 Raymond A. Clarke
Raymond A. Clarke Teri M. Furlong
Teri M. Furlong Valsamma Eapen
Valsamma Eapen- 1School of Psychiatry, University of New South Wales, Sydney, NSW, Australia
- 2Ingham Institute for Applied Medical Research, Liverpool, NSW, Australia
- 3School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
- 4South West Sydney Local Health District, Liverpool Hospital, Liverpool, NSW, Australia
Gilles de la Tourette syndrome (GTS) is a neurodevelopmental disorder characterized by motor and vocal tics with an estimated prevalence of 1% in children and adolescents. GTS has high rates of inheritance with many rare mutations identified. Apart from the role of the neurexin trans-synaptic connexus (NTSC) little has been confirmed regarding the molecular basis of GTS. The NTSC pathway regulates neuronal circuitry development, synaptic connectivity and neurotransmission. In this study we integrate GTS mutations into mitochondrial pathways that also regulate neuronal circuitry development, synaptic connectivity and neurotransmission. Many deleterious mutations in GTS occur in genes with complementary and consecutive roles in mitochondrial dynamics, structure and function (MDSF) pathways. These genes include those involved in mitochondrial transport (NDE1, DISC1, OPA1), mitochondrial fusion (OPA1), fission (ADCY2, DGKB, AMPK/PKA, RCAN1, PKC), mitochondrial metabolic and bio-energetic optimization (IMMP2L, MPV17, MRPL3, MRPL44). This study is the first to develop and describe an integrated mitochondrial pathway in the pathogenesis of GTS. The evidence from this study and our earlier modeling of GTS molecular pathways provides compounding support for a GTS deficit in mitochondrial supply affecting neurotransmission.
Introduction
Gilles de la Tourette Syndrome (GTS) is a neurodevelopmental disorder with an estimated prevalence of 1% in children and adolescents (1). Neuroanatomical evidence suggests that GTS pathology is related to abnormal brain development and the physiological involvement of the cortico–striato–thalamo– cortical (CSTC) circuitry connecting the cortex, basal ganglia and thalamus (1). Clinical evidence further suggests the involvement of neurotransmitters such as dopamine, glutamate and γ-aminobutyric acid (GABA) (1). Epidemiological, phenomenological and genetic evidence demonstrate broad overlap between GTS and autism spectrum disorder (ASD) (2, 3) with both exhibiting high incidence in first-degree relatives, high monozygotic to dizygotic concordance (4), and with both conditions beginning during childhood with a high male preponderance. Furthermore, GTS and ASD share associated clinical features of compulsive behaviors, obsessions, involuntary movements (tics in GTS and stereotypies in ASD), poor speech control and echolalia common in both conditions (5). Attention deficit hyperactivity disorder (ADHD) is also present in both ASD and GTS (5, 6). GTS is over represented in ASD, with 5% having GTS and up to 40% experiencing tics (5). Similarly, the rate of autism in GTS exceeds that expected by chance, with reports of ASD in around 22.8% of children and 8.7% in adults (7), subclinical autistic symptoms occurring in a third of GTS populations, and a further two-thirds showing social deficits relating to the autism spectrum (8). Pharmaco-therapeutic agents such as the α2-adrenergic agonists Clonidine and Guanfacine and the antipsychotics such as Risperidone and Aripiprazole are usually the first-line of therapy for moderate to severe GTS. However, side effects are particularly problematic during childhood years when the symptoms are most predominant and often affect compliance and hence there is a critical need for targeted therapeutic development based on a better understanding of the genetic etiology of the disorder.
GTS is one of the most heritable neuropsychiatric disorders of non-Mendelian inheritance, however, with the exception of the neurexin trans-synaptic connexus (NTSC) little is known regarding the molecular basis of GTS (9). One of the strongest mutation associations to date has been with neurexin 1 and the genes encoding the NTSC which regulate neuronal circuitry development, synaptic connectivity and neurotransmission (2, 3, 10, 11). Members of the NTSC family of synaptic proteins bind across the synapse in different combinations to facilitate trans-synaptic cell-adhesion that helps establish and maintain neural circuits and neurotransmission within the brain. All major gene families of the NTSC (Figure 1) have been repeatedly mutated or otherwise associated with GTS and ASD (2, 11). Moreover, the number of mutations identified in and associated with the NTSC has continued to grow to such an extent that the NTSC now represents a collective mutation hot spot for GTS and ASD (2, 11, 13–22). Moreover, the NTSC model for GTS (Figure 1) provides a reliable starting point for further mutation pathway analysis into the mitochondrial regulation of neuronal circuitry development, synaptic connectivity and neurotransmission as it relates to mutations in GTS.
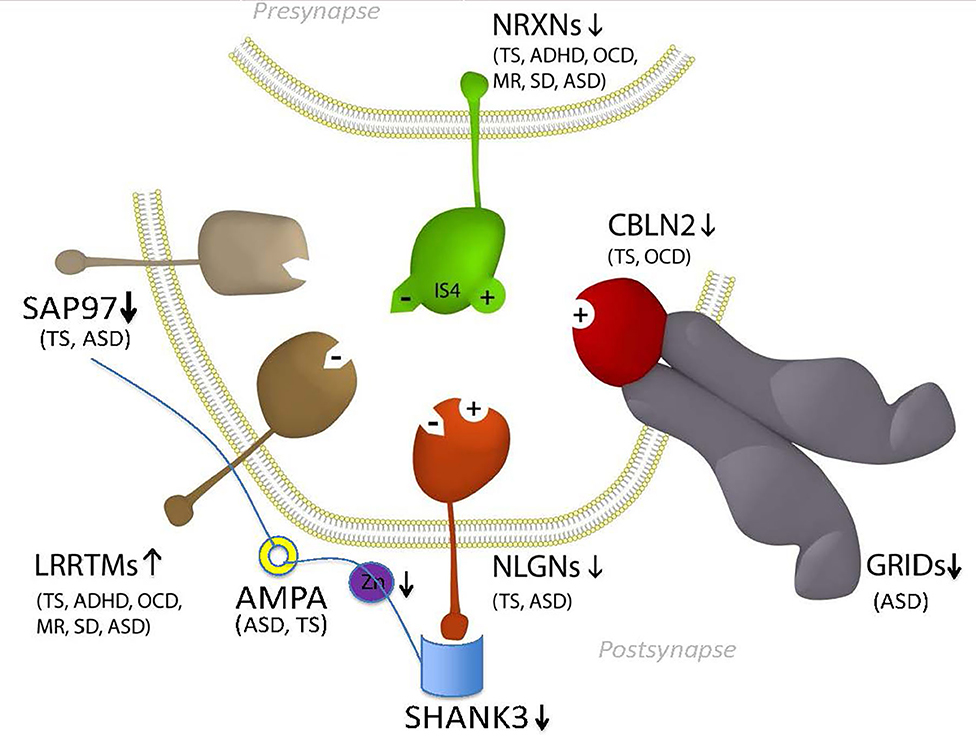
Figure 1. Model NTSC pathway to GTS (2)—implicates the full complement of known neurexins (NRXNs) and their trans-synaptic cell-adhesion ligand gene families through multiple means of enquiry: neuroligins (NLGNs); leucine-rich repeat transmembrane proteins (LRRTMs); cerebellin precursors (CBLNs), Glutamate ionotropic receptor delta type subunit (GRIDS) and the synapse associated proteins (SAPs/DLGs) (2, 11, 12) in addition to the ProSAP proteins that interact in the NTSC postsynaptic signaling pathway (2, 11, 13–24). Presynaptic NRXNs 1-3 form competitive trans-synaptic complexes with postsynaptic ligands NLGNs, LRRTMs and CBLN-GRID complexes in the formation and/or maintenance of neuronal circuitry within the brain. Vertical arrows indicate putative pathogenic dose effects. NRXN1 isoforms with (+) and without (–) the 30 amino-acid insert at splice site 4 (IS4) indicate competitive binding of NRXN1 between its ligands. NRXN4/CNTNAP2 binds across the synapse with SAP97/DLG1 (12, 25). Comorbidities listed are those associated with the gene mutations and copy number variations (CNVs) in ASD and GTS (2, 11, 12, 26–32). Zinc represents one environmental factor important for mitochondrial protein import/processing and function that is also known to affect SHANK3 function (23, 24). Moreover the Zinc transporter ZnT7 has been recurrently linked with GTS in GWAS (2).
Neuronal Mitochondria: Up to 20% of the total energy consumed by humans at rest is attributable to brain activity despite a brain-to-body mass ratio of only 2% (33, 34). This high energy consumption by the human brain is largely attributed to requirements for synaptic transmission (33, 34). Neurons require particularly large amounts of energy for synaptic vesicle release and to power the ion pumps that restore ion gradients in the synapse following the ion influx associated with neuronal firing (34). These high energy demands are largely met by neuronal mitochondria, which also power other important neurodevelopmental processes including neurite outgrowth (35–40) which ultimately provides for optimal synaptic connections and neurotransmission. Mitochondria have additional roles in the neuron including calcium buffering, which is of particular importance in mitochondrial dynamics and neurotransmission (40). Although mitochondria are essential in almost every cell type, the extended branching structure and specialized function of neurons comes with unique demands over extended distances that render neurons especially sensitive to deficits in mitochondrial dynamics, structure and function. The sensitivity of neuronal development and function to mitochondrial deficiencies is corroborated by the strong association between the mutation of mitochondrial component molecules and neurological disorders (41), and there is increasing evidence that mitochondrial dynamics and dysfunction contribute to neuropsychiatric disorders including Schizophrenia (SCZ) and Bipolar Disorder (42–44).
The higher brain functions affected in neurodevelopmental and psychiatric disorders are thought to require precise spatiotemporal regulation of neuronal circuitry development. In this developmental process the relationship between neuronal outgrowth, synaptogenesis and synaptic transmission is widely appreciated. However, the requirements that these neuro developmental processes have on mitochondria is still emerging. In our pathway analysis, we outline the importance of mitochondrial dynamics, structure and function to neuronal outgrowth and development, synaptogenesis and neurotransmission as the basis for understanding the genetic etiology of GTS.
