 Thomas Hartwig Siebner1*
Thomas Hartwig Siebner1* Karen Sandø Ambrosen1
Karen Sandø Ambrosen1 Cecilie Koldbæk Lemvigh1
Cecilie Koldbæk Lemvigh1 Christine Natasha Ryan2
Christine Natasha Ryan2 Mikkel Erlang Sørensen1María Hernández-Lorca1
Mikkel Erlang Sørensen1María Hernández-Lorca1 Birte Yding Glenthøj1,3
Birte Yding Glenthøj1,3 Kit Melissa Larsen4Michael-Robin Witt2
Kit Melissa Larsen4Michael-Robin Witt2 Bob Oranje1
Bob Oranje1 Bjørn Hylsebeck Ebdrup1,3
Bjørn Hylsebeck Ebdrup1,3- 1Center for Neuropsychiatric Schizophrenia Research (CNSR), Mental Health Center, Glostrup, Copenhagen University Hospital, Mental Health Services CPH, Copenhagen, Denmark
- 2Gabather AB, Södertälje, Sweden
- 3Department of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 4Danish Research Centre for Magnetic Resonance, Department of Radiology and Nuclear Medicine, Copenhagen University Hospital - Amager and Hvidovre, Copenhagen, Denmark
Background: Cognitive impairment remains a critical unmet treatment need in schizophrenia spectrum disorders (SSD). Disruption of cortical excitation/inhibition balance, involving dysfunction of the gamma-aminobutyric acid (GABA) system, leads to aberrant gamma oscillations and impaired brain network function. This disruption may manifest as hypofrontality, which is associated with deficits in basic information processing thought to underlie the cognitive impairments observed in SSD. GT-002 is a novel GABAA receptor partial positive allosteric modulator. Preclinical rodent studies have demonstrated GT-002’s potential to reduce hypofrontality, while three Phase I trials have established its safety and tolerability in healthy participants.
Aim: The TOTEMS Phase II trial examines acute effects of a single oral dose of GT-002 on psychophysiological measures of early information processing, including event-related electroencephalography (EEG), electromyography, and resting-state EEG in SSD patients.
Methods: A single-center, double-blind, placebo- and active comparator-controlled, randomized, four-way crossover challenge trial. We will recruit 20 clinically stable patients with SSD and 30 healthy controls. Participants will receive a single dose of GT-002 (1 mg and 2 mg, developed by Gabather AB), oxazepam (15 mg), and placebo across four study drug exposure days, separated by a washout period ≥7 days. Psychophysiological measures and cognitive assessments, including the Trail Making Test and selected subtests from the Brief Assessment of Cognition in Schizophrenia and Cambridge Neuropsychological Test Automated Battery, will be conducted following each administration.
Anticipated results: We hypothesize that GT-002 will improve prepulse inhibition of the startle reflex in patients relative to placebo and oxazepam, reflecting improved sensorimotor gating. Secondary hypotheses include improved mismatch negativity, selective attention, 40-Hz auditory steady-state response, and normalized resting-state EEG in SSD patients following GT-002. Exploratory endpoints include safety and tolerability of GT-002 as well as differential effects on cognition compared to oxazepam, particularly in processing speed, attention, reaction time, and working memory.
Perspectives: TOTEMS is the first trial to investigate acute effects of GABAA receptor modulation by GT-002 on early information processing in SSD. If successful, it will support further clinical trials of longer-term GT-002 treatment as a novel pharmacological approach to target impairments in information processing in SSD, potentially ameliorating cognitive impairments.
Clinical trial registration: EU CT number 2024-519389-28-00.
1 Introduction
Schizophrenia spectrum disorders (SSD) are severe and debilitating mental illnesses associated with substantial impairments in real-world functioning and markedly reduced quality of life (1–3). Deficits in basic information processing are thought to underlie cognitive impairments, which represent one of the core dimensions of SSD (4, 5) and has been linked to both real-world functional impairment (6–9), and lower quality of life (10). This has led the international scientific community to propose the term ‘Cognitive Impairment Associated with Schizophrenia’ (CIAS) (11). At present, no pharmacological agent has received regulatory approval specifically for the treatment of CIAS, nor is any medication currently recommended to improve CIAS in any international guidelines (12–16). Nevertheless, multiple compounds have been investigated or are undergoing evaluation for their potential efficacy in ameliorating CIAS (17, 18), targeting diverse neurobiological mechanisms that reflect the multifactorial etiology of CIAS. However, the treatment remains complex as many different neurobiological mechanisms likely underly CIAS, such as hypofrontality, excitatory and inhibitory (E/I) imbalance at a cortical level, altered neuronal functioning and neurotransmission, grey matter volume reduction and aberrant neural network organization (19–22).
Several neurotransmitter systems and neural circuits, including the gamma aminobutyric acid (GABA), glutamatergic, dopaminergic and muscarinic pathways, converge on the regulation of E/I balance within cortical circuits (5). In these cortical circuits, the excitatory output of cortical pyramidal cells is regulated by inhibition from GABAergic interneurons (5, 20). This finely tuned balance between excitatory glutamatergic pyramidal cells and inhibitory GABAergic interneurons generates synchronized neural oscillations. Particularly, the neural oscillations occurring at approximately 40 Hz, termed ‘gamma oscillations’, seem essential to the generation of slow fluctuations in neural activity, as observed with functional magnetic resonance imaging (fMRI), that underlie functional brain networks (23, 24). In healthy individuals, these neural oscillations and functional networks have been associated with various cognitive functions, including working memory (25, 26). Multimodal evidence indicates that this E/I balance is disrupted in schizophrenia, thereby resulting in aberrant gamma oscillations that lead to brain network dysfunction (5, 20) and may manifest as hypofrontality, reflected by reduced glucose metabolism and cerebral blood flow in the prefrontal cortex (19, 27, 28). In turn, hypofrontality has been associated with disturbances in basic information processing in schizophrenia, and both factors are thought to play a role in the observed cognitive impairments (19, 27–31), although the precise causal relationships remain incompletely understood. Notably, hypofrontality is not ameliorated by treatment with the currently available antipsychotics and may even be exacerbated by such treatment (28). Several studies have indicated that antagonists of N-methyl-D-aspartate (NMDA) receptors, such as phencyclidine (PCP), ketamine and dizocilpine (MK-801) induce states of hypofrontality (27, 28, 32). This induced hypofrontality is likely underlying the schizophrenia-like deficits in electrophysiological parameters of early information processing observed in our previous studies on the effects of ketamine in healthy controls. Specifically, we found that ketamine reduced the P300 amplitude as well as processing negativity (PN) and mismatch negativity (MMN) all of which are event-related potentials (ERPs) (33, 34). When the results from both studies were combined, we also observed reductions in sensory and sensorimotor gating (35, 36). Moreover, we demonstrated deficits in sensorimotor gating, as measured by prepulse inhibition of the startle reflex (PPI), in drug-naive first-episode patients with schizophrenia (37).
Psychophysiological measures, including electroencephalography (EEG) and electromyography (EMG), are widely used to quantify neural mechanisms underlying early information processing. Examples include sensorimotor gating by PPI (38), pre-attentive sensory discrimination by MMN (39), and selective attention (SA) by the P300 amplitude (40). Patients with schizophrenia show abnormalities in all three indices compared to healthy controls (38–40), whether treated with antipsychotics (41–44) or not (37, 41, 45–50). We previously showed that the electrophysiological phenomena are related to several higher-order cognitive functions, e.g., strategy formation, visual short-term memory, verbal fluency, and cognitive inhibition and flexibility (51). In spectral analysis of resting-state EEG recordings, schizophrenia is characterized by increased delta and theta activity (52), which has been associated with dysfunctional processing of sensory input (53). Additionally, patients with schizophrenia exhibit abnormalities in the alpha frequency band (52), indicative of the above-mentioned hypofrontality (54, 55). Abnormalities are also observed in the gamma frequency band (56), involved in neuronal synchronization in both local and large-scale neuronal networks underlying a large range of perceptual and higher-order cognitive functions commonly disrupted in schizophrenia (57–59).
Studies in rodents and in humans support the involvement of the GABAergic system in the regulation of sensorimotor gating, as measured by PPI (60–64), although the dopaminergic, serotonergic, and glutamatergic systems are also involved (65). Pre-attentive auditory processing, as indexed by MMN, is thought to reflect glutamatergic NMDA receptor function and E/I balance (66, 67). Rowland et al. further provided in vivo evidence supporting glutamatergic and GABAergic regulation of MMN and verbal working memory function in schizophrenia (67). The 40-Hz auditory steady-state response (ASSR) provides a noninvasive measure of the ability to generate neural synchrony in the gamma range within the auditory system. Emerging evidence suggests that GABAergic neurotransmission modulates 40-Hz ASSRs and is a sensitive marker for E/I balance alterations (68–70). Several studies have investigated 40-Hz ASSRs in patients with schizophrenia, with the majority reporting 40-Hz ASSR deficits with medium-level effect sizes (69, 71, 72). In resting-state EEG of healthy participants, the benzodiazepine oxazepam is known to reduce the power of low-frequency waves, i.e., delta, theta, and alpha bands (73–76), while increasing the activity in the higher frequency ranges, i.e., beta band (73, 74, 77). The latter effect was statistically significant in as few as five participants when a single dose of 30 mg oxazepam was administered (77). Besides this activity on resting-state EEG, oxazepam is also known to reduce amplitudes of ERPs, especially of the P300 amplitude (78, 79).
Dysfunction and/or loss of parvalbumin-positive GABAergic interneurons has been proposed to disrupt the E/I balance and contribute to a diminished capacity for the gamma-frequency synchronized neuronal activity in schizophrenia (5, 71, 80–83). Accordingly, GABAergic inhibitory neurons have been suggested as potential therapeutic targets for cognitive deficits (84). The GABAA receptors represent the most prevalent subtype in the central nervous system and regulate circuit activity through distinct modes of inhibition based on their localization (84–86). GABAA receptors are highly expressed on postsynaptic neuronal membranes opposite to GABA releasing presynaptic nerve terminals but are also present extrasynaptically along the dendritic membrane. Synaptic GABAA receptors respond to high concentrations of synaptically released GABA and mediate fast, short-lasting phasic inhibition. This form of inhibition provides timing-based signaling that defines the temporal window for neuronal network firing, thereby playing a key role in the generation and regulation of gamma or theta oscillations, as well as in maintaining network synchrony. In contrast, extrasynaptic GABAA receptors respond to consistent, low concentrations of GABA and mediate slow, long-lasting tonic inhibition, which regulates neuronal excitability by modulating the amplitude and duration of postsynaptic excitatory potentials (84–86).
The GABAA receptors are heteropentameric, ligand-gated chloride (Cl−) channels composed of different subunits that form a ring around a central ion-conducting pore in the membrane (84–86). The subunits of the GABAA receptor family, encoded by 19 known genes, include α1-6, β1-3, γ1-3, δ, ϵ, π, θ, and ρ1-3. GABAA receptors are generally composed of two α subunits, two β subunits, and either a γ2 or δ subunit, where the vast majority are believed to constitute of three receptor subtypes (α1β2/3γ2, α2β2/3γ2 and α3β2/3γ2) (84–86). The precise subunit composition is a major determinant of the functional characteristics of the receptor, including sensitivity to GABA, conductance, desensitisation, spatiotemporal distributions, and sensitivity to allosteric modulator (84–86).
