 Koen Deserranno
Koen Deserranno Laurentijn Tilleman
Laurentijn Tilleman Dieter Deforce
Dieter Deforce Filip Van Nieuwerburgh
Filip Van Nieuwerburgh- Laboratory of Pharmaceutical Biotechnology, Faculty of Pharmaceutical Sciences, Ghent University, Ghent, Belgium
Clinical pharmacogenomics (PGx) testing strategies are mainly based on targeted PCR, microarrays, or short-read sequencing. These methods perform well for detecting known single-nucleotide variants (SNVs), small insertions/deletions (indels), and certain copy number variants (CNVs), but they fall short in resolving complex structural variants (SVs), particularly in complex pharmacogenes such as CYP2D6. Therefore, we previously developed a targeted PGx test based on long-read Oxford Nanopore Technologies (ONT) sequencing. Harnessing adaptive sampling (AS) for in silico enrichment of a panel of PGx genes, we illustrated superior performance in star-allele calling compared to the Genetic Testing Reference Materials Program (GeT-RM) truth set. However, accurate diplotyping of CYP2D6 remained challenging. In this work, we adopted the latest basecalling, variant calling, phasing, and star-allele calling tools on our pre-existing data from the HG001, HG01190, NA19785, HG002, and HG005 reference samples. Additionally, we benchmarked the results to public data obtained using the long-read compatible Twist Alliance PGx panel. The re-analyzed ONT-AS data demonstrated correct CYP2D6 star-alleles compared to the GeT-RM truth set. Upon benchmarking to the Twist Alliance PGx panel, perfect star-allele matching was obtained between our panel and the Twist PGx panel for all included Clinical Pharmacogenomics Implementation Consortium (CPIC) Level A genes. However, our ONT-AS panel demonstrated superior variant phasing, resulting in three times more variants per phasing block. These findings confirm the robustness of ONT-AS for targeted long-read PGx applications and highlight its potential to support more accurate pharmacogenomic testing, particularly for structurally complex genes like CYP2D6.
1 Introduction
Pharmacogenomics (PGx) studies the impact of pre-existing DNA variation on the function of medicines. Missense or loss-of-function mutations in protein-coding genes involved in absorption, distribution, metabolization, and excretion (ADME) processes can have profound effects on the drug’s pharmacokinetic and pharmacodynamic parameters. Alternatively, mutations can impact the PGx gene’s expression levels (EMA, 2018). The results of the Pre-emptive Pharmacogenomic Testing for Preventing Adverse Drug Reactions (PREPARE) trial, an international implementation study that investigated the feasibility, acceptability, and effectiveness of panel-based PGx testing, found that 93.5% of all patients carried at least one actionable variant within a 12-gene PGx panel (Swen et al., 2023). Adopting the patient’s genetic information before initiating drug therapy would fully deliver the promise of personalized medicine to achieve better therapeutic outcomes and fewer adverse drug reactions.
PGx is slowly getting incorporated within population healthcare systems. The Estonian BioBank, encompassing both omics-data and health-related information, established PGx microarray genotyping for all of its participants. Based on phenotype translations, recommendations for drug therapy based on nine pharmacogenes for 211,257 individuals are available and will be adopted within the Estonian Health Insurance Fund (Milani et al., 2025). In the Netherlands, the P4Care study, that aimed to study whether DNA-passports result in better therapeutic outcomes for depression and anxiety medication, was recently funded and will include over 2,000 patients (ZonMw, 2025). Additionally, also within clinical guidelines, the recommendations of the Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenomics Working Group (DPWG) are getting adopted, as illustrated by the recommendation for CYP2C19 genotyping to guide clopidogrel usage by the National Institute for Health and Care Excellence (NICE) and the American Heart Association (Pereira et al., 2024; NICE, 2024).
Most of PGx genotyping is still performed using PCR-based techniques, microarrays, or panel-based short-read sequencing (SRS) (Bourgeois et al., 2024; Bourgeois et al., 2025; Knezevic et al., 2025; Tafazoli et al., 2021; Tayeh et al., 2022). Although these strategies offer high throughput at low cost and provide genotyping of single-nucleotide variants (SNVs), insertions/deletions (indels), and a set of copy number variants (CNVs), they are less suited to elucidate complex genes and to provide haplotype phasing information. Additionally, current PGx assays mostly do not query rare variants or are not able to resolve complex PGx genes that harbor complex structural variants (SVs), high sequence homology, or repeat regions. The CYP2D6 gene, involved in the metabolization of 25% of the commonly used drugs, is highly polymorphic and notoriously difficult to genotype, due to the presence of two neighboring pseudogenes CYP2D7 and CYP2D8 with high sequence homology and complex gene hybrids (Turner et al., 2023). Combined with the observation that only a part of the inherited variability in drug response, even within predicted identical metabolizer phenotypes, can be explained via currently known genetic polymorphisms, it is clear that there is still room for improvement in current genotyping strategies (Zhou et al., 2022; Lauschke et al., 2024; Tremmel et al., 2025).
We recently developed a long-read sequencing (LRS) test that enables the creation of personalized PGx passports, both for pre-emptive and reactive applications (Deserranno et al., 2023). This LRS strategy adopted the unique feature of Oxford Nanopore Technologies (ONT) to dynamically enrich for a prespecified list of target genes without additional steps during library preparation. Using adaptive sampling (AS), we enriched for 1,036 PGx genes extracted from the Pharmacogenomics Knowledge Base (PharmGKB) (Whirl-Carrillo et al., 2021). We demonstrated improved star-allele calling compared to the Genetic Testing Reference Materials Program (GeT-RM) truth set, findings that were recently corroborated by a group from Singapore (Gan et al., 2025). However, our CYP2D6 star-allele calls still proved discordant compared to the Get-RM truth set.
The unique advantage of ONT sequencing is that raw squiggle data can be rebasecalled. Our original data were obtained during 2023. Meanwhile, ONT adopted a new raw file format and a novel basecaller yielding increased base call accuracies. Simultaneously, improvements in bioinformatic approaches to variant calling and star-allele calling software were developed. We hypothesized that the combined improvements of rebasecalling and the updated analysis tools might improve the accuracy of CYP2D6 typing. Moreover, Twist Bioscience recently commercialized a PGx-specific hybridization capture panel compatible with LRS, for which improved CYP2D6 and NAT2 genotyping was demonstrated compared to SRS (Barthélémy et al., 2023). Therefore, we additionally performed a direct comparison of our re-analyzed ONT AS PGx data against public data from the Twist Alliance PGx panel.
