 Oliver Gruber
Oliver Gruber Antonella Chadha Santuccione
Antonella Chadha Santuccione Helmut Aach
Helmut Aach- 1Center for Translational Research in Systems Neuroscience and Psychiatry, Clinic for Psychiatry and Psychotherapy, University Medical Center Göttingen, Göttingen, Germany
- 2Medical Affairs – Psychiatry, Roche Pharma AG, Grenzach-Wyhlen, Germany
Schizophrenia is characterized by positive, negative, and cognitive symptoms. While positive symptoms occur periodically during psychotic exacerbations, negative and cognitive symptoms often emerge before the first psychotic episode and persist with low functional outcome and poor prognosis. This review article outlines the importance of modern functional magnetic resonance imaging techniques for developing a stratified therapy of schizophrenic disorders. Functional neuroimaging evidence on the neural correlates of positive and particularly negative symptoms and cognitive deficits in schizophrenic disorders is briefly reviewed. Acute dysregulation of dopaminergic neurotransmission is crucially involved in the occurrence of psychotic symptoms. However, increasing evidence also implicates glutamatergic pathomechanisms, in particular N-methyl-d-aspartate (NMDA) receptor dysfunction in the pathogenesis of schizophrenia and in the appearance of negative symptoms and cognitive dysfunctions. In line with this notion, several gene variants affecting the NMDA receptor’s pathway have been reported to increase susceptibility for schizophrenia, and have been investigated using the imaging genetics approach. In recent years, several attempts have been made to develop medications modulating the glutamatergic pathway with modest evidences for efficacy. The most successful approaches were those that aimed at influencing this pathway using compounds that enhance NMDA receptor function. More recently, the selective glycine reuptake inhibitor bitopertin has been shown to improve NMDA receptor hypofunction by increasing glycine concentrations in the synaptic cleft. Further research is required to test whether pharmacological agents with effects on the glutamatergic system can help to improve the treatment of negative symptoms in schizophrenic disorders.
Neuroimaging techniques have been developed as important tools to investigate brain dysfunctions that underlie psychiatric disorders. In particular, modern functional magnetic resonance imaging (fMRI) holds the promise to provide neurofunctional biomarkers for improved diagnosis, prognosis, and optimized treatment of mental disorders [e.g., Ref. (1–4)]. In this brief, not exhaustive review, we will exemplify this translational neuroimaging research by focusing on schizophrenia and current challenges to advancing therapeutic approaches for this heterogeneous diagnostic category. First, the importance of negative symptoms and cognitive deficits for successful treatment of schizophrenic disorders will be highlighted, and an overview will be given on the neural correlates of these symptom domains as revealed by fMRI studies. Second, neuroimaging studies of glutamate levels and genetic risk factors pointing to an important role of glutamate–dopamine interactions in the pathophysiology of schizophrenic disorders will be briefly reviewed and will be related to recent findings of pharmacological and animal studies. In the final part of this review, we will present current approaches to develop therapeutic strategies that target the glutamatergic pathway in schizophrenic disorders.
“Psychopathophysiology” of Schizophrenic Disorders and the Importance of Negative Symptoms and Cognitive Deficits
In traditional psychiatry, mental disorders are diagnosed and classified on the basis of more or less specific psychopathological symptoms in the course of the disorder. Numerous recent findings from modern systems neuroscience and molecular neuroscience strongly suggest that diagnoses made on the basis of psychopathological criteria do not represent “natural disease entities” in the sense of diseases with uniform pathogenesis and pathology [see Ref. (1, 3–8)]. Therefore, schizophrenia represents a group of mental disorders that share more or less characteristic psychopathological symptoms, such as verbal hallucinations, delusions, and formal thought disorders.
During the last two decades, modern brain imaging techniques have allowed for the first time scientific investigations into the neural correlates of different symptom dimensions that characterize schizophrenic disorders. These studies of pathophysiological processes that underlie different psychopathological symptoms and syndromes (an approach that may be termed “psychopathophysiology”) are briefly reviewed in the following.
Positive symptoms of schizophrenic disorders such as verbal hallucinations (“hearing voices”) and delusional symptoms are certainly the most impressive characteristics of schizophrenic disorders. Nevertheless, much evidence has been provided that negative symptoms and cognitive disturbances are more strongly associated with the long-term functional outcome of patients suffering from schizophrenia than positive symptoms (9–14). Cognitive deficits are even present at first episode and remain relatively constant over the course of illness (15–17). In contrast to the dopamine model of schizophrenia, glutamatergic theories of schizophrenia account for negative symptoms and cognitive dysfunction as well, and may therefore lead to new treatment approaches specifically targeting the unmet medical need to improve negative symptomatology and cognitive deficits (18).
Neural Correlates of Positive Symptoms in Schizophrenic Disorders
The positive symptoms of schizophrenic disorders particularly include auditory–verbal hallucinations (“hearing voices”) and delusional symptoms like paranoia. As regards auditory–verbal hallucinations, a number of functional brain imaging studies have been performed in the past. Overall, findings of these studies appear quite heterogeneous. However, a finding that has been replicated several times is overactivation of the superior temporal gyrus during experimental phases in which the patients exhibited the symptom of hearing voices [e.g., Ref. (19–21)]. In most of these studies, this activation of auditory association cortices was associated with additional activations in other brain regions. For instance, some of these studies also reported an increased brain activity in Broca’s area, the anterior cingulate cortex, the hippocampus, and the amygdala [e.g., Ref. (20)]. Some of these findings have also been confirmed on the meta-analytical level. Jardri et al. (22) confirmed that phases of “hearing voices” are associated with increased activity in Broca’s area, anterior insula, precentral gyrus, frontal opercular cortex, middle and superior temporal gyrus, inferior parietal lobule as well as in hippocampus and parahippocampal gyrus. It has to be noted, however, that patients suffering from this kind of intermittently occurring auditory–verbal hallucinations are probably not representative for most types of schizophrenic disorders in which the hallucinations persist for longer time periods. A second meta-analysis by Kühn and Gallinat (23) came to the conclusion that a current psychopathological state of experiencing auditory–verbal hallucinations may be associated with abnormal activation of brain regions that are also involved in speech production (e.g., Broca’s area), whereas a subgroup of schizophrenic patients that exhibits the symptom of auditory–verbal hallucinations (in comparison to a subgroup without “life-time diagnosis” of auditory–verbal hallucinations) may be particularly characterized by abnormal brain activation in areas involved in speech processing and, more generally, the processing of auditory stimuli. In another, qualitative and quantitative review, Goghari et al. (24) showed an association between positive symptoms, in particular ideas of persecution, with the activity in medial prefrontal cortex, amygdala, hippocampus, and parahippocampal gyrus.
Neural Correlates of Negative Symptoms in Schizophrenic Disorders
Negative symptoms of schizophrenic disorders are usually defined as symptoms representing a qualitative and/or quantitative reduction of mental capacities or qualities of experience. This class of symptoms is relatively heterogeneous and traditionally includes the five “A”s, which are affective flattening, apathy (reduced drive), anhedonia, asociality (social withdrawal), and alogia (impoverishment of thought) [e.g., Ref. (25)]. Early studies on the structural correlates of negative symptoms in schizophrenic disorders have shown gray matter reduction in temporal, medial frontal, insular, and hippocampal regions [e.g., Ref. (26)]. On the other hand, evidence on brain structural correlates of negative symptoms is very heterogeneous as there are also several studies that failed to find any correlation of brain volumes to negative symptoms in schizophrenia [e.g., Ref. (27–30)].
The development of functional neuroimaging techniques like PET and fMRI led to an increasing number of studies on the neurofunctional correlates of negative symptoms. Several studies have shown a reduced activation of the prefrontal cortex in schizophrenic patients with negative symptoms (25, 31–33). This principal finding of “hypofrontality” associated with negative symptoms has been confirmed by later studies though for different subregions of the prefrontal cortex. For example, in a study using memory retrieval tasks, Heckers et al. (34) found a significantly reduced recruitment of left frontal cortex (Brodmann area 44/9) in schizophrenic patients with deficit syndrome (i.e., patients with negative symptoms as primary and enduring features) as compared to both schizophrenic patients without deficit syndrome and healthy controls. Using auditory working memory tasks (n-back tasks), Menon et al. (35) found an inverse correlation of negative symptoms with activation in the frontal opercular cortex and in the right DLPFC. In contrast to that, another experiment using the n-back task (36) reported a correlation of activation deficits in the DLPFC with disorganization symptoms rather than with negative symptoms.
