 Gleb Yu. Kosovsky1
Gleb Yu. Kosovsky1 Galina V. Glazko
Galina V. Glazko Olga I. Skobel
Olga I. Skobel- 1Department of Biotechnology, Afanas‘ev Research Institute of Fur-Bearing Animal Breeding and Rabbit Breeding, Moscow, Russia
- 2Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR, United States
Background: Without identifying and studying the genomic characteristics associated with domestication, managing farm animal genetic resources becomes overwhelmingly difficult. Accumulated data confirm that mobile genetic elements participate in the domestication process and, in particular, generate widely abundant microRNAs.
Methods: The recombination products were compared in silico between the long interspersed nuclear element (LINE) and the endogenous retrovirus (ERV), forming the LINE/ERV/LINE sequence, located in a closely linked conserved block of 12 genes, as well as the microRNAs formed by these recombination products in domesticated-wild pairs of mammals. For this comparison, the reference genomes of domesticated cattle (Bos taurus) and its closely related wild species counterpart, bison (Bison bison), were used.
Results: It was found that the above-noted highly conserved recombination products (with more than 81.5% identity) were present in the corresponding block of 12 genes in bison. These recombination products served as sources of 51 microRNAs in bison and 129 microRNAs in cattle, including 50 microRNAs that were similar in both species. A total of 79 microRNAs were found only in cattle trinomial recombination products, with 98% belonging to the mir-30 family, including the cattle-specific bta-miR-30a-5p and bta-miR-30e-5p. The mir-30 family is closely associated with biological processes influencing the quantity and quality of agricultural products.
Conclusion: Trinomial retrotransposon recombination products were fixed in both the cattle genome and the genome of its closely related wild species, the bison. It was found that these products may be involved in the response to intensive artificial selection and the domestication process since interspecific differentiation of microRNAs is associated with regulatory networks that have a significant impact on the formation of economically important traits.
1 Introduction
Bos species (taurine cattle, zebu, yak, river buffalo, swamp buffalo, etc.) have complex patterns of domestication and have been subjects of strong artificial selection (1). The American bison (Bison bison) is one of the extant Bos species that has not been domesticated (2). Since domesticated and semi-domesticated Bos species are well known, cattle and bison represent two extremes on this scale. Therefore, the identification of genomic characteristics that distinguish highly specialized commercial cattle breeds from closely related wild species is of particular interest. However, the genetic factors underlying the domestication of Bos species remain unknown (3).
In mammals, mobile genetic elements affect the formation of new genes and their functional evolution. Increased activity of mobile genetic elements can contribute to the formation, subsequent selection, and fixation of new adaptive phenotypic traits during domestication (4, 5). In addition, these elements are capable of forming conserved and variable genomic domains (6–8) with unknown functional features.
Retrotransposons and their recombination products are known to be the main source of new microRNAs, which are an extensive class of single-stranded, short (19–24 bp) non-coding RNAs (9), and they are widely distributed throughout the genome (10–12). Many studies have highlighted the significant importance of microRNAs in the regulation of a wide range of biological processes in different mammalian species (13, 14).
The involvement of microRNA regulatory variants in the selection process plays an important role during domestication and subsequent artificial selection. The origin of modern taurine cattle is closely related to the presence of polymorphic 3′ UTR microRNA binding sites in 1,620 genes of modern-day cattle breeds, compared to its ancestral form, the wild aurochs (Bos primigenius). These sites influence neurobiological, metabolic, immunobiological, and reproductive phenotypes associated with domestication (15).
Earlier, we identified 511 domains in bovine chromosome 1 (13,436,028 bp) that were recombination products of the long interspersed nuclear element (LINE) and the endogenous retrovirus (ERV). A total of 30 RTE-BovB/BTLTR1/RTE-BovB clusters (hereinafter BovLTRBov) were found in 12 structural genes (kcne2, gart, tmem50b, il10rb, ifnar2, urb1, grik1, usp16, ltn1, cyyr1, app, and jam2). These genes form a large syntenic block that has been preserved during the evolution of mammals, starting with the platypus (7, 16). It was found that these BovLTRBov regions are preserved in the bovine genome with high identity, as they are part of its regulatory system containing different microRNAs. Some of these microRNAs are associated with milk and meat production (12).
To determine how these retrotransposon recombination products are involved in the response to intensive artificial selection and, presumably, the domestication process, we compared microRNA-containing homologous regions in cattle and bison. The analysis was conducted using open-source bison genomic sequence data and the sequenced genome of Hereford cattle.
2 Results
2.1 Bison conservative syntenic group
The functional roles of the 12 structural genes (kcne2, gart, tmem50b, il10rb, ifnar2, urb1, grik1, usp16, ltn1, cyyr1, app, and jam2) were mostly analyzed in humans (Homo sapiens) and laboratory mice (Mus musculus), suggesting their close connection with the central nervous system, particularly in relation to the occurrence of behavioral disorders (17, 18), Alzheimer's disease (19–21), and Down syndrome (22). They form an evolutionarily conserved block, which can be found in a number of mammals, including human chromosome 21, mouse chromosome 16, rabbit chromosome 14, and platypus chromosome 17 (16).
They are also present in bison chromosome 1, maintaining the same co-localization (Figure 1).
