
- Committee on the Neurobiology of Addictive Disorders, The Scripps Research Institute, La Jolla, CA, USA
Drug addiction can be defined by a three-stage cycle – binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation – that involves allostatic changes in the brain reward and stress systems. Two primary sources of reinforcement, positive and negative reinforcement, have been hypothesized to play a role in this allostatic process. The negative emotional state that drives negative reinforcement is hypothesized to derive from dysregulation of key neurochemical elements involved in the brain reward and stress systems. Specific neurochemical elements in these structures include not only decreases in reward system function (within-system opponent processes) but also recruitment of the brain stress systems mediated by corticotropin-releasing factor (CRF) and dynorphin-κ opioid systems in the ventral striatum, extended amygdala, and frontal cortex (both between-system opponent processes). CRF antagonists block anxiety-like responses associated with withdrawal, block increases in reward thresholds produced by withdrawal from drugs of abuse, and block compulsive-like drug taking during extended access. Excessive drug taking also engages the activation of CRF in the medial prefrontal cortex, paralleled by deficits in executive function that may facilitate the transition to compulsive-like responding. Neuropeptide Y, a powerful anti-stress neurotransmitter, has a profile of action on compulsive-like responding for ethanol similar to a CRF1 antagonist. Blockade of the κ opioid system can also block dysphoric-like effects associated with withdrawal from drugs of abuse and block the development of compulsive-like responding during extended access to drugs of abuse, suggesting another powerful brain stress system that contributes to compulsive drug seeking. The loss of reward function and recruitment of brain systems provide a powerful neurochemical basis that drives the compulsivity of addiction.
What is Addiction?
Addiction can be defined as a chronic, relapsing disorder that has been characterized by (i) a compulsion to seek and take drugs, (ii) loss of control over drug intake, and (iii) emergence of a negative emotional state (e.g., dysphoria, anxiety, and irritability) that defines a motivational withdrawal syndrome when access to the drug is prevented (1). The occasional, limited, recreational use of a drug is clinically distinct from escalated drug use, the loss of control over drug intake, and the emergence of compulsive drug-seeking behavior that characterize addiction.
Addiction has been conceptualized as a three-stage cycle – binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation – that worsens over time and involves allostatic changes in the brain reward and stress systems. Two primary sources of reinforcement, positive and negative reinforcement, have been hypothesized to play a role in this allostatic process. Positive reinforcement is defined as the process by which presentation of a stimulus increases the probability of a response; negative reinforcement is defined as the process by which removal of an aversive stimulus (or negative emotional state of withdrawal in the case of addiction) increases the probability of a response. Reward is operationally defined similarly to positive reinforcement as any stimulus that increases the probability of a response but also has a positive hedonic effect. Different theoretical perspectives from experimental psychology (positive and negative reinforcement frameworks), social psychology (self-regulation failure framework), and neurobiology (counteradaptation and sensitization frameworks) can be superimposed on the stages of the addiction cycle (1). These stages are thought to feed into each other, become more intense, and ultimately lead to the pathological state known as addiction (Figure 1). The neural substrates for the two sources of reinforcement that play a key role in the allostatic neuroadaptations derive from two key motivational systems required for survival: the brain reward and brain stress systems.
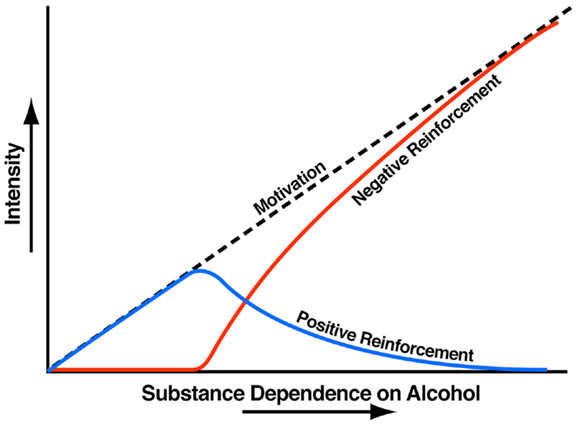
Figure 1. Theoretical framework relating addiction cycle to motivation for drug seeking. The figure shows the change in the relative contribution of positive and negative reinforcement constructs during the development of substance dependence [taken with permission from Ref. (61)].
Brain Reward Systems
Comprehension of a brain reward system was greatly facilitated by the discovery of electrical brain stimulation reward by Olds and Milner (2). Brain stimulation reward involves widespread neurocircuitry throughout the brain, but the most sensitive sites include the trajectory of the medial forebrain bundle that connects the ventral tegmental area with the basal forebrain [(2–4); Figure 2]. All drugs of abuse acutely decrease brain stimulation reward thresholds [i.e., increase or facilitate reward; (5)]. When drugs are administered chronically, withdrawal from drugs of abuse increases reward thresholds (decrease reward). Although much emphasis was initially placed on the role of ascending monoamine systems, particularly the dopamine system, in the medial forebrain bundle in mediating brain stimulation reward, other non-dopaminergic systems in the medial forebrain bundle clearly play a key role (6–8). Indeed, the role of dopamine is hypothesized to be more indirect. Many studies suggest that activation of the mesolimbic dopamine system attaches incentive salience to stimuli in the environment (9–11) to drive the performance of goal-directed behavior (12) or activation in general (13, 14), and work concerning the acute reinforcing effects of drugs of abuse supports this hypothesis.
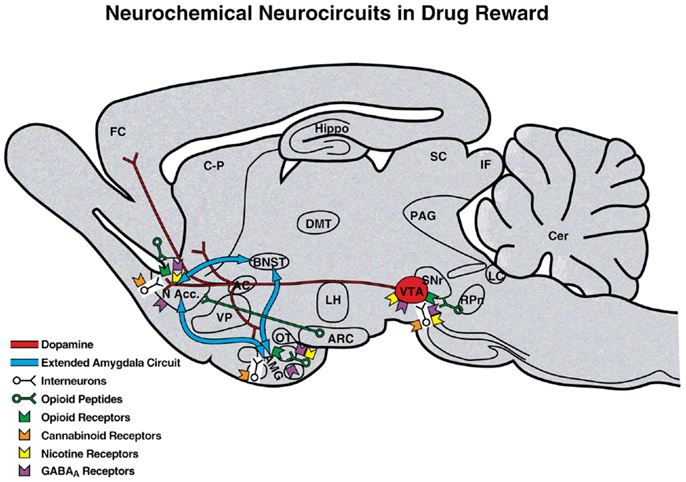
Figure 2. Neurotransmitter pathways and receptor systems implicated in the acute reinforcing effects of drugs of abuse within the medial forebrain bundle. A sagittal rodent brain section is shown. The medial forebrain bundle represents ascending and descending projections between the ventral forebrain (nucleus accumbens, olfactory tubercle, and septal area) and ventral midbrain (ventral tegmental area; not shown in figure for clarity). Cocaine and amphetamines increase dopamine levels in the nucleus accumbens and amygdala via direct actions on dopamine terminals. Opioids activate endogenous opioid receptors in the ventral tegmental area, nucleus accumbens, and amygdala. Opioids also facilitate the release of dopamine in the nucleus accumbens via actions either in the ventral tegmental area or nucleus accumbens but are also hypothesized to activate elements independent of the dopamine system. Alcohol activates GABAA receptors or enhances GABA release in the ventral tegmental area, nucleus accumbens, and amygdala. Alcohol is also hypothesized to facilitate the release of opioid peptides in the ventral tegmental area, nucleus accumbens, and central nucleus of the amygdala. Alcohol facilitates the release of dopamine in the nucleus accumbens via an action either in the ventral tegmental area or nucleus accumbens. Nicotine activates nicotinic acetylcholine receptors in the ventral tegmental area, nucleus accumbens, and amygdala either directly or indirectly via actions on interneurons. Cannabinoids activate cannabinoid CB1 receptors in the ventral tegmental area, nucleus accumbens, and amygdala. Cannabinoids facilitate the release of dopamine in the nucleus accumbens via an unknown mechanism, either in the ventral tegmental area or nucleus accumbens. The blue arrows represent the interactions within the extended amygdala system hypothesized to play a key role in psychostimulant reinforcement. AC, anterior commissure; AMG, amygdala; ARC, arcuate nucleus; BNST, bed nucleus of the stria terminalis; Cer, cerebellum; C-P, caudate-putamen; DMT, dorsomedial thalamus; FC, frontal cortex; Hippo, hippocampus; IF, inferior colliculus; LC, locus coeruleus; LH, lateral hypothalamus; MFB, medial forebrain bundle; N Acc., nucleus accumbens; OT, olfactory tract; PAG, periaqueductal gray; RPn, reticular pontine nucleus; SC, superior colliculus; SNr, substantia nigra pars reticulata; VP, ventral pallidum; VTA, ventral tegmental area [taken with permission from Ref. (183)].
Our knowledge of the neurochemical substrates that mediate the acute reinforcing effects of drugs of abuse has contributed significantly to our knowledge of the brain reward system. These substrates include connections of the medial forebrain bundle reward system with primary contributions from the ventral tegmental area, nucleus accumbens, and amygdala. Much evidence supports the hypothesis that psychostimulant drugs dramatically activate the mesolimbic dopamine system (projections from the ventral tegmental area to the nucleus accumbens) during limited-access drug self-administration and that this mechanism is critical for mediating the rewarding effects of cocaine, amphetamines, and nicotine. However, evidence supports both dopamine-dependent and dopamine-independent neural substrates for opioid and alcohol reward (15–17). Serotonin systems, particularly those involving serotonin 5-HT1B receptor activation in the nucleus accumbens, have also been implicated in the acute reinforcing effects of psychostimulant drugs, whereas μ-opioid receptors in both the nucleus accumbens and ventral tegmental area mediate the reinforcing effects of opioids. Opioid peptides in the ventral striatum and amygdala have been hypothesized to mediate the acute reinforcing effects of ethanol self-administration, largely based on the effects of opioid antagonists. Inhibitory γ-aminobutyric acid (GABA) systems are activated both pre- and postsynaptically in the amygdala by ethanol at intoxicating doses, and GABA receptor antagonists block ethanol self-administration [for comprehensive reviews, see (16, 17)].
For the binge/intoxication stage of the addiction cycle, studies of the acute reinforcing effects of drugs of abuse per se have identified key neurobiological substrates. Evidence is strong for a role for dopamine in the acute reinforcing actions of psychostimulants, opioid peptide receptors in the acute reinforcing effects of opioids, and GABA and opioid peptides in the acute reinforcing actions of alcohol. Important anatomical circuits include the mesocorticolimbic dopamine system that originates in the ventral tegmental area and local opioid peptide systems, both of which converge on the nucleus accumbens (17).
Brain Stress Systems
The brain stress systems can be defined as neurochemical systems that are activated during exposure to acute stressors or in a chronic state of stress and mediate species-typical behavioral responses. These behavioral responses in animals range from freezing to flight and typically have face and predictive validity for similar behavior responses in humans. For example, animals exposed to a stressor will show an enhanced freezing response to a conditioned fear stimulus, an enhanced startle response to a startle stimulus, avoidance of open areas, open arms, or height, and enhanced species-typical responses to an aversive stimulus (e.g., burying a shock probe in the defensive burying test). Key neuronal/neurochemical systems with circumscribed neurocircuitry that mediate behavioral responses to stressors include glucocorticoids, corticotropin-releasing factor (CRF), norepinephrine, and dynorphin, and key neurochemical systems that act in opposition to the brain stress systems include neuropeptide Y (NPY), nociceptin, and endocannabinoids [for reviews, see (18–20)]. For the purposes of this review, two brain stress systems with prominent roles in driving the dark side of addiction will be considered: CRF and dynorphin.
Corticotropin-Releasing Factor
Corticotropin-releasing factor is a 41-amino-acid polypeptide that controls hormonal, sympathetic, and behavioral responses to stressors (21, 22). Central administration of CRF mimics the behavioral response to activation and stress in rodents, and administration of competitive CRF receptor antagonists generally has anti-stress effects [for reviews, see (23–26)]. Two major CRF receptors have been identified, with CRF1 receptor activation associated with increased stress responsiveness (27) and CRF2 receptor activation associated with decreases in feeding and decreases in stress responsiveness (28, 29), although there is some controversy in this area (30). CRF neurons are present in the neocortex, the extended amygdala, the medial septum, the hypothalamus, the thalamus, the cerebellum, and autonomic midbrain and hindbrain nuclei (31). Extensive research has been performed on CRF neurons in the paraventricular nucleus of the hypothalamus (PVN), central nucleus of the amygdala (CeA), and bed nucleus of the stria terminalis (BNST), demonstrating a key role for PVN CRF neurons in controlling the pituitary adrenal response to stress (32) and a key role for BNST and CeA CRF in mediating the negative affective responses to stress and drug withdrawal (33).
The neuroanatomical entity termed the extended amygdala (34) may represent a common anatomical substrate that integrates brain arousal-stress systems with hedonic processing systems to produce the neuroadaptations associated with the development of addiction (see below). The extended amygdala is composed of the CeA, BNST, and a transition zone in the medial (shell) subregion of the nucleus accumbens. Each of these regions has cytoarchitectural and circuitry similarities (34). The extended amygdala receives numerous afferents from limbic structures, such as the basolateral amygdala and hippocampus, and sends efferents to the medial part of the ventral pallidum and a large projection to the lateral hypothalamus, thus further defining the specific brain areas that interface classical limbic (emotional) structures with the extrapyramidal motor system (35). CRF in the extended amygdala has long been hypothesized to play a key role not only in fear conditioning (36, 37) but also in the emotional component of pain processing (38).