A number of functional neuroimaging studies have also reported associations of negative symptoms with activation in other brain regions including temporal cortices and the ventral striatum. For instance, Tamminga et al. (37) found both limbic system abnormalities and neocortical alterations associated with the deficit syndrome. A later series of studies have demonstrated a significant correlation of activation in temporal cortices with negative symptoms (38–40). Using the monetary incentive delay task, which activates the reward system, Juckel et al. (41) found a correlation of diminished ventral striatal activation with negative symptoms in schizophrenic patients. In another study by Simon et al. (42), a ventral striatal activation during reward anticipation was negatively correlated with symptoms of apathy, while activation during receipt of reward was negatively correlated with severity of depressive symptoms. Finally, in a recent systematic review and meta-analysis of 25 fMRI studies on schizophrenic symptomatology, Goghari and colleagues (24) have confirmed a relationship of negative symptoms with the functioning of the ventrolateral prefrontal cortex and the ventral striatum.
Neural Correlates of Cognitive Dysfunctions in Schizophrenic Disorders
Traditionally, psychiatric diagnosis of schizophrenic disorders is based on psychopathological, particularly positive and negative symptoms. Over the last few decades, the advent of modern experimental neuropsychological and functional neuroimaging techniques has switched the focus of interest toward brain dysfunctions in the domains of cognitive, emotional, and motivational processes that are highly prevalent in schizophrenic disorders. Especially cognitive deficits are of major interest as they have been proposed to represent core deficits of schizophrenic disorders [e.g., Ref. (43, 44)]. These core cognitive deficits of schizophrenic disorders include deficits in working memory, executive functions, episodic memory, and social cognitions.
Deficits in working memory in schizophrenic disorders have been found to be associated with dysfunctions of prefrontal cortices, especially of the dorsolateral prefrontal cortex, of the deep fronto-opercular cortex, and of the anterior cingulate cortex [e.g., Ref. (45–50)]. In the last few years, there have also been several reports of a disturbed connectivity between these prefrontal areas and the medial temporal lobe, particularly the hippocampus [e.g., Ref. (51, 52)].
Executive function is a construct that encompasses a variety of sub-functions [see for example Ref. (53, 54)], among them selective attention, background monitoring of the environment for potentially significant sensory events [e.g., Ref. (55)], and the adaptation of behavior to changing environmental conditions [e.g., Ref. (56–60)]. Patients with schizophrenic disorders exhibit multiple dysfunctions in these areas of executive control mechanisms that are associated with reduced activity in the posterior frontal medial cortex and the inferior frontal junction area (IFJA; a cortical subregion at the intersection of the precentral sulcus and the inferior frontal sulcus) (61, 62) as well as with abnormally increased activations in brain stem nuclei and the ventral striatum (63).
Episodic memory deficits in schizophrenic disorders have been found to be associated firstly with dysfunction of the extended hippocampal formation (including the hippocampus proper and the surrounding medial temporal structures) [e.g., Ref. (64)], and secondly with dysfunctions of prefrontal cognitive control mechanisms that are involved in encoding and retrieval processes (65).
Disturbed social cognitions in schizophrenic disorders include particularly impaired recognition of facial emotional expressions and reduced theory-of-mind capacities. Neuroimaging studies have revealed that deficits in these domains of social cognitions are associated, firstly, with reduced activation in the amygdala and the fusiform gyrus (66) and, secondly, with reduced activity in the fronto-median cortex, in the temporo-parietal junction cortex, and the amygdala–hippocampus complex [e.g., Ref. (67)]. Disturbed functional coupling between prefrontal areas and the amygdala has also been reported as a correlate of impaired emotional regulation mechanisms (68).
Taken together, neuroimaging studies on the neural correlates of different (positive, negative, cognitive) symptom domains in schizophrenic disorders suggest the involvement of different lateral and medial prefrontal, temporal (particularly including hippocampus and amygdala), and subcortical (in particular ventral striatum as part of the dopaminergic reward system) brain regions in the occurrence of these symptoms.
Pathogenesis, Pathophysiology, and Treatment of Schizophrenic Disorders: The Role of Glutamate–Dopamine Interactions
It is well-established that acute dysregulation of dopaminergic (and glutamatergic) neurotransmission is crucially involved in the occurrence of psychotic symptoms, whereas more chronic cellular neuropathology may be responsible for the development of cognitive deficits and negative symptoms (69). In more recent years, classical elements of the dopamine hypothesis such as the overactive mesolimbic dopamine system and a reduced mesocortical dopamine turnover are considered rather as a “final common pathway” (70), i.e., as the expression of upstream pathophysiological changes. In particular, it is postulated that N-methyl-d-aspartate (NMDA) receptor dysfunction may lead to dopaminergic dysregulation in schizophrenic disorders through a complex interaction between glutamatergic and dopaminergic, but also GABA-ergic mechanisms. Schwartz and co-workers (71), for example, explain both positive and negative symptoms of schizophrenia with dysfunctions of NMDA-glutamatergic synapses. Via effects on various complex circuits including GABA-ergic interneurons, these dysfunctions ultimately result both in hyperfunction of the mesolimbic dopamine system leading to positive symptoms, and also in hypofunction of the mesocortical dopamine system associated with negative symptoms and cognitive dysfunctions (Figure 1). Such pathomechanisms in functional interactions between prefrontal cortices and brain stem nuclei, in particular the ventral tegmental area (VTA), the striatum, and the thalamus, may be influenced by predisposing and/or protective genetic variants that exert effects on the glutamatergic synapse. Thus, the glutamatergic hypothesis is also compatible with our knowledge about the effects of susceptibility genes of schizophrenia (see below).
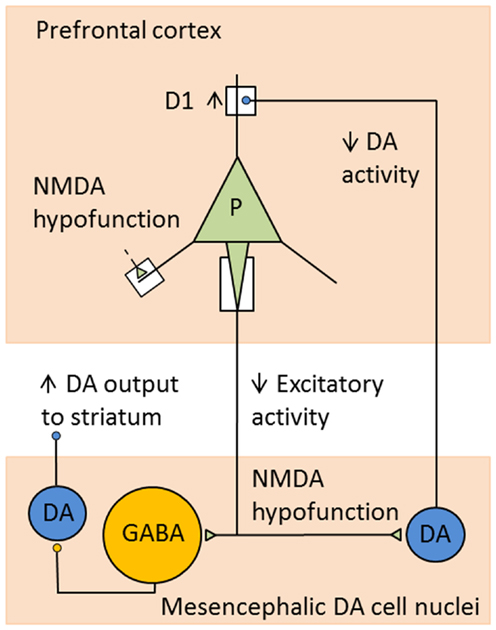
Figure 1. Example for consequences of NMDA receptor hypofunction in glutamatergic–dopaminergic circuits. Hypofunction of NMDA receptors mediating excitatory inputs to prefrontal pyramidal cells in schizophrenia leads to decreased activity in cortical excitatory projections to mesencephalic DA cell nuclei. This results in decreased activity of DA neurons projecting to the DLPFC and increased activity of DA cells projecting to the striatum, as a consequence of decreased stimulation of GABA interneurons. Reduced DA levels in DLPFC lead to compensatory, but functionally insufficient, upregulation of D1 receptors [adapted from Lewis and Gonzalez-Burgos (72)].
