 Zhenfei Ning
Zhenfei Ning John M. Williams
John M. Williams Romika Kumari
Romika Kumari Pavel V. Baranov
Pavel V. Baranov Tom Moore
Tom Moore- School of Biochemistry and Cell Biology, University College Cork, Cork, Ireland
Autism is a genetically complex neurobehavioral disorder with a population prevalence of more than 1%. Cerebellar abnormalities, including Purkinje cell deficits in the vermis, are consistently reported, and rodent models of cerebellar dysfunction exhibit features analogous to human autism. We previously analyzed the regulation and expression of the pseudoautosomal region 2 gene SPRY3, which is adjacent to X chromosome-linked TMLHE, a known autism susceptibility gene. SPRY3 is a regulator of branching morphogenesis and is strongly expressed in Purkinje cells. We previously showed that mouse Spry3 is not expressed in cerebellar vermis lobules VI–VII and X, regions which exhibit significant Purkinje cell loss or abnormalities in autism. However, these lobules have relatively high expression of p75NTR, which encodes a neurotrophin receptor implicated in autism. We propose a mechanism whereby inappropriate SPRY3 expression in these lobules could interact with TrkB and p75NTR signaling pathways resulting in Purkinje cell pathology. We report preliminary characterization of X and Y chromosome-linked regulatory sequences upstream of SPRY3, which are polymorphic in the general population. We suggest that an OREG-annotated region on chromosome Yq12 ∼60 kb from SPRY3 acts as a silencer of Y-linked SPRY3 expression. Deletion of a β-satellite repeat, or alterations in chromatin structure in this region due to trans-acting factors, could affect the proposed silencing function, leading to reactivation and inappropriate expression of Y-linked SPRY3. This proposed male-specific mechanism could contribute to the male bias in autism prevalence.
Introduction
Autism is a spectrum disorder whose core features include language delay, social deficits, and restricted interests and repetitive behaviours. In addition, there are significant co-morbidities including attention deficit hyperactivity disorder (ADHD), anxiety, intellectual disability, motor delay, and epilepsy, among others (1–3). Recently, the ESSENCE protocol was developed in response to the increasing diagnosis of autism associated with an expanding list of co-morbidities, including cases in which the co-morbidity may be the predominant clinical entity (1, 4). There is also a trend towards the increased diagnosis and inclusion of cases at the mild end of the spectrum (5). The combined effects of these trends may explain the current estimates of autism prevalence in school age children, exemplified by two recent reports, which found 1.68% prevalence in the USA and 2.85% in Northern Ireland (6, 7).
Heritability estimates for autism are high, ranging from 38% (8) to more than 80% (9, 10), and there is an emerging consensus that the majority of the genetic risk is attributable to common genetic variants of small effect size acting in combination with rare or de novo variants of larger effect size (11–15).
A striking and unexplained feature of autism is the preponderance of affected males, with a sex ratio of between 3 and 4 to 1 consistently reported, including in recent large studies (6, 7, 16, 17). Earlier studies often reported more extreme male biases, particularly in milder cases (so-called high functioning autism or Asperger’s syndrome), and there is continuing debate on the possibility of a female protective or “camouflage” effect that may result in their under-diagnosis (18–25). Currently, multiple genes and genomic variants are associated with autism with varying levels of confidence; however, the majority are autosomal and do not explain observed sex differences in prevalence (26). Rather than exhibiting sex-specific expression, autism genes may interact with normal regulatory pathways that are themselves sex-specifically regulated (27–29). This is conceptually similar to the proposal that autism genes operate against a background of sex-specific hormone profiles (30–32), and shifts the explanatory burden from the autism genes themselves to the normal sex-specific pathways with which they interact.
We previously analyzed the pseudoautosomal region 2 (PAR2)-linked SPRY3 gene in autism because it is highly expressed in the cerebellum (33), a region consistently implicated in autism pathogenesis (34–39). SPRY3 is expressed in Purkinje cells, a key cell type deficient in autism (40, 41), but we note that mouse Spry3 is not expressed in the cerebellar lobules (VI–VII, X) homologous to those most affected in human autism (33, 41). If this expression pattern is recapitulated in the human, as suggested by a human SPRY3 promoter–LacZ transgenic mouse strain (33), it suggests two alternative mechanisms by which SPRY3 could be implicated in loss of Purkinje cells preferentially in these lobules. First, the normal absence of SPRY3 expression in these lobules may increase their sensitivity to genetic or environmental “insults” that cause Purkinje cell loss. However, this would not explain the male bias. Second, the deregulation and inappropriate overexpression of SPRY3 in these lobules may be pathogenic, and could provide a male-specific mechanism, as described below. SPRY3 is a receptor tyrosine kinase (RTK) signaling inhibitor that interacts with the TrkB neurotrophin receptor pathway (42), which is implicated in autism and social behavior (43–49).
The X-linked copy of SPRY3 is adjacent to a known autism gene, TMLHE, and a proportion of SPRY3 transcripts arise from upstream promoters in the X-linked F8A3 and TMLHE regions (33). The F8A2–F8A3 region contains an inversion polymorphism that could potentially affect the expression of flanking genes, including SPRY3. The Y-linked copy of SPRY3 is epigenetically silenced in normal males (50), which could contribute to the male bias in autism due to X-linkage of the expressed gene copy. Alternatively, deregulation and reactivation of the silenced Y-linked copy could provide a male-specific pathological mechanism. A possible further mode of SPRY3 deregulation is suggested by the fact that SPRY3 is upregulated in the liver of piglets fed high levels of carnitine (51). Notably, the gene adjacent to SPRY3, TMLHE, encodes an enzyme in the carnitine biosynthesis pathway. As carnitine deficiency is implicated in autism causation (52), this suggests a mechanism whereby carnitine levels could impact on SPRY3 regulation and autism.
In this study, we examined the expression of SPRY3 and its functionally associated genes in cerebellum, and we analyzed genetic variation in predicted X and Y chromosome regulatory regions that may impact on SPRY3 expression. We propose a pathogenic mechanism in autism involving SPRY3 deregulation impacting on the BDNF–TrkB–p75NTR neurotrophin pathway.
Materials and Methods
Online Bioinformatics and Other Resources
The following databases and online resources were used in this study: UCSC genome browser (https://genome.ucsc.edu/); GENSAT Brain Atlas of gene expression in EGFP Transgenic Mice (http://gensat.org/index.html); Allen Brain Atlases (http://portal.brain-map.org/; 53); GTEx Portal, v7, updated 09/05/2017 (https://gtexportal.org/home/); SFARI (Simon Foundation Autism Research Initiative; https://www.sfari.org/); AGRE (Autism genetic Resource Exchange; https://research.agre.org/program/descr.cfm). Other websites are listed under “Analysis of PsychENCODE data.”
Whole Mount Immunohistochemistry of Mouse Cerebellum
All reagents were from Sigma, UK, unless otherwise stated. Adult male and female C57Bl/6J mice were humanely euthanized under permissions obtained following animal ethics and welfare review by UCC committees, under national and European legislation. Dissected mouse cerebellum was fixed in 4% Paraformaldehyde (PFA)-Phosphate-buffered saline (PBS) for 10 h, post-fixed in methanol–Dimethyl sulfoxide (DMSO) (4:1) overnight at 4ºC, and then bleached in methanol–DMSO–30% H2O2 (4:1:1) overnight at 4ºC. After 3 × 60 min wash in 100% methanol, it was frozen at −80ºC and thawed at room temperature (RT) for six cycles in 100% methanol. After rehydrating with 50% methanol, 15% methanol, and PBS for 2 h each, it was digested by proteinase K (10 mg/ml; Sigma, UK) in PBS for 3 min at RT, washed in PBS for 3 × 2 h at RT, and incubated in PBS with 10% goat serum and 0.1% Triton X-100 overnight at 4ºC. It was then incubated with anti-Spry3 primary antibody (Abcam, UK) in PBS containing 10% goat serum, 0.1% Triton X-100, 5% DMSO for 48 h at 4ºC and washed twice in PBS containing 10% goat serum, 0.1% Triton X-100 for 20 min each, followed by incubation with secondary antibody in PBS containing 10% goat serum, 0.1% Triton X-100, 5% DMSO for 24 h at 4ºC. It was then washed twice in PBS containing 10% goat serum, 0.1% Triton X-100 for 2 h each. Immunoreactivity was visualized by incubating the cerebellum in freshly prepared DAB solution (Sigma, UK) for 3 min at RT. The stained cerebellum was imaged with a Nikon SMZ1500 microscope and Nikon DXM1200 camera.
Droplet Polymerase Chain Reaction Analysis of F8A2–F8A3 Inversion Genotype
All reagents were from Sigma, UK, unless otherwise stated. Cultured cells were lysed in lysis buffer (0.1 M Tris, 0.2 M NaCl, 5 mM EDTA, 0.4% SDS, and 0.2 mg/ml proteinase K, pH 8.0) at 55ºC. Cell DNA was precipitated by adding isopropanol and washed with 70% ethanol. DNA pellet was dissolved in water and digested with NruI and BspEI (NEB, UK). Polymerase chain reactions (PCRs) were prepared in a total volume of 100 μl with 1× Go-taq buffer (Promega, UK), 25 mM MgCl2, 250 μM dNTPs, 1 μM primers (Eurofins Genomics, Germany): F8A2-F1-2 5′-CACATGATGAAAGTGGGAGGA-3′, F8A2-R2-2 5′-GAATGCAACAAATCAGCAAGA-3′, and F8A2-R3-2 5′-TTCAGACCCATATAGTATTACTGGTGA-3′, 30 nM primer F8A2-R1-2 5′-GCATACACTGCTAGGTGGGAATTCACAGCCACTGGAATGAC-3′, 200 ng digested genomic DNA, and 16 units Go-Taq DNA polymerase.
Emulsion step was carried out by adding PCR reaction dropwise over 30 s to 200 μl light mineral oil with 4.5% v/v Span 80, 0.4% v/v Tween 80, and 0.05% Triton X-100, in a 2 ml Corning Cryo-Tube stirring with a magnetic bar (8 × 3 mm with a pivot ring; VWR) at 1,000 rpm. Emulsions were stirred for 3 min before being overlaid with 30 μl mineral oil. The PCR conditions were 95°C for 120 s; 40 cycles of 95°C for 20 s, 60°C for 30 s, and 72°C for 15 s; 72°C for 5 min. Emulsions were disrupted using 600 μl hexane. Each clean PCR product (2 μl) was amplified in a total volume of 50 μl using primers F8A2-F1-2, F8A2-R2-2, and F8A2-R3-2 and reaction mix: 1× Go-taq buffer, 5 μl 25 mM MgCl2, 250 μM dNTPs, 300 nM primers, and 1 unit Go-Taq DNA polymerase.