Dynorphin-κ Opioid System
Dynorphins are opioid peptides that derive from the prodynorphin precursor and contain the leucine (leu)-enkephalin sequence at the N-terminal portion of the molecule and are the presumed endogenous ligands for the κ opioid receptor (39). Dynorphins are widely distributed in the central nervous system (40) and play a role in neuroendocrine regulation, pain regulation, motor activity, cardiovascular function, respiration, temperature regulation, feeding behavior, and stress responsivity (41). Dynorphins bind to all three opioid receptors but show a preference for κ receptors (39). Dynorphin-κ receptor system activation produces some actions that are similar to other opioids (analgesia) but others opposite to those of μ opioid receptors in the motivational domain. Dynorphins produce aversive dysphoric-like effects in animals and humans and have been hypothesized to mediate negative emotional states (42–45).
Dopamine receptor activation in the nucleus accumbens shell stimulates a cascade of events that ultimately lead to cyclic adenosine monophosphate response element-binding protein (CREB) phosphorylation and subsequent alterations in gene expression, notably the activation of the expression of prodynorphin mRNA. Subsequent activation of dynorphin systems has been hypothesized to feed back to decrease dopamine release in the mesolimbic dopamine system (46–50) and glutamate release in the nucleus accumbens (51, 52). Both of these changes may contribute to the dysphoric syndrome associated with cocaine dependence. In vivo microdialysis studies have also provided evidence that κ opioid receptors located in the prefrontal cortex (PFC) and ventral tegmental area also regulate the basal activity of mesocortical dopamine neurons (53, 54). In the extended amygdala, enhanced dynorphin action may also activate brain stress responses, such as CRF (55), or CRF in turn may activate dynorphin (56, 57).
Dynamic Changes in Reward: Opponent Process
Changes in reinforcement were inextricably linked with hedonic, affective, or emotional states in addiction in the context of temporal dynamics by Solomon’s opponent-process theory of motivation. Solomon and Corbit (58) postulated that hedonic, affective, or emotional states, once initiated, are automatically modulated by the central nervous system through mechanisms that reduce the intensity of hedonic feelings. The a-process includes affective or hedonic habituation (or tolerance), and the b-process includes affective or hedonic withdrawal (abstinence). The a-process in drug use consists of positive hedonic responses, occurs shortly after the presentation of a stimulus, correlates closely with the intensity, quality, and duration of the reinforcer, and shows tolerance. In contrast, the b-process in drug use appears after the a-process has terminated, consists of negative hedonic responses, and is sluggish in onset, slow to build up to an asymptote, slow to decay, and gets larger with repeated exposure. The thesis we have elaborated is that there is a neurocircuitry change in specific neurochemical systems that account for the b-process. Such opponent processes are hypothesized to begin early in drug taking, reflecting not only deficits in brain reward system function but also the recruitment of brain stress systems. Furthermore, we hypothesize that the recruitment of brain stress systems forms one of the major sources of negative reinforcement in addiction. Finally, we have hypothesized that such changes result not in a return to homeostasis of reward/stress function but in allostasis of reward/stress function that continues to drive the addiction process (Figure 3).
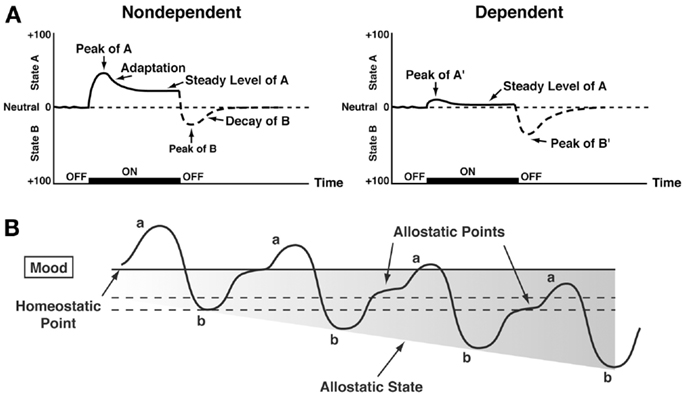
Figure 3. (A) The standard pattern of affective dynamics produced by (left) a relatively novel unconditioned stimulus (i.e., in a non-dependent state) and (right) a familiar, frequently repeated unconditioned stimulus (i.e., in a dependent state) [taken with permission from Ref. (184)]. (B) The changes in the affective stimulus (state) in an individual with repeated frequent drug use that may represent a transition to an allostatic state in the brain reward systems and, by extrapolation, a transition to addiction. Note that the apparent b-process never returns to the original homeostatic level before drug taking is reinitiated, thus creating a greater and greater allostatic state in the brain reward system. In other words, the counteradaptive opponent-process (b-process) does not balance the activational process (a-process) but in fact shows a residual hysteresis. While these changes are exaggerated and condensed over time in the present conceptualization, the hypothesis here is that even during post-detoxification, a period of “protracted abstinence,” the reward system is still bearing allostatic changes. In the non-dependent state, reward experiences are normal, and the brain stress systems are not greatly engaged. During the transition to the state known as addiction, the brain reward system is in a major underactivated state while the brain stress system is highly activated [taken with permission from Ref. (15)].
Allostasis, originally conceptualized to explain persistent morbidity of arousal and autonomic function, can be defined as “stability through change.” Allostasis involves a feed-forward mechanism rather than the negative feedback mechanisms of homeostasis, with continuous reevaluation of need and continuous readjustment of all parameters toward new set points. An allostatic state has been defined as a state of chronic deviation of the regulatory system from its normal (homeostatic) operating level (15). Allostatic load was defined as the “long-term cost of allostasis that accumulates over time and reflects the accumulation of damage that can lead to pathological states” (59).
Opponent process-like negative emotional states have been characterized in humans by acute and protracted abstinence from all major drugs of abuse (60–62). Similar results have been observed in animal models with all major drugs of abuse using intracranial self-stimulation (ICSS) as a measure of hedonic tone. Withdrawal from chronic cocaine (63), amphetamine (64), opioids (65), cannabinoids (66), nicotine (67), and ethanol (68) leads to increases in reward threshold during acute abstinence, and some of these elevations in threshold can last for up to 1 week (69). These observations lend credence to the hypothesis that opponent processes in the hedonic domain have an identifiable neurobiological basis and provide an impetus for defining the mechanisms involved. Understanding the mechanisms that drive this increase in reward thresholds is key to understanding the mechanisms that drive negative reinforcement in addiction.
Such elevations in reward threshold begin rapidly and can be observed within a single session of self-administration (70), bearing a striking resemblance to human subjective reports of acute withdrawal. Dysphoria-like responses also accompany acute opioid and ethanol withdrawal (71, 72). Here, naloxone administration following single injections of morphine increased reward thresholds, measured by ICSS, and increased thresholds with repeated morphine and naloxone-induced withdrawal experience (71). Similar results were observed during repeated acute withdrawal from ethanol (72).
Neuroadaptations Responsible for Opponent Process
One hypothesis is that drug addiction progresses from a source of positive reinforcement that may indeed involve a form of sensitization of incentive salience, as argued by Robinson and Berridge (9), to sensitization of opponent processes that set up a powerful negative reinforcement process. A further elaboration of this hypothesis is that there are both within- and between-system neuroadaptations to excessive activation of the reward system at the neurocircuitry level. Within-system neuroadaptations are defined as the process by which the primary cellular response element to the drug (circuit A) itself adapts to neutralize the drug’s effects. Persistence of the opposing effects after the drug disappears produces adaptation. A between-system neuroadaptation is a circuitry change, in which B circuits (i.e., the stress or anti-reward circuits) are activated by circuit A (i.e., the reward circuit). In the present treatise, within-system neuroadaptations can dynamically interact with a between-system neuroadaptation, in which circuit B (i.e., the anti-reward circuit) is activated either in parallel or in series to suppress the activity of circuit A (see below).
Animal Models of the Transition to an Addiction-Like State as Defined by Escalation in Drug Self-Administration with Prolonged Access
A progressive increase in the frequency and intensity of drug use is one of the major behavioral phenomena that characterize the development of addiction and has face validity with the criteria of the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV): “The substance is often taken in larger amounts and over a longer period than was intended” (American Psychological Association, 1994). A framework with which to model the transition from drug use to drug addiction can be found in recent animal models of prolonged access to intravenous cocaine self-administration. Historically, animal models of cocaine self-administration involved the establishment of stable behavior from day to day to allow the reliable interpretation of data provided by within-subject designs aimed at exploring the neuropharmacological and neurobiological bases of the reinforcing effects of acute cocaine. Up until 1998, after the acquisition of self-administration, rats were typically allowed access to cocaine for 3 h or less per day to establish highly stable levels of intake and patterns of responding between daily sessions. This was a useful paradigm for exploring the neurobiological substrates for the acute reinforcing effects of drugs of abuse.
However, in an effort to explore the possibility that differential access to drugs of abuse may have more face validity for the compulsive-like responding observed in addiction, animals have been allowed extended access to all major drugs of abuse (Figure 4). Increased intake was observed in the extended-access group for intravenous cocaine, methamphetamine, heroin, and nicotine and oral alcohol during extended access and dependence (73–79). For example, when animals were allowed access for 1 and 6 h to different doses of cocaine, after escalation, both the long-access (LgA) and short-access (ShA) animals titrated their cocaine intake, but LgA rats consistently self-administered almost twice as much cocaine at any dose tested, further suggesting an upward shift in the set point for cocaine reward in the escalated animals (80–82).
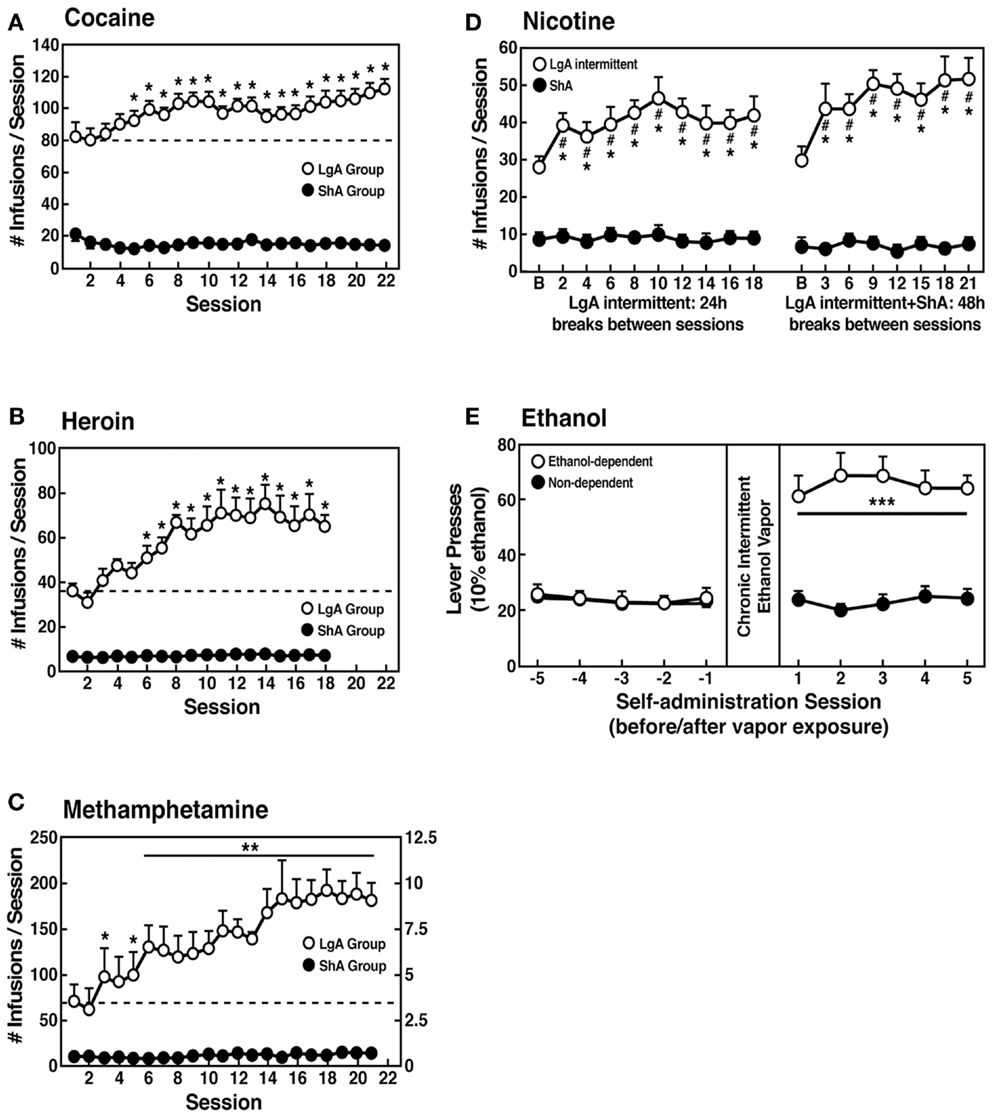
Figure 4. (A) Effect of drug availability on cocaine intake (mean ± SEM). In long-access (LgA) rats (n = 12) but not short-access (ShA) rats (n = 12), the mean total cocaine intake started to increase significantly from session 5 (p < 0.05; sessions 5–22 compared with session 1) and continued to increase thereafter (p < 0.05; session 5 compared with sessions 8–10, 12, 13, and 17–22) [taken with permission from Ref. (74)]. (B) Effect of drug availability on total intravenous heroin self-infusions (mean ± SEM). During the escalation phase, rats had access to heroin (40 μg per infusion) for 1 h (ShA rats, n = 5–6) or 11 h per session (LgA rats, n = 5–6). Regular 1 h (ShA rats) or 11 h (LgA rats) sessions of heroin self-administration were performed 6 days a week. The dotted line indicates the mean ± SEM number of heroin self-infusions in LgA rats during the first 11 h session. *p < 0.05, different from the first session (paired t-test) [taken with permission from Ref. (73)]. (C) Effect of extended access to intravenous methamphetamine on self-administration as a function of daily sessions in rats trained to self-administer 0.05 mg/kg/infusion of intravenous methamphetamine during the 6 h session. ShA, 1 h session (n = 6). LgA, 6 h session (0.05 mg/kg/infusion, n = 4). **p < 0.01, compared with day 1 [taken with permission from Ref. (75)]. (D) Nicotine intake (mean ± SEM) in rats that self-administered nicotine under a fixed-ratio (FR) 1 schedule in either 21 h (LgA) or 1 h (ShA) sessions. LgA rats increased their nicotine intake on an intermittent schedule with 24–48 h breaks between sessions, whereas LgA rats on a daily schedule did not. The left shows the total number of nicotine infusions per session when the intermittent schedule included 24 h breaks between sessions. The right shows the total number of nicotine infusions per session when the intermittent schedule included 48 h breaks between sessions. #p < 0.05, compared with baseline; *p < 0.05, compared with daily self-administration group. n = 10 per group [taken with permission from Ref. (185)]. (E) Ethanol self-administration in ethanol-dependent and non-dependent animals. The induction of ethanol dependence and correlation of limited ethanol self-administration before and excessive drinking after dependence induction following chronic intermittent ethanol vapor exposure is shown. ***p < 0.001, significant group × test session interaction. With all drugs, escalation is defined as a significant increase in drug intake within-subjects in extended-access groups, with no significant changes within-subjects in limited-access groups [taken with permission from Ref. (186)].