Methods
Data Mining and Mutation Pathway Analysis: In this study we integrated the findings from published and unpublished (database) sources including ASD brain gene expression profiling, GTS risk-gene mouse modeling of behavior, genome wide linkage studies, genome wide association studies, chromosomal translocations and copy number variations (CNVs), gene set analysis, haplotype sharing, cell modeling, whole exome sequencing (WES), and whole genome sequencing (WGS) of rare and common deleterious mutations in GTS and ASD (see sources in Table 1). In the case where a CNV spanned multiple genes only one gene was selected for inclusion in our pathway analysis (Table 2). In these cases, first priority was given to genes that directly regulate those pathways already implicated in GTS (9) including neuronal development (e.g., SLIT2), synaptic connectivity (e.g., NTSC pathway genes), synaptic function (e.g., SAP97) and neurotransmission (Figure 1) (9). Only then was the second priority exercised to assign a pathway to those genes that encode mitochondrial proteins. Following this selection process all mutations, deletions and duplications of mitochondrial protein genes in GTS were included within the mutation pathway analysis thereby eliminating any bias or cherry picking in our development of the first contiguous mitochondrial pathways to GTS (Figures 2–4).

Table 1. Data sources.

Table 2. GTS risk genes in mitochondrial dynamics, structure, and function.
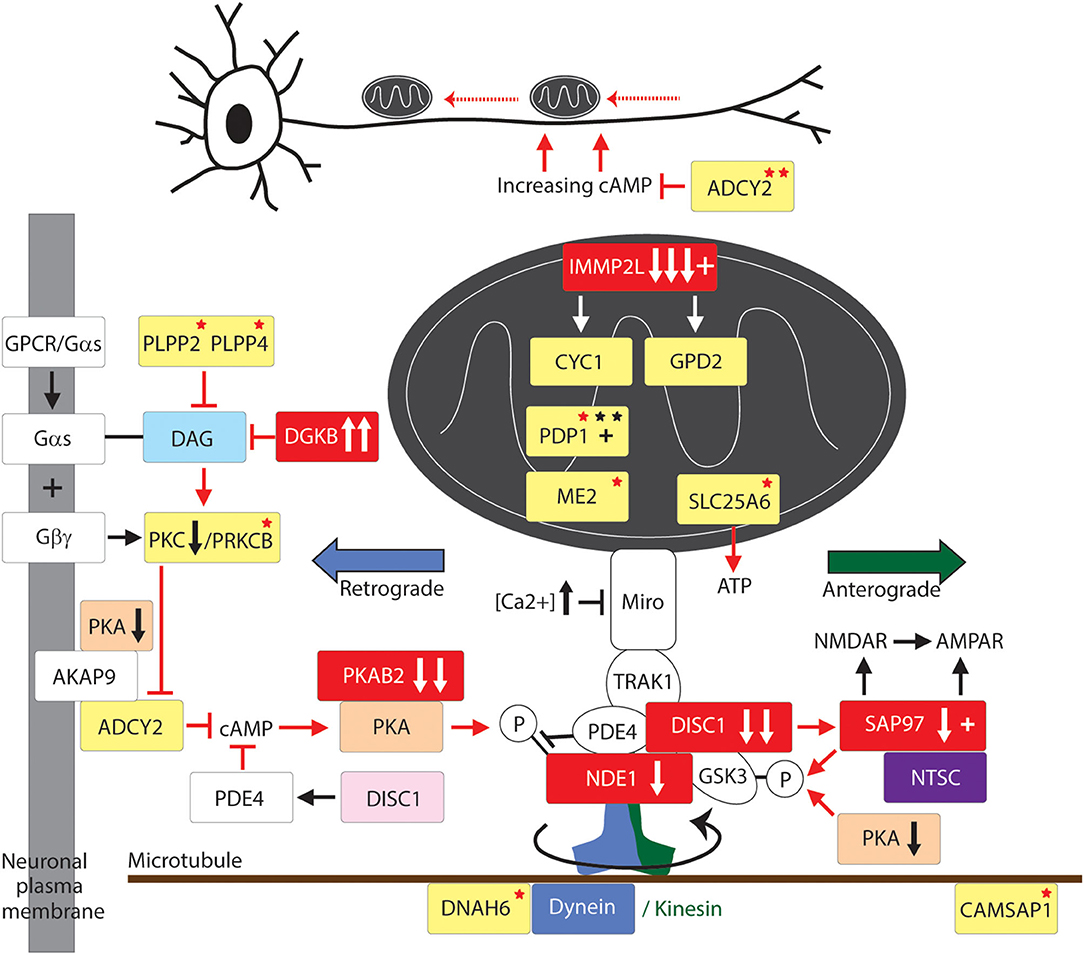
Figure 2. Mitochondrial transport pathway to GTS. Red fill indicates CNV deletion (arrows down) or duplication (arrows up); Yellow fill indicates GTS associated gene; Red asterisk* indicates deleterious mutation; Black asterisk indicates another GTS genetic association; + indicates linkage; Red arrow indicates GTS affected pathway. Vertical arrows beside PKA and PKC indicate putative pathogenic dose effects.
Results and Discussion
Mitochondrial Dynamics: Mitochondrial Transport
To optimize the development and function of neuronal circuitry, mitochondria need to be located at the right place at the right time in sufficient numbers and to be functioning at optimum efficiency. Given that neurons are often hyper-extended with a complex network of axonal and dendritic branches, such optimisation requires the active bidirectional transport of mitochondria to their required destinations along microtubule tracks (39, 86). This may involve long-distance transport of mitochondria from the soma, where the majority of mitochondrial biogenesis occurs, to the pre- and post-synaptic termini where demands for mitochondrial homeostasis is highest (39, 86). Mitochondrial transport is also thought to optimize mitochondrial fission, fusion and function (39, 86).
Dynein and Kinesin Motor Proteins
Transport of mitochondria is regulated by the dynein and kinesin motor proteins through their interaction with the microtubule cytoskeleton of the cell (Figure 2). The kinesins (KIFs) mediate anterograde mitochondrial transport away from the soma of the neuron while dynein complexes regulate retrograde transport (87). DNAH6, the dynein axonemal heavy chain 6 gene implicated in mitochondrial depletion syndrome is mutated in GTS as are DNAH5 and a number of kinesin genes including KIF6 and KIF7 (Figure 2) (58). Moreover, DYNC1H1 which encodes the dynein 1 heavy chain mitochondrial transporter is mutated in ASD and KIF1A which encodes the Kinesin 1A mitochondrial transporter is disrupted and duplicated in ASD (18, 20).
KIFs form homo and heteromeric complexes in the regulation of organelle transport including but not limited to mitochondria (88). The importance of the motor proteins, and the microtubule network they travel on, in nervous system development and function is evidenced from their strong association with neurological phenotypes. Mutations in KIF5-family members give rise to a range of dominant negative phenotypes including deficits in mitochondrial transport, structure and function and reduced activity of the electron transport chain (ETC) (89, 90), axonal degeneration and aberrant synaptic transmission (Figure 2) (58, 91). GTS mutations have also been identified in genes that regulate tubulin and microtubule dynamics including: duplication of the TUBB2A and TUBB2B tubulin genes and the mutation of TUBB3 (58); mutation of the TTLL1, TTLL2, and TTLL5 tubulin ligase genes (58); recurrent duplication of the microtubule polymerization gene KANK1 implicated in spastic paraplegia (58, 92); and mutation of the CAMSAP1 gene that regulates microtubule dynamics and neurite outgrowth (Table 2 and Figure 2) (58, 93).
Mitochondrial Transport Adaptor and Accessory Proteins
Mitochondrial transport provides for the site-specific requirements and function of the neuron (39, 86, 94–97) and it has been demonstrated that mitochondria directly regulate synaptic transmission (94). Furthermore, synapses with mitochondria can sustain repeated cycles of neurotransmitter release whereas the transport of mitochondria either in or out of the synapse dynamically modulates this synaptic strength (95–97). To halt the transport of mitochondria at a required destination such as the synapse requires a braking system. In this respect, synaptic firing renders synapses to be sites of high calcium influx. After synaptic firing the high (Ca2+) acts to halt mitochondrial transport through the action of the Ca2+ sensitive GTPase of MIRO which is embedded within the outer mitochondrial membrane which then inactivates the molecular motor kinesin (Figure 2). To summarize, the precise location and relocation of mitochondria closely matches the site-specific requirements of the neuron including a rich supply of energy, in the form of ATP, to power the synaptic calcium ion pumps that expel calcium from the cell and for direct mitochondrial buffering of Ca2+ (98–100). Furthermore, mitochondria directly regulate the strength of synaptic transmission (94).
Motor proteins interact with mitochondria through adaptor and accessory proteins (98, 101) that determine the direction of mitochondrial transport (Figure 2). The mitochondrial transport adaptor proteins TRAK1 and TRAK2 link mitochondria to the motor proteins kinesin and dynein (102). TRAK1 and TRAK2 interact with the mitochondria through the Ca2+ sensitive GTPases MIRO1 and MIRO2 embedded within the outer mitochondrial membrane (Figure 1) (102–106). MIRO and TRAK work in concert with the transport accessory proteins DISC1 and NDE1 that determine the direction of mitochondrial movement (Figure 2). While no mutations in MIRO or TRAK have been identified (107) both DISC1 and NDE1 display recurrent hemizygous deletion in GTS, ASD and SCZ (Table 2) (58–62). DISC1 is localized predominantly to mitochondria and in synapses, centrosomes, nuclei, endoplasmic reticulum and the Golgi (108, 109). DISC1 forms oligomers that interact with kinesin (110) and dynein (111), MIRO and TRAK, and the mitochondrial transport accessory proteins LIS1, GSK3β, NDE1 and its homolog NDEL1 (Figure 2) (112). DISC1 promotes anterograde and retrograde mitochondrial transport in a dose dependent manner in both axons and dendrites (112–114) possibly through blocking SNPH-mediated anchoring of mitochondria (115). DISC1 is also directly linked to anomalies in mitochondrial fission, fusion, structure and function (112–114).
NDE1 interacts with DISC1, TRAK1, NDEL1, LIS1, and with the dynein motor protein to promote retrograde axonal transport (Figure 2) (112). NDE1, and its close homolog NDEL1 form a complex with LIS1 that regulates neuronal proliferation, differentiation, and migration within the brain (116). NDE1 is a centrosomal protein with a crucial role in the growth of the cerebral cortex (116). Homozygous frame shift mutations in NDE1 are associated with extreme microlissencephaly (117, 118), whereas heterozygous deletions (LOH) in NDE1 are associated with GTS, ASD, and SCZ (62). Interestingly, the subcellular localization of NDE1, its protein-protein interactions and its regulation of retrograde mitochondrial transport, are all modulated through its phosphorylation by AMP-activated protein kinase A (AMPK/PKA) (Figure 2) (119). This regulatory association between NDE1 and PKA is note-worthy on many counts. Firstly, the gene encoding beta subunit 2 of PKA (PRKAB2) is recurrently deleted in GTS (Table 2) (58, 63, 120, 121) and PKA activity is greatly decreased in the frontal cortex of subjects with regressive autism (122). Secondly, cAMP mediated PKA phosphorylation of NDE1 at threonine residue 131 regulates NDE1's all-important interactions with NDEL1 and LIS1 that are thought to activate dynein and facilitate its ability to move high-load cargo like mitochondria (123, 124). Thirdly, PKA's activation by rising cAMP levels provides a mitochondrial transport switch that can be activated on depletion of cellular ATP. Finally, DISC1 modulates the phosphorylation of NDE1 by PKA through its regulation of PDE4, a cAMP-hydrolyzing enzyme which creates a co-complex with DISC1 and NDE1 (Figure 2) and LIS1 and NDEL1 (112, 119, 125).