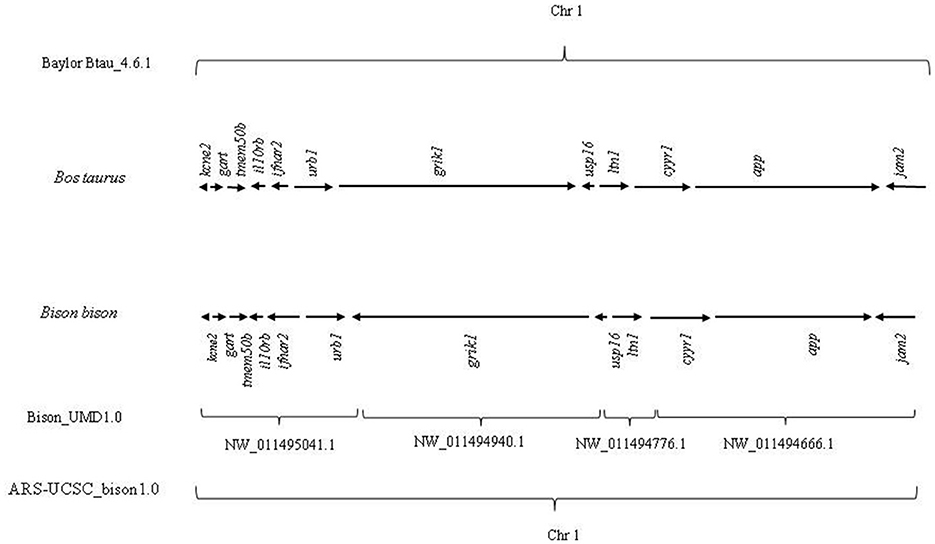
Figure 1. Genomic distribution of evolutionarily conserved 12 genes (kcne2, gart, tmem50b, il10rb, ifnar2, urb1, grik1, usp16, ltn1, cyyr1, app, and jam2) in Bos taurus and Bison bison.
Pairwise comparisons for 12 genes between bison and cattle demonstrated a high percentage of identity. The maximum percentage of identity was 99.00% (Bos taurus phosphoribosylglycinamide formyltransferase, gart), and the minimum percentage of identity was 91.00% (Bos taurus cysteine and tyrosine rich 1, cyyr1) (Table 1).
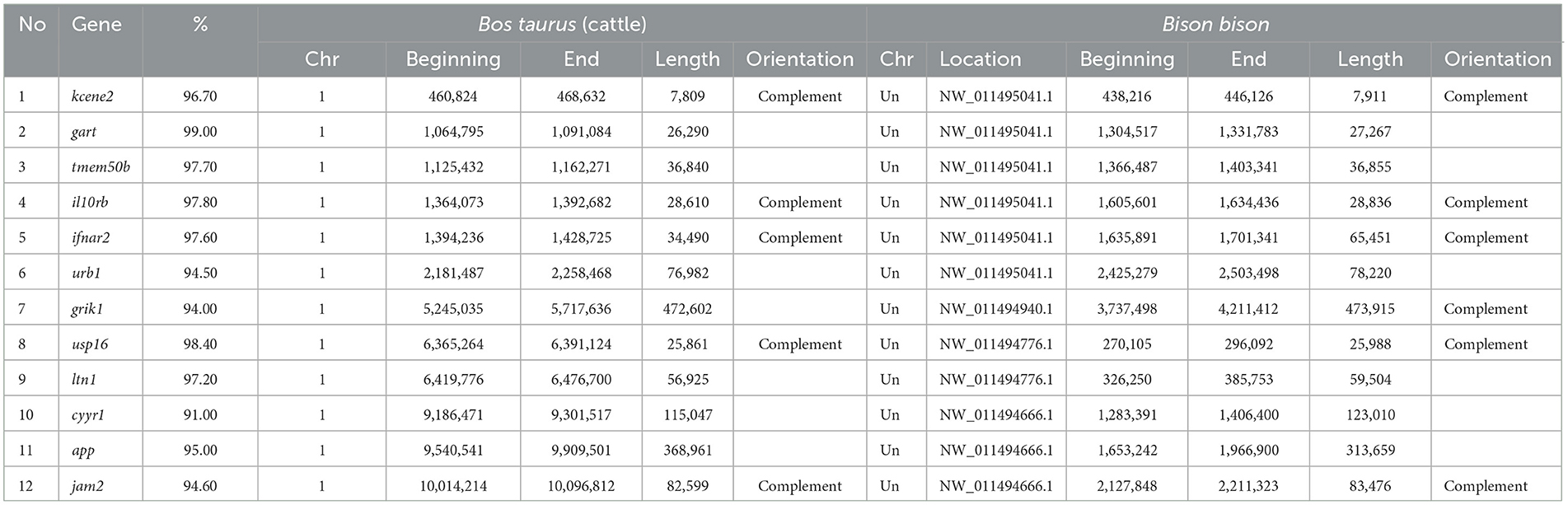
Table 1. Genomic characteristics for 12 evolutionarily conserved genes (kcne2, gart, tmem50b, il10rb, ifnar2, urb1, grik1, usp16, ltn1, cyyr1, app, jam2) in cattle (ver. 2011) and bison (ver. 2014).
Furthermore, intron-located regions of bovine chromosome 1, homologous to BovLTRBov, were also found in bison for these genes. The maximum percentage of identity was 99.63% (Bos taurus phosphoribosylglycinamide formyltransferase, gart), and the minimum was 81.5% (Bos taurus junctional adhesion molecule 2, jam2) (Table 2). The frequency of BovLTRBov was 32% higher in cattle than in bison.
