 Ryoko Kakehi1,2,3
Ryoko Kakehi1,2,3 Hiroaki Hori1*Fuyuko Yoshida1,4Mariko Itoh1,5Mingming Lin1Madoka Niwa1Megumi Narita1Keiko Ino1,6Risa Imai7
Hiroaki Hori1*Fuyuko Yoshida1,4Mariko Itoh1,5Mingming Lin1Madoka Niwa1Megumi Narita1Keiko Ino1,6Risa Imai7 Daimei Sasayama8Toshiko Kamo9
Daimei Sasayama8Toshiko Kamo9 Hiroshi Kunugi4,10Yoshiharu Kim1
Hiroshi Kunugi4,10Yoshiharu Kim1- 1Department of Behavioral Medicine, National Center of Neurology and Psychiatry, National Institute of Mental Health, Tokyo, Japan
- 2Department of Medical Science, Tohoku University Graduate School of Medicine, Sendai, Japan
- 3Department of Nursing, Wayō Women’s University, Chiba, Japan
- 4Department of Mental Disorder Research, National Center of Neurology and Psychiatry, National Institute of Neuroscience, Tokyo, Japan
- 5Center for Environmental and Health Sciences, Hokkaido University, Sapporo, Japan
- 6Department of Psychiatry and Cognitive-Behavioral Medicine, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan
- 7Risa Irinaka Mental Clinic, Nagoya, Japan
- 8Department of Psychiatry, Shinshu University School of Medicine, Nagano, Japan
- 9Wakamatsu-cho Mental and Skin Clinic, Tokyo, Japan
- 10Department of Psychiatry, Teikyo University School of Medicine, Tokyo, Japan
Accumulated evidence shows that psychological trauma and posttraumatic stress disorder (PTSD) are associated with dysfunction in the hypothalamic-pituitary-adrenal (HPA) axis. Besides the HPA axis hormones, recent evidence suggests that the renin-angiotensin-aldosterone (RAA) system and genetic factors may be involved in trauma/PTSD as well as in HPA axis regulation. This study attempted to better understand the HPA axis function in relation to PTSD and childhood maltreatment by simultaneously examining RAA system and genetic polymorphisms of candidate genes. Here we studied 69 civilian women with PTSD and 107 healthy control women without DSM-IV-based traumatic experience. Childhood maltreatment history was assessed with the Childhood Trauma Questionnaire. PTSD severity was assessed with the Posttraumatic Diagnostic Scale. Functional disability was assessed with the Sheehan Disability Scale. HPA axis was examined by measuring blood levels of cortisol, adrenocorticotropic hormone, and dehydroepiandrosterone-sulphate (DHEA-S). RAA system was examined by measuring blood renin and aldosterone levels. The FKBP5 rs1360780 and CACNA1C rs1006737 polymorphisms were genotyped. No significant differences were seen between patients and controls in any of the five hormone levels. DHEA-S levels were significantly negatively correlated with overall PTSD severity (p = 0.003) and functional disability (p = 0.008). A two-way analysis of variance with diagnostic groups and genotypes as fixed factors revealed that patients with the rs1006737 A-allele had significantly lower DHEA-S levels than patients with the GG genotype (p = 0.002) and controls with the A-allele (p = 0.006). Childhood maltreatment history was not significantly correlated with any of the five hormone levels. These results were generally unchanged after controlling for the potentially confounding effect of age, depression, and anxiety. Our findings suggest that lower DHEA-S levels could indicate more severe subtype of PTSD, the association of which might be partly modified by the CACNA1C polymorphism.
1. Introduction
Posttraumatic stress disorder (PTSD) is a serious psychiatric condition that can develop after a major traumatic event, often leading to a chronic course and severe functional impairment. While the etiology of PTSD remains unclear, this disorder likely involves alterations in stress response systems since it is associated with severe traumatic stress.
Among these systems, the hypothalamic-pituitary-adrenal (HPA) axis is crucially involved in the coordination of stress responses and the maintenance of homeostasis. The HPA axis activation results in an increased secretion of glucocorticoid (cortisol in humans) from the adrenal cortex, and glucocorticoid in turn regulates its own production through negative feedback by binding to glucocorticoid receptors (GRs) in the pituitary, hypothalamus, and hippocampus as well as mineralocorticoid receptors in the hippocampus. HPA axis dysfunction, including lower cortisol levels and greater GR sensitivity, is a well-known biological feature of PTSD (1, 2). However, findings to date have been controversial especially with respect to basal cortisol levels, such that lower levels in individuals with PTSD compared to controls have been shown in some (3, 4) but not all (5) meta-analyses. This inconsistency might be attributable to several methodological issues, such as possible effects of trauma exposure itself in control individuals and different paradigms of cortisol measurement (6).
In addition to cortisol, dehydroepiandrosterone (DHEA) and its more stable sulphated conjugated metabolite, DHEA-S [herein together referred to as “DHEA(S)”], have been implicated in the pathophysiology of PTSD (6, 7). Like cortisol, DHEA(S) is secreted by the adrenal cortex, at least partly in response to adrenocorticotropic hormone (ACTH). DHEA(S) is the most abundant adrenal steroid in humans. Compared to DHEA, DHEA-S shows much higher concentrations in serum, has longer half-life, and exhibits smaller diurnal variation. DHEA(S) has neuroprotective actions, and is also classified as a neurosteroid, in that it not only crosses the blood brain barrier to exert effects within the central nervous system but is synthesized de novo in the brain from its sterol precursor (8). DHEA(S) acts as a gamma-aminobutyric acid type A receptor antagonist and is involved in the regulation of neuronal survival and differentiation (9). It is also shown to act as a sigma-1 receptor agonist (10). Furthermore, DHEA(S) is thought to possess anti-glucocorticoid properties, as it antagonizes the effect of cortisol (11), inhibits glucocorticoid-induced enzyme activity (12), and rapidly reduces cortisol levels when acutely administered (13). Thus, DHEA(S) is assumed to be involved in stress responses (14) and, as such, investigated in various psychiatric disorders including PTSD (6, 7). However, the findings on DHEA(S) levels in PTSD patients relative to controls are mixed, with higher (15, 16), lower (17), and similar (18, 19) levels have all been reported.
Besides the HPA axis, the renin-angiotensin-aldosterone (RAA) system is activated in response to stress, resulting in increased levels of angiotensin II and aldosterone (20). The RAA system is an endocrine system centrally involved in the regulation of systemic blood pressure and blood fluid volume. This system has a bidirectional interaction with the HPA axis, such that angiotensin activates the HPA axis to increase circulating glucocorticoids (20) and that ACTH stimulates aldosterone production (21). Recently, several lines of research have suggested that the RAA system can be associated with PTSD. In clinical studies, the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers (ARBs) was found to be associated with significantly decreased PTSD symptoms (22, 23). In a neuroimaging study targeting highly anxious individuals, an ARB losartan affected emotional processing and amygdala activity (24). In rodent models, the RAA system has been shown to be involved in the regulation of fear memory (25, 26). Furthermore, biomarker studies have reported that circulating renin levels are increased in PTSD patients (27) and that aldosterone levels are increased in relation to childhood trauma (28, 29). In contrast to these findings, however, a recent longitudinal study reported that women with chronic PTSD had significantly lower plasma aldosterone levels averaged over time, compared to women unexposed to trauma (30). As there have been only a few studies investigating RAA system hormones in PTSD, more needs to be done to clarify the nature of trauma-related alterations in this system.
The HPA axis is affected by multiple genes as well as environmental factors; for example, basal cortisol is reported to have a heritability of around 60% by twin studies (31). Of these genetic factors, the FKBP5 (FK506 binding protein five) gene, whose protein acts as a co-chaperone of GR complex, plays a pivotal role in the regulation of HPA axis. The FKBP5 overexpression can decrease glucocorticoid binding affinity and nuclear translocation of GRs, thereby reducing GR sensitivity and negative feedback of HPA axis (32). FKBP5 expression is moderated by common genetic variants that are part of a haplotype including rs1360780, a single nucleotide polymorphism (SNP) considered as a functional variant of this gene (33). Studies have investigated the effect of rs1360780 on stress-related psychiatric disorders, demonstrating that it can interact with childhood trauma and be associated with PTSD (32, 33). Another gene that could be involved in both PTSD (34) and HPA axis (35) is the CACNA1C (calcium voltage-gated channel subunit alpha1C) gene. This gene encodes an alpha-1 subunit of the voltage-gated L-type calcium channel (Cav1.2), which mediates the influx of calcium ions into the cell upon membrane polarization and plays a role in dendritic development, neuronal survival, synaptic plasticity, and memory/learning (36). An intronic SNP rs1006737 of CACNA1C has been identified as a risk factor for multiple psychiatric disorders and their various phenotypes in many studies including our previous one (37–39). The G>A substitution of rs1006737 is shown to be associated with decreased expression of CACNA1C (40, 41). Importantly, a recent study showed that mice with global heterozygous loss of Cacna1c and mice with specific deletion of Cacna1c from dopamine D1-receptor-expressing neurons both exhibited exaggerated remote contextual fear, which corresponds to the re-experiencing symptom of PTSD (34). These findings together raise the possibility that the inter-individual differences in HPA axis in PTSD may be explained in part by the functional SNPs of FKBP5 and CACNA1C.
The present study aimed to examine HPA axis function by measuring blood levels of cortisol, ACTH, and DHEA-S, in PTSD and childhood maltreatment history. We also investigated RAA system by measuring blood renin and aldosterone levels. These hormones were also examined in relation to PTSD severity and functional disability. We further explored the effects of genetic variations in FKBP5 (rs1360780) and CACNA1C (rs1006737) on HPA axis hormones and PTSD symptomatology. Given the modest sample size, we restricted the genetic investigation to these two functional loci implicated in both HPA axis and PTSD. Another issue that needs to be considered is the paradigm of HPA axis hormone measurement. Although dysregulated negative feedback is shown to reflect an important aspect of HPA axis abnormalities in trauma/PTSD (42, 43), we only measured baseline levels of these hormones in the main sample because it was difficult to conduct laborious challenge tests in the clinical sample. Instead, prior to the main study, we conducted a dexamethasone (DEX) suppression test in a small independent sample of healthy individuals. This test provided information regarding to what extent the basal levels of HPA axis hormones would predict their levels after DEX challenge. While DEX-induced negative feedback of cortisol is well documented, this is not the case with DHEA-S, and therefore we attempted to know the physiological response of DHEA-S to the DEX challenge in a general population.