Mitochondria also localize to sites of neuronal branching. During development of neuronal circuitry the axons are guided to their target sites by extracellular guidance molecules like DSCAM, ROBO1, SLIT2 and SLIT3. SLIT3 has been recurrently associated with GTS (2, 45, 46) as has SLIT2 with ASD (2, 68, 69). Moreover, DSCAM and ROBO1 have been recurrently mutated in ASD as have DSCAM's DNA regulatory elements (21, 22).
Neuronal growth cone navigation also relies on intracellular changes to microtubule and F-actin architecture downstream of these guidance cues (126), for example CAMSAP1 which was mentioned above in relation to its regulation of microtubule dynamics and neurite outgrowth (Table 2 and Figure 2) (58, 93). Furthermore, AMPK/PKA regulates F-actin cytoskeletal dynamics (127). After extension to their target sites axons undergo local branching to establish the appropriate functional connections between pre- and postsynaptic neuronal termini. The intracellular mechanism that regulates this axonal branching also involves PKA through its regulation of mitochondrial transport and recruitment to sites of future axon branching (128). Here, neuronal depolarization-induced rebalance of mitochondrial motility between anterograde and retrograde transport underlies the formation of axonal branches (128). Axon branching is formed in an ATP-depletion dependent manner through an increase in activated/phosphorylated PKA—a function which can be recapitulated by the pharmacological activation of PKA (128). Following neuronal depolarization there is an increase in anterograde transport of mitochondria into axons thus providing a mechanism for mitochondrial relocation and recruitment to sites of high energy demand that would appear to include sites of future branching. Moreover, the continued localization of mitochondria at branch points correlates with the longevity of axonal branches indicating a probable role for mitochondrial localization in the maintenance of axon branches (128). To summarize, a role for mitochondria in neuronal function and neurological disease has been established. Moreover, the role of mitochondrial transport in GTS is greatly strengthened by the interacting roles of the dynein motor protein, DISC1, NDE1, and PKA in mitochondrial transport and the complementary nature of their deleterious mutations in GTS and ASD (Table 2) (61–63, 112).
The master regulator glycogen synthase kinase 3β (GSK3β) is phospho-deactivated by another master regulator AMPK/PKA (129–131). This is important given that GSK3β associates with both DISC1 and TRAK1 in the regulation of mitochondrial transport (Figure 2) (112). In the synapse GSK3β is also deactivated by SAP97 downstream of DISC1 (132) (Figure 2). SAP97 is linked to GTS (12) and downregulated in GTS and ASD (11) and forms part of the high-risk NTSC pathway to GTS (Figure 1) (2, 12). As such, SAP97 functions at the intersection of the NTSC, DISC1 and mitochondrial transport pathways to GTS (Figures 1, 2) (2, 11, 12). GSK3β is also translocated into the mitochondria where it regulates mitochondrial homeostasis (112, 129, 133, 134). In the mitochondria GSK3β regulates the structure and function of the inner mitochondrial membrane (134). This is noteworthy given that the LOH and/or downregulation of SAP97, DISC1, and PRKAB2 in GTS (2, 11, 12, 58, 61–63) are all consistent with stronger activation of GSK3β which is in turn consistent with the success of lithium chloride in the treatment of psychosis through its highly selective phospho-deactivation of GSK3 in both the cytosol and mitochondria (12, 134).
Mitochondrial Fusion and Fission
The constant optimisation of mitochondrial function requires mitochondria to undergo fusion and fission. Fusion of suboptimal mitochondria with healthy mitochondria creates larger healthier mitochondria where the damage is diluted (98, 135). Fission of mitochondria can rapidly increase the number of healthy mitochondria to allow for their wider distribution in the extended network of neuronal branches and boutons (98, 135). Fission can also help separate out damaged mitochondrial components for clearance by mitophagy (98, 102, 135). Conversely, if transport of mitochondria is retarded or otherwise defective mitochondria are less likely to merge thereby decreasing the clearance of damaged mitochondria and the overall health of the mitochondrial pool (39, 86).
Mitochondrial Fusion
Fusion of the outer mitochondrial membrane is coordinated by Mitofusins 1 and 2 (Mfn1/2) whereas fusion of the inner mitochondrial membrane is regulated by Optic Atrophy 1 (OPA1) (136, 137). This is most relevant as OPA1 is recurrently mutated in GTS (Table 2 and Figure 3) (58) and the levels of MFN1, MFN2, and OPA1 are decreased in the temporal lobe of the autistic brain, and there are deficits in MFN1 and MFN2 in Fragile X syndrome (Figure 3) (58, 77, 83). Mfn1/2 and OPA1 act through the formation of complexes both within and across the membranes (136, 137). Mfn2 coding mutations appear to inhibit mitochondrial fusion by forming a complex, in a dominant-negative fashion, with wild-type Mfn1 and Mfn2 (138). Mutations in Mfn2 cause Charcot Marie Tooth Disease Type 2A, a severe and early onset motor and sensory peripheral neuropathy with autosomal dominant inheritance (111, 139). These Mfn2 mutations promote mitochondrial fragmentation in dorsal root ganglion neurons and impair axonal mitochondrial transport which is suggestive of a link between mitochondrial fission/fusion equilibrium and mitochondrial transport (140). This link is supported further by the physical interaction between Mfn2 and the MIRO complex (141) and the finding that Purkinje-neuron-specific deletion of Mfn2 in mice (total knockout of Mfn2 is embryonic lethal) impairs mitochondrial fusion and the dendritic localization of mitochondria, dendrite development, degeneration of Purkinje neurons (142, 143). Similar to the situation in mice, Mfn2 loss-of-function in zebrafish reduces mitochondrial transport and depletes mitochondria from distal axons (144). Acting in a similar dominant-negative fashion to Mfn2, hypomorphic mutations in OPA1 cause dominant optic atrophy (DOA), the most common cause of hereditary blindness. Dominant optic atrophy is characterized by the early loss of retinal ganglion cells and degeneration of the optic nerve (145). Moreover, DOA patients often present with neurological disorders, including ataxia, myopathy, deafness and peripheral neuropathy, indicating an essential neurological role for OPA1 (146, 147). Like Mfn2, knockout of Opa1 in mice is embryonic lethal (141) whereas Opa1 LOH in mice recapitulates the DOA seen in patients, including early-onset degeneration of the optic nerve and vision loss (148). In vitro experiments indicate that Opa1 has a critical role in dendritogenesis and synaptogenesis. Knock-down of Opa1 in cultured rat cortical neurons promotes mitochondrial fragmentation, decreases expression of ETC components, mitochondrial DNA content, dendritic outgrowth and synapse formation (149). As such, the characterization of mitochondrial phenotypes in those GTS patients identified with recurrent OPA1 mutations is eagerly anticipated (Figure 3) (58).
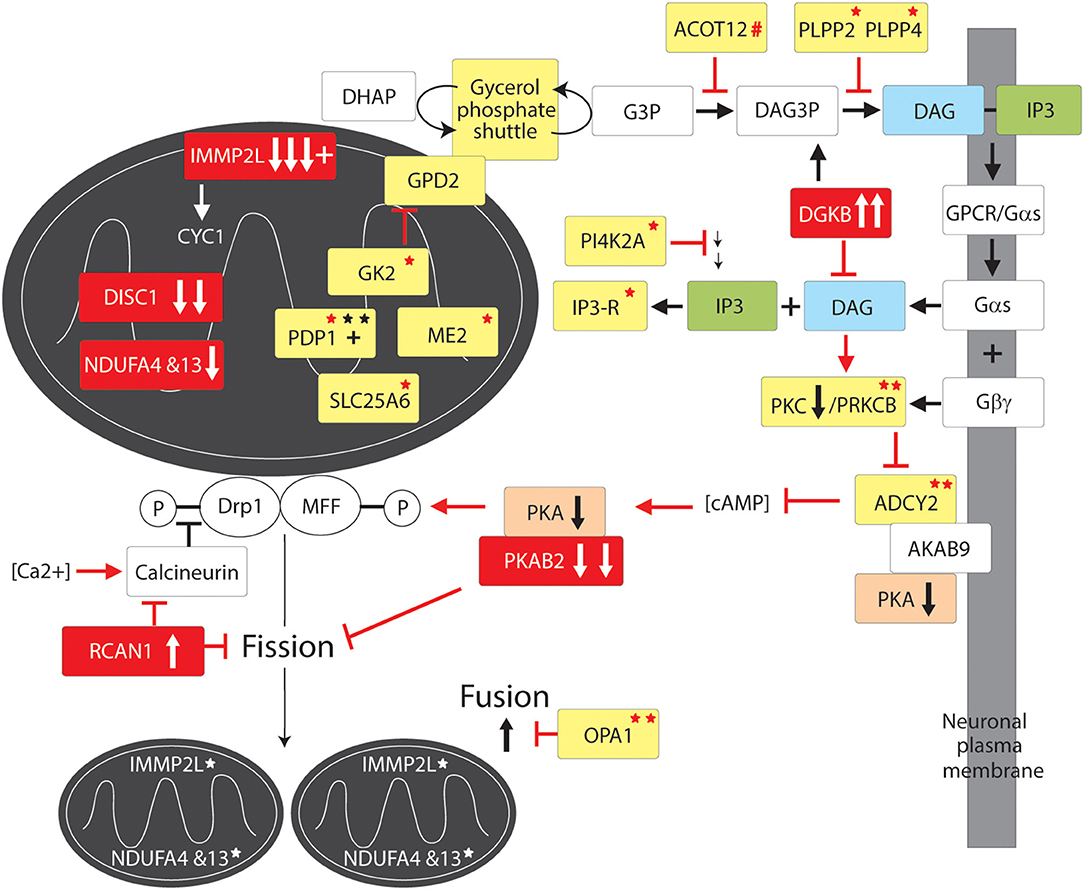
Figure 3. Mitochondrial fission and fusion pathway to GTS. Red fill indicates CNV deletion (arrows down) or duplication (arrows up); All other vertical arrows within boxes indicate putative pathogenic dose effects.Yellow fill indicates GTS associated gene; Red asterisk* indicates deleterious mutation; Black asterisk indicates another GTS genetic association; # indicates adjacent to deletion or duplication; + indicates linkage; Red arrow indicates GTS affected pathway. Vertical arrows indicate putative pathogenic dose effects.
