 Pingping Sun
Pingping Sun Qiang Ma
Qiang Ma Lei Zhang
Lei Zhang- College of Horticulture and Plant Protection, Inner Mongolia Agricultural University, Hohhot, China
Cytospora mali is an important factor for apple valsa canker, and Bacillus veleznesis L-1 is an effective biocontrol agent against apple valsa canker. Quantitative acetyl-proteomics is known to regulate transcriptional activity in different organisms; limited knowledge is available for acetylation modification in C. mali, and its response to biocontrol agents. In this study, using Tandem Mass tag proteomic strategies, we identified 733 modification sites on 416 proteins in C. mali, functions of these proteins were analyzed using GO enrichment and KEGG pathway. Some lysine acetylated proteins are found to be important to the fungal pathogenicity of C. mali, and also the response of fungi to biostress. B. velezensis L-1 suppressed the C. mali QH2 by causing the energy shortage and reduced virulence. Correspondingly, the C. mali QH2 could alleviate the suppression of biostress by upregulation of autophagy, peroxidase, cytochrome P450, ABC transporter and Heat shock protein 70. In summary, our results provided the first lysine acetylome of C. mali and its response to B. velezensis L-1.
Introduction
Protein acetylation is an evolutionarily conserved post-translational modification (Weinert et al., 2013), which is catalyzed by lysine acetyltrasferases and reversed by lysine deacetylases (Henriksen et al., 2012). Lysine acetylation has been found to occur in almost every compartment of a cell, and to play important roles in various cellular processes including protein-protein interactions (Hou et al., 2010), enzymatic activity (Starai and Escalante-Semerena, 2004; Nambi et al., 2013), metabolic pathways (Hou et al., 2010; Wang et al., 2010; Guan and Xiong, 2011; Nambi et al., 2013; Xie et al., 2015), cell morphology (Choudhary et al., 2009), calorie restriction (Imai and Guarente, 2010; Henriksen et al., 2012), protein-nucleic acid interactions (Arif et al., 2010; Ren et al., 2016), transcription factors, hormone receptors, signal transducers, chaperones and cytoskeleton (Singh et al., 2010; Zhang et al., 2013). Lysine acetylation is believed to be a main signaling modulator due to its widely occurrences and diverse cellular functions (Norvell and McMahon, 2010).
In recent years, with the advance of mass spectrometry (MS) and high affinity purification of acetylated peptides, acetylproteome studies have been successfully performed in some prokaryotic microorganisms, such as Escherichia coli (Kuhn et al., 2014), Salmonella enterica (Wang et al., 2010), Bacillus subtilis (Kim et al., 2013), Geobacillus kaustophilus (Lee et al., 2013), Erwinia amylovora (Wu et al., 2013), Thermus thermophilus (Okanishi et al., 2013) and Saccharopolyspora erythraea (Huang et al., 2015); the budding yeast: Saccharomyces cerevisiae (Henriksen et al., 2012), Oomycete: Phytophthora sojae (Li et al., 2016) and in filamentous fungi: Botrytis cinerea (Lv et al., 2016) and Fusarium graminearum (Zhou and Wu, 2019) et al. The knockout of some acetylated related genes caused the reduced mycelium growth, spore-production and virulence of fungi (Ding et al., 2010; Chen et al., 2018; Zhou and Wu, 2019), which indicated that lysine acetylation play important role in the growth and virulence of pathogens.
Apple valsa canker (AVC), caused by Cytospora sp., is one of the most destructive diseases of apple trees in eastern Asia (Cao et al., 2009; Ma et al., 2020). Bacillus velezensis L-1 was characterized as an effective biocontrol agent (BCA) against AVC in our previous work submitted to European Journal of Plant Pathology. Genome analysis revealed that there are a large number of acetyltransferase and deacetylase orthologs in the genomic of C. mali (Yin et al., 2015), which suggests that protein acetylation may play a critical role in the biological process, pathogenesis and the responses to BCA of C. mali. During the infection of AVC into apple barks, genes associated with plant cell wall degrading enzymes, toxic secondary metabolite biosynthesis were up-regulated, indicating their roles in the pathogenicity of C. mali (Ke et al., 2014), accordingly, BCA could inhibit the AVC by disrupting the cell wall-related genes of AVC, affecting the carbohydrate metabolic pathway of pathogens (Liu et al., 2018). On the other side, pathogens recruit numerous genes to resist the BCA. Sclerotinia sclerotiorum (the causing agent of sclerotinia stem rot) could up-regulate cell wall, cell membrane synthesis, antioxidants, glutathione S-transferase (GST), ABC transporter, and the autophagy pathway to alleviate the pressure of BCA (Yang et al., 2020). Study on the biological control mechanism of BCA and the response mechanism of AVC to biotic stress are in need for the further application of BCA in controlling AVC. In this study, a comparative acetyl-proteomics study based on Tandem Mass Tag (TMT) technology (McAlister et al., 2012) was carried out between C. mali QH2 and the C. mali QH2 suppressed by B. velezensis L-1. These data will complement current knowledge of the C. mali acetylproteome and provide an expanded platform for better understanding of the function of acetylation in the cell metabolites of pathogens and responses to the BCA stress.
Materials and methods
Mycelium preparation
Bacillus velezensis strain L-1 and Cytospora mali virulent strain QH2, were isolated by authors (Sun et al., 2017; Ma et al., 2020). C. mali QH2 was cultured on potato dextrose agar (PDA) plates and incubated at 28°C for 5 days, B. velezensis L-1 was cultured on Luria Bertani (LB) broth at 28°C, 180 rpm for 2 days before use. For the mycelium of C. mali QH2 suppressed by B. velezensis L-1 (TR), B. velezensis L-1 was first centrifuged at 8,000 rpm for 15 min, the supernatant was then filtered through 0.22 μm film, a total of 100 μL non-spore filtrate of L-1 was added into 100 mL PD broth, and a dash of C. mali QH2 mycelium was then inoculated into the PD medium. The PD medium inoculated with QH2 mycelium without the L-1 was treated as control (CK). The QH2 was cultured at 28°C, 180 rpm for 5 days, and the mycelium was collected by centrifuged at 8,000 rpm for 15 min, and washed with 0.1 M PBS solution for three times. Washed mycelium was frozen in liquid nitrogen, and stored at −80°C for protein extraction. Three biological replicates for each treatment were performed.
