B by 2.0 min, coupled to an AB Sciex API4000 mass spectrometer operating in positive MRM mode (372.0→176.1 for trazodone; 378.2→182.1 for D6; 289.2→109.0 for testosterone; 163.0→107.0 for 7-hydroxycoumarin).
CLint was calculated from the first-order elimination constant k, adjusted for the volume of incubation and hepatocyte count per reaction, and further normalized using a scaling factor for liver cells (expressed in million cells/g liver). Positive control reactions confirmed appropriate biotransformation activity in the hepatocytes, validating the experimental setup.
2.2 Metabolic profiling
The metabolic profile of trazodone was determined using in vitro incubations with rat and human liver microsomes, hepatocytes, and rat plasma samples. Trazodone was prepared at a final concentration of 8 μM. Microsomal incubations were conducted with pooled human and rat liver microsomes (Celsis IVT) using 0.6 mg/mL microsomal protein in 0.1 M phosphate buffer (pH 7.4). NADPH (1 mM) was added as a cofactor to initiate phase I metabolic reactions. Reactions were performed at 37 °C for 40 min with gentle shaking.
For hepatocyte incubations, pooled cryopreserved human hepatocytes (20-donor mix, Celsis) and male Sprague-Dawley rat hepatocytes (12-donor mix, Bioreclamation IVT) were used. Each incubation contained 1 million viable cells/mL in Celsis InVitro HI medium, at a pH of 7.4, and was conducted at 37 °C for 120 min under gentle agitation to facilitate metabolic activity.
In addition, rat plasma samples were collected at 0.25 and 6 h post-oral administration of trazodone at a dose of 30 mg/kg. Following incubation, all reactions were terminated by adding an equal volume of cold acetonitrile to precipitate proteins. Samples were then centrifuged at 13,000 x g for 10 min, and the supernatants were transferred to 96-well plates for analysis.
Processed supernatants (4 µL) were injected onto a Thermo U3000 UHPLC system coupled to a Thermo Q-Exactive Orbitrap MS equipped with a PDA detector. Separation was achieved on a Waters ACQUITY BEH C18 column (2.1 × 50 mm, 1.8 µm; 40 °C) at 0.50 mL min-1 using a gradient of 2 mM ammonium-formate (A) and acetonitrile (B): 95:5 (A:B) from 0 to 0.50 min, 50:50 at 3.00 min, 10:90 from 3.50 to 3.80 min, then back to 95:5 by 4.50 min. The Orbitrap operated in positive ESI mode (sheath/aux/sweep N2 55/8/4 units; capillary 3.0 kV, 370 °C; auxiliary heater 550 °C) with full-scan m/z 100–1,000 at 35,000 FWHM (m/z 200) and DDI-MS/MS at 17,500 FWHM (7 Hz acquisition; IT 100 m; HCD 40 eV). External mass calibration and a 5 ppm extraction window were applied, and data were processed in Xcalibur 3.0.63/Compound Discoverer 1.0.
2.3 Identification of CYP isoforms using selective inhibitors
The role of specific CYP isoforms in trazodone metabolism was examined using selective inhibitors in human and rat liver microsomes. Trazodone (4 μM) was incubated with liver microsomal protein (0.4 mg/mL) in 0.1 M phosphate buffer (pH 7.4), with NADPH (1 mM) added to initiate phase I metabolic reactions. The incubation was conducted at 37 °C for 20 min with gentle shaking. Optimized concentrations of selective CYP inhibitors were introduced based on published studies (Turpeinen et al., 2005; Walsky et al., 2005; Turpeinen et al., 2006; Perloff et al., 2009). A summary of the CYP isoforms, corresponding inhibitors, and concentrations used is provided in Table 1. Reactions were terminated by adding an equal volume of acetonitrile to precipitate proteins, followed by centrifugation at 13,000 × g for 10 min.
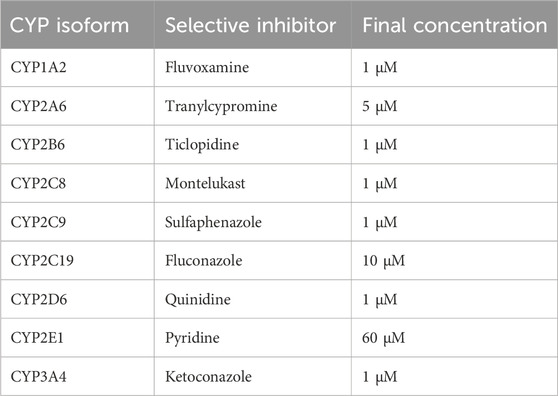
Table 1. Selective cytochrome P450 (CYP) inhibitors used for reaction phenotyping in human and rat liver microsomes. Final concentrations were selected based on literature-reported specificity and efficacy.
Metabolites were quantified under the same UHPLC-Orbitrap conditions described in § 2.2, ensuring consistent chromatography and MS performance across all inhibitor experiments. Metabolite levels in inhibitor-treated samples were compared to control samples (no inhibitor) to determine the extent of each CYP isoform involvement in metabolite production.
2.4 Identification of CYP isoforms using recombinant enzymes
To confirm the role of individual CYP isoforms in metabolizing trazodone, recombinant human and rat CYP enzymes were employed in separate incubations. Trazodone (4 μM) was incubated with each recombinant enzyme, including human CYP3A4, CYP3A5, CYP2D6, CYP2C19, CYP1A2, and FMO enzymes (FMO1 and FMO3), in a microsomal incubation buffer with NADPH as a cofactor at 37 °C for 40 min. Each reaction was terminated with acetonitrile, centrifuged, and the supernatant was analyzed by identical UHPLC-Orbitrap setup outlined in §2.2, allowing direct comparison of metabolite spectra across all in-vitro systems. The production of primary metabolites, such as M1, M2, M9, and M16, was monitored to determine which recombinant enzymes produced each metabolite. This procedure helped confirm the contributions of individual isoforms to specific biotransformation reactions.
2.5 Extrapolation to human using a predictive model
A predictive model was utilized to extrapolate the relative contributions of each CYP isoform in human liver to the overall metabolism of trazodone (Rostami-Hodjegan and Tucker, 2007). This model incorporated the known relative abundances of CYP isoforms in human liver tissue and was used to estimate the predominant pathways mediated by CYP3A4 and CYP3A5. The data from recombinant enzyme studies provided specific metabolite formation rates, which were adjusted for enzyme abundance to predict in vivo contributions. Additional extrapolation considered minor roles of CYP2C8, CYP2C19, CYP2D6, and FMO1/FMO3 based on enzyme activity data.
2.6 Inhibition of CYP enzymes by trazodone and mCPP
The last section of our study assessed the inhibitory effects of trazodone and its primary metabolite, mCPP, on the cytochrome P450 enzymes CYP1A2, CYP2A6, CYP2E1, and CYP3A4 in human and mouse hepatocytes. To evaluate the extent of enzyme inhibition, model probe substrates specific to each CYP enzyme were used: phenacetin for CYP1A2, coumarin for CYP2A6, chlorzoxazone for CYP2E1, and midazolam for CYP3A4. Known inhibitors served as positive controls, including furafylline for human CYP1A2, rutaecarpine for mouse CYP1A2, and tranylcypromine for CYP2A6, 4-methylpyrazole for CYP2E1, and ketoconazole for CYP3A4.