GABA binds to the GABAA receptors at the interface between the α and β subunits, enhancing Cl− conductance across the membrane (Figure 1). This leads to hyperpolarization of the postsynaptic membrane, hence reducing the probability that postsynaptic neurons will generate an action potential.
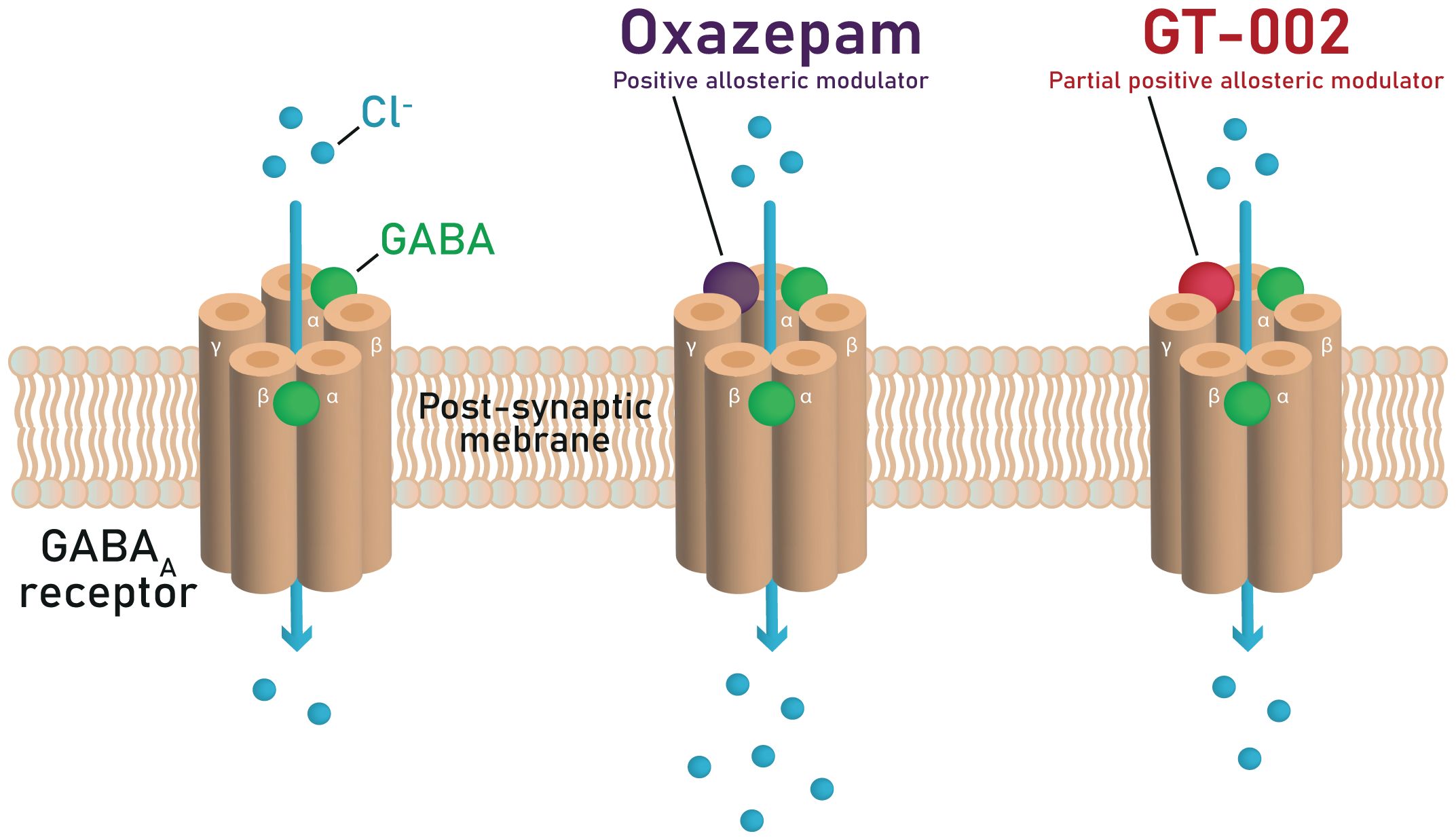
Figure 1. Mechanisms of GABA potentiation by oxazepam and GT-002 at the GABAA receptor, illustrating that GT-002 induces only a minor enhancement of GABA-elicited Cl− currents compared to oxazepam.
Benzodiazepines such as diazepam and oxazepam are non-selective positive allosteric modulators (PAMs) of GABAA receptors (84, 86, 87). They enhance GABA’s action at GABAA receptors by interacting with the allosteric modulatory benzodiazepine binding site formed by one of the α subunits (α1–3 or α5) and the γ2 subunit (Figure 1). This binding increases the frequency of the Cl- channel opening in the presence of GABA, thereby increasing Cl- conductance across the neuronal cell membrane and enhancing inhibitory neurotransmission. It has been suggested that benzodiazepines can mediate different effects depending upon the GABAA receptor subtype in distinct neuronal circuits targeted (87–91). For example, the sedative and anterograde amnestic actions of benzodiazepines are thought to be mediated by α1-containing GABAA receptors (87, 88), the anxiolytic activity by α2-containing GABAA receptors (89), and the muscle relaxant activity by α2, α3, and α5 containing GABAA receptors (90, 91).
Several GABAA receptor modulators with diverse subunit selectivity profiles, mechanisms of action, and therapeutic indications are currently in development, as recently summarized by Thompson et al. (86). Most of these agents target epilepsy, anxiety, or depression, with relatively few explicitly targeting CIAS. GT-002 distinguishes itself within this landscape as a partial PAM at the GABAA receptor, designed to potentiate GABAergic transmission to a limited extent while minimizing sedation, thereby providing a mechanistically novel approach to CIAS. Beyond GABAergic dysfunction, multiple neurotransmitter systems and pathways may be implicated, including glutamatergic, cholinergic, dopaminergic, and inflammatory mechanisms. Reflecting this complexity, compounds with distinct mechanisms are under investigation for CIAS, such as positive allosteric modulation of the α7 nicotinic acetylcholine receptor (e.g., galantamine), inhibition of d-amino acid oxidase (e.g., luvadaxistat), a combination of muscarinic agonism at M1, M4, and M5 receptors with peripheral muscarinic antagonism (e.g., xanomeline-trospium), anti-inflammatory agents (e.g., minocycline, N-acetylcysteine), trace amine-associated receptor 1 agonism (e.g., ulotaront), and selective glycine transporter 1 inhibition (e.g., iclepertin) (17, 18). Collectively, these efforts illustrate the breadth of mechanisms being explored to address CIAS and underscore the novelty of GT-002’s approach within the GABAergic domain.
The TOTEMS trial investigates the acute effects of GT-002, a novel GABAA receptor partial PAM that produces minimal potentiation of GABA-elicited Cl− currents, as described in the Investigator’s Brochure (IB), distinguishing it pharmacodynamically from traditional benzodiazepines (Figure 1). This limited potentiation may underlie the absence of sedation typically associated with benzodiazepines, while preserving GABAergic enhancement, thereby offering potential for clinical applications. The trial employs a single-dose design to assess whether GT-002 produces measurable effects on EEG and EMG without inducing sedation. Our chosen measures represent sensitive, well-established markers of early sensory and sensorimotor information processing and provide electrophysiological indicators to assess an individual’s level of hypofrontality. Acute changes in these measures may indicate engagement of neural circuits underlying hypofrontality, providing an initial signal of potential therapeutic effects. From an ethical perspective, initiating a single-dose design in this first patient trial is appropriate, as it allows preliminary pharmacodynamic evaluation while minimizing exposure. While this single-dose trial does not directly target cognitive impairments in patients with SSD, measurable effects on EEG and EMG following single dosing would provide necessary preliminary pharmacodynamic evidence to justify subsequent repeated-dose trials aimed at evaluating potential improvements in CIAS or broader cognitive function.
2 Methods and analysis
2.1 Objective of the trial
The overall objective of the TOTEMS clinical trial is to investigate the acute effects of partial GABAA receptor modulation by GT-002 on psychophysiological measures, including event-related EEG and EMG, as well as resting-state EEG, in patients with SSD. Collectively, these measures provide sensitive and well-established markers of early sensory and sensorimotor information processing deficits, which are central to the pathophysiology of SSD, and serve as electrophysiological proxies for evaluating GT-002’s effect on hypofrontality. The trial endpoints are described in detail in Table 1.
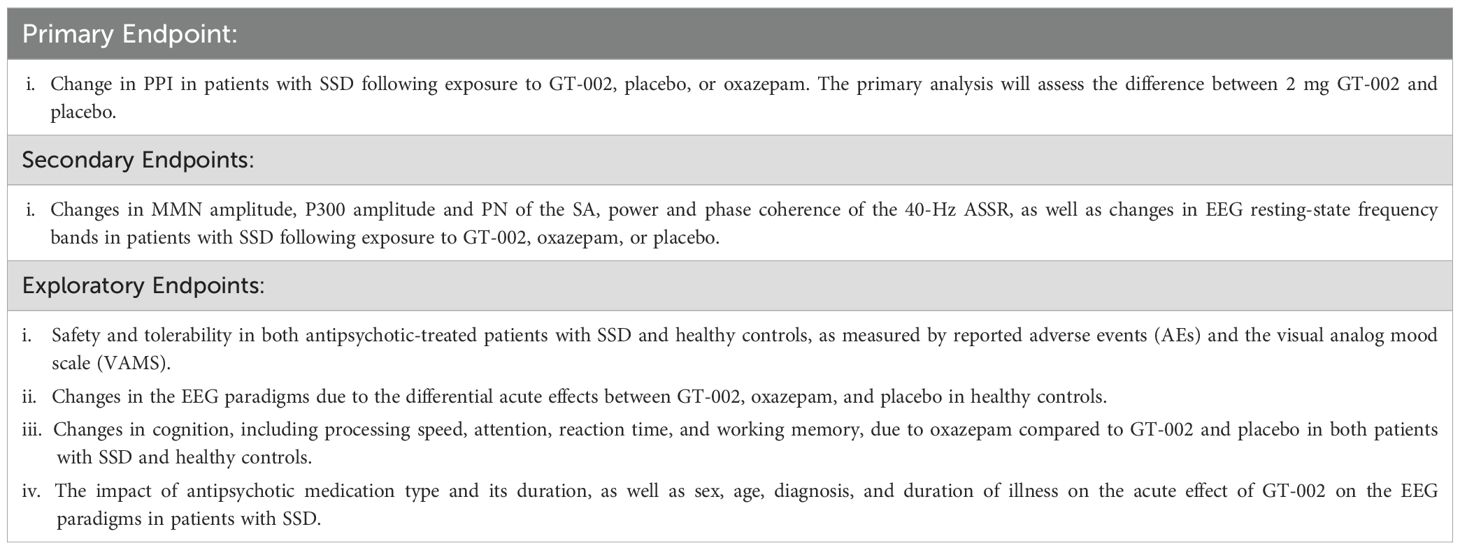
Table 1. Description of the endpoints of the trial.