Lymphoblastoid Cell Lines and DNA Samples
Cell lines and DNA samples were randomly selected from AGRE (https://research.agre.org/program/descr.cfm) and SFARI (https://www.sfari.org/) resources. AGRE samples are from multiplex families, and SFARI samples are from simplex families. Further details are available from provider websites using sample reference numbers listed below. Additional autism DNA samples were obtained from Prof. David Skuse, University College London. Control DNA samples were from the Caucasian DNA panel from the Coriell Institute for Medical Research, USA. Cells were grown in T25 suspension cell flasks with RPMI-1640 medium supplemented with 10% FBS (Sigma, UK) at 37°C, 5% CO2.
Cell lines used were as follows (double-underlining indicates samples with F8A2–F8A3 inversion; see Results section):
AGRE: 2095, 2325, 2396, 2479, 2609, 2615, 2659, 2664, 2718, 2815, 2838, 2853, 2880, 2883, 3126, 2742, 2831, 2327, 2628, 2678, 2326, 2791, 2487, 2328.
SFARI: SSC00317, SSC00591, SSC00636, SSC02727, SSC03440, SSC03459, SSC03537, SSC03774, SSC03989, SSC04232, SSC05124, SSC05350, SSC05435, SSC07444, SSC10172, SSC10210, SSC10777, SSC11067, SSC12271.
Long-Range PCR of β-Satellite Repeat
PCR reactions were prepared in a total volume of 50 μl with 25 μl 2× GoTaq Long PCR Master Mix (Promega, UK), 10 μl 300 nM primers (Eurofins Genomics, Germany) (Y-Chr BSR-Del-3F 5′-CACAGGCTGTAGTGCAGGTGATG-3′ and Y-Chr BSR-Del-4R 5′-CTGTGTTGTTGATCTGTCTAATGTTGACATTA-3′), and 500 ng genomic DNA. The PCR conditions were 95°C for 120 s; 40 cycles of 93°C for 20 s, 60°C for 16 min; final extension of 72°C for 20 min.
Analysis of PsychENCODE Data
We obtained paired-end RNA-seq libraries of cerebellar vermis from 33 autism and 38 controls from PsychENCODE (54). Individual libraries contained 50–200 million reads. Human transcriptome sequence was obtained from the RefSeq database (55), downloaded from NCBI (Annotation Release 108). Raw reads were aligned to the set of human RefSeq transcript sequences using bowtie2 short read alignment program (56). Default parameters were used for local alignments. Reads mapping to only one location in the transcriptome were selected by removing the alignments with “XS:i” bowtie2 tag, which represents reads having more than one possible mapping to the reference. SAMtools version 1.3.1 (57) was used to obtain the sorted BAM alignment files, which were further used to predict the heterozygosity in SPRY3 expressed sequences. SPRY3 had a total mapped read count range of 653–4033. SAMtools mpileup (57) and BCFtools (58) were used to characterize variations in mapped reads at each coordinate in the SPRY3 locus. The frequency of variants at each position was analyzed to estimate the likelihood of heterozygosity. For heterozygous genotypes, it is expected that the probability of finding a nucleotide matching the reference sequence at the single nucleotide polymorphism (SNP) position is 0.5, while for homozygous genotypes it is either 0 or 1.
Results
Cerebellar Lobule Gene Expression Screen Identifies Opposite Expression of Spry3 and p75NTR in Lobules VI–VII and X
We used whole mount immunohistochemistry of cerebellums from adult male and female C57Bl/6J strain mice and confirmed relatively low Spry3 expression in lobules VI–VII and X, as previously noted in mouse Allen Brain Atlas (ABA) and GENSAT data [Figure 1A, B, F; see also Ref. (33)]. We next sought to determine whether other genes share this expression pattern by visually inspecting the spatial expression patterns of genes in sagittal sections of the mouse ABA data as follows: i) 54 genes with biased expression in “Cerebellar cortex, Purkinje layer” under the “Fine Structure Search” option of the mouse ABA (Supplementary Table 1); ii) mouse homologues of 87 high-risk autism genes from SFARI (https://www.sfari.org/resource/sfari-gene/#bottom; Supplementary Table 2). Three of 54 cerebellar cortex-biased gene set (Abhd3, Lrp8, and Plcβ4) had lower expression in lobules VI–VII and X (Figure 1G, H, I), reminiscent of the Spry3 pattern, but none of 87 SFARI gene mouse homologues had this pattern; however, many of the latter had faint staining and were difficult to score.
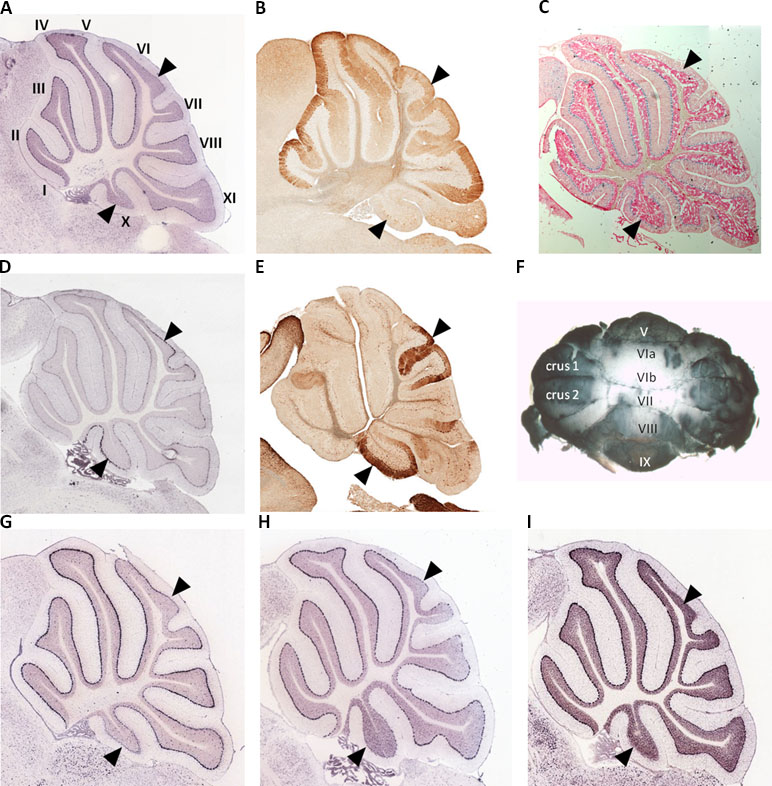
Figure 1 Lobular expression of genes in adult mouse cerebellum. (A, B)Spry3. (C) Human SPRY3 promoter–LacZ reporter transgenic mouse. (D, E)p75NTR. (F) Representative whole mount immumohistochemistry of adult female mouse cerebellum using anti-Spry3 antibody. (G–I)Abhd3, Lrp8, and Plcβ4. Images (A, D) and (G–I) were from Allen Brain Atlas; (B and E) were from GENSAT. Data for images (A–C) were previously published (31) and are included here for comparison with p75NTR expression. Roman numerals (I–X) in panel A indicate lobule identity. Arrowheads indicate lobules VI–VII and X with notable gene expression patterns.
Transcription factors (TFs) predicted to regulate human SPRY3 (ZNF263, MAZ, PURA, EGR1, PAX6) are expressed in mouse Purkinje cells (33), and GTEx data confirm relatively high expression of these factors in human adult cerebellum (Figure 2). However, limited data on these genes in ABA and GENSAT did not allow us to determine whether their spatial expression patterns coincide with Spry3 lobular expression.
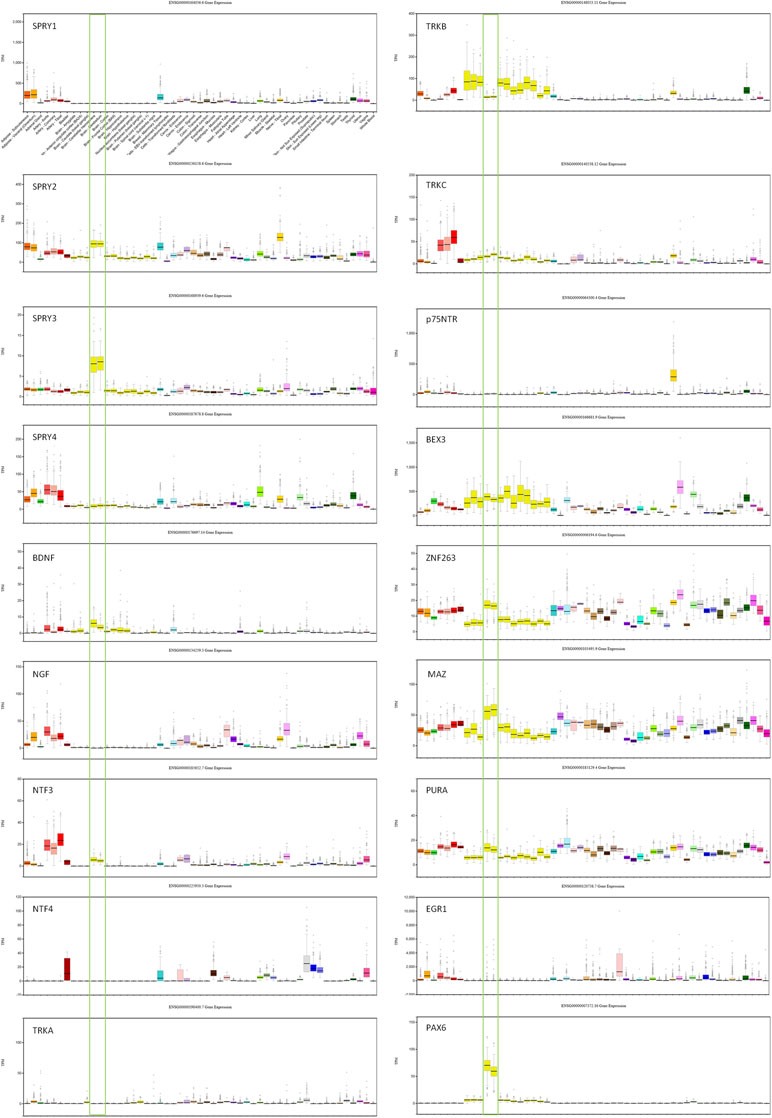
Figure 2 GTEx data for genes associated with SPRY3 expression and regulation. Cerebellum samples are boxed to highlight expression relative to other tissues: brain–cerebellar hemisphere; brain–cerebellum. Note different scales on TPM (transcripts per million) Y-axis for each gene.