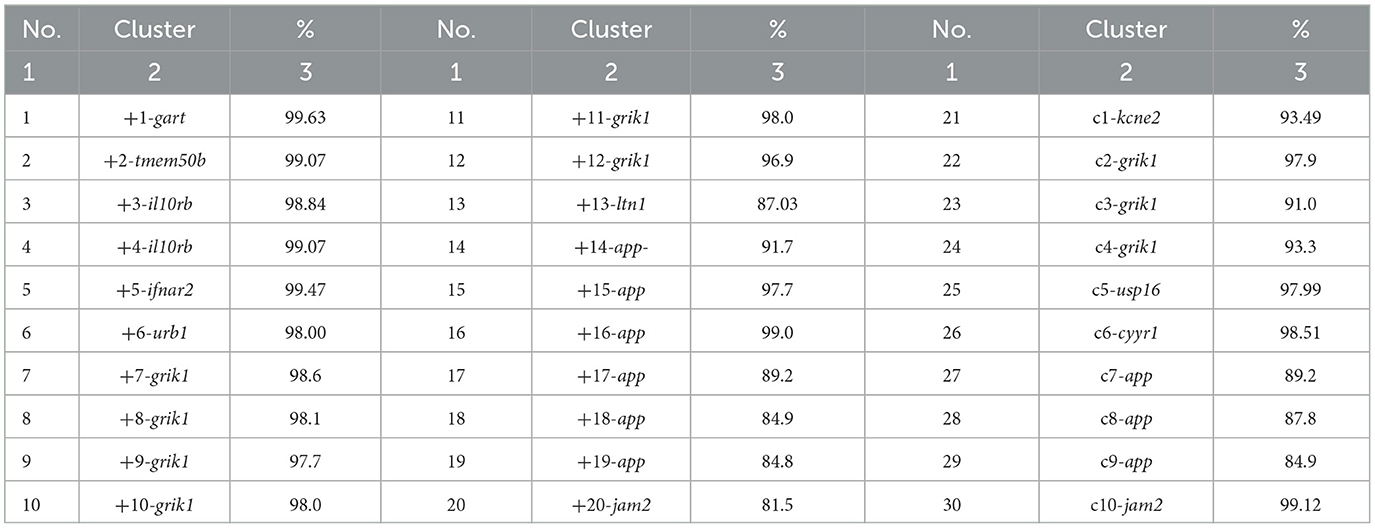
Table 2. Percent identity matrix of the RTE-BovB/BTLTR1/RTE-BovB recombination products in cattle and bison (pairwise alignment) (%).
It was found that regions that closely match six bovine BovLTRBov recombination products in Bos taurus amyloid beta precursor protein, app, were all present in the same bison sequence of 2,772 bp in length, which is also part of the app (the coordinates are shown in Table 3).
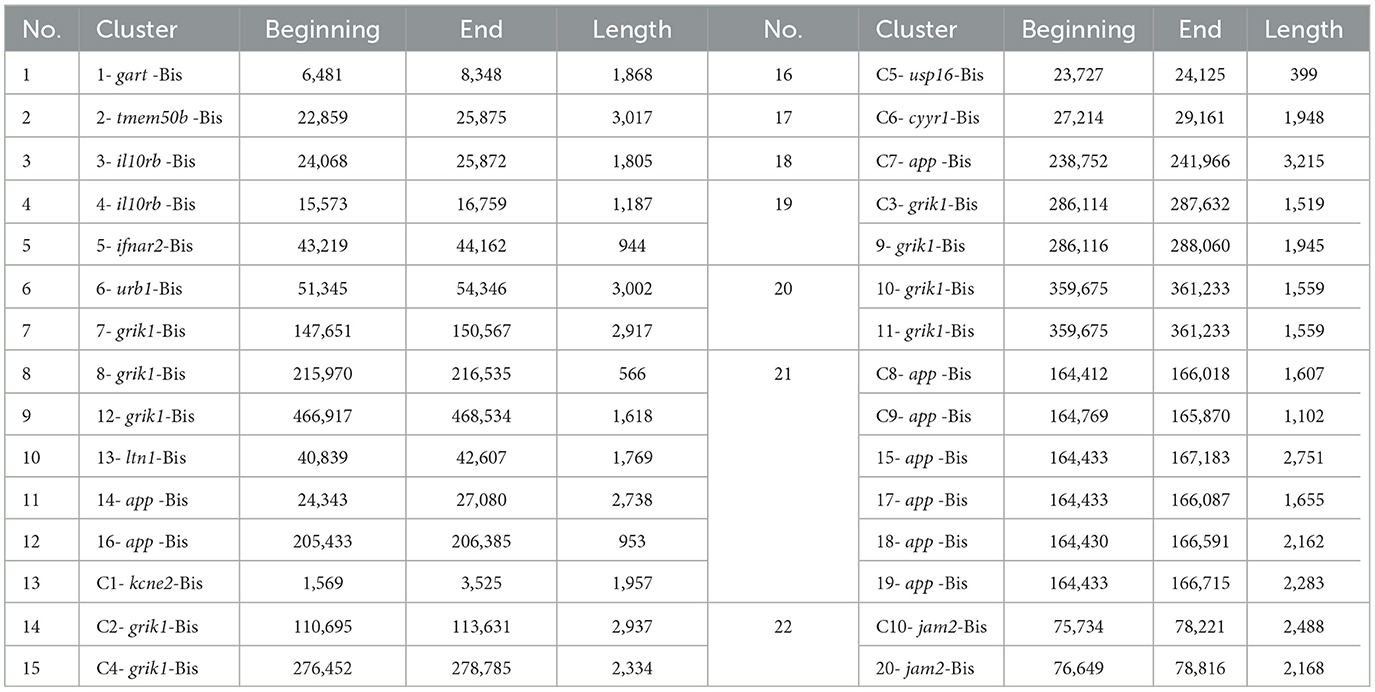
Table 3. Bison sequence coordinates (within the corresponding genes) with a high percentage of identity to the RTE-BovB/BTLTR1/RTE-BovB recombination products in the cattle.
One sequence from Bos taurus junctional adhesion molecule 2, jam 2, which is 3,083 bp long in bison, showed a high degree of homology to two bovine BovLTRBov regions located within the same gene (Table 3).
Two pairs of highly homologous bovine BovLTRBov regions from Bos taurus glutamate ionotropic receptor kainate type subunit 1, grik1, were found in two sections of the bison grik1 gene, measuring 1,559 bp and 1,947 bp in length (Table 3).
Multiple sequence alignments of all bison sequences with a high percentage of identity to bovine BovLTRBov showed a minimum percent identity of 90.48% (sequences in listerin E3 ubiquitin protein ligase 1, ltn1, and potassium voltage-gated channel subfamily E regulatory subunit 2, kcne2) and a maximum of 100% (between two sequences in amyloid beta precursor protein, app) (Figure 2).