2.2 Trial population
We aim to recruit a total of 50 participants aged 18–45 years, distributed as 30 healthy individuals with no current or past mental disorders or severe physical conditions, and 20 patients diagnosed with SSD without severe physical conditions. We aim for a balanced distribution of age and sex in both groups. Healthy participants will be recruited through advertisements. Patients will primarily be recruited through outpatient clinics in the Capital Region of Denmark and Region Zealand.
Detailed inclusion and exclusion criteria for participants are described in Table 2, and rules for concomitant treatments and medications before and during the trial are presented in Table 3.

Table 2. Description of the inclusion and exclusion criteria.

Table 3. Rules for concomitant treatments and medications before and during the trial.
2.3 Study design
The TOTEMS clinical Phase II trial follows a single-center, double-blind, placebo- and active-comparator-controlled, randomized four-way crossover design with single exposure.
Each participant will attend up to 8 visits at the Center for Neuropsychiatric Schizophrenia Research (CNSR), as outlined in Figure 2.

Figure 2. Overview of the eight visits conducted during the TOTEMS clinical trial, with time intervals indicated between each visit.
The visits include an information session, a screening visit (after which participants will be scheduled for the first study drug exposure within 3 to 42 days), followed by four study drug exposure days (each separated by a washout period of at least 7 days and no more than 56 days between them), and two final follow-ups. The first safety follow-up examination will take place 7 to 14 days after the last study drug exposure day (Visit 6), followed by a second safety follow-up telephone call 30–40 days after the first safety follow-up or study discontinuation.
We aim to schedule the first study drug exposure day following the availability of laboratory results from screening, as well as ensuring minimal intervals between subsequent study drug exposure days. The 56-day upper limit between study drug exposure days accommodates scheduling constraints, including holidays and participant availability. Consequently, we anticipate each trial subject’s participation to extend for a minimum of 9 weeks, with a theoretical maximum duration of 38 weeks. For a detailed description of each visit see Figures 3–6.

Figure 3. Overview of assessments and study procedures performed by each visit.

Figure 4. Timeline of Visit 2. Diagnostic codes include schizophrenia (ICD-10: F20.0), persistent delusional disorder (F22.x), acute and transient psychotic disorders (F23.x), induced delusional disorders (F24.x), schizoaffective disorders (F25.x), other non-organic psychotic disorders (F28), and unspecified non-organic psychosis (F29).

Figure 5. Timeline of Visits 3-6.
Data collection is scheduled to occur over a period of 27 months from the start of the trial.
Participants will receive a compensation of DKK 3,000 (≈ € 400) upon completion of the full study. If a participant chooses to withdraw before completion, reimbursement will cover only the visits completed. Patients will be provided with taxi transport to and from the study site for Visits 2-7, with the cost covered by the study. Alternatively, if patients prefer to use public transportation, these expenses will also be compensated.
2.4 Randomization and blinding procedure
Randomization of study drugs will be performed centrally at the clinical research unit of the Capital Region Pharmacy of Denmark and will be conducted separately for healthy participants and patients.
With four different study drug exposures and each participant receiving each study drug once, 24 possible administration sequences exist for randomization. The randomization codes for the study drug exposures will not be accessible to the TOTEMS investigators until data analysis for each group is complete.
Unblinding will occur in two phases: first, after all data from the healthy participants has been analyzed, and second, after all data from the patients has been analyzed. This approach accommodates the expected longer recruitment period for patients while maintaining blinding integrity. To ensure further objectivity in the data analysis, all data processing will be conducted automatically with batch jobs. This approach eliminates any potential subjectivity in the statistical interpretation or the scoring of the data.
Due to the potential alteration of GT-002’s pharmacokinetics, the GT-002 capsule cannot be encapsulated and differs significantly from oxazepam tablets. Additionally, producing a placebo identical to the 15 mg oxazepam tablet is not feasible given the formulation characteristics. Therefore, two types of placebos are required for this trial: the first placebo (for GT-002) is a soft gelatine capsule that is identical to the 1 mg GT-002 capsule, and the second (for oxazepam) is an encapsulated placebo tablet that is identical to the encapsulated oxazepam tablet. Both placebos will contain no active drug substance.
As GT-002 is supplied in 1 mg capsules, the 2 mg dose will be administered as two capsules. To ensure double blinding, each study drug exposure will include three capsules, as outlined in Figure 7: two yellow capsules (either GT-002 or matching placebo) and one brown-and-orange capsule (either encapsulated oxazepam tablet or matching encapsulated placebo tablet).

Figure 6. Timeline of Visits 7 and 8.
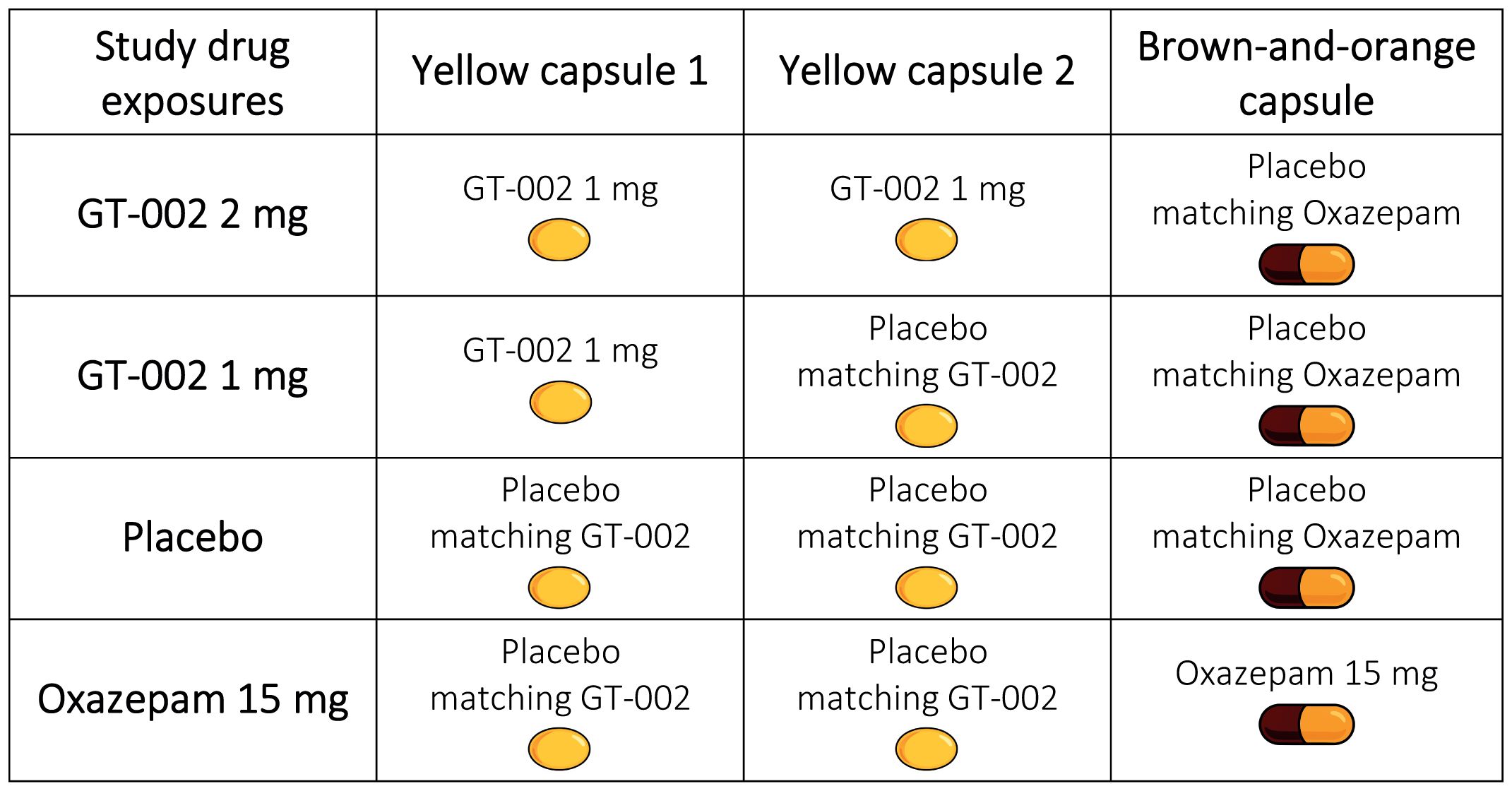
Figure 7. Composition of study drug exposures, each comprising of three capsules administered per exposure day, according to the randomized sequence assigned to each participant.
Gabather will supply GT-002 and its matching placebos, while the Capital Region Pharmacy of Denmark will provide oxazepam tablets (15 mg, Alternova), the corresponding placebo tablets, and the capsules used to encapsulate both.
2.5 Study drugs
2.5.1 GT-002
The preclinical and clinical data on GT-002 are based on the IB for GT-002 (Edition No. 4, dated 20 December 2024), as supplied by Gabather AB. GT-002 is a novel, orally administered drug candidate that targets the GABAA receptor and acts as a partial PAM. It is a selective α3-preferring PAM with limited α5 modulation, as demonstrated in electrophysiological patch-clamp studies (data not included in the IB), suggesting a unique pharmacological profile and potential translational value in clinical trials. In vitro competitive binding studies demonstrated dose-dependent displacement of the GABAA agonist radioligand [3H]-muscimol, while no displacement of the dopamine D3 antagonist ([3H]-methylspiperone) or D5 antagonist ([3H]-SCH 23390) radioligands was observed. Further characterization using radioligand competition binding assays showed that GT-002 also displaced [3H]-flunitrazepam, which binds to the benzodiazepine site. Thus, GT-002 demonstrated high-affinity binding to this site (Ki = 0.57 nM) and was ten times more potent than diazepam, with an IC50 of 0.68 nM compared to 7 nM for diazepam. Receptor binding selectivity profiling using an in vitro radioligand binding assay revealed that GT-002 did not significantly affect any of the selected panel of receptors (i.e. dopamine, glutamate, GABA, glycine, and serotonin) and transporters (i.e. GABA transporter). However, potential off-target interactions revealed that, aside from the GABAA receptor, GT-002 had significant effects on the dopamine transporter (DAT) at 1 μM and 10 μM but not at 0.1 μM. Since there is an ~800-fold difference in the IC50 values between DAT and the GABAA receptor (0.55 μM vs. 0.68 nM, respectively), significant pharmacological interaction with DAT is not expected. Thus, the risk of off-target dopaminergic effects is considered minimal. Ion channel profiling demonstrated that, in contrast to diazepam, GT-002 elicited only minor potentiation of GABA-elicited Cl− currents, as illustrated in Figure 1. GT-002 induced only 10-20% of the ion channel activation compared to diazepam, consistent with a profile of partial positive allosteric modulation at the GABAA receptor. Preclinical efficacy studies have demonstrated significant effects of GT-002 in animal models of schizophrenia using NMDA antagonists PCP and MK-801. GT-002 demonstrated beneficial effects on cognition, memory, and social interaction, as assessed by the Social Interaction Test and the Novel Object Recognition Test.