We next examined spatial expression of Spry1, Spry2, and Spry4; neurotrophins (Ngf, Bdnf, NTF3, and NTF4); neurotrophin receptors (p75NTR, TrkA, TrkB, and TrkC); and Bex3 (Ngfrap1), which encodes a p75NTR interacting protein (59), in cerebellum to determine possible lobular co-expression with Spry3. On ABA, Spry1 and Spry4 exhibit faint staining, whereas Spry2 is widely expressed, including in cerebellar Purkinje cells, but does not exhibit a specific lobular expression pattern like Spry3. This is consistent with GTEx data in which SPRY2 has relatively high expression in human cerebellum, whereas SPRY1 and SPRY4 exhibit no and low expression, respectively (Figure 2).
For the neurotrophins, expression data on GTEx indicated that BDNF and NTF3 are relatively highly expressed in cerebellum, compared to other brain regions, whereas there was no expression of NGF and NTF4 (Figure 2). However, on ABA, mouse Ntf4 is expressed in Purkinje cells, whereas there is no Bdnf or Ngf, and barely detectable Ntf3 expression. It is unclear if these species differences reflect biological differences or technical limitations.
The data for neurotrophin receptor expression were generally consistent between ABA (mouse) and GTEx (human) datasets. On ABA, TrkB is widely expressed in the brain, including in cerebellar Purkinje cells, and GTEx data also indicated its wide expression in brain including cerebellum (Figure 2). TRKA and TRKC exhibited low and moderate expression, respectively, on GTEx, and no expression was detected on ABA. Analysis of p75NTR expression in GTEx suggested that it is virtually absent from the central nervous system, apart from a marginal signal in cerebellum and high expression in the peripheral nervous system (spinal cord, tibial nerve; Figure 2). However, scrutiny of ABA and GENSAT data indicated wide expression of p75NTR in the mouse adult brain, but with restricted expression in cerebellar vermis Purkinje cells, largely restricted to lobules VI–VII and X (Figure 1D, E), the exact opposite of the Spry3 lobular expression pattern. Bex3 (and other Bex genes; data not shown) is expressed throughout the adult mouse brain (ABA), including in cerebellar Purkinje cells, and is highly expressed in human cerebellum (Figure 2).
Genetic Analysis of X Chromosome-Linked Regulatory Sequences Upstream of Human SPRY3
The unique genomic configuration of human SPRY3 due to the evolution of the PAR2 in the hominin lineage suggests that sex-linked upstream elements could regulate expression of X-linked SPRY3 and epigenetic silencing of Y-linked SPRY3 (33). X-linked SPRY3 transcription initiates in the F8A3–TMLHE region (33). F8A3 is associated with inversions involving F8A1 and F8A2 (60, 61). The F8A2–F8A3 interval contains regulatory elements approximately 20 and 60 kb centromeric of F8A3 (Figure 3). Inversion of this sequence might affect regulation of flanking genes including F8A3 region-associated SPRY3 transcription. We developed a single-molecule droplet PCR assay to determine the orientation of the F8A2–F8A3 interval using a modified version of Turner et al. (62, 63) (Figure 4). We analyzed DNA from autism cases comprising 20 individuals from SFARI resource and 24 individuals from AGRE, representing simplex and multiplex families, respectively (see Materials and Methods for sample identifiers). There were 2/20 and 4/24 inversions compared to the reference sequence (GRCh38/hg38 assembly, December 2013, UCSC browser), which is similar to the 20% frequency (4/20 samples) found in non-autistic F8 gene-associated hemophilia patients (64).

Figure 3 ENCODE/OREG annotations in Xq28 F8A2–F8A3 interval.
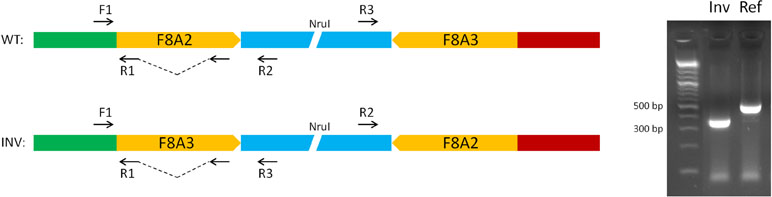
Figure 4 Strategy for single-molecule PCR genotyping of F8A2–F8A3 inversion polymorphism. Ref, orientation allele described in reference sequence on UCSC browser (HumanGRch37/hg19); Inv, opposite orientation to reference sequence.
Analysis of Y Chromosome-Linked Regulatory Sequences Upstream of Human SPRY3
We hypothesised that Y chromosome sequences proximal to the Yq–PAR2 boundary act as a silencer of Y-linked SPRY3. Scrutiny of this region using the UCSC browser identified a predicted regulatory region that begins 60 kb upstream of the SPRY3 PAR2 transcriptional start site (TSS) and extends a further 60 kb towards the centromere. This element comprises compositionally distinct regions of ∼25 and ∼35 kb and is flanked by a 50 kb sequence gap proximally, on the far side of which are the major Yq12 satellite sequences (Figure 5). The 25 kb region comprises ∼10 kb of simple CATTC and CACTC repeats, while the remaining ∼15 kb is a beta satellite repeat (BSR). The entire ∼60 kb region has multiple DNaseI sensitive sites and CTCF binding sites (data not shown), and the BSR, similarly to the SPRY3 core promoter AG-rich repeat, contains multiple ZNF263 binding sites. BLAST of the ∼15 kb Yq12 BSR identified related sequences on chrs. 4, 10, 14, and 18 with 100% coverage and identity scores of 78–83%. The BSR-containing allele of the FSHD gene locus on chr. 4q35 (65) is not represented on the UCSC browser; but the structurally similar 10q26.3 locus is (66, 67). This exhibits a different pattern of chromatin modifications to the Yq12 BSR, increasing our confidence in the attribution of Yq12 annotations (Figure 5).
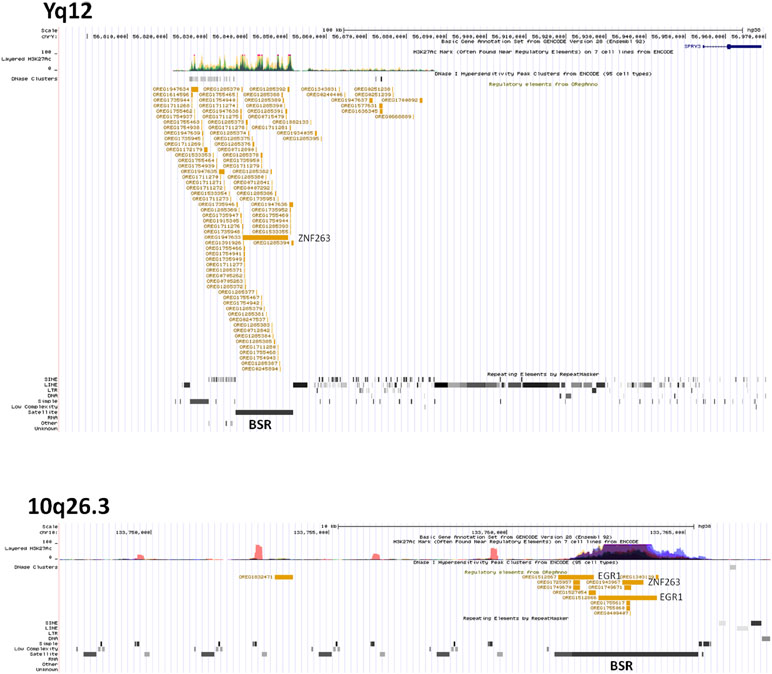
Figure 5 ENCODE/OREG annotations in Yq12 and 10q26.3 regions showing major DNA factors binding in BSR regions (ZNF263, EGR1). Note different scales in each panel.
Inspection of the Yq12 region in the database of structural variants (DSV) using the UCSC browser indicated significant variability of the BSR, including independently reported deletions and duplications (Figure 6A). We attempted to use long PCR of genomic DNA using primers flanking the BSR to determine whether structural variants or length polymorphisms are associated with autism. Primer design was severely restricted due to the genomic architecture of the region, and the selected primer pair amplified a ∼3.8 kb product from all male samples and no female samples (Figure 6B). Samples were as follows: 20 male autism (AGRE); 21 male autism (Skuse samples); 12 normal male and 4 normal female (Skuse and Coriell Caucasian panel). Cloning of this PCR product was problematic, but sequences obtained from both ends of a cloned partial product matched the expected 5′ and 3′ boundaries of the reference sequence (Figure 6C). However, this product was not amplified from BAC clone RP11-88F4, which covers the region, reducing confidence in the genomic location of the template for the ∼3.8 kb amplicon (data not shown).
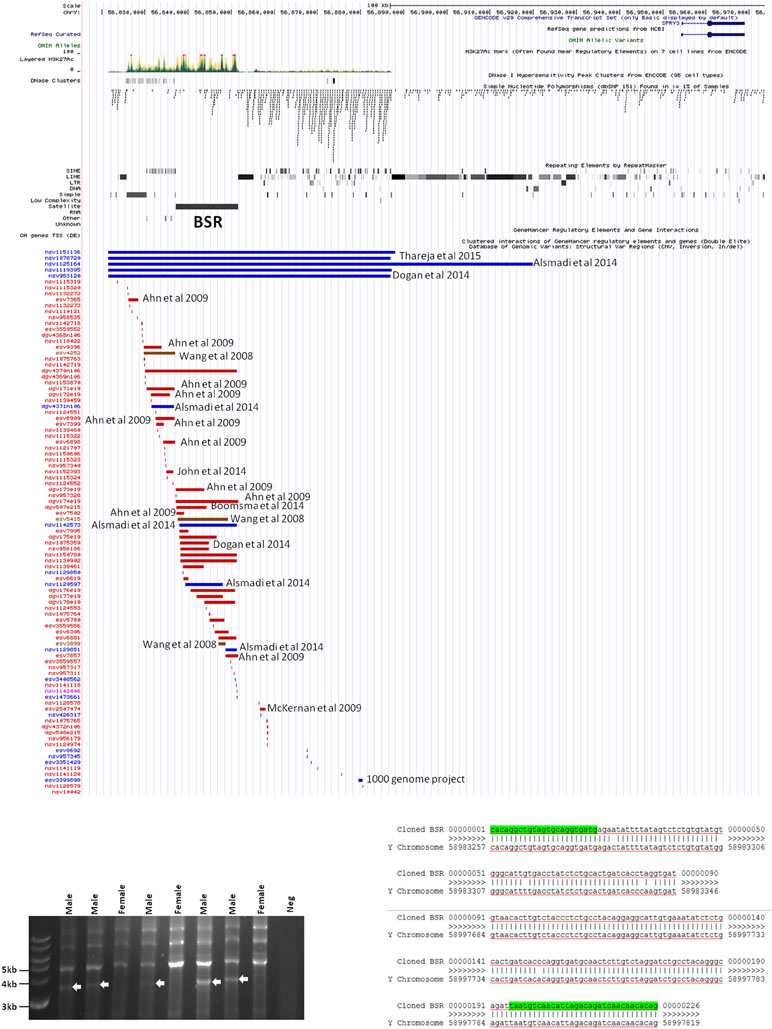
Figure 6 (A) Map of genomic variants in Yq12 region. Details of annotated publication references are available on UCSC browser. (B) Male-specific ∼3.5 kb PCR product amplified by BSR-specific primers (8 of 57 samples analyzed are shown). (C) Sequences of 5′ and 3′ ends of cloned PCR product.