2 Methods
2.1 Rebasecalling the ONT data
We re-used the raw sequencing data previously obtained from our AS PromethION sequencing runs (Deserranno et al., 2023). No additional sequencing was performed. In brief, we previously performed targeted ONT sequencing by enriching for 1,036 PGx genes in five Genome in a Bottle (GIAB) reference samples. Rather than physically designing a hybridization capture panel, we used ONT’s AS to selectively enrich library fragments corresponding to the genes of interest during sequencing. On a first PromethION R10.4.1 flow cell, the NA24385 (HG002) and NA24631 (HG005) samples were multiplexed. On a second PromethION flow cell, the NA12878 (HG001), HG01190, and NA19785 were sequenced. The raw .fast5 data were first converted to .pod5 using pod5 convert fast5 (Githup, 2024). Rebasecalling and demultiplexing were performed using Dorado (v0.9.0) (Oxford Nanopore Technologies, 2025). For the first flow cell with HG002 and HG005, the dna_r10.4.1_e8.2_400bps_sup@v4.1.0 model was used. As MinKNOW was upgraded to perform sequencing at 5 kHz instead of 4 kHz in between the sequencing of both flow cells, the second flow cell was rebasecalled using the dna_r10.4.1_e8.2_400bps_sup@v5.0.0 model.
2.2 Processing, variant calling, and phasing of the rebasecalled ONT data
From the rebasecalled .fastq files, we selected the reads corresponding to those read_ids enriched by AS. In particular, we filtered the adaptive_sampling.csv file obtained from MinKNOW by only retaining the read_ids with the value “unblock” in the decision column, i.e., retaining the read_ids corresponding to off-target reads. Using grep, we selected all read_ids from the rebasecalled .fastq files that did not correspond to this list of off-target reads. Alignment was performed using minimap2 (v2.28) (Li, 2021) with the GRCh38 reference. For variant calling, we used Clair3 (v1.0.10) (Zheng et al., 2022) with the corresponding pretrained model r1041_e82_400bps_sup_v410 (for HG002 and HG005) and r1041_e82_400bps_sup_v500 (for HG001, HG01190, and NA19785), obtained from Rerio (https://github.com/nanoporetech/rerio). Variant phasing and haplotagging were performed using WhatsHap (v2.4) (Patterson et al., 2015), with flags --include-homozygous --indels --distrust-genotypes --ignore-read-groups.
2.3 Star-allele calling using StarPhase
StarPhase (v1.4.0) (Holt et al., 2024) was used for star-allele calling. The phased variant calls and alignment .bam obtained from WhatsHap were used as input. Chromosome naming in the .bam files was adjusted to include the “chr” prefix. StarPhase was run using pbstarphase diplotype --database --bam {bam} –vcf {vcf} --reference {GRCh38} --include-set {list_of_interest} --output-calls PGx_passport.json --normalize-d6-only --output-debug {debug_dir}. The list of interest includes ABC0G2, CACNA1S, CFTR, CYP1A2, CYP2B6, CYP2C18, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5, CYP4F2, DPYD, G6PD, HLA-DRB1, HLA-B, IFNL3, MT-RNR1, NAT2, NUDT15, RYR1, SLCO1B1,TPMT, UGT1A1, and VKORC1.
2.4 Comparison to the Twist Alliance PGx and Dark Genes panel
We retrieved the alignment .bam files for the Twist Alliance PGx and Dark Genes panel from PacBio (https://downloads.pacbcloud.com/public/dataset/). For the Dark Genes panel, we downloaded the dataset for HG001 and HG002, sequenced on the PacBio Revio. For the PGx panel, we downloaded the dataset for HG002 and HG01190, sequenced on the PacBio Sequel II platform. Similarly, we retrieved the alignment .bam files for the Twist Alliance PGx panel sequenced on ONT of sample HG01190 (data available upon request to ONT). The corresponding .bed files listing the targeted gene regions were obtained from the Twist website. For consistency between variant callers, the retrieved .bam files were used as input for Clair3 and WhatsHap with the same settings as listed above. For Clair3 variant calling, the dedicated Clair3 models for Sequel II and Revio data were used.
2.5 Read-length and phasing success rate
QC-statistics N50s were obtained using NanoComp (De Coster and Rademakers, 2023). For calculation of the phasing success, the phased .vcf files were filtered to only include the 44 common genes between the ONT-AS PGx panel and the Twist PGx panel using bcftools view (v1.20) (Danecek et al., 2021) by providing a .bed file listing each gene’s start and stop positions. Subsequently, we used WhatsHap (v2.4) stats to obtain the phasing metrics, which were subsequently summarized using multiqc (v1.28) (Ewels et al., 2016). Mosdepth (v.0.3.10) was used for the sequencing depth calculations (Pedersen and Quinlan, 2018).
3 Results
3.1 Dorado rebasecalling and StarPhase deliver accurate CYP2D6 star-alleles
Holt et al. recently developed StarPhase, a tool specifically suited to perform diplotyping of PGx genes based on HiFi-sequencing data (Holt et al., 2024). StarPhase allows diplotyping for all genes with CPIC Level A annotation, except IFNL4, and a growing number of additional genes. In particular, for genes harboring complex structural variation such as the HLA-loci and CYP2D6, StarPhase generates consensus haplotypes from the alignment .bam file to call the diplotypes, without supplying a separate .vcf file containing the variant calls. Upon benchmarking the performance of StarPhase to the GeT-RM reference set for 12 common genes, the authors of StarPhase reported 73.8% accuracy. They concluded that the incongruent calls were due to the detection of alleles that were not assayed in the GeT-RM benchmark, updated star-allele nomenclature, or phasing errors in the truth set. These are all known limitations of the GeT-RM reference calls (van der Maas et al., 2025).
Therefore, while developed for PacBio HiFi reads, we tested the performance of StarPhase to call the CYP2D6 star-alleles based on our existing ONT-AS data. We first directly performed StarPhase on the existing alignment .bam files from our previous research (Table 1, “StarPhase not-rebasecalled ONT data”). Although the star-allele provided for NA19785 is correct, the calls for the other samples remained incorrect. We hypothesized that the lower quality of the provided ONT data confused StarPhase, as marked by the detection of two copies of the *68 allele.
