 Andre A. S. Goldani
Andre A. S. Goldani Susan R. Downs
Susan R. Downs Felicia Widjaja2
Felicia Widjaja2 Robert L. Hendren
Robert L. Hendren- 1Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Department of Psychiatry, University of California San Francisco, San Francisco, CA, USA
Autism spectrum disorders (ASDs) are complex, heterogeneous disorders caused by an interaction between genetic vulnerability and environmental factors. In an effort to better target the underlying roots of ASD for diagnosis and treatment, efforts to identify reliable biomarkers in genetics, neuroimaging, gene expression, and measures of the body’s metabolism are growing. For this article, we review the published studies of potential biomarkers in autism and conclude that while there is increasing promise of finding biomarkers that can help us target treatment, there are none with enough evidence to support routine clinical use unless medical illness is suspected. Promising biomarkers include those for mitochondrial function, oxidative stress, and immune function. Genetic clusters are also suggesting the potential for useful biomarkers.
Introduction
Several neurodevelopmental disorders have complex genetic and epigenetic features that lead to their phenotype and for some there is no single genetic marker for the diagnosis; therefore, the diagnosis is made phenotypically as in schizophrenia, ADHD, and autism spectrum disorder (ASD). While phenotypic characterization of neurodevelopmental disorders is an integral part of advances in clinical practice and research, a given phenotype may arise from a diverse set of biochemical processes (especially when the disorder is caused by numerous genetic and epigenetic factors). Therefore, the treatment of a “phenotypic diagnosis” with a specific drug or intervention might be extremely effective for one “phenotypically characterized” individual with a given set of genetic and/or epigenetic biomarkers, but completely ineffective for another with a different pattern of biomarkers. An important goal of ongoing research in ASD, therefore, is to more precisely identify the many different abnormal genetic and epigenetic processes that underlie the phenotype of the disorder. This might allow individuals with ASD to be characterized into subsets with certain biomarker profiles that would respond more favorably to specific treatments. It also has the potential to elucidate the abnormal physiology that leads to autism, which could improve the understanding of the disorder and lead to earlier diagnosis and more targeted treatments.
A significant challenge in identifying biomarkers in ASD is that biomarkers may reflect genetic and neurobiological changes or epigenetic (broadly defined, see below) processes that may be active only during particular periods of time and do not define the disorder, only the process that led to it. In addition, treatment research should ideally include biomarkers that are believed to predict improvements in clinical symptoms from clinical interventions (1) to know if an intervention is altering or targeting an active biomedical process that relates to response in the subject at that time. Indeed, the National Institute of Mental Health (NIMH) has changed how they fund clinical trials so that “trial proposals will need to identify a target or mediator; a positive result will require not only that an intervention ameliorated a symptom but also that it had a demonstrable effect on a target, such as a neural pathway implicated in the disorder or a key cognitive operation”(2).
Traditionally, research in psychiatry has been guided by DSM symptom based diagnoses and selection criteria for clinical trials were based on these symptom clusters. Biomarkers have not been reliable or valid markers of response to treatment in past trials, and this may be due to the wide variety of genetic and epigenetic processes that underlie the DSM-based diagnosis. Recently, progress in biomarker research has led to the commitment to the Research Domain Criteria project (RDoC) as a basis for future NIMH funding for biomarker based research (3, 4). The RDoC goal is to define basic dimensions of functioning to be studied across multiple units of analysis, from genes to neural circuits to behaviors, cutting across disorders as traditionally defined. The intent is to translate rapid progress in basic neurobiological and behavioral research to an improved integrative understanding of psychopathology and the development of new and/or optimally matched treatments for mental disorders (5).
In this article, we review the literature on biomarkers for ASD including genetic, epigenetic, brain based, and body metabolism biomarkers. This is a huge area and this review is not intended to be comprehensive. New potential biomarkers for ASD are being identified every day so the list needs to be updated frequently. We do extensively review the literature at the time of this writing, report on methodologically sounds studies, offer summary tables, and summarize what we know.
Genetic Biomarkers
The literature supports a hereditary component in the susceptibility to ASDs, there are much higher concordance rates of ASDs in monozygotic twins (92%) than dizygotic twins (10%), and a recent estimate of the sibling recurrence risk ratio (λs) is 22 for autism. Despite being highly heritable, ASDs show heterogeneous clinical symptoms and genetic architecture, which have hindered the identification of common genetic susceptibility factors. Although previous linkage studies, candidate gene association studies, and cytogenetic studies have implicated several chromosomal regions for the presence of autism susceptibility loci, they have not consistently identified and replicated common genetic variants that increase the risk of ASDs other than some clearly genetic disorders such as fragile X, tuberous sclerosis, and RASopathies whose phenotypes meet the ASD category description (5). As autism is not a single clinical entity, it can be viewed as a behavioral manifestation of tens or perhaps hundreds of genetic and genomic disorders (6). It has been estimated that there are over 500 distinct genetic loci that may be related to ASD (7) (Figure 1).
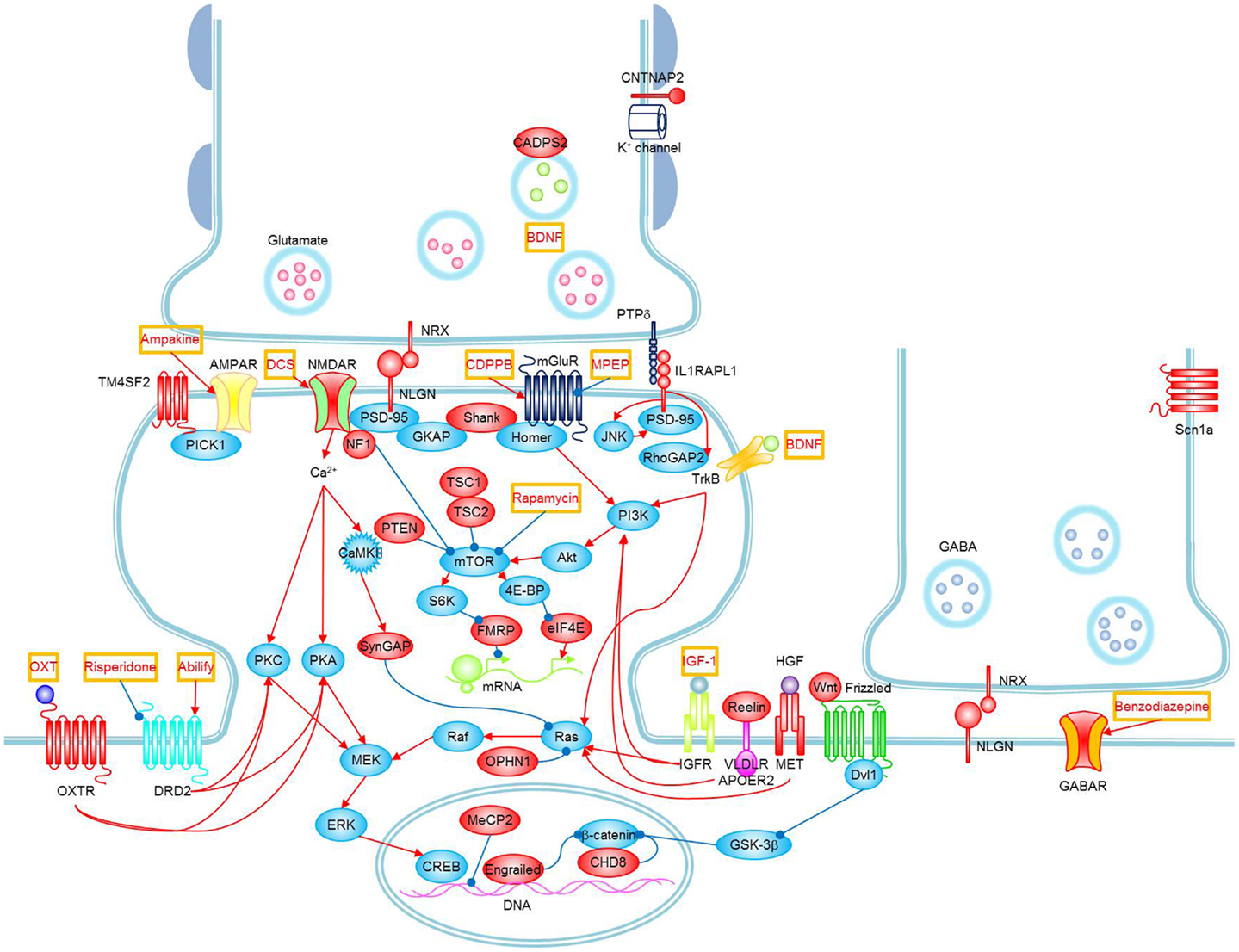
Figure 1. Signaling pathways and possible treatments associated with ASD. Molecules whose mutations or polymorphisms are associated with ASD are indicated in red. Stimulations and inhibitions are indicated by red and blue arrows, respectively. Possible treatments and their target molecules are indicated by red texts in orange boxes. SynGAP1, which directly interacts with PSD-95, could not be placed next to PSD-95 for simplicity. Figure as originally published in Won et al. (8).
In addition, recent research has shown that there are many epigenetic mechanisms that could account for hereditary influences. A study by Hallmayer et al. (9) reports that the environment may actually account for more of the etiology of autism than genetics. Their study, the largest population-based twin study of autism that used contemporary standards for the autism diagnosis, found that heritability estimated at 38%, while shared environmental component was 58% (9). Heritability of ASD and autistic disorder is estimated to be approximately 50% (10).