Analysis of SPRY3 Allele-Specific Expression in Cerebellum Using PsychENCODE Dataset
Genetic and structural analysis of the Yq12 putative regulatory region was inconclusive; therefore, we looked for loss of epigenetic silencing and reactivation of the Y-linked SPRY3 allele in a comparison of RNA-Seq data from male autism and control cerebellum samples. We obtained paired-end RNA-seq libraries of cerebellar vermis from 33 autism and 38 control samples from PsychENCODE (54). Genotypes for X and Y chromosome (including PAR2) markers are not available for these samples; therefore, we used a statistical approach to analyze the level of heterozygosity of transcripts of the SPRY3 and control genes. Heterozygous expression of SPRY3 would be indicative of expression of both X- and Y-linked alleles due to pathological reactivation of the Y-linked copy. Genes flanking SPRY3 were also examined. TMLHE was used as a negative control to estimate the level of variants due to technical noise (e.g., sequencing errors, substitutions during library preparation, and misalignments) since it is X-linked and no bona fide heterozygosity is expected. SYBL1 was analyzed because it flanks SPRY3 distally and is similar to SPRY3 in having a Y-linked copy; however, its silencing is associated with a methylated CpG island, and it may be regulated differently to SPRY3 (68). Autosomal genes (NPTN and MCM6) were used as positive controls, where we expected to identify heterozygosity. There was no significant difference in the heterozygosity plots of autism versus control samples for any of the genes analyzed (Figure 7; Supplementary Figure 1).
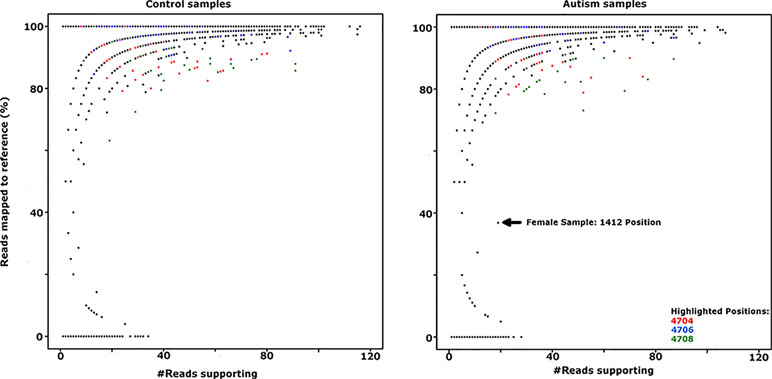
Figure 7 Comparison of SPRY3 (RefSeq ID: NM_005840) expression heterozygosity maps for autism and control samples. SNP positions 4704, 4706, and 4708 (highlighted in red, blue, and green, respectively) produced all possible genotypes in the majority of control and autism samples, suggesting a sequencing artefact at these positions, and were not counted as heterozygotes. Female sample 1412 position is included as indicative of a true heterozygous sample.
Discussion
We have extended our previous work implicating SPRY3 in autism (33) and provide evidence for a possible mechanism of chromosome Y-linked SPRY3 gene deregulation underpinning male susceptibility. At the cellular level, we propose that SPRY3 deregulation affects the functioning of the BDNF–TrkB–p75NTR neurotrophin pathway, leading to cerebellar Purkinje cell pathology. Our hypothesis can explain the specific lobular distribution of Purkinje cell loss in autism (lobules VI, VII, and X), as previously described (41). More speculatively, SPRY3 deregulation could explain a reported, although currently unconfirmed, lung branching abnormality in autism (69).
The male bias in autism prevalence is not explained by known DNA susceptibility variants because the majority are autosomal, and a major sex-linked gene effect has not been identified (10, 14, 17, 26, 70–76). This suggests that autosomal variants interact with one or more sex-specific developmental or regulatory pathways, which could include sex hormones, or X or Y chromosome-linked gene-encoded regulators. This hypothesis requires that the majority (perhaps hundreds) of individual susceptibility variants converge on sex-specific mechanisms that underpin either a female protective effect or male susceptibility effect (FPE or MSE) (27, 29). The plausibility of FPE/MSE mechanisms is supported by mouse mutants of known autism genes, which exhibit sex-specific phenotypes (77–79), the presence of X-linked regulators expressed differently in males and females (80), and the influence of sex hormones such as testosterone and estrogen on normal and abnormal brain development and function (30, 31, 81–83).
However, recent studies did not detect a predicted Carter effect in autism because an increase in disease aggregation in families with a female proband was not observed, as would be expected if affected females require a higher mutation burden to overcome an FPE threshold (73, 84–86). This suggests that sex-specific departures from normal physiology, rather than normal sex-specific physiology per se, may underlie the male bias in autism (87). An alternative hypothesis to FPE/MSE is therefore the existence of one or more male-specific disease mechanisms (MDM). Such MDM would have to occur at a high frequency to explain the large male prevalence bias.
There are potentially two general mechanisms of SPRY3 deregulation in autism. First, trans-acting effects of susceptibility variants at other loci encoding, for example, chromatin regulators could deregulate X- or Y-linked SPRY3. This category would also include environmental effects, for example, due to alterations in carnitine levels, to which SPRY3 may be responsive (see below). Second, cis-acting de novo mutations or common variants in regulatory regions could cause aberrant expression of X- or Y-linked SPRY3. Previous genetic studies have not associated the Xq28 or PAR2 regions with autism (88). However, both the X and Y chromosome regions upstream of PAR2 are structurally complex and difficult to analyze, and therefore, autism-associated variants may have been overlooked.
The F8A2–F8A3 interval has a common inversion polymorphism that may alter the orientation and distance from SPRY3 of ENCODE/OREG-predicted regulatory sequences. The major regulatory factors that bind in this region (TRIM28, SMARCA4) are associated with autism (89–91); therefore, inversions or other rearrangements of this region may impact on expression of F8A2/F8A3 region-associated transcription or on flanking genes (CLIC2, TMLHE, SPRY3), potentially contributing to autism risk. It is unknown how frequently de novo inversions occur, and inversion alleles are not tagged by known SNPs. Therefore, we used single-molecule analysis to determine orientation of this inversion in a small number of autism and control DNA samples. We observed similar allele frequencies to those reported from an analysis of 20 hemophilia patients (64), with no evidence of a strong association with autism.
The Yq12 PAR2 region is poorly characterized due to the absence of genetic recombination and the highly repetitive DNA sequences that comprise much of the Yq and PAR2 boundary regions. However, our identification of a putative regulatory region in distal Yq12, 60 kb upstream of the SPRY3 TSS, suggests a mechanism of silencing of Y-linked SPRY3. Similar to the SPRY3 core promoter AG-rich repeat (33), a BSR in this region has multiple ZNF263 binding sites, suggesting a possible regulatory interaction with the SPRY3 promoter. A BSR at chr. 10q26.3, also annotated in ENCODE, has a different pattern of chromatin modifications, increasing confidence in the Yq12 annotations. Interestingly, copy number variants (CNV) in the 10q26.3 region are associated with autism, although there is no evidence that this is due to the BSR (92; https://gene.sfari.org/database/cnv/10q26.3). Non-BSR sequences in the Yq12 region have abundant CTCF binding sites, a factor associated with gene imprinting and genome topology (93), further suggesting a regulatory function for this region in regulating Y-linked SPRY3.
There is extensive sequence and structural variation across the Yq12 putative regulatory region, including reported length polymorphisms of the BSR. However, BSRs are abundant in the genome (94, 95), and the majority are not annotated; therefore, caution is required when interpreting annotations arising from genome-wide studies underpinned by short sequence reads. We were unable to confirm Yq12 BSR variation using long PCR due to severe sequence constraints in primer design and instability of cloned PCR products. Other approaches, such as fiber FISH or single-molecule sequencing, anchored in unique sequences in PAR2, will be required to provide confirmation of BSR length alleles and to conduct genetic association studies. We also cannot exclude a role for somatic cell mutations, which are increasingly implicated in neurodegeneration (96). Somatic instability of the repeat-rich Yq12–PAR2 region could result in cell autonomous DNA rearrangements and deregulation of Y-linked SPRY3.
Notwithstanding significant technical difficulties in analyzing the Yq12–PAR2 boundary region, our data suggest a hypothetical mechanism whereby genetic (DNA sequence) or epigenetic (chromatin structure) variation could lead to reactivation and inappropriate lobular expression of Y-linked SPRY3. We sought to test this hypothesis by analyzing allelic expression of SPRY3 in PsychENCODE cerebellum expression data, but we did not detect biallelic expression in male samples as would be predicted if the Y-linked copy is active. However, we lacked the sample genotypes and information about the exact cerebellar lobules sampled. Also, the pathological mechanism we propose may not be detectable in RNA-Seq data if reactivation of Y-linked SPRY3 ultimately results in Purkinje cell death. Therefore, we do not consider this a definitive rejection of our hypothesis.
A possible further mode of SPRY3 deregulation is suggested by its linkage with TMLHE and their overlapping regulatory sequences (33). TMLHE is an enzyme in the carnitine biosynthesis pathway, and carnitine deficiency is associated with autism (52, 97, 98). Intriguingly, there is evidence that carnitine levels modulate SPRY3 expression in the pig (51). TMLHE mutations are rare but well-established autism susceptibility factors (52, 88, 97, 99, 100). Mouse Tmlhe is expressed in Purkinje cells; however, the lobular expression pattern is not restricted like Spry3. At least one mutation attributed to TMLHE-associated autism risk also affects SPRY3 sequences (33), suggesting a possible role for deregulation of SPRY3 in some reported cases.
