 Boutheïna Kerkeni
Boutheïna Kerkeni Malek Boukallaba2
Malek Boukallaba2 Mariem Hechmi
Mariem Hechmi Denis Duflot
Denis Duflot Céline Toubin
Céline Toubin- 1Université de La Manouba, Institut Supérieur des Arts Multimédia de La Manouba, La Manouba, Tunisia
- 2Département de Physique, Laboratoire Physique de La Matière Condensée (LPMC) Faculté des Sciences de Tunis, Université Tunis El Manar, Campus Universitaire, Tunis, Tunisia
- 3LERMA, Observatoire de Paris, PSL Research University, CNRS-UMR8112, Sorbonne Universités, UPMC Univ. Paris 06, Meudon, France
- 4Université de Lille, CNRS, UMR 8523—PhLAM–Physique des Lasers Atomes et Molécules, Lille, France
Formaldehyde has been widely observed in the icy mantle of interstellar grains. H2CO may be formed from successive hydrogenations of CO and may further contribute to the chemical complexity of the Interstellar medium (ISM) participating to heterogeneous reactions with colliding gas phase atoms. Within this context, Eley-Rideal and Langmuir-Hinshelwood rate constants of H2 formation on a formaldehyde doped amorphous water ice grain model of the ISM, were computed over a wide temperature range [15–2000 K]. We used classical molecular dynamics (MD) simulations to build the model of the H2CO doped ice surface. Then we studied theoretically by means of hybrid QM/MM ab initio and molecular mechanics methodology (ONIOM) H atoms abstraction from formaldehyde molecules and the H2 formation. Specifically, we investigate the reactivity of the gas phase H atom toward one formaldehyde molecule lying at one of the slab surfaces. The reaction path and the energetics are predicted, the mechanism is found to be exothermic by 14.89 kcal/mol and the barrier is 6.75 kcal/mol at the QM level CBS/DLPNO-CCSD(T)//ONIOM/aug-cc-pVTZ. We employ two approaches that take into account tunnelling and non-classical reflection effects by means of the Zero Curvature Tunnelling (ZCT), and the Small Curvature Tunnelling (SCT) which all provided comparable results to predict the kinetics of the reaction path. The rate constants show important quantum tunnelling effects at low temperatures when compared to rates obtained from the purely classical transition-state theory (TST) and from the canonical variational transition state theory (CVT). Corner cutting effects are highlighted in the SCT calculations by 4 to 5 orders of magnitude with respect to ZCT rate constants at low temperatures.
1 Introduction
H2 molecules are ubiquitous in the Universe and constitute the most abundant species and consequently are the main trigger of dust chemistry which is responsible for the formation of key molecules, such as H2O, NH3, and CH3OH. Detection of H2 was achieved in the interstellar medium (ISM) via a rocket flight in 1970 (Carruthers, 1970). It has been shown that almost certainly H2 forms at the surface of dust grains and icy mantles in a variety of interstellar environments (Hollenbach and Salpeter, 1970; Williams et al., 2003; Vidali, 2013; Wakelam et al., 2017). Extensive computational (Katz et al., 1999; Cuppen et al., 2010; Cazaux et al., 2011), and experimental studies (Pirronello et al., 1997; Creighan et al., 2006; Watanabe et al., 2010) over the 20 past years have been conducted. Many focused on testing the role of grain models on the energetics prediction of the chemical routes leading to H2 formation.
Among the grain models, there appear carbonaceous (Farebrother et al., 2000; Ivanovskaya et al., 2010), and silicate ones (Goumans, 2011; Kerkeni and Bromley, 2013). From the modeling point of view molecular clusters and also periodic slabs have served to mimic interstellar grains (Farebrother et al., 2000; Ivanovskaya et al., 2010; Goumans, 2011; Chaabouni et al., 2012; Navarro-Ruiz et al., 2014; Oueslati et al., 2014; Navarro-Ruiz et al., 2015; Oueslati et al., 2015; Navarro-Ruiz et al., 2016; Song and Kästner, 2017), upon which H2 formation was investigated. Several experimental techniques e.g., (Latimer et al., 2008; Wakelam et al., 2017) at cryogenic temperatures have been realized to investigate the efficiency of H2 formation in the laboratory. Given the very low temperatures ≈10 K in dense regions of space, dust grains are covered with layers of icy mantles (Ehrenfreund and Schutte, 2000). These serve as a reservoir for molecular accretion, that would subsequently allow the reagents to thermalize, diffuse and react (Congiu et al., 2014), and finally desorb in a later phase during star formation processes. These formed molecules are detected in warmer diffuse clouds of the interstellar medium. The essential enablers of this rich and complex reactivity at the surface of grains may rely in some cases on radical-radical recombination or the presence of UV irradiation and cosmic rays. Dense molecular clouds regions are however less exposed to radiations, suggesting that the grain may truly act as a catalyst reducing the reaction barriers.
Pure H2O ice was detected at 3 μm by Gillett and Forrest since 1973 (Gillett et al., 1973), it is the most abundant molecular ice in dense ISM regions. While formaldehyde absorption features in ices were observed in young stellar objects (YSOs) in the 5.7–6 μm region. These detections hint to abundances around 6% with respect to H2O (Gibb et al., 2004; Boogert et al., 2008).
Formaldehyde (CH2O) ices are believed to form upon water-CO mixtures hydrogenation (Hiraoka et al., 1994; Hiraoka et al., 2002; Watanabe and Kouchi, 2002; Fuchs et al., 2009; Pirim et al., 2010). Comets are also known to contain water related ices in their cores, and when heated by nearby sources, can participate to a complex chemistry network (Schutte et al., 1993; Bossa et al., 2009; Vinogradoff et al., 2011). Laboratory experiments have tackled thermal evolution of comets in order to understand chemical pathways towards Complex Organic Molecules (COMs) formation (Duvernay et al., 2014).
Despite the past extensive investigations, some uncertainties remain, such as the exact composition of the grain models, and the role of mixed ices in the efficiency of H2 formation. The chemical mechanisms of H2 formation and the underlying energetic pathways can be model-dependent, and can in some cases present hindering energetic barriers preventing H2 formation at the low temperatures of the ISM. It is thus timely to provide a detailed energetic description of the possible reaction mechanisms on each specific grain model. In order to be used by astrochemical models, diffusion barriers, adsorption energies and analytic expressions of rate constants should be predicted.
Hydrogen addition and abstraction from formaldehyde have been investigated computationally with DFT by Goumans (Goumans, 2011), and CCSD (T) (by one of the authors) (Siaï et al., 2016) rate constants predictions in gas phase, and on pure water ice (Song and Kästner, 2017). In Woon (2002), quantum chemical electronic structure calculations of H + H2CO reaction in gas phase and in the presence of water clusters were lately investigated, the catalytic activity of the ice models was analyzed as a function of the activation energies magnitude and without any kinetic modeling. Song and Kästner (2017), employing a QM/MM model, studied the H2 formation on an amorphous solid water (ASW) upon which one formaldehyde molecule was adsorbed, and rate constants for both Langmuir-Hinshelwood and Eley-Rideal mechanisms were evaluated with the instanton theory, over the temperature range 60–300 K. On the other hand, Monte Carlo simulations (Cuppen et al., 2009) of formaldehyde formation upon CO hydrogenation on interstellar ice surfaces show that H2CO molecules are found to predominantly exist in the deeper layers of their ice model. H abstraction and addition reactions were also studied experimentally (Chuang et al., 2016) in mixed CO, CH3OH, and H2CO ices, where the authors confirm the occurrence of the product channels leading to CH3OH through hydrogenation, however successive abstraction reactions lead to the formation of HCO and back to CO molecules.
This study may serve as a useful investigation for further model variants to be explored computationally, and for suggesting possible constraints on new experimental setups relevant to ice models and H2 formation on icy grains in general.
In the present paper we are specifically interested in studying H2 formation from a formaldehyde-doped ice sample prepared by Molecular Dynamics techniques. The H abstraction from ice formaldehyde by a free incoming gaseous H atom will be investigated considering both reaction mechanisms:
- The Eley-Rideal (ER) mechanism:
- The Langmuir-Hinshelwood (LH) mechanism:
Where H (gas), H2 (gas) are the free hydrogen atom and molecule respectively in gas phase, while H (ads), H2 (ads) are the adsorbed species on the grain model.
We use a slab model of formaldehyde doped water ice, and choose a reactive site where an accessible CH2O molecule can be reached upon reaction. We investigate the reaction energetics for H2 (gas) formation with QM/MM methodologies. Semi-classical rate constants at a large temperature range are predicted using sophisticated Variational Transition State Theory (VTST) (Fernandez-Ramos et al., 2007), that allow unlike other methods to compute rate constants for temperatures above the cross over temperature Tc and also at very low temperatures ≈15 K characteristic of dense regions in the ISM where molecules are believed to form. We compare our results to available data from the literature and investigate the role of the present model grain and computational methodologies on the prediction of the energetics and the rate constants. In Computational Details we describe the methodology to generate the doped ice slab, the adopted computational methods for electronic structure and rate constants calculations. Results and their implications are discussed in Results and Discussion. Finally, the main results are put into perspective in Conclusion.
