 Berit Kerner
Berit Kerner- Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, Los Angeles, CA, USA
Bipolar disorder is a common, complex psychiatric disorder characterized by mania and depression. The disease aggregates in families, but despite much effort, it has been difficult to delineate the basic genetic model or identify specific genetic risk factors. Not only single gene Mendelian transmission and common variant hypotheses but also multivariate threshold models and oligogenic quasi-Mendelian modes of inheritance have dominated the discussion at times. Almost complete sequence information of the human genome and falling sequencing costs now offer the opportunity to test these models in families in which the disorder is transmitted over several generations. Exome-wide sequencing studies have revealed an astonishing number of rare and potentially damaging mutations in brain-expressed genes that could have contributed to the disease manifestation. However, the statistical analysis of these data has been challenging, because genetic risk factors displayed a high degree of dissimilarity across families. This scenario is not unique to bipolar disorder, but similar results have also been found in schizophrenia, a potentially related psychiatric disorder. Recently, our group has published data which supported an oligogenic genetic model of transmission in a family with bipolar disorder. In this family, three affected siblings shared rare, damaging mutations in multiple genes, which were linked to stress response pathways. These pathways are also the target for drugs frequently used to treat bipolar disorder. This article discusses these findings in the context of previously proclaimed disease models and suggests future research directions, including biological confirmation and phenotype stratification as an approach to disease heterogeneity.
Introduction
“Manic-depressive illness magnifies common human experiences to larger-than-life proportions” (1). This opening sentence to Goodwin and Jamison’s acclaimed and comprehensive book on bipolar disorder places the often extreme and strange-appearing symptoms of mania and depression in a more comprehensible framework of shared human experiences. Bipolar disorder is a severe, complex psychiatric disorder, but still, it is so common that most people likely know a friend, a neighbor, or even a family member affected with this disease. After all, with an estimated prevalence rate of 2.4% (2) and a world population of 7 billion people, it is expected that several million patients might suffer from bipolar disorder worldwide. The core symptoms of bipolar disorder are episodes of abnormally elevated, expansive, and irritable mood accompanied by inflated self-esteem and grandiosity. Decreased need for sleep, increased talkativeness, and flight of ideas, could also be present, in addition to excessive goal-directed activity and extreme involvement in pleasurable activities, which frequently are associated with a high potential for painful consequences (3). To meet diagnostic criteria, the symptoms must have caused marked impairment in social and occupational functioning or required hospitalization to prevent harm to oneself or others. Symptoms of depression can precede or follow manic episodes, and sometimes even accompany manic episodes, although they are not required for making the diagnosis. Psychotic symptoms, such as hallucinations and/or delusions, occur in about 50% of bipolar disorder patients, suggesting some symptomatic and even pathophysiological overlap with schizophrenia (4). For many patients, the mood symptoms can be so agonizing that suicide seems to be the only escape (2). Bipolar disorder appears to have strong genetic risk factors. Twin studies have suggested a monozygotic concordance rate of 0.43, and population-based family risk studies have estimated a heritability rate of about 58% (5, 6). Environmental risk factors, such as trauma (7, 8), infection, and inflammation (9), have been found to contribute to a lesser degree. Despite the widespread occurrence, the cause of the disease remains elusive.
KEY CONCEPT 1. Single gene Mendelian transmission.
Many rare, Mendelian disorders, such as cystic fibrosis, are caused by mutations in one or a few genes. The disease manifests if one allele (dominant inheritance) or both alleles (recessive inheritance) of a gene carry the mutation and environmental influences are of minor importance.
KEY CONCEPT 2. Multivariate threshold model
This genetic model is concerned with quantitative traits that are conceptualized as being normally distributed in the population. A change in the phenotype occurs if a threshold of genetic or environmental influences has been reached. The relationship between traits and disease is often unclear.
KEY CONCEPT 3. Oligogenic quasi-Mendelian mode of inheritance
This genetic model conceptualizes disorders as being influenced by a small number of genetic mutations in genes that are potentially related to a specific biological function. A disorder occurs if the interacting mutations are present and environmental factors are considered of minor importance. The inheritance pattern of the disease follows Mendelian rules if all mutations are inherited jointly.