Deficits in cerebellar vermis structure and Purkinje cell number and morphology have been reported frequently in autism at post mortem, using MRI imaging, and in mouse models (39, 82, 101–110). In a histological study of human brain tissues from autism cases, Skefos et al. (41) reported that Purkinje cell loss predominantly affects crus I and II (lobule VIIa), and they also noted a possible male-specific deficit in lobule X of the flocculonodular lobe. Following our previous study (33), and arising from our current observations, we show that mouse Spry3 and p75NTR have opposite expression patterns in cerebellar vermis lobules VI–VII and X. Spry3 is not expressed in these lobules, whereas p75NTR is strongly expressed [see also Figure 2 in Ref. (111) and Figure 1F in Ref. (112)]. In a screen of 135 genes in the adult mouse (selected for high cerebellar expression or prior association with autism), we identified only three (Abhd3, Lrp8, and Plcβ4) with a somewhat similar lobular expression pattern to Spry3, and none that recapitulated the p75NTR expression pattern. This suggests that there are relatively few genes whose expression could explain the lobular pattern of abnormalities described by Skefos et al. (41). Our qualitative screen of the ABA mouse data was restricted to adult brain and may therefore lack sensitivity; however, we are reassured regarding its specificity by an independent report that Plcβ4 is not expressed in lobules VI–VII and IX–X (113).
The opposite expression patterns of Spry3 and p75NTR mirror the well-known opposite expression patterns of zebrin II/aldolase C and Hsp25/Hspb1 on the anterior–posterior (AP) axis (114–118). The mechanisms responsible for patterning the anterior–posterior axis of the cerebellum include the autism gene Engrailed-2 (En2; 118, 119), and intriguingly, En2 has specific functions in the development of lobules VI–VII and X (118, 120), suggesting a further mechanism underpinning involvement of these lobules in autism (after 39).
A recent report suggests that lobules VI–VII (Crus I) in rodents are homologous to Crus I and II in primates (121). Therefore, if mouse Spry3 and p75NTR expression patterns are conserved in human, as appears likely from a SPRY3 promoter–reporter transgenic mouse (33), and scrutiny of the ABA (human) and GTEx databases, we can propose an MDM in which aberrant expression of human SPRY3 in lobules VI–VII and X interferes with neurotrophin signaling, causing Purkinje cell pathology through a BDNF–TrkB–p75NTR mechanism. Spry1, 2, and 4 regulate receptor tyrosine kinase signaling including FGFR (122–125), whereas evidence from Xenopus and mouse indicates a regulatory loop involving BDNF- and TrkB-dependent expression of Spry3, and Spry3-mediated inhibition of BDNF–TrkB signaling (42). Therefore, it is possible that both SPRY3 and p75NTR proteins interact with BDNF–TrkB signaling, a pathway implicated in neuronal (including Purkinje) cell development and survival (126–130). Although p75NTR is not listed on the SFARI autism gene database, it is a compelling candidate for involvement in autism pathogenesis (128, 131, 132). There is extensive evidence of both pro- and anti-apoptotic functions for p75NTR, particularly in contexts with concomitant alteration of neurotrophin receptor signaling, including of TrkB (130, 133–138). SPRY3 regulates TrkB signaling (42); therefore, we speculate that inappropriate expression of SPRY3 in lobules VI–VII and X, in the context of TrkB and p75NTR expression, may affect Purkinje cell function or survival, although the exact mechanism would have to be established in relevant models, as neurotrophin signaling effects depend strongly on physiological context (136, 139).
We previously reported that a human SPRY3 promoter–LacZ reporter transgenic mouse substantially recapitulated the mouse Spry3 expression pattern (33). Similar to mouse Spry3, human SPRY3-LacZ is expressed in Purkinje cells throughout the cerebellum, except in lobules VI–VII. However, unlike mouse Spry3, human SPRY3-LacZ is expressed in lobule X. Therefore, sequences outside of the human core promoter, or trans-acting factors whose expression differs between mouse and human, may be required for regulation of SPRY3 in this lobule. Interestingly, the deficits in lobule X identified by Skefos et al. (41) may be male-specific, which, in the context of our proposed mechanism of pathological over-expression of SPRY3, could indicate a role for deregulation (reactivation) of the Y-linked copy in this lobule, which might have a different expression pattern compared to the X-linked copy due to the lack of cis-acting X-linked regulatory sequences, or the inappropriate influence of Y-linked sequences (Figure 8).
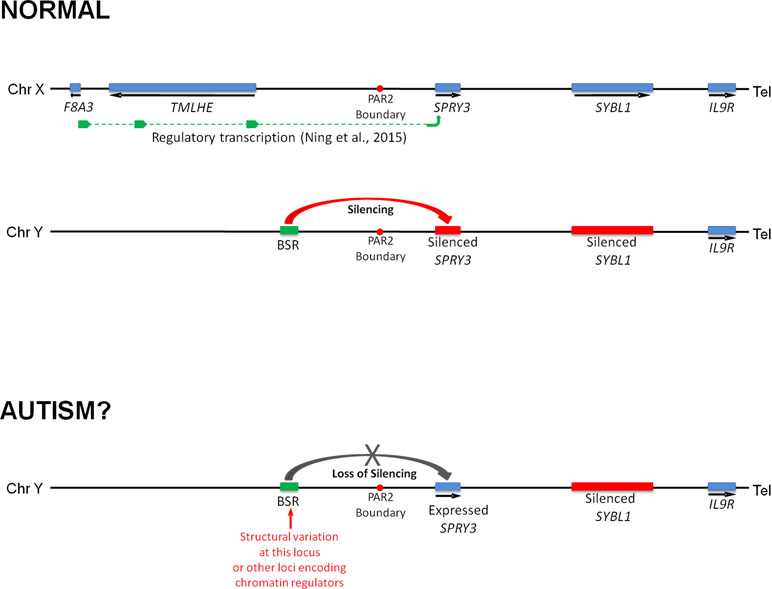
Figure 8 Model of regulation of human SPRY3 by X and Y chromosome-linked regulatory regions and deregulation in autism. We propose that regions including the F8A2–F8A3 interval and associated transcription spanning the Xq28–PAR2 boundary, and Y-linked ENCODE/OREG-annotated regions including the BSR, may be deregulated by cis- or trans-acting factors. More specifically, we propose that cis-acting Yq12 genomic variants, including BSR length variants, may result in variable reactivation and expression of Y-linked SPRY3, leading to inappropriate lobular expression. Y-linked transcription may have altered lobular expression due to lack of putative Xq28-linked regulatory elements associated with the normal pattern of lobular expression from X-linked SPRY3 or due to influence of Yq12 genomic elements.
Finally, we note that Sprouty was originally described based on a branching phenotype of the apical airways of Drosophila (140), and mouse Spry2 coordinates vascular and airway branching in the lung (141). Spry3 is expressed in the mouse lung bronchial tree (our unpublished data) and in human lung (GTEx), suggesting that deregulation of SPRY3 could potentially provide a mechanism underpinning a lung branching abnormality reported in autism patients (69).
In future work, we aim to deepen our understanding of SPRY3 and p75NTR expression and functional interactions during brain and lung development in the human and mouse, including in autism mouse models. Transgenic under- or over-expression of Spry3 in cerebellar lobules VI–VII and X in mice would provide an in vivo model of our proposed MDM in autism. Due to the unique genomic architecture and regulation of the human PAR2, and the difficulty in sourcing matched tissue samples from specific cerebellar lobules from normal and autism brains, and from other organs such as lung, the analysis of Y-linked SPRY3 deregulation in the cerebellum and lung will be challenging, particularly if the pathology results in cell death. However, advances in single-molecule DNA sequencing techniques will facilitate detection of genomic variants in this region that may be associated with autism.
Ethics Statement
Access to human genetic data and biomaterials for this study was carried out in accordance with the procedures and recommendations of the UCC Office of the Vice-President for Research and Innovation, specifically for accessing AGRE and SFARI datasets and biomaterials, under their respective procedures. All subjects gave written informed consent in accordance with AGRE and SFARI protocols and procedures, consistent with the Declaration of Helsinki. Access to animal tissues for this study was pursued in accordance with the recommendations of the UCC Animal Experimentation and Ethics Committee under licencing from the Irish Health Products Regulatory Authority (https://www.hpra.ie/).
Author Contributions
ZN and JW carried out all of the bench work and aspects of bioinformatics and data analysis. RK and PB carried out analysis of PsychENCODE data. TM conceived of, and supervised, the study and wrote most of the manuscript. All authors contributed to various aspects of manuscript preparation.
Funding
This work was funded by grants to TM from Science Foundation Ireland (TIDA Award 13/TIDA/B2659) and Higher Education Authority “Irish Transgenic Network” award.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge use of data and images from Allen Brain Atlas, GENSAT, GTEx, and UCSC Browser, as described in the main text. We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. AGRE is a program of Autism Q2 Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M. Lajonchere (PI). We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We are grateful to D. Skuse for DNA samples. We are grateful to D. Geschwind for access to PsychENCODE data. We thank J. Quinn for discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00416/full#supplementary-material
Supplementary Table 1 | Mouse Purkinje cell expressed genes whose spatial expression was examined in Allen Brain Atlas data. For consistency, the numbering scheme for the 54 genes identified as “cerebellum, Purkinje cell enhanced” in the Allen Brain Atlas “fine structure” search is maintained here, with inclusion of two blank rows (29 and 53).
Supplementary Table 2 | Human autism genes from SFARI “High confidence” and “Strong candidate” lists (https://www.sfari.org/resource/sfari-gene/#bottom) for which spatial expression of their mouse orthologues was examined in Allen Brain Atlas data.
Supplementary Figure 1 | Comparison of SPRY3 expression heterozygosity map with maps for TMLHE (X-linked), SYBL1 (PAR2-linked), and NPTN, MCM6 (autosomal) genes.