2 Computational Details
2.1 Classical Molecular Dynamics Calculations
The mixed formaldehyde/water ice was generated by classical molecular dynamics calculations using the Gromacs code (Abraham et al., 2015). The water molecules were described with the TIP4P/2005 model that proved to reproduce fairly well ice along the phase diagram (Abascal and Vega, 2005; Michoulier et al., 2018). OPLS force field (Jorgensen et al., 1983; Cornell et al., 1995) was employed for the formaldehyde molecules. The combination of OPLS and water models derived from TIP4P has been validated in Refs. (Collignon and Picaud, 2004; Ballenegger et al., 2006). The equations of motion were integrated by the leapfrog algorithm with a 1 fs time step. The Lennard-Jones and the short-range part of the Coulomb interactions were truncated at 10 Å. The long-range part of the Coulomb interaction was evaluated using the smooth particle-mesh Ewald method with a relative tolerance of 10–5, fourth-order cubic interpolation and a Fourier spacing parameter of 0.12. Water and formaldehyde molecules were treated as rigid bodies using the SETTLE algorithm.
Amorphous ice was doped with formaldehyde at the ratio 20 H2CO:1000 H2O (Pontoppidan et al., 2004). The slab is formed from 20 CH2O randomly distributed in a box containing 1000 H2O molecules. In order to favor diffusion of the molecules, an annealing procedure is carried out in the NPT ensemble using a Berendsen thermostat, heating the sample from 100 to 300 K and then cooling it back to 110 K for a total duration of 10 ns. The obtained box, of dimensions 26 × 30 × 35 Å, is then elongated along the z direction to form two interfaces exposed to vacuum. The resulting box cell dimensions are 26 × 30 × 100 Å. A final equilibration in the NVT ensemble was then performed at 110 K during 10 ns. A side view of the doped ice sample is shown in Figure 1A. The amorphous character of water was insured by checking the water density as shown in Figure 1B. Moreover, the compacity and low porosity of the ice are not affected by the presence of the organic molecules, the density of the ice slab is only changing from 0.955 g/cm3 for pure amorphous ice to 0.989 g/cm3 for our doped ice sample. H2CO molecules are distributed all over the slab but few of them remain at the surface. The abstraction reaction is subsequently studied from one of these surface formaldehyde molecules within a QM/MM scheme. It should be noted that at the low concentration considered here, the mutual interactions between H2CO molecules is screened by water molecules. The water network is also only slightly affected by the presence of the H2CO molecules.
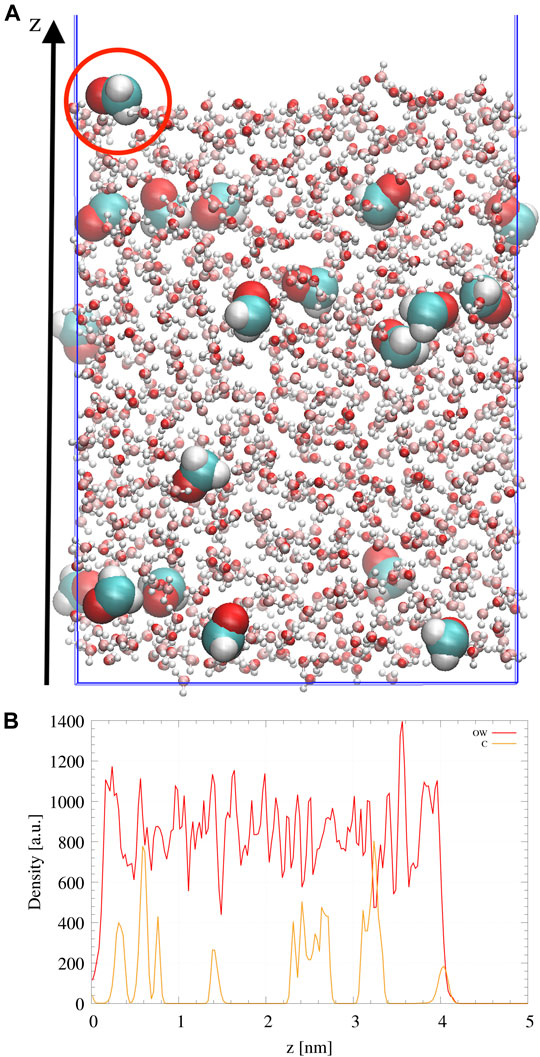
FIGURE 1. (A) Side view of the doped ice slab. Water is represented in light colors and CH2O by van der Waals spheres. The picked surface CH2O molecule is highlighted by a red circle. (B) Atomic distributions in arbitrary units along the z direction of the Oxygen atoms of H2O (OW) or Carbon atoms of CH2O (C). For a better visualization, Carbon density has been multiplied by a factor 10.
2.2 Quantum Chemical Calculations
Because of its amorphous nature and large molecular size, the whole system cannot be studied within a quantum mechanical treatment. Therefore, it has been divided into two parts. The first one, where the electronic energy of the system is calculated with an ab initio framework (QM), is located around the site where the reaction occurs. On the other hand, classical mechanics methods (MM) were used for the rest of the icy layer.
We employ the hybrid QM/MM ONIOM methodology (Chung et al., 2015) where the active species involved in the reaction process, i.e., H, CH2O and 10 H2O within a 4 Å radius of the CH2O molecule are studied with the hybrid meta-GGA functional M05-2X (Zhao et al., 2006) combined with an augmented correlation-consistent polarized valence basis set of Dunning namely, the aug-cc-pVTZ (Dunning, 1989; Kendall et al., 1992). This approach satisfactorily predicts the geometry and is particularly suitable for treating weak interactions as well as for providing accurate activation energy barriers (Hohenstein et al., 2008; Leverentz et al., 2013; Sirianni et al., 2018). M05-2X has been tested to perform in a similar way as M06-2X in the case of hydroxylated pyroxene clusters (Kerkeni et al., 2019). The rest of the system, i.e., the icy grain minus CH2O + 10H2O is treated with the Amber MM force field. The mechanical embedding scheme (Chung et al., 2015) was employed for all calculations. For open-shell structures, unrestricted wave functions were employed. All the DFT quantum chemical calculations were performed with the Gaussian 16 code (Frisch et al., 2016).
Initially the CH2O + 10H2O molecules in the QM region were allowed to relax in a QM/MM calculation, the final structure was afterwards used while freezing the 10 H2O molecules. Therefore, the reactive H and CH2O fragments were allowed to relax without any constraint during the reaction mechanism. The remainder of the grain model was kept frozen during all calculations. Geometry optimizations were carried out using the default Gaussian convergence thresholds. The nature of the stationary points of the potential energy surface (PES), minimum or saddle point, was checked by computing unscaled harmonic frequencies. Transition states (TSs) were searched for using the Berny algorithm (Schlegel, 1982). The Minimum Energy Path (MEP) was computed using the Intrinsic Reaction Coordinate (IRC) method (Fukui, 1981; Hratchian and Schlegel, 2005) with a gradient step size of 0.01 a0.(amu)1/2. For 46 points along each side of the MEP, the harmonic frequencies were computed at the M05-2X/aug-cc-pVTZ/Amber level. The MEP energies were finally corrected by adding the unscaled zero-point vibrational energy (ZPE), leading to the vibrational adiabatic potential energy
In order to obtain more accurate energies at an affordable cost, the DFT results were improved using a methodology recently used to obtain accurate binding energies of atoms and small molecules at the surface of both crystalline and amorphous ices (Duflot et al., 2021). To this end, the optimized geometries of the QM zone for all stationary and IRC points were recovered. The resulting cartesian coordinates were then used as inputs for a CBS/DLPNO-CCSD(T) (Guo et al., 2018) single point calculation, using the ORCA code (Neese, 2018; Neese et al., 2020). Resolution of the identity (RIJK) was used (Weigend et al., 2009) with the auxiliary basis set from Weigend (Weigend, 2008). For DLPNO-CCSD(T), the normal criteria were used as discussed by Liakos et al. (2015). The frozen core approximation was also used so carbon and oxygen 1 s orbitals were not included. The auxiliary basis set for correlation is taken from Hellweg et al. (2007). For Complete Basis Set (CBS) extrapolation, def2-TZVPP and def2-QVZPP basis sets were employed (Weigend and Ahlrichs, 2005). The Hartree-Fock and the correlation energies were extrapolated separately as shown in (Neese and Valeev, 2011) with α = 7.880 for Hartree-Fock and β = 2.970 for correlation. Interestingly, the T1 diagnostic (Lee et al., 1995) remains small along the path, with a maximum value of 1.2% at the TS geometry, thus confirming the absence of multireference character to the wavefunction. This diagnostic was slightly larger for gas phase species, peaking at 2.4% for the TS. By replacing the energy part of the total ONIOM calculation by the CBS/DLPNO-CCSD (T) energies, a new ONIOM extrapolated energy is obtained. On the other hand, the DFT frequencies and the resulting ZPE values were kept unmodified. This procedure was applied on the stationary and all the IRC points along the calculated reaction path.
2.3 Reaction Kinetics
Conventional Transition State Theory
The conventional transition-state-theory rate constant is the product of a symmetry number by kBT/h multiplied by the ratio of a partition function evaluated at the saddle point to one evaluated at the reactant, times an exponential decay of the classical barrier height as in Eq. 3. The partition functions are evaluated as products of electronic and vibrational partition functions. As we are dealing with surface reactions, we discard the relative translational partition function per unit volume and the rotational ones.