Modern pharmacological and animal research approaches are increasingly focusing on the interactions between the dopaminergic and glutamatergic system, especially within fronto-striato-thalamo-frontal loops and in the interactions between frontal cortex, hippocampus, nucleus accumbens, and VTA. Within and between these brain regions, the dopamine and the glutamate system interact in a very complex way, and dysfunctions in these interactions are a central pathomechanistic explanation for the development of schizophrenic disorders [e.g., Ref. (73, 74)]. Particularly, the hippocampus plays a major role in regulating the dopaminergic reward system. Animal studies have shown that increased activity of the ventral hippocampus (subiculum) leads to increased dopamine turnover in the ventral striatum (nucleus accumbens) (75, 76). Other recent findings suggest that this glutamatergically mediated effect of the subiculum on the nucleus accumbens further leads to increased GABA-ergic projection onto the ventral pallidum with reduced tonic activity of the pallidum and consecutive disinhibition of dopamine neurons in the VTA (77). In this way, hyperactivity of the ventral hippocampus observed in schizophrenic disorders could lead to overstimulation and hyperactivity of dopaminergic VTA neurons which may account for various symptoms, in particular for delusional phenomena. Further support for this theory is provided by studies using the MAM animal model of schizophrenia which have shown that the relevant pathophysiological changes such as VTA hyperactivity and increased response to amphetamines are no longer present after inactivation of the subiculum. This indicates that the subiculum is necessary to induce hyperdopaminergic states in this animal model (78).
The pathophysiological role of glutamatergic disbalances has also been investigated in vivo in patients with schizophrenia, for example using magnetic resonance spectroscopy (MRS). In very recent publications (79, 80), findings of such glutamatergic proton magnetic resonance spectroscopic imaging studies have been nicely reviewed, particularly with respect to their implications for drug discovery. Overall, neuroimaging studies support the current glutamate model of schizophrenia by suggesting a hypofunction of the NMDA receptor. In particular, proton magnetic resonance spectroscopic (1H-MRS) studies have provided evidence for altered levels of glutamate and glutamine in the medial prefrontal cortex and in the basal ganglia in early-stage, drug-naïve, or drug-free schizophrenia patients.
Some studies with unmedicated patients with schizophrenia have reported elevated glutamatergic levels in the medial prefrontal cortex as compared to healthy controls (81–84). More precisely, a recent meta-analysis by Marsman and colleagues (79) indicated that it is glutamine which is increased in the frontal cortex in schizophrenic patients, whereas glutamate is reduced. Such an elevated glutamine/glutamate ration may result from either a deficiency in glutaminase, which converts glutamine into glutamate, or from NMDA receptor hypofunction which has also been shown to increase glutamine levels and decrease glutamate levels (79). Further, glutamate levels in the medial prefrontal cortex have been found to be associated with negative symptoms and worse global functioning and to be decreased in remitted patients as compared to non-remitted patients (85). Consistent with that, most studies comparing medicated patients with healthy control subjects reported unchanged glutamate levels in the medial prefrontal cortex (81, 86–92). The meta-analysis by Marsman and colleagues (79) provided additional support for a progressive decrease of frontal glutamate and glutamine in patients with schizophrenia possibly indicating a progressive loss of synaptic activity. Finally, particularly in first episodes schizophrenic patients, increased glutamatergic levels have also been reported in the basal ganglia (93–95), and they appear to decrease to normal levels during antipsychotic treatment with risperidone (94).
Over the last 10–15 years, numerous potential susceptibility genes of schizophrenia have been identified, among them COMT, dysbindin-1, neuregulin-1, RGS4, GRM3, and DISC1. Many of these candidate genes have been shown to influence dopaminergic and/or glutamatergic neurotransmission, and effects on neuroplastic processes and particularly on synaptogenesis have also been reported. Imaging genetics is still a relatively novel approach that, however, has already made substantial contributions to our knowledge about genetic effects on brain structure and function. Early studies, for example, demonstrated the influence of variants in the COMT gene on working memory-related prefrontal activation (96) and on the functional interplay between dopamine synthesis in the midbrain and prefrontal function (97). Although the evidence for an association between the COMT gene and schizophrenia is not unequivocal, these findings nevertheless have high biological plausibility insofar as the influence of the COMT gene on the dopaminergic tone in the prefrontal cortex has been convincingly demonstrated (98). Further studies on the COMT genotype have shown more complex haplotype effects on prefrontal cerebral activations (99) and on gene–gene interactions between COMT and other genes such as RGS4, G72, DISC1, and GRM3 (49, 100, 101). Especially the latter finding is consistent with a role of glutamate–dopamine interactions in the pathophysiology and pathogenesis of schizophrenic disorders.
The number of genome-wide association studies between gene variants and diseases has markedly increased over the last few years due to the availability of modern chips. This has also inspired imaging genetics studies as genome-wide confirmed risk variants have also been investigated for their effects on brain structure and function. Two examples of this are the zinc finger protein 804A (ZNF804A), the function of which has not yet been more closely characterized, but which showed a genome-wide significant association with schizophrenia and also with bipolar disorder (102), as well as the CACNA1C gene, which was first discovered as a risk gene for bipolar disorder, but later also for schizophrenia (103). Studies on the ZNF804A polymorphism have shown an effect on the connectivity between the prefrontal cortex and the hippocampus (104–106). Effects of the CACNA1C gene have been reported with regard to activation of the hippocampus and the subgenual ACC (107) as well as activation of the amygdala during reward and fear recognition paradigms (108, 109).
Taken together, the studies summarized here support the important pathophysiological role of glutamate in schizophrenia and encourage further development of therapeutic strategies that target the glutamatergic pathway in schizophrenia.
Therapeutic Strategies Targeting the Glutamatergic Pathway in Schizophrenia
A valid treatment for positive symptoms, based on the use of antipsychotic agents and their main capacity to modulate the dopaminergic system, is currently available for schizophrenia. However, antipsychotics are less effective in reducing negative symptoms or in ameliorating cognitive dysfunctions (110–112). Based on the novel findings that the glutamatergic system plays an important role in the pathogenesis of schizophrenia, several attempts have been made to identify drugs which, by modulating this system, could improve negative symptoms and cognitive dysfunction (113). Pharmacological targets are different types of glutamate receptors, which interact in a complex not yet fully understood way within glutamatergic synapses. These receptors include both ionotropic receptors [NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors] and metabotropic glutamate receptors (mGluRs) [reviewed in Ref. (114)]. AMPA and kainate receptors are fast gating by glutamate binding and permeable for Na+ and K+ ions, whereas NMDA receptors exhibit a slow gating kinetic and need a predepolarization of the neuronal membrane for activation by glutamate binding. This predepolarization occurs when vicinal AMPA receptors are frequently activated and leads to the release of a blocking Mg2+ ion from the NMDA receptor. Activated NMDA receptor channels are not ion specific and pass Ca2+, Na+, and K+ ions, which leads not only to further depolarization but also intracellular processes making the synapse more sensible for signals from upstream and sending stronger signals downstream. This so-called long-term potentiation (LTP) provides the basis for synaptic and dendritic proliferation or pruning, learning, and memory (115).
Metabotropic glutamate receptors are G protein-coupled receptors influencing intracellular metabolic processes and are present on presynaptic and postsynaptic neurons as well as on glial cells near glutamatergic synapses. Currently, eight mGlu receptors are known of which the mGlu 2/3 receptors are investigated as targets for schizophrenia therapy, because they regulate presynaptic glutamate secretion.
After successful completion of preclinical trial programs of pharmacodynamic activity and safety, potential compounds are investigated in escalating clinical study programs. Phase 1 studies are open label, single to multiple dose trials with healthy volunteers, exploring pharmacokinetic and first safety characteristics. Phase 2 studies are explorative or proof-of-concept studies that are usually controlled and blinded with small to medium numbers of patients, and designed for dosis finding and first in patient proof of safety and efficacy. Phase 3 studies are large scale, double blinded controlled studies to confirm safety and efficacy. Some of the following described trial results are not yet published in peer-reviewed journals, and we had to quote congress presentations or company statements.