Acetylatic analysis
Protein extraction and FASP digestion
SDT buffer was used to extract the protein from each sample, the filtrate was quantified by BCA Protein Assay Kit (Bio-Rad, USA). A total of 20 μg protein was taken from each sample, and Filter-aided Sample Preparation (FASP) procedure was performed for protein digestion (Wiśniewski et al., 2009). The digested peptides of each sample were desalted on C18 Cartridges (Empore™ SPE, Sigma), concentrated by vacuum centrifugation and reconstituted in 40 μL of 0.1% (v/v) formic acid.
TMT labeling, enrichment of acetylated peptides, and fractionation
Hundred microgram peptide mixture of each sample was labeled using TMT reagent according to the manufacturer's instructions (Thermo Fisher Scientific) (McAlister et al., 2012). The labeled samples were reconstituted in 1.4 mL precooled IAP buffer, the pretreated Anti-Ac-K antibody beads [PTMScan Acetyl-Lysine Motif (Ac-K) Kit] were added, and then incubated at 4°C for 1.5 h, centrifuged at 2,000 ×g for 30 s. Anti-Ac-K antibody beads were washed with 1 mL precooled IAP buffer and then precooled water for 3 times. 40 μL 0.15% TFA was added to the washed beads, incubated for 10 min at room temperature, then added 0.15% TFA again, centrifuged at 2,000 ×g for 30 s, the supernatant was desalted by C18 STAGE Tips.
Pierce high pH reversed-phase fractionation kit (Thermo scientific) was used to fractionate labeled digest samples into 6 fractions by an increasing acetonitrile step-gradient elution according to instructions.
HPLC and LC-MS/MS analysis
LC-MS/MS analysis was performed on a Q Exactive HF mass spectrometer (Thermo Scientific) that was coupled to Easy nLC (Thermo Fisher Scientific) for 90 min. The peptides were loaded onto a reverse phase trap column (Thermo Scientific Acclaim PepMap100, 100 μm*2 cm, nanoViper C18) connected to the C18-reversed phase analytical column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (0.1% Formic acid) and separated with a linear gradient of buffer B (84% acetonitrile and 0.1% Formic acid) at a flow rate of 300 nL/min.
The mass spectrometer was operated in positive ion mode. MS data was acquired using a data-dependent top 10 method dynamically choosing the most abundant precursor ions from the survey scan (300–1,800 m/z) for HCD fragmentation. Automatic gain control (AGC) target was set to 3e6, and maximum inject time to 10 ms. Dynamic exclusion duration was 40 s. Survey scans were acquired at a resolution of 70,000 at m/z 200 and resolution for HCD spectra was set to 17,500 at m/z 200, and isolation width was 2 m/z. Normalized collision energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled.
Identification and quantitation of acetylated proteins
MS/MS spectra were searched using MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer 2.4. Trypsin was specified as cleavage enzyme allowing up to 2 missing cleavages, mass error was set to 20 ppm for precursor ions and 0.1 Da for fragment ions. The other parameters in MaxQuant were set to default values.
Bioinformatic analysis
Motifs, subcellular localization
The modified site and six upstream/downstream amino acids from the modified site (13 amino acid sites in total) were used for information analysis. The MEME (http://meme-suite.org/index.htm) and CELLO (http://cello.life.nctu.edu.tw/) were used to predict the motifs, subcellular localization, respectively. The protein sequences were locally searched using the NCBI BLAST+ client software and InterProScan to find homolog sequence. Blast2GO was used to map and annotate the GO terms, and online Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://geneontology.org/) was applied to retrieve the KEGG pathway annotations.
Functional enrichment analysis
Enrichment analysis was applied based on the Fisher' exact test, considering the whole quantified proteins as background dataset. Benjamini-Hochberg correction for multiple testing was further applied to adjust derived p-values. And only functional categories and pathways with p-values under a threshold of 0.05 were considered significant.
Correlation analysis of the proteome and transcriptome
The transcriptome study about the C. mali QH2 and its response to B. velezensis L-1 was conducted in our another work, and the related results were submitted to the European Journal of Plant Pathology. In our transcriptomic studies, B. velezensis L-1 impaired the membrane formation, cell stability, autophagy of C. mali QH2, reduced its expression of virulence related genes, and C. mali QH2 could enhance the antioxidation and detoxification to alleviate the biostress. The correlations of the differently expressed genes/proteins were created between RNA-seq results in our previous work and acetylated proteins in this study. If a gene and its cognate protein were both significantly differently expressed in the TR, they were defined as differently expressed genes and differently abundant proteins, respectively (Liu et al., 2019). The Spearman correlation coefficients were calculated to evaluate the concordance of the differences in abundance at the transcript and protein levels (De Winter et al., 2016; Warrens, 2017).
Data information
The mass spectrometry proteomics data is available from the ProteomeXchange Consortium (https://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the dataset identifier PXD035537. username: cHhkMDM1NTM3QGViaS5hYy51aw==, password: bPhHTbsj.
Results
Features of lysine acetylation sites and proteins
A total of 724 acetylated peptides spanning 416 proteins with 733 lysine acetylation sites were identified, among which, 475 peptides in 299 proteins were accurately quantified (Table 1).
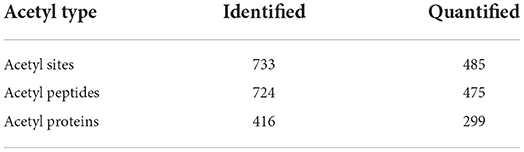
Table 1. Summary of quantified and identified lysine acetylation.
The number of lysine acetylation sites in identified acetylated proteins was diverse and distributed from 1 to 12. There were 156 proteins (37.35%) with more than two acetylation sites. Protein number A0A194VZB7 [encoding heatshock protein domain 70 (Hsp70)] had 12 actylation sites (Figure 1A). The average distribution of modification sites per amino acids was 0.59% (Figure 1B).