The conventional transition state-theory rate constant, kTST(T), is given by the following expression (Truhlar et al., 1985):
where
Canonical Variational Transition State Theory
Canonical variational transition-state theory (CVT) (Liu et al., 1993) has been used to estimate the rate constants. The canonical variational, transition state theory rate constant kCVT is obtained by finding the location of the minimum flux through the transition state at a given temperature. Note that in using a free energy of activation criterion to determine the location of the variational transition state, both entropic and energetic contributions are taken into account. We may write:
where
VMEP(s) is the classical potential along the minimum path (MEP) referenced to the reactant energy, s is the mass scaled reaction coordinate, and s*,CVT is the location of the canonical transition state on the reaction path, i.e., where the flux is minimal. s = 0 corresponds to the transition state; at the reactant s is a finite negative number, while for the product s is a finite positive number.
Semiclassical Reaction Probabilities
Light atoms such as hydrogen tunnel through barriers that cannot be surmounted classically. This quantum effect can be important at low temperatures. The exponential damping of wave tunnelling through a given barrier leads to the tunnelling probability P(E) at total energy E:
where θ(E) represents the amount of exponential decay. This function may be obtained in several ways.
Zero Curvature Tunnelling Method
When the reaction path curvature can be neglected, the proper tunnelling path is the MEP. For tunnelling along the MEP, θ(E) is the imaginary action integral given by:
VAG is the maximum of the ground state vibrational adiabatic potential energy:
where
In the ZCT approach, tunnelling is calculated from the semi-classical (i.e., WKB) ground-state transmission coefficient κ(T) (Skodje et al., 1981):
The transmission coefficient κ is temperature dependent, which takes into account tunnelling and non-classical reflection effects, κ(T) is given by
Small Curvature Tunnelling Method
Liu et al. (1993) have reported that tunnelling probabilities may increase because of the corner-cutting effect, which is not accounted for in the ZCT method. More advanced tunnelling methods are based on the inclusion of deviations between the tunnelling path and the reaction path (Vandeputte et al., 2010). In the present work, the centrifugal dominant small curvature semiclassical adiabatic ground state (CD-SCSAG) tunnelling method (Lu et al., 1992; Liu et al., 1993), is applied, where the effect of the reaction curvature is included by replacing the reduced mass μ by an effective mass μeff (Liu et al., 1993). The tunnelling probability at energy E is:
where θ(E) is the imaginary action integral along the tunnelling path as in Eq. 6. κ(T) is calculated as in Eq. 9 and the thermal rate constant is tunnelling corrected in the corresponding way as done for ZCT.
The TST, CVT, CVT/ZCT and the CVT/SCT rate constants are computed using the POLYRATE program, version 2010-A (Zheng et al., 2010). The CVT rates were fitted using the analytical expression proposed by Zheng and Truhlar (2010):
where α, β, γ and T0 are constants optimized to match the calculated rates.
3 Results and Discussion
3.1 Gas Phase Abstraction
In order to validate the methods employed in the ONIOM calculations, namely M05-2X/aug-cc-pVTZ and CBS/DLPNO-CCSD(T), we have employed them to calculate the gas phase reactions. It should be kept in mind that the CBS single point calculations were performed at the M05-2X geometries and ZPE is evaluated at this DFT level too. The results are compared to previous works (Goumans, 2011; Ruscic, 2015; Siaï et al., 2016; Song and Kästner, 2017) in Table 1. The computed energetics given in this table are the exothermicity (ΔE) and the vibrational adiabatic barrier (ΔVa) between the transition state and the reactants and between the products and the reactants, respectively. The corresponding ZPE-corrected values are labelled by a star symbol. It can be seen that the M05-2X/aug-cc-pVTZ overestimates the barrier height and underestimates the exothermicity when compared to the CBS/DLPNO-CCSD(T) values. The same behavior was also found previously at the MP2/cc-pVTZ level (Siaï et al., 2016). The CBS/DLPNO-CCSD(T) gas-phase barrier height (5.85 kcal/mol) is slightly below the previous CCSD(T)/cc-pVTZ//MP2/cc-pVTZ result of 6.69 kcal/mol (Siaï et al., 2016) and the CCSD(T)-F12a/cc-pVTZ-F12 value of 6.68 kcal/mol. The non-ZPE-corrected barriers can also be compared to the PWB6K-D3/def2-TZVP and (U)CCSD(T)-F12/def2-TZVP results of Song and Kästner (2017), i.e., 6.60 and 7.03 kcal/mol, respectively. As shown by these authors [see Table 4 in (Song and Kästner, 2017)], the barrier height is very sensitive to the choice of the functional and basis set, leading sometimes to negative values, but always below the (U)CCSD(T)-F12 result. Hence, our M05-2X/aug-cc-pVTZ value of 9.08 is too large. On the other hand, the present CBS/DLPNO-CCSD(T) value of 7.62 kcal/mol is only about 8% larger than the (U)CCSD(T)-F12 one. These results confirm that the present improved treatment, i.e., the composite methodology beyond DFT, ensures reliable results on the energetics of the reaction.
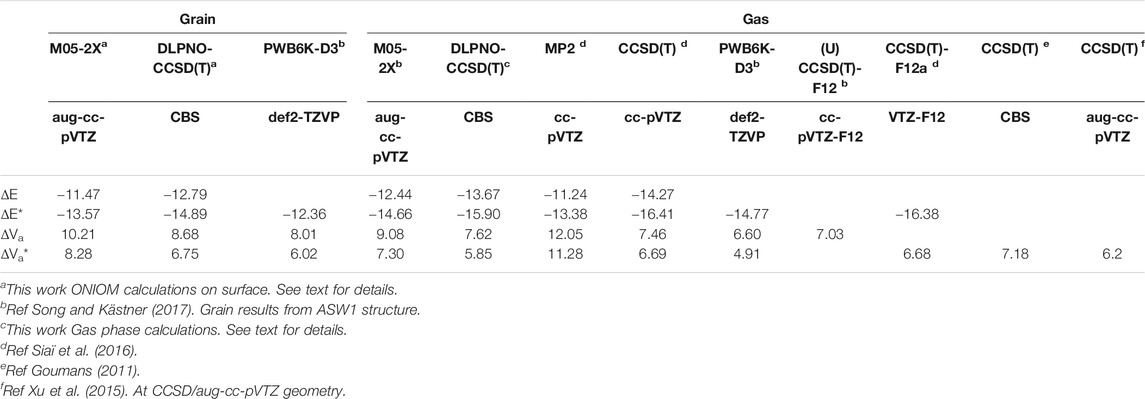
TABLE 1. Calculated exothermicity and barrier height compared to gas phase results (kcal/mol). The inclusion of ZPE is denoted by a star symbol.
3.2 Reaction Mechanisms on the Model Grain
3.2.1 Stationary Points
We initiated the reaction process by the reactive collision of one single gaseous H atom with an already adsorbed CH2O molecule dangling at the doped ice grain surface (Figure 2). The present geometry is similar to the most abundant one obtained by Song and Kästner on pure ASW ice (see Figure 1A in ref. (Song and Kästner, 2017). It is worth mentioning that in ref (Song and Kästner, 2017) a single formaldehyde molecule was adsorbed on their ice models. In the present model, the reactive formaldehyde oxygen atom forms a hydrogen bond with the nearest water hydrogen atom with a length of about 2.00 Å, close to the 1.90 Å value obtained by Song and Kästner. A value of 1.88 Å was also obtained at BHLYP/6-311++G (d,p) level on a 18-water cluster by Rimola et al. (Rimola et al., 2012). The search of the transition state corresponds to the geometrical configuration where the hydrogen in CH2O is abstracted by the incoming H atom (Figure 3 and Table 2). At the transition state the CO bond is shortened by ≈0.017 Å with respect to the reactant one (see Figure 3 and Table 2). Table 3 displays the calculated frequencies, showing the 6 low-value modes corresponding to the motion of the H2CO and TS structures with respect to the surface. When the H atom abstraction occurs, H2 forms with an equilibrium distance of 0.738 Å, and the distance from the carbon to the abstracted H is 2.324 Å, the CHH angle being ≈177°. The abstraction process is thus quasi-colinear, very much like in the gas phase case (Siaï et al., 2016) and on pure ASW ice (Song and Kästner, 2017). The relevant geometrical parameters are given in Figure 4 and Table 2 for all involved species in the reaction mechanism. The calculated H-H distance at the TS (1.068 Å) is slightly larger than the 1.022 Å obtained by Song and Kästner (2017).
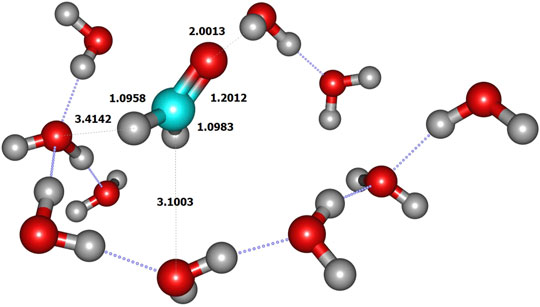
FIGURE 2. Reactant geometry (limited to QM zone).