Consistent with the hypothesis that extended access to drugs of abuse produces compulsive-like responding, in which animals will “continue to respond in the face of adverse consequences” (another DSM-IV criteria for Substance Dependence), animals with extended access that show escalation in self-administration also show increased responding on a progressive-ratio schedule of reinforcement [(83–85); Figure 5]. Changes in the reinforcing and incentive effects of drug intake that are consistent with the increases in progressive-ratio responding have been observed following extended access and include increased drug-induced reinstatement after extinction, a decreased latency to goal time in a runway model for drug reward, and responding in the face of punishment (86–92). Altogether, these results suggest that drug taking with extended-access changes the motivation to seek the drug. Some have argued that enhanced drug taking reflects a sensitization of reward (93), but studies of locomotor sensitization suggest that locomotor sensitization occurs independently of escalation (94–96). The increased brain reward thresholds and neuropharmacological studies outlined below argue for a reward deficit state that drives the increased drug taking during extended access.
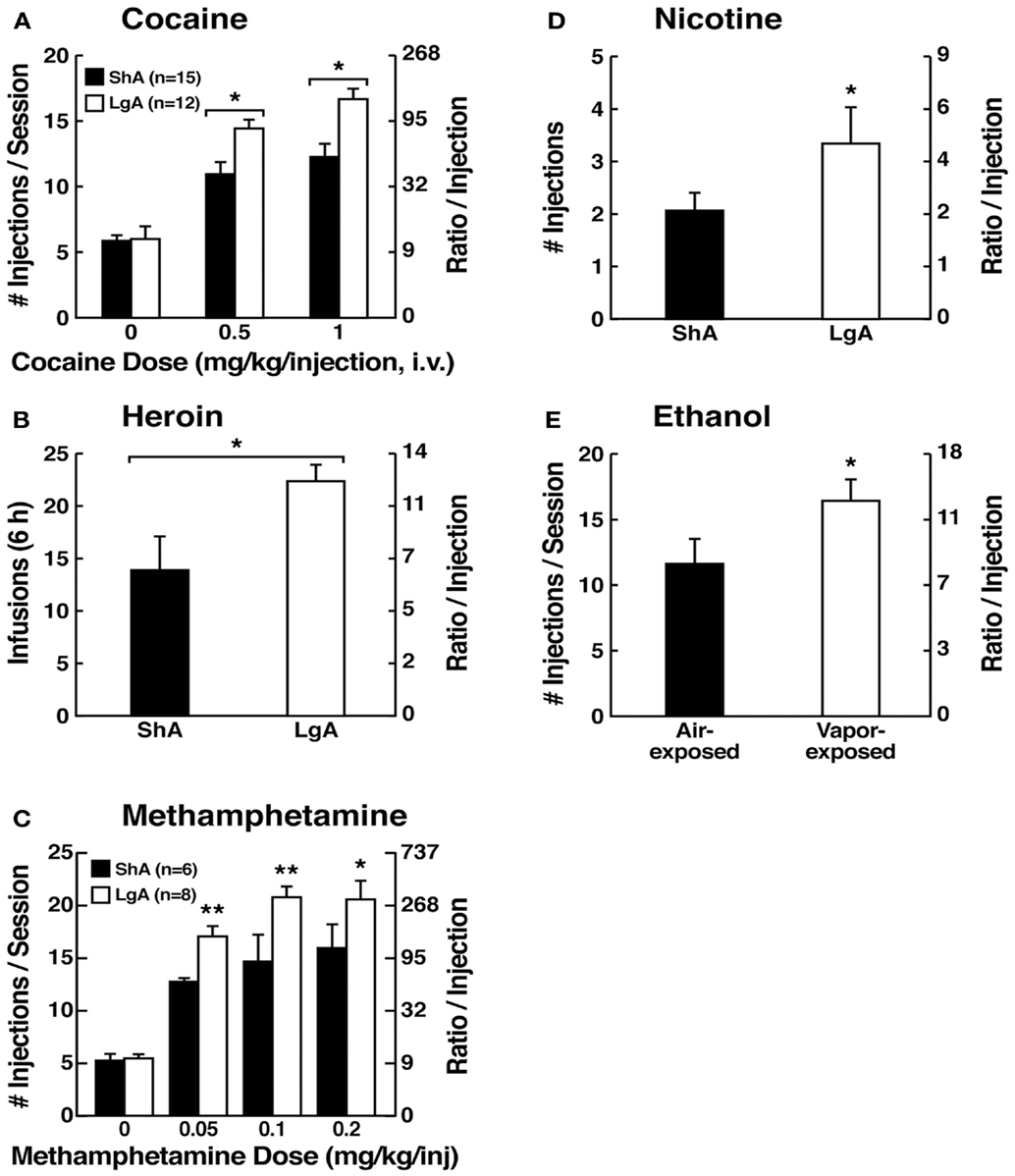
Figure 5. (A) Dose-response function of cocaine by rats responding under a progressive-ratio schedule. Test sessions under a progressive-ratio schedule ended when rats did not achieve reinforcement within 1 h. The data are expressed as the number of injections per session on the left axis and ratio per injection on the right axis. *p < 0.05, compared with ShA rats at each dose of cocaine [taken with permission from Ref. (84)]. (B) Responding for heroin under a progressive-ratio schedule of reinforcement in ShA and LgA rats. *p < 0.05, LgA significantly different from LgA [Modified with permission from Ref. (187)]. (C) Dose-response for methamphetamine under a progressive-ratio schedule. Test sessions under a progressive-ratio schedule ended when rats did not achieve reinforcement within 1 h. *p < 0.05, **p < 0.01, LgA significantly different from ShA [Modified from Ref. (188)]. (D) Breakpoints on a progressive-ratio schedule in long-access (LgA) rats that self-administered nicotine with 48 h abstinence between sessions. LgA rats on an intermittent schedule reached significantly higher breakpoints than LgA rats that self-administered nicotine daily. The data are expressed as mean ± SEM. *p < 0.05. n = 9 rats per group [taken with permission from Ref. (185)]. (E) Mean (±SEM) breakpoints for ethanol while in non-dependent and ethanol-dependent states. **p < 0.01, main effect of vapor exposure on ethanol self-administration [taken with permission from Ref. (85)].
Animals Escalate Their Intake of Drugs with Extended Access, with a Parallel Increase in Reward Thresholds
The hypothesis that compulsive cocaine use is accompanied by a chronic perturbation in brain reward homeostasis has been tested in animal models of escalation in drug intake with prolonged access combined with measures of brain stimulation reward thresholds. Animals implanted with intravenous catheters and allowed differential access to intravenous self-administration of cocaine showed increases in cocaine self-administration from day to day in the LgA group (6 h; LgA) but not in the ShA group (1 h; ShA). The differential exposure to cocaine self-administration had dramatic effects on reward thresholds that progressively increased in LgA rats but not ShA or control rats across successive self-administration sessions (97). Elevations in baseline reward thresholds temporally preceded and were highly correlated with escalation in cocaine intake (Figure 6). Post-session elevations in reward thresholds failed to return to baseline levels before the onset of each subsequent self-administration session, thereby deviating more and more from control levels. The progressive elevation in reward thresholds was associated with a dramatic escalation in cocaine consumption that was observed previously (97). Similar results have been observed with extended access to methamphetamine (98) and heroin (99). Rats allowed 6 h access to methamphetamine or 23 h access to heroin also showed a time-dependent increase in reward thresholds that paralleled the increases in heroin intake (Figure 6). Similar results of parallel increases in brain reward thresholds with escalation of nicotine intake have been observed with extended access to nicotine (100).
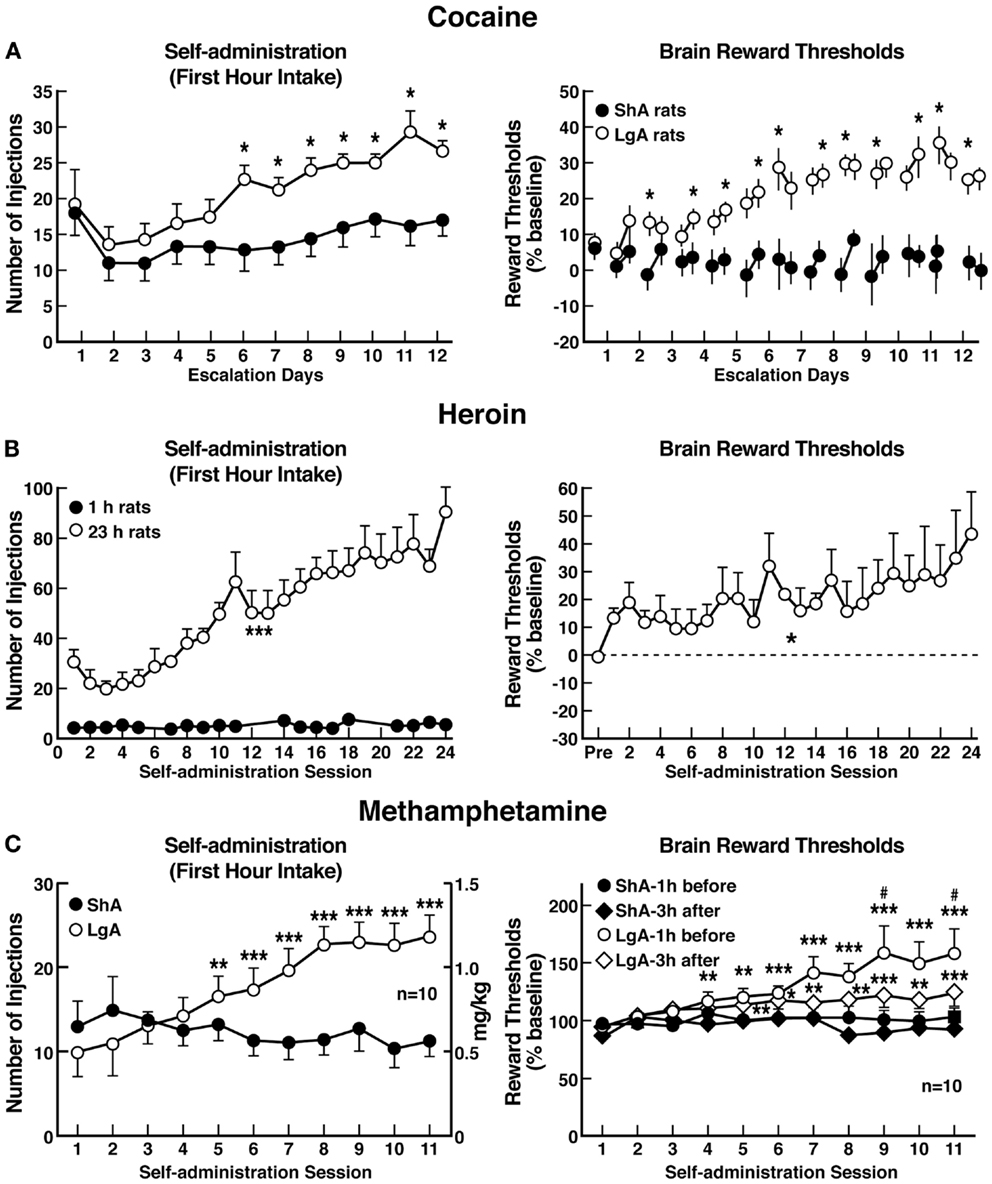
Figure 6. (A) Relationship between elevation in ICSS reward thresholds and cocaine intake escalation (Left). Percent change from baseline response latencies (3 h and 17–22 h after each self-administration session; first data point indicates 1 h before the first session) (Right). Percent change from baseline ICSS thresholds. *p < 0.05, compared with drug-naive and/or ShA rats (tests for simple main effects) [taken with permission from Ref. (97)]. (B) Unlimited daily access to heroin escalated heroin intake and decreased the excitability of brain reward systems (Left). Heroin intake (±SEM; 20 μg per infusion) in rats during limited (1 h) or unlimited (23 h) self-administration sessions. ***p < 0.001, main effect of access (1 or 23 h) (Right). Percent change from baseline ICSS thresholds (±SEM) in 23 h rats. Reward thresholds, assessed immediately after each daily 23 h self-administration session, became progressively more elevated as exposure to self-administered heroin increased across sessions. *p < 0.05, main effect of heroin on reward thresholds [taken with permission from Ref. (99)]. (C) Escalation in methamphetamine self-administration and ICSS in rats. Rats were daily allowed to receive ICSS in the lateral hypothalamus 1 h before and 3 h after intravenous methamphetamine self-administration with either 1 or 6 h access (Left). Methamphetamine self-administration during the first hour of each session (Right). ICSS measured 1 h before and 3 h after methamphetamine self-administration. *p < 0.05, **p < 0.01, ***p < 0.001, compared with session 1. #p < 0.05, compared with LgA 3 h after [taken with permission from Ref. (98)].