Compounds Enhancing NMDA Receptor Function
Since the activation of the NMDA receptor also requires glycine as a co-agonist, the glycine binding site at the NMDA receptor is regarded as a promising pharmacological target to enhance its activity, thereby minimizing the risk of excitotoxicity that is associated with direct overactivation of the glutamate binding site (116). A first review of clinical trials published until 2003 with agonists of the glycine binding site of the NMDA receptor including glycine, d-cycloserine, and d-serine reported moderate effect sizes for glycine and d-serine on negative symptoms, but no effect of d-cycloserine (117). It included 358 small randomized trials with 6–51 participants and a maximum duration of 12 weeks. Up to now, the amount of data has grown confirming these early results. In almost all trials, the compounds were used as adjunctive treatment to regular antipsychotic therapy, and generally no effect was reported in patients taking clozapine.
Several clinical trials have been conducted using glycine, a substance that is endogenously produced and that can act as a co-agonist of NMDA receptors binding at the glycine modulatory site. The results of these studies suggest that glycine can significantly improve symptoms of schizophrenia (118), including negative symptoms, although there are also negative and equivocal studies (119).
d-Cycloserine is a selective co-agonist of NMDA receptors containing the NR2C subunit, and these receptors are involved in fear conditioning and memory consolidation (120). When used as a single dosis in rodents, d-cycloserine led to enhanced memory consolidation of novel information (120). In an exploratory clinical trial, once weekly dosing of d-cycloserine augmentation over 8 weeks did not improve cognitive symptoms but reduced negative symptoms and delusion severity in stable schizophrenic patients medicated with a range of different antipsychotics excluding clozapine (121).
d-Serine, another agonist of the glycine modulatory site within the NMDA receptor, has been shown in clinical trials to be capable of ameliorating several symptom domains in schizophrenia (122–124).
Glycine Transporter-1 Inhibitors
Sarcosine is a potent natural glycine transporter-1 inhibitor (GlyT-1) (125). The inhibition of this transporter leads to increased levels of glycin in the synapsis and consequently enhanced NMDA receptor activation, which may represent a possible treatment mechanism for schizophrenic disorders in which a hypofunction of NMDA receptors is present (126). As recently shown, sarcosine may reduce negative symptoms in acutely ill schizophrenia patients receiving atypical antipsychotics, being more effective than the NMDA/glycine site agonist d-serine (125).
A meta-analysis including all abovementioned molecules found that, overall, the NMDA-enhancers were effective against most symptom domains of schizophrenia. Glycine, d-serine, and sarcosine significantly improved multiple symptom domains, whereas no symptom domain was improved by d-cycloserine. Furthermore, glycine, d-serine, and sarcosine were found to be superior to d-cycloserine in improving overall psychopathology [Ref. (118); see Figure 2]. However, these compounds have individual disadvantages to be developed to drugs licensed for long-term use. Glycine is fast metabolized and passes the blood–brain barrier poorly. So it has to be applied with daily doses up to 60 g. Although d-serine is substantially metabolized, daily doses of 30–120 mg/kg were effective. But there is concern about nephrotoxicity at higher doses, although no significant adverse events have yet been observed at doses of ≤4 g/day (127). According to Sreekumar et al. (128), sarcosine might play a role in aggravating prostate cancer progression.
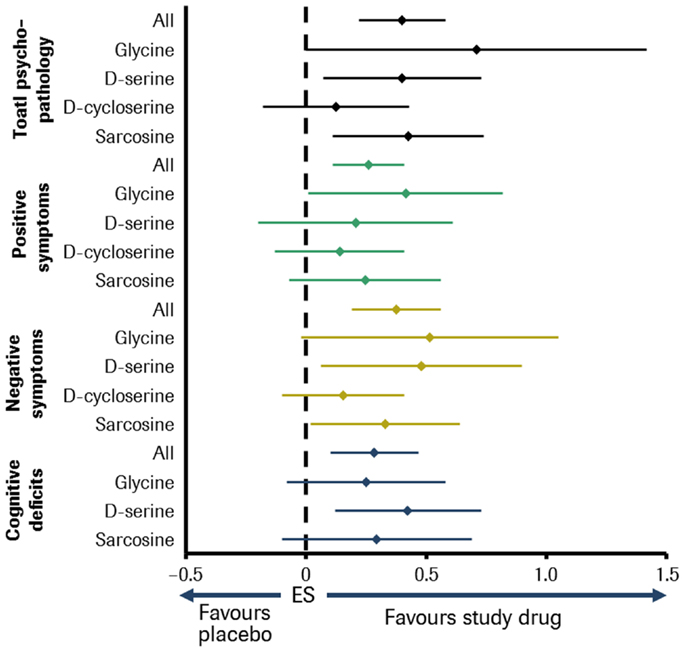
Figure 2. Meta-analysis of double-blind, placebo-controlled studies of small-molecule NMDA receptor enhancers in patients with schizophrenia. Sample size: 26 studies comprising about 800 patients. Effect size (ES) for glycine, d-serine, d-cycloserine, sarcosine, and all in different symptom domains of schizophrenia [adapted from Tsai and Lin (118)].
Bitopertin is a GlyT-1 inhibitor that increases levels of the glycine neurotransmitter by inhibiting its reuptake from the synaptic cleft. Preclinical evidence showed that this molecule is capable of ameliorating the symptoms of schizophrenia in animal models (129, 130). These preclinical findings encouraged a double blinded placebo-controlled phase IIb clinical trial which showed that adjunctive treatment with bitopertin in stable patients with predominant negative symptoms was capable of ameliorating negative symptoms and improving general clinical status (131). Currently, several phase III studies are underway with the hope that bitopertin may help in the treatment of currently unsatisfyingly responding stages of schizophrenia.
A major goal for future research combining psychopharmacology and modern functional neuroimaging techniques would be to understand how these molecules modulate the activity of pathophysiologically relevant neural structures as outlined above. Such studies could, for example, provide important information on whether these pharmacological agents can be successfully used to treat patient subgroups that are characterized by specific symptoms of schizophrenia.
AMPA Receptor Modulators
As described above, the fast trafficking of AMPA receptors in the synaptic cleft has an impact on NMDA receptor-mediated LTP and depression. These intracellular mechanisms influence synaptic strength and therefore constitute the basis of learning and memory (132). Therefore, modulation of AMPA activity could lead to amelioration of cognitive dysfunction in schizophrenia. To this aim and based on preclinical evidences, two molecules have been used in schizophrenia clinical trials: piracetam (133) and CX516 (121, 134).
Piracetam augmentation of haloperidol was capable of improving psychotic symptoms in schizophrenia, but had no effect on PANSS (133). Because only 30 patients (all receiving haloperidol) completed the placebo-controlled trial, more scientific evidence is needed to support such an effect. Trials with CX156 led to controversial results. In a small study, CX156 improved cognitive functions and negative symptoms in schizophrenic patients when compared to patients treated with clozapine (134). However, a larger study was unable to show any effect of CX516 on cognition or negative symptoms when compared to controls (135). Taken together, there is only little evidence about these molecules and their therapeutic effect.
mGlu Receptor Modulators
In contrast to the concept of improving symptoms of schizophrenia with ampakines, a line of evidence points to an overactivation of AMPA synapses in the prefrontal cortex downstream of NMDA receptor hypofunction (136). NMDA receptor blockade on GABA-ergic interneurons reduces inhibition of pyramidal cells and leads to excessive glutamate release in AMPA receptor synapses in the prefrontal cortex (137). Metabolic glutamate receptors 2 and 3 (mGlu 2/3) facilitate a feedback regulation of synaptic glutamate release (138). Consequently, mGlu 2/3 enhancing agents were developed to delimit pathologically enhanced glutamate release. In schizophrenia, clinical trials were conducted with the mGluR2/3 agonist, pomaglumetad methionil (LY2140023) and the mGlu2 positive allosteric modulator, ADX71149.
Pomaglumetad methionil was investigated as monotherapy in three clinical trials. At first, a phase 2 proof-of-concept trial showed significant improvement of positive and negative symptoms versus placebo (139). In a following phase 2 dose ranging trial, all of the four investigated dosing groups did not differ from placebo (140), which was also the case for a phase 2 trial comparing pomaglumetad methionil to olanzapine and a placebo group. In this trial, both active treatment groups did not separate regarding efficacy and safety parameters from the placebo group (141).