2. Materials and methods
2.1. Participants
The present study was conducted at three institutes: National Center of Neurology and Psychiatry, Tokyo Women’s Medical University, and Nagoya City University. This study was approved by the ethics committees of these institutes, and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants after they had received a detailed explanation of the study. The study procedures were identical across the three sites.
This study was conducted as part of an ongoing larger project. Data collection for the present study was conducted between 2015 and 2020. This study included only women for both patients and controls; the sole inclusion of women, or exclusion of men, was because the vast majority of the sample included in our entire project were women.
A total of 69 civilian women with PTSD (age range: 18–56 years) participated in this study. Of them, 64 were outpatients at these hospitals and clinics, and their attending doctors were asked to inform the researcher of all potentially eligible patients. The remaining five patients were outpatients at the nearby clinics and were recruited through advertisements on our website. All patients had already been diagnosed as having PTSD by attending clinicians. The experience of traumatic events and diagnosis of PTSD were confirmed by the validated Japanese version (44) of the Posttraumatic Diagnostic Scale (PDS) (45). In addition, the validated Japanese version (46) of the Mini International Neuropsychiatric Interview (M.I.N.I) (47) was administered by an expert clinician to identify any other Axis-I disorders as well as PTSD. Patients with comorbid schizophrenia and those with marked manic episodes of bipolar disorder were excluded from the study.
In addition, 107 healthy control women without PDS-based traumatic experience (20–59 years) were recruited mostly through advertisements in free local magazines and on our website, and a few of them were recruited by word of mouth. The PDS was administered to healthy controls in order to examine the presence/absence of traumatic experiences and, if present, they were excluded from this study. All healthy individuals were interviewed by a board-certified psychiatrist which included M.I.N.I and non-structured interview, and those who demonstrated current Axis-I disorders or apparent signs of past psychiatric disorders were excluded.
2.2. Psychological assessment
Psychological and clinical characteristics of the patients and controls were assessed using the following five self-report questionnaires:
2.2.1. Posttraumatic Diagnostic Scale (PDS) (45).
The PDS comprises 4 parts that evaluate traumatic experiences reflecting Criteria A of the DSM-IV (Parts 1 and 2), PTSD severity reflecting Criteria B-D (Part 3), and functional impairments associated with PTSD symptoms (Part 4). Part 3 comprises 17 items pertaining to PTSD symptomatology during the past month, and each item is scored on a 4-point scale of frequency from 0 to 3, with higher scores indicating more frequently occurring (or more severe) symptoms.
In the present study, we administered Parts 1 and 2 to all participants in order to determine the presence/absence of traumatic experiences, and if present, Parts 3 and 4 were administered for the assessment of diagnosis and severity of PTSD. We used the Japanese version of PDS that has demonstrated good reliability and validity (44, 48).
2.2.2. Childhood Trauma Questionnaire (CTQ) (49).
Childhood maltreatment history was assessed with our Japanese version (CTQ-JNIMH) (50) of the CTQ (49), a widely used questionnaire for assessing history of childhood maltreatment. The CTQ has demonstrated adequate psychometric properties as demonstrated by a good fit of the 5-factor structure (49, 51), internal consistency (49, 52), and test-retest reliability (53). The commonly used 28-item version of CTQ includes 25 clinical items and 3 validity items. The 25 items load onto five subscales that assess different types of childhood maltreatment, including emotional abuse, physical abuse, sexual abuse, emotional neglect, and physical neglect. All items start with the phrase “when I was growing u,p” and are rated on a 5-point scale ranging from 1 to 5 (1 = “never true” to 5 = “very often true”), with higher scores indicating greater maltreatment. Accordingly, the minimum/maximum possible score of each CTQ subscale, i.e., emotional abuse, physical abuse, sexual abuse, emotional neglect, and physical neglect, is 5–25. Scores of these five subscales are summed to yield a total CTQ score.
Cronbach α coefficients of the 5 CTQ subscales, namely emotional abuse, physical abuse, sexual abuse, emotional neglect, and physical neglect, in the present sample (i.e., combined sample of patients and controls) were 0.93, 0.86, 0.93, 0.92, and 0.70, respectively. There was one participant (patient) who did not complete the sexual abuse subsection.
2.2.3. Beck Depression Inventory-II (BDI-II) (54).
The BDI-II is a 21-item self-report questionnaire widely used to measure depression severity during the past 2 weeks (54). Each item is scored on a 4-point scale from 0 to 3, with higher scores indicating more severe depressive symptoms. We used the validated Japanese version (55) of BDI-II.
2.2.4. State-Trait Anxiety Inventory (STAI) (56).
The STAI is a self-report questionnaire widely used to assess anxiety (56). It consists of two subscales for trait (STAI-T) and state (STAI-S) anxiety, both of which comprise 20 items that are scored on a 4-point scale from 1 to 4; higher scores indicate greater anxiety. We used the validated Japanese version (57) of STAI.
2.2.5. Sheehan Disability Scale (SDISS) (58).
The SDISS is a discretized visual analog rating scale of functional disability in three domains, namely work/school, social, and family life (58). Each domain is rated from 0 (unimpaired) to 10 (extremely impaired), which are summed to yield a global disability score ranging from 0 to 30. In this study the validated Japanese version (59) of SDISS was administered to all participants. There were two participants (both patients) who did not complete this questionnaire.
2.3. Measurement of biomarkers
Blood sampling was performed on the same day as the psychological assessments. The samples were collected from each participant around noon before lunch, between 11:30 a.m. and 12:30 p.m., in order to minimize possible effects of the time of day and dietary intake.
Levels of cortisol, ACTH, DHEA-S, renin, and aldosterone were measured at a clinical laboratory (SRL Inc., Tokyo, Japan). Serum cortisol levels and plasma ACTH levels were measured by electro chemiluminescence immunoassay. Serum DHEA-S levels, plasma renin levels, and plasma aldosterone levels were measured by chemiluminescent enzyme immunoassay.
Measurements of cortisol, ACTH and DHEA-S were performed at the laboratory soon after the blood was collected from each participant, while measurements of renin and aldosterone were performed afterward at the same laboratory using extracted plasma samples that had been stored at −80°C in a deep freezer at our institute. Data of cortisol, ACTH, and DHEA-S were available for all participants, while those of renin and aldosterone were available for 61 patients (88.4%) and 96 controls (89.7%). This sample attrition was due to lack of residual plasma samples for a subset of participants.
The detection limit for cortisol was 0.06 μg/dl. There were no participants whose cortisol levels were below this detection limit. The intra- and inter-assay coefficients of variation for cortisol were both less than 2.7%. For ACTH, the detection limit was 1.5 pg/ml; only one participant showed an ACTH level below this limit. The intra- and inter-assay coefficients of variation for ACTH were both less than 2.5%. For DHEA-S, the detection limit was 2.0 μg/dl; one participant showed a DHEA-S level below this limit. The intra- and inter-assay coefficients of variation for DHEA-S were both less than 5.0%. For renin, the detection limit was 0.20 pg/ml; no participants showed renin levels below this limit. The intra- and inter-assay coefficients of variation for renin were both less than 3.2%. For aldosterone, the detection limit was 4.0 pg/ml; no participants showed aldosterone levels below this limit. The intra- and inter-assay coefficients of variation for aldosterone were both less than 2.8%. Values under the detection limits were treated as 0 (μg/dl or pg/ml).
2.4. Genotyping
A majority of the participants also participated in the genetic testing by blood sampling, and genotyping was performed for 51 patients and 94 controls. The reason for this sample attrition was that informed consent to the genetic study was not provided for a portion of participants. Genomic DNA was prepared from venous blood according to standard procedures. Rs1360780 of FKBP5 (assay ID: C_8852038_10) and rs1006737 of CACNA1C (C___2584015_10) were genotyped using the TaqMan SNP Genotyping Assays. The thermal cycling conditions for polymerase chain reaction were: 1 cycle at 95°C for 10 min followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The allele-specific fluorescence was measured with ABI PRISM 7,900 Sequence Detection Systems (Applied Biosystems, Foster City, CA). These genotyping data have been deposited to the ClinVar database with accession numbers SCV002587063 (for rs1360780) and SCV002588462 (for rs1006737).
2.5. Additional experiments for HPA axis negative feedback in an independent sample
Prior to the main study, we conducted a DEX suppression test to measure cortisol and DHEA-S levels before and after DEX administration as a complementary examination of HPA axis, using an independent sample of 33 women (age range: 20–67; mean 44.8 ± 12.8 years) recruited via advertisements in free local magazines and on our website. This study was approved by the ethics committee of National Center of Neurology and Psychiatry, and written informed consent was obtained from all participants. They were confirmed to be psychiatrically healthy by the M.I.N.I; while information on the presence/absence of traumatic experiences was not available for these individuals, as the PDS was not administered to them.
The test procedure was as follows: on the first day, participants visited our laboratory and blood samples were collected at 10:00 h, and they took 0.5 mg tablet of DEX at 23:00 h at home; on the next day, blood samples were collected at 10:00 h at our laboratory again. High correlation between the basal and post-DEX levels indicates that the post-DEX level can be predicted by its basal level, irrespective of the extent of DEX-induced negative feedback; while low correlation suggests that the negative feedback is independent of the basal level. We used 0.5 mg of DEX, given that the DEX suppression test with this dose of DEX has been widely employed in the studies of stress-related psychiatric disorders including PTSD (42, 43). We collected blood samples at 10:00 h, considering that the previous studies of DEX suppression test in relation to trauma and PTSD have conducted blood sampling in the morning (42, 43).
2.6. Statistical analysis
Averages are reported as “means ± SD,” or “median (25th–75th percentile)” where appropriate. Categorical variables were compared using the χ2 test. Group differences were examined using the t-test, Mann–Whitney U-test, Kruskal–Wallis test, or analysis of variance (ANOVA), according to the nature and distribution of data. Correlations were calculated using Pearson’s r or Spearman’s rho.