Being one of the most familial psychiatric disorders, autism has garnered inquiries about possible genetic biomarkers (11); however, progress has been slow until recently with the introduction of genome-wide association studies (GWAS) and microarrays (12). Research into the microbiological underpinnings of ASDs suggests that it is not a monogenic disorder following Mendelian tendencies, with a few studied individuals and families as notable exceptions (11). In fact, the literature suggests that the risk of developing autism is derived by variations across many genes, none of which have been conclusively, definitively responsible for ASDs although some individuals with single gene disorders such as fragile X also meet the criteria for ASD.
Genome-wide association studies have identified, with replication, de novo variations that are strongly associated (with sufficient power) with ASDs (Table 1): deletions at the Neurexin 1 (NRXN1) locus, duplications at 7q11.23, duplications at 15q11-13, and deletions and duplications at 16p11.2. Earlier studies found rare, functional mutations in genes encoding for NRXN1, SHANK3, and SHANK2, all of which are proteins that affect the functioning of synapses and have been linked to other, known genetic disorders (12). In addition, whole exome sequencing verified by four reports have found genetic mutations associated with autism including SNC2A, CHD8, DYRKIA, POG2, GRIN2B, and KATNAL2 (13).

Table 1. Genetic biomarkers in ASD (see text for references).
Studying particular genes in certain, recognized disorders with social deficits, such as fragile X syndrome and tuberous sclerosis, may shed light on the genetic underpinnings of ASDs. This strategy gives credence to the idea that ASD is the result of many variations among genes that converge to a similar phenotype. A prime example of implementation of such a strategy is with contactin 4 (CNTN4), and its association with social and intellectual disability in a recurrent deletion syndrome. Mutations in the respective genes are identified in idiopathic ASDs. Similarly, mutations in CNTNAP2 are linked to a variety of results, such as language delay, functional connectivity abnormalities, selective mutism, and anxiety. More importantly in the scope of ASDs, alterations in CNTNAP2 are noted in consanguineous pedigrees (12). Research shows an increased prevalence of ASDs in families that are consanguineous (11).
In a study published by Nature in 2009, Wang and colleagues completed a genetic analysis in a large number of ASD individuals and families, with a combined sample set of more than 10,000 subjects of European ancestry. They identified common genetic variants on 5p14.1 that are associated with susceptibility to ASDs and replicated these findings in separate analyses. The contribution of chromosome 5p14 to cell adhesion and its connection to autism susceptibility supports the conclusion that specific genes in this class help create the connectivity and structure of the brain that ultimately leads to ASD (14). Besides the potential role of the nearby CDH10 and CDH9 genes, pathway-based association analysis lend further support to neuronal cell-adhesion molecules in conferring susceptibility to ASDs, suggesting that specific genetic variants in this gene class may be involved in shaping the physical structure and functional connectivity of the brain that leads to the clinical manifestations of ASDs (14).
Among the common polymorphisms found to be associated with autism risk, the methylenetetrahydrofolate reductase (MTHFR) polymorphism is one of the most widely studied genetic correlations with autism. The MTHFR 677C > T polymorphism causes a reduction in enzyme activity, which results in higher production of 5-formyltetrahydrofolate (5-FTHF) necessary for DNA synthesis and repair along with lower 5-MTHF production. The MTHFR 677C > T polymorphism causes decline of normal enzyme activity to 35% (15). The MTHFR 677T-variant allele is correlated with a 2.79-fold increased risk for autism. However, this study also found that MTRR 66A and SHMT 1420T alleles demonstrated protective roles against autism risk (16). MTHFR also has a strong interaction with maternal folic acid intake before and during pregnancy, which is associated with autism risk. Children with high autism risk whose mothers carried MTHFR 677 TT allele and were reported taking prenatal vitamins had fewer diagnoses of autism than the children whose mothers with the same allele and did not take prenatal vitamins (17).
In several GWAS (14, 18–20), four genes have been associated with ASDs. These genes, cadherin (CDH9), cadherin 10 (CDH10), semaphorin 5A (SEMA5A), and taste receptor, type 2, member 1 (TAS2R1), are found on chromosome 5p14, which regulates axon growth and cell adhesion. While gene networks could not be established from the small number of genes, these findings do suggest that these genes and the dysregulation of synaptic connection may be a key feature in ASDs (21).
Griswold and coworkers found a significantly higher burden in the number and size of deletions carried by ASD individuals when compared with controls (22). Among the copy-number variations (CNVs) identified were several that overlapped with well-established autism-associated regions and candidate genes. They isolated four large, novel deletions on 2q22.1, 3p26.3, 4q12, and 14q23 that include new genes and regions linked to ASDs. Scattered findings related to NLGN4 and autism susceptibility occur across cultures. In the Chinese ASD cases, there were no significant findings regarding SNPs along NLGN4 gene and autism risk (23), yet in Greek ASD cases, nine nucleotide changes in NLGN4X are found to be associated with autism (24).
Copy-number variations has unveiled the overexpression of rare, de novo structural variations in the genome of simplex families (families which have one affected offspring) when compared to families with multiple affected offspring, and especially control families. Furthermore, these results have been replicated in later studies, bolstering the confidence in which discoveries can be made about genetic ties with common diseases and autism (12); however, de novo CNVs have been found in only 5–10% of researched subjects, and thus, do not make up the majority of affected, researched individuals. Despite this finding, it seems as though large (>100 kb), multigenic de novo CNVs are the most indicative of ASD risk at this time.
The genetic component of a disorder can be transmitted or acquired through de novo (“new”) mutations. A study based on a 343 family subset of the Simons Simplex collection did not find significantly greater numbers of de novo missense mutations in affected versus unaffected children, but gene-disrupting mutations (nonsense, splice site, and frame shifts) were twice as frequent (59 versus 28) (25). They found that the father is more frequently the parent of origin for de novo mutations than the mother (50/17) for single nucleotide variants (SNVs). Parental age also appears to play a role in mutation rate. A study published in Nature found that the rate of de novo SNVs increases with paternal age (p = 0.008) and that paternal and maternal ages are highly correlated (p < 0.0001) (26). Overall these data demonstrate that non-synonymous de novo SNVs, and particularly highly disruptive nonsense and splice-site de novo mutations, are associated with ASD.
Several companies are marketing genetic testing for autism based on clusters of genes with a strong clustering for ASD risk (27, 28). In the future, there may be biomarkers that can pinpoint for high risk for ASD diagnosis. For example, a mother who may be high risk for immune dysfunction leading to ASD in a second child once the first child has ASD (29) or the increase in the Akt-mTOR pathway, which can be seen in fragile X syndrome and in other ASD subtypes (30).
Epigenetics
Considerable symptom severity differences within ASD-concordant monozygotic twins, strongly implicates a role for non-genetic epigenetic factors (31). Epigenetics refers to the study of heritable changes in gene activity that are not caused by changes in the DNA sequence; it also can be used to describe the study of stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable. Epigenetic changes in ASD occur through methylation, histone modification (31), chromatin remodeling, transcriptional feedback loops, and RNA silencing (32). Processes in the gene × environment interaction that influence gene expression include metabolic processes such as oxidative stress, mitochondrial function, methylation, immune function, and inflammation that are byproducts of influences such as the mothers and fathers immune systems, environmental toxicants, and diet to name a few. This section will review these epigenetic influences associated with ASD.
Studies show that DNA methylation differences can occur in many loci including AFF2, AUTS2, GABRB3, NLGN3, NRXN1, SLC6A4, UBE3A (31), the oxytocin receptor (33), MeCP2 (a cause for most cases of Rett syndrome) in the frontal cortex (34), and changed chromatin structure in prefrontal cortex neurons at hundreds of loci (35). The severity of the autistic phenotype is related to DNA methylation at specific sites across the genome (31). Environmental and physiological influences are important factors accounting for interindividual DNA methylation differences, and these influences differ across the genome (36). The following sections describe markers for metabolic pathways and environmental influences that can effect epigenetic changes.
Metabolic Biomarkers
There are no autism-defining, metabolic biomarkers, but examining the biomarkers of pathways associated with ASD can point to potentially treatable metabolic abnormalities and provide a baseline that can be tracked over time. Each child may have different metabolic pathologies related to SNPs, nutrient deficiencies, and toxic exposures. Examples of metabolic disorders that can lead to an autistic-like presentation include phenylketonuria (PKU) (37), disorders of purine metabolism (38), biotinidase deficiency (39), cerebral folate deficiency (40), creatine deficiency (41), and excess propionic acid (which is produced by Clostridium) (42, 43).
A recent review assessed the research on physiological abnormalities associated with ASD (44). The authors identified four main mechanisms that have been increasingly studied during the past decade: immunologic/inflammation, oxidative stress, environmental toxicants, and mitochondrial abnormalities. In addition, there is accumulating research on the lipid, GI systems, microglial activation, and the microbiome, and how these can also contribute to generating biomarkers associated with ASD (45, 46).
Pathways are interconnected with a defect in one likely leading to dysfunction in others. Many metabolic disorders can lead to endpoints such as impaired methylation, sulfuration, and detoxification pathways and nutritional deficiencies. Mitochondrial dysfunction, environmental risk factors, metabolic imbalances, and genetic susceptibility can all lead to oxidative stress (47), which in turn leads to inflammation, damaged cell membranes, autoimmunity (48), impaired methylation (49), cell death (48), and neurological deficits (50). The brain is highly vulnerable to oxidative stress (51), particularly in children (52) during the early part of development (47). As environmental events and metabolic imbalances affect oxidative stress and methylation, they also can affect the expression of genes.