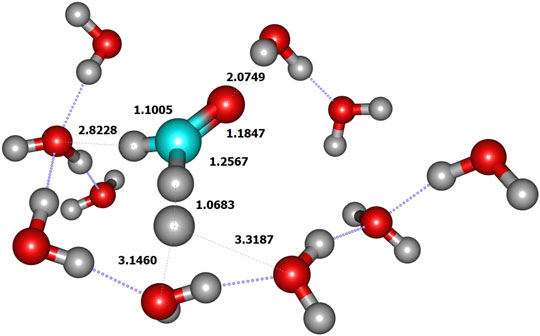
FIGURE 3. TS geometry (limited to QM zone).
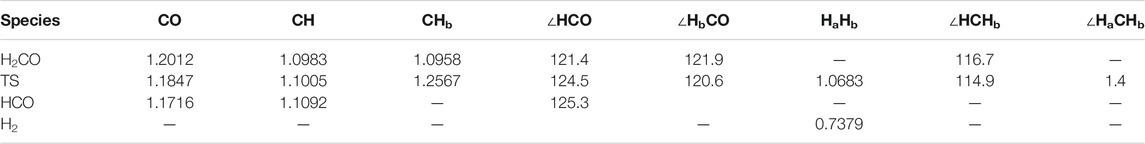
TABLE 2. Molecular geometry parameters of the fragments [bond lengths (Å) and angles (°)] at the M05-2X/aug-cc-pVTZ level. Hb is the abstracted atom, while Ha is the incoming atom.

TABLE 3. Calculated harmonic vibrational frequencies (in cm−1) and zero-point energies (in Hartrees) for the stationary points.
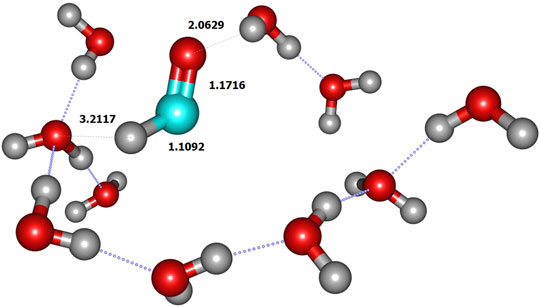
FIGURE 4. Product geometry (limited to QM zone).
All attempts to locate reactive and product Van der Waals complexes failed. Song and Kästner (2017) mention a complex with an energy very close to the reactants but no details are given about its geometry. In the gas phase case (Siaï et al., 2016), it was concluded that the product complex was electronically unstable after including BSSE effects. The reactant complex, with the incoming H atom at about 3.35 Å, lies very close to the reactant (-023 kcal/mol at CCSD(T) level) and did not play any significant role in the dynamics study of the abstraction reaction.
Figure 5 shows a comparison between the geometries of the stationary points and the last points of the IRC calculations. Beyond these geometries, the IRC calculation stops due to the stability of the energy. On the reactant side, at CBS level, the last IRC point lies 0.23 kcal/mol (with ZPE) above the energies of the isolated grain + H system. This suggests that the reaction barrier for the Eley-Rideal mechanism would be 0.23 kcal/mol higher than that for the Langmuir-Hinshelwood one (see Table 4). The H atoms is at ≈ 3.4 Å from the CH2 closest hydrogen. On the other side, the last IRC point is 1.71 kcal/mol above the isolated products (see also Figure 6). The closest H atom of H2 is ≈2.8 Å from the carbon atom. For both cases there remains a large difference between the orientation of the isolated H2CO (Figure 5A) or HCO (Figure 5B) molecules compared to the last IRC point, which proves that the convergence of the MEP calculation has already reached local minima in both sides. The same behavior is also found for the TS as shown in Figures 5C & 5D, where one can see the orientation of the reactant and product compared to the TS. Several attempts were made to determine whether isolated reactant and product geometries were in fact local minima and not connected to the IRC path. For example, the geometry optimization was restarted using the final IRC geometries by removing the H and H2 molecules. In all cases, the minima were found to be the same as obtained independently (as shown in Figures 2, 4). This suggests that the TS and the MEP are common to the ER and LH mechanisms. There are several possibilities to explain these results. First, the presence of an H atom or an H2 molecule even at rather large distances has a strong enough influence to re-orient the H2CO and HCO molecules with respect to the surface. Nevertheless, the PES is rather flat too. Finally, the fact that the surrounding water molecules are frozen may have also some minor impact on the geometries.
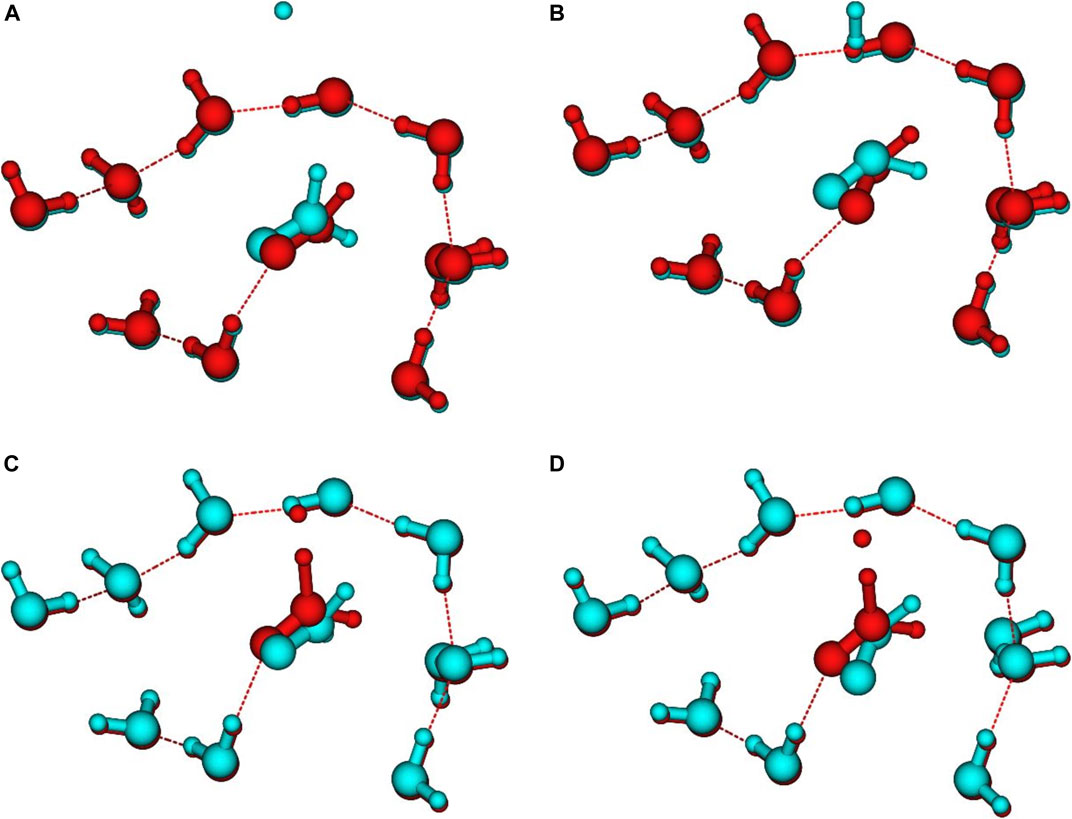
FIGURE 5. Comparison between: (A) reactant (red) and last VMEP point (blue) geometries, (B) product (blue) and last VMEP point (red) geometries, (C) reactant (blue) and TS (red) geometries, (D) product (blue) and TS (red) geometries.
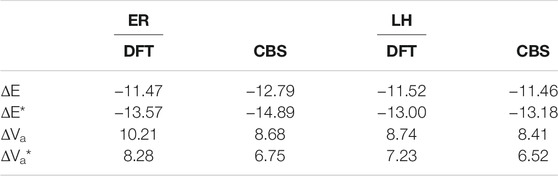
TABLE 4. Comparison between ER and LH ZPE-corrected energies (kcal/mol).
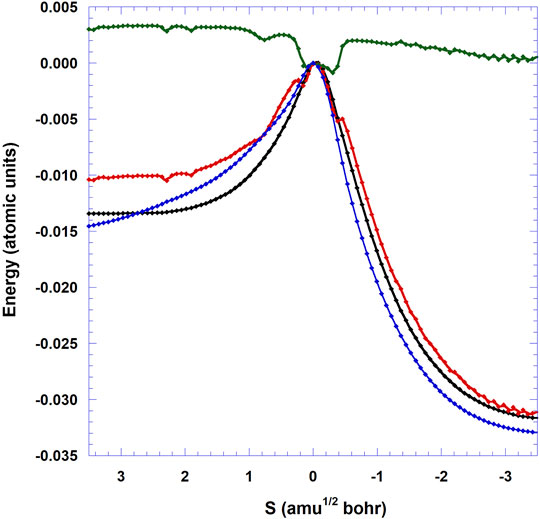
FIGURE 6. Minimum energy path VMEP (blue: ONIOM(M05-2X:Amber) level, black: CBS/DLPNO-CCSD(T)//ONIOM(M05-2X:Amber) level), vibrational adiabatic potential energy (VGa, red), and ΔZPE (green) of the 3N-6 vibrational modes with respect to TS as functions of (s) (amu1/2 a0). The energy of the TS is set to 0.