Mitochondrial Fission Regulatory Genes in GTS
The number and the complementary nature of the deleterious GTS mutations identified in mitochondrial fission pathways provide compelling evidence for mitochondrial fission deficiency in the etiology of GTS. The fission of the mitochondrial membranes is dependent on Dynamin-related protein1 (Drp1) (150–160) which is downregulated in the brain of patients with ASD (78–81). Drp1 also regulates peroxisomal fission and proliferation (161). Primarily a cytosolic enzyme, Drp1 translocates to the outer mitochondrial membrane when dephospho-activated by the Ca2+-activated phosphatase calcineurin/PPP3CA (Figure 3) (162, 163). Conversely, calcineurin is inhibited by RCAN1 which blocks Drp1 translocation to the mitochondria (164, 165) and hence it is of immense interest that RCAN1 has been duplicated in GTS and Calcineurin is downregulated in the cerebral cortex of patients with ASD (Figure 3) (47, 63, 80, 81, 166). Calcinueurin activated Drp1 is then recruited to the outer mitochondrial membrane by phospho-activated mitochondrial fission factor (MFF). MFF is phospho-activated in neurones by PKA in response to increasing levels of cAMP (Figure 3) (167, 168). Activated Drp1 then assembles into spirals around the mitochondrion, which constrict and ultimately divide the organelle in two while dynamin-2 catalyzes the final membrane scission event (169). As mentioned earlier, the gene for beta subunit 2 of cAMP activated PKA (PRKAB2) is recurrently deleted in GTS (58, 63). Furthermore, cAMP levels in the brain are largely regulated by ADCY2 (170), the adenylate cyclase gene recurrently mutated in GTS including the loss of an ADCY2 intron/exon splice site in GTS (58). ADCY2 is also mutated in bipolar disorder and a mutation in the ADCY2 binding site of the AKAP9 synaptic scaffolding protein has been found associated with SCZ (54, 170). This is fascinating since AKAP9 acts as a synaptic membrane anchor for ADCY2 and PKA which keeps this primary target of cAMP (PKA) in close proximity to the primary cAMP generator ADCY2 (170–173).
ADCY2 is activated by G-protein signaling through release of the alpha subunit of trimeric G protein (Gαs) (170, 174). Gαs is released when the G protein coupled receptor (GPCR) in the plasma membrane is bound by an extracellular regulatory molecule such as the neurotransmitter dopamine (Figure 3) (174, 175). When released, Gαs activates Phospholipase-C which in turn acts on phospholipid (phosphatidylinositol-triphosphate) within the plasma membrane cleaving off the inositol triphosphate (IP3) second messenger which frees yet another important second messenger diacylglycerol (DAG) (175). DAG activates Protein Kinase C (PKC), which in turn activates ADCY2 and other proteins (Figure 3) (175). Notably, the gene encoding PKC subunit B (PRKCB1) is mutated in GTS (Figure 3) (58) as well as being both linked and strongly associated with ASD, moreover, PKC activity is significantly reduced in the frontal cortex of subjects with regressive autism (55, 56, 82). PRKCB1 is also associated with nominal autistic-like traits in the general population (176). Another compelling finding that links the PKCB/ADCY2 pathway to GTS is the recurrent duplication of the DAG kinase gene in GTS (DGKB) (58). DGKB terminates DAG-based signals by reducing DAG levels by converting DAG to diacylglycerol-3-phosphate (DAG3P) (Figure 3). Moreover, the phosphatases PLPP2 and PLPP4 which convert DAG3P back to DAG are both mutated in GTS (Figure 3) (58). It is also worthy of mention here that PI4K2A, an enzyme in the synthesis pathway of phosphatidylinositol-triphosphate with the potential to limit the bioavailability of both the IP3 and DAG second messengers, is mutated in GTS. This together with a GTS mutation in the IP3 receptor ITPR3 (Figure 3 and Table 2) (58) suggests the potential for an IP3 signaling affect in GTS notwithstanding ambiguity with regards to the calcium sensitivity of ACDY2 (170, 174, 175). To summarize, there is an impressive number of GTS gene mutations with the potential to limit the activation of Drp1, or the MFF-mediated recruitment of Drp1, for mitochondrial fission (Figure 3).
Optimisation of Mitochondrial Supply
Many of the genes mutated in GTS regulate mitochondrial function. Most notable is the IMMP2L gene commonly disrupted/deleted in GTS (64, 72–75, 84). IMMP2L encodes a mitochondrial peptidase (inner mitochondrial membrane peptidase-2-like protein) which processes other mitochondrial proteins within the inner mitochondrial membrane (IMM) (Figure 4 and Table 2) (84). The IMMP2L association with GTS was first reported in a GTS family with a balanced t (7;18) (q22–q31; q22.3) translocation that disrupted the IMMP2L gene (72). More recently a Danish study reported 5′-end intragenic deletions in IMMP2L in seven out of a cohort of 188 GTS patients (3.7%) which was significantly higher than that of the control population (64). The IMMP2L gene has been repeatedly linked to ASD inheritance at the Autism 1 (AUTS1) locus (48–52). In addition, IMMP2L has demonstrated haplotype sharing in multiple ASD populations and deleterious exon deletions have been identified in ASD individuals and families (22, 52, 65, 66, 71, 74, 177) at significantly higher frequency than in control populations. Furthermore, reducing Immp2l dose in mice causes behavioral changes relevant to GTS behavioral deficits (85).
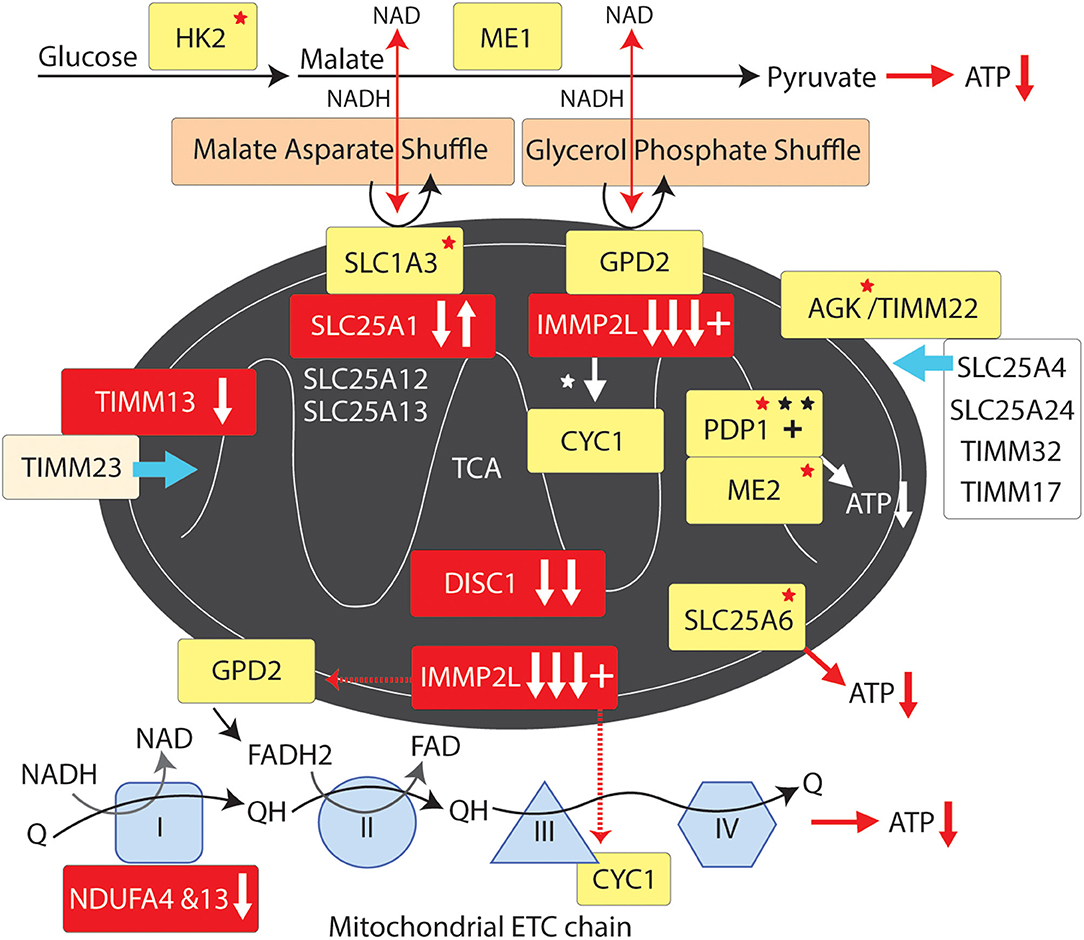
Figure 4. Mitochondrial supply pathway to GTS. Red fill indicates CNV deletion (arrows down) or duplication (arrows up); Yellow fill indicates GTS associated gene; Red asterisk* indicates deleterious mutation; Black asterisk indicates another GTS genetic association; + indicates linkage; Red arrow indicates GTS affected pathway.
IMMP2L cleaves IMM signature signal peptides from a number of IMM proteins including cytochrome C1 (CYC1) and mitochondrial glycerol-3-phosphate dehydrogenase (GPD2) (84). CYC1 (oxidative phosphorylation complex 111 subunit 4) is a heme-containing subunit of the cytochrome complex of the electron transport chain (ETC). CYC1 has an important role in accepting electrons from the Rieske protein and transferring them to Cytochrome C in the respiratory chain. On the other hand GPD2 functions as part of the glycerol phosphate shuttle (Figure 4). GPD2, which is activated by IMMP2L, is located on the outer surface of the inner mitochondrial membrane where it catalyzes the interconversion of glycerol-3-phosphate (G3P) to dihydroxyacetone phosphate. Interestingly, mitochondrial glycerol kinase (GK2) which generates G3P is also mutated in GTS (58). Together, GPD1 and GPD2 constitute the glycerol phosphate shuttle, which generates FADH2 for the mitochondrial ETC and NAD+ for glycolysis in the cytosol. The coordinated action of GPD1 and GPD2 results in the transfer of two reducing equivalents from G3P to the mobile electron carrier ubiquinone (Coenzyme Q10) which in turn passes these electrons to CYC1 located downstream in the ETC (80, 81, 178–180).