Several studies have detected altered levels of a large collection of substances in body-based fluids from ASD subjects compared to controls (e.g., serum, whole-blood, and CSF) (53). These findings encompass either of two main disease-provoking mechanisms: a CNS disorder that is being detected peripherally [e.g., serotonin and its metabolites, sulfate (54), low platelet levels of gamma-aminobutyric acid (GABA) (55), low oxytocin (which affects social affiliation) (56), and low vitamin D levels (57, 58)] or a systemic abnormality that has repercussions in the brain (59).
Serotonin in the brain promotes prosocial behavior and correct assessment of emotional, social cues (60) and can contribute to immune abnormalities (61). Oxytocin can affect social affiliation and social communication deficits (62). Vitamin D has many effects including regulating serotonin synthesis, reducing maternal antibodies that attack the fetal brain, modulating oxytocin synthesis, lowering GI inflammation by lowering gut serotonin (58), DNA repair, anti-inflammatory actions, anti-autoimmune activities, antiseizure activity, increase in regulatory T cells, mitochondrial protection, stimulation of antioxidant pathway (63), and increasing glutathione (64).
Oxidative Stress Markers
Oxidative stress can be detected by studying antioxidant status, antioxidant enzymes, lipid peroxidation, and protein/DNA oxidation, all of which have been found to be elevated in children with autism (Table 2). Different subgroups of children with ASD have different redox abnormalities, which may arise from various sources (65). A recent meta-analysis from 29 studies of blood samples from subjects with ASD shows that reduced levels of glutathione, glutathione peroxidase, methionine, and cysteine along with increased levels of oxidized glutathione are statistically different in ASD (66). The level of antioxidants excreted in urine was found to be significantly lower than normal in autistic children. These findings correlated with the severity of the ASD (67).

Table 2. Oxidative stress biomarkers in ASD (see text for references).
Measurements of antioxidant status include measurement of glutathione, the primary antioxidant in the protection against oxidative stress, neuroinflammation, and mitochondrial damage (68, 69). Glutathione is instrumental in regulating detoxification pathways and modulates the production of precursors to advanced glycation end products (AGEs) (70). Measuring reduced glutathione, oxidized glutathione, or the ratio of reduced glutathione to oxidized glutathione helps determine the patient’s oxidation status. In many patients with ASD, the ratio of reduced glutathione to oxidized glutathione is decreased, indicating a poor oxidation status (71).
The enzyme glutathione peroxidase has been used as a marker and is typically reduced. There are mixed results concerning the enzyme levels of superoxide dismutase (SOD) (72). Other markers for glutathione inadequacy include alpha hydroxybutyrate, pyroglutamate, and sulfate, which can be assessed in an organic acid test. Lipid peroxidation refers to the oxidative degradation of cell membranes. There is a significant correlation between the severity autism and urinary lipid peroxidation products (67), which are increased in patients with ASD.
Plasma F2t-Isoprostanes (F2-IsoPs) are the most sensitive indicator of redox dysfunction and are considered by some to be the gold standard measure of oxidative stress (73). They are increased in patients with ASD and are even higher when accompanied by gastrointestinal dysfunction (73). F2t-isoprostanes (F2-IsoPs) can be measured in the urine as well.
Urine 8-OHdG is biomarker for oxidative damage to DNA. It is commonly used although there are confounding factors and intra individual variations (74) and some researchers have reported that the increases in urine 8-OHdG in patients with ASD is not significant. The increases in urine 8-OHdG did not reach statistical significance (75).
Decreased levels of major antioxidant serum proteins transferrin (iron-binding protein) and ceruloplasmin (copper binding protein) have been observed in patients with ASD. The levels of reduction in these proteins correlate with loss of previously acquired language (47) although there are mixed reviews of the significance of this (66).
Plasma 3-chlortyrosine (3CT), a measure of reactive nitrogen species and myeloperoxidase activity, is an established biomarker of chronic inflammatory response. Plasma 3CT levels reportedly increased with age for those with ASD and mitochondrial dysfunction but not for those with ASD without mitochondrial dysfunction (65).
3-Nitrotyrosine (3NT) is a plasma measure of chronic immune activation and is a biomarker of oxidative protein damage and neuron death. This measure correlates with several measures of cognitive function, development, and behavior for subjects with ASD and mitochondrial dysfunction but not for subjects with ASD without a mitochondrial dysfunction (65).
Mitochondrial Dysfunction Markers
Mitochondrial dysfunction is marked by impaired energy production. Some children with ASD are reported to have a spectrum of mitochondrial dysfunction of differing severity (44) (Table 3). Mitochondrial dysfunction, most likely an early event in neurodegeneration (76), is one of the more common dysfunctions found in autism (77) and is more common than in typical controls (78). There is no reliable biomarker to identify all cases of mitochondrial dysfunction (79). It is possible that up to 80% of the mitochondrial dysfunction in patients with both ASD and a mitochondrial disorder are acquired rather than inherited (44).

Table 3. Mitochondrial function biomarkers in ASD (see text for references).
Mitochondrial dysfunction can be a downstream consequence of many proposed factors including dysreactive immunity and altered calcium (Ca2+) signaling (80), increased nitric oxide and peroxynitrite (68), propionyl CoA (81), malnutrition (82), vitamin B6 or iron deficiencies (83), toxic metals (83), elevated nitric acid (84, 85), oxidative stress (86), exposure to environmental toxicants, such as heavy metals (87–89), chemicals (90), polychlorinated biphenyls (PCBs) (91), pesticides (92, 93), persistent organic pollutants (POPs) (94), and radiofrequency radiation (95). Other sources of mitochondrial distress include medications such as valproic acid (VPA), which inhibits oxidative phosphorylation (96) and neuroleptics (97, 98).
Markers of mitochondrial dysfunction include lactate, pyruvate and lactate-to-pyruvate ratio, carnitine (free and total), quantitative plasma amino acids, ubiquinone, ammonia, CD, AST, ALT, CO2 glucose, and creatine kinase (CK) (44). Many studies of ASD report elevations in lactate and pyruvate, others report a decrease in carnitine, while others report abnormal alanine in ASD patients (44) or elevations in aspartate aminotransferase and serum CK (99). Increases in lactate are not specific and may only occur during illness, after exercise or struggling during a blood draw (100).
Rossignol and Frye (44) recommend a mitochondrial function screening algorithm. This includes fasting morning labs of lactate, pyruvate, carnitine (free and total), acyl carnitine panel, quantitative plasma amino acids, ubiquinone, ammonia, CK, AST/ALT, CO2, and glucose (44). The interpretation of such a panel and the indications for specific treatments has not yet been established.
Methylation
The methylation pathway provides methyl groups for many functions, including the methylation of genes, which can result in the epigenetic changes of turning genes on and off (Table 4). This transfer occurs when S-adenosylmethionine (SAM) donates a methyl group and is transformed to S-adenosylhomocysteine (SAH). SAH can be transferred to homocysteine, which can either be re-methylated to methionine or be transferred by the sulfuration pathway to cysteine to create glutathione. With increased oxidative stress, SAH might be diverted away from the methylation pathway to the sulfuration pathway in order to make more glutathione. This will result in less methionine and less methylation ability.

Table 4. Methylation biomarkers in ASD (see text for references).
Impaired methylation may reflect the effects of toxic exposure on sulfur metabolism. Oxidative stress initiated by environmental factors in genetically vulnerable individuals, can lead to impaired methylation and neurological deficits (49) both of which may contribute to the manifestation of autism (71).
A marker of methylation dysfunction is decreased SAM/SAH ratio in patients with ASD. Fasting plasma methionine decreases since through SAM it is the main methyl donor. Fasting plasma cysteine, a sulfur containing amino acid is the rate-limiting step in the production of glutathione and is significantly decreased. Plasma sulfate is decreased, which may impair detoxification pathways. Homocysteine is generally increased, but the studies are mixed (66). Vitamin B12 and folate are required for the methylation pathway. The MTHFR genetic SNP is reported to heavily influence the methylation pathway (66).
Immune Dysregulation
Cytokine evaluation
Chronic inflammation and microglia cell activation is present in autopsied brains of people with ASD (101, 102) (Table 5). Factors that increase the risk of activating brain microglia include traumatic brain injury (TBI) (103) reactive oxygen species (104) and a dysfunctional blood brain barrier (105). The blood brain barrier can be compromised by oxidative stress (106), acutely stressful situations (107), elevated homocysteine (108), diabetes (109), and hyperglycemia (110). Cytokines can pass through a permeable blood brain barrier and start this process (111). Hence, cytokines can serve as a marker of the immune dysregulation, which can further complicate ASD.

Table 5. Immune biomarkers in ASD (see text for references).
Irregular cytokines profiles are found in ASD (112, 113) and elevations in plasma cytokines are reportedly correlated with regressive onset and severity of autistic and behavioral symptoms (113). Altered pro-inflammatory cytokines, complement proteins, chemokines, adhesion molecules, and growth factors are correlated with ASD. More specifically, altered TGF-beta, CCL2, and CCL5, IgM and IgG classes of immunoglobulin circulating levels are linked with a worsening of behavioral scores (114). An imbalance in Th1/Th2 has are found as well, which may play a role in the pathogenesis of autism (115).
Neopertin as a urine marker of immune dysfunction and activation. Neopterin is associated with increased production of reactive oxygen systems and can be considered as a measurement of the oxidative stress elicited by the immune system. Neopterin levels are found to be significantly higher in children with autism than in the comparison subjects (116).