Brain Reward System Substrates for the Negative Reinforcement Associated with Addiction (Within-System Neuroadaptations)
The withdrawal/negative affect stage can be defined as the presence of motivational signs of withdrawal in humans, including chronic irritability, physical pain, emotional pain [i.e., hyperkatifeia; (101)], malaise, dysphoria, alexithymia, and loss of motivation for natural rewards. It is characterized in animals by increases in reward thresholds during withdrawal from all major drugs of abuse. More compelling, as noted above, in animal models of the transition to addiction, similar changes in brain reward thresholds occur that temporally precede and are highly correlated with escalation in drug intake (97–99). Such acute withdrawal is associated with decreased activity of the mesocorticolimbic dopamine system, reflected by electrophysiological recordings and in vivo microdialysis [(102–104); Figure 7].
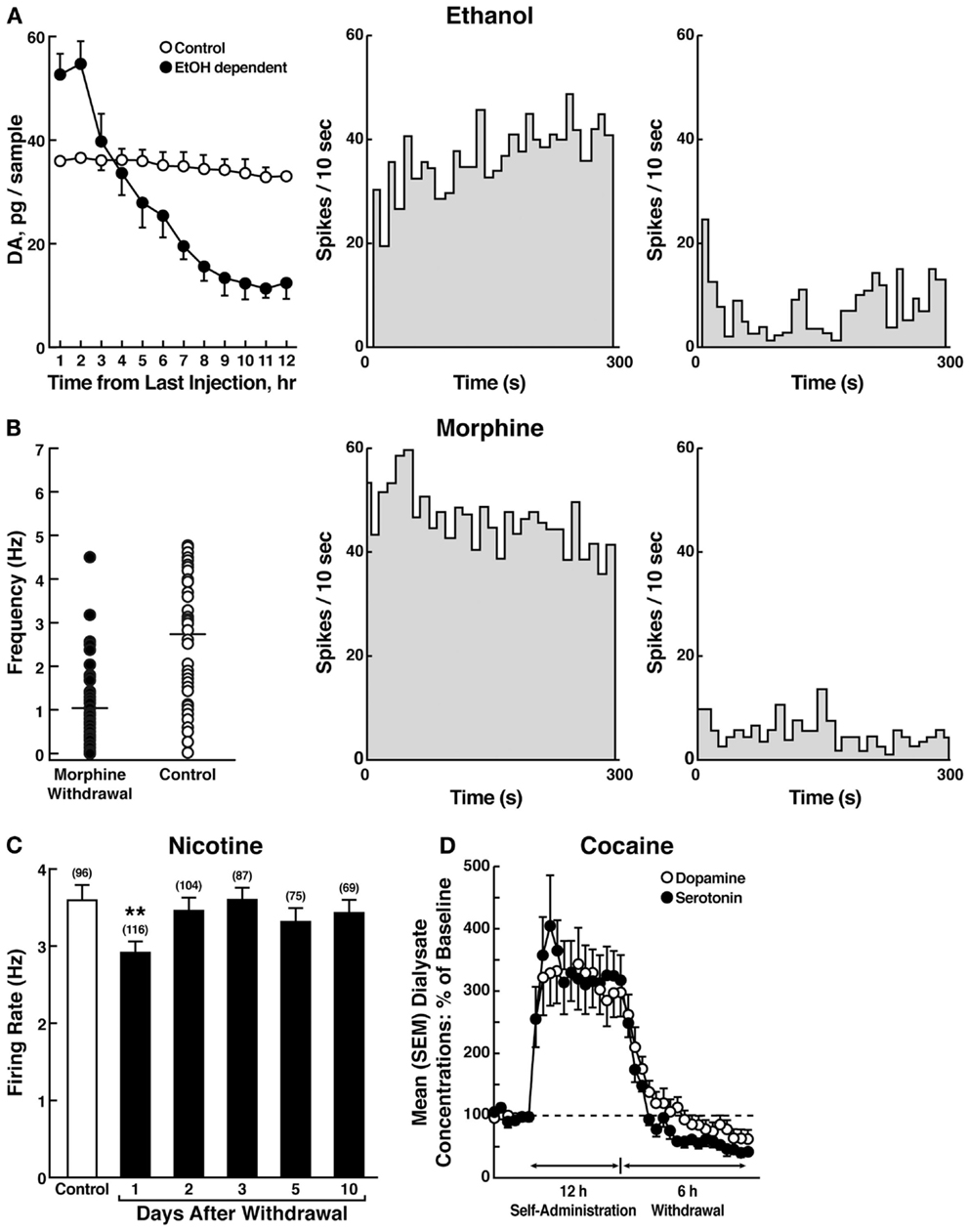
Figure 7. (A) The left panel shows the effect of ethanol withdrawal on absolute extracellular dopamine concentrations in the nucleus accumbens in ethanol-withdrawn rats. The middle and right panels show the spontaneous activity of antidromically identified ventral tegmental area-nucleus accumbens dopamine neurons in control (middle) and ethanol-withdrawn (right) rats [taken with permission from Ref. (102)]. (B) The left panel shows individual firing rates of antidromically identified ventral tegmental area-nucleus accumbens dopamine neurons recorded from morphine-withdrawn and control rats. Each circle represents the mean firing of at least a 5-min recording. Horizontal lines indicate the mean activity. The middle and right panels show the spontaneous activity of a selected number (4) or antidromically identified ventral tegmental area-nucleus accumbens dopamine neurons in control (middle) and morphine-withdrawn (right) rats. Each panel represents the neuronal activity of a single cell. Recordings in both cases were obtained 24 h after the last morphine and saline administration, respectively [taken with permission from Ref. (103)]. (C) Firing rates of dopamine cells in the ventral tegmental area following 1–10 days of withdrawal from chronic nicotine treatment (6 mg/kg/day for 12 days). The data are expressed as mean ± SEM. The number of dopamine cells recorded is given in parentheses. *p < 0.01, compared with control group [taken with permission from Ref. (189)]. (D) Profile of dialysate serotonin and dopamine concentrations during a 12-h extended-access cocaine self-administration session. The mean ± SEM presession baseline dialysate concentrations of serotonin and dopamine were 0.98 ± 0.1 nM and 5.3 ± 0.5 nM, respectively (n = 7) [taken with permission from Ref. (104)].
Human imaging studies of individuals with addiction during withdrawal or protracted abstinence have generated results that are consistent with animal studies. There are decreases in dopamine D2 receptors (hypothesized to reflect hypodopaminergic functioning), hyporesponsiveness to dopamine challenge (105), and hypoactivity of the orbitofrontal-infralimbic cortex system (105). These are hypothesized to be within-system neuroadaptations that may reflect presynaptic release or postsynaptic receptor plasticity.
In the context of chronic alcohol administration, multiple molecular mechanisms have been hypothesized to counteract the acute effects of ethanol that could be considered within-system neuroadaptations. For example, chronic ethanol decreases γ-aminobutyric acid (GABA) receptor function, possibly through downregulation of the α1 subunit (106, 107). Chronic ethanol also decreases the acute inhibition of adenosine reuptake [i.e., tolerance develops to the inhibition of adenosine by ethanol; (108)]. Perhaps more relevant to the present treatise, whereas acute ethanol activates adenylate cyclase, withdrawal from chronic ethanol decreases CREB phosphorylation in the amygdala and is linked to decreases in the function of NPY and anxiety-like responses observed during acute ethanol withdrawal (109, 110).
Brain Stress System Substrates for the Negative Reinforcement Associated with Addiction (Between-System Neuroadaptations)
Brain neurochemical systems involved in arousal-stress modulation have been hypothesized to be engaged within the neurocircuitry of the brain stress systems in an attempt to overcome the chronic presence of the perturbing drug and restore normal function despite the presence of drug (18). Both the hypothalamic-pituitary-adrenal (HPA) axis and extrahypothalamic brain stress system mediated by CRF are dysregulated by chronic administration of all major drugs with dependence or abuse potential, with a common response of elevated adrenocorticotropic hormone, corticosterone, and amygdala CRF during acute withdrawal (24, 69, 111–116). Indeed, activation of the HPA response may be an early dysregulation associated with excessive drug taking that ultimately “sensitizes” the extrahypothalamic CRF systems (33, 92).
As noted above, the excessive release of dopamine and opioid peptides produces subsequent activation of dynorphin systems, which has been hypothesized to feed back to decrease dopamine release and also contribute to the dysphoric syndrome associated with cocaine dependence (48). Dynorphins produce aversive dysphoric-like effects in animals and humans and have been hypothesized to mediate negative emotional states (42–45).
A common response to acute withdrawal and protracted abstinence from all major drugs of abuse is the manifestation of anxiety-like responses that are reversed by CRF antagonists. Withdrawal from repeated administration of cocaine produces an anxiogenic-like response in the elevated plus maze and defensive burying test, both of which are reversed by administration of CRF receptor antagonists (117, 118). Opioid dependence also produces irritability-like effects that are reversed by CRF receptor antagonists (119, 120). Ethanol withdrawal produces anxiety-like behavior that is reversed by intracerebroventricular administration of CRF1/CRF2 peptidergic antagonists (121) and small-molecule CRF1 antagonists (122–124) and intracerebral administration of a peptidergic CRF1/CRF2 antagonist into the amygdala (125). Thus, some effects of CRF antagonists have been localized to the CeA (125). Precipitated withdrawal from nicotine produces anxiety-like responses that are also reversed by CRF antagonists (77, 126). CRF antagonists injected intracerebroventricularly or systemically also block the potentiated anxiety-like responses to stressors observed during protracted abstinence from chronic ethanol (127–131).
Another measure of negative emotional states during drug withdrawal in animals is conditioned place aversion, in which animals avoid an environment previously paired with an aversive state. Such place aversions, when used to measure the aversive stimulus effects of withdrawal, have been observed largely in the context of opioids (132, 133). Systemic administration of a CRF1 receptor antagonist and direct intracerebral administration of a peptide CRF1/CRF2 antagonist also decreased opioid withdrawal-induced place aversions (134–136). These effects have been hypothesized to be mediated by actions in the extended amygdala. The selective CRF1 antagonist antalarmin blocked the place aversion produced by naloxone in morphine-dependent rats (134), and a CRF peptide antagonist injected into the CeA also reversed the place aversion produced by methylnaloxonium injected into the CeA (135). CRF1 knockout mice failed to show conditioned place aversion to opioid withdrawal and failed to show an opioid-induced increase in dynorphin mRNA in the nucleus accumbens (136).
A compelling test of the hypothesis that CRF-induced increases in anxiety-like responses during drug withdrawal has motivational significance in contributing to negative emotional states is the observation that CRF antagonists can reverse the elevation in reward thresholds produced by drug withdrawal. Nicotine and alcohol withdrawal-induced elevations in reward thresholds were reversed by a CRF antagonist (137, 138). These effects have been localized to both the CeA and nucleus accumbens shell (139).
Enhanced dynorphin action is hypothesized to mediate the depression-like, aversive responses to stress, and dysphoric-like responses during withdrawal from drugs of abuse (49, 56, 57, 140–145). For example, pretreatment with a κ-opioid receptor antagonist blocked stress-induced analgesia and stress-induced immobility (57), decreased anxiety-like behavior in the elevated plus maze and open field, decreased conditioned fear in fear-potentiated startle (145), and blocked depressive-like behavior induced by cocaine withdrawal (140).
Brain Stress Substrates That Mediate Drug Taking with Extended Access
Corticotropin-Releasing Factor, Compulsive-Like Drug Seeking, and the Extended Amygdala
The ability of CRF antagonists to block the anxiogenic-like and aversive-like motivational effects of drug withdrawal predicted motivational effects of CRF antagonists in animal models of extended access to drugs. CRF antagonists selectively blocked the increased self-administration of drugs associated with extended access to intravenous self-administration of cocaine (146), nicotine (77), and heroin [(147); Figure 8]. For example, systemic administration of a CRF1 antagonist blocked the increased self-administration of nicotine associated with withdrawal in extended-access (23 h) animals (77).