After demonstrating an augmentation of the efficacy of atypical antipsychotics in preclinical trials, a phase 1 study was conducted to prove the safety of the combination of pomaglumetad methionil with four different second generation antipsychotics in healthy subjects (142). A following placebo-controlled phase 2 study tested the substance as adjunctive to standard of care in patients with prominent negative symptoms of schizophrenia. This trial did not indicate a significantly greater reduction of negative symptoms or an improvement of secondary efficacy endpoints over placebo (143). Based on these results, a phase 3 study started in the meantime was stopped.
The mGluR2 selective positive allosteric modulator ADX71149 is co-developed by Addex Therapeutics (144) and Johnson & Johnson, who code it JNJ40411813. Results of a randomized placebo-controlled phase 2 study evaluating the safety, tolerability, and exploratory efficacy of the compound given in two different doses as adjunctive to an ongoing antipsychotic medication were reported at the 2013 annual meeting of the American Psychiatric Society. The study population comprised three groups: patients with residual negative symptoms, patients with residual positive symptoms, and patients with insufficient response to clozapine treatment. Tolerability results suggest that dose titration may be beneficial. An efficacy signal seen in the negative symptoms subgroup treated with the lower dose suggests this population warrants further evaluation in a formal proof-of-concept study (144, 145).
Conclusion and Future Perspectives
In this brief review article, we have summarized recent findings from genetic, animal, and functional neuroimaging studies that together point to an important role of glutamate–dopamine interactions within cortico-striato-thalamo-cortical (CSTC) loops, which are modulated by hippocampal and amygdala inputs, in the pathophysiology of schizophrenic disorders. These findings provide the empirical basis for the development of novel treatment approaches for schizophrenic disorders that target glutamatergic mechanisms. The new findings from animal studies may also inspire analogous clinical neuroimaging investigations of neurofunctional interactions within CSTC loops as well as of the dynamic effects of mediotemporal structures such as hippocampus and amygdala on these CSTC loops in the human brain. Here, the development of valid animal model-supported experimental paradigms is of major importance as it may allow for the targeted in vivo investigation of these pathomechanisms involved in the pathophysiology of schizophrenic disorders [e.g., Ref. (4)]. Such targeted investigations may enable a future stratification of the heterogeneous group of schizophrenic disorders into pathophysiologically more homogenous “natural disease entities” (146). Moreover, the development of neurofunctional MRI biomarkers for sub-classification of patient groups and prediction of individual treatment responses may generally play an important role in a future individualized medicine in psychiatry [e.g., Ref. (4)].
Conflict of Interest Statement
Oliver Gruber was honorary speaker for the following companies: Astra Zeneca, Bristol Myers Squibb, Janssen Cilag, Lilly, Lundbeck, Otsuka, Servier. Oliver Gruber has been invited to scientific congresses by Astra Zeneca, Janssen Cilag, Pfizer, Servier. Oliver Gruber has received a research grant from Servier and a publication grant from Roche. Antonella Chadha Santuccione and Helmut Aach are full employes of Roche Pharma AG, Grenzach, Germany.
Acknowledgments
This publication was supported by a grant of the Roche Pharma AG, Germany.
References
1. Insel TR, Wang PS. Rethinking mental illness. JAMA (2010) 303(19):1970–1. doi: 10.1001/jama.2010.555
2. Meyer-Lindenberg A. From maps to mechanisms through neuroimaging of schizophrenia. Nature (2010) 468(7321):194–202. doi:10.1038/nature09569
3. Gruber O, Falkai P. Von der Identifizierung neurofunktioneller Systeme zur Individualisierung der Therapie schizophrener Störungen. Nervenarzt (2009) 80:12–8. doi:10.1007/s00115-008-2615-y
5. Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry (2009) 66(11):988–9. doi:10.1016/j.biopsych.2009.10.008
6. Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry (2010) 167(7):748–51. doi:10.1176/appi.ajp.2010.09091379
8. Gruber O, Gruber E, Falkai P. Neural correlates of working memory deficits in schizophrenic patients. Ways to establish neurocognitive endophenotypes of psychiatric disorders. Radiologe (2005) 45(2):153–60.
9. Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry (1996) 153:321–30.
10. Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull (2000) 26:119–36. doi:10.1093/oxfordjournals.schbul.a033430
11. Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res (2004) 72:41–51. doi:10.1016/j.schres.2004.09.009
12. Kurtz MM, Moberg PJ, Ragland JD, Gur RC, Gur RE. Symptoms versus neurocognitive test performance as predictors of psychosocial status in schizophrenia: a 1- and 4-year prospective study. Schizophr Bull (2005) 31:167–74. doi:10.1093/schbul/sbi004
13. Fenton WS, McGlashan TH. Antecedents, symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry (1994) 151:351–6.
14. Kirkpatrick B, Buchanan RW, Ross DE, Carpenter WT Jr. A separate disease within the syndrome of schizophrenia. Arch Gen Psychiatry (2001) 58:165–71. doi:10.1001/archpsyc.58.2.165
15. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses (2010) 4:189–200. doi:10.3371/CSRP.4.3.6
16. Woodberry KA, Giuliano AJ, Seidman LJ. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry (2008) 165:579–87. doi:10.1176/appi.ajp.2008.07081242
17. Bilder RM, Goldman RS, Robinson D, Reiter G, Bell L, Bates JA, et al. Neuropsychology of first-episode schizophrenia: initial characterization and clinical correlates. Am J Psychiatry (2000) 157:549–59. doi:10.1176/appi.ajp.157.4.549
18. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2012) 37:4–15. doi:10.1038/npp.2011.181
19. Suzuki M, Yuasa S, Minabe Y, Murata M, Kurachi M. Left superior temporal blood flow increases in schizophrenic and schizophreniform patients with auditory hallucination: a longitudinal case study using 123I-IMP SPECT. Eur Arch Psychiatry Clin Neurosci (1993) 242(5):257–61. doi:10.1007/BF02190383
20. Dierks T, Linden DE, Jandl M, Formisano E, Goebel R, Lanfermann H, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron (1999) 22(3):615–21. doi:10.1016/S0896-6273(00)80715-1
21. Lennox BR, Park SBG, Medley I, Morris PG, Jones PB. The functional anatomy of auditory hallucinations in schizophrenia. Psychiatry Res (2000) 100:13–20. doi:10.1016/S0925-4927(00)00068-8
22. Jardri R, Pouchet A, Pins D, Thomas P. Cortical activations during auditory verbal hallucinations in schizophrenia: a coordinate-based meta-analysis. Am J Psychiatry (2011) 168:73–81. doi:10.1176/appi.ajp.2010.09101522
23. Kühn S, Gallinat J. Quantitative meta-analysis on state and trait aspects of auditory verbal hallucinations in schizophrenia. Schizophr Bull (2012) 38:779–86. doi:10.1093/schbul/sbq152
24. Goghari VM, Sponheim SR, MacDonald AW. The functional neuroanatomy of symptom dimensions in schizophrenia: a qualitative and quantitative review of a persistent question. Neurosci Biobehav Rev (2010) 34:468–86. doi:10.1016/j.neubiorev.2009.09.004
25. Andreasen NC, Olsen S. Negative vs positive schizophrenia. Definition and validation. Arch Gen Psychiatry (1982) 39:789–94. doi:10.1001/archpsyc.1982.04290070025006
26. Turetsky B, Cowell PE, Gur RC, Grossman RI, Shtasel DL, Gur RE. Frontal and temporal lobe brain volumes in schizophrenia. Relationship to symptoms and clinical subtype. Arch Gen Psychiatry (1995) 52:1061–70. doi:10.1001/archpsyc.1995.03950240079013
27. Bogerts B, Lieberman JA, Ashtari M, Bilder RM, Degreef G, Lerner G, et al. Hippocampus-amygdala volumes and psychopathology in chronic schizophrenia. Biol Psychiatry (1993) 33:236–46. doi:10.1016/0006-3223(93)90289-P
28. Buchanan RW, Breier A, Kirkpatrick B, Elkashef A, Munson RC, Gellad F, et al. Structural abnormalities in deficit and nondeficit schizophrenia. Am J Psychiatry (1993) 150:59–65.