Increased S100B protein, a calcium binding protein produced primarily by astrocytes, is a biomarker reflecting neurological/brain damage found elevated in ASD and correlated to autistic severity (117).
Autoimmunity and Maternal Antibodies
Autoimmune autistic disorder is proposed as a major subset of autism (118), and autoimmunity may play a role in the pathogenesis of language and social developmental abnormalities in a subset of children with these disorders (119). There are many autoantibodies found in the nervous system of children with ASD who have a high level of brain antibodies (120, 121). These can be measured as biomarkers in this subset of ASD patients. The anti ganglioside M1 antibodies (122), antineuronal antibodies (123), and serum anti-nuclear antibodies (123, 124) correlate with the severity of autism. Other autoantibodies postulated to play a pathological role in autism include: anti neuron-axon filament protein (anti-NAFP) and glial fibrillary acidic protein (anti-GFAP) (125), antibodies to brain endothelial cells and nuclei (119), antibodies against myelin basic protein (126, 127), and anti myelin associated glycoprotein, an index for autoimmunity in the brain (128). BDNF antibodies were found higher in ASD (129), and low BDNF levels may be involved in the pathophysiology of ASD (130).
Antibodies in patients with autism are found to cells in the caudate nucleus (131), cerebellum (132, 133), hypothalamus and thalamus (121), the cingulate gyrus (134), and to cerebral folate receptors (135). Children with cerebellar autoantibodies had lower adaptive and cognitive function as well as increased aberrant behaviors compared to children without these antibodies (132).
Mother’s Immune Status
Research studies indicate an association between viral or bacterial infections in expectant mothers and their ASD offspring (136, 137). Maternal antibodies cross the underdeveloped blood brain barrier of the fetus (138) leading to impaired fetal neurodevelopment and long-term neurodegeneration, neurobehavioral, and cognitive difficulties (139).
A maternal infection or immune response includes cytokines, which affect aspects of fetal neurogenesis, neuronal migration (140), synaptic plasticity, and stem cell fate (141). Elevated serum IFN-γ, IL-4, and IL-5 were more common in women who gave birth to a child subsequently diagnosed with ASD (142). Fetal IL-6 exposure, especially in late pregnancy, leads abnormalities of hippocampal structural and morphology, and decreased learning during adulthood (139).
Some of the antibodies that cross the fetal developing blood brain barrier recognize and attack the brain (138). The presence of fetal brain protein antibodies in ASD can result in an inappropriate approach to unfamiliar peers (143).
Braunschweig et al. developed a panel of clinically significant maternal autoantibody-related autoantibody biomarkers with over 99% specificity for autism risk (144). This panel is suggested to lead to an early diagnosis of maternal autoantibody-related autism, allow for interventions that limit fetal exposure to these antibodies and allow for early behavioral intervention.
Dysbiosis
When the gut becomes inflamed, it breaks down and becomes permeable, sometimes referred to as dysbiosis. Dysbiosis is reported to be an upstream contributing factor to autoimmune conditions and inflammation. Markers under consideration include circulating antibodies against tight junction proteins, LPS, actomyosin (145) calprotectin (146), and lactoferrin (147). Dysbiosis was found in 25.6% of patients with ASD (148). It is proposed to have a direct effect on the brain as it is a hypothesized source of inflammation (149–151) and autoimmunity (152, 153), possibly through molecular mimicry (154). Diet is one source of dysbiosis (155).
Amino Acids and Neuropeptides
Platelet hyperserotonemia is considered one of the most consistent neuromodulator findings in patients with ASD (Table 6). As for other neuropeptides, a recent review reported approximately 15 components that are altered in ASD compared to controls (53). Among them, interesting research has been done on glutamate, GABA, BDNF, and dopamine and noradrenaline systems. A recent study reported a positive correlation between severity of clinical symptoms and plasma GABA levels in patients with ASD, supporting the idea of a disrupted GABAergic system (156). Additionally, a similar grouping of substances measured in the urine is suggested as a more convenient and less invasive way to draw information on these patients (41).

Table 6. Other potential biomarkers in ASD.
Fatty Acid Analysis
Abnormal fatty acid metabolism may play a role in the pathogenesis of ASD and may suggest some metabolic or dietary abnormalities in the regressive form of autism (42, 157). There is evidence of a relationship between changes in brain lipid profiles and the occurrence of ASD-like behaviors using a rodent model of autism (42). Hyperactivity in patients was inversely related to the fluidity of the erythrocyte membrane and membrane polyunsaturated fatty acid (PUFA) levels (158). Imbalances of membrane fatty acid composition and PUFA loss can affect ion channels and opiate, adrenergic, insulin receptors (159) and the modulation of (Na + K)-ATPase activity (160). Analysis of red blood cell membrane fatty acids is a very sensitive indicator of tissue status and may reflect the brain fatty acid composition (161).
Seventeen percent of children with ASD manifest biomarkers of abnormal mitochondrial fatty acid metabolism, the majority of which are not accounted for by genetic mechanisms (162). Patients with ASD had reduced percentages of highly unsaturated fatty acids (163) and an increase in ω6/ω3 ratio (158).
Environmental Toxicants
For environmental toxicant biomarkers, it is difficult to interpret abnormal levels in ASD. For instance, a high burden of aluminum, cadmium, lead, mercury, and arsenic was found in a subgroup of a sample of over 500 patients with ASD (164). Other studies have described decreased levels of some of these heavy metals in urine and in hair samples, which may imply that the body is not excreting the heavy metals adequately (41).
A systematic review of toxicant-related studies in ASD found that pesticides, phthalates, PCBs, solvents, toxic waste sites, air pollutants, and heavy metals were implicated in ASD, with the strongest evidence found for air pollutants and pesticides (165).
Brain Focused Biomarkers
Magnetic Resonance Imaging
Like other areas in psychiatry, new approaches are being devised to tackle ASD in a “bottom-up paradigm” – that is, identifying genetic or biological alterations, which are associated with the clinical manifestations of symptoms. In neuroimaging, much progress has been made toward understanding the condition, but only very few observed biomarkers have sufficient evidence to suggest that they might hold diagnostic or treatment significance.
One of the best-replicated brain findings from subjects with ASD is an early-accelerated brain volume growth. The increase is usually around 10%, peaking between 2 and 4 years of age followed by a plateau (166). Head circumference (HC), an adequate proxy for brain size, is being investigated for diagnostic relevance for ASD (167). However, recent findings on HC in ASD show that there might be an unrelated growth in HC in both patients and controls. Thus, the abnormal overgrowth observed in older studies might be because of a biased Center for Disease Control (CDC) HC norm, which is commonly used as the control group (168).
Gray matter thickness and surface areas and white matter integrity are also being studied. A general trend demonstrating increased gray matter thickness in subjects with ASD compared to controls is observed with an age-dependent effect (166). Even though there are studies correlating symptom severity with altered thickness there are several limitations such as using a cross-sectional approach and a small number of subjects that hinder clinical application (169). Likewise, diffusion tensor imaging (DTI) studies on white matter connectivity are not yet conclusive across studies.
Early studies using functional magnetic resonance imaging (fMRI) focus on task specific cognitive networks (e.g., face recognition, theory of mind, imitation, language processing, and proxies for receptive behavior) (166). In these cognitive network studies, individuals with ASD and controls perform a task while the fMRI is monitored. More recently, researchers are investigating the connectivity between these network and resting-state methods where fMRI is obtained while a subject is at rest and not performing a task. These more recent studies reveal a pattern that suggests less activity in the brain areas that typically perform executive function tasks (such as organization or planning). This combination of activity patterns in ASD is often called a “high noise-information ratio,” supporting an excitatory/inhibitory imbalance theory of ASD (170). Conversely, even though all these fMRI findings shed light on the pathophysiology of ASD, they also are not mature enough to translate into a reliable biomarker that can be used in clinical practice.
Electroencephalography
Aligned with the notion that ASD is an abnormal connectivity disorder, studies using electroencephalography (EEG) have explored local changes in signal complexities in patients (171). Some studies were able to detect abnormalities as early as 6 months of age, suggesting an important tool for early detection and risk group assessment (172). However, despite findings like multi-scale entropy differences being proposed as an early diagnostic biomarker, EEG has not yet been established as a reliable tool for diagnosis or to document clinical changes (173).
Neurochemistry
Neuroimaging techniques also are used to monitor in vivo concentration of substances in the brain, and include positron emission tomography (PET), single photon emission tomography (SPECT), and magnetic resonance spectroscopy (MRS). So far, the majority of studies report abnormalities in several of neurotransmitter networks and their respective metabolites (e.g., dopamine, GABA, serotonin, glutamate, and N-acetyl-aspartate), varying from synthesis, transport, and receptor activity in different regions of the brain in the glutamate–glutamine system, in particular, there appears to be either hyper (174) or hypoglutamatergic (175) states depending on the brain region, which could be interpreted as an excitatory increase relative to inhibition in key neural circuits (176). In addition, studies pointing toward GABA alterations also are accumulating, with findings of reduced levels of GABA in the frontal lobes of subjects with ASD. Using MRS (177), corroborated the histopathologic research on altered density and distribution of the GABA receptors (178).
Biomedical Interventions
There are no published studies of interventions for ASD that use neuroimaging or genetic biomarkers in a prospective manner to guide treatment. Biomedical interventions based on body fluid/product biomarkers have been used in a small but growing numbers of well designed, published studies. Several recent reviews summarize these (179–181).