In three clinical trials in healthy volunteers, including a first-in-human single ascending dose study, a multiple ascending dose study, and an EEG/fMRI target engagement study, GT-002 was safe and well-tolerated with no serious adverse events reported (92, 93). No drug-related changes in cognitive function or mood were observed. There are no known contraindications to its administration. Pharmacokinetic analysis indicated that GT-002 reached peak plasma concentration (Tmax) approximately 2 hours post-administration, had an elimination half-life (T1/2) of around 20 hours, and was not associated with sedative effects. In the TOTEMS trial, participants will receive 1 mg and 2 mg doses, both within the established safe and well-tolerated range. For further details regarding the three clinical trials in healthy volunteers, see the Supplementary Material.
2.5.2 Oxazepam
Oxazepam (ATC-code: N05BA04) is an authorized medicinal product with extensive clinical use and a well-established safety profile, approved by the Danish Medicines Agency. For this trial, it will be sourced from the manufacturer Alternova and used as an active comparator to GT-002, selected for its GABAA receptor targeting and pharmacokinetic similarity. Oxazepam is approved for the treatment of anxiety and agitation but is used off-label in the TOTEMS trial as a tool compound. Participants will receive a single 15 mg dose, which is the minimum recommended by both the manufacturer and the Danish Medicines Agency. The tablets cannot be accurately divided to achieve a lower dose (e.g., 7.5 mg), as the scored line on the tablet is intended solely to facilitate swallowing rather than to provide precise fractional dosing. Administering an imprecise lower dose could introduce variability in oxazepam’s pharmacodynamic effects, which is why a 15 mg dose was selected for the trial. While sedation is a known effect of oxazepam, this characteristic is deliberately utilized as part of its role as an active comparator, enabling differentiation between a classical benzodiazepine profile with sedation and the intended non-sedating profile of GT-002.
All participants will be screened for contraindications to oxazepam. Exclusion criteria reflect those listed in the official summary of product characteristics, including hypersensitivity, myasthenia gravis, sleep apnea, severe hepatic impairment, and acute respiratory depression. Only individuals in overall good physical health will be enrolled, and relevant parameters will be monitored throughout the study.
2.6 Safety measures
The described inclusion and exclusion criteria (Table 2) will exclude those at higher risk for toxicities from the experimental compounds. Moreover, participants will undergo safety monitoring during the study, including assessment of the nature, frequency, and severity of adverse events (AEs). Safety monitoring will include on-site medical supervision during study drug administration days, with a physician physically present to manage any adverse events. Participants will have access to study personnel during working hours and emergency contact options outside office hours. Two safety follow-up visits will be conducted after completion of all four study drug exposures. An AE checklist, covering 46 symptoms across multiple organ systems, will be used at the end of each study drug exposure day and during follow-up visits to ensure comprehensive monitoring. The checklist includes common and compound-specific adverse events associated with both oxazepam and GT-002. Suspected unexpected serious adverse reaction (SUSAR) will be reported to the Danish Medicines Agency in accordance with applicable regulatory timelines. All patients will be clinically stable at enrollment and continue antipsychotic monotherapy throughout the study.
In case of medical emergency or SUSAR, the participant’s treatment sequence can be unblinded. Sealed envelopes with unblinding codes will be stored securely at the study site. The decision to unblind will be at the discretion of the TOTEMS investigators. Unblinding may also occur if required by local laws or regulations.
Women of childbearing potential must use highly effective contraception (failure rate <1% per year) during the study drug exposure period and for five days after the last dose, based on the elimination half-life of GT-002 (approximately 20 hours).
All procedures will be conducted with attention to participant comfort and well-being. Participants may withdraw at any time without reason.
2.7 Electroencephalography and electromyography
The EEG/EMG battery comprises the PPI paradigm, MMN paradigm, SA paradigm, 40-Hz ASSR paradigm, and resting-state. See a detailed description of the paradigms in Table 4. These paradigms were selected based on evidence demonstrating moderate-to-large group differences between patients with schizophrenia and healthy controls (71, 72, 94, 95), with the MMN, 40-Hz ASSR, and resting-state EEG additionally demonstrating good test–retest reliability across sites and feasibility for standardized, automated data acquisition (96). The EEG/EMG battery, followed by the cognitive test battery, will be conducted 2 hours after dosing, corresponding to the Tmax of both GT-002 and oxazepam. EEG recordings will be conducted using BioSemi® hardware (BioSemi, Netherlands) with a cap containing 64 Active Two electrodes, arranged according to the extended 10–20 system. All auditory stimuli will be presented by a computer running Presentation® software (Version 24.0, Neurobehavioral Systems, Inc., Berkeley, CA, USA), and delivered binaurally through stereo insert earphones (E-A-RTONE™ GOLD 3A Insert Earphones, 3M United Kingdom PLC, Bracknell, UK). The eye-blink component of the acoustic startle response in the PPI paradigm will be measured by recording EMG activity from the right orbicularis oculi muscle. For this purpose, two electrodes will be placed under the right eye for PPI and habituation assessment. The first of these will be aligned with the pupil, while the other will be positioned laterally in the direction of the outer canthus of the eye. The EMG recordings will also be assessed using BioSemi® hardware.

Table 4. Detailed description of the paradigms in the EEG/EMG battery.
During EEG/EMG recording, participants will be seated in a comfortable chair and instructed to maintain their gaze on a fixation cross positioned at eye level on a screen directly opposite from their seating position, approximately 2.5 meters away. The only exception will be the MMN paradigm, during which participants will be presented a muted nature documentary featuring animals and landscapes on the screen. The total duration of the EEG/EMG battery is approximately 70 minutes.
EEG and EMG data from each paradigm will undergo preprocessing prior to analysis. Preprocessing steps will generally include filtering (high- and low-pass), epoching, and artifact rejection. Artifact detection and rejection procedures may vary between paradigms and will be performed using established software tools, such as BESA®, Python, or MATLAB, according to the specific requirements of each analysis. The preprocessing pipeline will follow methodologies applied in our previous publications [e.g., Rydkjaer et al., 2020 (46); Bak et al., 2017 (51); Oranje et al., 2017 (49); Randau et al., 2019 (50); Larsen et al., 2018 (97)], ensuring consistency and reproducibility. Detailed preprocessing parameters, including filter settings, epoch lengths, and artifact rejection criteria, will be reported in the methods section of subsequent publications to ensure reproducibility and transparency.
2.8 Cognitive assessment
The cognitive tests have been selected based on literature demonstrating that acute benzodiazepine administration induces sedation, drowsiness, psychomotor slowing, anterograde amnesia, and impaired learning (98, 99), which represent potential effects of oxazepam at the administered dose in this trial. A single 2 mg oral dose of lorazepam (which has a similar Tmax and a slightly longer T1/2 compared to oxazepam) has been shown to significantly impair immediate recognition, reaction time, and delayed memory (100). Cognitive side effects have also been reported across multiple domains, including processing speed and memory, following the same dose (101). Moreover, two 2 mg oral doses administered 12 hours apart adversely affected cognitive performance, including domains such as attention, working memory, verbal memory, and executive functions, among others (102).
We include the Trail Making Test and selected subtasks from the Brief Assessment of Cognition in Schizophrenia (BACS), including the Digit Sequencing Test and Symbol Coding Test, as well as tasks from the Cambridge Neuropsychological Test Automated Battery (CANTAB), including the Motor Screening Task (MOT), Reaction Time (RTI), Spatial Working Memory (SWM), and Rapid Visual Information Processing (RVP). BACS was developed for repeated measurement in clinical trials of patients with schizophrenia and is sensitive to the cognitive deficits observed in this group (103). CANTAB is a well validated computerized neuropsychological test battery that has previously been used in a variety of clinical samples including patients with schizophrenia (104, 105).
In addition, during Visit 2, we will estimate intelligence (IQ) using two subtests from the Wechsler Adult Intelligence Fourth Edition (WAIS-IV), i.e., block design and matrix reasoning, as these have shown the strongest correlation with Full Scale IQ in the Danish reference population (106).
2.9 Clinical assessments
The Brief Psychiatric Rating Scale (BPRS) is an 18-item rating scale that assesses psychiatric symptoms occurring over the preceding three days, using a five-point Likert scale ranging from 0 to 4 (107). The Danish translation by Anne Marie Johansen will be used (108). Given the single-dose nature of the study, significant changes in patients’ symptom severity are not anticipated. Although included patients are considered clinically stable, fluctuations in symptom severity may occur during the study. At screening, the BPRS is administered alongside the Calgary Depression Scale for Schizophrenia to establish baseline psychiatric symptom severity. During the study drug exposure days, the BPRS will be administered immediately following drug administration. Thus, no treatment-related effects are expected at this time point given the Tmax of two hours for both GT-002 and oxazepam. Moreover, due to its retrospective assessment window of three days, the BPRS is not suited for detecting hyper-acute treatment effects, which are instead monitored using an adapted Visual Analog Mood Scale. Therefore, the BPRS is administered to patients only at screening and at each study drug exposure day to assess symptom stability across the trial, thereby providing the possibility to control for potential confounding influences of natural symptom variability on outcome measures between study drug exposure days.
The Calgary Depression Scale for Schizophrenia (CDSS) is a nine-item clinician rated outcome measure assessing symptoms experienced over the preceding two weeks and is the most widely used scale for assessing depression in schizophrenia. It has excellent psychometric properties, internal consistency, inter-rater reliability, sensitivity, specificity, and discriminant and convergent validity (109). The CDSS is administered to patients only at screening to identify the presence and severity of depressive symptoms at baseline.
The Personal and Social Performance Scale (PSP Scale) evaluates four domains: socially useful activities, personal and social relationships, self-care, and disturbing and aggressive behaviors (110, 111). Each domain is rated using a 6-point severity scale based on a structured clinical interview, resulting in a total score ranging from 1 to 100, where higher scores indicate better functioning. The PSP is administered to patients only at screening to assess social and personal functioning, and ratings are based on the participant’s functioning over the past month.
An adapted version of the Visual Analog Mood Scale (VAMS) is used to assess eight specific mood states: Afraid, Confused, Sad, Angry, Energetic, Tired, Happy, and Tense. Each mood is rated using a horizontal visual analog scale, anchored with a “neutral” descriptor at the left end and the target mood descriptor at the right end. Respondents mark the point along the scale that best represents their current emotional state. Scores range from 0 to 100, with 100 indicating the maximal intensity of the mood and 0 indicating minimal intensity or absence of that mood. The VAMS is administered to all participants repeatedly throughout each study drug exposure day (prior to study drug administration, before the EEG/EMG battery, prior to the cognitive test battery, and following completion of testing) to sensitively detect acute mood fluctuations and potential sedative effects during each study drug exposure day.
2.10 Patients’ involvement in the design of the trial
Following the finalization of the trial protocol, we consulted an advisory group comprised of individuals with lived experience of psychosis and their relatives, to obtain feedback, identify potential challenges, and gather suggestions to enhance the trial’s feasibility. Two panel members with SSD reviewed the protocol and provided practical recommendations, including flexible scheduling, procedures for support outside of office hours, transportation assistance through the option of taxi transport, and appropriate, ongoing monetary compensation. All suggestions were carefully considered and incorporated into the trial design.