KEY CONCEPT 4. Genetic risk factors
The genetic code contains information about the structure, function, and timely expression of proteins, which are the basic building blocks of the cell machinery. Changes in the base-pair sequence of this code (mutations) could lead to disease-causing changes in the structure and/or function of the encoded proteins. The protein expression level or timing might also be altered.
Early Models of Disease Transmission and Heritability in Bipolar Disorder
In some families, bipolar disorder has been transmitted over several generations, closely resembling a Mendelian disorder (10, 11). This observation had originally inspired researchers to study rare, large multi-generational pedigrees under the assumption of a single gene with large effect size and autosomal dominant, recessive, or X-linked inheritance (12, 13). After initial enthusiasm supported by strong genetic linkage signals, it was quickly discovered that these results could not be replicated (14). Incomplete penetrance, etiological heterogeneity, and recombination events might have contributed to the replication failure. However, it was also likely that the underlying disease model was not supported by the data. After all, not all segregation studies had supported a disease model built on a single major disease locus (15–18). The high frequency in the population of bipolar disorder also clearly distinguished the disease from rare Mendelian disorders. As a consequence, the idea of a single major risk locus was quickly rejected (19, 20).
Why is Bipolar Disorder so Common in the Population?
The question of why and how severe and debilitating disorders, such as bipolar disorder, could have persisted in the population at a relatively high rate of about 2–4% is among the leading questions of evolutionary epidemiology (21, 22). According to Darwinian Theory, common, positively selected traits provided an evolutionary advantage, but in the case of some traits, left almost all members of a population vulnerable to the disease (Figure 1) (23). Supporting evidence has come from comparisons of the human genome to the genome of the chimpanzee, which revealed evidence for positive selection in the opioid receptor genes (24) and immune response genes (25, 26). These studies provided support for a link between entire genes or even gene families and common human traits, such as creativity and novelty seeking, which might have not only provided an evolutionary advantage but also made all humankind susceptible to addiction and other psychiatric disorders (27).
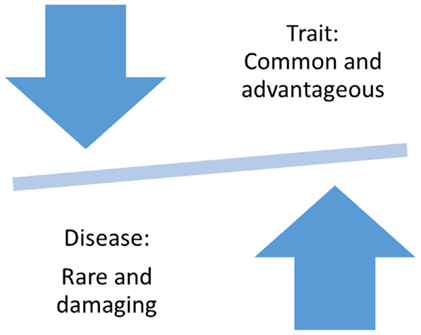
Figure 1. Evolution-based hypothesis about traits and diseases.
On the other hand, diseases are thought to be subject to negative selection. Only in rare cases has evolutionary selection seemed to have led to the accumulation of Mendelian disorders. An often cited text-book example for a disease with evolutionary advantage is sickle cell anemia, a Mendelian disorder with a population frequency of up to 0.16% in African Americans. Heterozygous mutations in a single disease-causing gene have provided a protective effect against malaria, a common environmental threat in Africa, leading to higher allele frequencies for the protective allele than expected based on mutation rates alone (28–30). However, examples of other disorders have not supported the theory of evolutionary advantages of common variants. The Mendelian disorder cystic fibrosis has reached relatively high prevalence in the population, but increased vulnerability to mutation at a specific location in the disease-causing gene, and not evolutionary advantage, likely contributed to the increased allele frequency of the disease-causing CFTRΔF508 mutation (31). Since more than 1,000 rare mutations in other parts of the gene have been identified as disease-causing alleles, an evolutionary advantage of a single mutation appears to be less likely. Last, but not least, population bottleneck could have resulted in disease aggregation in certain populations. For example, Tay–Sachs disease is a genetically heterogeneous Mendelian disorder with an increased prevalence of 0.04% in the Jewish population (32). The disease is caused by more than 30 different mutated alleles, but because of population isolation and selective mating, the disease could increase in prevalence. These examples demonstrate that disease-causing alleles are relatively rare, even in relatively common diseases and that, with a few exceptions, evolutionary advantages do not explain the increased population prevalence of severe disorders.