A recurrent functional variant in the glutamate aspartate transporter GLAST/SLC1A3 has been identified in GTS (181) (Table 2). SLC1A3 is also of interest as it imports glutamate from the cytosol into the mitochondrial matrix and exports aspartate from the matrix to the cytosol at varying levels in different cell types including astrocytes and neurons (178, 181). In addition to reducing glutamate signaling within the synapse the malate aspartate shuttle, like the glycerol phosphate shuttle, provides NADH to the ETC to generate ATP and NAD+ for another round of glycolysis (Figure 4) (178). In addition, the mitochondrial ADP/ATP exchange transporter SLC25A6 is mutated in GTS as is the mitochondrial ATPase ATP5B (Table 2) (58). SLC25A4 is a nuclear encoded protein located at the 4q35 GTS linkage locus (2, 46) (Table 1). SLC25A4 is transported into the mitochondria by the TIMM22 mitochondrial translocase complex inclusive of AGK, a vital component of TIMM22 involved in its assembly and function, and which is downregulated in the cerebral cortex of autism sufferers (80, 81). Importantly, AGK is mutated in GTS as is the TIMM13 (Table 2 and Figure 4) (58, 182, 183). TIMM13 facilitates translocation of the TIMM23 translocase into the IMM which in turn forms a translocase complex with TIMM17A/B, which itself is translocated into the IMM by TIMM22 (184). Importantly, TIMM17A/B facilitates the translocation of two additional glutamate/aspartate exchange transporters into the IMM, namely SLC25A12 and SLC25A13 (Figure 4) with the former being downregulated in the brain of patients with ASD (78, 79).
Mitochondrial Maintenance
The MPV17 channel protein (53, 58) that regulates the transmembrane potential of the IMM and mitochondrial DNA maintenance has been mutated in GTS (Table 2), moreover, the MPV17 gene is located at the 2p23.2 non-parametric linkage locus identified in GTS (53) (Table 2). MGME1 which also regulates mitochondrial DNA maintenance is located immediately adjacent to a genomic DNA duplication at 20p11 in GTS (Table 2) (58). RMRP, a gene which regulates mitochondrial RNA processing, is deleted in GTS as is the PTCD3 gene which regulates translation in the mitochondria (Table 2). In addition, a number of mitochondrial ribosomal protein genes are mutated in GTS: MRPL44 was found mutated in 3 unrelated GTS patients (Table 2) (58), a mutation in MRPL3 was found segregating with GTS in a large affected family (67) and MRPL48 was found mutated in another GTS patient (Table 2) (58). Together these findings are reminiscent of the MRPL19 gene association with ASD, Dyslexia and Reading Disorder (57).
Conclusion
The mitochondrial pathways involved in GTS overlap and interact making it possible to trace these pathways to common endpoints in mitochondrial dynamics and supply. While it is unlikely that all of the genes cited in this study are causative in GTS, or that they all act alone in GTS etiology, we present convincing weight of evidence that mitochondria are implicated in GTS. Notwithstanding, the deleterious mutation of genes directly involved in mitochondrial dynamics and supply (Figure 4) have the potential to limit neurodevelopment and neurotransmission during periods of peak demand. The exact mitochondrial mechanism implicated in GTS has not been identified as there is no evidence of mitochondrial mediated increases in ROS or ROS related neurodegeneration in GTS as is commonly the case in neurodegenerative disorders. A deficit in neuronal energy supply during development is one possible contributing factor in the etiology of GTS, however, the waning of tic severity in GTS over time appears more consistent with a deficit in neurotransmission possibly compensated for at later ages (185–189). We have no non-molecular evidence of a mitochondrial pathway to GTS at this time, notwithstanding, this is the 1st study to report a mitochondrial pathway to GTS and we are confident it will not be the last. The mitochondrial pathways identified in this study (Figures 2–4) have roles in neuronal circuitry development, synaptic connectivity and neurotransmission (Figure 1) (2, 11). The NTSC and DISC1 and mitochondrial pathways to GTS all intersect around the pivotal role of SAP97 in regulating synaptic signaling downstream of the NTSC and mitochondrial transport downstream of DISC1 thus providing compounding support for a GTS deficit in mitochondrial supply affecting neurotransmission.
Data Availability Statement
The original contributions generated in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Robertson MM, Eapen V, Singer HS, Martino D, Scharf JM, Paschou P, et al. Gilles de la Tourette syndrome. Nat Rev Dis Primers. (2017) 3:16097. doi: 10.1038/nrdp.2016.97
2. Clarke RA, Lee S, Eapen V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including Autism. Transl Psychiatry. (2012) 2:e158. doi: 10.1038/tp.2012.75
3. State MW. The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron. (2010) 68:254–69. doi: 10.1016/j.neuron.2010.10.004
4. O'Rourke JA, Scharf JM, Yu D, Pauls DL. The genetics of Tourette syndrome: a review. J Psychosom Res. (2009) 67:533–45. doi: 10.1016/j.jpsychores.2009.06.006
5. Burd L, Li Q, Kerbeshian J, Klug MG, Freeman RD. Tourette syndrome and comorbid pervasive developmental disorders. J Child Neurol. (2009) 24:170–5. doi: 10.1177/0883073808322666
6. Green JL, Rinehart N, Anderson V, Nicholson JM, Jongeling B, Sciberras E. Autism spectrum disorder symptoms in children with ADHD: a community-based study. Res Dev Disabil. (2015) 47:175–84. doi: 10.1016/j.ridd.2015.09.016
7. Darrow SM, Grados M, Sandor P, Hirschtritt ME, Illmann C, Osiecki L, et al. Autism spectrum symptoms in a Tourette's disorder sample. J Am Acad Child Adolesc Psychiatry. (2017) 56:610–7.e1. doi: 10.1016/j.jaac.2017.05.002
8. Kadesjo B, Gillberg C. Tourette's disorder: epidemiology and comorbidity in primary school children. J Am Acad Child Adolesc Psychiatry. (2000) 39:548–55. doi: 10.1097/00004583-200005000-00007
9. Eapen V, Robertson MM. Are there distinct subtypes in Tourette syndrome? Pure-Tourette syndrome versus Tourette syndrome-plus, and simple versus complex tics. Neuropsychiatr Dis Treat. (2015) 11:1431–6. doi: 10.2147/NDT.S72284
10. Clarke RA, Eapen V. Balance within the neurexin trans-synaptic connexus stabilizes behavioral control. Front Hum Neurosci. (2014) 8:52. doi: 10.3389/fnhum.2014.00052
11. Fang Z, Eapen V, Clarke RA. CTNNA3 discordant regulation of nested LRRTM3, implications for autism spectrum disorder and Tourette syndrome. Meta Gene. (2017) 11:43–8. doi: 10.1016/j.mgene.2016.11.003
12. Verkerk AJ, Cath DC, van der Linde HC, Both J, Heutink P, Breedveld G, et al. Genetic and clinical analysis of a large Dutch Gilles de la Tourette family. Mol Psychiatry. (2006) 11:954–64. doi: 10.1038/sj.mp.4001877
13. Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. (2012) 21:3513–23. doi: 10.1093/hmg/dds164
14. Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol Psychiatry. (2018) 23:2167–83. doi: 10.1038/s41380-018-0018-4
15. Gazzellone MJ, Zhou X, Lionel AC, Uddin M, Thiruvahindrapuram B, Liang S, et al. Copy number variation in Han Chinese individuals with autism spectrum disorder. J Neurodev Disord. (2014) 6:34. doi: 10.1186/1866-1955-6-34
16. He WZ, Liu WQ, Zhong XQ, Chen XL, Li SY, Zhang HM, et al. Analysis of de novo copy number variations in a family affected with autism spectrum disorders using high-resolution array-based comparative genomic hybridization. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2012) 29:266–9. doi: 10.3760/cma.j.issn.1003-9406.2012.03.004
17. Kalkan Z, Durasi IM, Sezerman U, Atasever-Arslan B. Potential of GRID2 receptor gene for preventing TNF-induced neurodegeneration in autism. Neurosci Lett. (2016) 620:62–9. doi: 10.1016/j.neulet.2016.03.043
18. RK CY, Merico D, Bookman M, Jennifer LH, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. (2017) 20:602–11. doi: 10.1038/nn.4524
19. Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, et al. Genomic patterns of de novo mutation in simplex autism. Cell. (2017) 171:710–22.e12. doi: 10.1016/j.cell.2017.08.047
20. Lintas C, Picinelli C, Piras IS, Sacco R, Brogna C, Persico AM. Copy number variation in 19 Italian multiplex families with autism spectrum disorder: importance of synaptic and neurite elongation genes. Am J Med Genet B Neuropsychiatr Genet. (2017) 174:547–56. doi: 10.1002/ajmg.b.32537
21. Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. (2016) 98:58–74. doi: 10.1016/j.ajhg.2015.11.023
22. Leblond CS, Cliquet F, Carton C, Huguet G, Mathieu A, Kergrohen T, et al. Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med. (2019) 4:1. doi: 10.1038/s41525-018-0075-2
23. Schoen M, Asoglu H, Bauer HF, Muller HP, Abaei A, Sauer AK, et al. Shank3 Transgenic and prenatal zinc-deficient autism mouse models show convergent and individual alterations of brain structures in MRI. Front Neural Circuits. (2019) 13:6. doi: 10.3389/fncir.2019.00006
24. Napoli E, Ross-Inta C, Song G, Wong S, Hagerman R, Gane LW, et al. Premutation in the fragile X mental retardation 1 (FMR1) gene affects maternal Zn-milk and perinatal brain bioenergetics and scaffolding. Front Neurosci. (2016) 10:159. doi: 10.3389/fnins.2016.00159
25. Clarke RA, Fang ZM, Diwan AD, Gilbert DL. Tourette syndrome and klippel-feil anomaly in a child with chromosome 22q11 duplication. Case Rep Med. (2009) 2009:361518. doi: 10.1155/2009/361518
26. Krishnan V, Stoppel DC, Nong Y, Johnson MA, Nadler MJ, Ozkaynak E, et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature. (2017) 543:507–12. doi: 10.1038/nature21678
27. Lam M, Moslem M, Bryois J, Pronk RJ, Uhlin E, Ellstrom ID, et al. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal functionality. Exp Cell Res. (2019) 383:111469. doi: 10.1016/j.yexcr.2019.06.014
28. Zhang P, Lu H, Peixoto RT, Pines MK, Ge Y, Oku S, et al. Heparan sulfate organizes neuronal synapses through neurexin partnerships. Cell. (2018) 174:1450–64.e23. doi: 10.1016/j.cell.2018.07.002
29. Kasem E, Kurihara T, Tabuchi K. Neurexins and neuropsychiatric disorders. Neurosci Res. (2018) 127:53–60. doi: 10.1016/j.neures.2017.10.012
30. Gauthier J, Siddiqui TJ, Huashan P, Yokomaku D, Hamdan FF, Champagne N, et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet. (2011) 130:563–73. doi: 10.1007/s00439-011-0975-z
31. Huang AY, Yu D, Davis LK, Sul JH, Tsetsos F, Ramensky V, et al. Rare copy number variants in NRXN1 and CNTN6 increase risk for Tourette syndrome. Neuron. (2017) 94:1101–11.e7.