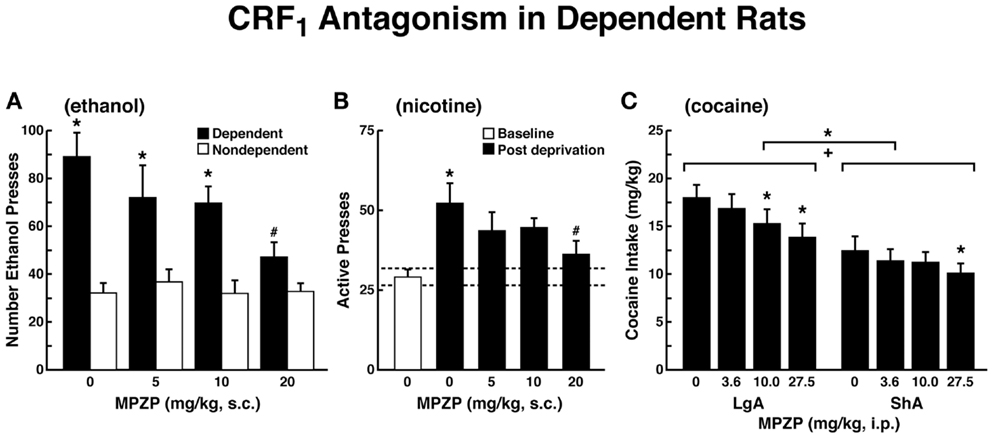
Figure 8. Effects of CRF1 antagonist on compulsive-like responding for drugs of abuse in rats with extended access to drug (A). The effect of the CRF1 receptor antagonist MPZP on operant self-administration of alcohol in dependent and non-dependent rats. Testing was conducted when dependent animals were in acute withdrawal (6–8 h after removal from vapors). Dependent rats self-administered significantly more than non-dependent animals, and MPZP dose-dependently reduced alcohol self-administration only in dependent animals. The data are expressed as mean + SEM lever presses for alcohol [taken with permission from Ref. (190)]. (B) Abstinence-induced escalation of nicotine intake is blocked by a CRF1 receptor antagonist. Effect of MPZP (s.c., −1 h) on nicotine self-administration during the active period in rats given extended access to nicotine. *p < 0.05, compared with baseline; #p < 0.05, compared with after-abstinence vehicle treatment; n = 8). The data are expressed as mean + SEM lever presses for nicotine [taken with permission from Ref. 77)]. (C) MPZP reduces cocaine intake in ShA and LgA rats. The data are expressed as mean + SEM cocaine intake (mg/kg). *p < 0.05, **p < 0.01, compared with vehicle [taken with permission from Ref. (146)].
Corticotropin-releasing factor antagonists also blocked the increased self-administration of ethanol in dependent rats [(124); Figure 8]. For example, exposure to repeated cycles of chronic ethanol vapor produced substantial increases in ethanol intake in rats during both acute withdrawal and protracted abstinence [2 weeks post-acute withdrawal; (76, 148)]. Intracerebroventricular administration of a CRF1/CRF2 antagonist blocked the dependence-induced increase in ethanol self-administration during both acute withdrawal and protracted abstinence (149). Systemic injections of small-molecule CRF1 antagonists also blocked the increased ethanol intake associated with acute withdrawal (124) and protracted abstinence (150). When administered directly into the CeA, a CRF1/CRF2 antagonist blocked ethanol self-administration in ethanol-dependent rats (151). These effects appear to be mediated by the actions of CRF on GABAergic interneurons within the CeA, and a CRF antagonist administered chronically during the development of dependence blocked the development of compulsive-like responding for ethanol (116). Altogether, these results suggest that CRF in the basal forebrain may also play an important role in the development of the aversive motivational effects that drive the increased drug-seeking associated with cocaine, heroin, nicotine, and alcohol dependence.
Dynorphin, Compulsive-Like Drug Seeking, and the Extended Amygdala
Recent evidence suggests that the dynorphin-κ opioid system also mediates compulsive-like drug responding (methamphetamine, heroin, and alcohol) with extended access and dependence. Evidence from our laboratory has shown a small-molecule κ antagonist selectively blocked responding on a progressive-ratio schedule for cocaine in rats with extended access (152). Even more compelling is that excessive drug self-administration can also be blocked by κ antagonists (152–155) and may be mediated by the shell of the nucleus accumbens (156). However, the neurobiological circuits involved in mediating the effects of activation of the dynorphin-κ opioid system on the escalation of methamphetamine intake with extended access, remain unknown.
NPY, Compulsive Drug Seeking, and the Extended Amgydala
Neuropeptide Y is a neuropeptide with dramatic anxiolytic-like properties localized to multiple brain regions but heavily innervating the amygdala. It is hypothesized to have effects opposite to CRF in the negative motivational state of withdrawal from drugs of abuse and as such increases in NPY function may act in opposition to the actions of increases in CRF (157). Significant evidence suggests that activation of NPY in the CeA can block the motivational aspects of dependence associated with chronic ethanol administration. NPY administered intracerebroventricularly blocked the increased drug intake associated with ethanol dependence (158, 159). NPY also decreased excessive alcohol intake in alcohol-preferring rats (160). Injection of NPY directly into the CeA (161) and viral vector-enhanced expression of NPY in the CeA also blocked the increased drug intake associated with ethanol dependence (162). At the cellular level, NPY, like CRF1 antagonists, blocks the increase in GABA release in the CeA produced by ethanol and also when administered chronically blocks the transition to excessive drinking with the development of dependence (163). The role of NPY in the actions of other drugs of abuse is limited, particularly with regard to dependence and compulsive drug seeking. NPY5 receptor knockout mice have a blunted response to the rewarding effects of cocaine (164, 165), and NPY knockout mice show hypersensitivity to cocaine self-administration (166). NPY itself injected intracerebroventricularly facilitated heroin and cocaine self-administration and induced reinstatement of heroin seeking in limited-access rats (167, 168). An NPY Y2 antagonist, possibly acting presynaptically to release NPY, blocked social anxiety associated with nicotine withdrawal (169), and NPY injected intracerebroventricularly blocked the somatic signs but not reward deficits associated with nicotine withdrawal (170). However, the role of NPY in compulsive drug seeking with extended-access remains to be studied. The hypothesis here would be that NPY is a buffer or homeostatic response to between-system neuroadaptations that can return the brain emotional systems to homeostasis (157, 171).
Corticotropin-releasing factor, stress, and the frontal cortex
Converging lines of evidence suggest that impairment of medial PFC (mPFC) cognitive function and overactivation of the CeA may be linked to the development of compulsive-like responding for drugs of abuse during extended access (172–174). Extended access to cocaine self-administration induced an escalated pattern of cocaine intake associated with an impairment of working memory and decrease in the density of dorsomedial PFC (dmPFC) neurons that lasted for months after cocaine cessation (172). Whereas LgA and ShA rats exhibited a high percentage of correct responses in the delayed non-matching-to-sample task under low cognitive demand (delay < 10 s), increasing the working memory load (i.e., close to the capacity limit of working memory) by increasing the delay from 10 to 70 and 130 s revealed a robust working memory deficit in LgA rats. Furthermore, the magnitude of escalation of cocaine intake was negatively correlated with working memory performance in ShA and LgA rats with the 70- and 130-s delays but not with the 10-s delay or with baseline performance during training, demonstrating that the relationship between the escalation of cocaine intake and behavioral performance in this task was restricted to working memory performance under high cognitive demand. The density of neurons and oligodendrocytes in the dmPFC was positively correlated with working memory performance. A lower density of neurons or oligodendrocytes in the dmPFC was associated with more severe working memory impairment. Working memory was also correlated with the density of oligodendrocytes in the orbitofrontal cortex (OFC), suggesting that OFC alterations after escalated drug intake may play a role in working memory deficits. However, no correlation was found between working memory performance and neuronal density in the OFC, suggesting that OFC neurons may be less vulnerable to the deleterious effects of chronic cocaine exposure than dmPFC neurons. Thus, PFC dysfunction may exacerbate the loss of control associated with compulsive drug use and facilitate the progression to drug addiction.
Similar results have been observed in an animal model of binge alcohol consumption, even before the development of dependence. Using an animal model of escalation of alcohol intake with chronic intermittent access to alcohol, in which rats are given continuous (24 h per day, 7 days per week) or intermittent (3 days per week) access to alcohol (20% v/v) using a two-bottle choice paradigm, FBJ murine osteosarcoma viral oncogene homolog (Fos) expression in the mPFC, CeA, hippocampus, and nucleus accumbens were measured and correlated with working memory and anxiety-like behavior (175). Abstinence from alcohol in rats with a history of escalation of alcohol intake specifically recruited GABA and CRF neurons in the mPFC and produced working memory impairments associated with excessive alcohol drinking during acute (24–72 h) but not protracted (16–68 days) abstinence. The abstinence from alcohol was associated with a functional disconnection of the mPFC and CeA but not mPFC or nucleus accumbens. These results show that recruitment of a subset of GABA and CRF neurons in the mPFC during withdrawal and disconnection of the PFC CeA pathway may be critical for impaired executive control over motivated behavior, suggesting that dysregulation of mPFC interneurons may be an early index of neuroadaptation in alcohol dependence.
Brain Stress Systems in Addiction: An Allostatic View
More importantly for the present thesis, as dependence and withdrawal develop, brain anti-reward systems, such as CRF and dynorphin, are recruited in the extended amygdala. We hypothesize that this brain stress neurotransmitter that is known to be activated during the development of excessive drug taking comprises a between-system opponent process, and this activation is manifest when the drug in removed, producing anxiety, hyperkatifeia, and irritability symptoms associated with acute and protracted abstinence. Notably, however, there is evidence of CRF immunoreactivity in the ventral tegmental area, and a CRF1 receptor antagonist injected directly into the ventral tegmental area blocked the social stress-induced escalation of cocaine self-administration (176). Altogether, these observations suggest between-system/within-system neuroadaptations that were originally hypothesized for dynorphin by Carlezon and Nestler (177), in which activation of CREB by excessive dopamine and opioid peptide receptor activation in the nucleus accumbens triggers the induction of dynorphin to feed back to suppress dopamine release. Thus, we hypothesize that anti-reward circuits are recruited as between-system neuroadaptations (178) during the development of addiction and produce aversive or stress-like states (179–181) via two mechanisms: direct activation of stress-like, fear-like states in the extended amygdala (CRF) and indirect activation of a depression-like state by suppressing dopamine (dynorphin).
A critical problem in drug addiction is chronic relapse, in which addicted individuals return to compulsive drug taking long after acute withdrawal. This corresponds to the preoccupation/anticipation stage of the addiction cycle outlined above. Koob and Le Moal also hypothesized that the dysregulations that comprise the “dark side” of drug addiction persist during protracted abstinence to set the tone for vulnerability to “craving” by activating drug-, cue-, and stress-induced reinstatement neurocircuits that are now driven by a reorganized and possibly hypofunctioning prefrontal system. The hypothesized allostatic, dysregulated reward, and sensitized stress state produces the motivational symptoms of acute withdrawal and protracted abstinence and provides the basis by which drug priming, drug cues, and acute stressors acquire even more power to elicit drug-seeking behavior (92). Thus, the combination of decreases in reward system function and recruitment of anti-reward systems provides a powerful source of negative reinforcement that contributes to compulsive drug-seeking behavior and addiction. A compelling argument can be made that the neuroplasticity that charges the CRF stress system may indeed begin much earlier that previously thought via stress actions in the PFC.
The overall conceptual theme argued here is that drug addiction represents an excessive and prolonged engagement of homeostatic brain regulatory mechanisms that regulate the response of the body to rewards and stressors. The dysregulation of the incentive salience systems may begin with the first administration of drug (182), and the dysregulation of the stress axis may begin with the binge and subsequent acute withdrawal, triggering a cascade of changes, from activation of the HPA axis to activation of CRF in the PFC to activation of CRF in the extended amygdala to activation of dynorphin in the ventral striatum (Figure 9). This cascade of overactivation of the stress axis represents more than simply a transient homeostatic dysregulation; it also represents the dynamic homeostatic dysregulation termed allostasis.
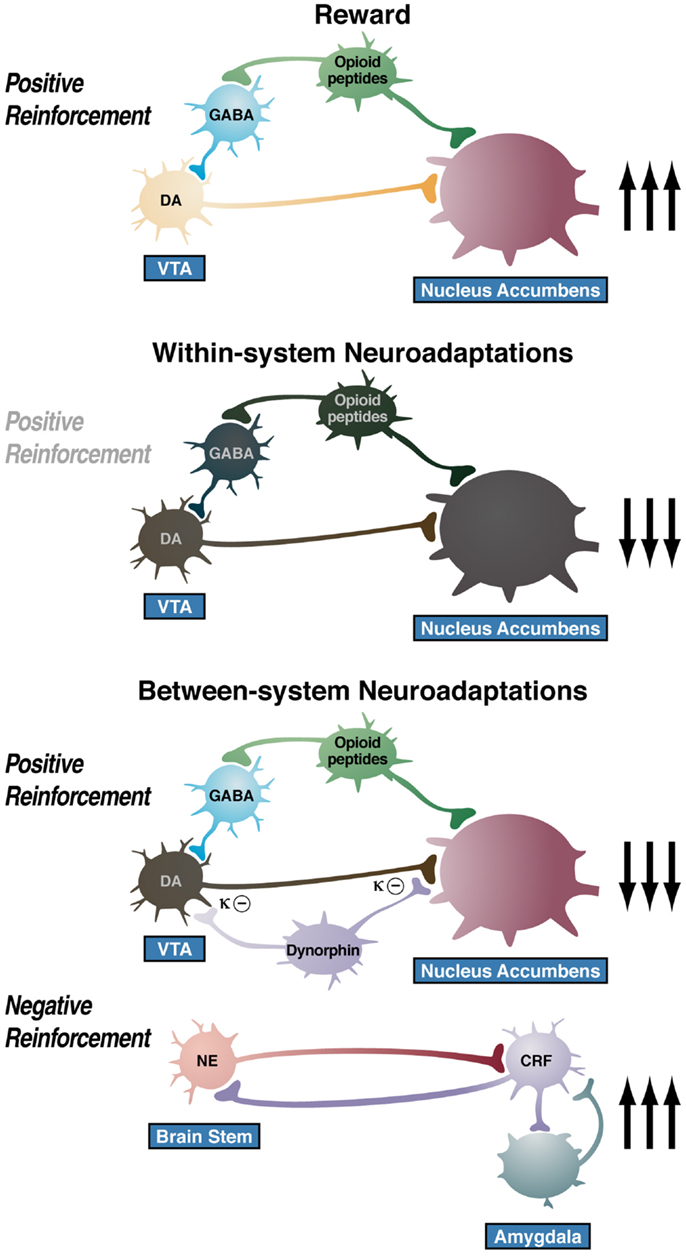
Figure 9. Diagram of the hypothetical “within-system” and “between-system” changes that lead to the “darkness within.” (Top) Circuitry for drug reward with major contributions from mesolimbic dopamine and opioid peptides that converge on the nucleus accumbens. During the binge/intoxication stage of the addiction cycle, the reward circuitry is excessively engaged, Middle. Such excessive activation of the reward system triggers “within-system” neurobiological adaptations during the withdrawal/negative affect stage, including activation of cyclic adenosine monophosphate (cAMP) and cAMP response element-binding protein (CREB), downregulation of dopamine D2 receptors, and decreased firing of ventral tegmental area (VTA) dopaminergic neurons, Bottom. As dependence progresses and the withdrawal/negative affect stage is repeated, two major “between-system” neuroadaptations occur. One is activation of dynorphin feedback that further decreases dopaminergic activity. The other is recruitment of extrahypothalamic norepinephrine (NE)-corticotropin-releasing factor (CRF) systems in the extended amygdala. Facilitation of the brain stress system in the prefrontal cortex is hypothesized to exacerbate the between-system neuroadaptations while contributing to the persistence of the dark side into the preoccupation/anticipation stage of the addiction cycle [taken with permission from Ref. (191)].