2.11 Power calculation and justification of the scheduled number of participants
Since this is the first trial investigating GT-002 in patients, there are no previous data on its effects on EEG, EMG, or cognition in patients with SSD. To provide a reference point, we considered our previous study using single doses of clonidine, which demonstrated a significant improvement in PPI, a highly sensitive measure of sensorimotor gating, with a Cohen’s d of 0.73 (42). While differences in drug mechanisms and study design preclude direct extrapolation, this informed a working estimate of Cohen’s d of 0.70 for GT-002. This assumed effect size is consistent with expectations for a compound progressing from a Phase II proof-of-concept trial to a subsequent repeated-dose Phase II trial and ultimately a Phase III trial, as smaller effects would indicate insufficient clinical efficacy to warrant further development of the compound.
The power calculation, based on a paired t-test with a significance level (α) of 0.05 and a desired power of 0.80, indicates that 18 patients would be required to detect the assumed effect. To provide a safety margin, we will include 20 patients. A sensitivity analysis indicates that this sample size achieves ~84% power for d = 0.70 and ~79% for d = 0.65, suggesting that the study retains acceptable power even if the true effect size proves slightly smaller than assumed.
In addition to the patient group, we will include 30 healthy controls for exploratory analyses. Prior research indicates that a significant effect of 30 mg oxazepam on resting-state EEG can be detected with as few as 5 healthy controls (77). To ensure a robust analysis and adequately evaluate the effects of GT-002 in comparison to oxazepam, we aim for a sample size sufficient to detect a moderate-to-large effect size (d = 0.55). Based on this, 28 participants would be required, which we rounded up to 30.
2.12 Planned statistical analyses
The primary analysis will consist of a single planned comparison using a paired t-test to evaluate changes in PPI between 2 mg GT-002 and placebo in patients with SSD. This statistical method allows for a direct assessment of the efficacy of GT-002 relative to placebo in the primary outcome measure. As only one statistical test is performed for the primary endpoint, no adjustment for multiplicity is required. A two-sided significance level of 0.05 will be applied throughout.
Secondary analyses will primarily employ linear mixed models (LMMs) to account for the study design, including multiple drug conditions, various EEG paradigms, and time intervals between measurements. LMMs will enable a comprehensive analysis of both fixed effects (e.g., drug conditions) and random effects (e.g., individual subject variability), thereby providing a more nuanced understanding of GT-002’s impact beyond PPI. This approach will enable direct comparisons between GT-002, oxazepam, and placebo across EEG paradigms. To account for multiple testing in these secondary and exploratory analyses, multiplicity will be controlled using the Holm–Bonferroni method or similar approach.
Exploratory analyses will be conducted to evaluate the impact of factors such as antipsychotic medication type, duration, sex, age, diagnosis, and duration of illness on the acute effects of GT-002 on the EEG paradigms in patients with SSD. Additionally, the differential acute effects of GT-002, oxazepam, and placebo on EEG paradigms and cognition in healthy controls will be assessed using appropriate statistical models. Baseline analyses will be conducted to assess participant characteristics and to facilitate exploratory correlation analyses.
Across all analyses, effect sizes (e.g., Cohen’s d for paired comparisons, partial η² for linear mixed models) with 95% confidence intervals will be reported alongside p-values. Statistical analyses will be performed using R software, and model assumptions (e.g., normality of residuals) will be assessed, with non-parametric alternatives applied as appropriate.
2.13 Biobank
A research biobank will be established for this trial to store blood samples collected from participants for the measurement of GT-002 plasma concentrations. The samples will be stored at the study site until the end of the data collection, after which they will be transferred to Gabather AB for batch analysis and destroyed immediately following the completion of the analyses. All procedures will comply with relevant regulations, including the General Data Protection Regulation (GDPR) and the Danish Data Protection Act, ensuring ethical handling and disposal of the biological material.
2.14 Data management, storage, and sharing
All personal data will be handled in full compliance with the General Data Protection Regulation (GDPR) and the Danish Data Protection Act. Patient data will be entered into an electronic Case Report Form (eCRF), depersonalized using subject numbers, and treated as confidential. The project is registered in Privacy, the research registry and of the Capital Region of Denmark (Approval no.: p-2024-15354). The sponsor/principal investigator will allow authorized access to trial data and relevant documents for monitoring, auditing, and inspection by relevant authorities. Upon completion of the TOTEMS trial and following peer-reviewed publication, the full trial results will be provided to Gabather AB in pseudonymized form.
3 Anticipated results and discussion
The TOTEMS clinical trial will compare the acute effects of GT-002 with those of the widely used benzodiazepine oxazepam, given that both compounds act on the GABAA receptor. The primary and secondary hypotheses are presented in Table 5. The expected differences in effect between GT-002 and oxazepam are likely attributable to GT-002’s high-affinity and selective binding to the GABAA receptor, which is currently its only identified high-affinity target, and to its action as a partial PAM. This is in contrast to oxazepam, which functions as a full PAM at the same receptor.

Table 5. Description of the primary and secondary hypotheses for both patients with SSD and healthy controls.
Schizophrenia spectrum disorders are characterized by considerable neurobiological and cognitive heterogeneity, which poses challenges for detecting pharmacodynamic effects. While enrichment based on specific baseline abnormalities (e.g., PPI deficits or cognitive impairment) could increase sensitivity to particular mechanistic effects, such an approach entails several limitations. First, selectively including patients with abnormal baseline values introduces a risk of regression to the mean, whereby extreme measurements naturally drift toward average levels upon retesting. This could potentially exaggerate apparent treatment effects or mask adverse pharmacodynamic responses. Second, enrichment may introduce a sample bias and reduce external validity, thereby limiting generalizability and potentially obscuring how the compound acts across the broader clinical population.
Evidence from meta-analytic and large multicenter studies supports the presence of robust neurophysiological abnormalities in schizophrenia. Meta-analyses of sensorimotor gating evaluated by PPI [San-Martín et al., 2020 (94)], MMN [Umbricht and Krljes, 2005 (95)], and 40-Hz ASSR [Thuné et al., 2016 (71); Zouaoui et al., 2023 (72)] consistently demonstrate moderate-to-large impairments in patients with schizophrenia compared with healthy controls. These findings were further corroborated by a large multicenter, industry-led study using standardized EEG acquisition and automated analysis pipelines [Cecchi et al., 2023 (96)], which confirmed that patients with schizophrenia exhibit deficits in MMN and 40-Hz ASSR consistent with prior literature. Collectively, this evidence indicates that neurophysiological deficits are prevalent and reliably measurable at the group level in patients with schizophrenia.
In this proof-of-concept trial, we will include the full schizophrenia spectrum, rather than only patients with schizophrenia, to capture the range of cognitive and neurophysiological variability relevant to GABAergic dysfunction, ensuring generalizability and allowing assessment of the overall pharmacodynamic effects of GT-002 in a clinically representative sample. As this is the first trial in a patient population, our main objective is to evaluate how GT-002 modulates early information processing across the full spectrum of schizophrenia-related neurophysiological variability, rather than focusing solely on individuals with marked baseline deficits. The crossover design helps account for interindividual variability, while the inclusion of healthy controls provides an external benchmark for interpreting pharmacodynamic effects. Exploratory analyses will examine whether baseline biomarker or cognitive measures moderate treatment response, thereby informing targeted or stratified approaches in subsequent confirmatory trials.
A potential limitation of this trial is that the sedative effects of oxazepam may be noticeable to some participants. To mitigate this, oxazepam and its matching placebo are both encapsulated to ensure they are visually indistinguishable, thereby preserving blinding and maintaining the integrity of the double-blind design.
While a primary endpoint is a prerequisite in randomized controlled trials, TOTEMS also includes multiple secondary endpoints reflecting other electrophysiological parameters of early information processing. Should the primary endpoint of this Phase II trial prove negative, findings from the secondary endpoints may still justify and encourage further clinical investigation of GT-002.
Although the trial is not designed to directly address CIAS, it aims to determine whether a pharmacological signal on EEG and EMG can be detected following a single dose of GT-002. Positive acute effects would provide preliminary evidence of engagement of the neural circuits underlying hypofrontality, offering a rationale for subsequent repeated-dose trials to evaluate the therapeutic potential of GT-002 as a novel pharmacological approach for alleviating hypofrontality and improving cognitive impairments in patients with SSD. The minor risks associated with trial participation are outweighed by considerable potential future benefits, including clinically significant improvements for patients with SSD and other disorders involving deficient basic information processing. If our hypotheses are confirmed, the trial will provide initial evidence for targeting hypofrontality in schizophrenia, with potential implications for improving treatment and quality of life. Gabather and CNSR will then evaluate initiation of follow-up studies aiming to determine the effectiveness of long-term GT-002 treatment on cognition and symptomatology in patients with SSD. Should a repeated-dose Phase II trial demonstrate efficacy, this would support progression to a larger, international Phase III trial. Ultimately, if development continues to demonstrate efficacy and safety, GT-002 could become globally available, potentially benefiting patients worldwide. Its therapeutic scope may also extend to other disorders where basic information processing is compromised, such as dementia and depression.
Author contributions
TS: Supervision, Methodology, Conceptualization, Writing – review & editing, Writing – original draft, Visualization, Project administration. KA: Supervision, Conceptualization, Writing – review & editing, Software, Writing – original draft, Methodology, Project administration. CL: Conceptualization, Writing – review & editing, Writing – original draft, Project administration, Supervision, Methodology. CR: Conceptualization, Writing – original draft, Funding acquisition, Resources, Writing – review & editing. MS: Writing – original draft, Methodology, Funding acquisition, Visualization, Software, Conceptualization, Writing – review & editing, Project administration. MH-L: Conceptualization, Methodology, Writing – review & editing, Writing – original draft. BG: Writing – review & editing, Supervision, Methodology, Writing – original draft, Conceptualization. KL: Writing – original draft, Writing – review & editing, Conceptualization, Supervision, Software, Methodology. M-RW: Resources, Funding acquisition, Writing – original draft, Conceptualization, Writing – review & editing. BO: Methodology, Conceptualization, Project administration, Writing – review & editing, Funding acquisition, Software, Supervision, Writing – original draft. BE: Resources, Writing – review & editing, Funding acquisition, Project administration, Writing – original draft, Methodology, Conceptualization, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The trial is initiated by Sponsor-Investigator Bjørn H. Ebdrup in collaboration with Gabather AB. The trial is financed by a Grand Solutions grant (6,115,854 DKK) from the Innovation Fund Denmark (IFD, grant ID: 3146-00002B) (granted to Sponsor). This grant is managed by Mental Health Services in the Capital Region of Denmark.