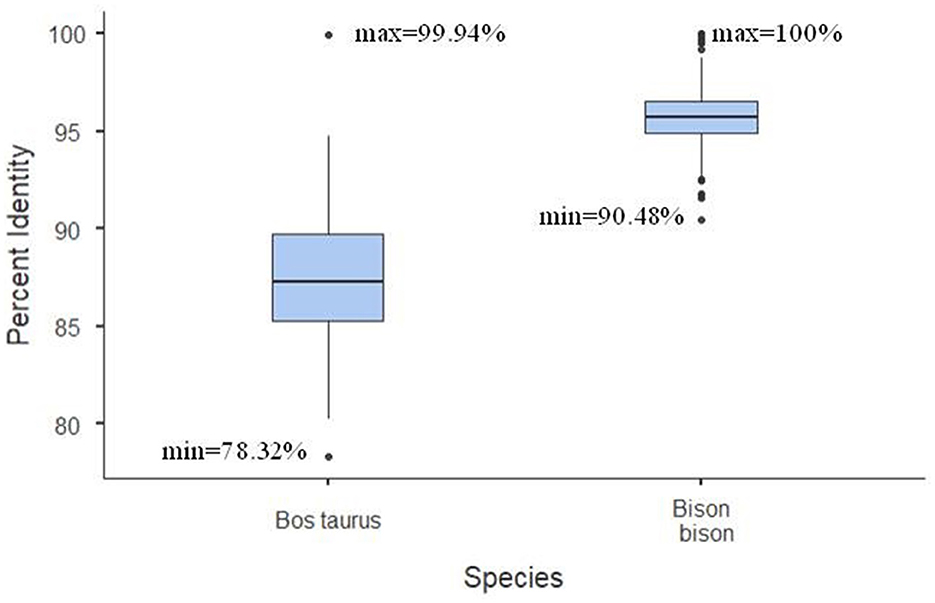
Figure 2. Percent identity matrix. The heatmap shows the bison sequences with a high percentage of identity to RTE-BovB/BTLTR1/RTE-BovB recombination products in cattle (multiple sequence alignment). The colors indicate the percent identity between the sequences. Green represents 90% identity, red represents 100% identity, and other colors represent values in between.
The percent identity of these bison regions was higher than that of the cattle recombination products (Figure 3).

Figure 3. The percent identity between all the detected regions in the gene block in the Bos taurus and Bison bison.
The high similarity between BovLTRBov regions within the genes from the studied wild-domesticated pair suggested their specific functional roles. This strong conservation may be due to the presence of regulatory network elements such as microRNAs. The regions, homologous to microRNAs, were further searched in the cattle and bison to test this assumption.
2.2 Analysis of microRNA presence
The following sequence (266 bp) was identified from the Bison regions homologous to bovine 30 BovLTRBov, with at least 95.29% similarity (Table 4):
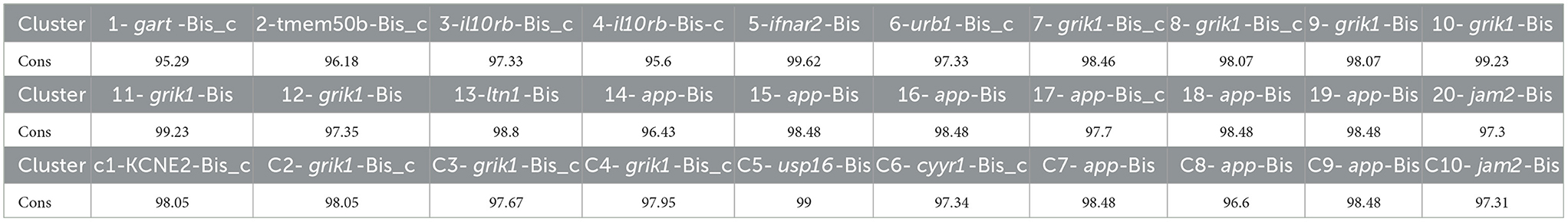
Table 4. Percent identity matrix of the bison conserved sequence (CS) with the identified nucleotide sequences (%).
TACTAGCGTGTGAGATGAGTGCAATTGTGC GGTAGTTTGAGCATTCTTTGGCATTGCCTTTC TTTGGGATTGGAATGAAAACTGACCTTTTCCAGTCCTGT GGCCACTGCTGAGTTTTCCAAATTTGCTGG CATATTGAGTGCAGCACTTTCACAGCATCAT CTTTCAGGATTTGAAATAGCTCAACTGG AATTCCATCACCTCCACTAGCTTTGTTC GTAGTGATGCTTCCTAAGGCCCACTTGACTTCACATTCCAGGATGTCT.
Earlier, we identified a conserved sequence of 266 bp in length from recombination products in cattle (12). The percent identity between the conserved sequences of cattle and bison was 99.2% (Figure 4).
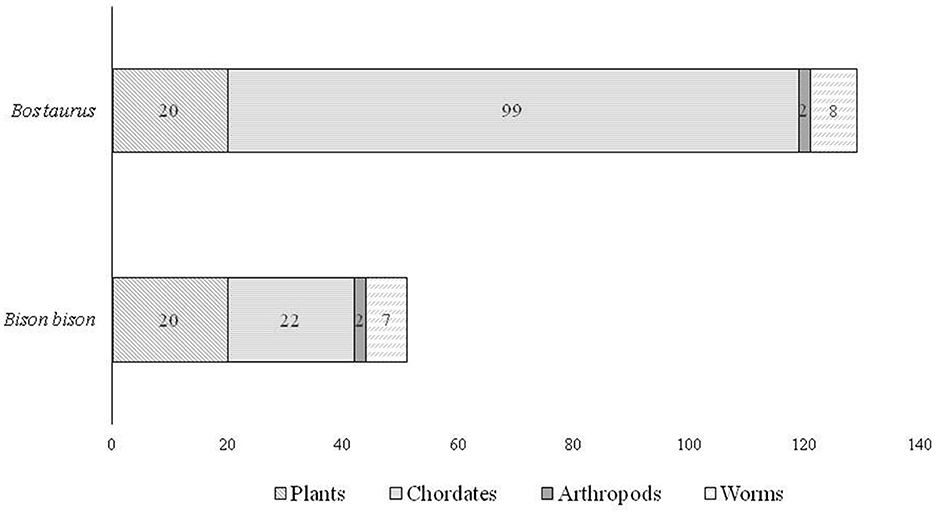
Figure 4. Alignment of the bison (Bison bison) conserved sequence with the cattle conserved sequence, as reported by Skobel et al. (12).