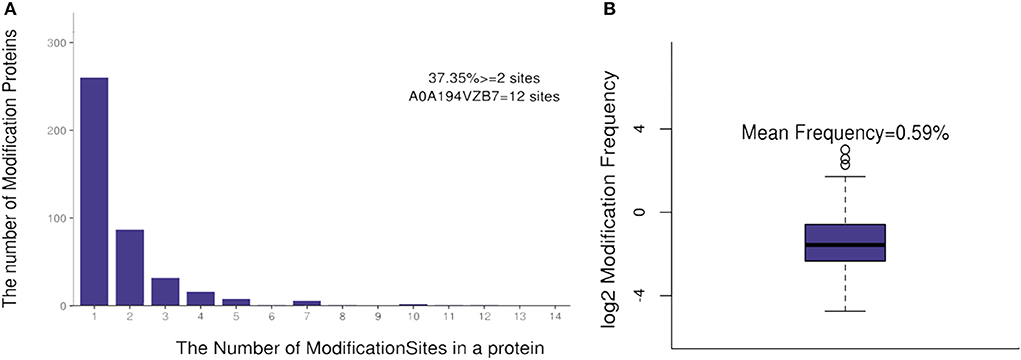
Figure 1. The distribution of acetylation sites in a protein (A) and the modification sites frequency (B).
Identification of differentially expressed acetylated proteins
A total of 195 identified acetylation proteins (DEAPs) were differently expressed at the level of|Fold change|>1.2 (1.2-fold over, or 0.83 less) and P < 0.05 in the TR, 101 proteins were up-regulated, and 94 proteins were down-regulated in the TR (Figure 2A). Heatmap of DEAPs were built, the similarity of the data patterns within each group was relatively high, whereas it was relatively low between the groups, and effectively distinguished the groups. The cluster analysis shows that the results of DEAPs identified in our data are believable (Figure 2B).
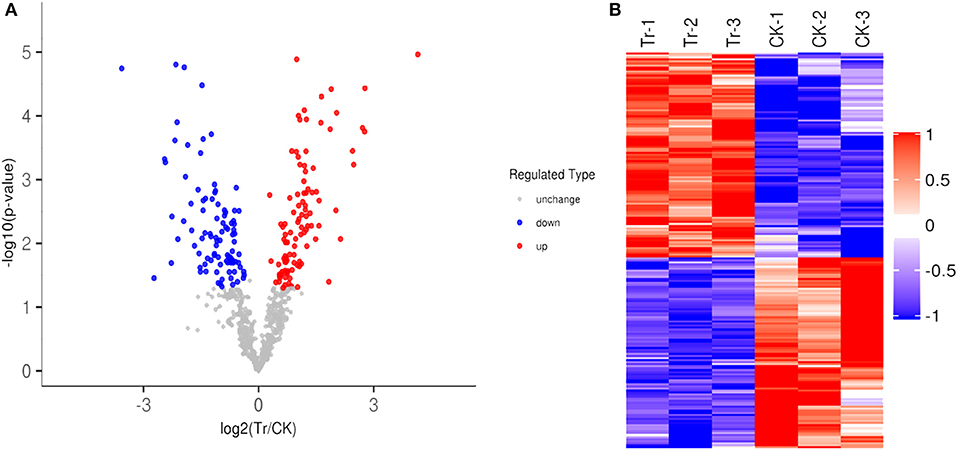
Figure 2. Volcano (A) and heatmap (B) of differently expressed acetylated proteins.
Correlation analysis of the transcriptome and proteome
The correlation analysis of differently expressed genes/proteins was conducted between the proteomic results in this study and the transcriptomic results of our previous study. The Spearman correlation coefficients was r = 0.134, P = 0.058, although the correlation was not significant, it could still be concluded from our work that DEGs/DEAPs between transcriptomic and proteomic results are positive related (Figure 3). The correlated differentlly abundant proteins and different genes were mainly involved in the ABC transporter, structural constituent of ribsomal, ATP binding, heat shock protein 70 (Hsp 70) and some are involved in oxidoreductase activity.
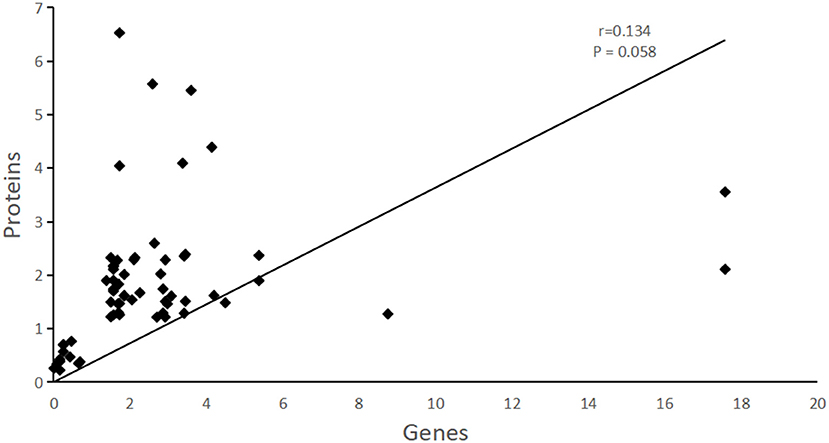
Figure 3. The correlation between the gene and protein expression level.
Subcellular localization of differentially expressed acetylated proteins
The CELLO algorithm analysis was used to analyze the subcellular localization of DEAPs. Results showed that 68, 49, 45, 5, and 2 of the acetylated proteins were located in the cytoplasmic, nuclear, mitochondrial, plasma membrane, and endoplasmic reticulum, respectively (Table 2).

Table 2. The subcellular localization of DEAPs.
Annotation of DEAPs
We analyzed the GO annotation and classified the identified proteins according to their biological processes (BP), molecular functions (MF) and cellular compartments (CC). Eighty proteins were involved in the BP category, and the acetylated proteins were mainly involved in the cellular process, metabolic process, cellular component organization of biogenesis, biological regulation, localization, regulation of biological process, and response to stimulus. One hundred twenty-four proteins were enriched in MF category, and these acetylated proteins were mainly involved in binding, catalytic activity, structural molecule activity, transporter activity, antioxidant activity, molecular function regulator. Seventy-one proteins were involved in the CC category, and the acetylated proteins were mainly involved in the cell, organelle, membrane, and membrane-enclosed lumen (Figure 4A).