Future Research Directions
A common feature of all prior studies of these putative biomarkers is that most consist of small samples of patients, and therefore, do not grasp the heterogeneity that characterizes ASD. Also, since they mainly compare subjects with ASD to typically developing controls, it is uncertain whether these biomarker profiles are unique to ASD – they may be present in other neurodevelopmental disorders. A promising new method that is designed to increase specificity of biomarkers in ASD is the multiplex immunoassay, a method that analyzes sets of biomarkers to create a diagnostic profile (182, 183). Furthermore, advances in chromatographic and proteomic techniques are also contributing to the progress of the field, allowing easier assessment of several substances (184, 185).
Thus far, numerous studies examining a diverse set of potential biomarkers have found a large number of genetic, imaging, and metabolic tests that are abnormal in children with ASD compared to control subjects. For most of these measures, it is not yet clear if the abnormal biomarker is a contributing factor to the development of ASD or a result of another underlying abnormality (i.e., causal or merely associated). Not surprisingly, the conclusion is that more studies are needed to further explore these possible mechanisms individually. However, the future in the ASD research might involve a broader view of these biomarkers, which might hold more value in combination than in isolation. As a result of new technological advances, it is possible to use a machine learning technique that is trained to identify complex patterns of data that can be applied to new individuals to make predictions (186). A recent study pooled regional white and gray matter volumes of whole-brain MRI scans in ASD subjects using this computer algorithm program, known as super vector machine. As a result, they could classify a new patient as having an ASD diagnosis or not with a high true positive rate (187). Although exemplified with neuroimaging, this approach could be generalized to other biomarkers (53, 188). In other words, individually insignificant biomarkers when analyzed together might generate a pattern of clinical relevance like diagnosis, severity staging, or response to treatment. These techniques might also be able to identify the most relevant or most predictive biomarkers among the many candidate biomarkers described above.
Although the maxim that “further studies are needed” still holds, ASDs may be witnessing the emergence of clinically relevant biomarkers in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hendren RL, Bertoglio K, Ashwood P, Sharp F. Mechanistic biomarkers for autism treatment. Med Hypotheses (2009) 73:950–4. doi:10.1016/j.mehy.2009.06.032
2. Insel T. Director’s Blog: A New Approach to Clinical Trials [Online] (2014). Available from: http://www.nimh.nih.gov/about/director/2014/a-new-approach-to-clinical-trials.shtml
3. Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry (2010) 167:748–51. doi:10.1176/appi.ajp.2010.09091379
4. Cuthbert BN, Insel TR. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med (2013) 11:126. doi:10.1186/1741-7015-11-126
5. Hagerman R, Hendren R. Treatments for Neurodevelopmental Disorders: Targeting Neurobiological Mechanisms. New York, NY: Oxford University Press (2014).
6. Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res (2011) 1380:42–77. doi:10.1016/j.brainres.2010.11.078
7. Stessman HA, Bernier R, Eichler EE. A genotype-first approach to defining the subtypes of a complex disease. Cell (2014) 156(5):872–7. doi:10.1016/j.cell.2014.02.002
8. Won H, Mah W, Kim E. Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front Mol Neurosci (2013) 6:19. doi:10.3389/fnmol.2013.00019
9. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry (2011) 68:1095–102. doi:10.1001/archgenpsychiatry.2011.76
10. Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA (2014) 311(17):1770–7. doi:10.1001/jama.2014.4144
11. El-Fishawy P, State MW. The genetics of autism: key issues, recent findings, and clinical implications. Psychiatr Clin North Am (2010) 33:83–105. doi:10.1016/j.psc.2009.12.002
12. State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci (2011) 14:1499–506. doi:10.1038/nn.2924
13. Murdoch JD, State MW. Recent developments in the genetics of autism spectrum disorders. Curr Opin Genet Dev (2013) 23:310–5. doi:10.1016/j.gde.2013.02.003
14. Wang K, Zhang HT, Ma DQ, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature (2009) 459:528–33. doi:10.1038/Nature07999
15. Chango A, Potier De Courcy G, Boisson F, Guilland JC, Barbe F, Perrin MO, et al. 5,10-Methylenetetrahydrofolate reductase common mutations, folate status and plasma homocysteine in healthy French adults of the Supplementation en Vitamines et Mineraux Antioxydants (SU.VI.MAX) cohort. Br J Nutr (2000) 84:891–6.
16. Mohammad NS, Jain JM, Chintakindi KP, Singh RP, Naik U, Akella RR. Aberrations in folate metabolic pathway and altered susceptibility to autism. Psychiatr Genet (2009) 19:171–6. doi:10.1097/YPG.0b013e32832cebd2
17. Schmidt RJ, Hansen RL, Hartiala J, Allayee H, Schmidt LC, Tancredi DJ, et al. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology (2011) 22:476–85. doi:10.1097/EDE.0b013e31821d0e30
18. Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet (2009) 73:263–73. doi:10.1111/j.1469-1809.2009.00523.x
19. Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature (2009) 461:802–8. doi:10.1038/nature08490
20. Ronald A, Butcher LM, Docherty S, Davis OS, Schalkwyk LC, Craig IW, et al. A genome-wide association study of social and non-social autistic-like traits in the general population using pooled DNA, 500 K SNP microarrays and both community and diagnosed autism replication samples. Behav Genet (2010) 40:31–45. doi:10.1007/s10519-009-9308-6
21. Lee TL, Raygada MJ, Rennert OM. Integrative gene network analysis provides novel regulatory relationships, genetic contributions and susceptible targets in autism spectrum disorders. Gene (2012) 496:88–96. doi:10.1016/j.gene.2012.01.020
22. Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet (2012) 21:3513–23. doi:10.1093/hmg/dds164
23. Liu Y, Du Y, Liu W, Yang C, Wang H, Gong X. Lack of association between NLGN3, NLGN4, SHANK2 and SHANK3 gene variants and autism spectrum disorder in a Chinese population. PLoS One (2013) 8:e56639. doi:10.1371/journal.pone.0056639
24. Volaki K, Pampanos A, Kitsiou-Tzeli S, Vrettou C, Oikonomakis V, Sofocleous C, et al. Mutation screening in the Greek population and evaluation of NLGN3 and NLGN4X genes causal factors for autism. Psychiatr Genet (2013) 23:198–203. doi:10.1097/YPG.0b013e3283643644
25. Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron (2012) 74:285–99. doi:10.1016/j.neuron.2012.04.009
26. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature (2012) 485:237–41. doi:10.1038/nature10945
27. Blue L. A Blood Test for Autism? (2012). Available from: http://healthland.time.com/2012/12/06/a-blood-test-for-autism/
28. Hughes V. Genetic Tests for Autism Debut Amid Concerns About Validity [Online] (2012). Available from: http://sfari.org/news-and-opinion/news/genetic-tests-for-autism-debut-amid-concerns-about-validity
29. Goines P, Van de Water J. The immune system’s role in the biology of autism. Curr Opin Neurol (2010) 23:111–7. doi:10.1097/WCO.0b013e3283373514
30. Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav (2012) 11:332–41. doi:10.1111/j.1601-183X.2012.00768.x
31. Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry (2014) 19:495–503. doi:10.1038/mp.2013.41
32. Ma DK, Marchetto MC, Guo JU, Ming GL, Gage FH, Song H. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci (2010) 13:1338–44. doi:10.1038/nn.2672
33. Gregory SG, Connelly JJ, Towers AJ, Johnson J, Biscocho D, Markunas CA, et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med (2009) 7:62. doi:10.1186/1741-7015-7-62
34. Nagarajan RP, Hogart AR, Gwye Y, Martin MR, Lasalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics (2006) 1:e1–11. doi:10.4161/epi.1.4.3514
35. Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry (2012) 69:314–24. doi:10.1001/archgenpsychiatry.2011.151
36. Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, et al. A longitudinal study of epigenetic variation in twins. Epigenetics (2010) 5:516–26. doi:10.4161/epi.5.6.12226
37. Lowe TL, Tanaka K, Seashore MR, Young JG, Cohen DJ. Detection of phenylketonuria in autistic and psychotic children. JAMA (1980) 243:126–8. doi:10.1001/jama.1980.03300280024022
38. Bottini N, De Luca D, Saccucci P, Fiumara A, Elia M, Porfirio MC, et al. Autism: evidence of association with adenosine deaminase genetic polymorphism. Neurogenetics (2001) 3:111–3. doi:10.1007/s100480000104
39. Zaffanello M, Zamboni G, Fontana E, Zoccante L, Tato L. A case of partial biotinidase deficiency associated with autism. Child Neuropsychol (2003) 9:184–8. doi:10.1076/chin.9.3.184.16457
40. Moretti P, Peters SU, Del Gaudio D, Sahoo T, Hyland K, Bottiglieri T, et al. Brief report: autistic symptoms, developmental regression, mental retardation, epilepsy, and dyskinesias in CNS folate deficiency. J Autism Dev Disord (2008) 38:1170–7. doi:10.1007/s10803-007-0492-z
41. Wang L, Angley MT, Gerber JP, Sorich MJ. A review of candidate urinary biomarkers for autism spectrum disorder. Biomarkers (2011) 16:537–52. doi:10.3109/1354750X.2011.598564
42. Thomas RH, Foley KA, Mepham JR, Tichenoff LJ, Possmayer F, Macfabe DF. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents: further development of a potential model of autism spectrum disorders. J Neurochem (2010) 113:515–29. doi:10.1111/j.1471-4159.2010.06614.x
43. Macfabe DF. Short-chain fatty acid fermentation products of the gut microbiome: implications in autism spectrum disorders. Microb Ecol Health Dis (2012) 23. doi:10.3402/mehd.v23i0.19260
44. Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry (2012) 17:290–314. doi:10.1038/mp.2010.136
45. El-Ansary A, Al-Ayadhi L. Lipid mediators in plasma of autism spectrum disorders. Lipids Health Dis (2012) 11:160. doi:10.1186/1476-511X-11-160
46. Woods AG, Sokolowska I, Taurines R, Gerlach M, Dudley E, Thome J, et al. Potential biomarkers in psychiatry: focus on the cholesterol system. J Cell Mol Med (2012) 16:1184–95. doi:10.1111/j.1582-4934.2012.01543.x
47. Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology (2006) 13:171–81. doi:10.1016/j.pathophys.2006.05.007
48. Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest (2003) 111:785–93. doi:10.1172/JCI18182
49. Deth R, Muratore C, Benzecry J, Power-Charnitsky VA, Waly M. How environmental and genetic factors combine to cause autism: a redox/methylation hypothesis. Neurotoxicology (2008) 29:190–201. doi:10.1016/j.neuro.2007.09.010
50. Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev (2006) 9:485–99. doi:10.1080/10937400600882079
51. Perry SW, Norman JP, Litzburg A, Gelbard HA. Antioxidants are required during the early critical period, but not later, for neuronal survival. J Neurosci Res (2004) 78:485–92. doi:10.1002/jnr.20272
52. Erden-Inal M, Sunal E, Kanbak G. Age-related changes in the glutathione redox system. Cell Biochem Funct (2002) 20:61–6. doi:10.1002/cbf.937
53. Ratajczak HV. Theoretical aspects of autism: biomarkers – a review. J Immunotoxicol (2011) 8:80–94. doi:10.3109/1547691X.2010.538749
54. Seneff S, Lauritzen A, Davidson RM, Lentz-Marino L. Is encephalopathy a mechanism to renew sulfate in autism? Entropy (2013) 15:372–406. doi:10.3390/e15010372
55. Baribeau DA, Anagnostou E. Social communication is an emerging target for pharmacotherapy in autism spectrum disorder – a review of the literature on potential agents. J Can Acad Child Adolesc Psychiatry (2014) 23:20–30.