Table 1 shows the comparison between the ONIOM energies of the stationary points using the two levels of calculations [M05-2X/aug-cc-pVTZ and CBS/DLPNO-CCSD(T)]. The same trends found in gas phase occur, namely that the exothermicity decreases when going from M05-2X to CBS (−11.47 to12.79 kcal/mol for ΔE). On the other hand, the barrier height is reduced, from 8.28 to 6.75 kcal/mol. It is also possible to compare the values of these properties to those obtained in gas phase with the same methods. As can be seen in Table 1, the exothermicity is larger in gas phase for both methods, while the barrier height is only slightly larger. This would certainly result in gas phase rate constants being somehow larger at small temperatures than those obtained from the grain model. Thus, the ice model yields a noticeable stabilization of the gas-phase TS with respect to the effect on the minima. Song and Kästner (2017), at the PWB6K-D3/def2-TZVP level, found the same behavior going from gas phase to pure ASW surface, i.e., an augmentation of the barrier height by about 1 kcal/mol and a reduction of the exothermicity (Table 1). This latter reduction is somewhat larger in Song and Kästner (2.41 kcal/mol) compared to the present result (1.01 kcal/mol).
Another interesting quantity which can be obtained from the present calculations is the binding energy (BE) of the reactants and products to the ice surface [see also (Duflot et al., 2021)]. We define the BE of a molecule M on the icy grain as the positive quantity:
where E (ice + M) is the energy of the adsorbed molecule M on the icy grain, E(M) is the energy of M in the gas phase and E(ice) is the energy of the bare icy grain. The calculations are performed with the QM/MM procedure detailed above, and include ZPE from DFT. For H2CO, BE is found to be 9.18 kcal/mol at DFT level and 8.38 kcal/mol at CBS level. At the PWB6K-D3/def2-TZVP/MM level (without ZPE), Song and Kästner (2017) found a broad distribution of BE’s on 81 structures obtained on pure ASW, between about 2.0 and 18.6 kcal/mol. The distribution was centered on BE’s between about 8.0 and 9.9 kcal/mol. These authors selected one ASW structure upon which they calculated the rates with a BE of about 9.14 kcal/mol. Finally, our QM/MM BE value lies within accordance with their BE distribution.
For HCO, the DFT and CBS values we computed are 8.08 and 7.37 kcal/mol, respectively. For this latter species, the CBS values can be compared to the largest BE’s obtained on pure crystalline (7.08 kcal/mol) and amorphous (5.67 kcal/mol) ices (Duflot et al., 2021). However, it should be kept in mind that in the present work, the ice surface was not allowed to relax in reactivity studies, and that in ref (Duflot et al., 2021) the ice model is either pure ASW, or perfect crystalline Ih ice. Relaxation of the surface during the reaction mechanism could alter the BE even in the case of physisorption.
3.2.2 Minimum Energy Path
Figure 6 displays the variation of the classical potential energy (VMEP) as a function of the reaction coordinate (s) for the reaction studied here on the ice grain. The CBS VMEP curve is flatter than the DFT one on the reactant side. The reduction of the barrier height is visualized via the crossing of the CBS and DFT curves. Unlike the gas phase case where the VMEP curve presents a discontinuity at s = −0.25 amu1/2 bohr [see Figure 1 in (Siaï et al., 2016)], due to the single point CCSD(T)//MP2 level, the present DFT and CBS VMEP profiles are more regular. The CBS
3.2.3 Kinetic Study: H2 Formation Mechanisms
Eley-Rideal Mechanism
Figure 7A displays the calculated rates for the Eley-Rideal mechanism, compared to the analytical functions fitted using Eq. 11 and the results from Song and Kästner (2017). It should be kept in mind that Song and Kästner used instanton theory (Zaverkin and Kästner, 2020) between 59 K and the so-called crossover temperature (Affleck, 1981) Tc = 366 K and variational TST for higher temperatures up to 1000 K. Also, in Eq. 11, the β parameter was set to 1.0 in ref. (Song and Kästner, 2017). In the present work, β was optimized and the temperature spans a broader interval (15–2000 K). The fitted parameters for CVT rates are shown in Table 5 and compared to Song and Kästner. From Figure 7A, it is clear that Eq. 11 fits very well both ZCT and SCT rates. At very high temperatures, tunnelling effects are small (e.g., κ ≈ 1.1 at 2000 K for both ZCT and SCT). However, at room temperature, tunneling effects are still important with κ(ZCT) = 16.8 and κ(SCT) = 34.0. As recently pointed out by Richardson (2018), when the TS imaginary frequency is larger than 1300 cm−1, deep-tunnelling still occurs at this temperature. This is indeed the case in the present work since the TS imaginary frequency is 1737 cm−1 (Table 3). This gives a cross-over temperature of Tc ≈ 400 K, which is very similar to that of Song and Kästner (2017). At low temperatures, the CVT/ZCT rates appear to be several orders of magnitude lower than the CVT/SCT, which is a common trend (Truhlar and Kuppermann, 1971; Liu et al., 1993), due to the corner cutting effects (Allison and Truhlar, 1998). Our CVT/SCT curve is thus closer to Song and Kästner’s one than the CVT/ZCT one. There remains however a difference of several orders of magnitude as can be seen in Table 6, between the present rates and the values provided by Song and Kästner for selected temperature values. The differences are due to the different barrier heights, IRC shape and theoretical models used to compute the rates. These differences are also reflected in Table 5: there is a large difference between the ZCT and SCT fitted parameters. Furhtermore, β is significantly different from 1 in our case.
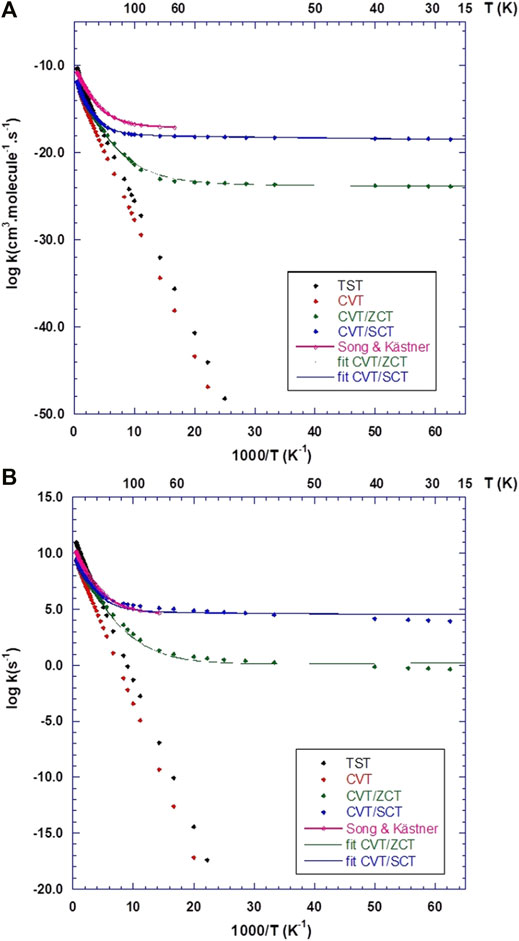
FIGURE 7. Calculated rate constants using CBS/DLPNO-CCSD(T)//ONIOM(M05-2X:Amber) results, compared to the analytical functions provided by Song and Kästner (2017). (A) Eley-Rideal (ER) rates. (B) Langmuir-Hinshelwood (LH) rates.
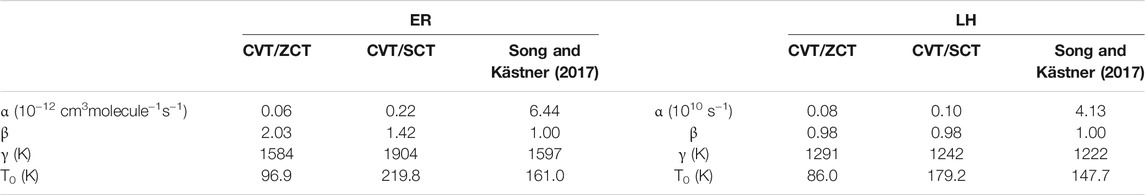
TABLE 5. Analytical parameters for the fitted rate constants.
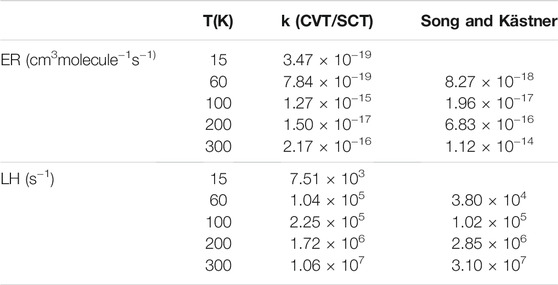
TABLE 6. Calculated rate constants at selected temperatures.
Langmuir-Hinshelwood Mechanism
In Figure 7B, the calculated rate constants obtained for the LH mechanism are shown together with Song and Kästner (2017)’s fitted rate function. As in the ER case, the CVT/ZCT and CVT/SCT curves were fitted using Eq. 11. The optimized parameters are given in Table 5. Most of the trends observed in the ER case, such as the importance of tunnelling at high and low temperatures and the difference between the ZCT and SCT results, are also observed. The main difference comes from the comparison between the present CVT/SCT rate and Song and Kästner's one: both curves appear to be very close, especially at low temperature. This is reflected in the fitted parameters given in Table 5: although our α value is about 40 times larger, the other parameters remain very similar. In contrast to our ER results, β is now very close to 1. Table 6 also shows that the rates differ by a factor of around 3 at all selected temperatures. The similarity with Song and Kästner's results may be rationalized by the closeness of our LH barrier (Table 4) compared to ref (Song and Kästner, 2017). Indeed, these authors calculated the LH rates using a complex with a barrier of 3030 K or 6.02 kcal/mol, virtually identical to the ER barrier. As shown in Table 4, our LH barrier is very close to this value (6.52 kcal/mol at the CBS level). Given the large rate coefficients predicted in the LH mechanism even at T = 15 K, it is anticipated that besides the exothermicity of the reaction process, the reaction mechanism is in favor of the formation of gaseous H2 molecules. However, even though our ice model is intended to mimic ISM conditions, we would expect that diffusion of the incoming H (gas) atoms, so that they meet one CH2O on the grain, will be the rate limiting step.