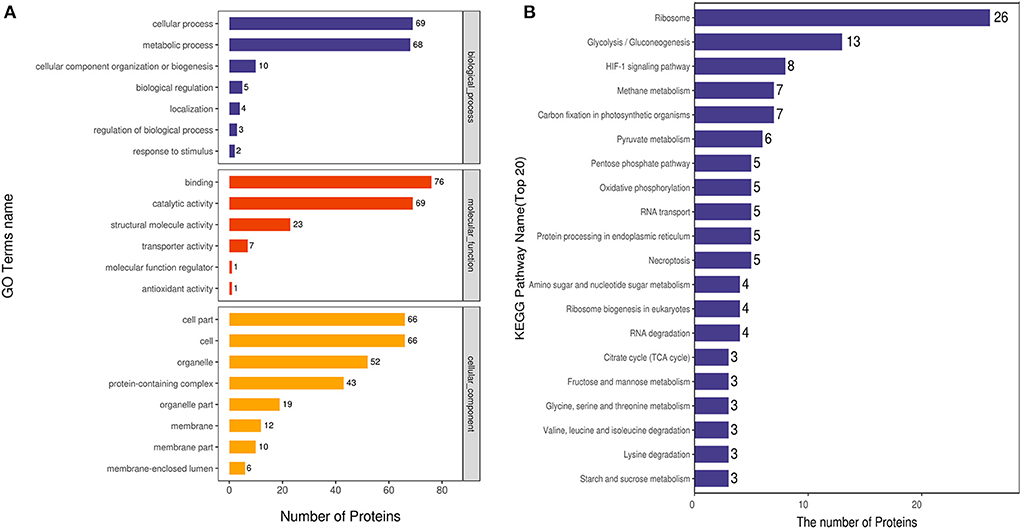
Figure 4. GO and KEGG pathway analyses of DEAPs. (A) GO classification of differentially expressed acetylated proteins. (B) KEGG pathway analysis of differently expressed acetylated proteins.
KEGG pathway analyses showed that 20 pathways were enriched for acetylated proteins (Figure 4B). The most abundant one was the ribosome pathway, which contains 26 acetylated proteins; other enriched pathways included glycolysis/gluconeogenesis pathways, HIF-1 signaling pathway, methane metabolism pathways, pyruvate metabolism, pentose phosphate pathways, oxidative phosphorylation, RNA transport, necroptosis, amino sugar and nucleotide sugar metablism. At the same time, acetylated proteins in HIF-1 signaling pathway, methane metabolism, and glycolysis/gluconeogenesis pathways were significantly up-regulated with P < 0.05.
DEAPs involved in the mycelium growth, pathogenicity of C. mali QH2, and its response to biostress
BCAs could inhibit the fungi by suppressing the normal growth of fungi or inhibiting the expression of their virulence-related metabolites, on the other side, the fungi could respond to the BCAs by improving the ability for detoxification and antioxidant (Wang et al., 2020). To investigate roles of the DEAPs in fungal pathogenicity and response to the biostress, we retrieved these proteins to gene repositories built from the literature annotations reported previously (Table 3; Figure 5).
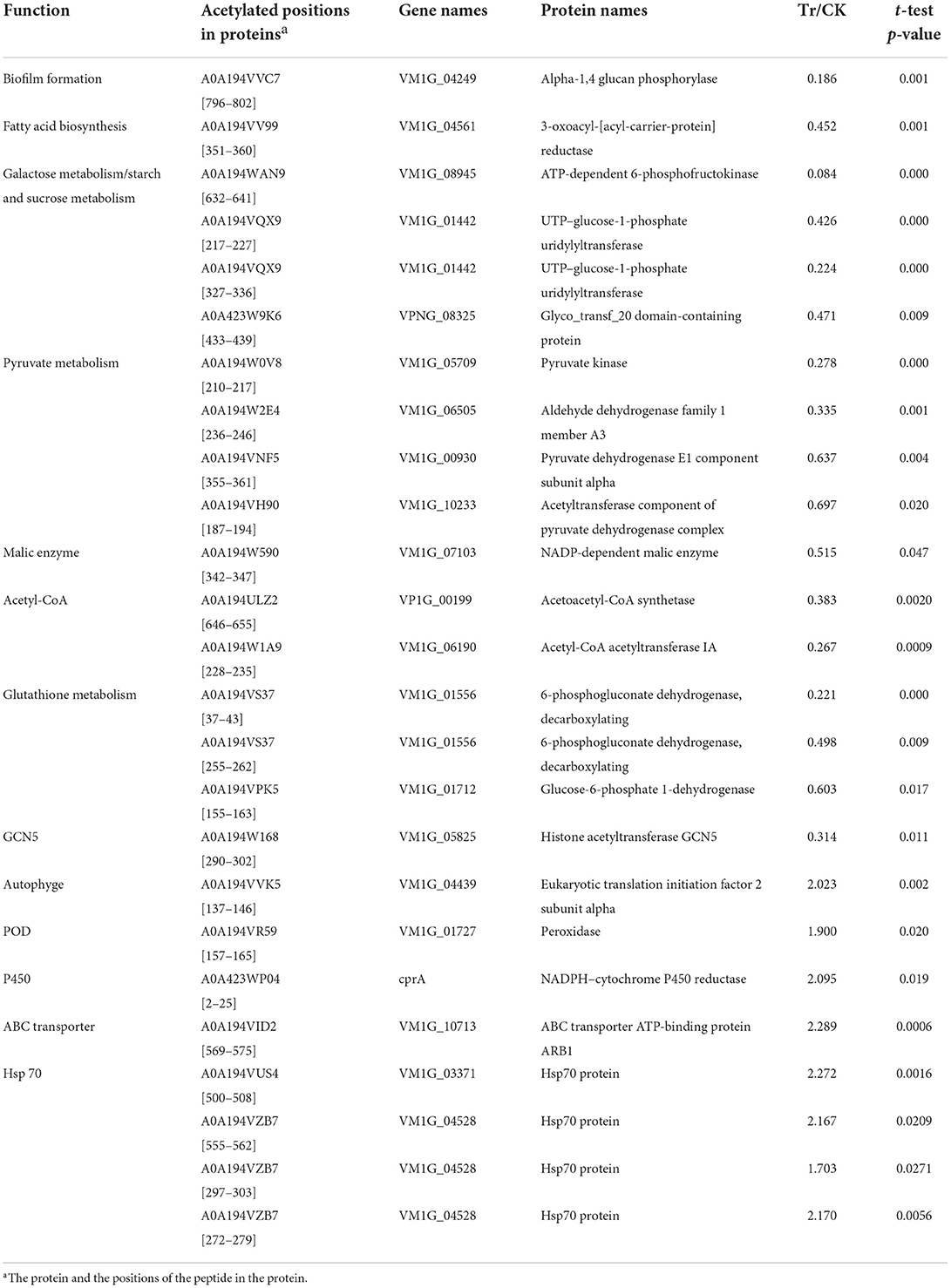
Table 3. List of acetylated proteins involved in the growth and pathogenicity of C. mali QH2, and its response to B. velezensis L-1.