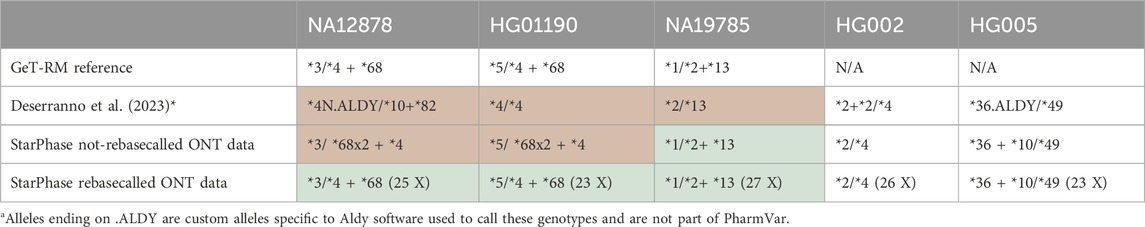
Table 1. Improvements to CYP2D6 star-allele calling for GeT-RM reference samples. CYP2D6 core star-allele calls for the ONT-AS PGx panel for five GIAB samples, before and after rebasecalling and processing with StarPhase. The values between parentheses specify the sequencing depth.
As the performance of ONT basecalling has improved in the last 2 years, the raw squiggle data were rebasecalled using the latest Dorado algorithm (Methods). Crucially, no new sequencing was performed. Rebasecalling shifted the median read quality Phred score significantly, as illustrated for the NA12878 data, resulting in a shift from 15.5 to 20.4 (Supplementary Figure S1). Supplying the newly basecalled alignment .bam files to StarPhase resulted in correct star-alleles of CYP2D6 in all samples (Table 1, bottom row). These detected CYP2D6 star-allele calls are identical to the Get-RM diplotypes and were further documented in the literature using orthogonal LR-sequencing workflows such as CRISPR–Cas9 target enrichment (Bettinotti et al., 2018; Gaedigk et al., 2019; Pratt et al., 2016; Pratt et al., 2010; Rubben et al., 2022). Therefore, together with the results of our previous research using Aldy (Hari et al., 2023) for the other PGx genes, the ONT-AS PGx panel results in an accurate complete long-read PGx passport for nearly all CPIC Level A genes. Only for HLA-A (currently not a part of the target file for AS, but can easily be added in our .bed file) and IFNL4 (currently not covered by StarPhase), calls are not yet included.
3.2 Benchmarking the ONT-AS PGx panel to the Twist hybridization panels
The Twist Alliance PGx Panel targets 49 genes, of which 44 are shared with our ONT-AS PGx panel. For future work, HLA-A can flexibly be included in our AS .bed file. The other four genes (CTBP2P2, NAGS, APOL1, and GBA) are absent in the clinical annotations from PharmGKB. Some genes are only partially covered in the Twist PGx panel. For ADD1, CACNA1S, CFTR, DPYD, F2, F5, GRIK4, HTR2C, POLG, and YEATS4, the Twist PGx capture probes only target specific parts of the gene, which limits the identification of haplotypes defined by SNVs outside these regions and the detection of novel variants. Focusing on DPYD, the Twist PGx panel covers 354 of the 434 annotated polymorphisms included in PharmVar 6.2.5 and, for example, does not include the rs773499329 (MAF 0.1% in the South Asian population) and rs762198241 variants (Pratt et al., 2024; Shrestha et al., 2018). Notably, the mitochondrial encoded MT-RNR1 gene, included within the CPIC Level A annotations in view of aminoglycoside-induced hearing loss, is not ordinarily included in the Twist PGx panel but may be spiked in, as was the case for the PacBio Twist PGx benchmark discussed below (McDermott et al., 2022). Similarly, it is not included in our ONT AS PGx panel, although it can be flexibly added by appending it to the enrichment .bed file for future experiments. Therefore, and because of the possibility of nuclear mitochondrial DNA segments being falsely aligned to the mitochondrial reference sequence without dedicated enrichment, we did not retain this gene in our analysis (Supplementary Note S1). Twist also supplies the Dark Genes panel, which captures fragments of 389 unique genes, of which 46 are shared with the AS panel list. The three panels share five genes (HLA-B, HLA-DRB1, IFNL3, CYP2D6, and VKORC1) (Supplementary Figure S2).
Of the common genes between the Twist Alliance PGx and ONT PGx panels, 25 (26 including MT-RNR1) can currently be diplotyped using StarPhase. The genes include all CPIC Level A genes (except for HLA-A and IFNL4 as discussed above) and CYP1A2, CYP2C8, CYP3A4, HLA-DRB1, HLA-DQA1, and NAT2. We therefore performed a direct comparison between both strategies using the GIAB reference sample HG01190. Additionally, for this sample, both ONT and PacBio data using the Twist PGx panel are available. A perfect star-allele match was observed across the three datasets, with consistent calls compared to the Get-RM truth set (Table 2). For CYP2B6, CYP2C9, and UGT1A1, the reference calls were improved, as discussed before (Deserranno et al., 2023). One exception seems to be HLA-DRB1, for which both strategies do not correspond to the reference call found by others (Thuesen et al., 2022). Of note, StarPhase was not able to call a diplotype for HLA-DRB1 based on the ONT sequencing data of the Twist PGx panel, probably due to the lower coverage of the first part of the gene (Supplementary Figure S3). According to the literature, due to the high complexity of the HLA locus, high sequencing depth is required (Thuesen et al., 2022). The sequencing depth for each of the genes profiled can be found in Supplementary Table S1. The median sequencing depths along HLA-DRB1 were 15.79 X and 19.62 X, for ONT-AS PGx and Twist PacBio, respectively, most probably explaining the discording call. Of note, for HLA-DQA1, a minor discrepancy regarding a difference in the non-coding region is found between ONT-AS PGX and PacBio Twist on one side and ONT Twist on the other side. As historical methods do not allow eight-digit HLA discrimination, we could not retrieve a comprehensive reference call for this sample; hence, we cannot accurately determine which one is correct.