Cryopreserved human and mouse hepatocytes were obtained from Xenotech and Celsis, pooled from multiple male and female donors to increase experimental reliability. Prior to use, hepatocytes were stored in vapor-phase liquid nitrogen and then thawed, counted, and diluted in William’s Medium E, supplemented with glutamine and Hepes to reach a final concentration of approximately 0.5 million cells/mL.
Trazodone and mCPP were tested across an eight-point concentration-response curve, ranging from 100 μM to 0.1 μM, in triplicate. Positive controls were tested similarly but in singlicate. Due to the exploratory nature of these studies and absence of biological replicates, standard deviations were not calculated. Each test compound was dissolved in methanol (except 4-methylpyrazole, which was dissolved in water to prevent interference with CYP2E1 activity), and further serial dilutions were prepared in the same solvent. Methanol was used as a control at the highest solvent concentration.
Incubation plates were prepared by adding 5 μL of each test compound per well, followed by 1 μL of the respective probe substrate (200× stock solution). Substrates were prepared in acetonitrile (phenacetin and chlorzoxazone) or methanol (coumarin and midazolam) to achieve the final reaction concentrations. Finally, hepatocytes were added (195 μL per well), bringing the total incubation volume to 200 μL per well. Plates were maintained at 37 °C with shaking to support optimal enzyme activity.
After incubation, CYP enzyme activities were monitored by measuring the formation or disappearance of specific metabolites via high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). IC50 values, representing the concentration at which enzyme activity was inhibited by 50%, were calculated for each test compound. Samples (100 µL) were protein-precipitated with 200 µL acetonitrile containing internal standard, vortexed, centrifuged (20 min, 2 800 rpm), diluted 1 : 2 with 18% acetonitrile and 10–15 µL injected onto an Agilent HP1100 LC fitted with a Varian ODS-3 column (33 × 3 mm, 3 µm) and coupled to an AB Sciex API4000 triple-quadrupole (Turbo Ion Spray 650 °C; gas 45/45/25; MRM mode). Mobile phases were water +0.1% formic acid (A) and acetonitrile +0.1% formic acid (B) with 3 min gradients optimised for each probe substrate; analyte transitions were 152→110 (paracetamol), 161→133 (7-OH-coumarin), 184→120 (6-OH-chlorzoxazone) and 342→203 (1′-OH-midazolam).
3 Results
3.1 Hepatic metabolic stability
The CLint values revealed notable interspecies differences. In rat hepatocytes, trazodone exhibited a high clearance rate of 16.2 mL/min/g liver, indicating rapid metabolism. In mouse hepatocytes, the clearance rate was moderate at 4.14 mL/min/g liver, while in human hepatocytes, trazodone displayed a much slower clearance rate of 1.31 mL/min/g liver (Table 2).
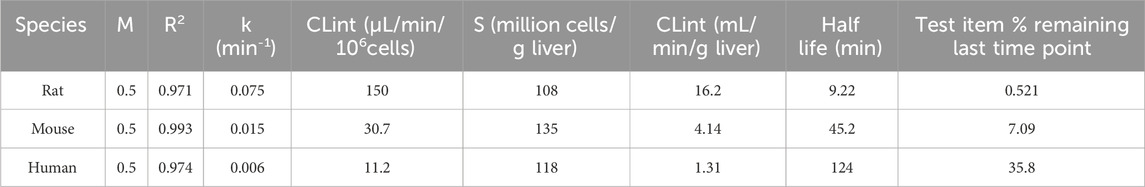
Table 2. Metabolic stability data for trazodone in rat, mouse and human cryopreserved hepatocytes.
3.2 Metabolic profiling
A comprehensive metabolic profiling study of trazodone identified 37 metabolites, labeled M1 through M37, across rat and human liver microsomes, hepatocytes, and rat plasma. Representative chromatograms are shown in Supplementary Figure S1, while the relative abundance (% of total LC-MS signal) for each metabolite across biological matrices is provided in Supplementary Table S1.
Key metabolites common to both species included the oxidation product of chlorophenyl-propylpiperazine (M1), the oxidation product of propylpiperazine (M2), the dioxidation and hydrogenation derivative of triazole-pyridinone (M9), and m-chlorophenylpiperazine (mCPP, M16), which is formed via dealkylation by loss of propyltriazolopyridinone (C9H9N3O). Additionally, conjugated forms of M1, such as glucuronide (M23) and sulfo-conjugates (M25), were also identified (Figure 1). Among these, mCPP is the only metabolite known to retain pharmacological activity, particularly through its agonist action on serotonin receptors (Otani et al., 1997).
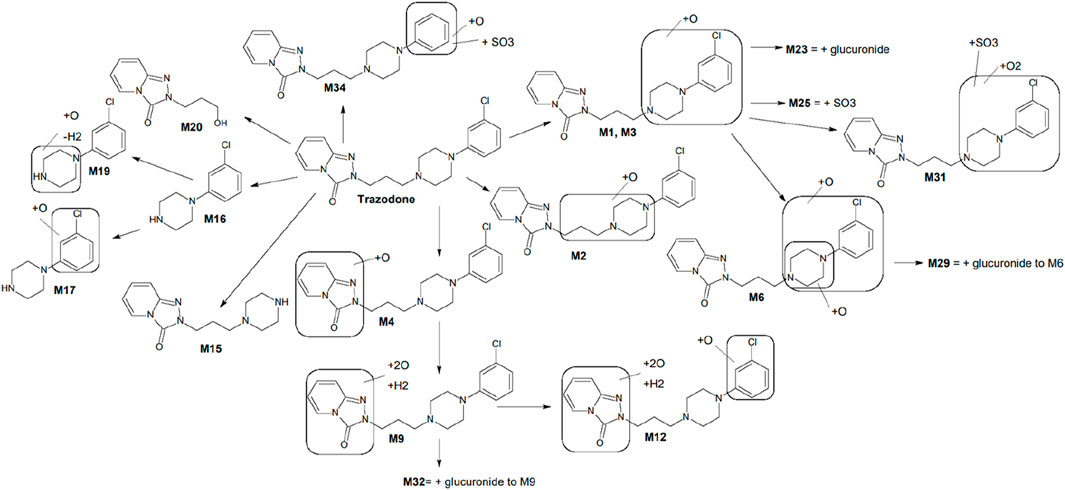
Figure 1. Proposed metabolic scheme of trazodone, constructed from in vitro incubations with human and rat liver microsomes, hepatocytes, and rat plasma. Key metabolites M1, M2, M9, M16, M17, and M20 were consistently observed in both human and rat systems. Metabolites such as M12, M23, and M25 showed markedly higher abundance in rat samples (particularly hepatocytes and plasma), whereas others displayed species-dependent variations in intensity. Full distribution and MS/MS characterization are detailed in Table 3 and Supplementary Figure S1.
Incubations with liver microsomes and hepatocytes yielded a high number of metabolites across species, with distinct similarities noted in metabolite profiles between human and rat systems. Of the 37 total metabolites, 26 were observed in liver microsomes, 32 in hepatocytes, and 24 in rat plasma. UPLC-QE-Orbitrap-MS analysis revealed that M1 and M2 were the most prominent metabolites in both human and rat liver microsomes. M1 (chlorophenyl-propylpiperazine oxide), was consistently the dominant metabolite across species, followed closely by M2 (propylpiperazine oxide). In hepatocytes, additional conjugated metabolites such as M23 (glucuronide of M1) and M25 (sulfate of M1) were detected in rat samples, particularly in plasma.