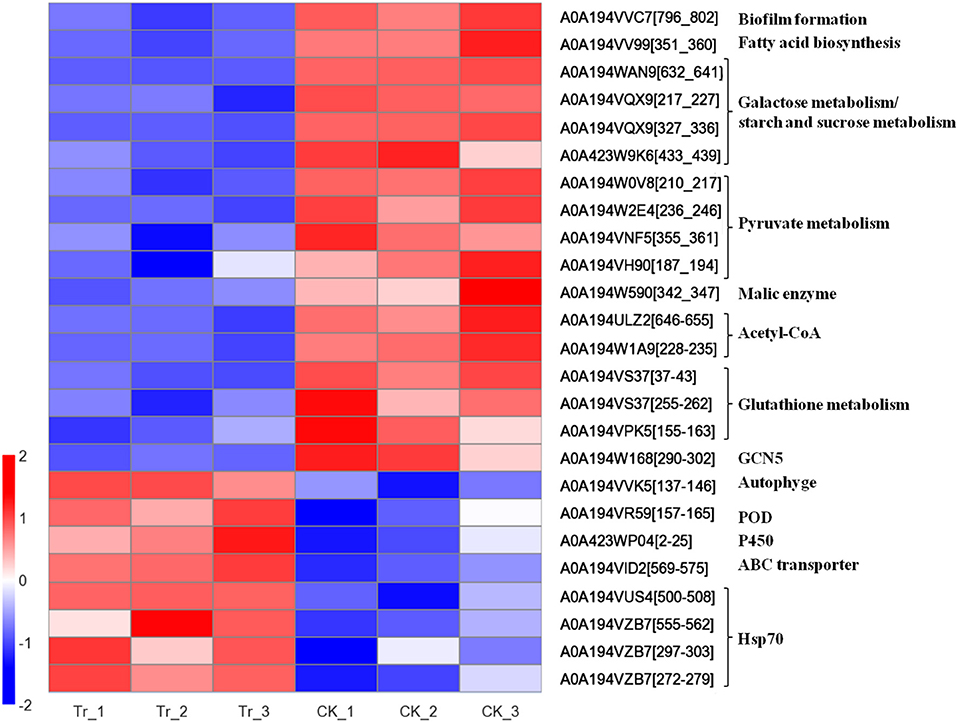
Figure 5. Heatmap of expression level of acetylated proteins involved in the growth and pathogenicity of C. mali QH2, and its response to B. velezensis L-1.
The DEAPs involved in the biofilm formation, energy production, and virulence of C. mali were significantly suppressed by B. velezensis L-1. The DEAP A0A194VV99, which encodes fatty acid biosynthesis, was significantly down regulated in the treatment, with TR/CK as 0.452. At the same time, A0A194VVC7, related to the biofilm formation was also significantly down regulated in TR, with TR/CK as 0.186, P = 0.001. DEAPs related to the energy production in C. mali were significantly down regulated: 4 DEAPs related to pyruvate metabolism were all significantly down regulated in the TR, and the acetylated protein A0A194W590, encodes NADP-dependent malic enzyme was significantly down regulated, with TR/CK = 0.515 and P = 0.047. Two acetylated proteins, encoding Acetoacetyl-CoA synthetase and Acetyl-CoA acetyltransferase IA, respectively, were both significantly down regulated. In addition, 4 DEAPs related to galactose metabolism/starch and sucrose metabolism were all significantly down regulated. Some virulence related DEAPs were suppressed by B. velezensis L-1: 3 DEAPS, related to the glutathione metabolism, were all signifiantly down regulated in the TR, and the GST (A0A194W4W5) was down regulated with TR/CK = 0.744, and P = 0.068. In addition, Histone acetyltransferase GCN5 was reported as key virulence factor in a variety fungus. In this treatment, the histone acetyltransferase GCN5 (A0A194W168) was significantly down regulated with TR/CK = 0.314, and P = 0.011 (Table 3; Figure 5).
Correspondingly, C. mali QH2 took measures to alleviate the biostress from B. velezensis L-1. The acetylated protein A0A194VVK5, related to the autophyge was significantly upregulated with TR/CK = 2.023, P = 0.002. A0A194VR59, encodes peroxidase (POD) was significantly upregulated with TR/CK = 1.9, P = 0.02. Acetylated protein, encodes NADPH-cytochrome P450 reductase was significantly upregulated, with TR/CK = 2.095, P = 0.019. A0A194VID2, encodes the ABC transporter ATP-binding protein ARB1 was significantly upregulated in the TR, with the TR/CK as 2.29, P < 0.001. In addition, 4 acetylated proteins encoding heat shock protein 70 (Hsp 70) were significantly upregulated in the TR (Table 3; Figure 5).
Discussion
In this study, we performed the different expression of lysine acetylated proteins in the C. mali QH2, during the suppresion of B. velezensis L-1. A total of 733 lysine acetylation sites in 416 proteins were identified, and 195 identified acetylation proteins were differently expressed at the level of|Fold change|>1.2 (1.2-fold over, or 0.83 less) and P < 0.05 in the TR. Among DEAPs, 101 proteins were up-regulated, and 94 proteins were down-regulated in the TR. Some DEAPs were predicted to be involved in the mycelium growth, pathogenicity of C. mali and its response to the biostress.
Two lysine acetylated proteins, located in the cytoplasm and related to the fatty acids and biofilm formation, respectively, were significantly down regulated in the TR. Fatty acids play important role in forming the plasma membrane and maintaining the fluidity of the cell membrane (Lattif et al., 2011). The formation of biofilm is closely related to the pahogenic and the drug resistance of the fungi (Wang et al., 2020). The significant down regulation of fatty acids and biofilm formation related acetylated proteins in the TR, indicating that lysine acetylated proteins are involved in the biofilm formation of C. mali QH2, and B. velezensis L-1 could inhibit the C. mali QH2 by inhibiting its synthesis of biofilms.