The search for microRNAs in the conserved bison sequence resulted in 51 microRNAs from 30 different species (both animals and plants). A total of 129 microRNAs from 63 different species were found in the Bos taurus conserved sequence (Figure 5).
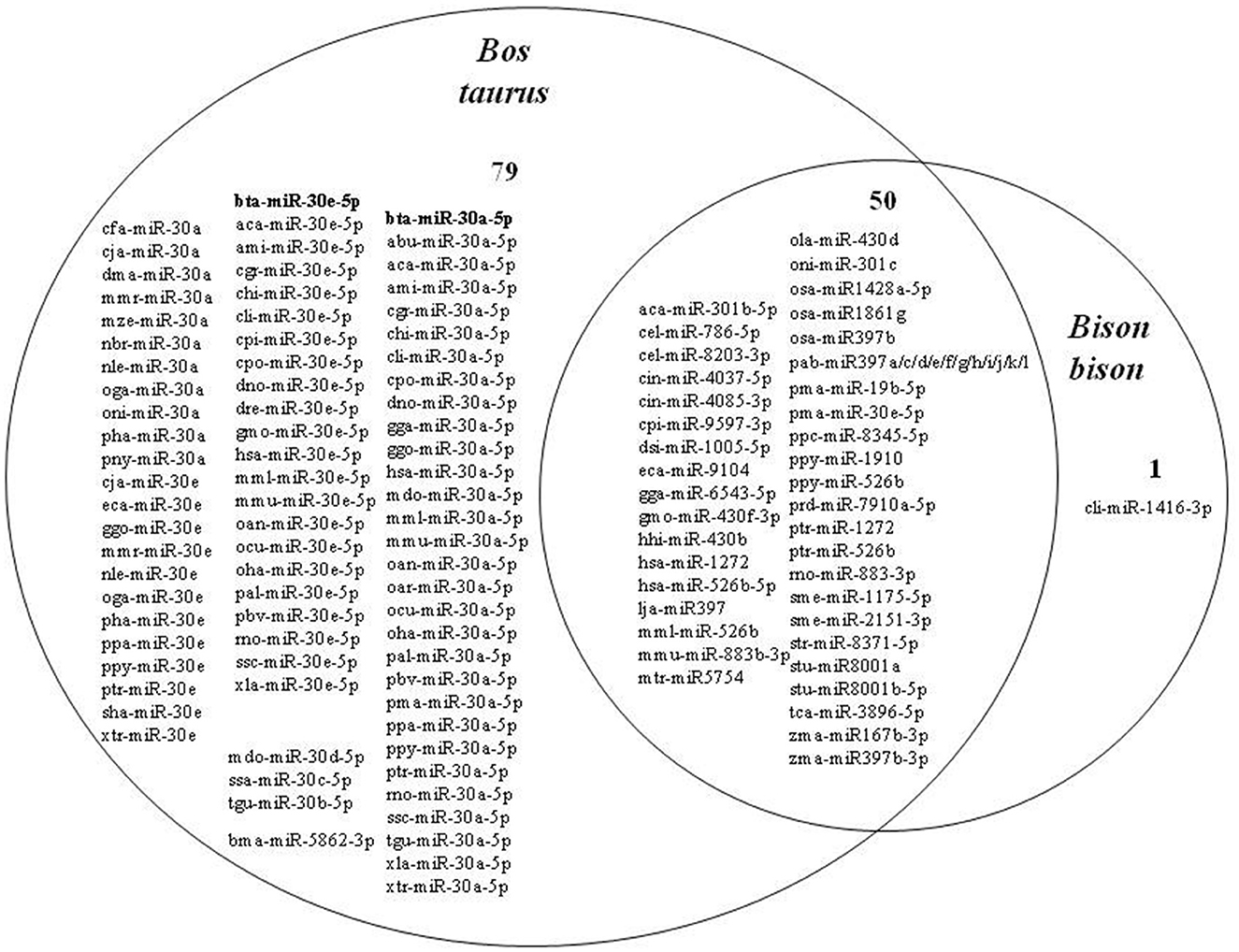
Figure 5. Distribution of microRNAs present in the conserved sequences in bison (Bison bison and Bos taurus).
A total of 50 microRNAs were identified as common between cattle and bison (Figure 6). For example, mtr-miR-5754 (Medicago truncatula, barrel clover), found in the conserved sequence of both bison and cattle, is known to decrease the stability of oncogenic target transcripts in humans, whose products promote cell proliferation (23).