4 Conclusion
In the present work, we have employed Density Functional Theory for geometry optimization, ZPE calculations, and CBS/DLPNO-CCSD(T)//ONIOM/aug-cc-pVTZ single point energies methods on a large-scale system derived from a classical molecular dynamics trajectory. These calculations serve indeed as a feasibility assessment of previously benchmarked DFT (and to some extent, CVT) methods for the study of chemical reactions.
We study the abstraction reaction between an incoming gas phase hydrogen atom and another hydrogen atom belonging to a formaldehyde molecule at the surface of a mixed formaldehyde/water icy grain. The size of the system required the use of hybrid QM/MM techniques to obtain the detailed shape of the full reaction pathway including the effect of the environment. The entire minimum energy path was computed with CBS and M05-2X zero-point energy inclusion for each stationary point.
These results were then used to obtain the rate constants of this reaction between 15 and 2000 K for both ER and LH mechanisms using methods more sophisticated than the usual transition state theory, including contributions from variational and tunnelling effects. This work provides new results with respect to the choice of the model grain, i.e., doped formaldehyde-H2O amorphous ice, to the application of CBS model chemistry as opposed to pure DFT treatments, and to the use of VTST over a larger temperature range than what was reported previously. Our calculations show that both reaction models (ER and LH) exhibit exothermic pathways.
The present work focused on the H abstraction of H2CO and formation of H2, but the H2CO + H reaction has actually several possible products, such as H3CO, H2COH. Using the proposed methodology to characterize the latter products formation through reactions at the grain surface would definitively be of interest and complementary to the present work.
In addition, the reaction was fully characterized for one configuration that was arbitrarily chosen along a classical molecular dynamics trajectory. It can be expected that various binding sites may exist on the surface characterized by different environments around the surface CH2O molecule. To confirm the trend, a sampling should be carried out over a variety of binding sites. However, a full characterization of the reaction path, as presented here, has to be carried out for each binding environment, such an investigation certainly requires a substantial computational effort.
Finally, the reaction was characterized on a mixed grain, the reacting CH2O molecule forming part of the doped ice model. This work proposes a methodology to account for the presence of impurities in interstellar ices. Further efforts are needed to quantify more precisely the influence of these dopants on reaction rate constants both in terms of molecular or atomic diversity and concentration of the species.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
DD did some of the quantum chemical calculations, data curating and wrote parts of the manuscript. BK performed all DFT calculations, and reaction rates, and provided a first draft of the paper. MB prepared the QM/MM input structures for Gaussian calculations. MH performed the MD calculations. CT supervised the MD calculations and participated in the data curating.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the PCMI (Programme National de Physique et Chimie du Milieu Interstellaire) for support and the Centre de Ressources Informatiques (CRI) of the Université of Lille for providing computing time. MH acknowledges Dr C. Fotsing-Kwetche and Dr E. Michoulier for their help on the Molecular Dynamics part. MH acknowledges the Ministry of Higher Education and Scientific Research Tunisia for 2 exchange scholarships with Laboratoire de Physique des Lasers, Atomes et Molécules, UMR CNRS 8523, Université de Lille. DD and CT acknowledge the support of the OVERSEE and CAPPA grants, managed by the Agence Nationale de la Recherche under the frame programs Investissements d’Avenir ANR-10-LABX-005 and I-SITE ULNE/ANR-16-IDEX-0004 ULNE, respectively. They also thank the Région Hauts de France, the Ministre de l’Enseignement Supérieur et de la Recherche (CPER Climibio) and the European Fund for Regional Economic Development for their support. This work used HPC resources from GENCI-TGCC (Grant No. 2020–A0050801859).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2022.807649/full#supplementary-material
Supplementary Figure S1 | Radial distribution functions (A) between the water oxygens in case of doped ice and pure ice at T=110K; (B) between the carbon atoms of the H2CO molecules
Supplementary Table S1 | Complete calculated reaction ER rate constants in cm3molecule-1s-1 for H2 formation on the grain model at CBS level.
Supplementary Table S2 | Complete calculated reaction LH rate constants in s-1 for H2 formation on the grain model at CBS level.
Supplementary Table S3 | Calculated absolute energies (a.u.).
References
Abascal, J. L. F., and Vega, C. (2005). A General Purpose Model for the Condensed Phases of Water: TIP4P/2005. J. Chem. Phys. 123, 234505. doi:10.1063/1.2121687
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 1-2, 19–25. doi:10.1016/j.softx.2015.06.001
Affleck, I. (1981). Quantum-Statistical Metastability. Phys. Rev. Lett. 46, 388–391. doi:10.1103/PhysRevLett.46.388
Allison, T. C., and Truhlar, D. G. (1998). “Testing the Accuracy of Practical Semiclassical Methods: Variational Transition State Theory with Optimized Multidimensional Tunneling,” in Modern Methods for Multidimensional Dynamics Computations in Chemistry (World Scientific), 618–712. doi:10.1142/9789812812162_0016
Ballenegger, V., Picaud, S., and Toubin, C. (2006). Molecular Dynamics Study of Diffusion of Formaldehyde in Ice. Chem. Phys. Lett. 432, 78–83. doi:10.1016/j.cplett.2006.10.014
Boogert, A. C. A., Pontoppidan, K. M., Knez, C., Lahuis, F., Kessler-Silacci, J., van Dishoeck, E. F., et al. (2008). The c2dSpitzerSpectroscopic Survey of Ices Around Low-Mass Young Stellar Objects. I. H2O and the 5-8 μm Bands1,2. Astrophysical J. 678, 985–1004. doi:10.1086/533425
Bossa, J. B., Theule, P., Duvernay, F., and Chiavassa, T. (2009). NH2CH2OH Thermal Formation in Interstellar Ices Contribution to the 5-8 μm Region Toward Embedded Protostars. Astrophysical J. 707, 1524–1532. doi:10.1088/0004-637X/707/2/1524
Carruthers, G. R. (1970). Rocket Observation of Interstellar Molecular Hydrogen. Astrophysical J. 161, L81. doi:10.1086/180575
Cazaux, S., Morisset, S., Spaans, M., and Allouche, A. (2011). When Sticking Influences H2formation. Astron. Astrophysics 535, A27. doi:10.1051/0004-6361/201117220
Chaabouni, H., Bergeron, H., Baouche, S., Dulieu, F., Matar, E., Congiu, E., et al. (2012). Sticking Coefficient of Hydrogen and Deuterium on Silicates under Interstellar Conditions. Astron. Astrophysics 538, A128. doi:10.1051/0004-6361/201117409
Chuang, K.-J., Fedoseev, G., Ioppolo, S., van Dishoeck, E. F., and Linnartz, H. (2016). H-atom Addition and Abstraction Reactions in Mixed CO, H2CO and CH3OH Ices - an Extended View on Complex Organic Molecule Formation. Monthly Notices R. Astronomical Soc. 455, 1702–1712. doi:10.1093/mnras/stv2288
Chung, L. W., Sameera, W. M. C., Ramozzi, R., Page, A. J., Hatanaka, M., Petrova, G. P., et al. (2015). The ONIOM Method and its Applications. Chem. Rev. 115, 5678–5796. doi:10.1021/cr5004419
Collignon, B., and Picaud, S. (2004). Comparison between Methanol and Formaldehyde Adsorption on Ice: a Molecular Dynamics Study. Chem. Phys. Lett. 393, 457–463. doi:10.1016/j.cplett.2004.06.085
Congiu, E., Minissale, M., Baouche, S., Chaabouni, H., Moudens, A., Cazaux, S., et al. (2014). Efficient Diffusive Mechanisms of O Atoms at Very Low Temperatures on Surfaces of Astrophysical Interest. Faraday Discuss. 168, 151–166. doi:10.1039/C4FD00002A
Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., et al. (1995). A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 117, 5179–5197. doi:10.1021/ja00124a002
Creighan, S. C., Perry, J. S. A., and Price, S. D. (2006). The Rovibrational Distribution of H2 and HD Formed on a Graphite Surface at 15-50 K. J. Chem. Phys. 124, 114701. doi:10.1063/1.2174878
Cuppen, H. M., Kristensen, L. E., and Gavardi, E. (2010). H2 Reformation in post-shock Regions. Monthly Notices R. Astronomical Soc. Lett. 406, no–L15. doi:10.1111/j.1745-3933.2010.00871.x
Cuppen, H. M., van Dishoeck, E. F., Herbst, E., and Tielens, A. G. G. M. (2009). Microscopic Simulation of Methanol and Formaldehyde Ice Formation in Cold Dense Cores. Astron. Astrophysics 508, 275–287. doi:10.1051/0004-6361/200913119
Duflot, D., Toubin, C., and Monnerville, M. (2021). Theoretical Determination of Binding Energies of Small Molecules on Interstellar Ice Surfaces. Front. Astron. Space Sci. 8, 645243. doi:10.3389/fspas.2021.645243
Dunning, T. H. (1989). Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms boron through Neon and Hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Duvernay, F., Rimola, A., Theule, P., Danger, G., Sanchez, T., and Chiavassa, T. (2014). Formaldehyde Chemistry in Cometary Ices: the Case of HOCH2OH Formation. Phys. Chem. Chem. Phys. 16, 24200–24208. doi:10.1039/C4CP03031A
Ehrenfreund, P., and Schutte, W. A. (2000). ISO Observations of Interstellar Ices: Implications for the Pristinity of Comets. Adv. Space Res. 25, 2177–2188. doi:10.1016/S0273-1177(99)01151-5
Farebrother, A. J., Meijer, A. J. H. M., Clary, D. C., and Fisher, A. J. (2000). Formation of Molecular Hydrogen on a Graphite Surface via an Eley-Rideal Mechanism. Chem. Phys. Lett. 319, 303–308. doi:10.1016/S0009-2614(00)00128-7
Fernandez-Ramos, A., Ellingson, B. A., Garrett, B. C., and Truhlar, D. G. (2007). Variational Transition State Theory with Multidimensional Tunneling - Fernandez‐Ramos - 2007 - Reviews in Computational Chemistry. Wiley Online Library. Available from: https://onlinelibrary-wiley-com.ressources-electroniques.univ-lille.fr/doi/10.1002/9780470116449.ch3 (accessed September 27, 2021).