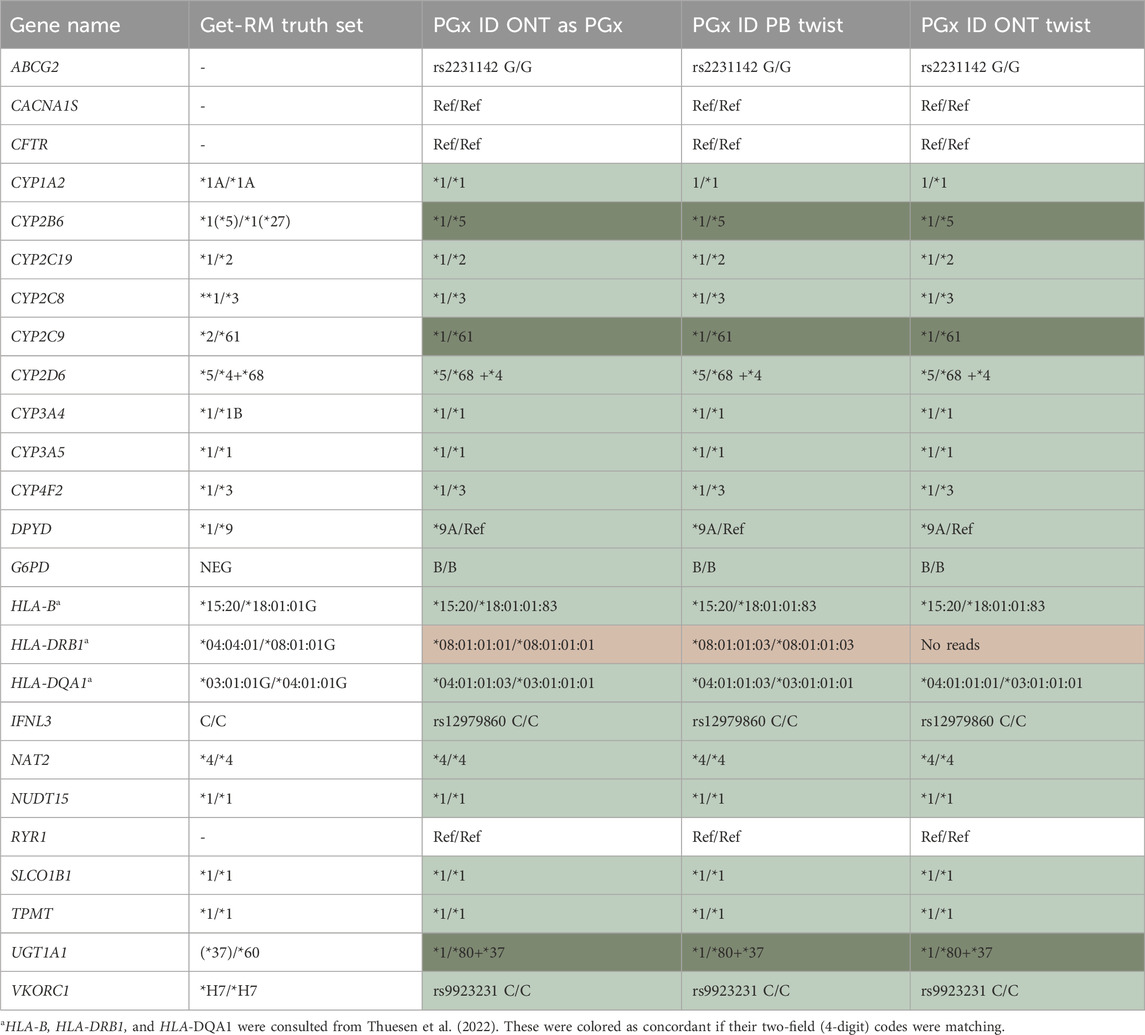
Table 2. PGx passport for HG01190, as established by ONT-AS PGx, and the Twist Alliance PGx panel sequenced on PacBio Sequel II and ONT. The results were compared to the those of the Get-RM Truth set, where possible. Dark green: improvement of the truth set; light green: concordance with the truth set; no fill: no truth set call available. Only the major star-alleles were considered. For CYP1A2 and CYP3A4, the minor allele suffix A or B is outdated.
Additionally, we repeated the star-allele calling benchmark for HG002 and found near-perfect agreement between both strategies (no Get-RM truth set available), except for the suballele of CYP2D6, the non-coding variant in HLA-DRB1 and one HLA-DQA1 genotype (Supplementary Table S2). CYP2D6 suballeles do not convey any additional functional impact compared to the major star-allele. The CYP2D6*4.014 suballele detected in the ONT-AS PGx panel is different from the CYP2D6*4.015 suballele detected using the Twist panel in the presence of rs1473203326 in the *4.014 allele. The presence of this SNV could not be deduced from manual inspection of the ONT-AS PGx reads, pointing to a possible artifact in StarPhase (Supplementary Table S2 and Supplementary Figure S4). The disagreeing HLA allele calls are discussed below.
Furthermore, we assessed the usability of the Twist Dark Genes panel for PGx as it shares five genes with the ONT-AS PGx panel and the Twist PGx Panel (Table 3, Supplementary Table S3). Strikingly, the star-allele call for CYP2D6 and IFNL3 for HG001 is wrong using the Twist Dark Genes panel. Upon closer examination, the capture region of CYP2D6 in this panel is too limited to reliably call the diplotype. The limited capture region is especially problematic for samples harboring complex CYP2D6 gene configurations, involving non-identical gene duplications and hybrids with neighboring pseudogenes, as is the case for HG001. Comparing the capture region of CYP2D6 between the Twist Dark Genes and Twist PGx panel, the PGx panel also captures the neighboring regions of CYP2D6, which might explain the wrong call with the Dark Genes panel (Supplementary Figure S5). For samples with non-complex CYP2D6 gene configurations such as HG002, our results indeed confirm that this limited capture region is less problematic (Table 3). Similarly, for IFNL3, the capture region included in the Dark Genes panel is too limited to reliably detect all possible star alleles (Supplementary Figure S6).
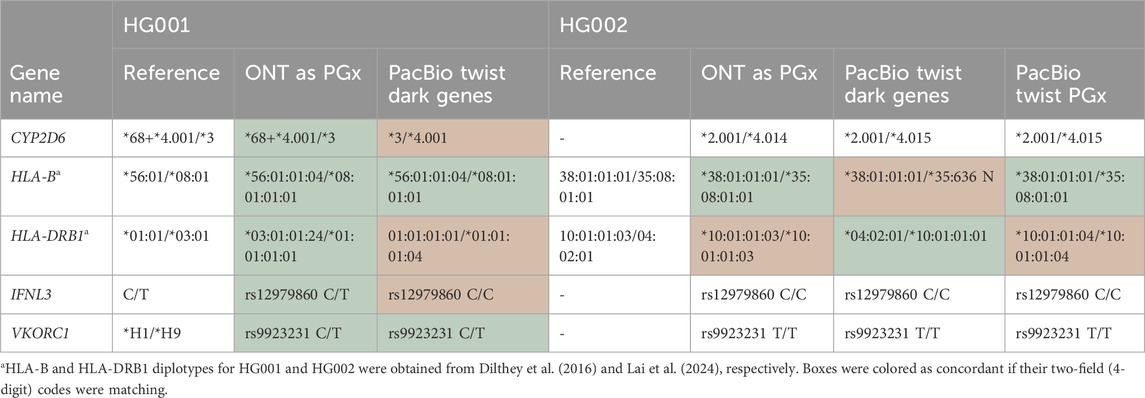
Table 3. Head-to-head comparison of the common and StarPhase supported genes between the ONT AS PGx, PacBio Twist Dark Genes, and PacBio Twist PGx panels. For HG002, no Get-RM reference data were available. Light green: concordant reference call; orange: incorrect call compared to reference.