29. Gur RE, Cowell P, Turetsky BI, Gallacher F, Cannon T, Bilker W, et al. A follow-up magnetic resonance imaging study of schizophrenia. Relationship of neuroanatomical changes to clinical and neurobehavioral measures. Arch Gen Psychiatry (1998) 55:145–52. doi:10.1001/archpsyc.55.2.145
30. Gur RE, Turetsky BI, Bilker WB, Gur RC. Reduced gray matter volume in schizophrenia. Arch Gen Psychiatry (1999) 56:905–11. doi:10.1001/archpsyc.56.10.905
31. Schröder J, Buchsbaum MS, Siegel BV, Geider FJ, Niethammer R. Structural and functional correlates of subsyndromes in chronic schizophrenia. Psychopathology (1995) 28:38–45. doi:10.1159/000284898
32. Schröder J, Buchsbaum MS, Siegel BV, Geider FJ, Lohr J, Tang C, et al. Cerebral metabolic activity correlates of subsyndromes in chronic schizophrenia. Schizophr Res (1996) 19:41–53. doi:10.1016/0920-9964(95)00043-7
33. Potkin SG, Alva G, Fleming K, Anand R, Keator D, Carreon D, et al. A PET study of the pathophysiology of negative symptoms in schizophrenia. Am J Psychiatry (2002) 159:227–37. doi:10.1176/appi.ajp.159.2.227
34. Heckers S, Goff D, Schacter DL, Savage CR, Fischman AJ, Alpert NM, et al. Functional imaging of memory retrieval in deficit vs nondeficit schizophrenia. Arch Gen Psychiatry (1999) 56:1117–23. doi:10.1001/archpsyc.56.12.1117
35. Menon V, Anagnoson RT, Mathalon DH, Glover GH, Pfefferbaum A. Functional neuroanatomy of auditory working memory in schizophrenia: relation to positive and negative symptoms. Neuroimage (2001) 13:433–46. doi:10.1006/nimg.2000.0699
36. Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry (2001) 158:1105–13. doi:10.1176/appi.ajp.158.7.1105
37. Tamminga CA, Thaker GK, Buchanan R, Kirkpatrick B, Alphs LD, Chase TN, et al. Limbic system abnormalities identified in schizophrenia using positron emission tomography with fluorodeoxyglucose and neocortical alterations with deficit syndrome. Arch Gen Psychiatry (1992) 49:522–30. doi:10.1001/archpsyc.1992.01820070016003
38. Ebmeier KP, Blackwood DH, Murray C, Souza V, Walker M, Dougall N, et al. Single-photon emission computed tomography with 99mTc-exametazime in unmedicated schizophrenic patients. Biol Psychiatry (1993) 33:487–95. doi:10.1016/0006-3223(93)90002-U
39. Kawasaki Y, Maeda Y, Sakai N, Higashima M, Yamaguchi N, Koshino Y, et al. Regional cerebral blood flow in patients with schizophrenia: relevance to symptom structures. Psychiatry Res (1996) 67:49–58. doi:10.1016/0925-4927(96)02685-6
40. Min SK, An SK, Jon DI, Lee JD. Positive and negative symptoms and regional cerebral perfusion in antipsychotic-naive schizophrenic patients: a high-resolution SPECT study. Psychiatry Res (1999) 90:159–68. doi:10.1016/S0925-4927(99)00014-1
41. Juckel G, Schlagenhauf F, Koslowski M, Wustenberg T, Villringer A, Knutson B, et al. Dysfunction of ventral striatal reward prediction in schizophrenia. Neuroimage (2006) 29:409–16. doi:10.1016/j.neuroimage.2005.07.051
42. Simon JJ, Biller A, Walther S, Roesch-Ely D, Stippich C, Weisbrod M, et al. Neural correlates of reward processing in schizophrenia – relationship to apathy and depression. Schizophr Res (2010) 118:154–61. doi:10.1016/j.schres.2009.11.007
43. Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol (2000) 14(1):1–21. doi:10.1615/CritRevNeurobiol.v14.i1.10
44. Silver H, Feldman P, Bilker W, Gur RC. Working memory deficit as a core neuropsychological dysfunction in schizophrenia. American Journal of Psychiatry (2003) 160(10):1809–16. doi:10.1176/appi.ajp.160.10.1809
45. Weinberger DR, Berman KF, Suddath R, Torrey EF. Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: a magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. Am J Psychiatry (1992) 149(7):890–7.
46. Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, et al. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex (2000) 10(11):1078–92. doi:10.1093/cercor/10.11.1078
47. Manoach DS, Gollub RL, Benson ES, Searl MM, Goff DC, Halpern E, et al. Schizophrenic subjects show aberrant fMRI activation of dorsolateral prefrontal cortex and basal ganglia during working memory performance. Biol Psychiatry (2000) 48(2):99–109. doi:10.1016/S0006-3223(00)00227-4
48. Perlstein WM, Dixit NK, Carter CS, Noll DC, Cohen JD. Prefrontal cortex dysfunction mediates deficits in working memory and prepotent responding in schizophrenia. Biol Psychiatry (2003) 53(1):25–38. doi:10.1016/S0006-3223(02)01675-X
49. Tan H-Y, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF, et al. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A (2007) 104:12536–41. doi:10.1073/pnas.0610125104
50. Henseler I, Falkai P, Gruber O. A systematic fMRI investigation of the brain systems subserving different working memory components in schizophrenia. Eur J Neurosci (2009) 30:693–702. doi:10.1111/j.1460-9568.2009.06850.x
51. Meyer-Lindenberg A, Olsen R, Kohn P. Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch Gen Psychiatry (2005) 62:379–86. doi:10.1001/archpsyc.62.4.379
52. Henseler I, Falkai P, Gruber O. Disturbed functional connectivity within brain networks subserving domain-specific subcomponents of working memory in schizophrenia: relation to performance and clinical symptoms. J Psychiatr Res (2010) 44:364–72. doi:10.1016/j.jpsychires.2009.09.003
53. Smith EE, Jonides J. Storage and executive processes in the frontal lobes. Science (1999) 283(5408):1657–61. doi:10.1126/science.283.5408.1657
54. Gruber O, Goschke T. Executive control emerging from dynamic interactions between brain systems mediating language, working memory and attentional processes. Acta Psychol (Amst). (2004) 115(2-3):105–21. doi:10.1016/j.actpsy.2003.12.003
55. Gruber O, Melcher T, Diekhof EK, Karch S, Falkai P, Goschke T. Brain mechanisms associated with background monitoring of the environment for potentially significant sensory events. Brain Cogn (2009) 69:559–64. doi:10.1016/j.bandc.2008.11.008
56. Montague PR, Berns GS. Neural economics and the biological substrates of valuation. Neuron (2002) 36(2):265–84. doi:10.1016/S0896-6273(02)00974-1
57. Cools R, Clark L, Owen AM, Robbins TW. Defining the neural mechanisms of probabilistic reversal learning using event-related functional magnetic resonance imaging. J Neurosci (2002) 22(11):4563–7.