BCAs could affect the carbohydrate metabolic pathway, and inhibit the energy production in fungi (Liu et al., 2018). In this study, the lysine acetylated proteins were involved in multiple processes of energy production. Galactose is widely utilized by organisms through its conversion to glucose-6-phosphate, which can be then metabolized in the cytoplasm of the cell through the glycolysis pathway to produce ATP (Martchenko et al., 2007). The galactose metabolism depicts the conversion of galactose into glucose, lactose, and other sugar intermediates that may be used for a range of metabolic process. Down regulation of galactose metabolism/starch and sucrose metabolism indicating the suppression of B. velezensis L-1 on the carbohydrate metabolism. Pyruvate is the end production of glycolysis, and plays an important role as the central metabolite in the branching point of the tricarboxylic acid (TCA) cycle as well as the synthesis of amino acids (Zhang et al., 2020). Specifically, the pyruvate can be converted into Acetyl-CoA, which was used as the primary energy source for the TCA cycle, or converted into oxaloacetate to replenish TCA cycle intermediates. In addition, pyruvate can also be used to synthesize carbohydrates, fatty acids, ketone bodies, alanine, and steroids. Malic acid synthase (MS) is one of the key enzymes in the glyoxylate cycle, down regulation of the NADP-dependent malic enzyme may also reduce the use of acetyl-CoA through the glyoxylate cycle, what's more, the acetoacetyl-CoA synthetase and acetyl-CoA acetyltransferase IA were significantly down regulated. Decreased carbohydrate metabolism, and the down production of acetyl-CoA all lead to the energy shortage in C. mali.
Glutathione is an endogenous component of cellular metabolism, also referred to as GSH, which functions in detoxification and antioxidant processes by catalyzing the binding GSH− to electrophilic centers on toxic substrates (Zettl et al., 1994; Marrs et al., 1995; Tian et al., 2016), and related to the virulence of fungi (Liu et al., 2018; Yang et al., 2020). Histone acetyltransferase GCN5 was proved as an important acetylated protein in a variety of fungi (Rösler et al., 2016; Chen et al., 2018). In our study, the B. velezensis L-1 suppressed C. mali showed the significantly down expression glutathione metabolism and GCN5. The down regulation of these virulence related proteins indicated that the acetylated proteins were involved in the virulence of C. mali QH2, and B. velezensis L-1 could reduce the virulence of C. mali QH2 by down expression of its virulence related genes.
C. mali has made multiple responses to the inhibition of BCAs. Autophagy pathway removes damaged organelles, and acts as an intracellular “scavengers” to maintain cellular homeostasis (Wang et al., 2019). BCAs cause oxidative damage to fungal cells, thus, antioxidant, hydrolase enzymes and other detoxification activities are regulated in the fungi to improve the tolerance (Van Acker et al., 2014). POD are enzymes that catalyze oxidation-reduction reaction by mechanism of free radical that transform several compounds into oxidized or polymerized products, they play important roles in eliminating reactive oxygen species damage, and are important enzymes involved in the biological antioxidant defense system (Horiguchi et al., 2001). In eukaryotes, CYP450 is membrane-binding protein, related to the metabolism of exogenous substance, and plays an important role in mytotoxin metabolism of necrotrophic fungus (Kelly et al., 1997; Syed et al., 2010; Ning and Wang, 2012). ABC transporters are important efflux transporters, are capable of translocating antifungal items out of cells and contribute to the development of tolerance to antifungals or biostress. Hsp 70 and other chaperones determine cell death and cell transformations; it has been implicated in the cellular stress initiation process (Pujhari et al., 2019). The upregulation of acetylated proteins in autophyge, POD, CYP450, ABC transporter and Hsp70 are the ways C. mali alleviate the suppression from biostress.
Taken together, these findings suggested that several lysine acetylated proteins related to fatty acids, biofilm formation, galactose metabolism/starch and sucrose metabolism pathway, TCA cycle, glutathione, and GCN5 were involved in the normal mycelium growth and pathogenicity of C. mali, and on the other side, the acetylated proteins involved in the autophagy, POD, CYP450, ABC transporter, and Hsp70 were expressed to release the stress from the biostress. Further verification of acetylated proteins involved in the interaction between B. velezensis L-1 and C. mali QH2 will be focused in our future work. It will be helpful to understand roles of the protein with acetylated lysine at the post-translational modification level in C. mali.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
Author contributions
PS and LZ designed the experiments, analyzed the data, and wrote the manuscript. PS and QM performed the experiments. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (Grant Nos. 32060594 and 31860540), partially supported by the Research Program of Science and Technology at Universities of Inner Mongolia Autonomous Region (NJZY21458), and Research Start-up for High-level Researchers in Inner Mongolia Agricultural University (Grant Nos. NDYB2018-14 and NDYB2019-1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arif, M., Selvi, B. R., and Kundu, T. K. (2010). Lysine acetylation: the tale of a modification from transcription regulation to Metab. Clin. Exp. 11, 1501–1504. doi: 10.1002/cbic.201000292
Cao, K., Guo, L. Y., Li, B., Sun, G., and Chen, H. (2009). Investigations on the occurrence and control of apple canker in China. Plant Protect 35, 114–116.