Conflict of interest
The TOTEMS trial is initiated by Sponsor-Investigator Bjørn H. Ebdrup at the Center for Neuropsychiatric Schizophrenia Research (CNSR), in collaboration with Gabather AB. The trial is funded by a Grand Solutions grant (6,115,854 DKK) from the public Innovation Fund Denmark (grant ID: 3146-00002B), awarded to the Sponsor and administered by Mental Health Services in the Capital Region of Denmark. CNSR is responsible for all phases of the study and holds medical responsibility, including adverse event reporting. Gabather AB provides the investigational compound GT-002 and placebo, along with clinical documentation and manufacturing support. Intellectual property arising from the study is jointly owned by CNSR and Gabather in proportion to contribution.
BE is part of the Advisory Board of Boehringer Ingelheim, Lundbeck Pharma, and Orion Pharma; and has received lecture fees from Boehringer Ingelheim, Otsuka Pharma Scandinavia AB, and Lundbeck Pharma. BYG has been the leader of a Lundbeck Foundation Centre of Excellence for Clinical Intervention and Neuropsychiatric Schizophrenia Research (CINS) (January 2009–December 2021), which was partially financed by an independent grant from the Lundbeck Foundation based on international review and partially financed by the Mental Health Services in the Capital Region of Denmark, the University of Copenhagen, and other foundations. All grants are the property of the Mental Health Services in the Capital Region of Denmark and administrated by them.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2025.1656792/full#supplementary-material
References
1. Harvey PD, Heaton RK, Carpenter WT, Green MF, Gold JM, and Schoenbaum M. Functional impairment in people with schizophrenia: focus on employability and eligibility for disability compensation. Schizophr Res. (2012) 140:1–8. doi: 10.1016/j.schres.2012.03.025, PMID: 22503642
2. Dong M, Lu L, Zhang L, Zhang YS, Ng CH, Ungvari GS, et al. Quality of life in schizophrenia: A meta-analysis of comparative studies. Psychiatr Q. (2019) 1590:519–32. doi: 10.1007/s11126-019-09633-4, PMID: 31119453
3. O’Keeffe D, Kinsella A, Waddington JL, and Clarke M. 20-year prospective, sequential follow-up study of heterogeneity in associations of duration of untreated psychosis with symptoms, functioning, and quality of life following first-episode psychosis. Am J Psychiatry. (2022) 179:288–97. doi: 10.1176/appi.ajp.2021.20111658, PMID: 35360921
4. Harvey PD, Bosia M, Cavallaro R, Howes OD, Kahn RS, Leucht S, et al. Cognitive dysfunction in schizophrenia: An expert group paper on the current state of the art. Schizophr Res Cogn. (2022) 29:100249. doi: 10.1016/j.scog.2022.100249, PMID: 35345598
5. McCutcheon RA, Keefe RSE, and McGuire PK. Cognitive impairment in schizophrenia: aetiology, pathophysiology, and treatment. Mol Psychiatry. (2023) 28:1902–18. doi: 10.1038/s41380-023-01949-9, PMID: 36690793
6. Harvey PD and Strassnig M. Predicting the severity of everyday functional disability in people with schizophrenia: cognitive deficits, functional capacity, symptoms, and health status. World Psychiatry. (2012) 11:73–9. doi: 10.1016/j.wpsyc.2012.05.004, PMID: 22654932
7. Galderisi S, Rucci P, Kirkpatrick B, Mucci A, Gibertoni D, Rocca P, et al. Interplay among psychopathologic variables, personal resources, context-related factors, and real-life functioning in individuals with schizophrenia: A network analysis. JAMA Psychiatry. (2018) 75:396–404. doi: 10.1001/jamapsychiatry.2017.4607, PMID: 29450447
8. Burton CZ, Vella L, Harvey PD, Patterson TL, Heaton RK, and Twamley EW. Factor structure of the MATRICS Consensus Cognitive Battery (MCCB) in schizophrenia. Schizophr Res. (2013) 146:244–8. doi: 10.1016/j.schres.2013.02.026, PMID: 23507359
9. Halverson TF, Orleans-Pobee M, Merritt C, Sheeran P, Fett AK, and Penn DL. Pathways to functional outcomes in schizophrenia spectrum disorders: Meta-analysis of social cognitive and neurocognitive predictors. Neurosci Biobehav Rev. (2019) 105:212–9. doi: 10.1016/j.neubiorev.2019.07.020, PMID: 31415864
10. Tolman AW and Kurtz MM. Neurocognitive predictors of objective and subjective quality of life in individuals with schizophrenia: a meta-analytic investigation. Schizophr Bull. (2012) 38:304–15. doi: 10.1093/schbul/sbq077, PMID: 20624752
11. Horan WP, Catalano LT, and Green MF. An update on treatment of cognitive impairment associated with schizophrenia. Curr Top Behav Neurosci. (2023) 63:407–36. doi: 10.1007/7854_2022_382, PMID: 35915386
12. Vita A, Gaebel W, Mucci A, Sachs G, Barlati S, Giordano GM, et al. European Psychiatric Association guidance on treatment of cognitive impairment in schizophrenia. Eur Psychiatry. (2022) 65:e57. doi: 10.1192/j.eurpsy.2022.2315, PMID: 36059103
13. Remington G, Addington D, Honer W, Ismail Z, Raedler T, and Teehan M. Guidelines for the pharmacotherapy of schizophrenia in adults. Can J Psychiatry. (2017) 62:604–16. doi: 10.1177/0706743717720448, PMID: 28703015
14. Keepers GA, Fochtmann LJ, Anzia JM, Benjamin S, Lyness JM, Mojtabai R, et al. The american psychiatric association practice guideline for the treatment of patients with schizophrenia. Am J Psychiatry. (2020) 177:868–72. doi: 10.1176/appi.ajp.2020.177901, PMID: 32867516
15. Galletly C, Castle D, Dark F, Humberstone V, Jablensky A, Killackey E, et al. Royal Australian and New Zealand College of Psychiatrists clinical practice guidelines for the management of schizophrenia and related disorders. Aust N Z J Psychiatry. (2016) 50:410–72. doi: 10.1177/0004867416641195, PMID: 27106681
16. Barnes TRE, Drake R, Paton C, Cooper SJ, Deakin B, Ferrier IN, et al. Evidence-based guidelines for the pharmacological treatment of schizophrenia: Updated recommendations from the British Association for Psychopharmacology. J Psychopharmacol. (2020) 34:3–78. doi: 10.1177/0269881119889296, PMID: 31829775
17. Calzavara-Pinton I, Nibbio G, Barlati S, Bertoni L, Necchini N, Zardini D, et al. Treatment of cognitive impairment associated with schizophrenia spectrum disorders: new evidence, challenges, and future perspectives. Brain Sci. (2024) 14:791. doi: 10.3390/brainsci14080791, PMID: 39199483
18. Vita A, Nibbio G, and Barlati S. Pharmacological treatment of cognitive impairment associated with schizophrenia: state of the art and future perspectives. Schizophr Bull Open. (2024) 5:sgae013. doi: 10.1093/schizbullopen/sgae013, PMID: 39144119
19. Hazlett EA, Buchsbaum MS, Jeu LA, Nenadic I, Fleischman MB, Shihabuddin L, et al. Hypofrontality in unmedicated schizophrenia patients studied with PET during performance of a serial verbal learning task. Schizophr Res. (2000) 43:33–46. doi: 10.1016/S0920-9964(99)00178-4, PMID: 10828413
20. McCutcheon RA, Reis Marques T, and Howes OD. Schizophrenia-an overview. JAMA Psychiatry. (2020) 77:201–10. doi: 10.1001/jamapsychiatry.2019.3360, PMID: 31664453
21. Luvsannyam E, Jain MS, Pormento MKL, Siddiqui H, Balagtas ARA, Emuze BO, et al. Neurobiology of schizophrenia: A comprehensive review. Cureus. (2022) 14:e23959. doi: 10.7759/cureus.23959, PMID: 35541299
22. Dietz AG, Goldman SA, and Nedergaard M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry. (2020) 7:272–81. doi: 10.1016/S2215-0366(19)30302-5, PMID: 31704113
23. Schirner M, McIntosh AR, Jirsa V, Deco G, and Ritter P. Inferring multi-scale neural mechanisms with brain network modelling. Elife. (2018) 7:e28927. doi: 10.7554/eLife.28927, PMID: 29308767
24. Niessing J, Ebisch B, Schmidt KE, Niessing M, Singer W, and Galuske RAW. Hemodynamic signals correlate tightly with synchronized gamma oscillations. Science. (2005) 309:948–51. doi: 10.1126/science.1110948, PMID: 16081740
25. Alekseichuk I, Turi Z, Amador de Lara G, Antal A, and Paulus W. Spatial working memory in humans depends on theta and high gamma synchronization in the prefrontal cortex. Curr Biol. (2016) 26:1513–21. doi: 10.1016/j.cub.2016.04.035, PMID: 27238283
26. van den Heuvel MP and Hulshoff Pol HE. Exploring the brain network: a review on resting-state fMRI functional connectivity. Eur Neuropsychopharmacol. (2010) 20:519–34. doi: 10.1016/j.euroneuro.2010.03.008, PMID: 20471808
27. Pratt JA, Winchester C, Egerton A, Cochran SM, and Morris BJ. Modelling prefrontal cortex deficits in schizophrenia: implications for treatment. Br J Pharmacol. (2008) 153 Suppl 1:S465–S470. doi: 10.1038/bjp.2008.24, PMID: 18311160
28. Krzystanek M and Pałasz A. NMDA receptor model of antipsychotic drug-induced hypofrontality. Int J Mol Sci. (2019) 20:1442. doi: 10.3390/ijms20061442, PMID: 30901926
29. Hazlett EA and Buchsbaum MS. Sensorimotor gating deficits and hypofrontality in schizophrenia. Front Biosci. (2001) 6:D1069–D1072. doi: 10.2741/hazlett, PMID: 11532605
30. Paulman RG, Devous MD, Gregory RR, Herman JH, Jennings L, Bonte FJ, et al. Hypofrontality and cognitive impairment in schizophrenia: Dynamic single-photon tomography and neuropsychological assessment of schizophrenic brain function. Biol Psychiatry. (1990) 27:377–99. doi: 10.1016/0006-3223(90)90549-H, PMID: 2106922
31. Carter CS, Perlstein W, Ganguli R, Brar J, Mintun M, and Cohen JD. Functional hypofrontality and working memory dysfunction in schizophrenia. Am J Psychiatry. (1998) 155:1285–7. doi: 10.1176/ajp.155.9.1285, PMID: 9734557
32. Adell A, Jiménez-Sánchez L, López-Gil X, and Romón T. Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr Bull. (2012) 38:9–14. doi: 10.1093/schbul/sbr133, PMID: 21965469