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16. Wallingford, CT: Rev. B.01.
Fuchs, H., Brauers, T., Häseler, R., Holland, F., Mihelcic, D., Müsgen, P., et al. (2009). Intercomparison of Peroxy Radical Measurements Obtained at Atmospheric Conditions by Laser-Induced Fluorescence and Electron Spin Resonance Spectroscopy. Atmos. Meas. Tech. 2, 55–64. doi:10.5194/amt-2-55-2009
Fukui, K. (1981). The Path of Chemical Reactions - the IRC Approach. Acc. Chem. Res. 14, 363–368. doi:10.1021/ar00072a001
Gibb, E. L., Whittet, D. C. B., Boogert, A. C. A., and Tielens, A. G. G. M. (2004). Interstellar Ice: The Infrared Space Observatory Legacy. Astrophys J. Suppl. S 151, 35–73. doi:10.1086/381182
Gillett, F. C., Forrest, W. J., and Merrill, K. M. (1973). 8-13 Micron Spectra of NGC 7027, BD +30 3639 and NGC 6572. Astrophysical J. 183, 87. doi:10.1086/152211
Goumans, T. P. M. (2011). Isotope Effects for Formaldehyde Plus Hydrogen Addition and Abstraction Reactions: Rate Calculations Including Tunnelling. Monthly Notices R. Astronomical Soc. 413, 2615–2620. doi:10.1111/j.1365-2966.2011.18329.x
Guo, Y., Riplinger, C., Becker, U., Liakos, D. G., Minenkov, Y., Cavallo, L., et al. (2018). Communication: An Improved Linear Scaling Perturbative Triples Correction for the Domain Based Local Pair-Natural Orbital Based Singles and Doubles Coupled Cluster Method [DLPNO-CCSD(T)]. J. Chem. Phys. 148, 011101. doi:10.1063/1.5011798
Hellweg, A., Hättig, C., Höfener, S., and Klopper, W. (2007). Optimized Accurate Auxiliary Basis Sets for RI-MP2 and RI-CC2 Calculations for the Atoms Rb to Rn. Theor. Chem. Acc. 117, 587–597. doi:10.1007/s00214-007-0250-5
Hiraoka, K., Ohashi, N., Kihara, Y., Yamamoto, K., Sato, T., and Yamashita, A. (1994). Formation of Formaldehyde and Methanol from the Reactions of H Atoms with Solid CO at 10-20 K. Chem. Phys. Lett. 229, 408–414. doi:10.1016/0009-2614(94)01066-8
Hiraoka, K., Sato, T., Sato, S., Sogoshi, N., Yokoyama, T., Takashima, H., et al. (2002). Formation of Formaldehyde by the Tunneling Reaction of H with Solid CO at 10 K Revisited. Astrophysical J. 577, 265–270. doi:10.1086/342132
Hohenstein, E. G., Chill, S. T., and Sherrill, C. D. (2008). Assessment of the Performance of the M05−2X and M06−2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules. J. Chem. Theor. Comput. 4, 1996–2000. doi:10.1021/ct800308k
Hollenbach, D., and Salpeter, E. E. (1970). Surface Adsorption of Light Gas Atoms. J. Chem. Phys. 53, 79–86. doi:10.1063/1.1673836
Hratchian, H. P., and Schlegel, H. B. (2005). Using Hessian Updating to Increase the Efficiency of a Hessian Based Predictor-Corrector Reaction Path Following Method. J. Chem. Theor. Comput. 1, 61–69. doi:10.1021/ct0499783
Ivanovskaya, V. V., Zobelli, A., Teillet-Billy, D., Rougeau, N., Sidis, V., and Briddon, P. R. (2010). EnhancedH2catalytic Formation on Specific Topological Defects in Interstellar Graphenic Dust Grain Models. Phys. Rev. B 82, 245407. doi:10.1103/PhysRevB.82.245407
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Katz, N., Furman, I., Biham, O., Pirronello, V., and Vidali, G. (1999). Molecular Hydrogen Formation on Astrophysically Relevant Surfaces. Astrophysical J. 522, 305–312. doi:10.1086/307642
Kendall, R. A., Dunning, T. H., and Harrison, R. J. (1992). Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 96, 6796–6806. doi:10.1063/1.462569
Kerkeni, B., Bacchus-Montabonel, M.-C., Shan, X., and Bromley, S. T. (2019). Understanding H2 Formation on Hydroxylated Pyroxene Nanoclusters: Ab Initio Study of the Reaction Energetics and Kinetics. J. Phys. Chem. A. 123, 9282–9291. doi:10.1021/acs.jpca.9b06713
Kerkeni, B., and Bromley, S. T. (2013). Competing Mechanisms of Catalytic H2 Formation and Dissociation on Ultrasmall Silicate Nanocluster Dust Grains. Monthly Notices R. Astronomical Soc. 435, 1486–1492. doi:10.1093/mnras/stt1389
Latimer, E. R., Islam, F., and Price, S. D. (2008). Studies of HD Formed in Excited Vibrational States from Atomic Recombination on Cold Graphite Surfaces. Chem. Phys. Lett. 455, 174–177. doi:10.1016/j.cplett.2008.02.105
Lee, T. J., and Scuseria, G. E. (1995). “Achieving Chemical Accuracy with Coupled-Cluster Theory,” in Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy. Editor S. R. Langhoff (Dordrecht: Springer Netherlands), 47–108. doi:10.1007/978-94-011-0193-6_2
Leverentz, H. R., Qi, H. W., and Truhlar, D. G. (2013). Assessing the Accuracy of Density Functional and Semiempirical Wave Function Methods for Water Nanoparticles: Comparing Binding and Relative Energies of (H2O)16 and (H2O)17 to CCSD(T) Results. J. Chem. Theor. Comput. 9, 995–1006. doi:10.1021/ct300848z
Liakos, D. G., Sparta, M., Kesharwani, M. K., Martin, J. M. L., and Neese, F. (2015). Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theor. Comput. 11, 1525–1539. doi:10.1021/ct501129s
Liu, Y. P., Lynch, G. C., Truong, T. N., Lu, D. H., Truhlar, D. G., and Garrett, B. C. (1993). Molecular Modeling of the Kinetic Isotope Effect for the [1,5]-sigmatropic Rearrangement of Cis-1,3-Pentadiene. J. Am. Chem. Soc. 115, 2408–2415. doi:10.1021/ja00059a041
Lu, D.-h., Truong, T. N., Melissas, V. S., Lynch, G. C., Liu, Y.-P., Garrett, B. C., et al. (1992). POLYRATE 4: A New Version of a Computer Program for the Calculation of Chemical Reaction Rates for Polyatomics. Comp. Phys. Commun. 71, 235–262. doi:10.1016/0010-4655(92)90012-N
Michoulier, E., Noble, J. A., Simon, A., Mascetti, J., and Toubin, C. (2018). Adsorption of PAHs on Interstellar Ice Viewed by Classical Molecular Dynamics. Phys. Chem. Chem. Phys. 20, 8753–8764. doi:10.1039/C8CP00593A
Navarro-Ruiz, J., Martínez-González, J. Á., Sodupe, M., Ugliengo, P., and Rimola, A. (2015). Relevance of Silicate Surface Morphology in Interstellar H2formation. Insights from Quantum Chemical Calculations. Mon. Not. R. Astron. Soc. 453, 914–924. doi:10.1093/mnras/stv1628
Navarro-Ruiz, J., Sodupe, M., Ugliengo, P., and Rimola, A. (2014). Interstellar H Adsorption and H2 Formation on the Crystalline (010) Forsterite Surface: a B3LYP-D2* Periodic Study. Phys. Chem. Chem. Phys. 16, 17447–17457. doi:10.1039/C4CP00819G
Navarro-Ruiz, J., Ugliengo, P., Sodupe, M., and Rimola, A. (2016). Does Fe2+ in Olivine-Based Interstellar Grains Play Any Role in the Formation of H2? Atomistic Insights from DFT Periodic Simulations. Chem. Commun. 52, 6873–6876. doi:10.1039/C6CC02313D
Neese, F. (2018). Software Update: the ORCA Program System, Version 4.0. Wires Comput. Mol. Sci. 8, e1327. doi:10.1002/wcms.1327
Neese, F., and Valeev, E. F. (2011). Revisiting the Atomic Natural Orbital Approach for Basis Sets: Robust Systematic Basis Sets for Explicitly Correlated and Conventional Correlated Ab Initio Methods. J. Chem. Theor. Comput. 7, 33–43. doi:10.1021/ct100396y
Neese, F., Wennmohs, F., Becker, U., and Riplinger, C. (2020). The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 152, 224108. doi:10.1063/5.0004608