The results for HLA typing across the different panels are more complex (Table 3). HLA-B and HLA-DRB1 reference calls for HG001 and HG002 were obtained from the literature (Lai et al., 2024; Chin et al., 2020). For HG001, the ONT-AS PGx panel demonstrates concordance with the reference, while HLA-DRB1 is most probably false for the Twist Dark Genes panel. Manual curation (Supplementary Figure S7) indicated that the increased aligned read length in the ONT-AS PGx panel compared to the Dark Genes panel might have contributed to the correct HLA-DRB1 gene assignment. Despite the full inclusion of HLA-B in the Dark Genes panel, the HLA-B call for HG002 with this panel is most likely wrong, probably due to the limited sequencing depth (Supplementary Figure S8). *35:636 N differs from *35:08:01:01 only by a C→T substitution in gDNA position 331 of exon 2 (Loginova et al., 2025). For HLA-DRB1, strikingly, only the Dark Genes panel seems to be concordant with the literature reference benchmark reported by Lai et al. (2024). Of note, one of the analysis tools used in the construction of their benchmark, i.e., HLA-VBseq, did output the HLA-DRB1*10:01:01:03/*10:01:01:03 diplotype retrieved by StarPhase for the ONT-AS PGx dataset. For HLA-DRB1, the Twist Dark Genes panel had a median sequencing depth of 30 X compared to 21 X both for the ONT-AS PGx and Twist PGx panels. Conversely, the Dark Genes panel had the lowest sequencing depth for HLA-B (median sequencing depth: 19 X) compared to the ONT-AS PGx panel (44 X) and Twist PGx panel (143 X), therefore most probably explaining the assumed wrong calls. Our results confirm that high coverage is crucial for HLA-typing, more than for other PGx genes, and longer read lengths might help in calling the correct allele.
Finally, we benchmarked the overall read length and variant phasing capabilities between the ONT-AS PGx and the Twist PGx panel data. Due to the nature of the hybridization capture library preparation, the aligned N50 read length for the hybridization panels is significantly smaller (ONT Twist PGx: 4,040 bp; PacBio Twist PGx: 5,370 bp) than the native ONT-AS PGx reads (8,047 bp), as profiled for HG01190. Corroborating the findings of previous research, this also impacted the level of the target region that can be phased. For the 44 common genes, the ONT-AS PGx panel had significantly larger proportions of the target gene loci phased (Figure 1A) than the Twist PGx panel. The percentage of the targeted region phased for the Twist PGx panel was similar to previously reported results (van der Lee et al., 2022). Additionally, the number of individual phasing blocks for the ONT-AS PGx panel was approximately half of the number of blocks in the Twist PGx data, and each phasing block included more variants (Figure 1B). For the ONT-AS PGx panel, the maximum read length that can be obtained theoretically only depends on the length of the input DNA molecules. For this research, no shearing was performed. However, longer DNA fragments tend to cause increased blocking of the nanopores, and DNA shearing to, for example, 10 kb has been proposed to address this. Although shearing might boast yield during ONT AS and allow more samples to be multiplexed on a single-flow cell, it limits the number of unique variants contained within a single read available for phasing.
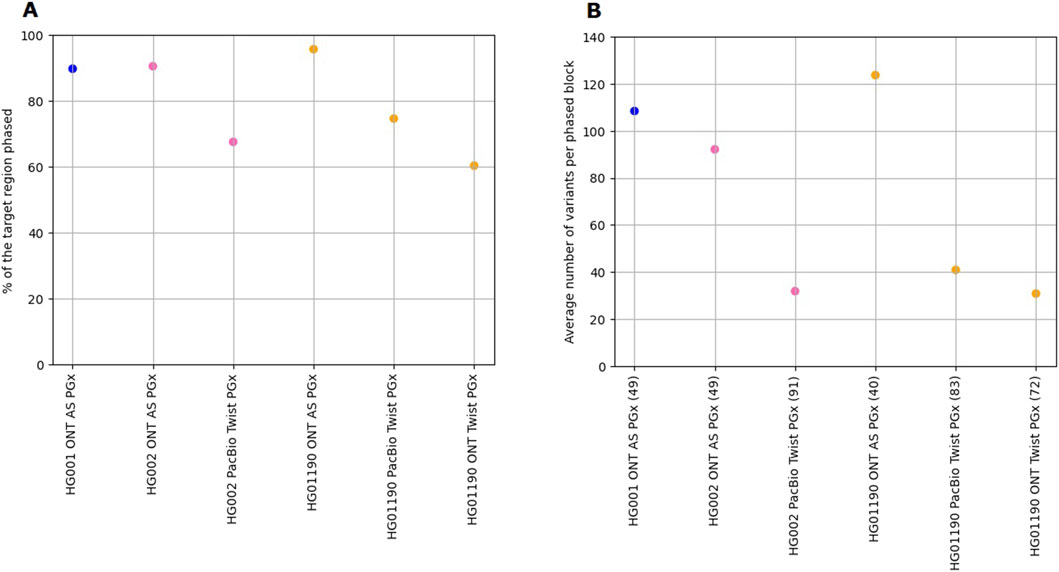
Figure 1. Benchmarking the level of phasing between the ONT-AS PGx panel and the Twist PGx panel across the HG001, HG002, and HG01190 GIAB samples. (A) The percentage of the target region that could be phased, calculated as the number of bases phased relative to the total number of bases targeted within the panel. (B) The average number of variants per phased block. The value between parentheses next to the sample names in the horizontal axis represents the number of phased blocks.
4 Discussion
The reanalysis of the ONT-AS PGx panel and the benchmarking results of the targeted Twist PGx panel illustrate the high potential of long-read PGx for clinical use. Both panels resulted in accurate or improved star-allele calls compared to the Get-RM truth. For the ONT-AS PGx data, reanalyzing the previously generated data with the latest software versions and leveraging StarPhase for star-allele calling now yielded correct CYP2D6 star-alleles compared to our previous research. Based on our comparison, the Twist Dark Genes panel is not suitable for pharmacogenomics research, due to the very limited inclusion of key gene regions. As discussed by others, another advantage of using LR sequencing is the phasing of variants to the allele-of-origin, which is particularly important for genes such as SLCO1B1 and CYP2B6 as they list star-alleles with overlapping variants which, without phasing information, can be wrongly assigned (Desta et al., 2021; Ramsey et al., 2023; van der Lee et al., 2020). Our results illustrate the superiority of the ONT-AS PGx panel in variant phasing and read length.