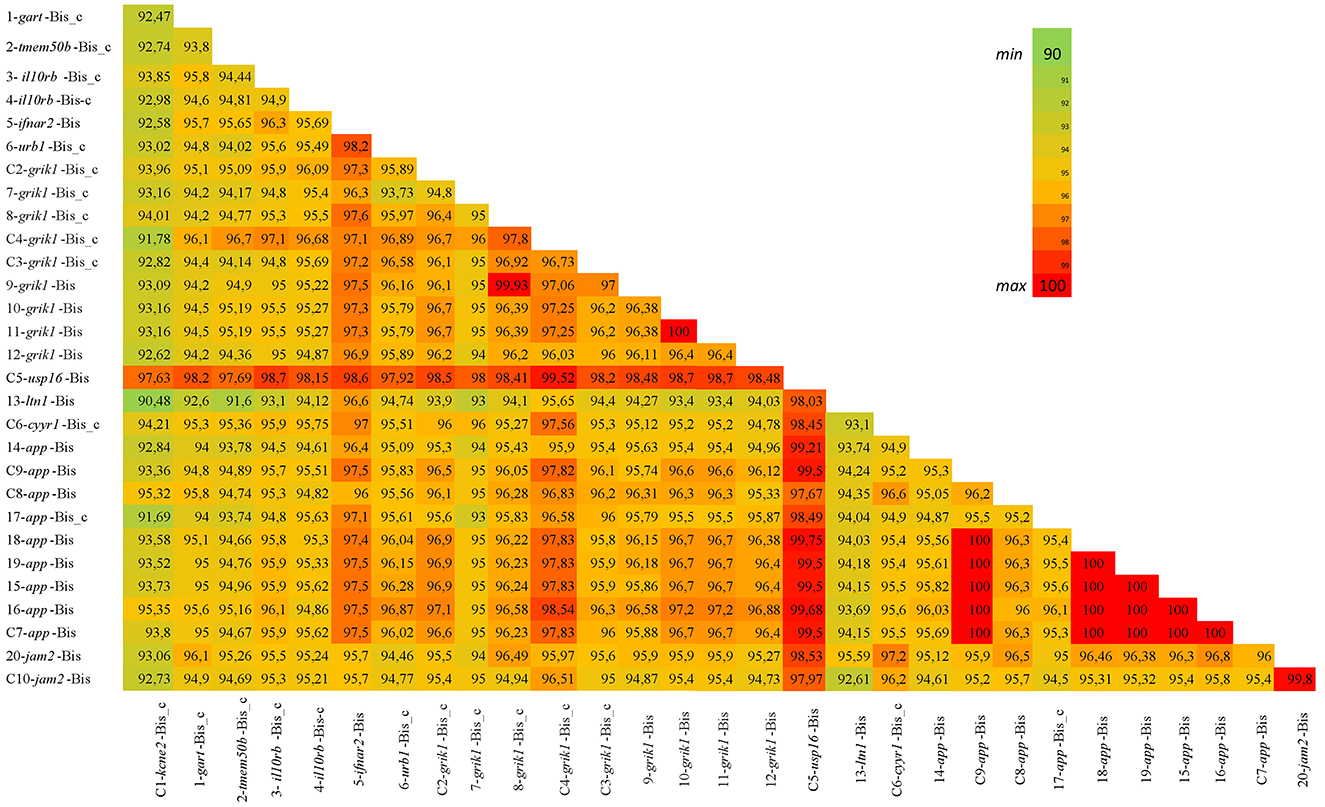
Figure 6. Comparison of microRNAs found in the Bos taurus and Bison bison syntenic gene block.
The most similar plant-specific microRNAs (14 out of 20) belonged to the miR-397 family. MiR-397 is involved in various biological processes, including cell growth, reproductive organ development, and plant resistance to external adverse stimulation. Additionally, it also is involved in regulating gene functions related to fatty acid metabolism (24).
MiR-526b overexpression is statistically significant in patients with bipolar disorder (25). MiR-526b is also associated with various types of oncological diseases, such as cervical cancer (26) and breast cancer (27). Moreover, MiR-1272 plays a crucial role in the regulation of immune signaling, cytokine production, and migration of immune cells in order to control visceral leishmaniasis infection in humans (28). Eca-miR-9104 is expressed in horses during equine herpesvirus 1 infection (29). However, for the majority of microRNAs homologous between cattle and bison, their functional roles remain to be studied.
At the same time, it was found that one microRNA, cli-miR-1416-3p, was absent in cattle but present in bison. This microRNA belongs to the miR-1416-3p family and is presumably involved in follicle atresia during specific stages of folliculogenesis in birds (30).
A total of 79 microRNAs were absent in bison but present in cattle, including 78 microRNAs belonging to the miR-30 family, with 2 bovine-specific microRNAs, namely bta-miR-30a-5p and bta-miR-30e-5p.
The accumulated evidence suggests that miR-30a-5p and miR-30e-5p are crucial for regulating key physiological systems in cattle. They play a key role in the response to heat stress (31), affect the development of the immune system and the immune response (32), are vital for milk production (85, 86), can influence milk composition (81, 82), inhibit the differentiation of muscle cells (33), and play an important regulatory role in the processes of fertilization and early development (34).
MiR-30e-5p and miR-30a-5p are also involved in the process of aging in humans (35) and play a role in the regulation of various diseases, including bacterial infections (36). They are considered potential biomarkers for neurodegenerative disorders (37), systemic lupus erythematosus (38), diabetes (39), various tumors (40), and heart diseases (83, 84).
3 Discussion
It is becoming clear that domesticated species differ from their closely related wild counterparts in terms of a high level of phenotypic variability (41). Understanding the evolution of the genetic mechanisms behind phenotypic traits has become possible by comparing closely related species with their wild counterparts and identifying key elements that regulate underlying variability (42).
Nonetheless, selecting the appropriate wild-domesticated pair has become a fundamental consideration. Along with the European bison (Bison bonasus) and the gaur (Bos gaurus), the American bison is one of the few remaining wild animals of the genus Bos (2). The gaur, on the other hand, has a domesticated form known as the gayal, and it has been noted that artificial selection has an impact on the gaur genome (43). In contrast, no domesticated form of bison is known, and evidence suggests that bovine alleles may only be present in a limited portion of the genome (44).
To date, a large amount of public data on the organization of the bovine genome has been accumulated (45–47). We used open-source Hereford cattle (Bos taurus) gene data based on the Baylor Btau_4.6.1/bosTau7 October 2011 assembly to maintain the logic and reproducibility of our previous research. It should be noted that for the purpose of this study, the use of the latest version of the cattle reference genome [The ARS-UCD1.3 (47)] did not affect the results as the sequences of the studied structural genes exhibited high similarity across different versions (Supplementary material). Modern sequencing technologies generate new high-quality assemblies, reducing possible errors in sequences enriched with elements such as CG repeats (48, 49). However, the type of animal used for genome obtaining is also of great importance. The reference scaffold-level bison genome, Bison_UMD1.0, is currently available in the NCBI database (50). A more recent genome assembly, ARS-UCSC_bison1.0, (51) exists, but this genome represents an F1 hybrid between a bison sire and a Simmental cow. To avoid potential hybridization effects (52, 53), we excluded this assembly from the analysis.