The microsomal profiling showed that human and rat liver microsomes generated similar metabolite patterns, indicating a high degree of cross-species consistency. In human liver microsomes, M1 was the most abundant metabolite, followed by M2, M9 (triazole-pyridinone dioxidation and hydrogenation), and M16 (mCPP). Rat liver microsomes exhibited a comparable profile, with M1 again showing the highest abundance, along with significant levels of M2, M9, M16, M17 (chlorobenzene hydroxylation product), and M20 (alcohol derivative resulting from dealkylation at the opposite end of the molecule).
Further profiling in hepatocytes confirmed the presence of these metabolites with some variations in relative abundance. M12, another significant metabolite related to trazodone N-dealkylation, showed a higher presence in rat hepatocytes than in humans. In addition, conjugated metabolites such as M23 and M25 were observed at higher levels in rat hepatocytes and plasma, which aligns with the trend of increased conjugation activity in rat hepatocytes compared to humans. Metabolites in rat plasma matched qualitatively with the profiles obtained in microsomes and hepatocytes, particularly for M1, M2, M9, M16, M23, and M25. A summary of the key metabolites identified in rat and human systems, including retention time, molecular formula, and characteristic MS/MS fragment ions, is provided in Table 3.
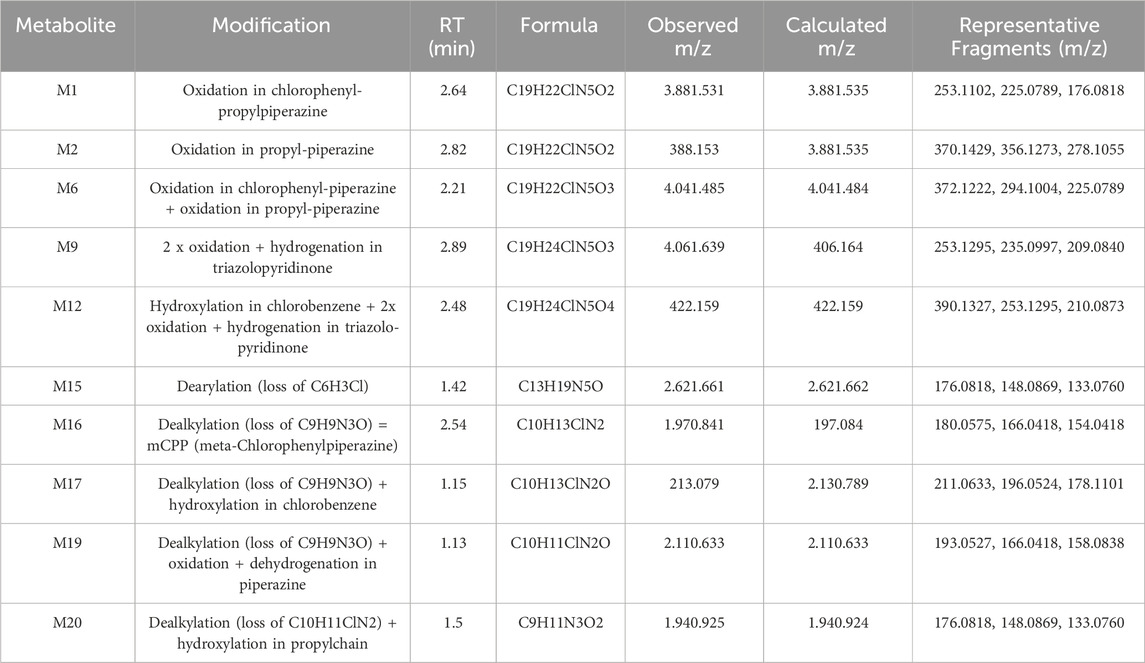
Table 3. Summary of selected trazodone metabolites identified in rat and human liver microsomes and hepatocytes. For each metabolite, retention time (RT), molecular formula, observed and calculated m/z, and key MS/MS fragment ions are reported. These include both phase I metabolites (e.g., mono- and di-oxidation, dealkylation, dearylation) and representative bioactivation products.
3.3 Identification of CYP isoforms using selective inhibitors
To identify specific CYP enzymes involved in the metabolism of trazodone, selective CYP inhibitors were applied in human and rat liver microsomal incubations. The inhibitors used included ketoconazole for CYP3A, fluconazole for CYP2C19, quinidine for CYP2D6, and fluvoxamine for CYP1A2, with each inhibitor employed at optimized concentrations to selectively inhibit its target enzyme (Turpeinen et al., 2005; Walsky et al., 2005; Turpeinen et al., 2006; Perloff et al., 2009).
In human liver microsomes (Table 4), the CYP3A inhibitor ketoconazole markedly reduced the formation of several key metabolites, notably M1 (chlorophenyl-propylpiperazine oxidation), M6 (mono-oxygenation product), M12 (N-oxidation product), M15 (hydroxylated derivative), M16 (mCPP), M17 (further chlorobenzene hydroxylation of mCPP), and M20 (alcohol product from dealkylation). Formation rates for these metabolites were reduced by 88%–100% compared to control samples without inhibitors, indicating that CYP3A4/5 plays a predominant role in their formation.
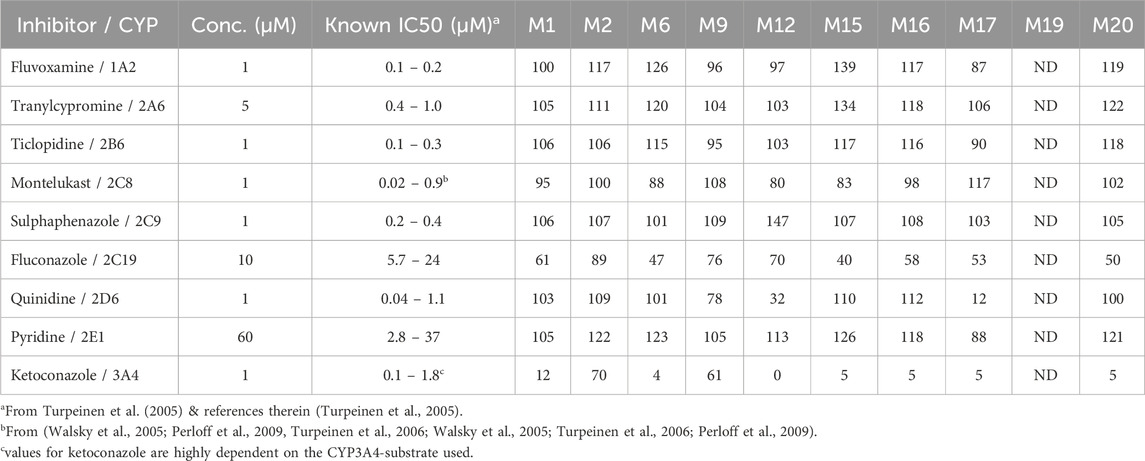
Table 4. The remaining percentage formation (as LC/MS peak areas) for the major trazodone metabolites in human liver microsomal incubations in the presence of CYP-selective inhibitors. ND = not detected.
The metabolite M9 (triazole-pyridinone dioxidation and hydrogenation) was less affected by ketoconazole but showed moderate inhibition by both fluconazole and quinidine, suggesting the involvement of multiple CYP enzymes in its formation, including CYP2C19, CYP2D6, and CYP3A4. This is similar to M12, which displayed a reduction down to 32% in formation with quinidine, indicating that CYP2D6 contributes alongside CYP3A4 to its production.
For M2 (propylpiperazine oxidation), ketoconazole produced a mild inhibition, reducing its formation to 70%, and other inhibitors had little to no effect, indicating that M2 formation may involve non-CYP pathways or other enzyme families, such as FMOs. This was consistent with the observed activity in recombinant FMO experiments (Table 5).