The “Common Disease–Common Variant” Hypothesis
Even though the rare nature of disease-causing mutations had been well accepted by Mendelian geneticists, genetic epidemiologists had been puzzled by the frequent occurrence of common disorders in the population, and also by the discovery of millions of common genetic polymorphisms across the genome that were not well explained by Darwinian Theory. It was tempting to claim that common genetic polymorphisms could be linked to common complex disorders (33). In addition, it had been noticed that association analyses in population samples provided increased statistical power over family based linkage analysis (34). Technical advantages in array-based approaches finally paved the way for genome-wide genotyping of common single-nucleotide polymorphisms (SNPs) and association testing with disease. Genome-wide association studies were widely disseminated across clinical and statistical fields, and thousands of publications followed without further questioning the biological foundation of the common disease-common variant hypothesis. Overall, these studies have revealed a complex genetic structure influencing almost all examined traits and disorders, including bipolar disorder (35–38), but the functional consequences of the common variants remained mostly elusive (39, 40). Overall, the results of these studies have not supported the assumption of a common genetic disease-causing risk factor in bipolar disorder or a link to positive adaptation. Instead, evidence is accumulating that founder effects and drift, but not Darwinian selection, might have caused common allele frequency variability, and a causal link between common variants and common disorders has not been substantiated for most disorders (21, 41, 42).
KEY CONCEPT 5. Common complex disorders.
Some common medical conditions, such as diabetes or high-blood pressure, are believed to be caused by genetic and environmental factors. Therefore, the transmission in families might not follow a simple Mendelian mode of transmission. According to this model, an individual might not manifest the disease, even though he or she carries a risk mutation, if the environmental exposure has not occurred.
Alternatives to the Common Disease–Common Variant Hypothesis
While a model of non-random, natural selection had dominated the search for genetic risk factors in traits that might have been related to psychiatric disorders, alternative explanations had also been considered (Figure 2). One hypothesis that had gained attention proposed a more complex polygenic, or even multifactorial, model of transmission (43). An example of polygenic transmission is eye color, which seems to be a purely genetic trait. According to this model, random mutations in many genes, some of which with a dominant effect, influence the expression of the trait in the population (44, 45). On the other hand, height is a trait that is influenced by a complex interplay of genetic and environmental factors (46). An adaptation of this model to psychiatric disorders was the liability threshold model (47). According to this theory, liability to psychiatric disorders or “traits” related to susceptibility follows a continuous distribution in the population. However, this model was contradicted by the finding that family risk did not follow this pattern (48–50). Furthermore, in many families, the disease was transmitted through the paternal and the maternal lineage. This pattern of transmission, also known as assortative mating, contributed to the aggregation of risk factors in a few families with multiple affected family members, whereas in most families the risk was low (51). In general, family studies have not supported a multifactorial threshold model of disease for bipolar disorder (52, 53). Instead, mathematical model fitting in families with bipolar disorder suggested an oligogenic, quasi-Mendelian mode of inheritance with significant locus heterogeneity (54).
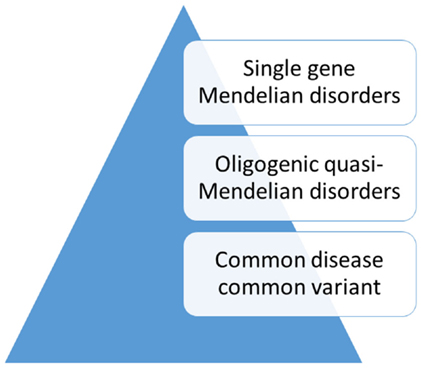
Figure 2. Genetic models of disease transmission in bipolar disorder.
Closing the Circle or is it a Spiral?