The Twist PGx panel and the ONT-AS PGx panel both have their unique properties. Considering the number of genes, the Twist PGx panel is limited to 50 genes, some not fully covered, compared to the ONT-AS PGx panel, currently covering 1,036 full-length genes with any level of evidence in the PharmGKB clinical annotation list. However, the genes included in the Twist PGx panel cover all CPIC Level A evidence genes, except for IFNL4. The ONT-AS PGx panel offers the additional advantage of flexibly adding genes to the .bed file, considering that, for successful performance of AS, the target panel should be large enough to prevent the nanopores from wearing out quickly. Regarding the throughput, PacBio states that 72 Twist PGx Panel-enriched samples could be sequenced at once on a single Revio SMRT cell, whereas ONT reports that up to 48 samples could be multiplexed on a PromethION flow cell, compared to three samples using AS (Deserranno et al., 2023; Whirl-Carrillo et al., 2021; Oxord Nanopore Technologies, 2025; PacBio, 2023). However, ONT-AS requires no additional reagents, hands-on time, cost, and can be performed completely PCR-free, while the hybridization procedure for Twist involves a multi-day protocol for library preparation, 16 h overnight incubation, and pre- and post-capture long-range PCR-amplification steps. The use of PCR steps also limits the potential of using 5-methylcytosine and 5-hydroxymethylcytosine detection, which can directly be retrieved from the ONT AS PGx data.
The ONT-AS PGx data have been demonstrated to be future-proof. Rebasecalling of already existing data provided increased accuracy and combined with the latest algorithms for phasing, variant calling, and star-allele calling have now yielded a complete PGx passport. However, for HLA typing, increased sequencing depth or improved analysis pipelines are needed. Currently, the provided star-alleles can be used as input, for example, PharmCAT to provide phenotypes and dosing recommendations. However, for some star-alleles, such as, CYP2D6, the impact is unknown. Modeling the enzymatic activity on a continuous scale, rather than the categorical star-allele classification based on long-read sequencing data, has shown promising results for CYP2D6 (van der Lee et al., 2021).
In conclusion, we demonstrate that our ONT-AS PGx strategy enables accurate diplotyping for common and actionable PGx genes. In particular, for the CYP2D6 gene, correct star-alleles are now retrieved. We compared the performance of our strategy against the Twist Alliance PGx panel, in combination with PacBio or ONT LRS, and demonstrated concordant star-allele calls for both panels. For HLA-typing, increased sequencing depth and long read lengths contribute to accurate genotyping. Although it is not the primary objective, we also assessed the performance of the Twist Dark Genes panel for PGx profiling and found it to be less suited for PGx purposes. Furthermore, we identified unique advantages for each strategy. Although the hybridization panels claim higher multiplexing of samples, the ONT-AS strategy demonstrates more comprehensive gene profiling in a less labor-intensive workflow and superior variant phasing information. Overall, we highlight the potential of targeted long-read PGx applications and its potential to support more accurate clinical PGx testing.
Data availability statement
Publicly available datasets were analyzed in this study. These data can be found here: The datasets analyzed for this study can be found in Array Express under accession E-MTAB-15248. The datasets analyzed for our previous study can be found on https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1003794.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
KD: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review and editing. LT: Data curation and Writing – review and editing. DD: Funding acquisition, Resources, Supervision, Writing – review and editing. FV: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. KD and this research are supported by the Special Research Fund (Bijzonder Onderzoeksfonds, BOF, University Ghent, BOF21/DOC/042) website: https://www.ugent.be/en/research/funding/bof.
Acknowledgments
The authors thank Sarah De Keulenaer, Ellen De Meester, and Sylvie Decraene of the Ghent University NXTGNT sequencing core facility for their expertise and help in performing the library preparation steps.
Conflict of interest
KD has received travel funding from Oxford Nanopore Technologies (ONT) to present his findings at a scientific meeting.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1653999/full#supplementary-material
References
Barthélémy, D., Belmonte, E., Pilla, L. D., Bardel, C., Duport, E., Gautier, V., et al. (2023). Direct comparative analysis of a pharmacogenomics panel with PacBio hifi® long-read and illumina short-read sequencing. J. Personalized Med. 13 (12), 1655. doi:10.3390/jpm13121655
Bettinotti, M. P., Ferriola, D., Duke, J. L., Mosbruger, T. L., Tairis, N., Jennings, L., et al. (2018). Characterization of 108 genomic DNA reference materials for 11 human leukocyte antigen loci: a GeT-RM collaborative project. J. Mol. Diagnostics 20 (5), 703–715. doi:10.1016/j.jmoldx.2018.05.009
Bourgeois, J., Costa, E., Devos, C., Hulstaert, F., Luyten, J., Ombelet, S., et al. (2024). Gebruik van farmacogenetische tests in belgië. Health Services Research (HSR). Brussels, Belgium: Federaal Kenniscentrum voor de Gezondheidszorg. Report No.: 382AS.
Bourgeois, J., Costa, E., Devos, C., Luyten, J., Ombelet, S., Thiry, N., et al. (2025). Unravelling the implementation of pharmacogenetic testing in Belgium. Eur. J. Clin. Pharmacol. 81 (5), 711–718. doi:10.1007/s00228-025-03816-8
Chin, C.-S., Wagner, J., Zeng, Q., Garrison, E., Garg, S., Fungtammasan, A., et al. (2020). A diploid assembly-based benchmark for variants in the major histocompatibility complex. Nat. Commun. 11 (1), 4794. doi:10.1038/s41467-020-18564-9
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10 (2), giab008. doi:10.1093/gigascience/giab008
De Coster, W., and Rademakers, R. (2023). NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 39 (5), btad311. doi:10.1093/bioinformatics/btad311
Deserranno, K., Tilleman, L., Rubben, K., Deforce, D., and Van Nieuwerburgh, F. (2023). Targeted haplotyping in pharmacogenomics using Oxford nanopore Technologies’ adaptive sampling. Front. Pharmacol. 14, 1286764. doi:10.3389/fphar.2023.1286764
Desta, Z., El-Boraie, A., Gong, L., Somogyi, A. A., Lauschke, V. M., Dandara, C., et al. (2021). PharmVar GeneFocus: CYP2B6. Clin. Pharmacol. Ther. 110 (1), 82–97. doi:10.1002/cpt.2166
Dilthey, A. T., Gourraud, P. -A, Mentzer, A. J., Cereb, N., Iqbal, Z., McVean, G., et al. (2016). High-accuracy HLA type inference from whole-genome sequencing data using population reference graphs. PLoS Comput. Biol. 12 (10), e1005151. doi:10.1371/journal.pcbi.1005151
EMA (2018). Guideline on good pharmacogenomic practice. London, UK: European Medicines Agency. First version.