References
1. Gillberg C. The ESSENCE in child psychiatry: early symptomatic syndromes eliciting neurodevelopmental clinical examinations. Res Dev Disabil (2010) 31:1543–51. doi: 10.1016/j.ridd.2010.06.002
2. Constantino JN, Charman T. Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet Neurol (2016) 15(3):279–91. doi: 10.1016/S1474-4422(15)00151-9
3. Posserud M, Hysing M, Helland W, Gillberg C, Lundervold AJ. Autism traits: the importance of “co-morbid” problems for impairment and contact with services. Res Dev Disabil (2018) 72:275–83. doi: 10.1016/j.ridd.2016.01.002
4. Gillberg C, Fernell E. Autism plus versus autism pure. J Autism Dev Disord (2014) 44(12):3274–6. doi: 10.1007/s10803-014-2163-1
5. Whitehouse AJ, Cooper MN, Bebbington K, Alvares G, Lin A, Wray J, et al. Evidence of a reduction over time in the behavioral severity of autistic disorder diagnoses. Autism Res (2017) 10(1):179–87. doi: 10.1002/aur.1740
6. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ (2018) 67(6):1–23. doi: 10.15585/mmwr.ss6706a1
7. Waugh I. The prevalence of autism (including Asperger syndrome) in school age children in Northern Ireland 2017. (2017) https://dera.ioe.ac.uk/29786/1/asd-children-ni-2017_Redacted.pdf.
8. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry (2011) 68(11):1095–102. doi: 10.1001/archgenpsychiatry.2011.76
9. Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry (2016) 57(5):585–95. doi: 10.1111/jcpp.12499
10. Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The heritability of autism spectrum disorder. JAMA (2017) 318(12):1182–4. doi: 10.1001/jama.2017.12141
11. Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet (2012) 131:565–79. doi: 10.1007/s00439-011-1094-6
12. Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders, Annu Rev Genom Hum Genet (2013) 14:191–213. doi: 10.1146/annurev-genom-091212-153431
13. Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet (2017) 49(7):978–85. doi: 10.1038/ng.3863
14. Constantino JN. Deconstructing autism: from unitary syndrome to contributory developmental endophenotypes. Int Rev Psychiatry (2018) 30(1):18–24. doi: 10.1080/09540261.2018.1433133
15. Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol (2016) 339(7-8):300–7. doi: 10.1016/j.crvi.2016.05.004
16. Loomes R, Hull L, Mandy WPL. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry (2017) 56(6):466–74. doi: 10.1016/j.jaac.2017.03.013
17. Palmer N, Beam A, Agniel D, Eran A, Manrai A, Spettell C, et al. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr (2017) 171(11):1107–12. doi: 10.1001/jamapediatrics.2017.2832
18. Scott FJ, Baron-Cohen S, Bolton P, Brayne C. Brief report: prevalence of autism spectrum conditions in children aged 5-11 years in Cambridgeshire, UK. Autism (2002) 6(3):231–7. doi: 10.1177/1362361302006003002
19. Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol (2011) 9(6):e1001081. doi: 10.1371/journal.pbio.1001081
20. Gould J, Ashton-Smith J. Missed diagnosis or misdiagnosis? Girls and women on the autism spectrum. Good Autism Practice (2011) 12(1):34–41.
21. Mandy W, Chilvers R, Chowdhury U, Salter G, Seigal A, Skuse D. Sex differences in autism spectrum disorder: evidence from a large sample of children and adolescents. J Autism Dev Disord (2012) 42(7):1304–13. doi: 10.1007/s10803-011-1356-0
22. Dworzynski K, Ronald A, Bolton P, Happé F. How different are girls and boys above and below the diagnostic threshold for autism spectrum disorders? J Am Acad Child Adolesc Psychiatry (2012) 51(8):788–97. doi: 10.1016/j.jaac.2012.05.018
23. Adviento B, Corbin IL, Widjaja F, Desachy G, Enrique N, Rosser T, et al. Autism traits in the RASopathies. J Med Genet (2014) 51(1):10–20. doi: 10.1136/jmedgenet-2013-101951
24. Westman Andersson G, Miniscalco C, Gillberg C. Autism in preschoolers: does individual clinician’s first visit diagnosis agree with final comprehensive diagnosis? Sci World J (2013) 2013:716267. doi: 10.1155/2013/716267
25. Lai MC, Baron-Cohen S, Buxbaum JD. Understanding autism in the light of sex/gender. Mol Autism (2015) 6:24. doi: 10.1186/s13229-015-0021-4
26. Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron (2015) 87(6):1215–33. doi: 10.1016/j.neuron.2015.09.016
27. Werling DM, Parikshak NN, Geschwind DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun (2016) 7:10717. doi: 10.1038/ncomms10717
28. Ecker C, Andrews DS, Gudbrandsen CM, Marquand AF, Ginestet CE, Daly E, et al. Association between the probability of autism spectrum disorder and normative sex-related phenotypic diversity in brain structure. JAMA Psychiatry (2017) 74(4):329–410. doi: 10.1001/jamapsychiatry.2016.3990
29. Floris DL, Lai MC, Nath T, Milham MP, Di Martino A. Network-specific sex differentiation of intrinsic brain function in males with autism. Mol Autism (2018) 9:17. doi: 10.1186/s13229-018-0192-x
30. Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci (2002) 6:248–54. doi: 10.1016/S1364-6613(02)01904-6
31. Baron-Cohen S, Auyeung B, Nørgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L, et al. Elevated fetal steroidogenic activity in autism. Mol Psychiatry (2014) 20:369–76. doi: 10.1038/mp.2014.48
32. Werling DM. The role of sex-differential biology in risk for autism spectrum disorder. Biol Sex Differ. (2016) 7:58. doi: 10.1186/s13293-016-0112-8
33. Ning Z, McLellan AS, Ball M, Wynne F, O’Neill C, Mills W, et al. Regulation of SPRY3 by X chromosome and PAR2-linked promoters in an autism susceptibility region. Hum Mol Genet (2015) 24(18):5126–41. Erratum in: Hum Mol Genet. 2015 Dec 20;24(25):7450. doi: 10.1093/hmg/ddv231
34. Becker EB, Stoodley CJ. Autism spectrum disorder and the cerebellum. Int Rev Neurobiol (2013) 113:1–34. doi: 10.1016/B978-0-12-418700-9.00001-0
35. Rogers TD, McKimm E, Dickson PE, Goldowitz D, Blaha CD, Mittleman G. Is autism a disease of the cerebellum? An integration of clinical and pre-clinical research. Front Syst Neurosci (2013) 7:15. doi: 10.3389/fnsys.2013.00015
36. Jeong J-W, Tiwari VN, Behen ME, Chugani HT, Chugani DC. In vivo detection of reduced Purkinje cell fibers with diffusion MRI tractography in children with autistic spectrum disorders. Front Hum Neurosci (2014) 8:110. doi: 10.3389/fnhum.2014.00110
37. Hampson DR, Blatt GJ. Autism spectrum disorders and neuropathology of the cerebellum. Front Neurosci (2015) 9:420. doi: 10.3389/fnins.2015.00420
38. Stoodley CJ, D’Mello AM, Ellegood J, Jakkamsetti V, Liu P, Nebel MB, et al. Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat Neuroscience (2017) 20:1744–51. doi: 10.1038/s41593-017-0004-1
39. Zhao G, Walsh K, Long J, Gui W, Denisova K. Reduced structural complexity of the right cerebellar cortex in male children with autism spectrum disorder. PLoS One (2018) 13(7):e0196964. doi: 10.1371/journal.pone.0196964
40. Whitney ER, Kemper TL, Rosens DL, Bauman ML, Blatt GJ. Density of cerebellar basket and stellate cells in autism: evidence for a late developmental loss of Purkinje cells. J Neurosci Res (2009) 87:2245–54. doi: 10.1002/jnr.22056
41. Skefos J, Cummings C, Enzer K, Holiday J, Weed K, Levy E, et al. Regional alterations in purkinje cell density in patients with autism. PLoS One (2014) 9(2):e81255. doi: 10.1371/journal.pone.0081255
42. Panagiotaki N, Dajas-Bailador F, Amaya E, Papalopulu N, Dorey K. Characterisation of a new regulator of BDNF signalling, Sprouty3, involved in axonal morphogenesis in vivo. Development (2010) 137(23):4005–15. doi: 10.1242/dev.053173
43. Correia CT, Coutinho AM, Sequeira AF, Sousa IG, Lourenço Venda L, Almeida JP, et al. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav (2010) 9(7):841–8. doi: 10.1111/j.1601-183X.2010.00627.x
44. Castrén ML, Castrén E. BDNF in fragile X syndrome. Neuropharmacology (2014) 76 Pt C:729–36. doi: 10.1016/j.neuropharm.2013.05.018
45. Koh JY, Lim JS, Byun HR, Yoo MH. Abnormalities in the zinc-metalloprotease-BDNF axis may contribute to megalencephaly and cortical hyperconnectivity in young autism spectrum disorder patients. Mol Brain (2014) 7:64. doi: 10.1186/s13041-014-0064-z
46. Zheng Z, Zhang Li, Zhu T, Huang J, Qu Y, Mu D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: a systematic review and meta-analysis. Sci Rep (2016) 6:31241. doi: 10.1038/srep31241
47. Kang MS, Choi TY, Ryu HG, Lee D, Lee SH, Choi SY, et al. Autism-like behavior caused by deletion of vaccinia-related kinase 3 is improved by TrkB stimulation. J Exp Med (2017) 214(10):2947–66. doi: 10.1084/jem.20160974
48. Ka M, Kim WY. ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol Dis (2018) 111:138–52. doi: 10.1016/j.nbd.2017.12.008
49. Maynard KR, Hobbs JW, Phan BN, Gupta A, Rajpurohit S, Williams C, et al. BDNF-TrkB signaling in oxytocin neurons contributes to maternal behaviour. eLife. (2018) 7:e33676. doi: 10.7554/eLife.33676