33. Oranje B, Gispen-De Wied CC, Westenberg HGM, Kemner C, Verbaten MN, and Kahn RS. Haloperidol counteracts the ketamine-induced disruption of processing negativity, but not that of the P300 amplitude. Int J Neuropsychopharmacol. (2009) 12:823–32. doi: 10.1017/S1461145708009814, PMID: 19154656
34. Oranje B, Van Berckel BNM, Kemner C, van Ree JM, Kahn RS, and Verbaten MN. The effects of a sub-anaesthetic dose of ketamine on human selective attention. Neuropsychopharmacology. (2000) 22:293–302. doi: 10.1016/S0893-133X(99)00118-9, PMID: 10693157
35. Oranje B, Gispen-De Wied CC, Verbaten MN, and Kahn RS. Modulating sensory gating in healthy volunteers: The effects of ketamine and haloperidol. Biol Psychiatry. (2002) 52:887–95. doi: 10.1016/S0006-3223(02)01377-X, PMID: 12399142
36. van Berckel BN, Oranje B, van ree JM, Verbaten MN, and Kahn RS. The effects of low dose ketamine on sensory gating, neuroendocrine secretion and behavior in healthy human subjects. Psychopharmacology (Berl). (1998) 137:271–81. doi: 10.1007/s002130050620, PMID: 9683005
37. Mackeprang T, Kristiansen KT, and Glenthoj BY. Effects of antipsychotics on prepulse inhibition of the startle response in drug-naïve schizophrenic patients. Biol Psychiatry. (2002) 52:863–73. doi: 10.1016/S0006-3223(02)01409-9, PMID: 12399139
38. Braff DL, Geyer MA, and Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) (2001) 156:234–58. doi: 10.1007/s002130100810, PMID: 11549226
39. Näätänen R, Pakarinen S, Rinne T, and Takegata R. The mismatch negativity (MMN): towards the optimal paradigm. Clin Neurophysiol. (2004) 115:140–4. doi: 10.1016/j.clinph.2003.04.001, PMID: 14706481
40. Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol. (2007) 118:2128–48. doi: 10.1016/j.clinph.2007.04.019, PMID: 17573239
41. Kruiper C, Glenthøj BY, and Oranje B. Effects of clonidine on MMN and P3a amplitude in schizophrenia patients on stable medication. Neuropsychopharmacology. (2019) 44:1062–7. doi: 10.1038/s41386-019-0351-6, PMID: 30797222
42. Oranje B and Glenthøj BY. Clonidine normalizes levels of P50 gating in patients with schizophrenia on stable medication. Schizophr Bull. (2014) 40:1022–9. doi: 10.1093/schbul/sbt144, PMID: 24106334
43. Oranje B, Van Oel CJ, Gispen-de Wied CC, Verbaten MN, and Kahn RS. Effects of typical and atypical antipsychotics on the prepulse inhibition of the startle reflex in patients with schizophrenia. J Clin Psychopharmacol. (2002) 22:359–65. doi: 10.1097/00004714-200208000-00005, PMID: 12172334
44. Bak N, Rostrup E, Larsson HB, Glenthøj BY, and Oranje B. Concurrent functional magnetic resonance imaging and electroencephalography assessment of sensory gating in schizophrenia. Hum Brain Mapp. (2014) 35:3578–87. doi: 10.1002/hbm.22422, PMID: 24375687
45. Lemvigh CK, Jepsen JRM, Fagerlund B, Pagsberg AK, Glenthøj BY, Rydkjær J, et al. Auditory sensory gating in young adolescents with early-onset psychosis: a comparison with attention deficit/hyperactivity disorder. Neuropsychopharmacology. (2020) 45:649–55. doi: 10.1038/s41386-019-0555-9, PMID: 31649298
46. Rydkjaer J, Jepsen JRM, Pagsberg AK, Fagerlund B, Glenthoej BY, and Oranje B. Do young adolescents with first-episode psychosis or ADHD show sensorimotor gating deficits? Psychol Med. (2020) 50:607–15. doi: 10.1017/S0033291719000412, PMID: 30873927
47. Kruiper C, Fagerlund B, Nielsen MO, Düring S, Jensen MH, Ebdrup BH, et al. Associations between P3a and P3b amplitudes and cognition in antipsychotic-naïve first-episode schizophrenia patients. Psychol Med. (2019) 49:868–75. doi: 10.1017/S0033291718001575, PMID: 29914589
48. Rydkjær J, Møllegaard Jepsen JR, Pagsberg AK, Fagerlund B, Glenthøj BY, and Oranje B. Mismatch negativity and P3a amplitude in young adolescents with first-episode psychosis: a comparison with ADHD. Psychol Med. (2017) 47:377–88. doi: 10.1017/S0033291716002518, PMID: 27776572
49. Oranje B, Aggernaes B, Rasmussen H, Ebdrup BH, and Glenthoj BY. Selective attention and mismatch negativity in antipsychotic-naïve, first-episode schizophrenia patients before and after 6 months of antipsychotic monotherapy. Psychol Med. (2017) 47:2155–65. doi: 10.1017/S0033291717000599, PMID: 28443529
50. Randau M, Oranje B, Miyakoshi M, Makeig S, Fagerlund B, Glenthøj B, et al. Attenuated mismatch negativity in patients with first-episode antipsychotic-naive schizophrenia using a source-resolved method. NeuroImage Clin. (2019) 22:101760. doi: 10.1016/j.nicl.2019.101760, PMID: 30927608
51. Bak N, Mann J, Fagerlund B, Glenthøj BY, Jepsen JRM, and Oranje B. Testing a decades’ old assumption: Are individuals with lower sensory gating indeed more easily distracted? Psychiatry Res. (2017) 255:387–93. doi: 10.1016/j.psychres.2017.05.048, PMID: 28666245
52. Newson JJ and Thiagarajan TC. EEG frequency bands in psychiatric disorders: A review of resting state studies. Front Hum Neurosci. (2019) 12:512. doi: 10.3389/fnhum.2018.00521, PMID: 30687041
53. Harmony T. The functional significance of delta oscillations in cognitive processing. Front Integr Neurosci. (2013) 7:83. doi: 10.3389/fnint.2013.00083, PMID: 24367301
54. Gattaz WF, Mayer S, Ziegler P, Platz M, and Gasser T. Hypofrontality on topographic EEG in schizophrenia. Correlations with neuropsychological and psychopathological parameters. Eur Arch Psychiatry Clin Neurosci. (1992) 241:328–32. doi: 10.1007/BF02191956, PMID: 1504108
55. Knyazeva MG, Jalili M, Meuli R, Hasler M, De Feo O, and Do KQ. Alpha rhythm and hypofrontality in schizophrenia. Acta Psychiatr Scand. (2008) 118:188–99. doi: 10.1111/j.1600-0447.2008.01227.x, PMID: 18636993
56. McNally JM and McCarley RW. Gamma band oscillations: A key to understanding schizophrenia symptoms and neural circuit abnormalities. Curr Opin Psychiatry. (2016) 29:202–10. doi: 10.1097/YCO.0000000000000244, PMID: 26900672
57. Andreou C, Nolte G, Leicht G, Polomac N, Hanganu-Opatz IL, Lambert M, et al. Increased resting-state gamma-band connectivity in first-episode schizophrenia. Schizophr Bull. (2015) 41:930–9. doi: 10.1093/schbul/sbu121, PMID: 25170031
58. Grützner C, Uhlhaas PJ, Genc E, Kohler A, Singer W, and Wibral M. Neuroelectromagnetic correlates of perceptual closure processes. J Neurosci. (2010) 30:8342–52. doi: 10.1523/JNEUROSCI.5434-09.2010, PMID: 20554885
59. Grent-‘t-Jong T, Rivolta D, Sauer A, Grube M, Singer W, Wibral M, et al. MEG-measured visually induced gamma-band oscillations in chronic schizophrenia: Evidence for impaired generation of rhythmic activity in ventral stream regions. Schizophr Res. (2016) 176:177–85. doi: 10.1016/j.schres.2016.06.003, PMID: 27349815
60. Swerdlow NR, Braff DL, and Geyer MA. GABAergic projection from nucleus accumbers to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res. (1990) 532:146–50. doi: 10.1016/0006-8993(90)91754-5, PMID: 2282510
61. Kodsi MH and Swerdlow NR. Prepulse inhibition in the rat is regulated by ventral and caudodorsal striato-pallidal circuitry. Behav Neurosci. (1995) 109:912–28. doi: 10.1037/0735-7044.109.5.912, PMID: 8554715
62. Yeomans JS, Bosch D, Alves N, Daros A, Ure RJ, and Schmid S. GABA receptors and prepulse inhibition of acoustic startle in mice and rats. Eur J Neurosci. (2010) 31:2053–61. doi: 10.1111/j.1460-9568.2010.07236.x, PMID: 20497471
63. Frau R, Bini V, Pillolla G, Malherbe P, Pardu A, Thomas AW, et al. Positive allosteric modulation of GABAB receptors ameliorates sensorimotor gating in rodent models. CNS Neurosci Ther. (2014) 20:679–84. doi: 10.1111/cns.12261, PMID: 24703381
64. Inui K, Takeuchi N, Sugiyama S, Motomura E, and Nishihara M. GABAergic mechanisms involved in the prepulse inhibition of auditory evoked cortical responses in humans. PloS One. (2018) 13:e0190481. doi: 10.1371/journal.pone.0190481, PMID: 29298327
65. Geyer MA, Krebs-Thomson K, Braff DL, and Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: A decade in review. Psychopharmacology (Berl) (2001) 156:117–54. doi: 10.1007/s002130100811, PMID: 11549216
66. Ford TC, Woods W, Enticott PG, and Crewther DP. Cortical excitation-inhibition ratio mediates the effect of pre-attentive auditory processing deficits on interpersonal difficulties. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 98:109769. doi: 10.1016/j.pnpbp.2019.109769, PMID: 31676468
67. Rowland LM, Summerfelt A, Wijtenburg SA, Du X, Chiappelli JJ, Krishna N, et al. Frontal glutamate and γ-aminobutyric acid levels and their associations with mismatch negativity and digit sequencing task performance in schizophrenia. JAMA Psychiatry. (2016) 73:166–74. doi: 10.1001/jamapsychiatry.2015.2680, PMID: 26720179
68. Vohs JL, Andrew Chambers R, Krishnan GP, O’Donnell BF, Berg S, and Morzorati SL. GABAergic modulation of the 40 Hz auditory steady-state response in a rat model of schizophrenia. Int J Neuropsychopharmacol. (2010) 13:487–97. doi: 10.1017/S1461145709990307, PMID: 19627651
69. Grent-’t-Jong T, Brickwedde M, Metzner C, and Uhlhaas PJ. 40-hz auditory steady-state responses in schizophrenia: toward a mechanistic biomarker for circuit dysfunctions and early detection and diagnosis. Biol Psychiatry. (2023) 94:550–60. doi: 10.1016/j.biopsych.2023.03.026, PMID: 37086914
70. Toso A, Wermuth AP, Arazi A, Braun A, Grent-‘t Jong T, Uhlhaas PJ, et al. 40 hz steady-state response in human auditory cortex is shaped by gabaergic neuronal inhibition. J Neurosci. (2024) 44:e2029232024. doi: 10.1523/JNEUROSCI.2029-23.2024, PMID: 38670804
71. Thuné H, Recasens M, and Uhlhaas PJ. The 40-Hz auditory steady-state response in patients with schizophrenia a meta-Analysis. JAMA Psychiatry. (2016) 73:1145–53. doi: 10.1001/jamapsychiatry.2016.2619, PMID: 27732692