Primary analyses were conducted as follows. We first tested the difference in blood biomarker levels between patients and controls using the Mann–Whitney U-test. Next, correlations of clinical/psychological characteristics with biomarkers were calculated in patients and in controls (data of PTSD severity was available only for patients), using the Spearman’s rho. Then, the correlation coefficient in patients was compared with that in controls in order to further examine whether the correlation is more apparent in patients than in controls. This analysis was done by converting Spearman’s correlation coefficients by Fisher’s z-transformation based on a standard procedure. For this purpose, Spearman’s coefficients were treated as though they were Pearson’s coefficients since the robustness of this assumption is demonstrated in a simulation study (60). Lastly, relationship of the two genetic polymorphisms with PTSD symptom severity was examined in patients using t-test. Relations of these polymorphisms with log-transformed cortisol and DHEA-S levels were investigated using a two-way ANOVA with diagnostic groups (patients vs. controls) and genotypes (minor allele carriers vs. non-carriers) as fixed factors.
Additionally, we performed a set of sensitivity analyses to control for the possible confounding effects of age, depression, and anxiety; we first controlled for age and then controlled for age, depression, and anxiety. Specifically, group comparisons were made by an analysis of covariance (ANCOVA) with these variables as covariates. Correlations were examined using unstandardized residuals of the biomarker levels corrected for these variables.
Statistical significance was set at 2-tailed p < 0.05 unless otherwise specified. For the correlation between biomarkers and psychological measures, a Bonferroni-corrected p < 0.01 (i.e., 0.05/5) was considered as statistically significant, given that five biomarkers were tested. For the SNP analyses, a Bonferroni-corrected p < 0.025 (i.e., 0.05/2) was considered as statistically significant, given that two biomarkers (i.e., cortisol and DHEA-S) were tested. All statistical analyses were performed using the Statistical Package for the Social Sciences version 27 (IBM Corp., Tokyo, Japan).
3. Results
3.1. Demographic, clinical, and psychological characteristics of the sample
Demographic/clinical/psychological variables in patients with PTSD and healthy controls are summarized in Table 1. The participants were all women, and age was well matched between patients and controls. They did not significantly differ in education level, smoking status, or body mass index. Most patients developed PTSD after experiencing interpersonal violence such as physical and/or sexual violence during adulthood, and had been ill for more than 6 months at the time of study participation. Many of them had psychiatric comorbidities, and were receiving psychotropic medications. They were on average moderately to severely ill, as indexed by the mean PDS total score. Compared to controls, patients reported significantly more childhood experiences of maltreatment (assessed with the CTQ), depression and anxiety symptoms (BDI and STAI, respectively), and functional disability (SDISS). For full correlations among all continuous variables in the total sample, see Supplementary Table 1.
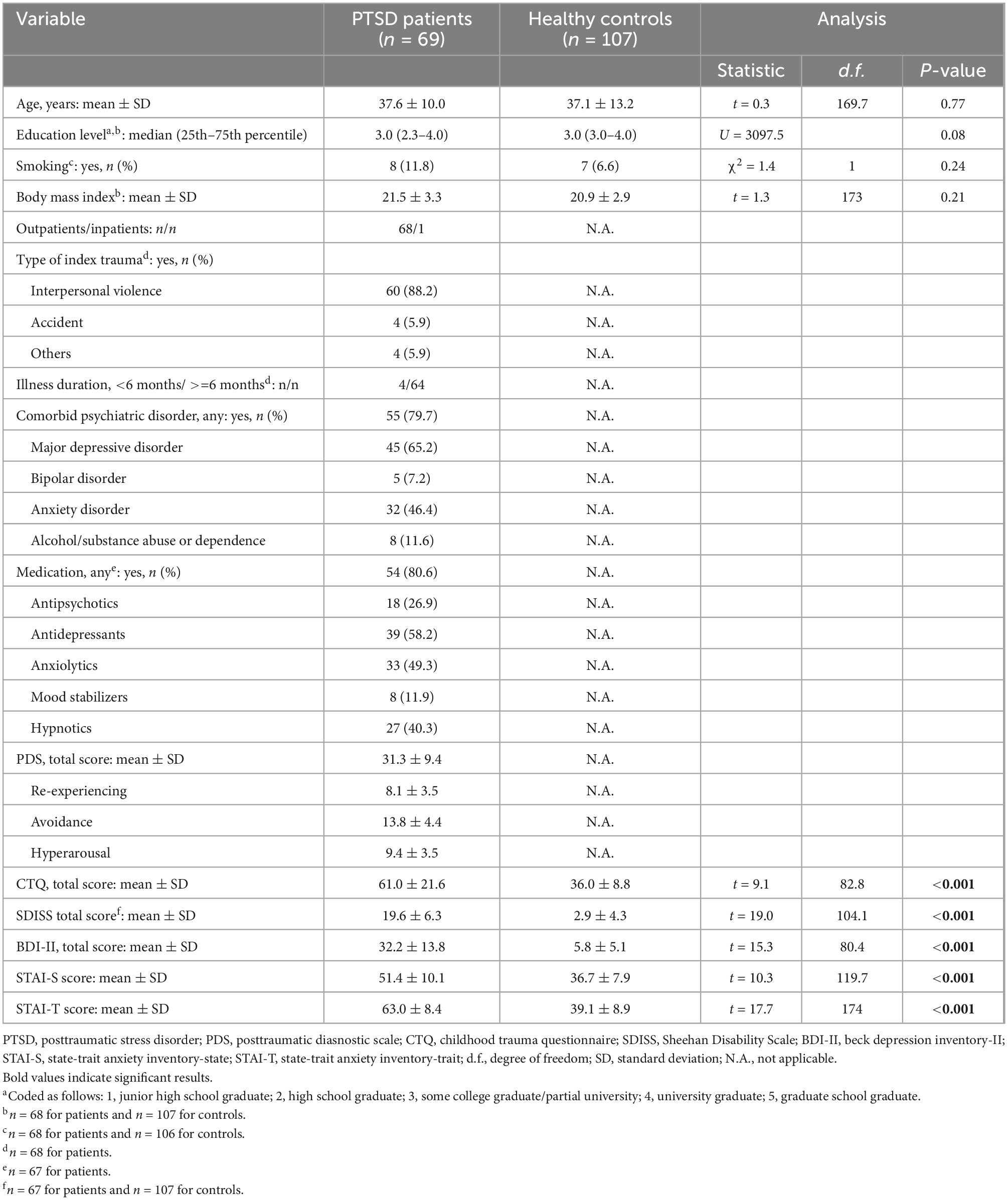
Table 1. Demographic/clinical/psychological characteristics in PTSD patients and healthy controls.
3.2. Potentially confounding variables
Age was significantly correlated negatively with ACTH, DHEA-S, and renin levels in patients, and significantly correlated positively with cortisol and negatively with DHEA-S, renin, and aldosterone levels in controls (all p < 0.05). Age was not correlated with PTSD severity (including the PDS total score and three subscale scores) in patients or with childhood maltreatment history (CTQ total score) in patients or in controls (all p > 0.8). Levels of the five hormones did not significantly differ between patients treated with and those without any class of medication, including antipsychotics, antidepressants, anxiolytics, mood stabilizers, and hypnotics (all p > 0.05), except for significantly higher cortisol levels in patients treated with mood stabilizers than those without (U = 383.5, p = 0.004). The five hormonal levels did not significantly differ between patients with comorbid major depressive disorder and those without (all p > 0.05). For menstrual cycles, the numbers of participants who endorsed “before menstruation (within 1 week),” “during menstruation,” “after menstruation (within 1 week),” and “after menopause” did not significantly differ between patients and controls (χ2 = 6.5, p = 0.09). In addition, five participants (all patients) were taking oral contraceptives. Patients taking oral contraceptives showed significantly higher cortisol levels than those without (U = 259.0, p = 0.014), while no significant differences were seen in the other four hormone levels (all p > 0.1). We therefore excluded cortisol data for these five patients in subsequent analyses; these five patients did not differ from the other 64 patients in any of the psychological characteristics including CTQ total, PDS total and three subscales, SDISS total, BDI-II total, and STAI-S/-T (all p > 0.2 by Mann–Whitney U-test). Levels of cortisol, ACTH, DHEA-S, renin, and aldosterone were not significantly different between the three study sites (all p > 0.05 by the Kruskal–Wallis test).
3.3. Biomarkers in patients vs. controls and their relation with clinical/psychological characteristics
As shown in Table 2, no significant differences were seen between patients and controls in any of the five markers, including cortisol, ACTH, DHEA-S, renin, and aldosterone.
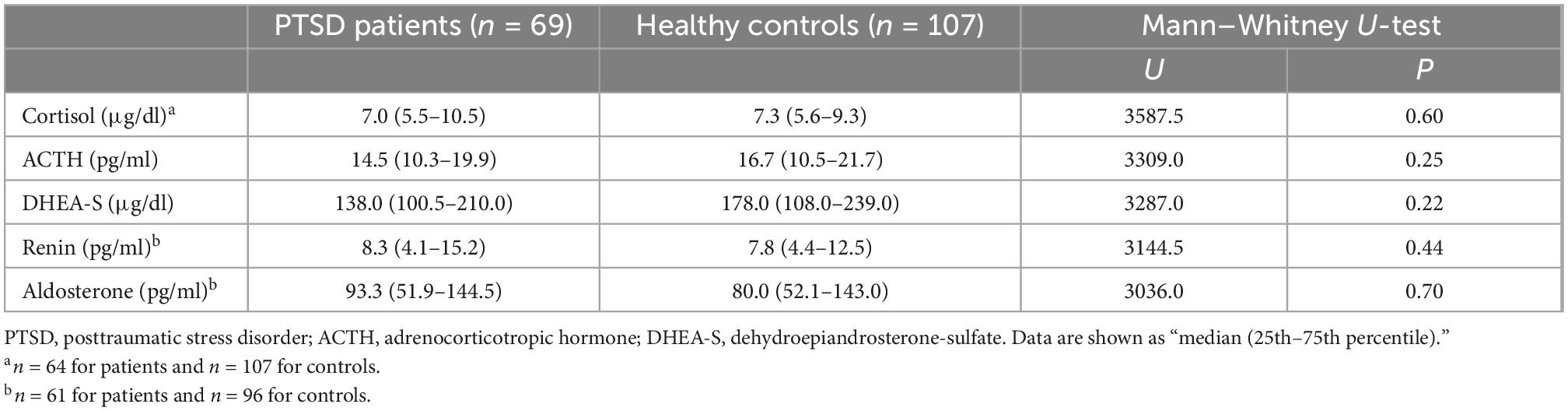
Table 2. Blood levels of HPA axis and RAA system hormones in patients and controls.