In 1990s, when large, multi-generational families were first studied, neither the analytical tools nor the biological knowledge were available to solve the problem of a complex, oligogenic inheritance. However, with almost complete information on the human genome available and rapidly falling sequencing costs, the time seemed to be right to revisit disease models proposed more than 20 years ago. Since bipolar disorder is inherited in families, pedigrees seemed to be a natural choice to test the hypothesis of a quasi-Mendelian, oligogenic model of disease transmission (Figure 3). According to this model, it was expected that a few rare and likely functional variants were shared among the affected family members with both parents contributing to the disease risk. Likewise, unaffected family members would not carry the damaging variants. Not only to avoid biases that could be introduced by selection of candidate genes but also to keep the focus on gene-coding regions for which functional information was available, we favored an exome-wide sequencing approach (55). The results of our study suggested that multiple, very rare, and likely protein-damaging mutations in highly conserved gene regions had affected genes that were linked in a single pathophysiological pathway regulated by MAP kinases. All mutations in this family had likely affected a specific signaling pathway known to be involved in the response to mood stabilizing medications. This finding supported the oligogenic hypothesis of genetic risk in bipolar disorder. While a statistical proof of disease association will require larger data sets, these results, nevertheless, point to genes and signaling pathways in which the functional consequences of the mutations could be tested in cell culture and animal models (Figure 3). Since the data have been published, several groups have completed candidate gene sequencing in population and family samples, and exome-wide sequencing in Old Order Amish families (40, 56–58). These studies have found significant haplotype and locus heterogeneity, and further rejected the hypothesis of a single major risk gene for bipolar disorder.

Figure 3. Identification and confirmation of genetic risk variants in bipolar disorder.
While exome-wide and genome-wide sequencing studies in bipolar disorder are still rare, we have tried to find further support for our hypothesis in studies on a potentially related psychiatric disorder, schizophrenia. These studies have revealed a high degree of de novo mutations and rare protein-damaging genomic variants in patients with schizophrenia (59–69). The largest exome-wide study available to date is a population-based Swedish study of 5,079 cases and controls (70, 71). The results of this study supported the hypothesis of significant locus heterogeneity in schizophrenia. Despite an increased burden of potentially damaging rare mutations in cases, no locus-specific associations reached genome-wide statistical significance. The apparent controversy that bipolar disorder appears to be quasi-Mendelian in families, but still very common in the population, could be due to rare mutations in hundreds or thousands of contributing genes, a disease model that has become apparent in intellectual disability and autism (72). Therefore, it will be essential to collect and annotate all identified genetic variants in psychiatric patients and to create a comprehensive searchable database to facilitate genetic testing and personalized genomic medicine (73).
Where to go from Here?
In bipolar disorder and schizophrenia, increasing evidence supports the role of rare, disease-causing mutations in brain-expressed genes. As a single major risk gene has become highly unlikely, the need for new analytical and statistical approaches has grown. Locus heterogeneity and private mutations challenge hypothesis testing with established statistical methods. However, even for statistically significant associations, the translation into clinical applications will ultimately require the demonstration of biological significance. So far, genome-wide approaches have only scratched the surface of genomic variability. Rare mutations in gene-coding regions certainly constitute only the tip of the iceberg and do not capture the full spectrum of potential disease-causing genomic changes. For example, researchers have only begun to explore the vast functional diversity of non-coding DNA. Functional exploration of micro RNAs (miRNAs), small nuclear RNAs (snoRNAs), and long non-coding RNAs (lncRNAs) could reveal their role in pathway regulation and other cellular processes (74). Furthermore, a growing number of investigators have requested a balanced approach to DNA-based and protein-based studies (75). The field of proteomics has already uncovered the complexities of context-specific and cell-type-specific protein function in a complex network of potential interactions. Posttranslational modifications and their consequences on the structure, function, and intracellular location of the modified proteins provide ever increasing possibilities of variability and interaction. Disease mechanisms might also involve cell-derived membrane vesicles (CVM), which play a role in cell–cell communication. Interdisciplinary approaches will be necessary to clearly elucidate the functional consequences of mutations and protein modifications in the context of intracellular events and pathways, as well as cell networks and developmental processes.