Chen, Y., Wang, J., Yang, N., Wen, Z., Sun, X., Chai, Y., et al. (2018). Wheat microbiome bacteria can reduce virulence of a plant pathogenic fungus by altering histone acetylation. Nat. Commun. 9, 3429–3429. doi: 10.1038/s41467-018-05683-7
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
De Winter, J. C., Gosling, S. D., and Potter, J. (2016). Comparing the Pearson and Spearman correlation coefficients across distributions and sample sizes: A tutorial using simulations and empirical data. Psychol. Methods 21, 273–290. doi: 10.1037/met0000079
Ding, S. L., Liu, W., Iliuk, A., Ribot, C., Vallet, J., Tao, A., et al. (2010). The tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus Magnaporthe oryzae. Plant Cell 22, 2495–2508. doi: 10.1105/tpc.110.074302
Guan, K. L., and Xiong, Y. (2011). Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116. doi: 10.1016/j.tibs.2010.09.003
Henriksen, P., Wagner, S. A., Weinert, B. T., Sharma, S., Bacinskaja, G., Rehman, M., et al. (2012). Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol. Cell. Proteomics 11, 1510–1522. doi: 10.1074/mcp.M112.017251
Horiguchi, H., Yurimoto, H., Goh, T.-K., Nakagawa, T., Kato, N., and Sakai, Y. (2001). Peroxisomal catalase in the methylotrophic yeast Candida boidini: transport efficiency and metabolic significance. J. Bacteriol. 183, 6372–6383. doi: 10.1128/JB.183.21.6372-6383.2001
Hou, J., Cui, Z., Xie, Z., Xue, P., Wu, P., Chen, X., et al. (2010). Phosphoproteome analysis of rat L6 myotubes using reversed-phase C18 prefractionation and titanium dioxide enrichment. J. Proteome Res. 9, 777–788. doi: 10.1021/pr900646k
Huang, D., Li, Z. H., You, D., Zhou, Y., and Ye, B. C. (2015). Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea. Appl. Microbiol. Biotechnol. 99, 1399–1413. doi: 10.1007/s00253-014-6144-2
Imai, S.-I., and Guarente, L. (2010). Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol. Sci. 31, 212–220. doi: 10.1016/j.tips.2010.02.003
Ke, X., Yin, Z., Song, N., Dai, Q., Voegele, R. T., Liu, Y., et al. (2014). Transcriptome profiling to identify genes involved in pathogenicity of Valsa mali on apple tree. Fungal Genet. Biol. 68, 31–38. doi: 10.1016/j.fgb.2014.04.004
Kelly, S. L., Lamb, D. C., Baldwin, B. C., Corran, A. J., and Kelly, D. E. (1997). Characterization of Saccharomyces cerevisiae CYP61, sterol Δ22-desaturase, and inhibition by azole antifungal agents. J. Biol. Chem. 272, 9986–9988. doi: 10.1074/jbc.272.15.9986
Kim, D., Yu, B. J., Kim, J. A., Lee, Y. J., Choi, S. G., Kang, S., et al. (2013). The acetylproteome of Gram-positive model bacterium Bacillus subtilis. Proteomics 13, 1726–1736. doi: 10.1002/pmic.201200001
Kuhn, M. L., Zemaitaitis, B., Hu, L. I., Sahu, A., Sorensen, D., Minasov, G., et al. (2014). Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS ONE 9, e94816. doi: 10.1371/journal.pone.0094816
Lattif, A. A., Mukherjee, P. K., Chandra, J., Roth, M. R., Welti, R., Rouabhia, M., et al. (2011). Lipidomics of Candida albicans biofilms reveals phase-dependent production of phospholipid molecular classes and role for lipid rafts in biofilm formation. Microbiology 157, 3232–3242. doi: 10.1099/mic.0.051086-0
Lee, D. W., Kim, D., Lee, Y. J., Kim, J. A., Choi, J. Y., Kang, S., et al. (2013). Proteomic analysis of acetylation in thermophilic Geobacillus kaustophilus. Proteomics 13, 2278–2282. doi: 10.1002/pmic.201200072
Li, D., Lv, B., Tan, L., Yang, Q., and Liang, W. (2016). Acetylome analysis reveals the involvement of lysine acetylation in diverse biological processes in Phytophthora sojae. Sci. Rep. 6, 29897. doi: 10.1038/srep29897
Liu, C., Fan, D., Li, Y., Chen, Y., Huang, L., and Yan, X. (2018). Transcriptome analysis of Valsa mali reveals its response mechanism to the biocontrol actinomycete Saccharothrix yanglingensis Hhs.015. BMC Microbiol. 18, 90. doi: 10.1186/s12866-018-1225-5
Liu, Z., Lv, J., Zhang, Z., Li, H., Yang, B., Chen, W., et al. (2019). Integrative transcriptome and proteome analysis identifies major metabolic pathways involved in pepper fruit development. J. Proteome Res. 18, 982–994. doi: 10.1021/acs.jproteome.8b00673
Lv, B., Yang, Q., Li, D., Liang, W., and Song, L. (2016). Proteome-wide analysis of lysine acetylation in the plant pathogen Botrytis cinerea. Sci. Rep. 6, 29313. doi: 10.1038/srep29313
Ma, Q., Ju, M., Liu, Q., Zhang, L., Li, Z., and Sun, P. (2020). Identification of the pathogenic fungi associated with apple valsa canker in Inner Mongolia, China. J. Fruit Sci. 37, 102–110. doi: 10.13925/j.cnki.gsxb.20190474
Marrs, K. A., Alfenito, M. R., Lloyd, A. M., and Walbot, V. (1995). A glutathione S-transferase involved in vacuolar transfer encoded by the maize gene Bronze-2. Nature 375, 397–400. doi: 10.1038/375397a0
Martchenko, M., Levitin, A., Hogues, H., Nantel, A., and Whiteway, M. (2007). Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr. Biol. 17, 1007–1013. doi: 10.1016/j.cub.2007.05.017
McAlister, G. C., Huttlin, E. L., Haas, W., Ting, L., Jedrychowski, M. P., Rogers, J. C., et al. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 84, 7469–7478. doi: 10.1021/ac301572t
Nambi, S., Gupta, K., Bhattacharyya, M., Ramakrishnan, P., Ravikumar, V., Siddiqui, N., et al. (2013). Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J. Biol. Chem. 288, 14114–14124. doi: 10.1074/jbc.M113.463992
Ning, D., and Wang, H. (2012). Involvement of cytochrome P450 in pentachlorophenol transformation in a white rot fungus Phanerochaete chrysosporium. PLoS ONE 7, e45887. doi: 10.1371/journal.pone.0045887
Norvell, A., and McMahon, S. B. (2010). Cell biology. Rise of the rival. Science 327, 964–965. doi: 10.1126/science.1187159