50. De Bonis ML, Cerase A, Matarazzo MR, Ferraro M, Strazzullo M, Hansen RS, et al. Maintenance of X- and Y-inactivation of the pseudoautosomal (PAR2) gene SPRY3 is independent from DNA methylation and associated to multiple layers of epigenetic modifications. Hum Mol Genet (2006) 15:1123–32. doi: 10.1093/hmg/ddl027
51. Keller J, Ringseis R, Priebe S, Guthke R, Kluge H, Eder K. Effect of L-carnitine on the hepatic transcript profile in piglets as animal model. Nutr Metab (Lond) (2011) 8:76. doi: 10.1186/1743-7075-8-76
52. Beaudet AL. Brain carnitine deficiency causes nonsyndromic autism with an extreme male bias: a hypothesis. Bioessays (2017) 39(8):1–26. doi: 10.1002/bies.201700012
53. Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature (2007) 445:168–76. doi: 10.1038/nature05453
54. Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature (2016) 540 :423–7. Erratum in: Nature. 2018 Aug;560 :E30. doi: 10.1038/nature20612
55. O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res (2016) 44(D1):D733–45. doi: 10.1093/nar/gkv1189
56. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol (2009) 10(3):R25. doi: 10.1186/gb-2009-10-3-r25
57. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
58. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, Depristo MA, et al. The variant call format and VCFtools. Bioinformatics (2011) 27(15):2156–8. doi: 10.1093/bioinformatics/btr330
59. Cabral KM, Raymundo DP, Silva VS, Sampaio LA, Johanson L, Hill LF, et al. Biophysical studies on BEX3, the p75NTR-associated cell death executor, reveal a high-order oligomer with partially folded regions. PLoS One (2015) 10(9):e0137916. doi: 10.1371/journal.pone.0137916
60. El-Hattab AW, Schaaf CP, Fang P, Roeder E, Kimonis VE, Church JA, et al. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review. BMC Med Genet (2015) 16:12. doi: 10.1186/s12881-015-0157-2
61. Bagnall RD, Ayres KL, Green PM, Giannelli F. Gene conversion and evolution of Xq28 duplicons involved in recurring inversions causing severe hemophilia A. Genome Res (2005) 15(2):214–23. doi: 10.1101/gr.2946205
62. Turner DJ, Shendure J, Porreca G, Church G, Green P, Tyler-Smith C, et al. Assaying chromosomal inversions by single-molecule haplotyping. Nat Methods (2006) 3(6):439–45. doi: 10.1038/nmeth881
63. Turner DJ, Hurles ME. High-throughput haplotype determination over long distances by haplotype fusion PCR and ligation haplotyping. Nat Protoc (2009) 4(12):1771–83. doi: 10.1038/nprot.2009.184
64. Bagnall RD, Giannelli F, Green PM. Int22h-related inversions causing hemophilia A: a novel insight into their origin and a new more discriminant PCR test for their detection. J Thromb Haemost (2006) 4(3):591–8. doi: 10.1111/j.1538-7836.2006.01840.x
65. Kim E, Rich J, Karoutas A, Tarlykov P, Cochet E, Malysheva D, et al. ZNF555 protein binds to transcriptional activator site of 4qA allele and ANT1: potential implication in Facioscapulohumeral dystrophy. Nucleic Acids Res. (2015) 43(17):8227–42. doi: 10.1093/nar/gkv721
66. van der Maarel SM, Frants RR. The D4Z4 repeat-mediated pathogenesis of facioscapulohumeral muscular dystrophy. Am J Hum Genet (2005) 76(3):375–86. Epub 2005 Jan 24. Review. No abstract available. doi: 10.1086/428361
67. Krom YD, Thijssen PE, Young JM, Hamer B, Balog J, Yao Z, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet (2013) 9(4):e1003415. doi: 10.1371/journal.pgen.1003415
68. Matarazzo MR, De Bonis ML, Gregory RI, Vacca M, Hansen RS, Mercadante G, et al. Allelic inactivation of the pseudoautosomal gene SYBL1 is controlled by epigenetic mechanisms common to the X and Y chromosomes. Hum Mol Genet (2002) 11(25):3191–8. doi: 10.1093/hmg/11.25.3191
69. Stewart BA, Klar AJ. Can bronchoscopic airway anatomy be an indicator of autism? J Autism Dev Disord (2013) 43(4):911–6. doi: 10.1007/s10803-012-1635-4
70. Hartley SL, Sikora DM. Sex differences in autism spectrum disorder: an examination of developmental functioning, autistic symptoms, and coexisting behaviour problems in toddlers. J Autism Dev Disord (2009) 39(12):1715–22. doi: 10.1007/s10803-009-0810-8
71. Virkud YV, Todd RD, Abbacchi AM, Zhang Y, Constantino JN. Familial aggregation of quantitative autistic traits in multiplex versus simplex autism. Am J Med Genet B Neuropsychiatr Genet (2009) 150B(3):328–34. doi: 10.1002/ajmg.b.30810
72. Constantino JN, Zhang Y, Frazier T, Abbacchi AM, Law P. Sibling recurrence and the genetic epidemiology of autism. Am J Psychiatry (2010) 167(11):1349–56. doi: 10.1176/appi.ajp.2010.09101470
73. Constantino JN. Data from the Baby Siblings Research Consortium confirm and specify the nature of the female protective effect in autism: a commentary on Messinger et al. Mol Autism (2016) 7:32. doi: 10.1186/s13229-016-0092-x
74. Szatmari P, Liu XQ, Goldberg J, Zwaigenbaum L, Paterson AD, Woodbury-Smith M, et al. Sex differences in repetitive stereotyped behaviors in autism: implications for genetic liability. Am J Med Genet B Neuropsychiatr Genet (2012) 159B(1):5–12. doi: 10.1002/ajmg.b.31238
75. Robinson EB, Lichtenstein P, Anckarsater H, Happe F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci U S A (2013) 110(13):5258–62. doi: 10.1073/pnas.1211070110
76. Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet (2014) 46(8):881–5. doi: 10.1038/ng.3039
77. Binder MS, Lugo JN. NS-Pten knockout mice show sex- and age-specific differences in ultrasonic vocalizations. Brain Behav (2017) 7(11):e00857. doi: 10.1002/brb3.857
78. Berkel S, Eltokhi A, Fröhlich H, Porras-Gonzalez D, Rafiullah R, Sprengel R, et al. Sex hormones regulate SHANK expression. Front Mol Neurosci (2018) 11:337. doi: 10.3389/fnmol.2018.00337
79. Weinhard L, Neniskyte U, Vadisiute A, di Bartolomei G, Aygün N, Riviere L, et al. Sexual dimorphism of microglia and synapses during mouse postnatal development. Dev Neurobiol (2018) 78(6):618–26. doi: 10.1002/dneu.22568
80. Raznahan A, Parikshak NN, Chandran V, Blumenthal JD, Clasen LS, Alexander-Bloch AF, et al. Sex-chromosome dosage effects on gene expression in humans. Proc Natl Acad Sci U S A (2018) 115(28):7398–403. doi: 10.1073/pnas.1802889115
81. Hedges VL, Ebner TJ, Meisel RL, Mermelstein PG. The cerebellum as a target for estrogen action. Front Neuroendocrinol (2012) 33(4):403–11. doi: 10.1016/j.yfrne.2012.08.005
82. Hoffman JF, Wright CL, McCarthy MM. A critical period in Purkinje cell development is mediated by local estradiol synthesis, disrupted by inflammation, and has enduring consequences only for males. J Neurosci (2016) 36(39):10039–49. doi: 10.1523/JNEUROSCI.1262-16.2016
83. Greenberg DM, Warrier V, Allison C, Baron-Cohen S. Testing the empathizing-systemizing theory of sex differences and the extreme male brain theory of autism in half a million people. Proc Natl Acad Sci U S A (2018) 115(48):12152–7. doi: 10.1073/pnas.1811032115
84. Carter CO, Evans KA. Inheritance of congenital pyloric stenosis. J Med Genet (1969) 6:233–54. doi: 10.1136/jmg.6.3.233
85. Goin-Kochel RP, Abbacchi A, Constantino JN, Autism Genetic Resource Exchange Consortium. Lack of evidence for increased genetic loading for autism among families of affected females: a replication from family history data in two large samples. Autism (2007) 11(3):279–86. doi: 10.1177/1362361307076857
86. Messinger D, Young GS, Ozonoff S, Dobkins K, Carter A, Zwaigenbaum L, et al. Beyond autism: a baby siblings research consortium study of high-risk children at three years of age. J Am Acad Child Adolesc Psychiatry (2013) 52(3):300–8.e1. doi: 10.1016/j.jaac.2012.12.011
87. Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC. Genomic patterns of de novo mutation in simplex autism. Cell (2017) 171(3):710–22.e12. doi: 10.1016/j.cell.2017.08.047
88. Celestino-Soper PB, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci U S A (2012) 109(21):7974–81. doi: 10.1073/pnas.1120210109
89. Zhang Z, Cao M, Chang CW, Wang C, Shi X, Zhan X, et al. Autism-associated chromatin regulator Brg1/SmarcA4 is required for synapse development and myocyte enhancer factor 2-mediated synapse remodeling. Mol Cell Biol (2015) 36(1):70–83. doi: 10.1128/MCB.00534-15
90. Lee N, Park SJ, Haddad G, Kim DK, Park SM, Park SK, et al. Interactomic analysis of REST/NRSF and implications of its functional links with the transcription suppressor TRIM28 during neuronal differentiation. Sci Rep (2016) 6:39049. doi: 10.1038/srep39049
91. Watanabe M, Hatakeyama S. TRIM proteins and diseases. J Biochem (2017) 161(2):135–44. doi: 10.1093/jb/mvw087
92. Ghasemi Firouzabadi S, Vameghi R, Kariminejad R, Darvish H, Banihashemi S, Firouzkouhi Moghaddam M, et al. Analysis of copy number variations in patients with autism using cytogenetic and MLPA techniques: report of 16p13.1p13.3 and 10q26.3 duplications. Int J Mol Cell Med (2016) 5(4):236–45.