58. Kringelbach ML, Rolls ET. Neural correlates of rapid reversal learning in a simple model of human social interaction. Neuroimage (2003) 20(2):1371–83. doi:10.1016/S1053-8119(03)00393-8
59. O’Doherty J, Winston J, Critchley H, Perrett D, Burt DM, Dolan RJ. Beauty in a smile: the role of medial orbitofrontal cortex in facial attractiveness. Neuropsychologia (2003) 41(2):147–55. doi:10.1016/S0028-3932(02)00145-8
60. Gruber O, Diekhof EK, Kirchenbauer L, Goschke T. A neural system for evaluating the behavioural relevance of salient events outside the current focus of attention. Brain Res (2010) 1351:212–21. doi:10.1016/j.brainres.2010.06.056
61. Minzenberg MJ, Laird AR, Thelen S, Carter CS, Glahn DC. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch Gen Psychiatry (2009) 66:811–22. doi:10.1001/archgenpsychiatry.2009.91
62. Krabbendam L, O’Daly O, Morley LA, van Os J, Murray RM, Shergill SS. Using the Stroop task to investigate the neural correlates of symptom change in schizophrenia. Br J Psychiatry (2009) 194:373–4. doi:10.1192/bjp.bp.108.055459
63. Murray GK, Corlett PR, Clark L, Pessiglione M, Blackwell AD, Honey G, et al. Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Mol Psychiatry (2008) 13:267–76. doi:10.1038/sj.mp.4002058
64. Achim AM, Lepage M. Episodic memory-related activation in schizophrenia: meta-analysis. Br J Psychiatry (2005) 187:500–9. doi:10.1192/bjp.187.6.500
65. Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology (2011) 36:316–38. doi:10.1038/npp.2010.156
66. Li H, Chan RCK, McAlonan GM, Gong Q. Facial emotion processing in schizophrenia: a meta-analysis of functional neuroimaging data. Schizophr Bull (2010) 36:1029–39. doi:10.1093/schbul/sbn190
67. Brunet-Gouet E, Decety J. Social brain dysfunctions in schizophrenia: a review of neuroimaging studies. Psychiatry Res (2006) 148:75–92. doi:10.1016/j.pscychresns.2006.05.001
68. Rasetti R, Mattay VS, Wiedholz LM, Kolachana BS, Hariri AR, Callicott JH, et al. Evidence that altered amygdala activity in schizophrenia is related to clinical state and not genetic risk. Am J Psychiatry (2009) 166:216–25. doi:10.1176/appi.ajp.2008.08020261
69. Straub RE, Weinberger DR. Schizophrenia genes – famine to feast. Biol Psychiatry (2006) 60:81–3. doi:10.1016/j.biopsych.2006.002
70. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull (2009) 35(3):549–62. doi:10.1093/schbul/sbp006
71. Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol (2012) 3:195. doi:10.3389/fphar.2012.00195
72. Lewis DA, Gonzalez-Burgos G. Pathophysiologically based treatment interventions in schizophrenia. Nat Med (2006) 12:1016–22. doi:10.1038/nm1478
73. O’Donnell P, Grace AA. Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull (1998) 24(2):267–83. doi:10.1093/oxfordjournals.schbul.a033325
74. Tost H, Meyer-Lindenberg A. Dopamine-glutamate interactions: a neural convergence mechanism of common schizophrenia risk variants. Biol Psychiatry (2011) 69(10):912–3. doi:10.1016/j.biopsych.2011.03.013
75. Blaha CD, Yang CR, Floresco SB, Barr AM, Phillips AG. Stimulation of the ventral subiculum of the hippocampus evokes glutamate receptor-mediated changes in dopamine efflux in the rat nucleus accumbens. Eur J Neurosci (1997) 9:902–11. doi:10.1111/j.1460-9568.1997.tb01441.x
76. Legault M, Wise RA. Injections of N-methyl-D-aspartate into the ventral hippocampus increase extracellular dopamine in the ventral tegmental area and nucleus accumbens. Synapse (1999) 31:241–9. doi:10.1002/(SICI)1098-2396(19990315)31:4<241::AID-SYN1>3.0.CO;2-#
77. Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration alters proteomic and metabolomic markers of hippocampal glutamatergic transmission. Neuropsychopharmacology (2012) 37:319–20. doi:10.1038/npp.2011.255
78. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci (2007) 27:11424–30. doi:10.1523/JNEUROSCI.2847-07.2007
79. Marsman A, van den Heuvel MP, Klomp DWJ, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull (2013) 39(1):120–9. doi:10.1093/schbul/sbr069
80. Poels EMP, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry (2013) 19(1):20–9. doi:10.1038/mp.2013.136
81. Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, et al. Elevated prefrontal cortex γ-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry (2012) 69:449–59. doi:10.1001/archgenpsychiatry.2011.1519
82. Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry (2002) 159:1944–6. doi:10.1176/appi.ajp.159.11.1944
83. Bartha R, Williamson PC, Drost DJ, Malla A, Carr TJ, Cortese L, et al. Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry (1997) 54:959–65. doi:10.1001/archpsyc.1997.01830220085012
84. Théberge J, Williamson KE, Aoyama N, Drost DJ, Manchanda R, Malla AK, et al. Longitudinal grey-matter and glutamatergic losses in first-episode schizophrenia. Br J Psychiatry (2007) 191:325–34. doi:10.1192/bjp.bp.106.033670
85. Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology (2012) 37:2515–21. doi:10.1038/npp.2012.113
86. Ohrmann P, Kugel H, Bauer J, Siegmund A, Kölkebeck K, Suslow T, et al. Learning potential on the WCST in schizophrenia is related to the neuronal integrity of the anterior cingulate cortex as measured by proton magnetic resonance spectroscopy. Schizophr Res (2008) 106:156–63. doi:10.1016/j.schres.2008.08.005
87. Bustillo JR, Chen H, Gasparovic C, Mullins P, Caprihan A, Qualls C, et al. Glutamate as a marker of cognitive function in schizophrenia: a proton spectroscopic imaging study at 4 Tesla. Biol Psychiatry (2011) 69:19–27. doi:10.1016/j.biopsych.2010.08.024
88. Ongür D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM, et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry (2008) 64:718–26. doi:10.1016/j.biopsych.2008.05.014
89. Wood SJ, Yücel M, Wellard RM, Harrison BJ, Clarke K, Fornito A, et al. Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res (2007) 94:328–31. doi:10.1016/j.schres.2007.05.008
90. Kraguljac NV, Reid MA, White DM, den Hollander J, Lahti AC. Regional decoupling of N-acetyl-aspartate and glutamate in schizophrenia. Neuropsychopharmacology (2012) 37:2635–42. doi:10.1038/npp.2012.126
91. Bustillo JR, Rowland LM, Mullins P, Jung R, Chen H, Qualls C, et al. 1H-MRS at 4 Tesla in minimally treated early schizophrenia. Mol Psychiatry (2010) 15:629–36. doi:10.1038/mp.2009.121
92. Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS, et al. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry (2010) 68:625–33. doi:10.1016/j.biopsych.2010.04.013
93. de la Fuente-Sandoval C, León-Ortiz P, Favila R, Stephano S, Mamo D, Ramírez-Bermúdez J, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology (2011) 36:1781–91. doi:10.1038/npp.2011.65
94. de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, Stephano S, Favila R, Díaz-Galvis L, et al. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry (2013) 70:1057–66. doi:10.1001/jamapsychiatry.2013.289
95. Goto N, Yoshimura R, Kakeda S, Nishimura J, Moriya J, Hayashi K, et al. Six-month treatment with atypical antipsychotic drugs decreased frontal-lobe levels of glutamate plus glutamine in early-stage first-episode schizophrenia. Neuropsychiatr Dis Treat (2012) 8:119–22. doi:10.2147/NDT.S25582
96. Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, et al. Effect of COMT Val(108/158) Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A (2001) 98:6917–22. doi:10.1073/pnas.111134598
97. Meyer-Lindenberg A, Kohn PD, Kolachana B, Kippenhan S, McInerney-Leo A, Nussbaum R, et al. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat Neurosci (2005) 8:594–6. doi:10.1038/nn1438
98. Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci (2006) 7:818–27. doi:10.1038/nrn1993
99. Meyer-Lindenberg A, Nichols T, Callicott JH, Ding J, Kolachana B, Buckholtz J, et al. Impact of complex genetic variation in COMT on human brain function. Mol Psychiatry (2006) 11:867–77. doi:10.1038/sj.mp.4001860
100. Nixon DC, Prust MJ, Sambataro F, Tan H-Y, Mattay VS, Weinberger DR, et al. Interactive effects of DAOA (G72) and catechol-O-methyltransferase on neurophysiology in prefrontal cortex. Biol Psychiatry (2011) 69:1006–8. doi:10.1016/j.biopsych.2010.10.031