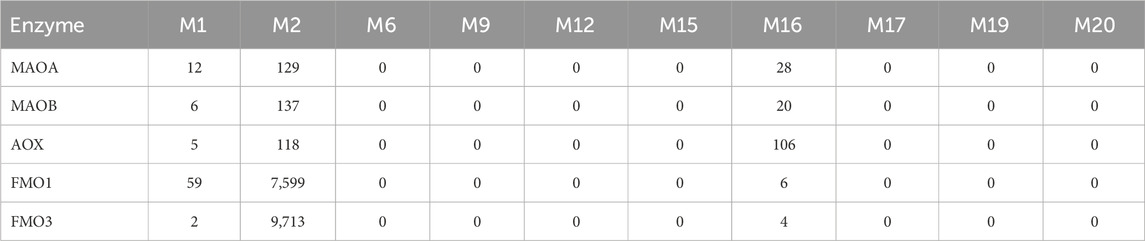
Table 5. Formation of trazodone major phase I metabolites catalyzed by recombinant human MAO, AOX and FMO enzymes (as LC/MS peak areas).
In rat liver microsomes (Table 6), the inhibition pattern was largely similar to that in humans, with ketoconazole reducing the formation of all major metabolites except M2, suggesting that CYP3A isoforms in rats play a similarly dominant role in metabolizing trazodone. However, unlike in human microsomes, quinidine had no significant effect on the formation of M9 and M12 in rats, which suggests that the rat CYP2D family may not contribute significantly to these pathways.
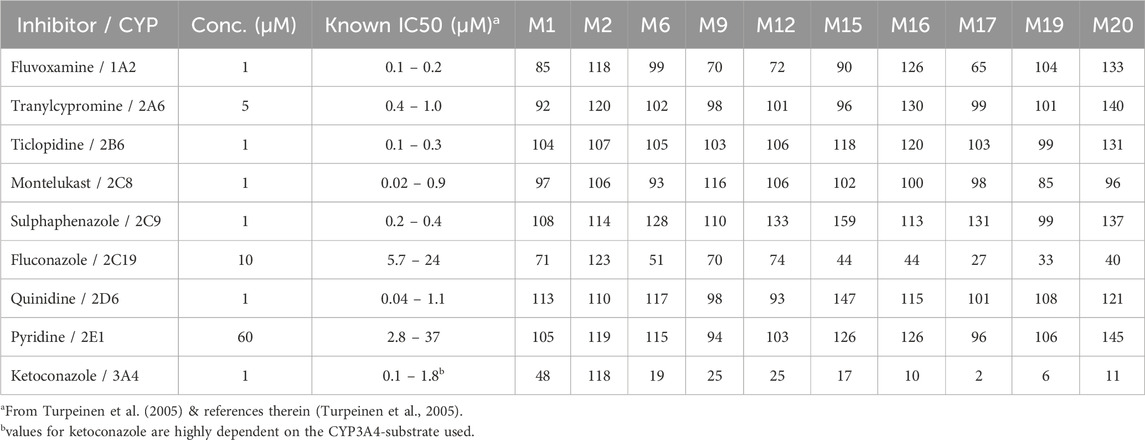
Table 6. The remaining percentage formation (as LC/MS peak areas) for the major trazodone metabolites in rat liver microsomal incubations in the presence of CYP-selective inhibitors.
Fluvoxamine, a CYP1A2 inhibitor, had a mild inhibitory effect on M9, M12, and M18 in rat liver microsomes, reducing their formation to 65%–72% of control levels. This suggests a minor role for CYP1A2 in these pathways, specifically in rats. Notably, M2 formation in rat liver microsomes was unaffected by any of the CYP inhibitors tested, aligning with findings in human microsomes and further supporting the hypothesis that M2 may form via an alternative enzyme pathway, potentially involving FMOs or non-CYP mechanisms. M19, a minor downstream metabolite of mCPP, was undetectable in human liver microsomal incubations under all tested conditions (Table 4). In contrast, M19 was consistently observed in rat liver microsomes, and its formation was strongly inhibited by ketoconazole, suggesting a primary role for CYP3A enzymes in its biosynthesis in rodents. Fluconazole also moderately reduced M19 levels, indicating a possible secondary contribution from CYP2C19. Other CYP inhibitors had negligible effects (Table 6).
3.4 Identification of CYP isoforms using recombinant enzymes
To further elucidate the CYP isoforms involved in trazodone metabolism, recombinant CYP enzymes from both human and rat sources were employed, focusing on the production of major metabolites M1, M2, M9, and M16. Incubations with recombinant human CYP3A4 and CYP3A5 yielded the highest levels primarily of M1 and M16 and secondarily of M9 and M15, indicating a strong role for these isoforms in metabolizing trazodone. Among these, M1 formation was particularly prominent, following the order of CYP3A5 > CYP2D6 > CYP3A4 > CYP2C9 (Table 7).
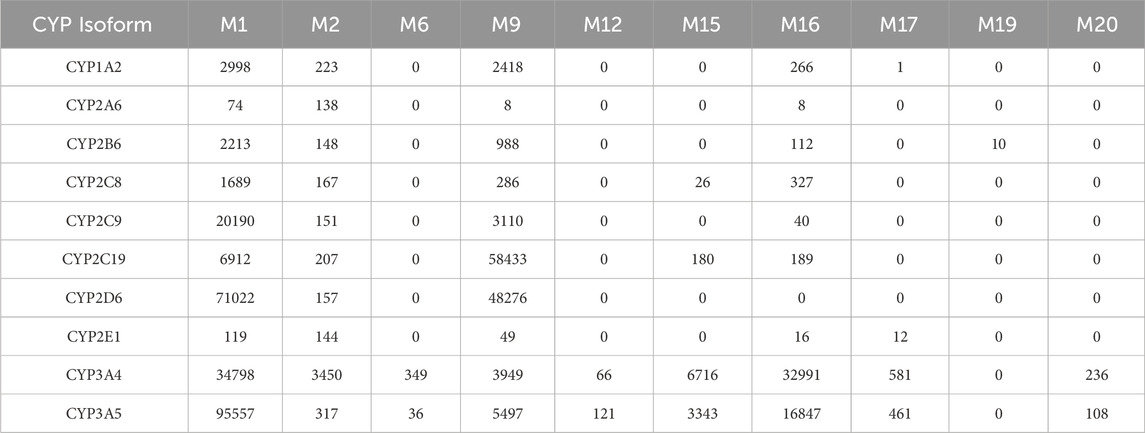
Table 7. Formation of trazodone major phase I metabolites catalyzed by recombinant human CYP enzymes (as LC/MS peak areas).
For M2, the primary enzyme contributing to its formation was CYP3A4, though both human FMO1 and FMO3 also facilitated M2 formation (Tables 5, 7), suggesting that its metabolic pathway may involve N-oxidation. This finding correlates with the low inhibition observed in microsomal studies when ketoconazole was used, indicating that CYP3A4 alone does not fully account for M2 formation. M16 (mCPP), resulting from N-dealkylation, was again most abundantly formed by CYP3A4, with secondary contribution from CYP3A5, in line with published evidence (Wen et al., 2008).