Correlations of the five biomarkers with PTSD symptomatology (PDS) and functional disability (SDISS) in patients are presented in Table 3. DHEA-S levels were significantly negatively correlated with overall PTSD severity (rho = −0.350, p = 0.003; Figure 1A), hyperarousal symptom (rho = −0.386, p = 0.001), and functional disability (rho = −0.322, p = 0.008; Figure 1B). Comparisons of correlations in patients vs. controls revealed that the association between lower DHEA-S levels and greater functional disability was significantly different between the groups, indicating that this association was specific to patients (Table 4). Cortisol, ACTH, renin, and aldosterone levels were not significantly correlated with any symptoms or functional disability in patients or in controls (Tables 3, 4).
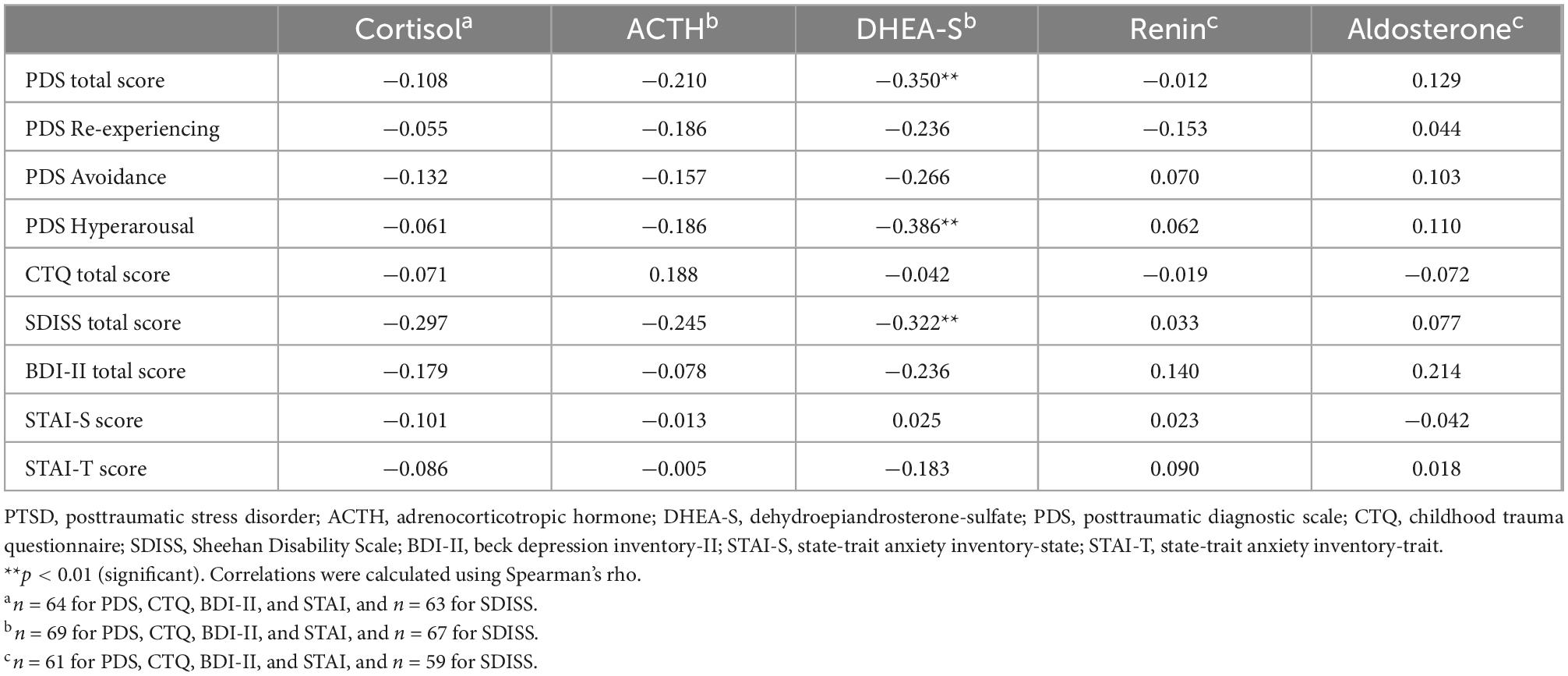
Table 3. Correlations of psychological characteristics with HPA axis and RAA system hormones in PTSD patients.
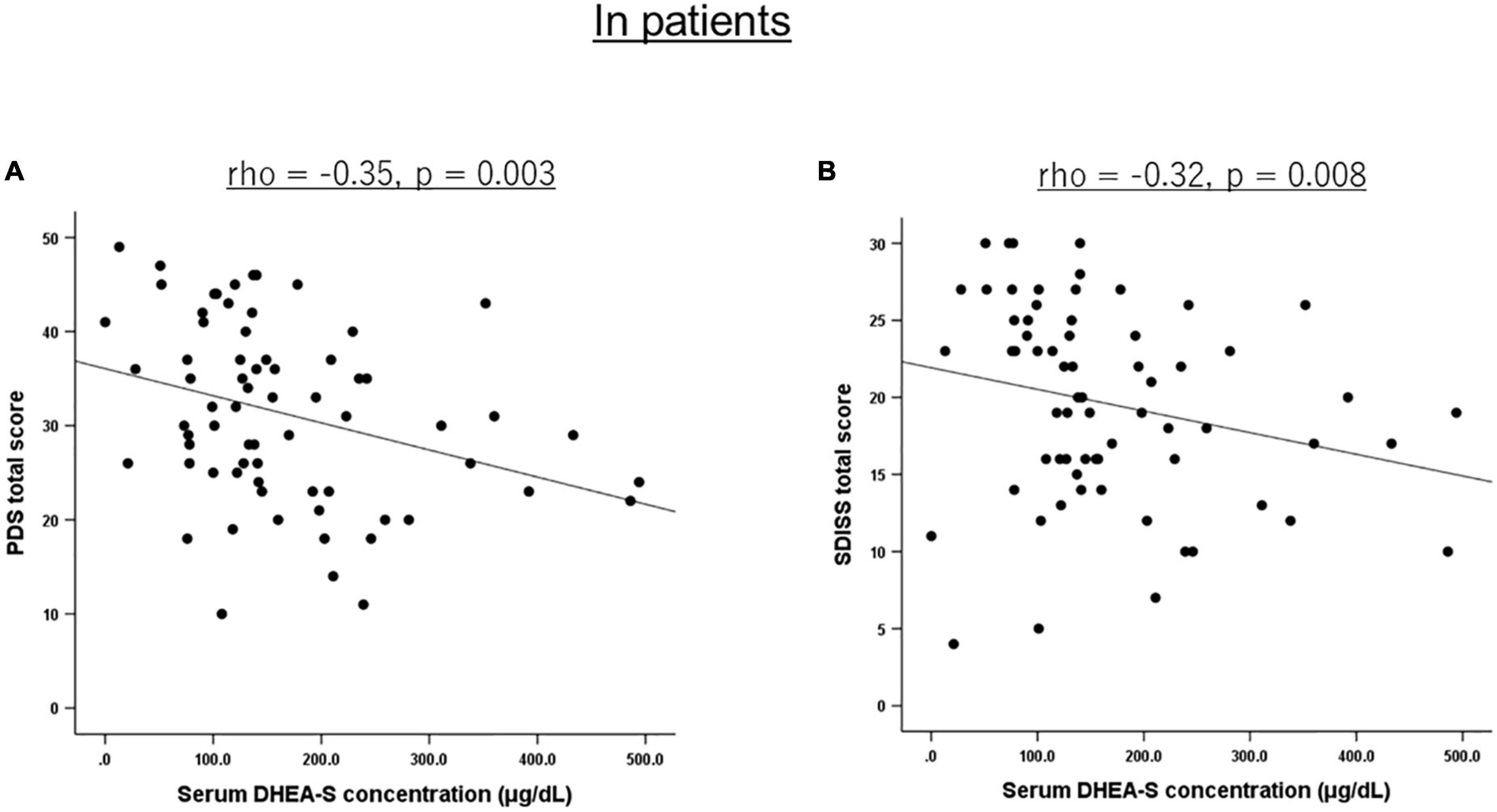
Figure 1. Scatterplot showing the relationship of serum DHEA-S levels with PTSD severity and functional disability in patients. In PTSD patients, serum DHEA-S levels were significantly negatively correlated with (A) overall PTSD severity assessed with the PDS total score and (B) functional disability assessed with the SDISS total score. Correlation was calculated by the Spearman’s rank order correlation.
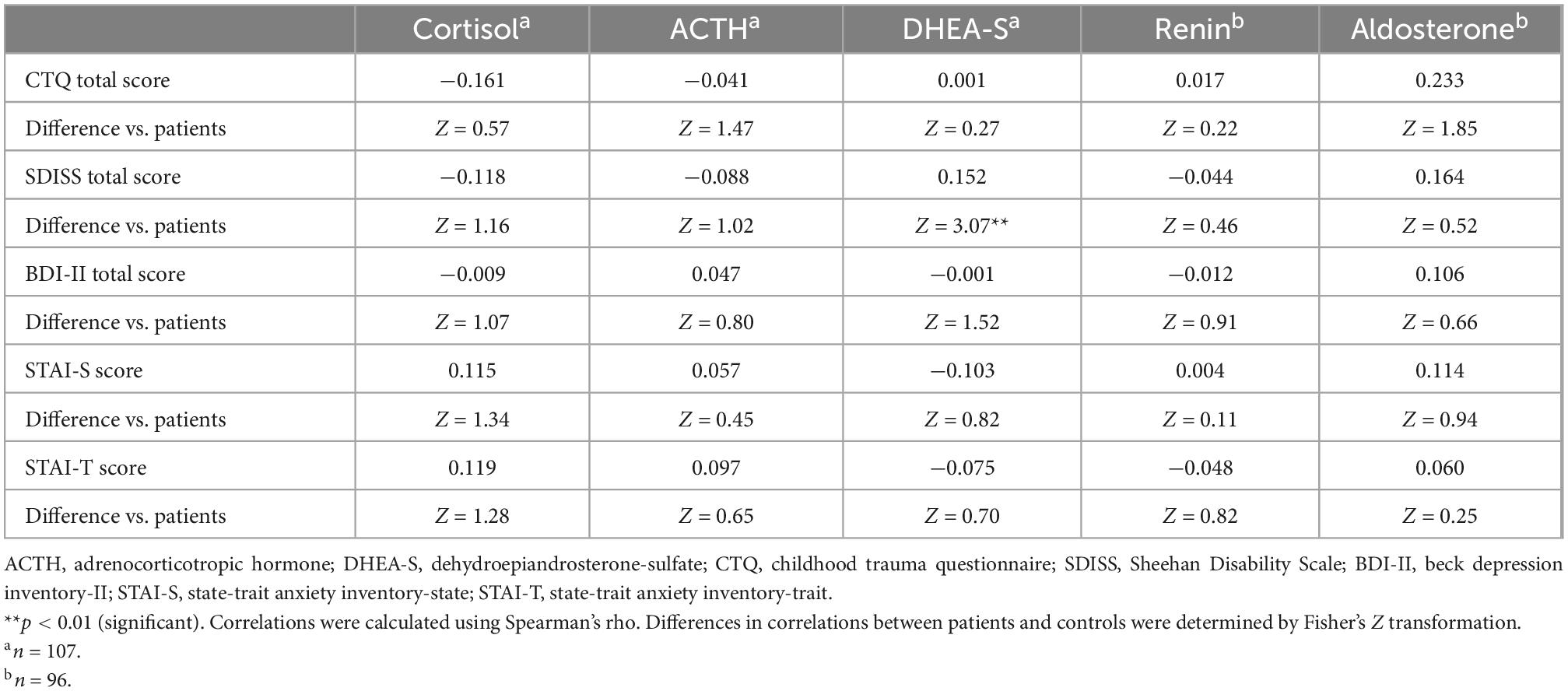
Table 4. Correlations of psychological characteristics with HPA axis and RAA system hormones in healthy controls (including differences in correlations between patients and controls).