Summary and Recommendations
To tackle the complexity of psychiatric disorders, we will need a balanced and broad approach to biological and social risk factors in which competing and complementary ideas could receive equal financial support. In a scientific culture that is reflected in phrases, such as “Go big or go home,” the focus is on large heterogeneous population-based samples. While this approach might be useful for studying common traits that are shared by most members of a population, it is less suitable for diseases that are transmitted in families and in which each family may carry a unique combination of susceptibility genes. Rare genomic risk factors with moderately strong effect could be best approached through exome-wide or genome-wide sequencing of multi-generational families in which the disease is transmitted in a Mendelian or “quasi-Mendelian” mode. Based on recent results, it should be recognized that these approaches are at least complementary in studies of schizophrenia and other neuropsychiatric disorders. Rigorous hypothesis testing and rejection of unsupported ideas, as well as transparency and replication of results, will ultimately lead to progress in our understanding of disease processes and risk factors. Reporting of positive, as well as negative, results will increase transparency and reduce redundancy of efforts.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by National Institutes of Health (NIH) grant R01 MH085744 and a NARSAD Young Investigator award to BK. I would like to thank Jacob Carpenter for editorial assistance.
Author Biography

References
2. Merikangas KR, Jin R, He J, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the World Mental Health Survey Initiative. Arch Gen Psychiatry (2011) 68(3):241–51. doi: 10.1001/archgenpsychiatry.2011.12
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). 5th ed. Washington, DC: American Psychiatric Association (2013). doi:10.1176/appi.books.9780890425596
4. Coryell W, Leon AC, Turvey C, Akiskal HS, Mueller T, Endicott J. The significance of psychotic features in manic episodes: a report from the NIMH collaborative study. J Affect Disord (2001) 67(1–3):79–88. doi:10.1016/S0165-0327(99)00024-5
5. Kieseppä T, Partonen T, Haukka J, Kaprio J, Lönnqvist J. High concordance of bipolar I disorder in a nationwide sample of twins. Am J Psychiatry (2004) 161:1814–21. doi:10.1176/appi.ajp.161.10.1814
6. Song J, Bergen SE, Kuja-Halkola R, Larsson H, Landén M, Lichtenstein P. Bipolar disorder and its relation to major psychiatric disorders: a family-based study in the Swedish population. Bipolar Disord (2015) 17(2):184–93. doi:10.1111/bdi.12242
7. Etain B, Aas M, Andreassen OA, Lorentzen S, Dieset I, Gard S, et al. Childhood trauma is associated with severe clinical characteristics of bipolar disorders. J Clin Psychiatry (2013) 74(10):991–8. doi:10.4088/JCP.13m08353
8. Bratlien U, Øie M, Haug E, Møller P, Andreassen OA, Lien L, et al. Environmental factors during adolescence associated with later development of psychotic disorders – a nested case-control study. Psychiatry Res (2014) 215(3):579–85. doi:10.1016/j.psychres.2013.12.048
9. Severance EG, Gressitt KL, Yang S, Stallings CR, Origoni AE, Vaughan C, et al. Seroreactive marker for inflammatory bowel disease and associations with antibodies to dietary proteins in bipolar disorder. Bipolar Disord (2014) 16(3):230–40. doi:10.1111/bdi.12159
10. Rice J, Reich T, Andreasen NC, Endicott J, Van Eerdewegh M, Fishman R, et al. The familial transmission of bipolar illness. Arch Gen Psychiatry (1987) 44(5):441–7. doi:10.1001/archpsyc.1987.01800170063009
11. Spence MA, Flodman PL, Sadovnick AD, Bailey-Wilson JE, Ameli H, Remick RA. Bipolar disorder: evidence for a major locus. Am J Med Genet (1995) 60(5):370–6. doi:10.1002/ajmg.1320600505
12. Egeland JA, Gerhard DS, Pauls DL, Sussex JN, Kidd KK, Allen CR, et al. Bipolar affective disorders linked to DNA markers on chromosome 11. Nature (1987) 325(6107):783–7. doi:10.1038/325783a0
13. Hodgkinson S, Sherrington R, Gurling H, Marchbanks R, Reeders S, Mallet J, et al. Molecular genetic evidence for heterogeneity in manic depression. Nature (1987) 325(6107):805–6. doi:10.1038/325805a0