When recombinant rat CYP enzymes were used, M1 and M16 were again the most abundant metabolites (Table 8). CYP2C11, CYP2D4, and CYP3A9 produced M1, with CYP2C11 being the primary contributor, followed by CYP3A9 and CYP2D4. For M16, the highest formation was observed with CYP3A2 and CYP3A9, aligning with findings from human recombinant enzymes, further supporting the central role of CYP3A isoforms in N-dealkylation processes across species. These experiments confirmed the involvement of CYP3A4, CYP3A5, and CYP2D6 in the formation of major trazodone metabolites, with contributions from FMOs (Table 5).
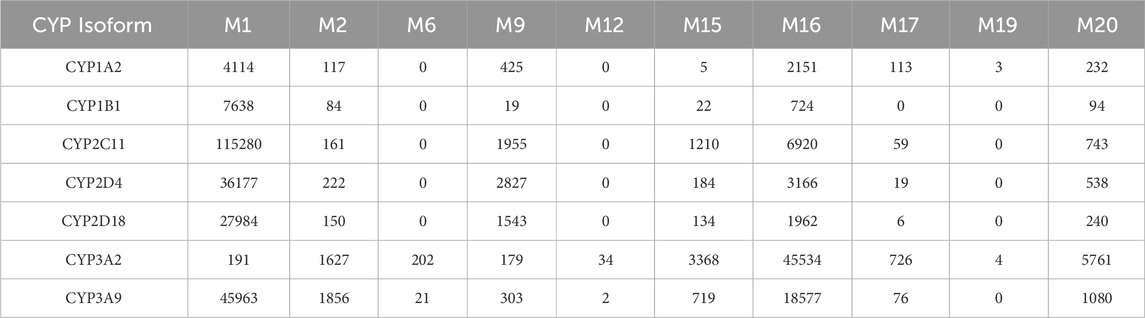
Table 8. Formation of trazodone major phase I metabolites catalyzed by recombinant rat CYP enzymes (as LC/MS peak areas).
3.5 Extrapolation to human using a predictive model
A predictive model was used to extrapolate the contributions of each CYP isoform to trazodone metabolism in human liver, incorporating enzyme abundances based on established literature values for human liver tissue (Rostami-Hodjegan and Tucker, 2007). This model indicated that CYP3A4 and CYP3A5 would mediate the majority of trazodone metabolism, especially for the formation of M1 and M16 (mCPP). These findings are consistent with results obtained in rats, where ketoconazole markedly reduced the formation of M16 in liver microsomal incubations, confirming the predominant role of CYP3A isoforms in both species (Tables 4, 6).
The model further suggested that CYP2C8 might contribute to M1 formation as a minor pathway, while M9 was predicted to involve multiple CYP isoforms, including CYP2C9, CYP2C19, CYP2D6, and CYP3A, with CYP2C19 and CYP3A identified as the most relevant enzymes for this pathway. Additionally, M2 formation was projected to involve FMO1 and FMO3, consistent with the observed high abundance of M2 in recombinant FMO incubations (Table 5).
3.6 Inhibition of CYP enzymes by trazodone and mCPP
The inhibition potential of trazodone and mCPP on CYP1A2, CYP2A6, CYP2E1, and CYP3A4 was examined across a concentration-response curve in human and mouse hepatocytes.
In human hepatocytes, trazodone selectively inhibited CYP2E1 with an IC50 of 6.18 μM, demonstrating no inhibitory effects on CYP1A2, CYP2A6, or CYP3A4 up to the maximum concentration tested (100 μM). Furthermore, mCPP exhibited inhibitory activity in human hepatocytes, inhibiting both CYP2A6 and CYP2E1, with IC50 values of 29.8 μM and 6.46 μM, respectively. Notably, mCPP remained inactive against CYP1A2 and CYP3A4 in human hepatocytes, mirroring trazodone inactivity for these isoforms. These findings highlight a specific inhibitory action of trazodone and mCPP on CYP2E1, with mCPP also affecting CYP2A6 (Table 9), which do not directly impact in vivo activity, as the IC50 values measured in vitro are not reached by the unbound concentrations achieved in clinical practice (Fagiolini et al., 2023).
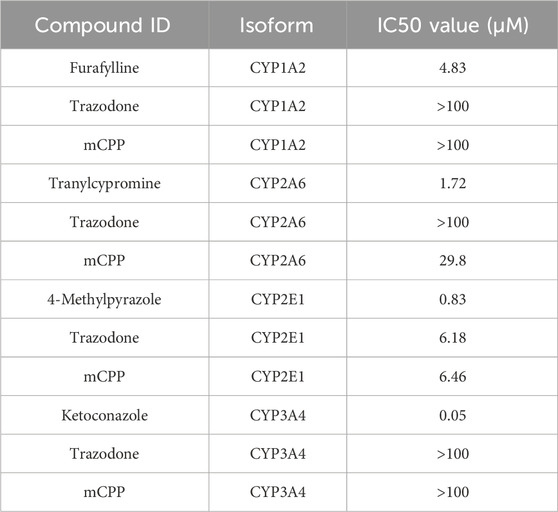
Table 9. Inhibition of cytochrome P450 enzymes by trazodone, mCPP and positive controls in human hepatocytes.
In mouse hepatocytes, trazodone inhibited CYP2E1 and CYP2A6, yielding IC50 values of 14.6 μM and 12.7 μM, respectively, indicating effective inhibition within this concentration range, while it resulted to be inactive up to 100 µM against CYP1A2 and CYP3A4. mCPP displayed significant potency against CYP2E1, with an IC50 of 1.88 μM, demonstrating a notably stronger inhibition than trazodone in this isoform (Table 10). In contrast, mCPP showed no activity against CYP1A2, CYP2A6, or CYP3A4 up to 100 μM in mouse hepatocytes. These results underscore a species-dependent variation in CYP inhibition profiles, with both trazodone and mCPP exhibiting stronger effects on CYP2E1 in mouse hepatocytes compared to human cells.
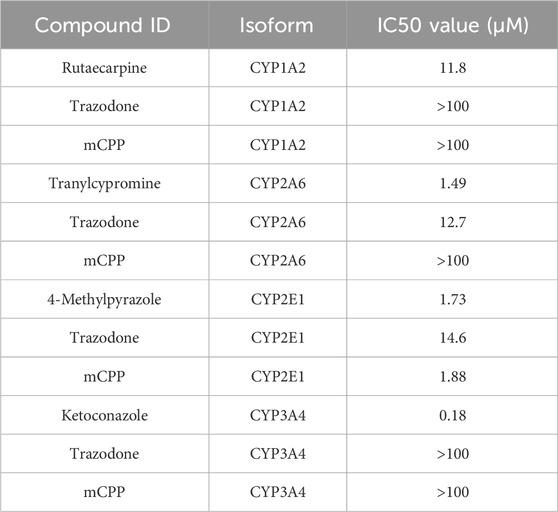
Table 10. Inhibition of cytochrome P450 enzymes by trazodone, mCPP and positive controls in CD1 mouse hepatocytes.
Control experiments with known CYP inhibitors—furafylline, rutaecarpine, tranylcypromine, 4-methylpyrazole, and ketoconazole—yielded IC50 values consistent with literature and in-house data, affirming the assay validity. The observed inhibition of CYP2E1 by trazodone and mCPP in mouse hapatocytes suggests a potential involvement of this isoform in species-specific metabolic pathways.
4 Discussion
Despite trazodone’s widespread clinical use, detailed understanding of its biotransformation—particularly the generation and pharmacological relevance of mCPP—remains limited. This study provides a systematic characterization of hepatic metabolism across species, using hepatocytes, microsomes, and recombinant enzyme systems.