Correlations between the CTQ total score and five biomarkers are shown in Table 3 for patients and Table 4 for controls. Childhood maltreatment history (CTQ total score) was not significantly correlated with any of the five biomarkers in patients or in controls. Considering that patients and controls did not differ in the five biomarker levels, we additionally examined the correlations between the CTQ total score and these markers in the total sample (i.e., patients and controls were combined) and found no significant correlation for any of the five markers (all p > 0.1; Supplementary Table 1).
Depression (BDI-II) and anxiety (STAI-S/-T) were not significantly correlated with any of the five biomarkers either in patients (Table 3) or in controls (Table 4).
3.4. Effects of genetic variations on biomarkers
Numbers of participants with the FKBP5 rs1360780 CC, CT, and TT genotypes were 28 (54.9%), 19 (37.3%), and 4 (7.8%) in patients (respectively) and 57 (60.6%), 30 (31.9%), and 7 (7.4%) in controls (respectively). Those with the CACNA1C rs1006737 GG, GA, and AA genotypes were 44 (86.3%), 7 (13.7%), and 0 (0.0%) in patients (respectively) and 77 (81.9%), 17 (18.1%), and 0 (0.0%) in controls (respectively). For both SNPs, genotype frequencies did not deviate from Hardy-Weinberg equilibrium either in patients or in controls (all p > 0.1). Due to the small number of the rs1360780 TT genotype, we combined the TT genotype with the CT genotype in all analyses, as in our previous study (61); thus, all comparisons were made with the two groups, i.e., CC vs. CT + TT genotypes. Likewise, as there were no participants with the rs1006737 AA genotype, all comparisons were made with the remaining two groups, i.e., GG vs. GA genotypes.
PTSD severity as assessed with the PDS total score in patients was not significantly different between the FKBP5 rs1360780 genotype groups (t = 0.56, p = 0.58) or between the CACNA1C rs1006737 genotype groups (t = 0.34, p = 0.74).
For the FKBP5 polymorphism, the two-way ANOVA for log-transformed cortisol levels showed no significant main effect of diagnostic group (p = 0.25) or rs1360780 genotype (p = 0.13) or their interaction (p = 0.26). Similarly, the two-way ANOVA for log-transformed DHEA-S levels showed no significant main effect of diagnostic group (p = 0.52) or rs1360780 genotype (p = 0.81) or their interaction (p = 0.23).
For the CACNA1C polymorphism, the two-way ANOVA for log-transformed cortisol levels showed no significant main effect of diagnostic group (p = 0.42) or rs1006737 genotype (p = 0.51) or their interaction (p = 0.93). However, the two-way ANOVA for log-transformed DHEA-S levels showed a significant main effect of diagnostic group (p = 0.013), rs1006737 genotype (p = 0.014), and their interaction (p = 0.009); this analysis further revealed that patients with the GG genotype and those with the GA genotype significantly differed in DHEA-S levels (p = 0.002) while controls with the GG genotype and those with the GA genotype did not differ in DHEA-S levels (p = 0.89) and that patients with the GG genotype and controls with the GG genotype did not differ in DHEA-S levels (p = 0.87) while patients with the GA genotype and controls with the GA genotype significantly differed in DHEA-S levels (p = 0.006); patients with the GA genotype had lower DHEA-S levels than patients with the GG genotype and controls with the GA genotype (Figure 2). To confirm this result using the raw DHEA-S data (i.e., without log-transformation), we further used the Mann–Whitney U-test to compare DHEA-S levels between patients with the GG genotype and those with the GA genotype and between patients with the GA genotype and controls with the GA genotype, which yielded similar results (p = 0.017 and p = 0.034, respectively).
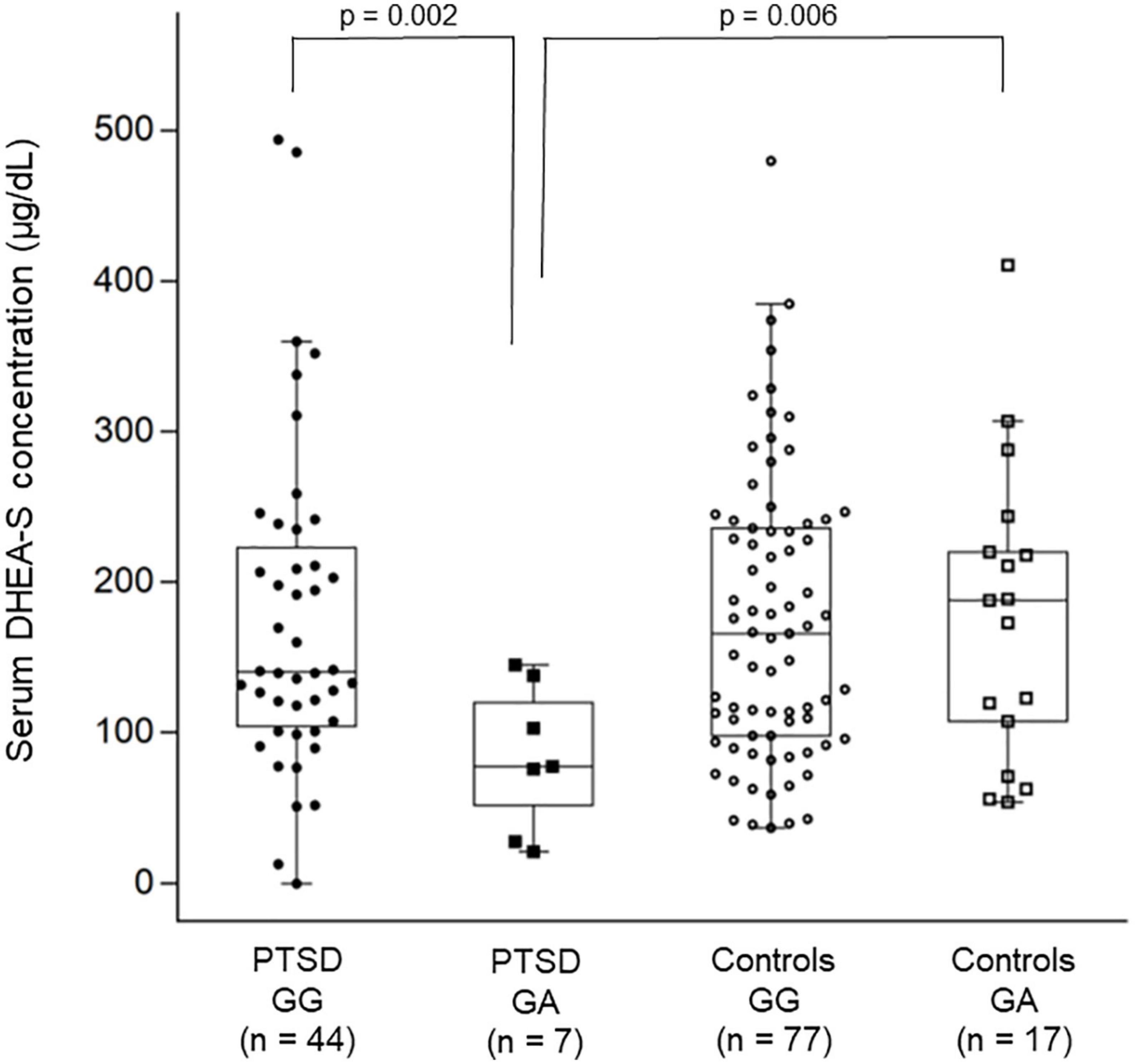
Figure 2. Combined dot- and box-plot showing the comparison of serum DHEA-S levels as a function of diagnostic groups and the CACNA1C rs1006737 genotype groups. Comparison of log-transformed DHEA-S levels was made by the two-way ANOVA. In this figure, raw data of DHEA-S levels without log-transformation is presented.
3.5. Sensitivity analysis to control for possible confounding effects of age, depression, and anxiety
An ANCOVA was used to compare log-transformed levels of the five hormones between patients and controls with age as a covariate, which confirmed no significant difference between groups in any of the five hormones (all p > 0.3), as in the original analysis. An additional age-, depression- (BDI-II), and anxiety- (STAI-S and –T) corrected ANCOVA also confirmed no significant difference between groups in any of the five hormones (all p > 0.2).
We also examined whether the main significant findings were robust to the effect of potential confounders. Specifically, Spearman’s correlations between the PDS/SDISS scores and DHEA-S levels in patients were calculated using unstandardized residuals of the DHEA-S levels corrected for age. This analysis confirmed significant correlations for all the PDS/SDISS indices, including PDS total (rho = −0.435, p < 0.001), re-experiencing (rho = −0.273, p = 0.023), avoidance (rho = −0.333, p = 0.005), hyperarousal (rho = −0.502, p < 0.001), and SDISS total (rho = −0.352, p = 0.003). An additional correlation analysis between the PDS/SDISS scores and DHEA-S levels in patients corrected for age, depression (BDI-II), and anxiety (STAI-S and –T) confirmed similar results for PDS total (rho = −0.366, p = 0.002), re-experiencing (rho = −0.250, p = 0.038), avoidance (rho = −0.257, p = 0.033), hyperarousal (rho = −0.434, p < 0.001), and SDISS total (rho = −0.259, p = 0.035).