93. Ghirlando R, Felsenfeld G. CTCF: making the right connections. Genes Dev (2016) 30(8):881–91. doi: 10.1101/gad.277863.116
94. Waye JS, Willard HF. Human beta satellite DNA: genomic organization and sequence definition of a class of highly repetitive tandem DNA. Proc Natl Acad Sci U S A (1989) 86(16):6250–4. doi: 10.1073/pnas.86.16.6250
95. Cardone MF, Ballarati L, Ventura M, Rocchi M, Marozzi A, Ginelli E, et al. Evolution of beta satellite DNA sequences: evidence for duplication-mediated repeat amplification and spreading. Mol Biol Evol (2004) 21(9):1792–9. doi: 10.1093/molbev/msh190
96. Leija-Salazar M, Piette C, Proukakis C. Somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol (2018) 44(3):267–85. doi: 10.1111/nan.12465
97. Ziats MN, Comeaux MS, Yang Y, Scaglia F, Elsea SH, Sun Q, et al. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation. Am J Med Genet (2015) 167A:2162–7. Note: Erratum: Am. J. Med. Genet. 167A: 2496 only. doi: 10.1002/ajmg.a.37144
98. Krsička D, Geryk J, Vlčková M, Havlovicová M, Macek M Jr, Pourová R. Identification of likely associations between cerebral folate deficiency and complex genetic- and metabolic pathogenesis of autism spectrum disorders by utilization of a pilot interaction modeling approach. Autism Res (2017) 10(8):1424–35. doi: 10.1002/aur.1780
99. Celestino-Soper PB, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet (2011) 20(22):4360–70. doi: 10.1093/hmg/ddr363
100. Nava C, Lamari F, Héron D, Mignot C, Rastetter A, Keren B, et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry (2012) 2:e179. doi: 10.1038/tp.2012.102
101. Bauman M, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology (1985) 35:866–74. doi: 10.1212/WNL.35.6.866
102. Courchesne E, Saitoh O, Yeung-Courchesne R, Press GA, Lincoln AJ, Haas RH, et al. Abnormality of cerebellar vermian lobules VI and VII in patients with infantile autism: identification of hypoplastic and hyperplastic subgroups with MR imaging. AJR Am J Roentgenol (1994) 162:123–30. doi: 10.2214/ajr.162.1.8273650
103. Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology (1995) 45:1581–6. doi: 10.1212/WNL.45.8.1581
104. Allen G, Courchesne E. Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: an fMRI study of autism. Am J Psychiatry (2003) 160:262–73. doi: 10.1176/appi.ajp.160.2.262
105. Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum (2012) 11:777–807. doi: 10.1007/s12311-012-0355-9
106. Riva D, Annunziata S, Contarino V, Erbetta A, Aquino D, Bulgheroni S. Gray matter reduction in the vermis and CRUS-II is associated with social and interaction deficits in low-functioning children with autistic spectrum disorders: a VBM-DARTEL Study. Cerebellum (2013) 12(5):676–85. doi: 10.1007/s12311-013-0469-8
107. Wang SSH, Kloth AD, Badura A. The cerebellum, sensitive periods, and autism. Neuron (2014) 83(3):518–32. doi: 10.1016/j.neuron.2014.07.016
108. Sundberg M, Sahin M. Cerebellar development and autism spectrum disorder in tuberous sclerosis complex. J Child Neurol (2015) 30(14):1954–62. doi: 10.1177/0883073815600870
109. Kim KC, Gonzales EL, Lázaro MT, Choi CS, Bahn GH, Yoo HJ, et al. Clinical and neurobiological relevance of current animal models of autism spectrum disorders. Biomol Ther (Seoul) (2016) 24(3):207–43. doi: 10.4062/biomolther.2016.061
110. Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol Psychiatry (2018) 23(11):2167–83. doi: 10.1038/s41380-018-0018-4
111. Carter AR, Berry EM, Segal RA. Regional expression of p75NTR contributes to neurotrophin regulation of cerebellar patterning. Mol Cell Neurosci (2003) 22(1):1–13. doi: 10.1016/S1044-7431(02)00015-5
112. Rahimi Balaei M, Jiao X, Ashtari N, Afsharinezhad P, Ghavami S, Marzban H. Cerebellar expression of the neurotrophin receptor p75 in naked-ataxia mutant mouse. Int J Mol Sci (2016) 17(1):E115. doi: 10.3390/ijms17010115
113. Marzban H, Chung S, Watanabe M, Hawkes R. Phospholipase Cbeta4 expression reveals the continuity of cerebellar topography through development. J Comp Neurol (2007) 502(5):857–71. doi: 10.1002/cne.21352
114. Brochu G, Maler L, Hawkes R. Zebrin II: a polypeptide antigen expressed selectively by Purkinje cells reveals compartments in rat and fish cerebellum. J Comp Neurol (1990) 291(4):538–52. doi: 10.1002/cne.902910405
115. Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of Purkinje cells in the adult mouse cerebellar cortex. J Comp Neurol (2000) 416(3):383–97. doi: 10.1002/(SICI)1096-9861(20000117)416:3<383::AID-CNE9>3.0.CO;2-M
116. Sillitoe RV, Chung SH, Fritschy JM, Hoy M, Hawkes R. Golgi cell dendrites are restricted by Purkinje cell stripe boundaries in the adult mouse cerebellar cortex. J Neurosci (2008a) 28(11):2820–6. doi: 10.1523/JNEUROSCI.4145-07.2008
117. Sillitoe RV, Gopal N, Joyner AL. Embryonic origins of ZebrinII parasagittal stripes and establishment of topographic Purkinje cell projections. Neuroscience (2008b) 162(3):574–88. doi: 10.1016/j.neuroscience.2008.12.025
118. Sillitoe RV, Stephen D, Lao Z, Joyner AL. Engrailed homeobox genes determine the organization of Purkinje cell sagittal stripe gene expression in the adult cerebellum. J Neurosci (2008c) 28(47):12150–62. doi: 10.1523/JNEUROSCI.2059-08.2008
119. Sgaier SK, Lao Z, Villanueva MP, Berenshteyn F, Stephen D, Turnbull RK, et al. Genetic subdivision of the tectum and cerebellum into functionally related regions based on differential sensitivity to Engrailed proteins. Development (2007) 134:2325–35. doi: 10.1242/dev.000620
120. White JJ, Sillitoe RV. Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol (20122013) 2(1):149–64. doi: 10.1002/wdev.65
121. Sugihara I. Crus I in the rodent cerebellum: its homology to Crus I and II in the primate cerebellum and its anatomical uniqueness among neighboring lobules. Cerebellum (2018) 17(1):49–55. doi: 10.1007/s12311-017-0911-4
122. Dikic I, Giordano S. Negative receptor signalling. Curr Opin Cell Biol (2003) 15(2):128–35. doi: 10.1016/S0955-0674(03)00004-8
123. Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol (2009) 203:191–202. doi: 10.1677/JOE-09-0110
124. Yu T, Yaguchi Y, Echevarria D, Martinez S, Basson MA. Sprouty genes prevent excessive FGF signalling in multiple cell types throughout development of the cerebellum. Development (2011) 138(14):2957–68. doi: 10.1242/dev.063784
125. Du W, Du W, Yu H. The role of fibroblast growth factors in tooth development and incisor renewal. Stem Cells Int (2018) 2018:14. doi: 10.1155/2018/7549160
126. Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron (1997) 19(2):269–81. doi: 10.1016/S0896-6273(00)80938-1
127. Bosman LW, Hartmann J, Barski JJ, Lepier A, Noll-Hussong M, Reichardt LF, et al. Requirement of TrkB for synapse elimination in developing cerebellar Purkinje cells. Brain Cell Biol (2006) 35:87–101. doi: 10.1007/s11068-006-9002-z
128. Lotta LT, Conrad K, Cory-Slechta D, Schor NF. Cerebellar Purkinje cell p75 neurotrophin receptor and autistic behavior. Transl Psychiatry (2014) 4:e416. doi: 10.1038/tp.2014.55
129. Choo M, Miyazaki T, Yamazaki M, Kawamura M, Nakazawa T, Zhang J, et al. Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat Commun (2017) 8(1):195. doi: 10.1038/s41467-017-00260-w
130. Cheng I, Jin L, Rose LC, Deppmann CD. Temporally restricted death and the role of p75NTR as a survival receptor in the developing sensory nervous system. Dev Neurobiol (2018) 78(7):701–17. doi: 10.1002/dneu.22591
131. Segura M, Pedreño C, Obiols J, Taurines R, Pàmias M, Grünblatt E, et al. Neurotrophin blood-based gene expression and social cognition analysis in patients with autism spectrum disorder. Neurogenetics (2015) 16(2):123–31. doi: 10.1007/s10048-014-0434-9
132. Ohja K, Gozal E, Fahnestock M, Cai L, Cai J, Freedman JH, et al. Neuroimmunologic and neurotrophic interactions in autism spectrum disorders: relationship to neuroinflammation. Neuromolecular Med (2018) 20(2):161–73. doi: 10.1007/s12017-018-8488-8
133. Florez-McClure ML, Linseman DA, Chu CT, Barker PA, Bouchard RJ, Le S, et al. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J Neurosci (2004) 24(19):4498–509. doi: 10.1523/JNEUROSCI.5744-03.2004
134. Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci (2006) 26(29):7756–66. doi: 10.1523/JNEUROSCI.1560-06.2006
135. Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci (2010) 30(46):15608–15. doi: 10.1523/JNEUROSCI.2581-10.2010
136. Vicario A, Kisiswa L, Tann JY, Kelly CE, Ibáñez CF. Neuron-type-specific signaling by the p75NTR death receptor is regulated by differential proteolytic cleavage. J Cell Sci (2015) 128(8):1507–17. doi: 10.1242/jcs.161745
137. Pathak A, Carter BD. Retrograde apoptotic signaling by the p75 neurotrophin receptor. Neuronal Signal (2017) 1:NS20160007. doi: 10.1042/NS20160007
138. Kisiswa L, Fernández-Suárez D, Sergaki MC, Ibáñez CF. RIP2 gates TRAF6 interaction with death receptor p75NTR to regulate cerebellar granule neuron survival. Cell Rep (2018) 24(4):1013–24. doi: 10.1016/j.celrep.2018.06.098
139. Chen Y, Zeng J, Cen L, Chen Y, Wang X, Yao G, et al. Multiple roles of the p75 neurotrophin receptor in the nervous system. J Int Med Res (2009) 37(2):281–8. Review. Erratum in: J Int Med Res. 2009 May-Jun;37(3):974. Cen, L [added]. doi: 10.1177/147323000903700201
140. Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell (1998) 92:253–63. doi: 10.1016/S0092-8674(00)80919-8
Keywords: autism, cerebellum, SPRY3, p75NTR, pseudoautosomal region, TMLHE, carnitine
Citation: Ning Z, Williams JM, Kumari R, Baranov PV and Moore T (2019) Opposite Expression Patterns of Spry3 and p75NTR in Cerebellar Vermis Suggest a Male-Specific Mechanism of Autism Pathogenesis. Front. Psychiatry 10:416. doi: 10.3389/fpsyt.2019.00416
Received: 20 December 2018; Accepted: 24 May 2019;
Published: 18 June 2019.
Edited by:
Yu-Qiang Ding, Tongji University, ChinaReviewed by:
Gabriëlla A M Blokland, Maastricht University, NetherlandsCurtis Kimball Deutsch, Eunice Kennedy Shriver Center, United States
Weihua Yue, Peking University Sixth Hospital, China
Copyright © 2019 Ning, Williams, Kumari, Baranov and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tom Moore, dC5tb29yZUB1Y2MuaWU=
†Present address: Romika Kumari, Institute for Molecular Medicine Finland, FIMM, University of Helsinki, Helsinki, Finland