CLint data revealed notable interspecies differences, with rat hepatocytes showing over 12-fold higher clearance rates than human hepatocytes (Table 2). These findings are consistent with historical in vivo data and suggest a faster metabolic turnover in rodents, particularly for CYP3A-mediated pathways such as M1 and M16, as confirmed by inhibition data (Tables 7, 8). In contrast, clearance in human hepatocytes was significantly slower, supporting the potential for sustained plasma levels and highlighting the need for human-specific in vitro systems to predict pharmacokinetics (Jansson-Lofmark et al., 2020). For other metabolites formed via CYP2C19 and CYP2D6, the interspecies variation was less marked. The observed differences may help contextualize the broad range in trazodone plasma levels, ranging from 21.5 to over 2,200 ng/mL under standard dosing conditions (Ding et al., 2022).
The metabolic profiling of trazodone—performed through incubations with human and rat liver microsomes, hepatocytes, and rat plasma—provided a comprehensive view of its biotransformation pathways. A total of 37 distinct metabolites were detected, involving hydroxylation, N-dealkylation, and conjugation. This range of transformations supports a metabolic cascade where primary oxidative and dealkylation reactions generate intermediates subsequently subjected to phase II conjugation (Figure 1). The active metabolite mCPP was consistently identified in both human and rat systems. Other major metabolites, such as M1 and M9, were also present across species, indicating conserved oxidative pathways. However, differences in relative abundance—such as increased M12 levels in rat hepatocytes—highlighted species-specific trends for some metabolites. Thesedifferences highlight the importance of considering species-specific patways when interpreting preclinical pharmacokinetic data, as rodent models may exhibit faster metabolism or alternative pathways compared to humans. The proposed metabolic scheme integrates these findings, outlining sequential steps leading to mCPP and other key derivatives. Oxidative processes dominate the early stages, with both N-dealkylation and hydroxylation contributing to mCPP formation. Conjugated derivatives such as M23 and M25 were primarily observed in rat hepatocytes and plasma, reflecting enhanced phase II capacity in rodents. The concordance between rat plasma and in vitro systems supports the relevance of these models for characterizing metabolic pathways involved in trazodone disposition.
The use of selective CYP inhibitors in human and rat liver microsomes confirmed CYP3A4 as the primary enzyme responsible for converting trazodone to mCPP. Ketoconazole markedly reduced the formation of mCPP and related metabolites in both species, consistent with the established central role of CYP3A4 in trazodone metabolism (Caccia et al., 1981; Rotzinger et al., 1998a; Zalma et al., 2000; Kalgutkar et al., 2005). In human liver microsomes, partial inhibition of other metabolites by fluconazole and quinidine suggested secondary contributions from CYP2C19 and CYP2D6. This multi-enzyme involvement underscores the importance of pharmacogenetic variability: CYP2C19 and CYP2D6 polymorphisms may influence trazodone metabolic patterns, with potential implications for interindividual variability. Indeed, steady-state studies show that CYP2D6 poor metabolizers accumulate higher mCPP/trazodone ratios compared to extensive metabolizers (Mihara et al., 1997; Wen et al., 2008). A recent case report described hepatotoxicity in a patient with a CYP2D6 loss-of-function genotype, reinforcing the relevance of CYP2D6 in mediating mCPP exposure (Ferdinande et al., 2024). Moreover, a scoping review suggested that while CYP2D6, CYP3A4, and CYP1A2 variants have limited impact on trazodone pharmacokinetics, polymorphisms in the ABCB1 transporter may significantly affect drug exposure and clearance (Meulman et al., 2023). In rat liver microsomes, ketoconazole inhibition produced similar results, confirming that CYP3A isoforms also mediate trazodone metabolism in rodents. However, quinidine had negligible effects on mCPP formation in rats, likely reflecting species differences in CYP2D function or inhibitor sensitivity. Fluvoxamine slightly reduced the formation of M17 (a metabolite derived from mCPP), suggesting a minor role for CYP1A2 in this transformation. In contrast, M19 formation appeared unaffected. These species-dependent variations highlight the complexity of extrapolating preclinical metabolism data to humans, underling the importance of integrated approaches.
Recombinant enzyme studies confirmed that CYP3A4 and CYP3A5 exhibit the highest activity in forming mCPP. Alongside them, CYP2D6 and CYP2C19 also contributed to the formation of M9 and hydroxylated products. Consistent with prior literature, CYP2D6 was shown to mediate para-hydroxylation of mCPP to p-hydroxy-mCPP, a pathway inhibited by quinidine in microsomal assays (Rotzinger et al., 1998b). Notably, the M2 metabolite—only mildly affected by ketoconazole—was produced by both CYP3A4 and the flavin-containing monooxygenases FMO1 and FMO3, supporting an N-oxidation pathway. This dual enzyme involvement explains the weak effect of CYP inhibitors on M2 formation in microsomes. To further investigate species-specific contributions, Table 8 reports the formation of major phase I metabolites in rat recombinant CYP systems, expressed as LC/MS peak areas. Among the isoforms tested, CYP3A2 showed the most robust formation of M1, M4, and M16—supporting the key role of rat CYP3A orthologs in oxidative metabolism of trazodone. In contrast, CYP2D2 and CYP2C11 catalyzed minor metabolites such as M17 and M9 with lower efficiency, suggesting more selective or limited roles.
Finally, inhibition assays showed that trazodone and mCPP were found to inhibit CYP2E1 in vitro, with mCPP also showing inhibition of CYP2A6. However, the in vitro IC50 values were substantially higher than unbound plasma concentrations achieved clinically. Given trazodone’s established safety profile over decades of use,the observed in vitro inihibition occurred at concentrations unlikely to be reached in vivo, suggesting low clinical relevance. Nonetheless, this highlights the challenges associated with in vitro-to-in vivo extrapolation, particularly when estimating drug interaction potential based on IC50 values alone (Jansson-Lofmark et al., 2020). These findings suggest a low potential for CYP2E1-or CYP2A6 mediated interactions under therapeutic conditions. However, combined with the interspecies variability observed in mCPP clearance and formation, these findings further support the use of human-specific models in drug interaction risk assessment.
5 Conclusion
This study clarifies the complex biotransformation of trazodone, identifying CYP3A isoforms as the primary enzymes responsible for its metabolism, with auxiliary contributions from CYP2D6, CYP2C19, and FMOs. Interspecies differences were evident in overall metabolic clearance, as highlighted by markedly faster hepatic metabolism in rats compared to humans, and in the formation of CYP3A-derived metabolites such as mCPP. In contrast, the metabolic patterns associated with CYP2D6 and CYP2C19 were relatively consistent across species. While FMOs (FMO1 and FMO3) also contributed to specific metabolic steps, such as M2 formation, their activity was only assessed in recombinant human systems and thus cannot be compared across species. These findings indicate that cross-species extrapolation is most limited for CYP3A-mediated pathways and may affect the extrapolation of exposure estimates from preclinical models to humans.
Human-specific models and dosing treatment personalization remain open points for future research, considering administration in patients with multiple concurrent disorders and genetic CYP polymorphisms. Collectively, this study advances the understanding of trazodone metabolic pathways, and highlights the role of CYP-mediated metabolism in contributing to pharmacological profile of trazodone in the treatment of MDD.
6 Limitations
This study did not include a direct assessment of cytotoxicity or nonspecific enzyme inhibition in hepatocytes, microsomes, or recombinant systems, as such evaluations were beyond the study’s scope. Although the tested concentrations fall within commonly used in vitro ranges, the absence of dedicated viability assays or controls precludes a definitive assessment of potential off-target or cytotoxic effects, particularly at the upper end of the concentration range. These findings are derived exclusively from in vitro models and should be regarded as exploratory; these in vitro findings are exploratory and cannot be directly translated to clinical safety outcomes.