101. Nicodemus KK, Kolachana BS, Vakkalanka R, Straub RE, Giegling I, Egan MF, et al. Evidence for statistical epistasis between catechol-O-methyltransferase (COMT) and polymorphisms in RGS4, G72 (DAOA), GRM3, and DISC1: influence on risk of schizophrenia. Hum Genet (2007) 120:889–906. doi:10.1007/s00439-006-0257-3
102. O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet (2008) 40:1053–5. doi:10.1038/ng.201
103. Ferreira MAR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet (2008) 40:1056–8. doi:10.1038/ng.209
104. Esslinger C, Walter H, Kirsch P, Erk S, Schnell K, Arnold C, et al. Neural mechanisms of a genome-wide supported psychosis variant. Science (2009) 324:605. doi:10.1126/science.1167768
105. Paulus FM, Krach S, Bedenbender J, Pyka M, Sommer J, Krug A, et al. Partial support for ZNF804A genotype-dependent alterations in prefrontal connectivity. Hum Brain Mapp (2013) 34:304–13. doi:10.1002/hbm.21434
106. Rasetti R, Sambataro F, Chen Q, Callicott JH, Mattay VS, Weinberger DR. Altered cortical network dynamics a potential intermediate phenotype for schizophrenia and association with ZNF804A RID E-3426-2010. Arch Gen Psychiatry (2011) 68:1207–17. doi:10.1001/archgenpsychiatry.2011.103
107. Erk S, Meyer-Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P, et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry (2010) 67:803–11. doi:10.1001/archgenpsychiatry.2010.94
108. Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry (2010) 67:939–45. doi:10.1001/archgenpsychiatry.2010.96
109. Wessa M, Linke J, Witt SH, Nieratschker V, Esslinger C, Kirsch P, et al. The CACNA1C risk variant for bipolar disorder influences limbic activity. Mol Psychiatry (2010) 15:1126–7. doi:10.1038/mp.2009.103
110. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res (2010) 122:1–23. doi:10.1016/j.schres.2010.05.025
111. Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet (2009) 373:31–41. doi:10.1016/S0140-6736(08)61764-X
112. Hill SK, Bishop JR, Palumbo D, Sweeney JA. Effect of second-generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother (2010) 10:43–57. doi:10.1586/ern.09.143
113. Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci (2013) 263(5):367–77. doi:10.1007/s00406-013-0399-y
114. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev (2010) 62:405–96. doi:10.1124/pr.109.002451
115. Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol (2007) 23:613–43. doi:10.1146/annurev.cellbio.23.090506.123516
116. Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature (1987) 325:529–31. doi:10.1038/325529a0
117. Tuominen HJ, Tiihonen J, Wahlbeck K. Glutamatergic drugs for schizophrenia. Cochrane Database Syst Rev (2006) (2):CD003730. doi:10.1002/14651858.CD003730.pub2
118. Tsai GE, Lin P-Y. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des (2010) 16:522–37. doi:10.2174/138161210790361452
119. Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, et al. The cognitive and negative symptoms in schizophrenia trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry (2007) 164:1593–602. doi:10.1176/appi.ajp.2007.06081358
120. Goff DC. D-cycloserine: an evolving role in learning and neuroplasticity in schizophrenia. Schizophr Bull (2012) 38:936–41. doi:10.1093/schbul/sbs012
121. Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, et al. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr Res (2008) 106:320–7. doi:10.1016/j.schres.2008.08.012
122. Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry (1998) 44:1081–9. doi:10.1016/S0006-3223(98)00279-0
123. Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry (2005) 57:577–85. doi:10.1016/j.biopsych.2004.12.037
124. Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res (2010) 121:125–30. doi:10.1016/j.schres.2010.05.012
125. Lane H-Y, Lin C-H, Huang Y-J, Liao C-H, Chang Y-C, Tsai GE. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol (2010) 13:451–60. doi:10.1017/S1461145709990939
126. Javitt DC. Glycine transport inhibitors in the treatment of schizophrenia. Handb Exp Pharmacol (2012) 213:367–99. doi:10.1007/978-3-642-25758-2_12
127. Javitt DC, Zukin SR, Heresco-levy U, Umbricht D. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull (2012) 38:958–66. doi:10.1093/schbul/sbs069
128. Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature (2012) 12(457):910–4. doi:10.1038/nature07762
129. Pinard E, Alanine A, Alberati D, Bender M, Borroni E, Bourdeaux P, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin- 2-yl)piperazin-1-yl][5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methyletho xy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Med Chem (2010) 53:4603–14. doi:10.1021/jm100210p
130. Alberati D, Moreau J-L, Lengyel J, Hauser N, Mory R, Borroni E, et al. Glycine reuptake inhibitor RG1678: a pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology (2012) 62:1152–61. doi:10.1016/j.neuropharm.2011.11.008
131. Roche Global Web Site. Phase II Study with First-in-Class Investigational Drug Demonstrates Improvement in Negative Symptoms in Patients with Schizophrenia. (2010). Available from: http://www.roche.com/investors/ir_update/inv-update-2010-12-06b.htm
132. Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature (1993) 361:31–9. doi:10.1038/361031a0
133. Noorbala AA, Akhondzadeh S, Davari-Ashtiani R, Amini-Nooshabadi H. Piracetam in the treatment of schizophrenia: implications for the glutamate hypothesis of schizophrenia. J Clin Pharm Ther (1999) 24:369–74. doi:10.1046/j.1365-2710.1999.00238.x
134. Goff DC, Leahy L, Berman I, Posever T, Herz L, Leon AC, et al. A placebo-controlled pilot study of the ampakine CX516 added to clozapine in schizophrenia. J Clin Psychopharmacol (2001) 21:484–7. doi:10.1097/00004714-200110000-00005
135. Goff DC, Lamberti JS, Leon AC, Green MF, Miller AL, Patel J, et al. A placebo-controlled add-on trial of the ampakine CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology (2008) 33:465–72. doi:10.1038/sj.npp.1301444
136. Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci (1997) 17(8):2921–7.
137. Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol (2012) 213:267–95. doi:10.1007/978-3-642-25758-2_10
138. Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol (2010) 50:295–322. doi:10.1146/annurev.pharmtox.011008.145533
139. Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med (2007) 13(9):1102–7. doi:10.1038/nm1007-1264
140. Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol (2011) 31(3):349–55. doi:10.1097/JCP.0b013e318218dcd5
141. Adams DH, Kinon BJ, Baygani S, Millen BA, Velona I, Kollack-Walker S, et al. A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry (2013) 13(1):143. doi:10.1186/1471-244X-13-143
142. Stauffer VL, Ayan-Oshodi M, Wondmagegnehu ET, Witcher J, Shen T, Yuen ESM, et al. A phase 1B study investigating the potential interaction between LY2140023 and second generation antipsychotics in subjects with schizophrenia or schizoaffective disorder. Schizophr Res (2012) 136:S169–70. doi:10.1016/S0920-9964(12)70533-9
143. Stauffer VL, Millen BA, Andersen S, Kinon BJ, Lagrandeur L, Lindenmayer JP, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res (2013) 150:434–41. doi:10.1016/j.schres.2013.08.020
144. Addex Therapeutics. Addex Reports Top-line Data from a Successful Phase 2a Clinical Study with ADX71149 in Schizophrenia Patients. Addex Therapeutics Press Release (2012). Available from: http://www.addextherapeutics.com/about/about-addex/
Keywords: schizophrenia, glutamate, negative symptoms, cognitive deficits, neuroimaging biomarkers, stratified therapy
Citation: Gruber O, Chadha Santuccione A and Aach H (2014) Magnetic resonance imaging in studying schizophrenia, negative symptoms, and the glutamate system. Front. Psychiatry 5:32. doi: 10.3389/fpsyt.2014.00032
Received: 01 October 2013; Accepted: 14 March 2014;
Published online: 03 April 2014.
Edited by:
André Schmidt, University of Basel, SwitzerlandReviewed by:
Alice Egerton, King’s College London, UKNaomi Robin Driesen, Yale University Medical School, USA
Copyright: © 2014 Gruber, Chadha Santuccione and Aach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oliver Gruber, Clinic for Psychiatry and Psychotherapy, University Medical Center Göttingen, Von-Siebold-Str. 5, Göttingen 37075, Germany e-mail:b2dydWJlckBnd2RnLmRl