32. Roppongi RT, Karimi B, Siddiqui TJ. Role of LRRTMs in synapse development and plasticity. Neurosci Res. (2017) 116:18–28. doi: 10.1016/j.neures.2016.10.003
33. Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. (2012) 75:762–77. doi: 10.1016/j.neuron.2012.08.019
34. Raichle ME, Gusnard DA. Appraising the brain's energy budget. Proc Natl Acad Sci USA. (2002) 99:10237–9. doi: 10.1073/pnas.172399499
35. Bogan N, Cabot JB. Light and electron microscopic analyses of intraspinal axon collaterals of sympathetic preganglionic neurons. Brain Res. (1991) 541:241–51. doi: 10.1016/0006-8993(91)91024-U
36. Mutsaers SE, Carroll WM. Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol. (1998) 96:139–43. doi: 10.1007/s004010050873
37. Fabricius C, Berthold CH, Rydmark M. Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J Neurocytol. (1993) 22l:941–54. doi: 10.1007/BF01218352
38. Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3–>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. (1998) 18:8300–10. doi: 10.1523/JNEUROSCI.18-20-08300.1998
39. Flippo KH, Strack S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophr Res. (2017) 187:26–32. doi: 10.1016/j.schres.2017.05.003
40. Kimura T, Murakami F. Evidence that dendritic mitochondria negatively regulate dendritic branching in pyramidal neurons in the neocortex. J Neurosci. (2014) 34:6938–51. doi: 10.1523/JNEUROSCI.5095-13.2014
41. Chaturvedi RK, Flint Beal M. Mitochondrial diseases of the brain. Free Radic Biol Med. (2013) 63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018
42. Adzic M, Brkic Z, Bulajic S, Mitic M, Radojcic MB. Antidepressant action on mitochondrial dysfunction in psychiatric disorders. Drug Dev Res. (2016) 77:400–6. doi: 10.1002/ddr.21332
43. Bergman O, Ben-Shachar D. Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatry. (2016) 61:457–69. doi: 10.1177/0706743716648290
44. Machado AK, Pan AY, da Silva TM, Duong A, Andreazza AC. Upstream pathways controlling mitochondrial function in major psychosis: a focus on bipolar disorder. Can J Psychiatry. (2016) 61:446–56. doi: 10.1177/0706743716648297
45. Curtis D, Brett P, Dearlove AM, McQuillin A, Kalsi G, Robertson MM, et al. Genome scan of Tourette syndrome in a single large pedigree shows some support for linkage to regions of chromosomes 5, 10 and 13. Psychiatr Genet. (2004) 14:83–7. doi: 10.1097/01.ypg.0000107927.32051.f5
46. Zhang H, Leckman JF, Pauls DL, Tsai CP, Kidd KK, Campos MR, et al. Genomewide scan of hoarding in sib pairs in which both sibs have Gilles de la Tourette syndrome. Am J Hum Genet. (2002) 70:896–904. doi: 10.1086/339520
47. Simonic I, Gericke GS, Ott J, Weber JL. Identification of genetic markers associated with Gilles de la Tourette syndrome in an Afrikaner population. Am J Hum Genet. (1998) 63:839–46. doi: 10.1086/302002
48. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International molecular genetic study of autism consortium. Hum Mol Genet. (1998) 7:571–8. doi: 10.1093/hmg/7.3.571
49. Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. Am J Med Genet. (1999) 88:609–15. doi: 10.1002/(SICI)1096-8628(19991215)88:6<609::AID-AJMG7>3.0.CO;2-L
50. Shao Y, Wolpert CM, Raiford KL, Menold MM, Donnelly SL, Ravan SA, et al. Genomic screen and follow-up analysis for autistic disorder. Am J Med Genet. (2002) 114:99–105. doi: 10.1002/ajmg.10153
51. Schellenberg GD, Dawson G, Sung YJ, Estes A, Munson J, Rosenthal E, et al. Evidence for multiple loci from a genome scan of autism kindreds. Mol Psychiatry. (2006) 11:1049–60. doi: 10.1038/sj.mp.4001874
52. Maestrini E, Pagnamenta AT, Lamb JA, Bacchelli E, Sykes NH, Sousa I, et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol Psychiatry. (2010) 15:954–68. doi: 10.1038/mp.2009.34
53. Tourette Syndrome Association International Consortium for G. Genome scan for Tourette disorder in affected-sibling-pair and multigenerational families. Am J Hum Genet. (2007) 80:265–72. doi: 10.1086/511052
54. Suarez-Rama JJ, Arrojo M, Sobrino B, Amigo J, Brenlla J, Agra S, et al. Resequencing and association analysis of coding regions at twenty candidate genes suggest a role for rare risk variation at AKAP9 and protective variation at NRXN1 in schizophrenia susceptibility. J Psychiatr Res. (2015) 66, 67:38–44. doi: 10.1016/j.jpsychires.2015.04.013
55. Lintas C, Sacco R, Garbett K, Mirnics K, Militerni R, Bravaccio C, et al. Involvement of the PRKCB1 gene in autistic disorder: significant genetic association and reduced neocortical gene expression. Mol Psychiatry. (2009) 14:705–18. doi: 10.1038/mp.2008.21
56. Philippi A, Roschmann E, Tores F, Lindenbaum P, Benajou A, Germain-Leclerc L, et al. Haplotypes in the gene encoding protein kinase c-beta (PRKCB1) on chromosome 16 are associated with autism. Mol Psychiatry. (2005) 10:950–60. doi: 10.1038/sj.mp.4001704
57. Eicher JD, Gruen JR. Language impairment and dyslexia genes influence language skills in children with autism spectrum disorders. Autism Res. (2015) 8:229–34. doi: 10.1002/aur.1436
58. Wang S, Mandell JD, Kumar Y, Sun N, Morris MT, Arbelaez J, et al. De novo sequence and copy number variants are strongly associated with tourette disorder and implicate cell polarity in pathogenesis. Cell Rep. (2018) 25:3544. doi: 10.1016/j.celrep.2018.12.024
59. Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. (2012) 148:1223–41. doi: 10.1016/j.cell.2012.02.039
60. Johnstone M, Maclean A, Heyrman L, Lenaerts AS, Nordin A, Nilsson LG, et al. Copy number variations in DISC1 and DISC1-interacting partners in major mental illness. Mol Neuropsychiatry. (2015) 1:175–90. doi: 10.1159/000438788
61. Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry. (2012) 71:392–402. doi: 10.1016/j.biopsych.2011.09.034
62. McGrath LM, Yu D, Marshall C, Davis LK, Thiruvahindrapuram B, Li B, et al. Copy number variation in obsessive-compulsive disorder and tourette syndrome: a cross-disorder study. J Am Acad Child Adolesc Psychiatry. (2014) 53:910–9. doi: 10.1016/j.jaac.2014.04.022
63. Sundaram SK, Huq AM, Wilson BJ, Chugani HT. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology. (2010) 74:1583–90. doi: 10.1212/WNL.0b013e3181e0f147
64. Bertelsen B, Melchior L, Jensen LR, Groth C, Glenthoj B, Rizzo R, et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur J Hum Genet. (2014) 22:1283–9. doi: 10.1038/ejhg.2014.24
65. Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. (2010) 15:637–46. doi: 10.1038/mp.2009.57
66. Jang W, Kim Y, Han E, Park J, Chae H, Kwon A, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann Lab Med. (2019) 39:299–310. doi: 10.3343/alm.2019.39.3.299
67. Sundaram SK, Huq AM, Sun Z, Yu W, Bennett L, Wilson BJ, et al. Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann Neurol. (2011) 69:901–4. doi: 10.1002/ana.22398
68. Wittkowski KM, Sonakya V, Bigio B, Tonn MK, Shic F, Ascano M, et al. A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism. Transl Psychiatry. (2014) 4:e354. doi: 10.1038/tp.2013.124
69. Wang L, Jia P, Wolfinger RD, Chen X, Zhao Z. Gene set analysis of genome-wide association studies: methodological issues and perspectives. Genomics. (2011) 98:1–8. doi: 10.1016/j.ygeno.2011.04.006
70. de Leeuw C, Goudriaan A, Smit AB, Yu D, Mathews CA, Scharf JM, et al. Involvement of astrocyte metabolic coupling in Tourette syndrome pathogenesis. Eur J Hum Genet. (2015) 23:1519–22. doi: 10.1038/ejhg.2015.22
71. Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet. (2012) 131:565–79. doi: 10.1007/s00439-011-1094-6
72. Boghosian-Sell L, Comings DE, Overhauser J. Tourette syndrome in a pedigree with a 7;18 translocation: identification of a YAC spanning the translocation breakpoint at 18q22.3. Am J Hum Genet. (1996) 59:999–1005.