Additionally, replicate experiments were not performed at a scale sufficient to provide statistical error estimates for IC50 or CLint determinations. As a result, the reported values are presented without standard deviations and should be interpreted as qualitative or semi-quantitative indicators of metabolic activity and inhibition potential.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
VP: Validation, Visualization, Data curation, Resources, Methodology, Formal Analysis, Project administration, Writing – review and editing, Investigation, Conceptualization, Software, Supervision, Funding acquisition. PD: Writing – review and editing, Funding acquisition, Project administration, Resources, Formal Analysis, Methodology, Visualization, Conceptualization, Software, Supervision, Data curation, Validation, Investigation. MCL: Investigation, Resources, Software, Formal Analysis, Funding acquisition, Visualization, Data curation, Conceptualization, Validation, Writing – review and editing, Methodology, Project administration, Supervision. LO: Supervision, Data curation, Methodology, Formal Analysis, Software, Visualization, Conceptualization, Resources, Investigation, Validation, Funding acquisition, Writing – review and editing, Project administration. VZ: Writing – review and editing, Supervision, Investigation, Conceptualization, Software, Methodology, Resources, Funding acquisition, Visualization, Project administration, Formal Analysis, Validation, Data curation. AC: Methodology, Supervision, Formal Analysis, Software, Writing – review and editing, Data curation, Conceptualization, Resources, Visualization, Project administration, Funding acquisition, Validation, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article.
Acknowledgments
The authors acknowledge Éthos S.r.l. for editorial and technical support, and Dr Andrea Ravelli for medical writing assistance.
Conflict of interest
Authors VP, PD, MCL and AC are full-time employees of Angelini Pharma S.p.A. Author VZ is a full-time employee of Angelini Pharma France SASU.
Author LO was a full time employee of Angelini Pharma S.p.A. at the time of the experiments conduct.
The authors declare that this study was sponsored by Angelini Pharma S.p.A. The funder had the following involvement in the study: study design, analysis, interpretation of data, the writing/reviewing of this article, and the decision to submit it for publication.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1636919/full#supplementary-material
References
Bihan, K., Sauzay, C., Goldwirt, L., Charbonnier-Beaupel, F., Hulot, J. S., Funck-Brentano, C., et al. (2015). Development and validation of a rapid and simple LC-MS/MS method for quantification of vemurafenib in human plasma: application to a human pharmacokinetic study. Ther. Drug Monit. 37 (1), 132–136. doi:10.1097/FTD.0000000000000110
Bromek, E., Haduch, A., and Daniel, W. A. (2010). The ability of cytochrome P450 2D isoforms to synthesize dopamine in the brain: an in vitro study. Eur. J. Pharmacol. 626 (2-3), 171–178. doi:10.1016/j.ejphar.2009.09.062
Bromek, E., Haduch, A., Golembiowska, K., and Daniel, W. A. (2011). Cytochrome P450 mediates dopamine formation in the brain in vivo. J. Neurochem. 118 (5), 806–815. doi:10.1111/j.1471-4159.2011.07339.x
Caccia, S., Ballabio, M., Fanelli, R., Guiso, G., and Zanini, M. G. (1981). Determination of plasma and brain concentrations of trazodone and its metabolite, 1-m-chlorophenylpiperazine, by gas-liquid chromatography. J. Chromatogr. 210 (2), 311–318. doi:10.1016/s0021-9673(00)97841-5
Cuomo, A., Ballerini, A., Bruni, A. C., Decina, P., Di Sciascio, G., Fiorentini, A., et al. (2019). Clinical guidance for the use of trazodone in major depressive disorder and concomitant conditions: pharmacology and clinical practice. Riv. Psichiatr. 54 (4), 137–149. doi:10.1708/3202.31796
Cuomo, A., Bianchetti, A., Cagnin, A., De Berardis, D., Di Fazio, I., Incalzi, R. A., et al. (2021). Trazodone: a multifunctional antidepressant. Evaluation of its properties and real-world use. J. Gerontology Geriatrics 69 (2), 120–129. doi:10.36150/2499-6564-N320
Ding, J., Zhang, Y., Cui, X., Zhao, C., and Zhang, S. (2022). Development of a novel analytical method for determining trazodone in human plasma by liquid chromatography coupled with mass spectrometry coupled with automatic 2-dimensional liquid chromatograph-mass spectrometer coupler 9500 and its application to therapeutic drug monitoring. Ther. Drug Monit. 44 (3), 465–473. doi:10.1097/FTD.0000000000000921
Fagiolini, A., Comandini, A., Catena Dell'Osso, M., and Kasper, S. (2012). Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs 26 (12), 1033–1049. doi:10.1007/s40263-012-0010-5
Fagiolini, A., Gonzalez-Pinto, A., Miskowiak, K. W., Morgado, P., Young, A. H., and Vieta, E. (2023). Role of trazodone in treatment of major depressive disorder: an update. Ann. Gen. Psychiatry 22 (1), 32. doi:10.1186/s12991-023-00465-y
Ferdinande, K., Declerck, L., Melis, C., Missiaen, L., Seynhaeve, L., Vloo, C., et al. (2024). CYP2D6 polymorphism may contribute to Trazodone-induced hepatotoxicity: a rare case of drug-drug-gene induced liver injury. Acta Gastro Enterol. Belg. 87, 332–335. doi:10.51821/87.2.12034
Fiorella, D., Rabin, R. A., and Winter, J. C. (1995). The role of the 5-HT2A and 5-HT2C receptors in the stimulus effects of m-chlorophenylpiperazine. Psychopharmacol. Berl. 119 (2), 222–230. doi:10.1007/BF02246164
Gartlehner, G., Hansen, R. A., Morgan, L. C., Thaler, K., Lux, L., Van Noord, M., et al. (2011). Comparative benefits and harms of second-generation antidepressants for treating major depressive disorder: an updated meta-analysis. Ann. Intern Med. 155 (11), 772–785. doi:10.7326/0003-4819-155-11-201112060-00009
Ghosh, C., Marchi, N., Desai, N. K., Puvenna, V., Hossain, M., Gonzalez-Martinez, J., et al. (2011). Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia 52 (3), 562–571. doi:10.1111/j.1528-1167.2010.02956.x
Haduch, A., Bromek, E., Sadakierska-Chudy, A., Wojcikowski, J., and Daniel, W. A. (2013). The catalytic competence of cytochrome P450 in the synthesis of serotonin from 5-methoxytryptamine in the brain: an in vitro study. Pharmacol. Res. 67 (1), 53–59. doi:10.1016/j.phrs.2012.10.009
Haduch, A., Bromek, E., Kot, M., Kaminska, K., Golembiowska, K., and Daniel, W. A. (2015). The cytochrome P450 2D-mediated formation of serotonin from 5-methoxytryptamine in the brain in vivo: a microdialysis study. J. Neurochem. 133 (1), 83–92. doi:10.1111/jnc.13031