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. doi:10.1093/bioinformatics/btw354
Gaedigk, A., Turner, A., Everts, R. E., Scott, S. A., Aggarwal, P., Broeckel, U., et al. (2019). Characterization of reference materials for genetic testing of CYP2D6 alleles: a GeT-RM collaborative Project. J. Mol. Diagnostics 21 (6), 1034–1052. doi:10.1016/j.jmoldx.2019.06.007
Gan, H. P. P., Han Lin, Y., Irfan Bin Hajis, M., Maulana, Y., Ng Qi Hui, A., Nathanael Ramanto, K., et al. (2025). Targeted adaptive sampling enables clinical pharmacogenomics testing and genome-wide genotyping. medRxiv Preprint. doi:10.1101/2025.05.05.25326970
Hari, A., Zhou, Q., Gonzaludo, N., Harting, J., Scott, S. A., Qin, X., et al. (2023). An efficient genotyper and star-allele caller for pharmacogenomics. Genome Res. 33 (1), 61–70. doi:10.1101/gr.277075.122
Holt, J. M., Harting, J., Chen, X., Baker, D., Saunders, C. T., Kronenberg, Z., et al. (2024). StarPhase: comprehensive phase-aware pharmacogenomic diplotyper for long-read sequencing data. bioRxiv Preprint., 2024.12.10.627527. 2024.12.10.627527. doi:10.1101/2024.12.10.627527
Knezevic, C. E., Stevenson, J. M., Merran, J., Snyder, I., Restorick, G., Waters, C., et al. (2025). Implementation of integrated clinical pharmacogenomics testing at an academic Medical center. J. Appl. Laboratory Med. 10 (2), 259–273. doi:10.1093/jalm/jfae128
Lai, S.-K., Luo, A. C., Chiu, I.-H., Chuang, H.-W., Chou, T.-H., Hung, T.-K., et al. (2024). A novel framework for human leukocyte antigen (HLA) genotyping using probe capture-based targeted next-generation sequencing and computational analysis. Comput. Struct. Biotechnol. J. 23, 1562–1571. doi:10.1016/j.csbj.2024.03.030
Lauschke, V. M., Zhou, Y., and Ingelman-Sundberg, M. (2024). Pharmacogenomics beyond single common genetic variants: the way forward. Annu. Rev. Pharmacol. Toxicol. 64 (1), 33–51. doi:10.1146/annurev-pharmtox-051921-091209
Li, H. (2021). New strategies to improve minimap2 alignment accuracy. Bioinformatics 37 (23), 4572–4574. doi:10.1093/bioinformatics/btab705
Loginova, M., Makhova, O., and Paramonov, I. (2025). Discovery of an HLA-B*35 null variant, HLA-B*35:636N in two unrelated individuals. HLA 105 (4), e70214. doi:10.1111/tan.70214
McDermott, J. H., Wolf, J., Hoshitsuki, K., Huddart, R., Caudle, K. E., Whirl-Carrillo, M., et al. (2022). Clinical pharmacogenetics implementation consortium guideline for the use of aminoglycosides based on MT-RNR1 genotype. Clin. Pharmacol. Ther. 111 (2), 366–372. doi:10.1002/cpt.2309
Milani, L., Alver, M., Laur, S., Reisberg, S., Haller, T., Aasmets, O., et al. (2025). The Estonian Biobank’s journey from biobanking to personalized medicine. Nat. Commun. 16 (1), 3270. doi:10.1038/s41467-025-58465-3
NICE (2024). CYP2C19 genotype testing to guide clopidogrel use after ischaemic stroke or transient ischaemic attack. London, UK. National Institute for Health Care Excellence. Available online at: https://www.nice.org.uk/guidance/dg59/chapter/1-Recommendations.
Oxord Nanopore Technologies (2025). Pharmacogenomic Oxford nanopore sequencing of the twist alliance long-read PGx panel. Oxord, UK: Oxord Nanopore Technologies.
PacBio (2023). Scalable HiFi sequencing with twist biosciences long read alliance panels. Menlo Park, California: PacBio.