14. Risch N, Botstein D. A manic depressive history. Editorial. Nat Genet (1996) 12:351–3. doi:10.1038/ng0496-351
15. Rüdin E. Zur Vererbung und Neuentstehung der Dementia praecox. Berlin: Springer Verlag OHG (1916).
16. Sham PC, Morton NE, Rice JP. Segregation analysis of the NIMH Collaborative study. Family data on bipolar disorder. Psychiatr Genet (1991) 2:175–84. doi:10.1097/00041444-199203000-00002
17. Bucher KD, Elston RC, Green R, Whybrow P, Helzer J, Reich T, et al. The transmission of manic depressive illness – II. Segregation analysis of three sets of family data. J Psychiatr Res (1981) 16(1):65–78. doi:10.1016/0022-3956(81)90014-5
18. Goldin LR, Gershon ES, Targum SD, Sparkes RS, McGinniss M. Segregation and linkage analyses in families of patients with bipolar, unipolar, and schizoaffective mood disorders. Am J Hum Genet (1983) 35(2):274–87.
19. Akiskal HS. The bipolar spectrum: research and clinical perspectives. Encephalocele (1995) 6:3–11.
20. Craddock N, Van Eerdewegh P, Reich T. Single major locus models for bipolar disorder are implausible. Am J Med Genet (1997) 74:18. doi:10.1002/(SICI)1096-8628(19970221)74:1<18::AID-AJMG4>3.0.CO;2-R
21. Wilson DR. Evolutionary epidemiology and manic depression. Br J Med Psychol (1998) 71(Pt 4):375–95. doi:10.1111/j.2044-8341.1998.tb00999.x
23. Wilson E. Sociobiology: The New Synthesis. Cambridge, MA: Belknap/Harvard University Press (1975).
24. Cruz-Gordillo P, Fedrigo O, Wray GA, Babbitt CC. Extensive changes in the expression of the opioid genes between humans and chimpanzees. Brain Behav Evol (2010) 76(2):154–62. doi:10.1159/000320968
25. Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, et al. Identifying recent adaptations in large-scale genomic data. Cell (2013) 152(4):703–13. doi:10.1016/j.cell.2013.01.035
26. Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science (2014) 345(6202):1254665. doi:10.1126/science.1254665
27. Jamison KR. Great wits and madness: more near allied? Br J Psychiatry (2011) 199(5):351–2. doi:10.1192/bjp.bp.111.100586
28. Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A (2010) 107(3):961–8. doi:10.1073/pnas.0912629107
29. Livingstone FB. The wave of advance of an advantageous gene: the sickle cell gene in Liberia. Hum Biol (1960) 32:197–202.
30. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun (2010) 1:104. doi:10.1038/ncomms1104
31. Collins F. Cystic fibrosis: molecular biology and therapeutic implication. Science (1992) 256:774–80. doi:10.1126/science.1375392
32. Bray SM, Mulle JG, Dodd AF, Pulver AE, Wooding S, Warren ST. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proc Natl Acad Sci U S A (2010) 107(37):16222–7. doi:10.1073/pnas.1004381107
33. Lander ES. The new genomics: global views of biology. Science (1996) 274(5287):536–9. doi:10.1126/science.274.5287.536
34. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science (1996) 273(5281):1516–7. doi:10.1126/science.273.5281.1516
35. Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet (2008) 40(9):1056–8. doi:10.1038/ng.209
36. Kloiber S, Czamara D, Karbalai N, Müller-Myhsok B, Hennings J, Holsboer F, et al. ANK3 and CACNA1C – missing genetic link for bipolar disorder and major depressive disorder in two German case-control samples. J Psychiatr Res (2012) 46(8):973–9. doi:10.1016/j.jpsychires.2012.04.017