Repeated challenges, such as with drugs of abuse, lead to attempts of the brain stress systems at the molecular, cellular, and neurocircuitry levels to maintain stability but at a cost. For the drug addiction framework elaborated here, the residual decrease in the brain reward systems and activation of the brain stress systems to produce the consequent negative emotional state is termed an allostatic state (15). This state represents a combination of recruitment of anti-reward systems and consequent chronic decreased function of reward circuits, both of which lead to the compulsive drug seeking and loss of control over intake. How these systems are modulated by other known brain emotional systems localized to the basal forebrain, where the ventral striatum and extended amygdala project to convey emotional valence, how frontal cortex dysregulations in the cognitive domain are linked to impairments in executive function to contribute to the dysregulation of the extended amygdala, and how individuals differ at the molecular-genetic level of analysis to convey loading on these circuits remain challenges for future research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Michael Arends and Mellany Santos for their assistance with the preparation of this manuscript. Research was supported by National Institutes of Health grants AA006420, AA020608, AA012602, and AA008459 from the National Institute on Alcohol Abuse and Alcoholism, DA010072, DA004043, DA023597, and DA004398 from the National Institute on Drug Abuse, and DK26741 from the National Institute of Diabetes and Digestive and Kidney Diseases. Research also was supported by the Pearson Center for Alcoholism and Addiction Research. This is publication number 24002 from The Scripps Research Institute.
References
1. Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science (1997) 278:52–8. doi:10.1126/science.278.5335.52
2. Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol (1954) 47:419–27. doi:10.1037/h0058775
3. Koob GF, Winger GD, Meyerhoff JL, Annau Z. Effects of d-amphetamine on concurrent self-stimulation of forebrain and brain stem loci. Brain Res (1977) 137:109–26. doi:10.1016/0006-8993(77)91015-0
4. Simon H, Stinus L, Tassin JP, Lavielle S, Blanc G, Thierry AM, et al. Is the dopaminergic mesocorticolimbic system necessary for intracranial self-stimulation? Biochemical and behavioral studies from A10 cell bodies and terminals. Behav Neural Biol (1979) 27:125–45. doi:10.1016/S0163-1047(79)91745-X
5. Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc (1979) 38:2473–6.
6. Hernandez G, Hamdani S, Rajabi H, Conover K, Stewart J, Arvanitogiannis A, et al. Prolonged rewarding stimulation of the rat medial forebrain bundle: neurochemical and behavioral consequences. Behav Neurosci (2006) 120:888–904. doi:10.1037/0735-7044.120.4.888
7. Garris PA, Kilpatrick M, Bunin MA, Michael D, Walker QD, Wightman RM. Dissociation of dopamine release in the nucleus accumbens from intracranial self-stimulation. Nature (1999) 398:67–9. doi:10.1038/18019
8. Miliaressis E, Emond C, Merali Z. Re-evaluation of the role of dopamine in intracranial self-stimulation using in vivo microdialysis. Behav Brain Res (1991) 46:43–8. doi:10.1016/S0166-4328(05)80095-6
9. Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev (1993) 18:247–91. doi:10.1016/0165-0173(93)90013-P
10. Robbins TW. Relationship between reward-enhancing and stereotypical effects of psychomotor stimulant drugs. Nature (1976) 264:57–9. doi:10.1038/264057a0
11. Miliaressis E, Le Moal M. Stimulation of the medial forebrain bundle: behavioral dissociation of its rewarding and activating effects. Neurosci Lett (1976) 2:295–300. doi:10.1016/0304-3940(76)90163-4
12. Salamone JD, Correa M, Farrar A, Mingote SM. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology (Berl) (2007) 191:461–82. doi:10.1007/s00213-006-0668-9
13. Le Moal M, Simon H. Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev (1991) 71:155–234.
14. Robbins TW, Everitt BJ. A role for mesencephalic dopamine in activation: commentary on Berridge (2006). Psychopharmacology (Berl) (2007) 191:433–7. doi:10.1007/s00213-006-0528-7
15. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology (2001) 24:97–129. doi:10.1016/S0893-133X(00)00195-0
16. Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction (2006) 101:23–30. doi:10.1111/j.1360-0443.2006.01586.x
17. Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci (2005) 8:1445–9. doi:10.1038/nn1578
18. Koob GF. A role for brain stress systems in addiction. Neuron (2008) 59:11–34. doi:10.1016/j.neuron.2008.06.012
19. Sidhpura N, Parsons LH. Endocannabinoid-mediated synaptic plasticity and addiction-related behavior. Neuropharmacology (2011) 61:1070–87. doi:10.1016/j.neuropharm.2011.05.034
20. Heilig M. The NPY system in stress, anxiety and depression. Neuropeptides (2004) 38:213–24. doi:10.1016/j.npep.2004.05.002
21. Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci (2004) 24:3471–9. doi:10.1523/JNEUROSCI.5740-03.2004
22. Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, et al. Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature (2012) 490:402–6. doi:10.1038/nature11436
23. Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Rev (1990) 15:71–100. doi:10.1016/0165-0173(90)90012-D
24. Koob GF, Heinrichs SC, Menzaghi F, Pich EM, Britton KT. Corticotropin releasing factor, stress and behavior. Semin Neurosci (1994) 6:221–9. doi:10.1006/smns.1994.1029
25. Koob GF, Bartfai T, Roberts AJ. The use of molecular genetic approaches in the neuropharmacology of corticotropin-releasing factor. Int J Comp Psychol (2001) 14:90–110.
26. Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev (2001) 53:209–43.
27. Koob GF, Heinrichs SC. A role for corticotropin-releasing factor and urocortin in behavioral responses to stressors. Brain Res (1999) 848:141–52. doi:10.1016/S0006-8993(99)01991-5
28. Spina M, Merlo-Pich E, Chan RKW, Basso AM, Rivier J, Vale W, et al. Appetite-suppressing effects of urocortin, a CRF-related neuropeptide. Science (1996) 273:1561–4. doi:10.1126/science.273.5281.1561
29. Pelleymounter MA, Joppa M, Carmouche M, Cullen MJ, Brown B, Murphy B, et al. Role of corticotropin-releasing factor (CRF) receptors in the anorexic syndrome induced by CRF. J Pharmacol Exp Ther (2000) 293:799–806.
30. Takahashi LK, Ho SP, Livanov V, Graciani N, Arneric SP. Antagonism of CRF2 receptors produces anxiolytic behavior in animal models of anxiety. Brain Res (2001) 902:135–42. doi:10.1016/S0006-8993(01)02405-2
31. Swanson LW, Sawchenko PE, Rivier J, Vale W. The organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology (1983) 36:165–86. doi:10.1159/000123454
32. Turnbull AV, Rivier C. Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc Natl Acad Sci U S A (1997) 215:1–10.
33. Koob GF, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry (2007) 164:1149–59. doi:10.1176/appi.ajp.2007.05030503
34. Heimer L, Alheid G. Piecing together the puzzle of basal forebrain anatomy. In: Napier TC, Kalivas PW, Hanin I editors. The Basal Forebrain: Anatomy to Function (series title: Advances in Experimental Medicine and Biology (Vol. 295), New York: Plenum Press (1991). p. 1–42.
35. Alheid GF, De Olmos JS, Beltramino CA. Amygdala and extended amygdala. In: Paxinos G editor. The Rat Nervous System. San Diego, CA: Academic Press (1995). p. 495–578.
36. Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev (2005) 29:829–41. doi:10.1016/j.neubiorev.2005.03.021
37. Sink KS, Walker DL, Freeman SM, Flandreau EI, Ressler KJ, Davis M. Effects of continuously enhanced corticotropin releasing factor expression within the bed nucleus of the stria terminalis on conditioned and unconditioned anxiety. Mol Psychiatry (2013) 18:308–19. doi:10.1038/mp.2011.188
38. Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist (2004) 10:221–34. doi:10.1177/1073858403261077
39. Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the κ opioid receptor. Science (1982) 215:413–5. doi:10.1126/science.6120570
40. Watson SJ, Khachaturian H, Akil H, Coy DH, Goldstein A. Comparison of the distribution of dynorphin systems and enkephalin systems in brain. Science (1982) 218:1134–6. doi:10.1126/science.6128790
41. Fallon JH, Leslie FM. Distribution of dynorphin and enkephalin peptides in the rat brain. J Comp Neurol (1986) 249:293–336. doi:10.1002/cne.902490302
42. Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther (2007) 116:306–21. doi:10.1016/j.pharmthera.2007.06.011
43. Wee S, Koob GF. The role of the dynorphin-κ opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) (2010) 210:121–35. doi:10.1007/s00213-010-1825-8
44. Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology (Berl) (1985) 86:274–80. doi:10.1007/BF00432213
45. Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by κ opiate receptors. Science (1986) 233:774–6. doi:10.1126/science.3016896
46. Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA Jr. Effects of κ-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology (Berl) (2004) 172:463–70. doi:10.1007/s00213-003-1680-y
47. Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA Jr. Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci (2001) 21:7397–403.
48. Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci (2004) 25:210–8. doi:10.1016/j.tips.2004.02.005
49. Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr., et al. Antidepressant-like effects of κ-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther (2003) 305:323–30. doi:10.1124/jpet.102.046433
50. Knoll AT, Carlezon WA Jr. Dynorphin, stress, and depression. Brain Res (2010) 1314:56–73. doi:10.1016/j.brainres.2009.09.074
51. Hjelmstad GO, Fields HL. Kappa opioid receptor inhibition of glutamatergic transmission in the nucleus accumbens shell. J Neurophysiol (2001) 85:1153–8.
52. Gray AM, Rawls SM, Shippenberg TS, McGinty JF. The kappa-opioid agonist, U-69593, decreases acute amphetamine-evoked behaviors and calcium-dependent dialysate levels of dopamine and glutamate in the ventral striatum. J Neurochem (1999) 73:1066–74. doi:10.1046/j.1471-4159.1999.0731066.x
53. Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A (2006) 103:2938–42. doi:10.1073/pnas.0511159103
54. Tejeda HA. Modulation of Extracellular Dopamine in the Prefrontal Cortex by Local and Ventral Tegmental Area Kappa-Opioid Receptors. Society for Neuroscience Meeting, Chicago (2009). Abstract no: 751.7.
55. Valdez GR, Platt DM, Rowlett JK, Rüedi-Bettschen D, Spealman RD. κ Agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J Pharmacol Exp Ther (2007) 323:525–33. doi:10.1124/jpet.107.125484
56. Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci (2008) 28:407–14. doi:10.1523/JNEUROSCI.4458-07.2008
57. McLaughlin JP, Marton-Popovici M, Chavkin C. κ Opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci (2003) 23:5674–83.
58. Solomon RL, Corbit JD. An opponent-process theory of motivation: 1. Temporal dynamics of affect. Psychol Rev (1974) 81:119–45.
59. McEwen BS. Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology (2000) 22:108–24. doi:10.1016/S0893-133X(99)00129-3
60. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington DC: American Psychiatric Press (1994).
61. Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction: alcohol addiction as a reward deficit disorder. In: Sommer WH, Spanagel R editors. Behavioral Neurobiology of Alcohol Addiction (Series Title: Current Topics in Behavioral Neuroscience (Vol. 13), Berlin: Springer-Verlag (2013). p. 3–30.
62. Khantzian EJ. The self-medication hypothesis of substance use disorders: a reconsideration and recent applications. Harvard Rev Psychiatry (1997) 4:231–44. doi:10.3109/10673229709030550
63. Markou A, Koob GF. Post-cocaine anhedonia: an animal model of cocaine withdrawal. Neuropsychopharmacology (1991) 4:17–26.
64. Paterson NE, Myers C, Markou A. Effects of repeated withdrawal from continuous amphetamine administration on brain reward function in rats. Psychopharmacology (Berl) (2000) 152:440–6. doi:10.1007/s002130000559
65. Schulteis G, Markou A, Gold LH, Stinus L, Koob GF. Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose-response analysis. J Pharmacol Exp Ther (1994) 271:1391–8.