Patterson, M., Marschall, T., Pisanti, N., van Iersel, L., Stougie, L., Klau, G. W., et al. (2015). WhatsHap: weighted haplotype assembly for future-generation sequencing reads. J. Comput. Biol. 22 (6), 498–509. doi:10.1089/cmb.2014.0157
Pedersen, B. S., and Quinlan, A. R. (2018). Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34 (5), 867–868. doi:10.1093/bioinformatics/btx699
Pereira, N. L., Cresci, S., Angiolillo, D. J., Batchelor, W., Capers, Q., Cavallari, L. H., et al. (2024). CYP2C19 genetic testing for oral P2Y12 inhibitor therapy: a scientific statement from the American heart association. Circulation 150 (6), e129–e150. doi:10.1161/CIR.0000000000001257
Pratt, V. M., Zehnbauer, B., Wilson, J. A., Baak, R., Babic, N., Bettinotti, M., et al. (2010). Characterization of 107 genomic DNA reference materials for CYP2D6, CYP2C19, CYP2C9, VKORC1, and UGT1A1: a GeT-RM and association for molecular pathology collaborative project. J. Mol. diagnostics 12 (6), 835–846. doi:10.2353/jmoldx.2010.100090
Pratt, V. M., Everts, R. E., Aggarwal, P., Beyer, B. N., Broeckel, U., Epstein-Baak, R., et al. (2016). Characterization of 137 genomic DNA reference materials for 28 pharmacogenetic genes: a GeT-RM collaborative project. J. Mol. diagnostics 18 (1), 109–123. doi:10.1016/j.jmoldx.2015.08.005
Pratt, V. M., Cavallari, L. H., Fulmer, M. L., Gaedigk, A., Hachad, H., Ji, Y., et al. (2024). DPYD genotyping recommendations: a joint consensus recommendation of the association for molecular pathology, American college of medical genetics and genomics, clinical pharmacogenetics implementation consortium, college of American pathologists, Dutch pharmacogenetics working group of the royal Dutch pharmacists association, European society for pharmacogenomics and personalized therapy, Pharmacogenomics knowledgebase, and pharmacogene variation consortium. J. Mol. Diagnostics 26, 851–863. doi:10.1016/j.jmoldx.2024.05.015
Ramsey, L. B., Gong, L., Lee, S., Wagner, J. B., Zhou, X., Sangkuhl, K., et al. (2023). PharmVar GeneFocus: SLCO1B1. Clin. Pharmacol. Ther. 113 (4), 782–793. doi:10.1002/cpt.2705
Rubben, K., Tilleman, L., Deserranno, K., Tytgat, O., Deforce, D., and Van Nieuwerburgh, F. (2022). Cas9 targeted nanopore sequencing with enhanced variant calling improves CYP2D6-CYP2D7 hybrid allele genotyping. PLoS Genet. 18 (9), e1010176. doi:10.1371/journal.pgen.1010176
Shrestha, S., Zhang, C., Jerde, C. R., Nie, Q., Li, H., Offer, S. M., et al. (2018). Gene-specific variant classifier (DPYD-Varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin. Pharmacol. Ther. 104 (4), 709–718. doi:10.1002/cpt.1020
Swen, J. J., van der Wouden, C. H., Manson, L. E. N., Abdullah-Koolmees, H., Blagec, K., Blagus, T., et al. (2023). A 12-gene pharmacogenetic panel to prevent adverse drug reactions: an open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 401 (10374), 347–356. doi:10.1016/S0140-6736(22)01841-4
Tafazoli, A., Guchelaar, H.-J., Miltyk, W., Kretowski, A. J., and Swen, J. J. (2021). Applying next-generation sequencing platforms for pharmacogenomic testing in clinical practice. Front. Pharmacol. 12, 693453. doi:10.3389/fphar.2021.693453
Tayeh, M. K., Gaedigk, A., Goetz, M. P., Klein, T. E., Lyon, E., McMillin, G. A., et al. (2022). Clinical pharmacogenomic testing and reporting: a technical standard of the American college of medical genetics and genomics (ACMG). Genet. Med. 24 (4), 759–768. doi:10.1016/j.gim.2021.12.009
Thuesen, N. H., Klausen, M. S., Gopalakrishnan, S., Trolle, T., and Renaud, G. (2022). Benchmarking freely available HLA typing algorithms across varying genes, coverages and typing resolutions. Front. Immunol., 13–2022. doi:10.3389/fimmu.2022.987655
Tremmel, R., Pirmann, S., Zhou, Y., and Lauschke, V. M. (2025). Translating pharmacogenomic sequencing data into drug response predictions—How to interpret variants of unknown significance. Br. J. Clin. Pharmacol. 91 (2), 252–263. doi:10.1111/bcp.15915
Turner, A. J., Nofziger, C., Ramey, B. E., Ly, R. C., Bousman, C. A., Agúndez, J. A., et al. (2023). PharmVar tutorial on CYP2D6 structural variation testing and recommendations on reporting. Clin. Pharmacol. Ther. 114 (6), 1220–1237. doi:10.1002/cpt.3044
van der Lee, M., Allard, W. G., Bollen, S., Santen, G. W., Ruivenkamp, C. A., Hoffer, M. J., et al. (2020). Repurposing of diagnostic whole exome sequencing data of 1,583 individuals for clinical pharmacogenetics. Clin. Pharmacol. Ther. 107 (3), 617–627. doi:10.1002/cpt.1665
van der Lee, M., Allard, W. G., Vossen, R. H., Baak-Pablo, R. F., Menafra, R., Deiman, B. A., et al. (2021). Toward predicting CYP2D6-mediated variable drug response from CYP2D6 gene sequencing data. Sci. Transl. Med. 13 (603), eabf3637. doi:10.1126/scitranslmed.abf3637
van der Lee, M., Busscher, L., Menafra, R., Zhai, Q., van den Berg, R. R., Kingan, S. B., et al. (2022). Design and performance of a long-read sequencing panel for pharmacogenomics. bioRxiv Preprint. doi:10.1101/2022.10.25.513646
van der Maas, S., Denil, S., Maes, B., Ertaylan, G., and Volders, P.-J. (2025). Dynamic star allele definitions in Pharmacogenomics: impact on diplotype calls, phenotype predictions and Statin therapy recommendations. Front. Pharmacol. 16, 1584658. doi:10.3389/fphar.2025.1584658
Whirl-Carrillo, M., Huddart, R., Gong, L., Sangkuhl, K., Thorn, C. F., Whaley, R., et al. (2021). An evidence-based framework for evaluating pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 110 (3), 563–572. doi:10.1002/cpt.2350
Zheng, Z., Li, S., Su, J., Leung, A. W.-S., Lam, T.-W., and Luo, R. (2022). Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat. Comput. Sci. 2 (12), 797–803. doi:10.1038/s43588-022-00387-x
Zhou, Y., Tremmel, R., Schaeffeler, E., Schwab, M., and Lauschke, V. M. (2022). Challenges and opportunities associated with rare-variant pharmacogenomics. Trends Pharmacol. Sci. 43 (10), 852–865. doi:10.1016/j.tips.2022.07.002
ZonMw (2025). Gehonoreerde projecten personalised medicine. Den Haag, Netherlands: ZonMw. Available online at: https://www.zonmw.nl/nl/artikel/gehonoreerde-projecten-personalised-medicine.
Keywords: pharmacogenomics, long-read sequencing, adaptive sampling, CYP2D6, Twist alliance PGx panel
Citation: Deserranno K, Tilleman L, Deforce D and Van Nieuwerburgh F (2025) Comparative evaluation of Oxford Nanopore Technologies’ adaptive sampling and the Twist long-read PGx panel for pharmacogenomic profiling. Front. Pharmacol. 16:1653999. doi: 10.3389/fphar.2025.1653999
Received: 25 June 2025; Accepted: 12 August 2025;
Published: 09 September 2025.
Edited by:
Simran D. S. Maggo, Shenandoah University, United StatesReviewed by:
Martin A. Kennedy, University of Otago, New ZealandYusmiati Cen Liau, Bragato Research Institute, New Zealand
Copyright © 2025 Deserranno, Tilleman, Deforce and Van Nieuwerburgh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filip Van Nieuwerburgh, ZmlsaXAudmFubmlldXdlcmJ1cmdoQHVnZW50LmJl