In the majority of cases of speciation, genetic information cannot be traced through the evolution of particular genes but only through gene clusters (54). Synteny analysis, that is, studying the conserved blocks of genes found in different species, is one of the comparative genomics methods used for understanding evolutionary relationships, including those during domestication (55–57). Genomic studies typically rely on closely linked and conserved gene clusters and also require an understanding of the functional features of the genes included in these clusters (58, 59).
The 12 genes mentioned in this study form a large syntenic block, maintained during the evolution of mammals, and are notable for their well-conserved synteny (7, 12). These genes are involved in social activity, which is a crucial component of the domestication process (60). Our results also support the accumulated evidence that evolutionarily conserved syntenic blocks have a higher density of genes involved in the formation of anatomical characteristics and the development of the central nervous system (61). Hence, these genes are also believed to be associated with animal socialization and, therefore, domestication (62, 63). Gart, il10rb, ifnar2, urb1, and ltn1 are also considered candidates for bovine artificial selection (64–66). In cattle, tmem50B and app are candidate domestication genes according to the presence of divergent microRNA binding sites (15).
The pairwise comparison of the full-length genes indicated they are highly conserved in both Bos genus species. Therefore, the identified differences in the presence and genomic position of the microRNAs, which are involved in the regulatory networks of gene expression, are intriguing.
The Bison regions, homologous to bovine BovLTRBov, were never found in exons of either bison or cattle. This may be because the consensus sequence is too long to be present in exons without interfering with the genes' functions. In addition, they had a lower frequency but higher pairwise identity compared to cattle. It probably reflects the differences between the two species in phenotypic variability and the width of species distribution (1). In addition, the higher proportion of retrotransposons in domesticated animals compared to their wild counterparts is likely related to the domestication process (67).
Retrotransposons and their genome distribution are very species-specific. For example, LINE/RTE-BovB is frequently found only in Bos species, despite being involved in horizontal genetic information transfer (68). This situation is similar to the presence of SINE/Alu in primate genomes and SINE/tRNA-Core-RTE in cattle genomes. Even very different transposons, such as the LINE, SINE, and long terminal repeat (LTR), can sometimes contain the same regulatory elements (69).
The in silico microRNA identification strategy is used as an accurate, fast, and reliable method for predicting microRNA homologs in different species (70, 71). The proportion of microRNAs derived from transposons in humans is higher than in other vertebrates, especially non-mammal vertebrates (72). We provide evidence that the number of microRNAs derived from transposons also increases during the domestication process (Figure 6). It is important to note that multiple hybridization events between Bos taurus and Bison bison have taken place over the last 200 years (44). Nevertheless, our study revealed interspecific differences in microRNA presence. Considering the high identity of the two conserved sequences, a number of microRNAs were found in both cattle and bison. The likely diversity of species-specific microRNAs has been proven by existing scientific data. Recent evidence suggests that microRNAs can move from plants to animals via the gastrointestinal tract and access cellular targets, affecting the physiological and pathological conditions of their recipients (73).
The most interesting result of our study is that some microRNAs were different between the genomes of cattle and bison. This occurred because of the variations in the conserved sequences of the two species in three nucleotides at the beginning and the presence of a single-nucleotide polymorphism (SNP) in the bison conserved sequence at 251 bp (Figure 4). Of these, 78 microRNAs were from the miR-30 family, including two bovine-specific microRNAs: bta-miR-30a-5p and bta-miR-30e-5p. Members of this family are known to increase milk fat content (74), contribute to the development of muscle tissue in cattle (75), and play a role in the development of stress and immune responses (32). Since the microRNAs that differed between the two species were related to important agricultural differences between cattle and bison, we assumed that these differences result from intensive artificial selection.
In general, the obtained results indicate that the accumulation of retrotransposons and their recombination products may be a source of microRNA regulatory networks. Our comparative analysis of the LINE and ERV sequences in a domesticated species (Bos taurus) and a closely related wild species (Bison bison) at 12 loci, where synteny has been maintained since the early stages of evolution, suggests the identification of molecular genetic pathways underlying the response to intensive artificial selection and, presumably, the domestication of Bov species. It should be noted that these gene products are likely to be involved in higher central nervous system activity in mammals.
Frequently, genome assemblies are insufficient for comparative genomic analysis as they may limit further interpretation by failing to capture the entire range of genetic diversity within a species (76, 77). However, in our study, we analyzed the sequences located in structural genes that preserve genetic linkage during evolution in different species. The clear difference we found in microRNA presence between the cattle and bison and the existing data on the formation of new binding sites during bovine domestication (15) both support our hypothesis that microRNAs could be involved in the process of artificial selection.
4 Materials and methods
The bison annotation release (IDs: 237421 [UID] 1351428 [GenBank] 1426508 [RefSeq]) (50) was retrieved from the NCBI GenBank (GenBank, RRID:SCR_002760). We used the Bison chromosome-scale genome, ARS-UCSC_bison1.0, to identify gene locations (51).
All gene data of Hereford cattle (Bos taurus) were based on the Baylor Btau_4.6.1/bosTau7 October 2011 assembly available in the Integrated Genome Browser (IGB, RRID:SCR_011792) to maintain the logic and reproducibility of our previous research. The ARS-UCD1.3 reference bovine genome was used to confirm the reliability and accuracy of the sequences utilized (47). The results are presented in Supplementary material.
The available cow RepeatMasker genomic dataset was used for obtaining information on the distribution of mobile genetic elements and their positioning in the cattle genome (78, 79). We identified 511 trinomial recombination products RTE-BovB/BTLTR1/RTE-BovB between the endogenous retrovirus (ERV) containing the LTR BTLTR1 and the non-LTR long interspersed nuclear element (LINE) RTE-BovB in the 13,436,028 bp nucleotide sequences of bovine chromosome 1. For further analysis, we took 30 RTE-BovB/BTLTR1/RTE-BovB (hereinafter BovLTRBov) recombination products detected in the 12 structural genes (kcne2, gart, tmem50b, il10rb, ifnar2, urb1, grik1, usp16, ltn1, cyyr1, app, and jam2), while the rest were found in intergenic spaces (7).