66. Gardner EL, Vorel SR. Cannabinoid transmission and reward-related events. Neurobiol Dis (1998) 5:502–33. doi:10.1006/nbdi.1998.0219
67. Epping-Jordan MP, Watkins SS, Koob GF, Markou A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature (1998) 393:76–9. doi:10.1038/30001
68. Schulteis G, Markou A, Cole M, Koob GF. Decreased brain reward produced by ethanol withdrawal. Proc Natl Acad Sci U S A (1995) 92:5880–4. doi:10.1073/pnas.92.13.5880
69. Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology (2009) 56:18–31. doi:10.1016/j.neuropharm.2008.07.043
70. Kenny PJ, Polis I, Koob GF, Markou A. Low dose cocaine self-administration transiently increases but high dose cocaine persistently decreases brain reward function in rats. Eur J Neurosci (2003) 17:191–5. doi:10.1046/j.1460-9568.2003.02443.x
71. Liu J, Schulteis G. Brain reward deficits accompany naloxone-precipitated withdrawal from acute opioid dependence. Pharmacol Biochem Behav (2004) 79:101–8. doi:10.1016/j.pbb.2004.06.006
72. Schulteis G, Liu J. Brain reward deficits accompany withdrawal (hangover) from acute ethanol in rats. Alcohol (2006) 39:21–8. doi:10.1016/j.alcohol.2006.06.008
73. Ahmed SH, Walker JR, Koob GF. Persistent increase in the motivation to take heroin in rats with a history of drug escalation. Neuropsychopharmacology (2000) 22:413–21. doi:10.1016/S0893-133X(99)00133-5
74. Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science (1998) 282:298–300. doi:10.1126/science.282.5387.298
75. Kitamura O, Wee S, Specio SE, Koob GF, Pulvirenti L. Escalation of methamphetamine self-administration in rats: a dose-effect function. Psychopharmacology (Berl) (2006) 186:48–53. doi:10.1007/s00213-006-0353-z
76. O’Dell LE, Roberts AJ, Smith RT, Koob GF. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res (2004) 28:1676–82. doi:10.1097/01.ALC.0000145781.11923.4E
77. George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH, et al. CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci U S A (2007) 104:17198–203. doi:10.1073/pnas.0707585104
78. Quadros IM, Miczek KA. Two modes of intense cocaine bingeing: increased persistence after social defeat stress and increased rate of intake due to extended access conditions in rats. Psychopharmacology (Berl) (2009) 206:109–20. doi:10.1007/s00213-009-1584-6
79. Vendruscolo L, Schlosburg JE, Misra KK, Chen SA, Greenwell TN, Koob GF. Escalation patterns of varying periods of heroin access. Pharmacol Biochem Behav (2011) 98:570–4. doi:10.1016/j.pbb.2011.03.004
80. Ahmed SH, Koob GF. Long-lasting increase in the set point for cocaine self-administration after escalation in rats. Psychopharmacology (Berl) (1999) 146:303–12. doi:10.1007/s002130051121
81. Deroche V, Le Moal M, Piazza PV. Cocaine self-administration increases the incentive motivational properties of the drug in rats. Eur J Neurosci (1999) 11:2731–6. doi:10.1046/j.1460-9568.1999.00696.x
82. Mantsch JR, Yuferov V, Mathieu-Kia AM, Ho A, Kreek MJ. Effects of extended access to high versus low cocaine doses on self-administration, cocaine-induced reinstatement and brain mRNA levels in rats. Psychopharmacology (Berl) (2004) 175:26–36. doi:10.1007/s00213-004-1778-x
83. Paterson NE, Markou A. Increased motivation for self-administered cocaine after escalated cocaine intake. Neuroreport (2003) 14:2229–32. doi:10.1097/00001756-200312020-00019
84. Wee S, Mandyam CD, Lekic DM, Koob GF. α1-Noradrenergic system role in increased motivation for cocaine intake in rats with prolonged access. Eur Neuropsychopharmacol (2008) 18:303–11. doi:10.1016/j.euroneuro.2007.08.003
85. Walker BM, Koob GF. The γ-aminobutyric acid-B receptor agonist baclofen attenuates responding for ethanol in ethanol-dependent rats. Alcohol Clin Exp Res (2007) 31:11–8. doi:10.1111/j.1530-0277.2006.00259.x
86. Deroche V, Le Moal M, Piazza PV. Cocaine self-administration increases the incentive motivational properties of the drug in rats. Eur J Neurosci (1999) 11:2731–6. doi:10.1046/j.1460-9568.1999.00696.x
87. Jonkman S, Pelloux Y, Everitt BJ. Drug intake is sufficient, but conditioning is not necessary for the emergence of compulsive cocaine seeking after extended self-administration. Neuropsychopharmacology (2012) 37:1612–9. doi:10.1038/npp
88. Ben-Shahar O, Posthumus EJ, Waldroup SA, Ettenberg A. Heightened drug-seeking motivation following extended daily access to self-administered cocaine. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32:863–9. doi:10.1016/j.pnpbp.2008.01.002
89. Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science (2004) 305:1017–9. doi:10.1126/science.1098975
90. Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science (2004) 305:1014–7. doi:10.1126/science.1099020
91. Pelloux Y, Everitt BJ, Dickinson A. Compulsive drug seeking by rats under punishment: effects of drug taking history. Psychopharmacology (Berl) (2007) 194:127–37. doi:10.1007/s00213-007-0805-0
92. Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield T Jr., Logrip ML, et al. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci (2012) 32:7563–71. doi:10.1523/JNEUROSCI.0069-12.2012
93. Vezina P. Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci Biobehav Rev (2004) 27:827–39. doi:10.1016/j.neubiorev.2003.11.001
94. Ben-Shahar O, Ahmed SH, Koob GF, Ettenberg A. The transition from controlled to compulsive drug use is associated with a loss of sensitization. Brain Res (2004) 995:46–54. doi:10.1016/j.brainres.2003.09.053
95. Knackstedt LA, Kalivas PW. Extended access to cocaine self-administration enhances drug-primed reinstatement but not behavioral sensitization. J Pharmacol Exp Ther (2007) 322:1103–9. doi:10.1124/jpet.107.122861
96. Ahmed SH, Cador M. Dissociation of psychomotor sensitization from compulsive cocaine consumption. Neuropsychopharmacology (2006) 31:563–71. doi:10.1038/sj.npp.1300834
97. Ahmed SH, Kenny PJ, Koob GF, Markou A. Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci (2002) 5:625–6.
98. Jang CG, Whitfield T, Schulteis G, Koob GF, Wee S. Sensitization of a negative emotional-like state during repeated withdrawal from extended access methamphetamine self-administration in rats. Psychopharmacology (Berl) (2013) 225:753–63. doi:10.1007/s00213-012-2864-0
99. Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF. Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci (2006) 26:5894–900. doi:10.1523/JNEUROSCI.0740-06.2006
100. Harris AC, Pentel PR, Burroughs D, Staley MD, Lesage MG. A lack of association between severity of nicotine withdrawal and individual differences in compensatory nicotine self-administration in rats. Psychopharmacology (Berl) (2011) 217:153–66. doi:10.1007/s00213-011-2273-9
101. Shurman J, Koob GF, Gutstein HB. Opioids, pain, the brain, and hyperkatifeia: a framework for the rational use of opioids for pain. Pain Med (2010) 11:1092–8. doi:10.1111/j.1526-4637.2010.00881.x
102. Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc Natl Acad Sci U S A (1993) 90:7966–9. doi:10.1073/pnas.90.17.7966
103. Diana M, Pistis M, Muntoni A, Gessa G. Profound decrease of mesolimbic dopaminergic neuronal activity in morphine withdrawn rats. J Phamacol Exp Ther (1995) 272:781–5.
104. Parsons LH, Koob GF, Weiss F. Serotonin dysfunction in the nucleus accumbens of rats during withdrawal after unlimited access to intravenous cocaine. J Pharmacol Exp Ther (1995) 274:1182–91.
105. Volkow ND, Fowler JS, Wang GJ. The addicted human brain: insights from imaging studies. J Clin Invest (2003) 111:1444–51. doi:10.1172/JCI18533
106. Mhatre MC, Pena G, Sieghart W, Ticku MK. Antibodies specific for GABAA receptor alpha subunits reveal that chronic alcohol treatment down-regulates 〈-subunit expression in rat brain regions. J Neurochem (1993) 61:1620–5. doi:10.1111/j.1471-4159.1993.tb09795.x
107. Devaud LL, Fritschy JM, Sieghart W, Morrow AL. Bidirectional alterations of GABAA receptor subunit pepetide levels in rat cortex during chronic ethanol consumption and withdrawal. J Neurochem (1997) 69:126–30. doi:10.1046/j.1471-4159.1997.69010126.x
108. Sapru MK, Diamond I, Gordon AS. Adenosine receptors mediate cellular adaptation to ethanol in NG108-15 cells. J Pharmacol Exp Ther (1994) 271:542–8.
109. Chance WT, Sheriff S, Peng F, Balasubramaniam A. Antagonism of NPY-induced feeding by pretreatment with cyclic AMP response element binding protein antisense oligonucleotide. Neuropeptides (2000) 34:167–72. doi:10.1054/npep.2000.0807
110. Pandey SC. The gene transcription factor cyclic AMP-responsive element binding protein: role in positive and negative affective states of alcohol addiction. Pharmacol Ther (2004) 104:47–58. doi:10.1016/j.pharmthera.2004.08.002
111. Rivier C, Bruhn T, Vale W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: role of corticotropin-releasing factor (CRF). J Pharmacol Exp Ther (1984) 229:127–31.
112. Merlo-Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, et al. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci (1995) 15:5439–47.
113. Rasmussen DD, Boldt BM, Bryant CA, Mitton DR, Larsen SA, Wilkinson CW. Chronic daily ethanol and withdrawal: 1. Long-term changes in the hypothalamo-pituitary-adrenal axis. Alcohol Clin Exp Res (2000) 24:1836–49. doi:10.1111/j.1530-0277.2000.tb01988.x
114. Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav (2002) 72:213–20. doi:10.1016/S0091-3057(01)00748-1
115. Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature (2000) 403:430–4. doi:10.1038/35000212
116. Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry (2010) 67:831–9. doi:10.1016/j.biopsych.2009.11.007
117. Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G. Brain corticotropin-releasing factor mediates “anxiety-like” behavior induced by cocaine withdrawal in rats. Brain Res (1995) 675:89–97. doi:10.1016/0006-8993(95)00043-P
118. Basso AM, Spina M, Rivier J, Vale W, Koob GF. Corticotropin-releasing factor antagonist attenuates the “anxiogenic-like” effect in the defensive burying paradigm but not in the elevated plus-maze following chronic cocaine in rats. Psychopharmacology (Berl) (1999) 145:21–30. doi:10.1007/s002130051028
119. Navarro-Zaragoza J, Núñez C, Laorden ML, Milanés MV. Effects of corticotropin-releasing factor receptor-1 antagonists on the brain stress system responses to morphine withdrawal. Mol Pharmacol (2010) 77:864–73. doi:10.1124/mol.109.062463
120. Iredale PA, Alvaro JD, Lee Y, Terwilliger R, Chen YL, Duman RS. Role of corticotropin-releasing factor receptor-1 in opiate withdrawal. J Neurochem (2000) 74:199–208. doi:10.1046/j.1471-4159.2000.0740199.x
121. Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl) (1991) 103:227–32. doi:10.1007/BF02244208
122. Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol (2004) 32:101–11. doi:10.1016/j.alcohol.2003.08.007
123. Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav (2004) 77:405–13. doi:10.1016/j.pbb.2003.11.010
124. Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry (2007) 61:78–86. doi:10.1016/j.biopsych.2006.03.063
125. Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res (1993) 605:25–32. doi:10.1016/0006-8993(93)91352-S
126. Tucci S, Cheeta S, Seth P, File SE. Corticotropin releasing factor antagonist, α-helical CRF9-41, reverses nicotine-induced conditioned, but not unconditioned, anxiety. Psychopharmacology (Berl) (2003) 167:251–6.
127. Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology (2005) 30:1662–9. doi:10.1038/sj.npp.1300706
128. Valdez GR, Zorrilla EP, Roberts AJ, Koob GF. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted ethanol abstinence. Alcohol (2003) 29:55–60. doi:10.1016/S0741-8329(03)00020-X
129. Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M, et al. Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. J Pharmacol Exp Ther (2010) 332:298–307. doi:10.1124/jpet.109.159186
130. Overstreet DH, Knapp DJ, Breese GR. Drug challenges reveal differences in mediation of stress facilitation of voluntary alcohol drinking and withdrawal-induced anxiety in alcohol-preferring P rats. Alcohol Clin Exp Res (2007) 31:1473–81. doi:10.1111/j.1530-0277.2007.00445.x
131. Wills TA, Knapp DJ, Overstreet DH, Breese GR. Sensitization, duration, and pharmacological blockade of anxiety-like behavior following repeated ethanol withdrawal in adolescent and adult rats. Alcohol Clin Exp Res (2009) 33:455–63. doi:10.1111/j.1530-0277.2008.00856.x
132. Hand TH, Koob GF, Stinus L, Le Moal M. Aversive properties of opiate receptor blockade: evidence for exclusively central mediation in naive and morphine-dependent rats. Brain Res (1988) 474:364–8. doi:10.1016/0006-8993(88)90452-0
133. Stinus L, Le Moal M, Koob GF. Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience (1990) 37:767–73. doi:10.1016/0306-4522(90)90106-E
134. Stinus L, Cador M, Zorrilla EP, Koob GF. Buprenorphine and a CRF1 antagonist block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology (2005) 30:90–8. doi:10.1038/sj.npp.1300487
135. Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L. Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol (1995) 6:74–80. doi:10.1097/00008877-199501000-00011
136. Contarino A, Papaleo F. The corticotropin-releasing factor receptor-1 pathway mediates the negative affective states of opiate withdrawal. Proc Natl Acad Sci U S A (2005) 102:18649–54. doi:10.1073/pnas.0506999102
137. Bruijnzeel AW, Zislis G, Wilson C, Gold MS. Antagonism of CRF receptors prevents the deficit in brain reward function associated with precipitated nicotine withdrawal in rats. Neuropsychopharmacology (2007) 32:955–63. doi:10.1038/sj.npp.1301192
138. Bruijnzeel AW, Small E, Pasek TM, Yamada H. Corticotropin-releasing factor mediates the dysphoria-like state associated with alcohol withdrawal in rats. Behav Brain Res (2010) 210:288–91. doi:10.1016/j.bbr.2010.02.043
139. Marcinkiewcz CA, Prado MM, Isaac SK, Marshall A, Rylkova D, Bruijnzeel AW. Corticotropin-releasing factor within the central nucleus of the amygdala and the nucleus accumbens shell mediates the negative affective state of nicotine withdrawal in rats. Neuropsychopharmacology (2009) 34:1743–52. doi:10.1038/npp.2008.231
140. Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA. Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology (2012) 62:1167–76. doi:10.1016/j.neuropharm.2011.06.014
141. Schindler AG, Li S, Chavkin C. Behavioral stress may increase the rewarding valence of cocaine-associated cues through a dynorphin/kappa-opioid receptor-mediated mechanism without affecting associative learning or memory retrieval mechanisms. Neuropsychopharmacology (2010) 35:1932–42. doi:10.1038/npp
142. Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, et al. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A (2009) 106:19168–73. doi:10.1073/pnas.0910705106
143. Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) (2008) 200:59–70. doi:10.1007/s00213-008-1122-y
144. McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C. Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology (2006) 31:1241–8.
145. Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA Jr. Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther (2007) 323:838–45. doi:10.1124/jpet.107.127415
146. Specio SE, Wee S, O’Dell LE, Boutrel B, Zorrilla EP, Koob GF. CRF1 receptor antagonists attenuate escalated cocaine self-administration in rats. Psychopharmacology (Berl) (2008) 196:473–82. doi:10.1007/s00213-007-0983-9
147. Greenwell TN, Funk CK, Cottone P, Richardson HN, Chen SA, Rice K, et al. Corticotropin-releasing factor-1 receptor antagonists decrease heroin self-administration in long-, but not short-access rats. Addict Biol (2009) 14:130–43. doi:10.1111/j.1369-1600.2008.00142.x
148. Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J (2002) 16:27–35. doi:10.1096/fj.01-0593com
149. Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, et al. Increased ethanol self-administration and anxiety-like behavior during acute withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res (2002) 26:1494–501. doi:10.1111/j.1530-0277.2002.tb02448.x
150. Gehlert DR, Cippitelli A, Thorsell A, Le AD, Hipskind PA, Hamdouchi C, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci (2007) 27:2718–26. doi:10.1523/JNEUROSCI.4985-06.2007
151. Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci (2006) 26:11324–32. doi:10.1523/JNEUROSCI.3096-06.2006
152. Wee S, Orio L, Ghirmai S, Cashman JR, Koob GF. Inhibition of kappa opioid receptors attenuated increased cocaine intake in rats with extended access to cocaine. Psychopharmacology (Berl) (2009) 205:565–75. doi:10.1007/s00213-009-1563-y
153. Walker BM, Zorrilla EP, Koob GF. Systemic κ-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol (2010) 16:116–9. doi:10.1111/j.1369-1600.2010.00226.x
154. Whitfield TW Jr., Wee S, Gould A, Schlosburg J, Vendruscolo L, Koob GF. Kappa Receptor Activation Underlies Compulsive Methamphetamine Intake. Society for Neuroscience Meeting Abstracts, Washington DC (2011). Abstract no: 797.19.
155. Schlosburg JE, Vendruscolo LF, Park PE, Whitfield TW Jr., Koob GF. Long-Term Antagonism of Kappa Opioid Receptors Prevents Escalation of, and Increased Motivation for, Heroin Intake. Society for Neuroscience Meeting Abstracts, Washington DC (2011). Abstract no: 16.07.
156. Nealey KA, Smith AW, Davis SM, Smith DG, Walker BM. Kappa-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol- dependent rats. Neuropharmacology (2011) 61:35–42. doi:10.1016/j.neuropharm.2011.02.012
157. Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci (2007) 30:399–406. doi:10.1016/j.tins.2007.06.006
158. Thorsell A, Slawecki CJ, Ehlers CL. Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake in Wistar rats: interaction with chronic ethanol exposure. Behav Brain Res (2005) 161:133–40. doi:10.1016/j.bbr.2005.01.016
159. Thorsell A, Slawecki CJ, Ehlers CL. Effects of neuropeptide Y on appetitive and consummatory behaviors associated with alcohol drinking in wistar rats with a history of ethanol exposure. Alcohol Clin Exp Res (2005) 29:584–90. doi:10.1097/01.ALC.0000160084.13148.02
160. Gilpin NW, Stewart RB, Murphy JM, Li TK, Badia-Elder NE. Neuropeptide Y reduces oral ethanol intake in alcohol-preferring (P) rats following a period of imposed ethanol abstinence. Alcohol Clin Exp Res (2003) 27:787–94. doi:10.1097/01.ALC.0000065723.93234.1D
161. Gilpin NW, Misra K, Koob GF. Neuropeptide Y in the central nucleus of the amygdala suppresses dependence-induced increases in alcohol drinking. Pharmacol Biochem Behav (2008) 90:475–80. doi:10.1016/j.pbb.2008.04.006
162. Thorsell A, Rapunte-Canonigo V, O’Dell L, Chen SA, King A, Lekic D, et al. Viral vector-induced amygdala NPY overexpression reverses increased alcohol intake caused by repeated deprivations in Wistar rats. Brain (2007) 130:1330–7. doi:10.1093/brain/awm033
163. Gilpin NW, Misra K, Herman MA, Cruz MT, Koob GF, Roberto M. Neuropeptide Y opposes alcohol effects on gamma-aminobutyric acid release in amygdala and blocks the transition to alcohol dependence. Biol Psychiatry (2011) 69:1091–9. doi:10.1016/j.biopsych.2011.02.004
164. Sørensen G, Wortwein G, Fink-Jensen A, Woldbye DPD. Neuropeptide Y Y5 receptor antagonism causes faster extinction and attenuates reinstatement in cocaine-induced place preference. Pharmacol Biochem Behav (2013) 105:151–6. doi:10.1016/j.pbb.2013.02.010
165. Sørensen G, Jensen M, Weikop P, Dencker D, Christiansen SH, Loland CJ, et al. Neuropeptide Y Y5 receptor antagonism attenuates cocaine-induced effects in mice. Psychopharmacology (Berl) (2012) 222:565–77. doi:10.1007/s00213-012-2651-y
166. Sørensen G, Woldbye DP. Mice lacking neuropeptide Y show increased sensitivity to cocaine. Synapse (2012) 66:840–3. doi:10.1002/syn.21568
167. Maric T, Tobin S, Quinn T, Shalev U. Food deprivation-like effects of neuropeptide Y on heroin self-administration and reinstatement of heroin seeking in rats. Behav Brain Res (2008) 194:39–43. doi:10.1016/j.bbr.2008.06.023
168. Maric T, Cantor A, Cuccioletta H, Tobin S, Shalev U. Neuropeptide Y augments cocaine self-administration and cocaine-induced hyperlocomotion in rats. Peptides (2009) 30:721–6. doi:10.1016/j.peptides.2008.11.006
169. Aydin C, Oztan O, Isgor C. Effects of a selective Y2R antagonist, JNJ-31020028, on nicotine abstinence-related social anxiety-like behavior, neuropeptide Y and corticotropin releasing factor mRNA levels in the novelty-seeking phenotype. Behav Brain Res (2011) 222:332–41. doi:10.1016/j.bbr.2011.03.067
170. Rylkova D, Boissoneault J, Isaac S, Prado M, Shah HP, Bruijnzeel AW. Effects of NPY and the specific Y1 receptor agonist [D-His26]-NPY on the deficit in brain reward function and somatic signs associated with nicotine withdrawal in rats. Neuropeptides (2008) 42:215–27. doi:10.1016/j.npep.2008.03.004
171. Valdez GR, Koob GF. Allostasis and dysregulation of corticotropin-releasing factor and Neuropeptide Y systems: implications for the development of alcoholism. Pharmacol Biochem Behav (2004) 79:671–89. doi:10.1016/j.pbb.2004.09.020
172. George O, Mandyam CD, Wee S, Koob GF. Extended access to cocaine self-administration produces long-lasting prefrontal cortex-dependent working memory impairments. Neuropsychopharmacology (2008) 33:2474–82. doi:10.1038/sj.npp.1301626
173. Briand LA, Flagel SB, Garcia-Fuster MJ, Watson SJ, Akil H, Sarter M, et al. Persistent alterations in cognitive function and prefrontal dopamine D2 receptors following extended, but not limited, access to self-administered cocaine. Neuropsychopharmacology (2008) 33:2969–80. doi:10.1038/npp.2008.18
174. Briand LA, Gross JP, Robinson TE. Impaired object recognition following prolonged withdrawal from extended-access cocaine self-administration. Neuroscience (2008) 155:1–6. doi:10.1016/j.neuroscience.2008.06.004
175. George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, et al. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A (2012) 109:18156–61. doi:10.1073/pnas.1116523109
176. Boyson CO, Miguel TT, Quadros IM, Debold JF, Miczek KA. Prevention of social stress-escalated cocaine self-administration by CRF-R1 antagonist in the rat VTA. Psychopharmacology (Berl) (2011) 218:257–69. doi:10.1007/s00213-011-2266-8
177. Carlezon WA Jr., Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, et al. Regulation of cocaine reward by CREB. Science (1998) 282:2272–5. doi:10.1126/science.282.5397.2272
178. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science (1988) 242:715–23. doi:10.1126/science.2903550
179. Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci (2001) 2:119–28. doi:10.1038/35053570
180. Koob GF. Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur Neuropsychopharmacology (2003) 13:442–52. doi:10.1016/j.euroneuro.2003.08.005
181. Aston-Jones G, Delfs JM, Druhan J, Zhu Y. The bed nucleus of the stria terminalis: a target site for noradrenergic actions in opiate withdrawal. In: McGinty JF editor. Advancing from the Ventral Striatum to the Extended Amygdala: Implications for Neuropsychiatry and Drug Abuse (series title: Annals of the New York Academy of Sciences (Vol. 877), New York: New York Academy of Sciences (1999). p. 486–98.
182. Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature (2001) 411:583–7. doi:10.1038/35079077
183. Edwards S, Koob GF. Neurobiology of dysregulated motivational systems in drug addiction. Future Neurol (2010) 5:393–410. doi:10.2217/fnl.10.14
184. Solomon RL. The opponent-process theory of acquired motivation: the costs of pleasure and the benefits of pain. Am Psychol (1980) 35:691–712. doi:10.1037/0003-066X.35.8.691
185. Cohen A, Koob GF, George O. Robust escalation of nicotine intake with extended access to nicotine self-administration and intermittent periods of abstinence. Neuropsychopharmacology (2012) 37:2153–60. doi:10.1038/npp.2012.67
186. Edwards S, Guerrero M, Ghoneim OM, Roberts E, Koob GF. Evidence that vasopressin V1b receptors mediate the transition to excessive drinking in ethanol-dependent rats. Addict Biol (2011) 17:76–85. doi:10.1111/j.1369-1600.2010.00291.x
187. Barbier E, Vendruscolo LF, Schlosburg JE, Edwards S, Juergens N, Park PE, et al. The NK1 receptor antagonist L822429 reduces heroin reinforcement. Neuropsychopharmacology (2013). doi:10.1038/npp.2012.261 (in press)
188. Wee S, Wang Z, Woolverton WL, Pulvirenti L, Koob GF. Effect of aripiprazole, a partial D2 receptor agonist, on increased rate of methamphetamine self-administration in rats with prolonged access. Neuropsychopharmacology (2007) 32:2238–47. doi:10.1038/sj.npp.1301353
189. Liu ZH, Jin WQ. Decrease of ventral tegmental area dopamine neuronal activity in nicotine withdrawal rats. Neuroreport (2004) 15:1479–81. doi:10.1097/01.wnr.0000126218.25235.b6
190. Richardson HN, Zhao Y, Fekete EM, Funk CK, Wirsching P, Janda KD, et al. MPZP: a novel small molecule corticotropin-releasing factor type 1 receptor (CRF1) antagonist. Pharmacol Biochem Behav (2008) 88:497–510. doi:10.1016/j.pbb.2007.10.008
Keywords: opponent process, extended amygdala, corticotropin-releasing factor, dynorphin, reward, compulsive, withdrawal, prefrontal cortex
Citation: Koob GF (2013) Addiction is a reward deficit and stress surfeit disorder. Front. Psychiatry 4:72. doi: 10.3389/fpsyt.2013.00072
Received: 23 April 2013; Accepted: 08 July 2013;
Published online: 01 August 2013.
Edited by:
Nicholas W. Gilpin, LSUHSC-New Orleans, USAReviewed by:
Benjamin Boutrel, Lausanne University Hospital, SwitzerlandGlenn Valdez, Grand Valley State University, USA
Copyright: © 2013 Koob. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George F. Koob, Committee on the Neurobiology of Addictive Disorders, The Scripps Research Institute, 10550 North Torrey Pines Road, SP30-2400, La Jolla, CA 92037, USA e-mail:Z2tvb2JAc2NyaXBwcy5lZHU=