37. Hattori E, Toyota T, Ishitsuka Y, Iwayama Y, Yamada K, Ujike H, et al. Preliminary genome-wide association study of bipolar disorder in the Japanese population. Am J Med Genet B Neuropsychiatr Genet (2009) 150B(8):1110–7. doi:10.1002/ajmg.b.30941
38. Kuo PH, Chuang LC, Liu JR, Liu CM, Huang MC, Lin SK, et al. Identification of novel loci for bipolar I disorder in a multi-stage genome-wide association study. Prog Neuropsychopharmacol Biol Psychiatry (2014) 51:58–64. doi:10.1016/j.pnpbp.2014.01.003
39. Doyle GA, Lai AT, Chou AD, Wang MJ, Gai X, Rappaport EF, et al. Re-sequencing of ankyrin 3 exon 48 and case-control association analysis of rare variants in bipolar disorder type I. Bipolar Disord (2012) 14(8):809–21. doi:10.1111/bdi.12002
40. Fiorentino A, O’Brien NL, Locke DP, McQuillin A, Jarram A, Anjorin A, et al. Analysis of ANK3 and CACNA1C variants identified in bipolar disorder whole genome sequence data. Bipolar Disord (2014) 16(6):583–91. doi:10.1111/bdi.12203
41. Nesse RM. Ten questions for evolutionary studies of disease vulnerability. Evol Appl (2011) 4(2):264–77. doi:10.1111/j.1752-4571.2010.00181.x
42. Kimura M. Stochastic processes and distribution of gene frequencies in natural selection. Cold Spring Harb Symp Quant Biol (1955) 22:33–53. doi:10.1101/SQB.1955.020.01.006
43. Smeraldi E, Negri F, Heimbuch RC, Kidd KK. Familial patterns and possible modes of inheritance of primary affective disorders. J Affect Disord (1981) 3(2):173–82. doi:10.1016/0165-0327(81)90042-2
44. Sturm RA, Larsson M. Genetics of human iris colour and patterns. Pigment Cell Melanoma Res (2009) 22(5):544–62. doi:10.1111/j.1755-148X.2009.00606.x
45. White D, Rabago-Smith M. Genotype-phenotype associations and human eye color. J Hum Genet (2011) 56(1):5–7. doi:10.1038/jhg.2010.126
46. Dubois L, Ohm Kyvik K, Girard M, Tatone-Tokuda F, Pérusse D, Hjelmborg J, et al. Genetic and environmental contributions to weight, height, and BMI from birth to 19 years of age: an international study of over 12,000 twin pairs. PLoS One (2012) 7(2):e30153. doi:10.1371/journal.pone.0030153
47. Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet (1965) 29:51–76. doi:10.1111/j.1469-1809.1965.tb00500.x
49. Gottesman II, Shields J, (with the assistance of D. R. Hanson). Schizophrenia: The Epigenetic Puzzle. Cambridge, MA: Cambridge University Press (1982).
50. Gottesman II, McGuffin P, Farmer AE. Clinical genetics as clues to the “real” genetics of schizophrenia (a decade of modest gains while playing for time). Schizophr Bull (1987) 13(1):23–47. doi:10.1093/schbul/13.1.23
51. Mathews CA, Reus VI. Assortative mating in the affective disorders: a systematic review and meta-analysis. Compr Psychiatry (2001) 42(4):257–62. doi:10.1053/comp.2001.24575
52. Baron M, Klotz J, Mendlewicz J, Rainer J. Multiple-threshold transmission of affective disorders. Arch Gen Psychiatry (1981) 38(1):79–84. doi:10.1001/archpsyc.1981.01780260081009
53. Price RA, Kidd KK, Pauls DL, Gershon ES, Prusoff BA, Weissman MM, et al. Multiple threshold models for the affective disorders: the Yale-NIMH collaborative family study. J Psychiatr Res (1985) 19(4):533–46. doi:10.1016/0022-3956(85)90071-8
54. Craddock N, Khodel V, Van Eerdewegh P, Reich T. Mathematical limits of multilocus models: the genetic transmission of bipolar disorder. Am J Hum Genet (1995) 57:690–702.