72. Zouaoui I, Dumais A, Lavoie ME, and Potvin S. Auditory steady-state responses in schizophrenia: an updated meta-analysis. Brain Sci. (2023) 13:1722. doi: 10.3390/brainsci13121722, PMID: 38137170
73. Cittadini A and Lader M. Lack of effect of a small dose of flumazenil in reversing short-term tolerance to benzodiazepines in normal subjects. J Psychopharmacol. (1991) 5:220–7. doi: 10.1177/026988119100500307, PMID: 22282559
74. McDevitt D, Currie D, Nicholson A, Wright N, and Zetlein M. Central effects of the diuretic, bendrofluazide. Br J Clin Pharmacol. (1994) 38:249–56. doi: 10.1111/j.1365-2125.1994.tb04349.x, PMID: 7826827
75. Nicholson A, Wright N, Zetlein M, Currie D, and McDevitt D. Central effects of beta-adrenoceptor antagonists. II–Electroencephalogram and body sway. Br J Clin Pharmacol. (1988) 26:129–41. doi: 10.1111/j.1365-2125.1988.tb03379.x, PMID: 2905149
76. McDevitt DG, Currie D, Nicholson AN, Wright NA, and Zetlein MB. Central effects of repeated administration of atenolol and captopril in healthy volunteers. Eur J Clin Pharmacol. (1994) 46:23–8. doi: 10.1007/BF00195911, PMID: 8005182
77. Ansseau M, Doumont A, Cerfontaine JL, Mantanus H, Rousseau JC, and Timsit-Berthier M. Self-reports of anxiety level and EEG changes after a single dose of benzodiazepines. Double-blind comparison of two forms of oxazepam. Neuropsychobiology. (1984) 12:255–9. doi: 10.1159/000118148, PMID: 6398863
78. van Leeuwen TH, Verbaten MN, Koelega HS, Camfferman G, van der Gugten J, and Slangen JL. Effects of oxazepam on performance and event-related brain potentials in vigilance tasks with static and dynamic stimuli. Psychopharmacology (Berl). (1994) 116:499–507. doi: 10.1007/BF02247484, PMID: 7701055
79. Johannes S, Wieringa BM, Nager W, Dengler R, and Münte TF. Oxazepam alters action monitoring. Psychopharmacology (Berl). (2001) 155:100–6. doi: 10.1007/s002130100680, PMID: 11374327
80. Lewis DA, Curley AA, Glausier JR, and Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. (2012) 35:57–67. doi: 10.1016/j.tins.2011.10.004, PMID: 22154068
81. Dienel SJ and Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis. (2019) 131:104208. doi: 10.1016/j.nbd.2018.06.020, PMID: 29936230
82. Vid Prkačin M, Banovac I, Petanjek Z, and Hladnik A. Cortical interneurons in schizophrenia - cause or effect? Croat Med J. (2023) 64:110–22. doi: 10.3325/cmj.2023.64.110, PMID: 37131313
83. Lewis DA, Hashimoto T, and Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. (2005) 6:312–24. doi: 10.1038/nrn1648, PMID: 15803162
84. Xu MY and Wong AHC. GABAergic inhibitory neurons as therapeutic targets for cognitive impairment in schizophrenia. Acta Pharmacol Sin. (2018) 39:733–53. doi: 10.1038/aps.2017.172, PMID: 29565038
85. Absalom NL, Lin SXN, Liao VWY, Chua HC, Møller RS, Chebib M, et al. GABAA receptors in epilepsy: Elucidating phenotypic divergence through functional analysis of genetic variants. J Neurochem. (2023) 168:3831–3852. doi: 10.1111/jnc.15932, PMID: 37621067
86. Thompson SM. Modulators of GABA(A) receptor-mediated inhibition in the treatment of neuropsychiatric disorders: past, present, and future. Neuropsychopharmacology. (2024) 49:83–95. doi: 10.1038/s41386-023-01728-8, PMID: 37709943
87. Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. (1999) 401:796–800. doi: 10.1038/44579, PMID: 10548105
88. McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. (2000) 3:587–92. doi: 10.1038/75761, PMID: 10816315
89. Löw K, Crestani F, Keist R, Benke D, Brünig I, Benson JA, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. (2000) 290:131–4. doi: 10.1126/science.290.5489.131, PMID: 11021797
90. Crestani F, Löw K, Keist R, Mandelli M, Möhler H, and Rudolph U. Molecular targets for the myorelaxant action of diazepam. Mol Pharmacol. (2001) 59:442–5. doi: 10.1016/S0026-895X(24)12233-X, PMID: 11179437
91. Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, et al. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U.S.A. (2002) 99:8980–5. doi: 10.1073/pnas.142288699, PMID: 12084936
92. Witt M, Ryan C, Rehnmark S, and Nielsen M. Preclinical and clinical studies with GT-002, a novel GABAA receptor ligand with putative antipsychotic and procognitive effects. In: Program No. 645.15. 2019 Neuroscience Meeting Planner. Chicago, IL: Society of neuroscience (2019).
93. Ryan C, Witt M, Lorenc-Koci E, Rogoíż Z, Rehnmark S, and Nielsen M. The GABAA receptor positive allosteric modulator, GT-002 has procognitive effects in rats. In: Program No. 285.02. 2021 Society of Neuroscience, Global Connectome. Washington, DC: Society for Neuroscience (2021).
94. San-Martin R, Castro LA, Menezes PR, Fraga FJ, Simões PW, and Salum C. Meta-analysis of sensorimotor gating deficits in patients with schizophrenia evaluated by prepulse inhibition test. Schizophr Bull. (2020) 46:1482–97. doi: 10.1093/schbul/sbaa059, PMID: 32506125
95. Umbricht D and Krljesb S. Mismatch negativity in schizophrenia: A meta-analysis. Schizophr Res. (2005) 76:1–23. doi: 10.1016/j.schres.2004.12.002, PMID: 15927795
96. Cecchi M, Adachi M, Basile A, Buhl DL, Chadchankar H, Christensen S, et al. Validation of a suite of ERP and QEEG biomarkers in a pre-competitive, industry-led study in subjects with schizophrenia and healthy volunteers. Schizophr Res. (2023) 254:178–89. doi: 10.1016/j.schres.2023.02.018, PMID: 36921403
97. Larsen KM, Pellegrino G, Birknow MR, Kjær TN, Baaré WFC, Didriksen M, et al. 22q11.2 deletion syndrome is associated with impaired auditory steady-state gamma response. Schizophr Bull. (2018) 44:388–97. doi: 10.1093/schbul/sbx058, PMID: 28521049
98. Stewart SA. The effects of benzodiazepines on cognition. J Clin Psychiatry. (2005) 66 Suppl 2:9–13.
99. Buffett-Jerrott SE, Stewart SH, Bird S, and Teehan MD. An examination of differences in the time course of oxazepam’s effects on implicit vs explicit memory. J Psychopharmacol. (1998) 12:338–47. doi: 10.1177/026988119801200403, PMID: 10065907
100. Loring DW, Marino SE, Drane DL, Parfitt D, Finney GR, and Meador KJ. Lorazepam effects on Word Memory Test performance: a randomized, double-blind, placebo-controlled, crossover trial. Clin Neuropsychol. (2011) 25:799–811. doi: 10.1080/13854046.2011.583279, PMID: 21756210
101. Loring DW, Marino SE, Parfitt D, Finney GR, and Meador KJ. Acute lorazepam effects on neurocognitive performance. Epilepsy Behav. (2012) 25:329–33. doi: 10.1016/j.yebeh.2012.08.019, PMID: 23103305
102. Meador KJ, Gevins A, Leese PT, Otoul C, and Loring DW. Neurocognitive effects of brivaracetam, levetiracetam, and lorazepam. Epilepsia (2011) 52:264–72. doi: 10.1111/j.1528-1167.2010.02746.x, PMID: 20887370
103. Keefe RSE, Goldberg TE, Harvey PD, Gold JM, Poe MP, and Coughenour L. The Brief Assessment of Cognition in Schizophrenia: Reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophr Res. (2004) 68:283–97. doi: 10.1016/j.schres.2003.09.011, PMID: 15099610
104. Sahakian BJ and Owen AM. Computerized assessment in neuropsychiatry using CANTAB: discussion paper. J R Soc Med. (1992) 85:399–402., PMID: 1629849
105. Levaux MN, Potvin S, Sepehry AA, Sablier J, Mendrek A, and Stip E. Computerized assessment of cognition in schizophrenia: Promises and pitfalls of CANTAB. Eur Psychiatry. (2007) 22:104–15. doi: 10.1016/j.eurpsy.2006.11.004, PMID: 17227707
106. Wechsler D. Wechsler adult intelligence scale – fourth edition (WAIS-IV). San Antonio, TX: Pearson (2008).
107. Overall JE and Gorham DR. The brief psychiatric rating scale. Psychol Rep. (1962) 10:799–812. doi: 10.2466/pr0.1962.10.3.799
108. Johansen AM. Brief psychiatric rating scale (BPRS). In: Elsass P, Ivanouw J, Mortensen EL, Poulsen S, and Rosenbaum B, editors. Assessmentmetoder - Håndbog for psykologer og psykiatere. Copenhagen, Denmark: Dansk Psykologisk Forlag (2006). p. 423–37.
109. Addington J, Shah H, Liu L, and Addington D. Reliability and validity of the calgary depression scale for schizophrenia (CDSS) in youth at clinical high risk for psychosis. Schizophr Res. (2014) 153:64. doi: 10.1016/j.schres.2013.12.014, PMID: 24439270
110. White S, Dominise C, Naik D, and Killaspy H. The reliability of the Personal and Social Performance scale - informing its training and use. Psychiatry Res. (2016) 243:312–7. doi: 10.1016/j.psychres.2016.06.047, PMID: 27428085
Keywords: schizophrenia, schizophrenia spectrum disorders, cognitive impairment associated with schizophrenia (CIAS), cognitive impairment (CI), hypofrontality, GABA, GABA receptor A, positive allosteric modulator (PAM)
Citation: Siebner TH, Ambrosen KS, Lemvigh CK, Ryan CN, Sørensen ME, Hernández-Lorca M, Glenthøj BY, Larsen KM, Witt M-R, Oranje B and Ebdrup BH (2025) Acute effects of partial positive allosteric GABAA receptor modulation by GT-002 on psychophysiological and cognitive measures: protocol for the TOTEMS phase II trial targeting cognitive impairment associated with schizophrenia. Front. Psychiatry 16:1656792. doi: 10.3389/fpsyt.2025.1656792
Received: 30 June 2025; Accepted: 16 October 2025;
Published: 25 November 2025.
Edited by:
Guglielmo Lucchese, Universitätsmedizin Greifswald, GermanyReviewed by:
Bruce J. Kinon, Karuna Therapeutics, Inc., United StatesWilliam Z. Potter, Consultant, Philadelphia, United States
Chenyang Yao, Capital Medical University, China
Copyright © 2025 Siebner, Ambrosen, Lemvigh, Ryan, Sørensen, Hernández-Lorca, Glenthøj, Larsen, Witt, Oranje and Ebdrup. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Hartwig Siebner, dGhvbWFzLmhhcnR3aWcuc2llYm5lci4wMkByZWdpb25oLmRr