56. Modahl C, Green L, Fein D, Morris M, Waterhouse L, Feinstein C, et al. Plasma oxytocin levels in autistic children. Biol Psychiatry (1998) 43:270–7. doi:10.1016/S0006-3223(97)00439-3
57. Mostafa GA, Al-Ayadhi LY. Reduced serum concentrations of 25-hydroxy vitamin D in children with autism: relation to autoimmunity. J Neuroinflammation (2012) 9:201. doi:10.1186/1742-2094-9-201
58. Patrick RP, Ames BN. Vitamin D hormone regulates serotonin synthesis. Part 1: relevance for autism. FASEB J (2014) 28(6):2398–413. doi:10.1096/fj.13-246546
59. Herbert MR, Ziegler DA, Deutsch CK, O’Brien LM, Kennedy DN, Filipek PA, et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain (2005) 128:213–26. doi:10.1093/brain/awh330
60. Crockett MJ. The neurochemistry of fairness: clarifying the link between serotonin and prosocial behavior. Ann N Y Acad Sci (2009) 1167:76–86. doi:10.1111/j.1749-6632.2009.04506.x
61. Burgess NK, Sweeten TL, McMahon WM, Fujinami RS. Hyperserotoninemia and altered immunity in autism. J Autism Dev Disord (2006) 36:697–704. doi:10.1007/s10803-006-0100-7
62. Watanabe T, Abe O, Kuwabara H, Yahata N, Takano Y, Iwashiro N, et al. Mitigation of sociocommunicational deficits of autism through oxytocin-induced recovery of medial prefrontal activity: a randomized trial. JAMA Psychiatry (2014) 71:166–75. doi:10.1001/jamapsychiatry.2013.3181
63. Cannell JJ, Grant WB. What is the role of vitamin D in autism? Dermatoendocrinol (2013) 5:199–204. doi:10.4161/derm.24356
64. Jain SK, Micinski D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem Biophys Res Commun (2013) 437:7–11. doi:10.1016/j.bbrc.2013.06.004
65. Frye RE, Delatorre R, Taylor H, Slattery J, Melnyk S, Chowdhury N, et al. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Transl Psychiatry (2013) 3:e273. doi:10.1038/tp.2013.51
66. Frustaci A, Neri M, Cesario A, Adams JB, Domenici E, Dalla Bernardina B, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med (2012) 52:2128–41. doi:10.1016/j.freeradbiomed.2012.03.011
67. Damodaran LP, Arumugam G. Urinary oxidative stress markers in children with autism. Redox Rep (2011) 16:216–22. doi:10.1179/1351000211Y.0000000012
68. Vali S, Mythri RB, Jagatha B, Padiadpu J, Ramanujan KS, Andersen JK, et al. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson’s disease: a dynamic model. Neuroscience (2007) 149:917–30. doi:10.1016/j.neuroscience.2007.08.028
69. Ghanizadeh A, Akhondzadeh S, Hormozi M, Makarem A, Abotorabi-Zarchi M, Firoozabadi A. Glutathione-related factors and oxidative stress in autism, a review. Curr Med Chem (2012) 19:4000–5. doi:10.2174/092986712802002572
70. Maher P. Methylglyoxal, advanced glycation end products and autism: is there a connection? Med Hypotheses (2012) 78:548–52. doi:10.1016/j.mehy.2012.01.032
71. James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr (2004) 80:1611–7.
72. Sogut S, Zoroglu SS, Ozyurt H, Yilmaz HR, Ozugurlu F, Sivasli E, et al. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin Chim Acta (2003) 331:111–7. doi:10.1016/S0009-8981(03)00119-0
73. Gorrindo P, Lane CJ, Lee EB, McLaughlin B, Levitt P. Enrichment of elevated plasma F2t-isoprostane levels in individuals with autism who are stratified by presence of gastrointestinal dysfunction. PLoS One (2013) 8:e68444. doi:10.1371/journal.pone.0068444
74. Sajous L, Botta A, Sari-Minodier I. [Urinary 8-hydroxy-2’-deoxyguanosine: a biomarker of environmental oxidative stress?]. Ann Biol Clin (2008) 66:19–29. doi:10.1684/abc.2008.0188
75. Ming X, Stein TP, Brimacombe M, Johnson WG, Lambert GH, Wagner GC. Increased excretion of a lipid peroxidation biomarker in autism. Prostaglandins Leukot Essent Fatty Acids (2005) 73:379–84. doi:10.1016/j.plefa.2005.06.002
76. Fernandez-Checa JC, Fernandez A, Morales A, Mari M, Garcia-Ruiz C, Colell A. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: lessons from mouse models. CNS Neurol Disord Drug Targets (2010) 9:439–54. doi:10.2174/187152710791556113
77. Oliveira G, Diogo L, Grazina M, Garcia P, Ataide A, Marques C, et al. Mitochondrial dysfunction in autism spectrum disorders: a population-based study. Dev Med Child Neurol (2005) 47:185–9. doi:10.1017/S0012162205000332
78. Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, et al. Mitochondrial dysfunction in autism. JAMA (2010) 304:2389–96. doi:10.1001/jama.2010.1706
79. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics (2007) 120:1326–33. doi:10.1542/peds.2007-0391
80. Palmieri L, Persico AM. Mitochondrial dysfunction in autism spectrum disorders: cause or effect? Biochim Biophys Acta (2010) 1797:1130–7. doi:10.1016/j.bbabio.2010.04.018
81. Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, Van Den Heuvel LP, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J (2006) 398:107–12. doi:10.1042/BJ20060221
82. Morava E, Rodenburg R, Van Essen HZ, De Vries M, Smeitink J. Dietary intervention and oxidative phosphorylation capacity. J Inherit Metab Dis (2006) 29:589. doi:10.1007/s10545-006-0227-x
83. Atamna H, Killilea DW, Killilea AN, Ames BN. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc Natl Acad Sci U S A (2002) 99:14807–12. doi:10.1073/pnas.192585799
84. Bolanos JP, Peuchen S, Heales SJ, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem (1994) 63:910–6. doi:10.1046/j.1471-4159.1994.63030910.x
85. Husain M, Bourret TJ, McCollister BD, Jones-Carson J, Laughlin J, Vazquez-Torres A. Nitric oxide evokes an adaptive response to oxidative stress by arresting respiration. J Biol Chem (2008) 283:7682–9. doi:10.1074/jbc.M708845200
86. Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, et al. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol (1997) 273:G7–17.