Okanishi, H., Kim, K., Masui, R., and Kuramitsu, S. (2013). Acetylome with structural mapping reveals the significance of lysine acetylation in Thermus thermophilus. J. Proteome Res. 12, 3952–3968. doi: 10.1021/pr400245k
Pujhari, S., Brustolin, M., Macias, V. M., Nissly, R. H., Nomura, M., Kuchipudi, S. V., et al. (2019). Heat shock protein 70 (Hsp70) mediates Zika virus entry, replication, and egress from host cells. Emerg. Microbes Infect. 8, 8–16. doi: 10.1080/22221751.2018.1557988
Ren, J., Sang, Y., Tan, Y., Tao, J., Ni, J., Liu, S., et al. (2016). Acetylation of lysine 201 inhibits the DNA-binding ability of PhoP to regulate Salmonella Virulence. PLoS Pathog. 12, e1005458. doi: 10.1371/journal.ppat.1005458
Rösler, S. M., Kramer, K., Finkemeier, I., Humpf, H. U., and Tudzynski, B. (2016). The SAGA complex in the rice pathogen Fusarium fujikuroi: structure and functional characterization. Mol. Microbiol. 102, 951–974. doi: 10.1111/mmi.13528
Singh, B., Zhang, G., Hwa, Y., Li, J., Dowdy, S., and Jiang, S.-W. (2010). Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 10, 935–954. doi: 10.1586/era.10.62
Starai, V. J., and Escalante-Semerena, J. C. (2004). Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 340, 1005–1012. doi: 10.1016/j.jmb.2004.05.010
Sun, P., Cui, J., Jia, X., and Wang, W. (2017). Isolation and characterization of Bacillus amyloliquefaciens L-1 for biocontrol of pear ring rot. Horticultural Plant J. 3, 183–189. doi: 10.1016/j.hpj.2017.10.004
Syed, K., Doddapaneni, H., Subramanian, V., Lam, Y. W., and Yadav, J. S. (2010). Genome-to-function characterization of novel fungal P450 monooxygenases oxidizing polycyclic aromatic hydrocarbons (PAHs). Biochem. Biophys. Res. Commun. 399, 492–497. doi: 10.1016/j.bbrc.2010.07.094
Tian, X.-L., Zhang, K., Wang, G.-L., and Liu, W.-D. (2016). Comparative transcriptome analysis of Setosphaeria turcica revealed its responses mechanisms to the biological control agent Bacillus amyloliquefaciens. Scientia Sinica Vitae 46, 627–636. doi: 10.1360/N052016-00166
Van Acker, H., Van Dijck, P., and Coenye, T. (2014). Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 22, 326–333. doi: 10.1016/j.tim.2014.02.001
Wang, D., Junfan, F., Rujun, Z., Zibo, L., Yueling, H., and Xinran, L. (2019). Transcriptome analysis of Sclerotinia ginseng and comparative analysis with the genome of Sclerotinia sclerotiorum. Physiol. Mol. Plant Pathol. 106, 30–41. doi: 10.1016/j.pmpp.2018.11.003
Wang, Q., Zhang, Y., Yang, C., Xiong, H., Lin, Y., Yao, J., et al. (2010). Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. doi: 10.1126/science.1179687
Wang, Q.-H., Ji, Y.-P., Qu, Y.-Y., Qi, Y.-K., Li, D.-W., Liu, Z.-Y., et al. (2020). The response strategies of Colletotrichum gloeosporioides s.s. due to the stress caused by biological control agent Bacillus amyloliquefaciens deciphered by transcriptome analyses. Biol. Control 150, 104372. doi: 10.1016/j.biocontrol.2020.104372
Warrens, M. J. (2017). Transforming intraclass correlation coefficients with the Spearman–Brown formula. J. Clin. Epidemiol. 85, 14–16. doi: 10.1016/j.jclinepi.2017.03.005
Weinert, B. T., Schölz, C., Wagner, S. A., Iesmantavicius, V., Su, D., Daniel, J. A., et al. (2013). Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 4, 842–851. doi: 10.1016/j.celrep.2013.07.024
Wiśniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Wu, X., Vellaichamy, A., Wang, D., Zamdborg, L., Kelleher, N. L., Huber, S. C., et al. (2013). Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J. Proteomics 79, 60–71. doi: 10.1016/j.jprot.2012.12.001
Xie, L., Wang, X., Zeng, J., Zhou, M., Duan, X., Li, Q., et al. (2015). Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int. J. Biochem. Cell Biol. 59, 193–202. doi: 10.1016/j.biocel.2014.11.010
Yang, X., Zhang, L., Xiang, Y., Du, L., Huang, X., and Liu, Y. (2020). Comparative transcriptome analysis of Sclerotinia sclerotiorum revealed its response mechanisms to the biological control agent, Bacillus amyloliquefaciens. Sci. Rep. 10, 12576. doi: 10.1038/s41598-020-69434-9
Yin, Z., Liu, H., Li, Z., Ke, X., Dou, D., Gao, X., et al. (2015). Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 208, 1202–1216. doi: 10.1111/nph.13544
Zettl, R., Schell, J., and Palme, K. (1994). Photoaffinity labeling of Arabidopsis thaliana plasma membrane vesicles by 5-azido-[7-3H]indole-3-acetic acid: identification of a glutathione S-transferase. Proc. Nat. Acad. Sci. 91, 689–693. doi: 10.1073/pnas.91.2.689
Zhang, K., Zheng, S., Yang, J. S., Chen, Y., and Cheng, Z. (2013). Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 12, 844–851. doi: 10.1021/pr300912q
Zhang, S., Wakai, S., Sasakura, N., Tsutsumi, H., Hata, Y., Ogino, C., et al. (2020). Pyruvate metabolism redirection for biological production of commodity chemicals in aerobic fungus Aspergillus oryzae. Metab. Eng. 61, 225–237. doi: 10.1016/j.ymben.2020.06.010
Keywords: acetylated proteins, Cytospora mali, Bacillus velezensis, apple valsa canker, biological control
Citation: Sun P, Ma Q and Zhang L (2022) Comprehensive acetyl-proteomic analysis of Cytospora mali provides insight into its response to the biocontrol agent Bacillus velezensis L-1. Front. Sustain. Food Syst. 6:999510. doi: 10.3389/fsufs.2022.999510
Received: 21 July 2022; Accepted: 12 August 2022;
Published: 31 August 2022.
Edited by:
Zhen He, Yangzhou University, ChinaReviewed by:
Mo Zhu, Henan Normal University, ChinaXingan Hao, Northwest Agriculture & Forest University, China
Copyright © 2022 Sun, Ma and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, bGVpLnpoYW5nQGltYXUuZWR1LmNu