In addition, we performed an age-corrected two-way (i.e., diagnostic group*rs1006737 genotype) ANCOVA to compare log-transformed DHEA-S levels, and confirmed the significant main effect of genotype (p = 0.020), while main effect of group (p = 0.077) and interaction (p = 0.11) were not significant. An additional age-, depression- (BDI-II), and anxiety- (STAI-S and –T) corrected two-way ANCOVA for log-transformed DHEA-S levels also confirmed the significant main effect of genotype (p = 0.014), but not main effect of group (p = 0.79) or interaction (p = 0.076).
3.6. DEX-induced HPA axis negative feedback in an independent sample
We performed the DEX suppression test in the independent sample of 33 healthy women. Median (25th–75th percentile) levels of basal and post-DEX cortisol (μg/dl) were 9.7 (8.1–12.5) and 1.2 (0.0–1.9), respectively; their correlation failed to reach statistical significance (rho = 0.34, p = 0.054). Basal and post-DEX DHEA-S levels (μg/dl) were 128.0 (95.5–171.5) and 92.0 (66.5–117.0), respectively; their correlation was highly significant (rho = 0.94, p < 0.001). These results indicated that post-DEX cortisol levels could be only weakly predicted by its basal levels, whereas post-DEX DHEA-S levels could be almost totally predicted by basal levels.
4. Discussion
HPA axis in individuals with trauma and PTSD has been extensively investigated for more than three decades, and several important aspects of stress hormone dysregulation associated with these conditions have been identified. However, a substantial proportion of studies have not replicated these findings, and the controversy also exists even in meta-analytic studies (3–5). To overcome this, studies have used various approaches, including different paradigms of hormone measurement (e.g., baseline, pharmacological, and psychosocial challenge), different samples (e.g., blood, saliva, urine, and hair), and different participant characteristics (e.g., controls with and without trauma). Still, the nature of HPA axis alterations in PTSD and trauma remains somewhat elusive.
The present study attempted to better understand the HPA axis (dys) function in PTSD by simultaneously examining RAA system and genetic polymorphisms of candidate genes. The main finding of this study was that DHEA-S levels were significantly negatively correlated with PTSD severity and functional disability. Furthermore, patients with the CACNA1C rs1006737 GA genotype had significantly lower DHEA-S levels than patients with the GG genotype and controls with the GA genotype. Childhood maltreatment history was not significantly associated with the five hormonal levels.
None of the HPA axis hormones measured were significantly different between PTSD patients and healthy controls. This negative result is not surprising given the mixed findings of previous studies on HPA axis in PTSD, some of which did not find significant case-control difference like the present study [e.g., (5)]. Nonetheless, DHEA-S levels were significantly negatively correlated with PTSD severity and functional disability. The comparison of correlations in patients vs. controls revealed that this association between lower DHEA-S levels and greater functional disability was specific to patients. This suggests that DHEA-S may have protective effects in these individuals, being in line with previous findings (11, 14). Consistently, a recent study reported significant negative association between blood DHEA-S levels and PTSD severity via personality traits (62). Our finding may also be related to the fact that circulating DHEA-S is relatively stable and that the basal DHEA-S level corresponds well to negative feedback inhibition to the DEX suppression test, given that such a biomarker that is relatively robust to intra- and inter-daily variations and measurement paradigms, compared to the one strongly affected by these factors, can better reflect psychopathology.
HPA axis is involved in stress response in concert with the immune/inflammatory system and autonomic nervous system, and indeed, these systems have also been implicated in the pathophysiology of PTSD (2, 63). Moreover, DHEA(S) has been shown to attenuate inflammatory responses (64, 65). It is therefore possible that alterations in DHEA(S) and HPA axis function may be involved in the biological mechanism of PTSD by interacting with the immune and inflammatory system. From a clinical viewpoint, there are currently no established biomarkers for the diagnosis or prognosis of PTSD. In this regard, our finding suggests that DHEA-S may serve as a marker for severity and disability of PTSD, although this possibility needs to be further investigated in future studies.
Renin and aldosterone levels did not significantly differ between patients and controls or correlate with childhood maltreatment history. On the other hand, a preceding study reported that plasma levels of renin, but not aldosterone, were increased in persons with PTSD than controls without trauma (27). In addition, another study found that childhood maltreatment was associated with increased aldosterone but not renin levels (28). This inconsistency may be due to the relatively small sample in the present study. It may also be possible that medications for our patients have normalized these hormone levels. As there have been only a few studies investigating the RAA system in relation to PTSD or childhood trauma, more research is needed to understand the role of this system in these conditions.
CACNA1C rs1006737 genotype was significantly associated with DHEA-S levels in patients, suggesting that the observed relation between DHEA-S and PTSD symptomatology could be at least in part influenced by this SNP. This observation is in accordance with a previous finding that this SNP can interact with early life stress to affect HPA axis reactivity (35). Rodent studies have shown that chronic stress and glucocorticoids can affect Cav1.2 mRNA expression encoded by Cacna1c (66–68). We found that minor A-allele carriers had lower DHEA-S levels than G-allele homozygotes. This pattern of unfavorable phenotypes related to the A-allele compared to G-allele accords with previous findings obtained in various populations (35, 39). In addition, the A-allele is shown to be associated with decreased CACNA1C expression (40, 41). On the other hand, the CACNA1C rs1006737 was not significantly associated with PTSD symptomatology. Moreover, the FKBP5 rs1360780 was not significantly associated with any hormonal levels or symptomatology. It should be noted here that there are considerable ethnic differences in minor allele frequencies of these SNPs. The minor T-allele frequency of FKBP5 rs1360780 and the minor A-allele frequency of CACNA1C rs1006737 in our sample were 0.245 and 0.083 (respectively), being consistent with the frequency of 0.227 and 0.059 (respectively) reported in a representative genome variation database of Japanese individuals (69). In contrast, the frequency of rs1360780 T-allele and that of rs1006737 A-allele are reported to be around 0.30 and 0.35 (respectively) among many other populations such as Europeans, according to the Genome Aggregation Database (gnomAD). Thus, our non-significant results may be attributable to the lower minor allele frequencies of these SNPs in Japanese, as well as the small sample size.
Our findings that the CACNA1C rs1006737 risk A-allele is associated with lower DHEA-S levels and that lower DHEA-S levels are related to more severe PTSD collectively suggest that genetic risk factors affect the stress response system, namely HPA axis, and as a result influence the disorder. In line with this possibility, it is widely accepted that both genetic factors and environmental stimuli, and the resultant gene-environment interaction, are involved in the etiology of PTSD (70). The present findings also suggest that rs1006737 and DHEA-S levels contribute to subtypes (or outcomes) within PTSD rather than the development of this disorder, considering that DHEA-S levels did not differ between patients and controls. Supporting this, it is postulated that PTSD is a heterogeneous disorder with distinct subtypes (71). While our findings suggest a role of CACNA1C and HPA axis, further studies are necessary to get a bigger picture of the gene-environment interaction in PTSD and subtypes of this disorder.
This study has several limitations. First, the sample size was not very large, particularly for the genetic analyses. For example, recent genome-wide association studies of PTSD and trauma have employed tens of thousands of participants (72, 73). It is therefore possible that some of our non-significant results might have been type II errors. It may be worth noting, however, that the present study adopted a hypothesis-based, candidate gene approach focusing on symptoms and biomarkers rather than case-control comparison. As a result, we observed a significant association between the CACNA1C risk allele and DHEA-S levels. Second, this cross-sectional study did not provide information as to the causality between trauma/PTSD and hormonal alterations. Future longitudinal studies are needed to draw any conclusion as to the causal relationship. Third, the hormonal measurement was conducted only once without any stimulations, although our additional DEX suppression test indicated that DEX-stimulated DHEA-S levels showed very high correlation with its basal levels. Still, given that HPA axis hormones, particularly cortisol, exhibit significant diurnal variations, it would be more informative to collect samples at multiple time points. In addition, caution should be exercised when extrapolating the DEX suppression test results obtained in the healthy sample to the PTSD sample. Fourth, this study included only female participants, which may have affected the findings. For instance, HPA axis function is shown to be different between men and women (74) and the heritability of PTSD is suggested to be different between sexes (75). Thus, our findings might represent some unique features of women with PTSD. Fifth, data of several participants were unavailable or excluded for some biomarkers and SNPs, which might have affected the results. Sixth, while the amount of liquid consumption can potentially influence the RAA system, such data were not collected in this study. Seventh, we used the DSM-IV-based scale (i.e., PDS), but not an updated DSM-5-based one, for the diagnosis of PTSD. While this was because we were not aware of any Japanese version of DSM-5-based scale for the diagnosis of PTSD at the time of the study initiation, differences in diagnostic criteria such as the absence and presence of the “negative alterations in cognitions and mood” criterion between DSM-IV and DSM-5 (respectively) might have affected the results. Finally, since most of our patients were receiving psychotropics, the possibility cannot be ruled out that such medication may have influenced the HPA axis and/or RAA system, even though we did not observe any significant relationship between medication and hormonal levels except for the relation between the use of mood stabilizers and cortisol levels.
In summary, this study shows that lower DHEA-S levels could indicate more severe subtype of PTSD, the association of which might be partly modified by the CACNA1C genetic polymorphism. Our findings suggest an involvement of DHEA-S in the pathophysiology of PTSD and further point to the importance of considering genetic factors in investigating stress response alterations in trauma and PTSD. Since neuroendocrine responses to stress depend on developmental timing, duration, time of day, and nature of stressors (76), future studies that address these issues would be necessary to get the bigger picture of HPA axis dysfunction in trauma-related conditions.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/clinvar/, SCV002587063 (for rs1360780) and SCV002588462 (for rs1006737).
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of National Center of Neurology and Psychiatry, Ethics Committee of Tokyo Women’s Medical University, and Ethics committee of Nagoya City University. The patients/participants provided their written informed consent to participate in this study.