87. Goyer RA. Toxic and essential metal interactions. Annu Rev Nutr (1997) 17:37–50. doi:10.1146/annurev.nutr.17.1.37
88. Shenker BJ, Guo TL, O I, Shapiro IM. Induction of apoptosis in human T-cells by methyl mercury: temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol Appl Pharmacol (1999) 157:23–35. doi:10.1006/taap.1999.8652
89. Pourahmad J, Mihajlovic A, O’Brien PJ. Hepatocyte lysis induced by environmental metal toxins may involve apoptotic death signals initiated by mitochondrial injury. Adv Exp Med Biol (2001) 500:249–52. doi:10.1007/978-1-4615-0667-6_38
90. Hiura TS, Li N, Kaplan R, Horwitz M, Seagrave JC, Nel AE. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. J Immunol (2000) 165:2703–11. doi:10.4049/jimmunol.165.5.2703
91. Wong PW, Garcia EF, Pessah IN. Ortho-substituted PCB95 alters intracellular calcium signaling and causes cellular acidification in PC12 cells by an immunophilin-dependent mechanism. J Neurochem (2001) 76:450–63. doi:10.1046/j.1471-4159.2001.00022.x
92. Yamano T, Morita S. Effects of pesticides on isolated rat hepatocytes, mitochondria, and microsomes II. Arch Environ Contam Toxicol (1995) 28:1–7. doi:10.1007/BF00213961
93. Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, et al. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem (2007) 100:1469–79. doi:10.1111/j.1471-4159.2006.04333.x
94. Lim S, Cho YM, Park KS, Lee HK. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome. Ann N Y Acad Sci (2010) 1201:166–76. doi:10.1111/j.1749-6632.2010.05622.x
95. Xu S, Zhou Z, Zhang L, Yu Z, Zhang W, Wang Y, et al. Exposure to 1800 MHz radiofrequency radiation induces oxidative damage to mitochondrial DNA in primary cultured neurons. Brain Res (2010) 1311:189–96. doi:10.1016/j.brainres.2009.10.062
96. Haas R, Stumpf DA, Parks JK, Eguren L. Inhibitory effects of sodium valproate on oxidative phosphorylation. Neurology (1981) 31:1473–6. doi:10.1212/WNL.31.11.1473
97. Casademont J, Garrabou G, Miro O, Lopez S, Pons A, Bernardo M, et al. Neuroleptic treatment effect on mitochondrial electron transport chain: peripheral blood mononuclear cells analysis in psychotic patients. J Clin Psychopharmacol (2007) 27:284–8. doi:10.1097/JCP.0b013e318054753e
98. Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem (2008) 283:21134–44. doi:10.1074/jbc.M801954200
99. Poling JS, Frye RE, Shoffner J, Zimmerman AW. Developmental regression and mitochondrial dysfunction in a child with autism. J Child Neurol (2006) 21:170–2. doi:10.1177/08830738060210021401
100. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab (2008) 94:16–37. doi:10.1016/j.ymgme.2007.11.018
101. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57:67–81. doi:10.1002/ana.20315
102. Blaylock RL. A possible central mechanism in autism spectrum disorders, part 1. Altern Ther Health Med (2008) 14:46–53.
103. Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol (2000) 28:28–30. doi:10.1177/019262330002800104
104. Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics (2010) 7:354–65. doi:10.1016/j.nurt.2010.05.014
105. Denieffe S, Kelly RJ, McDonald C, Lyons A, Lynch MA. Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain Behav Immun (2013) 34:86–97. doi:10.1016/j.bbi.2013.07.174
106. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res (2009) 43:348–64. doi:10.1080/10715760902751902
107. Esposito P, Gheorghe D, Kandere K, Pang X, Connolly R, Jacobson S, et al. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res (2001) 888:117–27. doi:10.1016/S0006-8993(00)03026-2
108. Kamath AF, Chauhan AK, Kisucka J, Dole VS, Loscalzo J, Handy DE, et al. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood (2006) 107:591–3. doi:10.1182/blood-2005-06-2506
109. Yi CX, Gericke M, Kruger M, Alkemade A, Kabra DG, Hanske S, et al. High calorie diet triggers hypothalamic angiopathy. Mol Metab (2012) 1:95–100. doi:10.1016/j.molmet.2012.08.004
110. Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke (2007) 38:1044–9. doi:10.1161/01.STR.0000258041.75739.cb
111. Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun (2004) 18:407–13. doi:10.1016/j.bbi.2004.01.004
112. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol (2009) 207:111–6. doi:10.1016/j.jneuroim.2008.12.002
113. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van De Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun (2011) 25:40–5. doi:10.1016/j.bbi.2010.08.003
114. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun (2012) 26:383–92. doi:10.1016/j.bbi.2011.08.007
115. Ashwood P, Wills S, Van De Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol (2006) 80:1–15. doi:10.1189/jlb.1205707
116. Sweeten TL, Posey DJ, McDougle CJ. High blood monocyte counts and neopterin levels in children with autistic disorder. Am J Psychiatry (2003) 160:1691–3. doi:10.1176/appi.ajp.160.9.1691
117. Al-Ayadhi LY, Mostafa GA. A lack of association between elevated serum levels of S100B protein and autoimmunity in autistic children. J Neuroinflammation (2012) 9:54. doi:10.1186/1742-2094-9-54
118. Singh VK. Phenotypic expression of autoimmune autistic disorder (AAD): a major subset of autism. Ann Clin Psychiatry (2009) 21:148–61.
119. Connolly AM, Chez MG, Pestronk A, Arnold ST, Mehta S, Deuel RK. Serum autoantibodies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. J Pediatr (1999) 134:607–13. doi:10.1016/S0022-3476(99)70248-9
120. Ashwood P, Van de Water J. Is autism an autoimmune disease? Autoimmun Rev (2004) 3:557–62. doi:10.1016/j.autrev.2004.07.036
121. Cabanlit M, Wills S, Goines P, Ashwood P, Van De Water J. Brain-specific autoantibodies in the plasma of subjects with autistic spectrum disorder. Ann N Y Acad Sci (2007) 1107:92–103. doi:10.1196/annals.1381.010
122. Mostafa GA, Al-Ayadhi LY. Increased serum levels of anti-ganglioside M1 auto-antibodies in autistic children: relation to the disease severity. J Neuroinflammation (2011) 8:39. doi:10.1186/1742-2094-8-39
123. Mostafa GA, Al-Ayadhi LY. The relationship between the increased frequency of serum antineuronal antibodies and the severity of autism in children. Eur J Paediatr Neurol (2012) 16:464–8. doi:10.1016/j.ejpn.2011.12.010
124. Mostafa GA, Kitchener N. Serum anti-nuclear antibodies as a marker of autoimmunity in Egyptian autistic children. Pediatr Neurol (2009) 40:107–12. doi:10.1016/j.pediatrneurol.2008.10.017
125. Singh VK, Warren R, Averett R, Ghaziuddin M. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr Neurol (1997) 17:88–90. doi:10.1016/S0887-8994(97)00045-3
126. Singh VK, Warren RP, Odell JD, Warren WL, Cole P. Antibodies to myelin basic protein in children with autistic behavior. Brain Behav Immun (1993) 7:97–103. doi:10.1006/brbi.1993.1010
127. Mostafa GA, Al-Ayadhi LY. A lack of association between hyperserotonemia and the increased frequency of serum anti-myelin basic protein auto-antibodies in autistic children. J Neuroinflammation (2011) 8:71. doi:10.1186/1742-2094-8-71
128. Mostafa GA, El-Sayed ZA, El-Aziz MM, El-Sayed MF. Serum anti-myelin-associated glycoprotein antibodies in Egyptian autistic children. J Child Neurol (2008) 23:1413–8. doi:10.1177/0883073808319321
129. Connolly AM, Chez M, Streif EM, Keeling RM, Golumbek PT, Kwon JM, et al. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol Psychiatry (2006) 59:354–63. doi:10.1016/j.biopsych.2005.07.004
130. Taurines R, Segura M, Schecklmann M, Albantakis L, Grunblatt E, Walitza S, et al. Altered peripheral BDNF mRNA expression and BDNF protein concentrations in blood of children and adolescents with autism spectrum disorder. J Neural Transm (2014). doi:10.1007/s00702-014-1162-x
131. Singh VK, Rivas WH. Prevalence of serum antibodies to caudate nucleus in autistic children. Neurosci Lett (2004) 355:53–6. doi:10.1016/j.neulet.2003.10.026
132. Goines P, Haapanen L, Boyce R, Duncanson P, Braunschweig D, Delwiche L, et al. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav Immun (2011) 25:514–23. doi:10.1016/j.bbi.2010.11.017
133. Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral DG, Van De Water J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav Immun (2009) 23:64–74. doi:10.1016/j.bbi.2008.07.007
134. Singer HS, Morris CM, Williams PN, Yoon DY, Hong JJ, Zimmerman AW. Antibrain antibodies in children with autism and their unaffected siblings. J Neuroimmunol (2006) 178:149–55. doi:10.1016/j.jneuroim.2006.05.025
135. Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol Psychiatry (2013) 18:369–81. doi:10.1038/mp.2011.175
136. Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord (2010) 40:1423–30. doi:10.1007/s10803-010-1006-y
137. Atladottir HO, Henriksen TB, Schendel DE, Parner ET. Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics (2012) 130:e1447–54. doi:10.1542/peds.2012-1107
138. Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol (2009) 9:449–56. doi:10.1038/nri2529
139. Samuelsson AM, Jennische E, Hansson HA, Holmang A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol (2006) 290:R1345–56. doi:10.1152/ajpregu.00268.2005