55. Kerner B, Rao AR, Christensen B, Dandekar S, Yourshaw M, Nelson SF. Rare genomic variants link bipolar disorder with anxiety disorders to CREB-regulated intracellular signaling pathways. Front Psychiatry (2013) 4:154. doi:10.3389/fpsyt.2013.00154
56. Georgi B, Craig D, Kember RL, Liu W, Lindquist I, Nasser S, et al. Genomic view of bipolar disorder revealed by whole genome sequencing in a genetic isolate. PLoS Genet (2014) 10(3):e1004229. doi:10.1371/journal.pgen.1004229
57. Strauss KA, Markx S, Georgi B, Paul SM, Jinks RN, Hoshi T, et al. A population-based study of KCNH7 p.Arg394His and bipolar spectrum disorder. Hum Mol Genet (2014) 23(23):6395–406. doi:10.1093/hmg/ddu335
58. Ament SA, Szelinger S, Glusman G, Ashworth J, Hou L, Akula N, et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc Natl Acad Sci U S A (2015) 112(11):3576–81. doi:10.1073/pnas.1424958112
59. Need AC, McEvoy JP, Gennarelli M, Heinzen EL, Ge D, Maia JM, et al. Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. Am J Hum Genet (2012) 91(2):303–12. doi:10.1016/j.ajhg.2012.06.018
60. Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, Jouan L, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet (2011) 43(9):860–3. doi:10.1038/ng.886
61. Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet (2011) 43(9):864–8. doi:10.1038/ng.902
62. Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet (2012) 44(12):1365–9. doi:10.1038/ng.2446
63. Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron (2014) 82(4):773–80. doi:10.1016/j.neuron.2014.04.043
64. Guipponi M, Santoni FA, Setola V, Gehrig C, Rotharmel M, Cuenca M, et al. Exome sequencing in 53 sporadic cases of schizophrenia identifies 18 putative candidate genes. PLoS One (2014) 9(11):e112745. doi:10.1371/journal.pone.0112745
65. Ionita-Laza I, Xu B, Makarov V, Buxbaum JD, Roos JL, Gogos JA, et al. Scan statistic-based analysis of exome sequencing data identifies FAN1 at 15q13.3 as a susceptibility gene for schizophrenia and autism. Proc Natl Acad Sci U S A (2014) 111(1):343–8. doi:10.1073/pnas.1309475110
66. McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry (2014) 19(6):652–8. doi:10.1038/mp.2014.29
67. Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell (2013) 154(3):518–29. doi:10.1016/j.cell.2013.06.049
68. Timms AE, Dorschner MO, Wechsler J, Choi KY, Kirkwood R, Girirajan S, et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry (2013) 70(6):582–90. doi:10.1001/jamapsychiatry.2013.1195
69. Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature (2014) 506(7487):179–84. doi:10.1038/nature12929
70. Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature (2014) 506(7487):185–90. doi:10.1038/nature12975
71. Ruderfer DM, Lim ET, Genovese G, Moran JL, Hultman CM, Sullivan PF, et al. No evidence for rare recessive and compound heterozygous disruptive variants in schizophrenia. Eur J Hum Genet (2015) 23(4):555–7. doi:10.1038/ejhg.2014.228
72. Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet (2014) 46(9):944–50. doi:10.1038/ng.3050
73. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet (2014) 133(1):1–9. doi:10.1007/s00439-013-1358-4
74. Geaghan M, Cairns MJ. MicroRNA and posttranscriptional dysregulation in psychiatry. Biol Psychiatry (2015) 78(4):231–9. doi:10.1016/j.biopsych.2014.12.009
Keywords: bipolar disorder, deep sequencing, genetic models of transmission, rare variants, common genomic polymorphisms
Citation: Kerner B (2015) Toward a deeper understanding of the genetics of bipolar disorder. Front. Psychiatry 6:105. doi: 10.3389/fpsyt.2015.00105
Received: 31 March 2015; Accepted: 08 July 2015;
Published: 03 August 2015
Edited by:
Ming D. Li, University of Virginia, USAReviewed by:
Chamindi Seneviratne, University of Maryland, USAJiekun Yang, University of Virginia, USA
Copyright: © 2015 Kerner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Ymtlcm5lckBtZWRuZXQudWNsYS5lZHU=