73. Petek E, Windpassinger C, Vincent JB, Cheung J, Boright AP, Scherer SW, et al. Disruption of a novel gene (IMMP2L) by a breakpoint in 7q31 associated with Tourette syndrome. Am J Hum Genet. (2001) 68:848–58. doi: 10.1086/319523
74. Zhang Y, Liu Y, Zarrei M, Tong W, Dong R, Wang Y, et al. Association of IMMP2L deletions with autism spectrum disorder: a trio family study and meta-analysis. Am J Med Genet B Neuropsychiatr Genet. (2018) 177:93–100. doi: 10.1002/ajmg.b.32608
75. Patel C, Cooper-Charles L, McMullan DJ, Walker JM, Davison V, Morton J. Translocation breakpoint at 7q31 associated with tics: further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur J Hum Genet. (2011) 19:634–9. doi: 10.1038/ejhg.2010.238
76. Robertson MM, Shelley BP, Dalwai S, Brewer C, Critchley HD. A patient with both Gilles de la Tourette's syndrome and chromosome 22q11 deletion syndrome: clue to the genetics of Gilles de la Tourette's syndrome? J Psychosom Res. (2006) 61:365–8. doi: 10.1016/j.jpsychores.2006.06.011
77. Tang G, Gutierrez Rios P, Kuo SH, Akman HO, Rosoklija G, Tanji K, et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis. (2013) 54:349–61. doi: 10.1016/j.nbd.2013.01.006
78. Anitha A, Nakamura K, Thanseem I, Matsuzaki H, Miyachi T, Tsujii M, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. (2013) 23:294–302. doi: 10.1111/bpa.12002
79. Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism. (2012) 3:12. doi: 10.1186/2040-2392-3-12
80. Voineagu I. Gene expression studies in autism: moving from the genome to the transcriptome and beyond. Neurobiol Dis. (2012) 45:69–75. doi: 10.1016/j.nbd.2011.07.017
81. Schwede M, Nagpal S, Gandal MJ, Parikshak NN, Mirnics K, Geschwind DH, et al. Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post-mortem autism cerebral cortex. J Neurodev Disord. (2018) 10:18. doi: 10.1186/s11689-018-9237-x
82. Ji L, Chauhan A, Chauhan V. Reduced activity of protein kinase C in the frontal cortex of subjects with regressive autism: relationship with developmental abnormalities. Int J Biol Sci. (2012) 8:1075–84. doi: 10.7150/ijbs.4742
83. Shen M, Wang F, Li M, Sah N, Stockton ME, Tidei JJ, et al. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat Neurosci. (2019) 22:386–400. doi: 10.1038/s41593-019-0338-y
84. Lu B, Poirier C, Gaspar T, Gratzke C, Harrison W, Busija D, et al. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol Reprod. (2008) 78:601–10. doi: 10.1095/biolreprod.107.065987
85. Kreilaus F, Chesworth R, Eapen V, Clarke R, Karl T. First behavioural assessment of a novel Immp2l knockdown mouse model with relevance for Gilles de la Tourette syndrome and Autism spectrum disorder. Behav Brain Res. (2019) 374:112057. doi: 10.1016/j.bbr.2019.112057
86. Norkett R, Modi S, Kittler JT. Mitochondrial roles of the psychiatric disease risk factor DISC1. Schizophr Res. (2017) 187:47–54. doi: 10.1016/j.schres.2016.12.025
87. Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. (2010) 68:610–38. doi: 10.1016/j.neuron.2010.09.039
88. Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. (2000) 20:6374–84. doi: 10.1523/JNEUROSCI.20-17-06374.2000
89. Iworima DG, Pasqualotto BA, Rintoul GL. Kif5 regulates mitochondrial movement, morphology, function and neuronal survival. Mol Cell Neurosci. (2016) 72:22–33. doi: 10.1016/j.mcn.2015.12.014
90. Duis J, Dean S, Applegate C, Harper A, Xiao R, He W, et al. KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann Neurol. (2016) 80:633–7. doi: 10.1002/ana.24744
91. Campbell PD, Shen K, Sapio MR, Glenn TD, Talbot WS, Marlow FL. Unique function of Kinesin Kif5A in localization of mitochondria in axons. J Neurosci. (2014) 34:14717–32. doi: 10.1523/JNEUROSCI.2770-14.2014
92. Guo Q, Liao S, Zhu Z, Li Y, Li F, Xu C. Structural basis for the recognition of kinesin family member 21A (KIF21A) by the ankyrin domains of KANK1 and KANK2 proteins. J Biol Chem. (2018) 293:557–66. doi: 10.1074/jbc.M117.817494
93. King MD, Phillips GW, Bignone PA, Hayes NV, Pinder JC, Baines AJ. A conserved sequence in calmodulin regulated spectrin-associated protein 1 links its interaction with spectrin and calmodulin to neurite outgrowth. J Neurochem. (2014) 128:391–402. doi.org/10.1111/jnc.12462
94. Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. (2010) 2:139. doi: 10.3389/fnsyn.2010.00139
95. Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. (2013) 4:413–9. doi: 10.1016/j.celrep.2013.06.040
96. Picard M, McEwen BS. Mitochondria impact brain function and cognition. Proc Natl Acad Sci USA. (2014) 111:7–8. doi: 10.1073/pnas.1321881111
97. Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci USA. (2014) 111:486–91. doi: 10.1073/pnas.1311310110
98. Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. (2013) 5. doi: 10.1101/cshperspect.a011304
99. Kang M, Akbarali HI. Denitration of L-type calcium channel. FEBS Lett. (2008) 582:3033–6. doi: 10.1016/j.febslet.2008.07.042
100. Cai Q, Sheng ZH. Moving or stopping mitochondria: miro as a traffic cop by sensing calcium. Neuron. (2009) 61:493–6. doi: 10.1016/j.neuron.2009.02.003
101. Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. (2012) 125:2095–104. doi: 10.1242/jcs.053850
102. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. (2013) 20:31–42. doi: 10.1038/cdd.2012.81
103. MacAskill AF, Brickley K, Stephenson FA, Kittler JT. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci. (2009) 40:301–12. doi: 10.1016/j.mcn.2008.10.016
104. van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. (2013) 77:485–502. doi: 10.1016/j.neuron.2012.11.027
105. Loss O, Stephenson FA. Localization of the kinesin adaptor proteins trafficking kinesin proteins 1 and 2 in primary cultures of hippocampal pyramidal and cortical neurons. J Neurosci Res. (2015) 93:1056–66. doi: 10.1002/jnr.23549
106. Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. (2011) 286:18079–92. doi: 10.1074/jbc.M111.236018
107. Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci USA. (2014) 111:E3631–40. doi: 10.1073/pnas.1402449111
108. Millar JK, James R, Christie S, Porteous DJ. Disrupted in schizophrenia 1 (DISC1): subcellular targeting and induction of ring mitochondria. Mol Cell Neurosci. (2005) 30:477–84. doi: 10.1016/j.mcn.2005.08.021
109. Park YU, Jeong J, Lee H, Mun JY, Kim JH, Lee JS, et al. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc Natl Acad Sci USA. (2010) 107:17785–90. doi: 10.1073/pnas.1004361107
110. Tsuboi D, Kuroda K, Tanaka M, Namba T, Iizuka Y, Taya S, et al. Disrupted-in-schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosci. (2015) 18:698–707. doi: 10.1038/nn.3984
111. Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. (2005) 116:23–7. doi: 10.1007/s00439-004-1199-2
112. Ogawa F, Murphy LC, Malavasi EL, O'Sullivan ST, Torrance HS, Porteous DJ, et al. NDE1 and GSK3beta associate with TRAK1 and regulate axonal mitochondrial motility: identification of cyclic AMP as a novel modulator of axonal mitochondrial trafficking. ACS Chem Neurosci. (2016) 7:553–64. doi: 10.1021/acschemneuro.5b00255
113. Atkin TA, MacAskill AF, Brandon NJ, Kittler JT. Disrupted in Schizophrenia-1 regulates intracellular trafficking of mitochondria in neurons. Mol Psychiatry. (2011) 16:122–4. doi: 10.1038/mp.2010.110
114. Norkett R, Modi S, Birsa N, Atkin TA, Ivankovic D, Pathania M, et al. DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J Biol Chem. (2016) 291:613–29. doi: 10.1074/jbc.M115.699447
115. Amiott EA, Lott P, Soto J, Kang PB, McCaffery JM, DiMauro S, et al. Mitochondrial fusion and function in Charcot-Marie-Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol. (2008) 211:115–27. doi: 10.1016/j.expneurol.2008.01.010
116. Feng Y, Walsh CA. Protein-protein interactions, cytoskeletal regulation and neuronal migration. Nat Rev Neurosci. (2001) 2:408–16. doi: 10.1038/35077559
117. Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. (2004) 44:279–93. doi: 10.1016/j.neuron.2004.09.023
118. Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, et al. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet. (2011) 88:536–47. doi: 10.1016/j.ajhg.2011.04.003
119. Bradshaw NJ, Ogawa F, Antolin-Fontes B, Chubb JE, Carlyle BC, Christie S, et al. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun. (2008) 377:1091–6. doi: 10.1016/j.bbrc.2008.10.120
120. Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. (2009) 16:256–70. doi: 10.1016/j.devcel.2009.01.005
121. Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci USA. (2011) 108:5849–54. doi: 10.1073/pnas.1013660108
122. Ji L, Chauhan V, Flory MJ, Chauhan A. Brain region-specific decrease in the activity and expression of protein kinase A in the frontal cortex of regressive autism. PLoS ONE. (2011) 6:e23751. doi: 10.1371/journal.pone.0023751
123. Cianfrocco MA, DeSantis ME, Leschziner AE, Reck-Peterson SL. Mechanism and regulation of cytoplasmic dynein. Annu Rev Cell Dev Biol. (2015) 31:83–108. doi: 10.1146/annurev-cellbio-100814-125438
124. Reddy BJ, Mattson M, Wynne CL, Vadpey O, Durra A, Chapman D, et al. Load-induced enhancement of Dynein force production by LIS1-NudE in vivo and in vitro. Nat Commun. (2016) 7:12259. doi: 10.1038/ncomms12259
125. Bradshaw NJ, Soares DC, Carlyle BC, Ogawa F, Davidson-Smith H, Christie S, et al. PKA phosphorylation of NDE1 is DISC1/PDE4 dependent and modulates its interaction with LIS1 and NDEL1. J Neurosci. (2011) 31:9043–54. doi: 10.1523/JNEUROSCI.5410-10.2011
126. Slater PG, Cammarata GM, Samuelson AG, Magee A, Hu Y, Lowery LA. XMAP215 promotes microtubule-F-actin interactions to regulate growth cone microtubules during axon guidance. J Cell Sci. (2019) 132:1–14. doi: 10.1242/jcs.224311
127. Sears JC, Choi WJ, Broadie K. Fragile X Mental Retardation Protein positively regulates PKA anchor Rugose and PKA activity to control actin assembly in learning/memory circuitry. Neurobiol Dis. (2019) 127:53–64. doi: 10.1016/j.nbd.2019.02.004
128. Tao K, Matsuki N, Koyama R. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev Neurobiol. (2014) 74:557–73. doi: 10.1002/dneu.22149
129. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
130. Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. (2003) 14:2415–9. doi: 10.1097/00001756-200312190-00025
131. Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, et al. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. (2007) 43:564–70. doi: 10.1016/j.yjmcc.2007.08.010
132. Boccitto M, Doshi S, Newton IP, Nathke I, Neve R, Dong F, et al. Opposing actions of the synapse-associated protein of 97-kDa molecular weight (SAP97) and Disrupted in Schizophrenia 1 (DISC1) on Wnt/beta-catenin signaling. Neuroscience. (2016) 326:22–30. doi: 10.1016/j.neuroscience.2016.03.048
133. Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. (2003) 87:1427–35. doi: 10.1046/j.1471-4159.2003.02113.x
134. King TD, Clodfelder-Miller B, Barksdale KA, Bijur GN. Unregulated mitochondrial GSK3beta activity results in NADH: ubiquinone oxidoreductase deficiency. Neurotox Res. (2008) 14:367–82. doi: 10.1007/BF03033861
135. Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. (2009) 18:R169–76. doi: 10.1093/hmg/ddp326
136. Ishihara N, Otera H, Oka T, Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal. (2013) 19:389–99. doi: 10.1089/ars.2012.4830
137. Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to Metabolism. Mol Cell. (2016) 61:683–94. doi: 10.1016/j.molcel.2016.02.022
138. Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. (2009) 218:268–73. doi: 10.1016/j.expneurol.2009.05.003
139. Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. (2004) 36:449–51. doi: 10.1038/ng1341
140. Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. (2007) 27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007
141. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. (2010) 30:4232–40. doi: 10.1523/JNEUROSCI.6248-09.2010
142. Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. (2007) 130:548–62. doi: 10.1016/j.cell.2007.06.026
143. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
144. Chapman AL, Bennett EJ, Ramesh TM, De Vos KJ, Grierson AJ. Axonal transport defects in a mitofusin 2 loss of function model of charcot-marie-tooth disease in Zebrafish. PLoS ONE. (2013) 8:e67276. doi: 10.1371/journal.pone.0067276
145. Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, et al. Dominant optic atrophy. Orphanet J Rare Dis. (2012) 7:46. doi: 10.1186/1750-1172-7-46
146. Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. (2010) 133:771–86. doi: 10.1093/brain/awq007
147. Chao de la Barca JM, Prunier-Mirebeau D, Amati-Bonneau P, Ferre M, Sarzi E, Bris C, et al. OPA1-related disorders: diversity of clinical expression, modes of inheritance and pathophysiology. Neurobiol Dis. (2016) 90:20–6. doi: 10.1016/j.nbd.2015.08.015
148. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
149. Bertholet AM, Millet AM, Guillermin O, Daloyau M, Davezac N, Miquel MC, et al. OPA1 loss of function affects in vitro neuronal maturation. Brain. (2013) 136:1518–33. doi: 10.1093/brain/awt060
150. Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. (2013) 1833:1256–68. doi: 10.1016/j.bbamcr.2013.02.002
151. Sheffer R, Douiev L, Edvardson S, Shaag A, Tamimi K, Soiferman D, et al. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am J Med Genet A. (2016) 170:1603–7. doi: 10.1002/ajmg.a.37624
152. Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. (2007) 356:1736–41. doi: 10.1056/NEJMoa064436
153. Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A. (2016) 170:2002–11. doi: 10.1002/ajmg.a.37721
154. Nasca A, Legati A, Baruffini E, Nolli C, Moroni I, Ardissone A, et al. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum Mutat. (2016) 37:898–903. doi: 10.1002/humu.23033
155. Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur J Hum Genet. (2016) 24:1084–8. doi: 10.1038/ejhg.2015.243
156. Yoon G, Malam Z, Paton T, Marshall CR, Hyatt E, Ivakine Z, et al. Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J Pediatr. (2016) 171:313–6.e1-2. doi: 10.1016/j.jpeds.2015.12.060
157. Oettinghaus B, Schulz JM, Restelli LM, Licci M, Savoia C, Schmidt A, et al. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. (2016) 23:18–28. doi: 10.1038/cdd.2015.39
158. Shields LY, Kim H, Zhu L, Haddad D, Berthet A, Pathak D, et al. Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. (2015) 6:e1725. doi: 10.1038/cddis.2015.94
159. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. (2009) 11:958–66. doi: 10.1038/ncb1907
160. Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. (2009) 186:805–16. doi: 10.1083/jcb.200903065
161. Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M. Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem. (2003) 278:8597–605. doi: 10.1074/jbc.M211761200
162. Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. (2007) 282:21583–7. doi: 10.1074/jbc.C700083200
163. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
164. Wong H, Levenga J, Cain P, Rothermel B, Klann E, Hoeffer C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer's disease. Acta Neuropathol. (2015) 130:829–43. doi: 10.1007/s00401-015-1499-8
165. Parra V, Altamirano F, Hernandez-Fuentes CP, Tong D, Kyrychenko V, Rotter D, et al. Down syndrome critical region 1 gene, Rcan1, helps maintain a more fused mitochondrial network. Circ Res. (2018) 122:e20–33. doi: 10.1161/CIRCRESAHA.117.311522
166. Mizuguchi T, Nakashima M, Kato M, Okamoto N, Kurahashi H, Ekhilevitch N, et al. Loss-of-function and gain-of-function mutations in PPP3CA cause two distinct disorders. Hum Mol Genet. (2018) 27:1421–33. doi: 10.1093/hmg/ddy052
167. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. (2016) 351:275–81. doi: 10.1126/science.aab4138
168. Yu R, Liu T, Jin SB, Ning C, Lendahl U, Nister M, et al. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci Rep. (2017) 7:880. doi: 10.1038/s41598-017-00853-x
169. Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. (2016) 540:139–43. doi: 10.1038/nature20555
170. Piggott LA, Bauman AL, Scott JD, Dessauer CW. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci USA. (2008) 105:13835–40. doi: 10.1073/pnas.0712100105
171. Sanderson JL, Scott JD, Dell'Acqua ML. Control of homeostatic synaptic plasticity by AKAP-anchored kinase and phosphatase regulation of Ca(2+)-Permeable AMPA receptors. J Neurosci. (2018) 38:2863–76. doi: 10.1523/JNEUROSCI.2362-17.2018
172. Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, et al. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. (2014) 7:1577–88. doi: 10.1016/j.celrep.2014.04.027
173. Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. (2011) 17:321–36. doi: 10.1177/1073858410384740
174. Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. (1987) 56:615–49. doi: 10.1146/annurev.bi.56.070187.003151
175. Glovaci I, Chapman CA. Activation of phosphatidylinositol-linked dopamine receptors induces a facilitation of glutamate-mediated synaptic transmission in the lateral entorhinal cortex. PLoS ONE. (2015) 10:e0131948. doi: 10.1371/journal.pone.0131948
176. Jones RM, Cadby G, Melton PE, Abraham LJ, Whitehouse AJ, Moses EK. Genome-wide association study of autistic-like traits in a general population study of young adults. Front Hum Neurosci. (2013) 7:658. doi: 10.3389/fnhum.2013.00658
177. Pagnamenta AT, Bacchelli E, de Jonge MV, Mirza G, Scerri TS, Minopoli F, et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. (2010) 68:320–8. doi: 10.1016/j.biopsych.2010.02.002
178. McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. (2006) 71:399–407. doi: 10.1016/j.bcp.2005.10.011
179. Sica V, Kroemer G. IMMP2L: a mitochondrial protease suppressing cellular senescence. Cell Res. (2018) 28:607–8. doi: 10.1038/s41422-018-0051-5
180. Yuan L, Zhai L, Qian L, Huang, Ding Y, Xiang H, et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. (2018) 28:625–43. doi: 10.1038/s41422-018-0043-5
181. Adamczyk A, Gause CD, Sattler R, Vidensky S, Rothstein JD, Singer H, et al. Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr Genet. (2011) 21:90–7. doi: 10.1097/YPG.0b013e328341a307
182. Jackson TD, Palmer CS, Stojanovski D. Mitochondrial diseases caused by dysfunctional mitochondrial protein import. Biochem Soc Trans. (2018) 46:1225–38. doi: 10.1042/BST20180239
183. Martensson CU, Becker T. Acylglycerol Kinase: mitochondrial protein transport meets lipid biosynthesis. Trends Cell Biol. (2017) 27:700–2. doi: 10.1016/j.tcb.2017.08.006
184. Curran SP, Leuenberger D, Schmidt E, Koehler CM. The role of the Tim8p-Tim13p complex in a conserved import pathway for mitochondrial polytopic inner membrane proteins. J Cell Biol. (2002) 158:1017–27. doi: 10.1083/jcb.200205124
185. Kadenbach B. Regulation of mammalian 13-subunit cytochrome c oxidase and binding of other proteins: role of NDUFA4. Trends Endocrinol Metab. (2017) 28:761–70. doi: 10.1016/j.tem.2017.09.003
186. Simonic I, Nyholt DR, Gericke GS, Gordon D, Matsumoto N, Ledbetter DH, et al. Further evidence for linkage of Gilles de la Tourette syndrome (GTS) susceptibility loci on chromosomes 2p11, 8q22 and 11q23-24 in South African Afrikaners. Am J Med Genet. (2001) 105:163–7. doi: 10.1002/ajmg.1192
187. Gokhale A, Hartwig C, Freeman AAH, Bassell JL, Zlatic SA, Sapp Savas C, et al. Systems analysis of the 22q11.2 microdeletion syndrome converges on a mitochondrial interactome necessary for synapse function and behavior. J Neurosci. (2019) 39:3561–81. doi: 10.1523/JNEUROSCI.1983-18.2019
188. Knight S, Coon H, Johnson M, Leppert MF, Camp NJ, McMahon WM, et al. Linkage analysis of Tourette syndrome in a large Utah pedigree. Am J Med Genet B Neuropsychiatr Genet. (2010) 153B:656–62. doi: 10.1002/ajmg.b.31035
Keywords: Tourette syndrome genes, Tourette syndrome cause, Tourette syndrome etiology, mitochondrial fission, mitochondrial supply
Citation: Clarke RA, Furlong TM and Eapen V (2021) Tourette Syndrome Risk Genes Regulate Mitochondrial Dynamics, Structure, and Function. Front. Psychiatry 11:556803. doi: 10.3389/fpsyt.2020.556803
Received: 28 April 2020; Accepted: 23 November 2020;
Published: 10 March 2021.
Edited by:
Hanna E. Stevens, The University of Iowa, United StatesReviewed by:
A. Gulhan Ercan-Sencicek, Masonic Medical Research Institute (MMRI), United StatesPiotr Janik, Medical University of Warsaw, Poland
Copyright © 2021 Clarke, Furlong and Eapen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond A. Clarke, ci5jbGFya2VAdW5zdy5lZHUuYXU=