140. Zhu Y, Yu T, Zhang XC, Nagasawa T, Wu JY, Rao Y. Role of the chemokine SDF-1 as the meningeal attractant for embryonic cerebellar neurons. Nat Neurosci (2002) 5:719–20. doi:10.1038/nn881
141. Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci (2007) 8:221–32. doi:10.1038/nrn2054
142. Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R, et al. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol Autism (2011) 2:13. doi:10.1186/2040-2392-2-13
143. Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM, et al. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry (2013) 3:e278. doi:10.1038/tp.2013.47
144. Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry (2013) 3:e277. doi:10.1038/tp.2013.50
145. Vojdani A, Lambert J. The onset of enhanced intestinal permeability and food sensitivity triggered by medication used in dental procedures: a case report. Case Rep Gastrointest Med (2012) 2012:265052. doi:10.1155/2012/265052
146. Erbayrak M, Turkay C, Eraslan E, Cetinkaya H, Kasapoglu B, Bektas M. The role of fecal calprotectin in investigating inflammatory bowel diseases. Clinics (Sao Paulo) (2009) 64:421–5. doi:10.1590/S1807-59322009000500009
147. Gisbert JP, McNicholl AG, Gomollon F. Questions and answers on the role of fecal lactoferrin as a biological marker in inflammatory bowel disease. Inflamm Bowel Dis (2009) 15:1746–54. doi:10.1002/ibd.20920
148. de Magistris L, Picardi A, Siniscalco D, Riccio MP, Sapone A, Cariello R, et al. Antibodies against food antigens in patients with autistic spectrum disorders. Biomed Res Int (2013) 2013:729349. doi:10.1155/2013/729349
149. Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev (2011) 91:151–75. doi:10.1152/physrev.00003.2008
150. Frazier TH, Dibaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr (2011) 35:14S–20S. doi:10.1177/0148607111413772
151. Bengmark S. Gut microbiota, immune development and function. Pharmacol Res (2013) 69:87–113. doi:10.1016/j.phrs.2012.09.002
152. Fasano A. Intestinal permeability and its regulation by zonulin: diagnostic and therapeutic implications. Clin Gastroenterol Hepatol (2012) 10:1096–100. doi:10.1016/j.cgh.2012.08.012
153. Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci (2012) 1258:25–33. doi:10.1111/j.1749-6632.2012.06538.x
155. Brown K, Decoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients (2012) 4:1095–119. doi:10.3390/nu4081095
156. Alabdali A, Al-Ayadhi L, El-Ansary A. Association of social and cognitive impairment and biomarkers in autism spectrum disorders. J Neuroinflammation (2014) 11:4. doi:10.1186/1742-2094-11-4
157. Bu B, Ashwood P, Harvey D, King IB, Water JV, Jin LW. Fatty acid compositions of red blood cell phospholipids in children with autism. Prostaglandins Leukot Essent Fatty Acids (2006) 74:215–21. doi:10.1016/j.plefa.2006.02.001
158. Ghezzo A, Visconti P, Abruzzo PM, Bolotta A, Ferreri C, Gobbi G, et al. Oxidative stress and erythrocyte membrane alterations in children with autism: correlation with clinical features. PLoS One (2013) 8:e66418. doi:10.1371/journal.pone.0066418
159. Murphy MG. Dietary fatty acids and membrane protein function. J Nutr Biochem (1990) 1:68–79. doi:10.1016/0955-2863(90)90052-M
160. Rodrigo R, Bachler JP, Araya J, Prat H, Passalacqua W. Relationship between (Na + K)-ATPase activity, lipid peroxidation and fatty acid profile in erythrocytes of hypertensive and normotensive subjects. Mol Cell Biochem (2007) 303:73–81. doi:10.1007/s11010-007-9457-y
161. Makrides M, Neumann MA, Byard RW, Simmer K, Gibson RA. Fatty acid composition of brain, retina, and erythrocytes in breast- and formula-fed infants. Am J Clin Nutr (1994) 60:189–94.
162. Frye RE, Melnyk S, Macfabe DF. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl Psychiatry (2013) 3:e220. doi:10.1038/tp.2012.143
163. Bell JG, Sargent JR, Tocher DR, Dick JR. Red blood cell fatty acid compositions in a patient with autistic spectrum disorder: a characteristic abnormality in neurodevelopmental disorders? Prostaglandins Leukot Essent Fatty Acids (2000) 63:21–5. doi:10.1054/plef.2000.0186
164. Yasuda H, Yasuda Y, Tsutsui T. Estimation of autistic children by metallomics analysis. Sci Rep (2013) 3:1199. doi:10.1038/srep01199
165. Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry (2014) 4:e360. doi:10.1038/tp.2014.4
166. Anagnostou E, Taylor MJ. Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Mol Autism (2011) 2:4. doi:10.1186/2040-2392-2-4
167. Samango-Sprouse CA, Stapleton EJ, Aliabadi F, Graw R, Vickers R, Haskell K, et al. Identification of infants at risk for autism spectrum disorder and developmental language delay prior to 12 months. Autism (2014). doi:10.1177/1362361314521329
168. Raznahan A, Wallace GL, Antezana L, Greenstein D, Lenroot R, Thurm A, et al. Compared to what? Early brain overgrowth in autism and the perils of population norms. Biol Psychiatry (2013) 74:563–75. doi:10.1016/j.biopsych.2013.03.022
169. Doyle-Thomas KA, Duerden EG, Taylor MJ, Lerch JP, Soorya LV, Wang AT, et al. Effects of age and symptomatology on cortical thickness in autism spectrum disorders. Res Autism Spectr Disord (2013) 7:141–50. doi:10.1016/j.rasd.2012.08.004
170. Minshew NJ, Keller TA. The nature of brain dysfunction in autism: functional brain imaging studies. Curr Opin Neurol (2010) 23:124–30. doi:10.1097/WCO.0b013e32833782d4
171. Bosl W, Tierney A, Tager-Flusberg H, Nelson C. EEG complexity as a biomarker for autism spectrum disorder risk. BMC Med (2011) 9:18. doi:10.1186/1741-7015-9-18
172. Wang J, Barstein J, Ethridge LE, Mosconi MW, Takarae Y, Sweeney JA. Resting state EEG abnormalities in autism spectrum disorders. J Neurodev Disord (2013) 5:24. doi:10.1186/1866-1955-5-24
173. Griffin R, Westbury C. Infant EEG activity as a biomarker for autism: a promising approach or a false promise? BMC Med (2011) 9:61. doi:10.1186/1741-7015-9-61
174. Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, et al. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS One (2012) 7:e38786. doi:10.1371/journal.pone.0038786
175. Bernardi S, Anagnostou E, Shen J, Kolevzon A, Buxbaum JD, Hollander E, et al. In vivo 1H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res (2011) 1380:198–205. doi:10.1016/j.brainres.2010.12.057
176. Uzunova G, Hollander E, Shepherd J. The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr Neuropharmacol (2014) 12:71–98. doi:10.2174/1570159X113116660046
177. Kubas B, Kulak W, Sobaniec W, Tarasow E, Lebkowska U, Walecki J. Metabolite alterations in autistic children: a 1H MR spectroscopy study. Adv Med Sci (2012) 57:152–6. doi:10.2478/v10039-012-0014-x
178. Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat Rec (2011) 294:1646–52. doi:10.1002/ar.21252
179. Lofthouse N, Hendren R, Hurt E, Arnold LE, Butter E. A review of complementary and alternative treatments for autism spectrum disorders. Autism Res Treat (2012) 2012:870391. doi:10.1155/2012/870391
180. Cheng J, Widjaja F, Choi J, Hendren R. Considering biomedical/CAM treatments. Adolesc Med (2013) 024:446–64.
181. Hendren R. Autism: biomedical complementary treatment approaches. Child Adolesc Psychiatr Clin N Am (2013) 22:443–56. doi:10.1016/j.chc.2013.03.002
182. Mizejewski GJ, Lindau-Shepard B, Pass KA. Newborn screening for autism: in search of candidate biomarkers. Biomark Med (2013) 7:247–60. doi:10.2217/bmm.12.108
183. Ramsey JM, Guest PC, Broek JA, Glennon JC, Rommelse N, Franke B, et al. Identification of an age-dependent biomarker signature in children and adolescents with autism spectrum disorders. Mol Autism (2013) 4:27. doi:10.1186/2040-2392-4-27
184. Dudley E, Hassler F, Thome J. Profiling for novel proteomics biomarkers in neurodevelopmental disorders. Expert Rev Proteomics (2011) 8:127–36. doi:10.1586/epr.10.97
185. Zurawicz E, Kaluzna-Czaplinska J, Rynkowski J. Chromatographic methods in the study of autism. Biomed Chromatogr (2013) 27:1273–9. doi:10.1002/bmc.2911
186. Orru G, Pettersson-Yeo W, Marquand AF, Sartori G, Mechelli A. Using support vector machine to identify imaging biomarkers of neurological and psychiatric disease: a critical review. Neurosci Biobehav Rev (2012) 36:1140–52. doi:10.1016/j.neubiorev.2012.01.004
187. Ecker C, Spooren W, Murphy DG. Translational approaches to the biology of autism: false dawn or a new era? Mol Psychiatry (2013) 18:435–42. doi:10.1038/mp.2012.102
Keywords: biomarker, autism spectrum disorders, epigenetics, treatment targets, neuroimaging, genetics
Citation: Goldani AAS, Downs SR, Widjaja F, Lawton B and Hendren RL (2014) Biomarkers in autism. Front. Psychiatry 5:100. doi: 10.3389/fpsyt.2014.00100
Received: 18 December 2013; Accepted: 22 July 2014;
Published online: 12 August 2014.
Edited by:
Paul Croarkin, Mayo Clinic, USAReviewed by:
Randi Hagerman, UC Davis Medical Center, USARichard Eugene Frye, Harvard University, USA
Copyright: © 2014 Goldani, Downs, Widjaja, Lawton and Hendren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert L. Hendren, University of California San Francisco, 401 Parnassus Avenue, San Francisco, CA 94143-0984, USA e-mail:cm9iZXJ0LmhlbmRyZW5AdWNzZi5lZHU=