Author contributions
RK and HH designed the study, undertook the statistical analyses, and wrote the draft of the manuscript. RK, HH, MI, ML, MaN, MeN, KI, RI, DS, and YK collected the data. FY performed the genotyping. DS, TK, HK, and YK gave critical comments on the manuscript. All authors contributed to the article and approved the final manuscript.
Funding
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI (20K07937 to HH and 19H01047 to YK), a grant from Innovative Research Program on Suicide Countermeasures (3–2 and 2-1 to YK), and Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (30-1 to HK and 2-2 to YK).
Acknowledgments
We thank all participants who took part in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.967779/full#supplementary-material
References
1. Daskalakis N, Cohen H, Nievergelt C, Baker D, Buxbaum J, Russo S, et al. New translational perspectives for blood-based biomarkers of PTSD: From glucocorticoid to immune mediators of stress susceptibility. Exp Neurol. (2016) 284:133–40. doi: 10.1016/j.expneurol.2016.07.024
2. Hori H, Kim Y. Inflammation and post-traumatic stress disorder. Psychiatry Clin Neurosci. (2019) 73:143–53. doi: 10.1111/pcn.12820
3. Meewisse M, Reitsma J, De Vries G, Gersons B, Olff M. Cortisol and post-traumatic stress disorder in adults: systematic review and meta-analysis. Br J Psychiatry. (2007) 191:387–92. doi: 10.1192/bjp.bp.106.024877
4. Morris M, Compas B, Garber J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: a systematic review and meta-analysis. Clin Psychol Rev. (2012) 32:301–15. doi: 10.1016/j.cpr.2012.02.002
5. Klaassens E, Giltay E, Cuijpers P, van Veen T, Zitman F. Adulthood trauma and HPA-axis functioning in healthy subjects and PTSD patients: a meta-analysis. Psychoneuroendocrinology. (2012) 37:317–31. doi: 10.1016/j.psyneuen.2011.07.003
6. Schumacher S, Niemeyer H, Engel S, Cwik J, Laufer S, Klusmann H, et al. HPA axis regulation in posttraumatic stress disorder: a meta-analysis focusing on potential moderators. Neurosci Biobehav Rev. (2019) 100:35–57. doi: 10.1016/j.neubiorev.2019.02.005
7. van Zuiden M, Haverkort S, Tan Z, Daams J, Lok A, Olff M. DHEA and DHEA-S levels in posttraumatic stress disorder: a meta-analytic review. Psychoneuroendocrinology. (2017) 84:76–82. doi: 10.1016/j.psyneuen.2017.06.010
8. Baulieu E, Robel P. Dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) as neuroactive neurosteroids. Proc Natl Acad Sci USA. (1998) 95:4089–91. doi: 10.1073/pnas.95.8.4089
9. Regelson W, Kalimi M. Dehydroepiandrosterone (DHEA)–the multifunctional steroid. II. Effects on the CNS, cell proliferation, metabolic and vascular, clinical and other effects. Mechanism of action? Ann NY Acad Sci. (1994) 719:564–75. doi: 10.1111/j.1749-6632.1994.tb56860.x
10. Maurice T, Grégoire C, Espallergues J. Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol Biochem Behav. (2006) 84:581–97. doi: 10.1016/j.pbb.2006.07.009
11. Maninger N, Wolkowitz O, Reus V, Epel E, Mellon S. Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front Neuroendocrinol. (2009) 30:65–91. doi: 10.1016/j.yfrne.2008.11.002
12. Browne E, Wright B, Porter J, Svec F. Dehydroepiandrosterone: antiglucocorticoid action in mice. Am J Med Sci. (1992) 303:366–71. doi: 10.1097/00000441-199206000-00003
13. Wolf O, Köster B, Kirschbaum C, Pietrowsky R, Kern W, Hellhammer D, et al. A single administration of dehydroepiandrosterone does not enhance memory performance in young healthy adults, but immediately reduces cortisol levels. Biol Psychiatry. (1997) 42:845–8. doi: 10.1016/s0006-322300323-5
14. Charney D. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry. (2004) 161:195–216. doi: 10.1176/appi.ajp.161.2.195
15. Yehuda R, Brand S, Golier J, Yang R. Clinical correlates of DHEA associated with post-traumatic stress disorder. Acta Psychiatr Scand. (2006) 114:187–93. doi: 10.1111/j.1600-0447.2006.00801.x
16. Kellner M, Muhtz C, Peter F, Dunker S, Wiedemann K, Yassouridis A. Increased DHEA and DHEA-S plasma levels in patients with post-traumatic stress disorder and a history of childhood abuse. J Psychiatr Res. (2010) 44:215–9. doi: 10.1016/j.jpsychires.2009.08.009
17. Kanter ED, Wilkinson C, Radant A, Petrie E, Dobie D, McFall M, et al. Glucocorticoid feedback sensitivity and adrenocortical responsiveness in posttraumatic stress disorder. Biol Psychiatry. (2001) 50:238–45. doi: 10.1016/s0006-322301158-1
18. Olff M, Güzelcan Y, de Vries G, Assies J, Gersons B. HPA- and HPT-axis alterations in chronic posttraumatic stress disorder. Psychoneuroendocrinology. (2006) 31:1220–30. doi: 10.1016/j.psyneuen.2006.09.003
19. Bremner D, Vermetten E, Kelley M. Cortisol, dehydroepiandrosterone, and estradiol measured over 24 hours in women with childhood sexual abuse-related posttraumatic stress disorder. J Nerv Ment Dis. (2007) 195:919–27. doi: 10.1097/NMD.0b013e3181594ca0
20. Grippo A, Johnson A. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress Amst Neth. (2009) 12:1–21. doi: 10.1080/10253890802046281
21. El Ghorayeb N, Bourdeau I, Lacroix A. Role of ACTH and other hormones in the regulation of aldosterone production in primary aldosteronism. Front Endocrinol. (2016) 7:72. doi: 10.3389/fendo.2016.00072
22. Khoury N, Marvar P, Gillespie C, Wingo A, Schwartz A, Bradley B, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. (2012) 73:17451. doi: 10.4088/JCP.11m07316
23. Seligowski A, Duffy L, Merker J, Michopoulos V, Gillespie C, Marvar P, et al. The renin-angiotensin system in PTSD: a replication and extension. Neuropsychopharmacol. (2021) 46:750–5. doi: 10.1038/s41386-020-00923-1
24. Reinecke A, Browning M, Klein Breteler J, Kappelmann N, Ressler K, Harmer C, et al. Angiotensin regulation of amygdala response to threat in high-trait-anxiety individuals. Biol Psychiatry Cogn Neurosci Neuro. (2018) 3:826–35. doi: 10.1016/j.bpsc.2018.05.007
25. Parrish J, Bertholomey M, Pang H, Speth R, Torregrossa M. Estradiol modulation of the renin–angiotensin system and the regulation of fear extinction. Transl Psychiatry. (2019) 9:1–12. doi: 10.1038/s41398-019-0374-0
26. Yu Z, Swiercz A, Moshfegh C, Hopkins L, Wiaderkiewicz J, Speth R, et al. Angiotensin II type 2 receptor-expressing neurons in the central amygdala influence fear-related behavior. Biol Psychiatry. (2019) 86:899–909. doi: 10.1016/j.biopsych.2019.05.027
27. Terock J, Hannemann A, Janowitz D, Freyberger H, Felix S, Dörr M, et al. Associations of trauma exposure and post-traumatic stress disorder with the activity of the renin-angiotensin-aldosterone-system in the general population. Psychol Med. (2019) 49:843–51. doi: 10.1017/S0033291718001496
28. Terock J, Hannemann A, Janowitz D, Van der Auwera S, Bahls M, Völzke H, et al. Differential activation of the renin-angiotensin-aldosterone-system in response to childhood and adulthood trauma. Psychoneuroendocrinology. (2019) 107:232–40. doi: 10.1016/j.psyneuen.2019.05.026
29. Terock J, Hannemann A, Klinger-König J, Janowitz D, Grabe H, Murck H. The neurobiology of childhood trauma—aldosterone and blood pressure changes in a community sample. World J Biol Psychiatry. (2021) 0:1–9. doi: 10.1080/15622975.2021.2018724
30. Nishimi K, Adler G, Roberts A, Sumner J, Jung S, Chen Q, et al. Associations of trauma and posttraumatic stress disorder with aldosterone in women. Psychoneuroendocrinology. (2021) 132:105341. doi: 10.1016/j.psyneuen.2021.105341
31. Bartels M, Van den Berg M, Sluyter F, Boomsma D, de Geus E. Heritability of cortisol levels: review and simultaneous analysis of twin studies. Psychoneuroendocrinology. (2003) 28:121–37. doi: 10.1016/s0306-453000003-3
32. Binder E. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. (2009) 34:S186–95. doi: 10.1016/j.psyneuen.2009.05.021
33. Matosin N, Halldorsdottir T, Binder E. Understanding the molecular mechanisms underpinning gene by environment interactions in psychiatric disorders: the FKBP5 model. Biol Psychiatry. (2018) 83:821–30. doi: 10.1016/j.biopsych.2018.01.021
34. Bavley C, Kabir Z, Walsh A, Kosovsky M, Hackett J, Sun H, et al. Dopamine D1R-neuron cacna1c deficiency: a new model of extinction therapy-resistant post-traumatic stress. Mol Psychiatry. (2021) 26:2286–98. doi: 10.1038/s41380-020-0730-8
35. Klaus K, Butler K, Gutierrez H, Durrant S, Pennington K. Interactive effects of early life stress and CACNA1C genotype on cortisol awakening response. Biol Psychol. (2018) 136:22–8. doi: 10.1016/j.biopsycho.2018.05.002
36. Bhat S, Dao D, Terrillion C, Arad M, Smith R, Soldatov N, et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. (2012) 99:1–14. doi: 10.1016/j.pneurobio.2012.06.001
37. Ferreira M, O’Donovan M, Meng Y, Jones I, Ruderfer D, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. (2008) 40:1056–8. doi: 10.1038/ng.209
38. Green E, Grozeva D, Jones I, Jones L, Kirov G, Caesar S, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry. (2010) 15:1016–22. doi: 10.1038/mp.2009.49
39. Hori H, Yamamoto N, Fujii T, Teraishi T, Sasayama D, Matsuo J, et al. Effects of the CACNA1C risk allele on neurocognition in patients with schizophrenia and healthy individuals. Sci Rep. (2012) 2:634. doi: 10.1038/srep00634
40. Roussos P, Mitchell A, Voloudakis G, Fullard J, Pothula V, Tsang J, et al. A role for non-coding variation in schizophrenia. Cell Rep. (2014) 9:1417–29. doi: 10.1016/j.celrep.2014.10.015
41. Gershon E, Grennan K, Busnello J, Badner J, Ovsiew F, Memon S, et al. A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brain. Mol Psychiatry. (2014) 19:890–4. doi: 10.1038/mp.2013.107
42. Grossman R, Yehuda R, New A, Schmeidler J, Silverman J, Mitropoulou V, et al. Dexamethasone suppression test findings in subjects with personality disorders: associations with posttraumatic stress disorder and major depression. Am J Psychiatry. (2003) 160:1291–8. doi: 10.1176/appi.ajp.160.7.1291
43. Yehuda R, Golier J, Halligan S, Meaney M, Bierer L. The ACTH response to dexamethasone in PTSD. Am J Psychiatry. (2004) 161:1397–403. doi: 10.1176/appi.ajp.161.8.1397
44. Nagae N, Hirohata S, Shimura Y, Yamada S, Foa E, Nedate K, et al. Development of the Japanese version of the posttraumatic diagnostic scale: ascertaining its reliability and validity among university students [in Japanese]. JPN J Trauma Stress. (2007) 5:51–6.