Ingelman-Sundberg, M., Persson, A., and Jukic, M. M. (2014). Polymorphic expression of CYP2C19 and CYP2D6 in the developing and adult human brain causing variability in cognition, risk for depression and suicide: the search for the endogenous substrates. Pharmacogenomics 15 (15), 1841–1844. doi:10.2217/pgs.14.151
Jansson-Lofmark, R., Hjorth, S., and Gabrielsson, J. (2020). Does in vitro potency predict clinically efficacious concentrations? Clin. Pharmacol. Ther. 108 (2), 298–305. doi:10.1002/cpt.1846
Kalgutkar, A. S., Henne, K. R., Lame, M. E., Vaz, A. D., Collin, C., Soglia, J. R., et al. (2005). Metabolic activation of the nontricyclic antidepressant trazodone to electrophilic quinone-imine and epoxide intermediates in human liver microsomes and recombinant P4503A4. Chem. Biol. Interact. 155 (1-2), 10–20. doi:10.1016/j.cbi.2005.03.036
Karhu, D., Gossen, E. R., Mostert, A., Cronje, T., and Fradette, C. (2011). Safety, tolerability, and pharmacokinetics of once-daily trazodone extended-release caplets in healthy subjects. Int. J. Clin. Pharmacol. Ther. 49 (12), 730–743. doi:10.5414/cp201546
Khouzam, H. R. (2017). A review of trazodone use in psychiatric and medical conditions. Postgrad. Med. 129 (1), 140–148. doi:10.1080/00325481.2017.1249265
Meulman, J., Visacri, M. B., Moriel, P., and Pincinato, E. C. (2023). Influence of genetic polymorphisms on the pharmacokinetics of trazodone hydrochloride: a scoping review and future perspective. Ther. Drug Monit. 45 (4), 479–486. doi:10.1097/FTD.0000000000001049
Mihara, K., Otani, K., Suzuki, A., Yasui, N., Nakano, H., Meng, X., et al. (1997). Relationship between the CYP2D6 genotype and the steady-state plasma concentrations of trazodone and its active metabolite m-chlorophenylpiperazine. Psychopharmacol. Berl. 133 (1), 95–98. doi:10.1007/s002130050376
Mihara, K., Yasui-Furukori, N., Kondo, T., Ishida, M., Ono, S., Ohkubo, T., et al. (2002). Relationship between plasma concentrations of trazodone and its active metabolite, m-chlorophenylpiperazine, and its clinical effect in depressed patients. Ther. Drug Monit. 24 (4), 563–566. doi:10.1097/00007691-200208000-00016
Niazi Shahabi, H., Bergquist, F., and Nissbrandt, H. (2003). An investigation of dopaminergic metabolites in the striatum and in the substantia nigra in vivo utilising radiolabelled L-DOPA and high performance liquid chromatography: a new approach in the search for transmitter metabolites. Neuroscience 120 (2), 425–433. doi:10.1016/s0306-4522(03)00330-0
Nissbrandt, H., Bergquist, F., Jonason, J., and Engberg, G. (2001). Inhibition of cytochrome P450 2E1 induces an increase in extracellular dopamine in rat substantia nigra: a new metabolic pathway? Synapse 40 (4), 294–301. doi:10.1002/syn.1052
Otani, K., Mihara, K., Yasui, N., Ishida, M., Kondo, T., Tokinaga, N., et al. (1997). Plasma concentrations of trazodone and m-chlorophenylpiperazine at steady state can be predicted from those after an initial dose of trazodone. Prog. Neuropsychopharmacol. Biol. Psychiatry 21 (1), 239–244. doi:10.1016/s0278-5846(96)00140-6
Penas-Lledo, E., Guillaume, S., Naranjo, M. E., Delgado, A., Jaussent, I., Blasco-Fontecilla, H., et al. (2015). A combined high CYP2D6-CYP2C19 metabolic capacity is associated with the severity of suicide attempt as measured by objective circumstances. Pharmacogenomics J. 15 (2), 172–176. doi:10.1038/tpj.2014.42
Perloff, E. S., Mason, A. K., Dehal, S. S., Blanchard, A. P., Morgan, L., Ho, T., et al. (2009). Validation of cytochrome P450 time-dependent inhibition assays: a two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 39 (2), 99–112. doi:10.1080/00498250802638155
Rostami-Hodjegan, A., and Tucker, G. T. (2007). Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat. Rev. Drug Discov. 6 (2), 140–148. doi:10.1038/nrd2173
Rotzinger, S., Fang, J., and Baker, G. B. (1998a). Trazodone is metabolized to m-chlorophenylpiperazine by CYP3A4 from human sources. Drug Metab. Dispos. 26 (6), 572–575. doi:10.1124/dmd.26.6.572
Rotzinger, S., Fang, J., Coutts, R. T., and Baker, G. B. (1998b). Human CYP2D6 and metabolism of m-chlorophenylpiperazine. Biol. Psychiatry 44 (11), 1185–1191. doi:10.1016/s0006-3223(97)00483-6
Sheng, Y., Yang, H., Wu, T., Zhu, L., Liu, L., and Liu, X. (2021). Alterations of cytochrome P450s and UDP-glucuronosyltransferases in brain under diseases and their clinical significances. Front. Pharmacol. 12, 650027. doi:10.3389/fphar.2021.650027
Stahl, S. M. (2009). Mechanism of action of trazodone: a multifunctional drug. CNS Spectr. 14 (10), 536–546. doi:10.1017/s1092852900024020
Turpeinen, M., Uusitalo, J., Jalonen, J., Pelkonen, O., Jorma, J., et al. (2005). Multiple P450 substrates in a single run: rapid and comprehensive in vitro interaction assay. Eur. J. Pharm. Sci. 24 (1), 123–132. doi:10.1016/j.ejps.2004.10.006
Turpeinen, M., Korhonen, L. E., Tolonen, A., Uusitalo, J., Juvonen, R., Raunio, H., et al. (2006). Cytochrome P450 (CYP) inhibition screening: comparison of three tests. Eur. J. Pharm. Sci. 29 (2), 130–138. doi:10.1016/j.ejps.2006.06.005
Walsky, R. L., Obach, R. S., Gaman, E. A., Gleeson, J. P., and Proctor, W. R. (2005). Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab. Dispos. 33 (3), 413–418. doi:10.1124/dmd.104.002766
Wen, B., Ma, L., Rodrigues, A. D., and Zhu, M. (2008). Detection of novel reactive metabolites of trazodone: evidence for CYP2D6-mediated bioactivation of m-chlorophenylpiperazine. Drug Metab. Dispos. 36 (5), 841–850. doi:10.1124/dmd.107.019471
Yamato, C., Takahashi, T., and Fujita, T. (1976). Studies on metabolism of trazodone. III Species differences. Xenobiotica 6 (5), 295–306. doi:10.3109/00498257609151641
Keywords: trazodone metabolism, cytochrome P450, drug interactions, pharmacokinetics, drug metabolism
Citation: Petrucci V, Dragone P, Laurenti MC, Oggianu L, Zabela V and Cattaneo A (2025) Characterization of trazodone metabolic pathways and species-specific profiles. Front. Pharmacol. 16:1636919. doi: 10.3389/fphar.2025.1636919
Received: 28 May 2025; Accepted: 20 August 2025;
Published: 30 September 2025.
Edited by:
Guru Raghavendra Valicherla, Pharmaron (Exton) Lab Services LLC, United StatesReviewed by:
Kannapiran Ponraj, Washington State University, United StatesGajanan Jadhav, University of Alabama at Birmingham, United States
Mohamed Elgawish, Korea University, Republic of Korea
Riyazuddin Mohammed, University of Pittsburgh, United States
Copyright © 2025 Petrucci, Dragone, Laurenti, Oggianu, Zabela and Cattaneo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnese Cattaneo, YWduZXNlLmNhdHRhbmVvQGFuZ2VsaW5pcGhhcm1hLmNvbQ==