Oueslati, I., Kerkeni, B., Spielfiedel, A., Tchang-Brillet, W.-Ü. L., and Feautrier, N. (2014). Ab Initio Investigation of the Abstraction Reactions by H and D from Tetramethylsilane and its Deuterated Substitutions. J. Phys. Chem. A. 118, 791–802. doi:10.1021/jp407310c
Oueslati, I., Kerkeni, B., Tchang-Brillet, W.-Ü. L., and Feautrier, N. (2015). Quantum Scattering Study of the Abstraction Reaction of H Atoms from Tetramethylsilane. Chem. Phys. Lett. 624, 29–36. doi:10.1016/j.cplett.2015.02.002
Pirim, C., Krim, L., Laffon, C., Parent, P., Pauzat, F., Pilmé, J., et al. (2010). Preliminary Study of the Influence of Environment Conditions on the Successive Hydrogenations of CO. J. Phys. Chem. A. 114, 3320–3328. doi:10.1021/jp909600q
Pirronello, V., Biham, O., Liu, C., Shen, L., and Vidali, G. (1997). Efficiency of Molecular Hydrogen Formation on Silicates. Astrophysical J. 483, L131–L134. doi:10.1086/310746
Pontoppidan, K. M., van Dishoeck, E. F., and Dartois, E. (2004). Mapping Ices in Protostellar Environments on 1000 AU Scales. Astron. Astrophysics 426, 925–940. doi:10.1051/0004-6361:20041276
Richardson, J. O. (2018). Perspective: Ring-Polymer Instanton Theory. J. Chem. Phys. 148, 200901. doi:10.1063/1.5028352
Rimola, A., Sodupe, M., and Ugliengo, P. (2012). Computational Study of Interstellar Glycine Formation Occurring at Radical Surfaces of Water-Ice Dust Particles. Astrophysical J. 754, 24. doi:10.1088/0004-637x/754/1/24
Ruscic, B. (2015). Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A. 119, 7810–7837. doi:10.1021/acs.jpca.5b01346
Schlegel, H. B. (1982). Optimization of Equilibrium Geometries and Transition Structures. J. Comput. Chem. 3, 214–218. doi:10.1002/jcc.540030212
Schutte, W. A., Allamandola, L. J., and Sandford, S. A. (1993). Formaldehyde and Organic Molecule Production in Astrophysical Ices at Cryogenic Temperatures. Science 259, 1143–1145. doi:10.1126/science.11540093
Siaï, A., Oueslati, I., and Kerkeni, B. (2016). Quantum Chemistry and Dynamics of the Abstraction Reaction of H Atoms from Formaldehyde. Chem. Phys. 474, 44–51. doi:10.1016/j.chemphys.2016.05.007
Sirianni, D. A., Alenaizan, A., Cheney, D. L., and Sherrill, C. D. (2018). Assessment of Density Functional Methods for Geometry Optimization of Bimolecular van der Waals Complexes. J. Chem. Theor. Comput. 14, 3004–3013. doi:10.1021/acs.jctc.8b00114
Skodje, R. T., Truhlar, D. G., and Garrett, B. C. (1981). A General Small-Curvature Approximation for Transition-State-Theory Transmission Coefficients. J. Phys. Chem. 85, 3019–3023. doi:10.1021/j150621a001
Song, L., and Kästner, J. (2017). Tunneling Rate Constants for H2CO+H on Amorphous Solid Water Surfaces. Astrophysical J. 850, 118. doi:10.3847/1538-4357/aa943e
Truhlar, D. G., Issacson, A., and Garrett, B. (1985). “Theory of Chemical Reaction Dynamics,” in The Theory of Chemical Reaction Dynamics (Boca Raton: CRC Press).
Truhlar, D. G., and Kuppermann, A. (1971). A Test of Transition State Theory against Exact Quantum Mechanical calculations11This Work Was Supported in Part by the United States Atomic Energy Commission, Report Code No. CALT-767P4-74. Chem. Phys. Lett. 9, 269–272. doi:10.1016/0009-2614(71)85049-2
Vandeputte, A. G., Reyniers, M.-F., and Marin, G. B. (2010). Theoretical Study of the thermal Decomposition of Dimethyl Disulfide. J. Phys. Chem. A. 114, 10531–10549. doi:10.1021/jp103357z
Vidali, G. (2013). H2 Formation on Interstellar Grains. Chem. Rev. 113, 8762–8782. doi:10.1021/cr400156b
Vinogradoff, V., Duvernay, F., Danger, G., Theulé, P., and Chiavassa, T. (2011). New Insight into the Formation of Hexamethylenetetramine (HMT) in Interstellar and Cometary Ice Analogs. Astron. Astrophysics 530, A128. doi:10.1051/0004-6361/201116688
Wakelam, V., Bron, E., Cazaux, S., Dulieu, F., Gry, C., Guillard, P., et al. (2017). H 2 Formation on Interstellar Dust Grains: The Viewpoints of Theory, Experiments, Models and Observations. Mol. Astrophysics 9, 1–36. doi:10.1016/j.molap.2017.11.001
Watanabe, N., Kimura, Y., Kouchi, A., Chigai, T., Hama, T., and Pirronello, V. (2010). Direct Measurements of Hydrogen Atom Diffusion and the Spin Temperature of Nascent H2 Molecule on Amorphous Solid Water.. Astrophysical J. 714, L233–L237. doi:10.1088/2041-8205/714/2/L233
Watanabe, N., and Kouchi, A. (2002). Efficient Formation of Formaldehyde and Methanol by the Addition of Hydrogen Atoms to CO in H[TINF]2[/TINF]O-CO Ice at 10 K. Astrophysical J. 571, L173–L176. doi:10.1086/341412
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/B508541A
Weigend, F. (2008). Hartree-Fock Exchange Fitting Basis Sets for H to Rn. J. Comput. Chem. 29, 167–175. doi:10.1002/jcc.20702
Weigend, F., Kattannek, M., and Ahlrichs, R. (2009). Approximated Electron Repulsion Integrals: Cholesky Decomposition versus Resolution of the Identity Methods. J. Chem. Phys. 130, 164106. doi:10.1063/1.3116103
Williams, D. A. (2003). “The Interstellar Medium: An Overview,” in Solid State Astrochemistry. Editors V. Pirronello, J. Krelowski, and G. Manicò (Dordrecht: Springer Netherlands), 1–20. doi:10.1007/978-94-010-0062-8_1
Woon, D. E. (2002). Modeling Gas-Grain Chemistry with Quantum Chemical Cluster Calculations. I. Heterogeneous Hydrogenation of CO and H2CO on Icy Grain Mantles. Astrophysical J. 569, 541–548. doi:10.1086/339279
Xu, Z. F., Raghunath, P., and Lin, M. C. (2015). Ab Initio Chemical Kinetics for the CH3 + O(3P) Reaction and Related Isomerization-Decomposition of CH3O and CH2OH Radicals. J. Phys. Chem. A. 119, 7404–7417. doi:10.1021/acs.jpca.5b00553
Zaverkin, V., and Kästner, J. (2020). “Chapter 7. Instanton Theory to Calculate Tunnelling Rates and Tunnelling Splittings,” in Tunnelling in Molecules, 245–260. doi:10.1039/9781839160370-00245
Zhao, Y., Schultz, N. E., and Truhlar, D. G. (2006). Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theor. Comput. 2, 364–382. doi:10.1021/ct0502763
Zheng, J., and Truhlar, D. G. (2010). Kinetics of Hydrogen-Transfer Isomerizations of Butoxyl Radicals. Phys. Chem. Chem. Phys. 12, 7782–7793. doi:10.1039/B927504E
Keywords: astrochemistry, ices, formaldehyde, quantum and classical modelling, reaction rates
Citation: Kerkeni B, Boukallaba M, Hechmi M, Duflot D and Toubin C (2022) QM/MM Study of the H2 Formation on the Surface of a Water Ice Grain Doped With Formaldehyde: Molecular Dynamics and Reaction Kinetics. Front. Astron. Space Sci. 9:807649. doi: 10.3389/fspas.2022.807649
Received: 02 November 2021; Accepted: 10 January 2022;
Published: 14 March 2022.
Edited by:
Ankan Das, Indian Centre for Space Physics, IndiaReviewed by:
Albert Rimola, Universitat Autònoma de Barcelona, SpainGerman Molpeceres De Diego, University of Stuttgart, Germany
Copyright © 2022 Kerkeni, Boukallaba, Hechmi, Duflot and Toubin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Céline Toubin, Y2VsaW5lLnRvdWJpbkB1bml2LWxpbGxlLmZy