45. Foa E. Posttraumatic Diagnostic Scale (PDS) Manual. Minneapolis, MN: National Computer Systems (1995).
46. Otsubo T, Tanaka K, Koda R, Shinoda J, Sano N, Tanaka S, et al. Reliability and validity of Japanese version of the mini-international neuropsychiatric interview. Psychiatry Clin Neurosci. (2005) 59:517–26. doi: 10.1111/j.1440-1819.2005.01408.x
47. Sheehan D, Lecrubier Y, Sheehan K, Amorim P, Janavs J, Weiller E, et al. The mini-international neuropsychiatric interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. (1998) 59:22–33.
48. Itoh M, Ujiie Y, Nagae N, Niwa M, Kamo T, Lin M, et al. The Japanese version of the posttraumatic diagnostic scale: validity in participants with and without traumatic experiences. Asian J Psychiatry. (2017) 25:1–5. doi: 10.1016/j.ajp.2016.09.006
49. Bernstein D, Stein J, Newcomb M, Walker E, Pogge D, Ahluvalia T, et al. Development and validation of a brief screening version of the childhood trauma questionnaire. Child Abuse Negl. (2003) 27:169–90. doi: 10.1016/S0145-213400541-0
50. Nakajima M, Hori H, Itoh M, Lin M, Kawanishi H, Narita M, et al. Validation of childhood trauma questionnaire-short form in Japanese clinical and nonclinical adults. Psychiatry Res Commun. (2022) 2:100065. doi: 10.1016/j.psycom.2022.100065
51. Scher C, Stein M, Asmundson G, McCreary D, Forde D. The childhood trauma questionnaire in a community sample: psychometric properties and normative data. J Trauma Stress. (2001) 14:843–57. doi: 10.1023/A:1013058625719
52. Gerdner A, Allgulander C. Psychometric properties of the Swedish version of the childhood trauma questionnaire-short form (CTQ-SF). Nord J Psychiatry. (2009) 63:160–70. doi: 10.1080/08039480802514366
53. Bernstein D, Fink L. Childhood trauma questionnaire: a retrospective self-report. San Antonio, TX: The Psychological Corporation (1998).
54. Beck A, Steer R, Brown G. BDI-II, beck depression inventory: manual. San Antonio, TX: Psychological Corp (1996).
55. Kojima M, Furukawa T, Takahashi H, Kawai M, Nagaya T, Tokudome S. Cross-cultural validation of the beck depression inventory-II in Japan. Psychiatry Res. (2002) 110:291–9. doi: 10.1016/S0165-178100106-3
56. Spielberger C, Gorsuch R, Lushene R. Manual for the State-Trait Anxiety Inventory. Palo Alto: Consulting Psychologist Press (1970).
57. Nakazato K, Mizuguchi T. Development and validation of Japanese version of state-trait anxiety inventory. JPN J Psychosom Intern Med. (1982) 22:107–12.
58. Sheehan D, Harnett-Sheehan K, Raj B. The measurement of disability. Int Clin Psychopharmacol. (1996) 11:89–95. doi: 10.1097/00004850-199606003-00015
59. Yoshida T, Ohtsubo T, Tsuchida H, Wada Y, Kamijima K, Fukui K. Study of reliability and validity of a Japanese version of Sheehan Disability Scale (SDISS) (in Japanese). JPN J Clin Psychopharmacol. (2004) 7:1645–53.
60. Myers L., Sirois M. Spearman correlation coefficients, differences between. Encyclopedia of statistical sciences. Hoboken, NJ: Wiley (2006).
61. Fujii T, Hori H, Ota M, Hattori K, Teraishi T, Sasayama D, et al. Effect of the common functional FKBP5 variant (rs1360780) on the hypothalamic-pituitary-adrenal axis and peripheral blood gene expression. Psychoneuroendocrinology. (2014) 42:89–97. doi: 10.1016/j.psyneuen.2014.01.007
62. Savic D, Knezevic G, Matic G, Damjanovic S. PTSD and depressive symptoms are linked to DHEAS via personality. Psychoneuroendocrinology. (2018) 92:29–33. doi: 10.1016/j.psyneuen.2018.03.017
63. Michopoulos V, Powers A, Gillespie C, Ressler K, Jovanovic T. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. (2017) 42:254–70. doi: 10.1038/npp.2016.146
64. Cao J, Li Q, Shen X, Yao Y, Li L, Ma H. Dehydroepiandrosterone attenuates LPS-induced inflammatory responses via activation of Nrf2 in RAW264.7 macrophages. Mol Immunol. (2021) 131:97–111. doi: 10.1016/j.molimm.2020.12.023
65. Zhang M, Zhang M, Wang W, Chen H, Wang X, Zhao K, et al. Dehydroepiandrosterone inhibits vascular proliferation and inflammation by modulating the miR-486a-3p/NLRP3 axis. Am J Transl Res. (2022) 14:6123–36.
66. Karst H, Nair S, Velzing E, Rumpff-van Essen L, Slagter E, Shinnick-Gallagher P, et al. Glucocorticoids alter calcium conductances and calcium channel subunit expression in basolateral amygdala neurons. Eur J Neurosci. (2002) 16:1083–9. doi: 10.1046/j.1460-9568.2002.02172.x
67. Qin Y, Karst H, Joëls M. Chronic unpredictable stress alters gene expression in rat single dentate granule cells. J Neurochem. (2004) 89:364–74. doi: 10.1111/j.1471-4159.2003.02332.x
68. van Gemert N, Joëls M. Effect of chronic stress and mifepristone treatment on voltage-dependent Ca2+ currents in rat hippocampal dentate gyrus. J Neuroendocrinol. (2006) 18:732–41. doi: 10.1111/j.1365-2826.2006.01472.x
69. Yamaguchi-Kabata Y, Nariai N, Kawai Y, Sato Y, Kojima K, Tateno M, et al. iJGVD: an integrative Japanese genome variation database based on whole-genome sequencing. Hum Genome Var. (2015) 2:1–4. doi: 10.1038/hgv.2015.50
70. Koenen K, Nugent N, Amstadter A. Gene-environment interaction in posttraumatic stress disorder: review, strategy and new directions for future research. Eur Arch Psychiatry Clin Neurosci. (2008) 258:82–96. doi: 10.1007/s00406-007-0787-2
71. Siegel C, Laska E, Lin Z, Xu M, Abu-Amara D, Jeffers M, et al. Utilization of machine learning for identifying symptom severity military-related PTSD subtypes and their biological correlates. Transl Psychiatry. (2021) 11:227. doi: 10.1038/s41398-021-01324-8
72. Gelernter J, Sun N, Polimanti R, Pietrzak R, Levey D, Bryois J, et al. Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nat Neurosci. (2019) 22:1394–401. doi: 10.1038/s41593-019-0447-7
73. Stein M, Levey D, Cheng Z, Wendt F, Harrington K, Pathak G, et al. Genome-wide association analyses of post-traumatic stress disorder and its symptom subdomains in the million veteran program. Nat Genet. (2021) 53:174–84. doi: 10.1038/s41588-020-00767-x
74. Rampp C, Eichelkraut A, Best J, Czamara D, Rex-Haffner M, Uhr M, et al. Sex-related differential response to dexamethasone in endocrine and immune measures in depressed in-patients and healthy controls. J Psychiatr Res. (2018) 98:107–15. doi: 10.1016/j.jpsychires.2017.12.020
75. Sartor C, McCutcheon V, Pommer N, Nelson E, Grant J, Duncan A, et al. Common genetic and environmental contributions to post-traumatic stress disorder and alcohol dependence in young women. Psychol Med. (2011) 41:1497–505. doi: 10.1017/S0033291710002072
Keywords: posttraumatic stress disorder (PTSD), childhood maltreatment, hypothalamic-pituitary-adrenal (HPA) axis, renin-angiotensin-aldosterone (RAA) system, dehydroepiandrosterone-sulphate (DHEA-S), gene, polymorphism
Citation: Kakehi R, Hori H, Yoshida F, Itoh M, Lin M, Niwa M, Narita M, Ino K, Imai R, Sasayama D, Kamo T, Kunugi H and Kim Y (2023) Hypothalamic-pituitary-adrenal axis and renin-angiotensin-aldosterone system in adulthood PTSD and childhood maltreatment history. Front. Psychiatry 13:967779. doi: 10.3389/fpsyt.2022.967779
Received: 13 June 2022; Accepted: 16 December 2022;
Published: 09 January 2023.
Edited by:
Philip Gold, National Institute of Mental Health (NIH), United StatesReviewed by:
T. John Wu, Uniformed Services University of the Health Sciences, United StatesNathaniel Harnett, McLean Hospital, United States
Juliann Purcell, McLean Hospital, United States
Copyright © 2023 Kakehi, Hori, Yoshida, Itoh, Lin, Niwa, Narita, Ino, Imai, Sasayama, Kamo, Kunugi and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroaki Hori,  aG9yaUBuY25wLmdvLmpw
aG9yaUBuY25wLmdvLmpw