The coordinates of the 30 cattle RTE-BovB/BTLTR1/RTE-BovB recombination products according to the Baylor Btau_4.6.1/bosTau7 October 2011 assembly are indicated in Supplementary material.
Our previous studies (7, 12) have provided detailed methods for detecting trinomial recombination products between the LINE and LTR ERV, analyzing their localization in relation to structural genes and identifying the RTE-BovB/BTLTR1/RTE-BovB conserved sequence.
Open-source software provided by the European Institute of Bioinformatics was used to identify regions of similarity between the nucleotide sequences of cattle (Bos taurus) and bison (Bison bison). We used Clustal Omega (Clustal Omega, RRID:SCR_001591) and Kalign (Kalign, RRID:SCR_011810) for multiple sequence alignments with default settings. The EMBOSS Matcher (EMBOSSMatcher, RRID:SCR_017252) option during the pairwise sequence alignment was changed. We set the maximum value of alternative matches to 20 to ensure that possible additional alignments were not missed, while keeping all other parameters at their default settings.
The conserved sequence from the bison regions homologous to bovine was identified manually based on the results obtained from Kalign (Kalign, RRID:SCR_011810).
To check the presence of microRNAs in the conserved sequences of both bison and cattle, we used the microRNA database (v20), sorted by E-value, with the maximum possible number of results set to be displayed (80).
5 Conclusion
It can be assumed that the trinomial recombination products between the LINE and the ERV are fixed in the genomes of cattle and the closely related wild species, bison. These products could be actively involved in the response to intensive artificial selection and the domestication process by serving as sources of microRNAs that have a significant impact on agriculturally important cattle traits. Consequently, regulatory networks could change significantly under intensive artificial selection and probably domestication, not only due to the origin of new microRNA binding sites (15) but also due to the formation of new microRNAs.
Future studies are needed to validate these results by examining other wild-domesticated pairs of vertebrates and verifying the functional association with the observed differentiation of microRNA.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
GG: Formal analysis, Writing – original draft, Writing – review & editing. GK: Conceptualization, Formal analysis, Methodology, Supervision, Writing – review & editing. OS: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Ministry of Science and Higher Education of the Russian Federation for government assignments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1516731/full#supplementary-material
Abbreviations
ERV, endogenous retrovirus; LTR, long terminal repeat; LINE, long interspersed nuclear element; BovLTRBov, the recombination products between the long interspersed nuclear element and endogenous retrovirus, such as RTE-BovB/BTLTR1/RTE-BovB; kcne2, Bos taurus potassium voltage-gated channel subfamily E regulatory subunit 2; gart, Bos taurus phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimidazole synthetase; tmem50b, Bos taurus transmembrane protein 50B; il10rb, Bos taurus interleukin 10 receptor subunit beta; ifnar2, Bos taurus interferon alpha and beta receptor subunit 2; urb1, Bos taurus URB1 71 ribosome biogenesis 1 homolog (S. cerevisiae); grik1, Bos taurus glutamate ionotropic receptor kainate type subunit 1; usp16, Bos taurus ubiquitin specifc peptidase 16; ltn1, Bos taurus listerin E3 ubiquitin protein ligase 1; cyyr1, Bos taurus cysteine and tyrosine rich 1; app, Bos taurus amyloid beta precursor protein; jam2, Bos taurus junctional adhesion molecule 2.
References
1. Zhang K, Lenstra JA, Zhang S, Liu W, Liu Evolution J. and domestication of the Bovini species. Anim Genet. (2020) 51:637–57. doi: 10.1111/age.12974
2. Wu DD, Ding XD, Wang S, Wójcik JM, Zhang Y, Tokarska M, et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat Ecol Evolut. (2018) 2:1139–45. doi: 10.1038/s41559-018-0562-y
3. Scherf B, Pilling D. The second report on the state of the World's animal genetic resources for food and agriculture. In: FAO Commission on Genetic Resources for Food and Agriculture Assessments 2. Rome, Italy: FAO. (2015).
4. Carelli FN, Hayakawa T, Go Y, Imai H, Warnefors M, Kaessmann H. The life history of retrocopies illuminates the evolution of new mammalian genes. Genome Res. (2016) 26:301–14. doi: 10.1101/gr.198473.115
5. Schrader L, Schmitz J. The impact of transposable elements in adaptive evolution. Mol Ecol. (2019) 28:1537–49. doi: 10.1111/mec.14794
6. Adelson DL, Raison JM, Edgar RC. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome. Proc Natl Acad Sci U S A. (2009) 106:12855–60. doi: 10.1073/pnas.0901282106
7. Glazko VI, Skobel OI, Kosovsky GYu, Glazko TT. Domain distribution of mobile genetic elements in the bovine genome Sel'skokhozyaistvennaya Biologiya (2017) 52:658–68. doi: 10.15389/agrobiology.2017.4.658eng
8. Adelson DL, Raison JM, Garber M, et al. Interspersed repeats in the horse (Equus caballus); spatial correlations highlight conserved chromosomal domains. Anim Genet. (2010) 41:91–9. doi: 10.1111/j.1365-2052.2010.02115.x
9. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–854. doi: 10.1016/0092-8674(93)90529-Y
10. Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE. (2007) 2:e203. doi: 10.1371/journal.pone.0000203
11. Roberts JT, Cardin SE, Borchert GM. Burgeoning evidence indicates that microRNAs were initially formed from transposable element sequences. Mob Genet Elements. (2014) 4:e29255. doi: 10.4161/mge.29255
12. Skobel OI, Glazko VI, Kosovsky GYu, Glazko TT. Mobile genetic elements recombinations as the source of microRNA. Izvestiya Timiryazevskoj sel‘skoxozyajstvennoj akademii (2017) 4:70–98. doi: 10.26897/0021-342X-2017-4-70-98
13. Malvisi M, Palazzo F, Morandi N, Lazzari B, Williams JL, Pagnacco G, et al. Responses of bovine innate immunity to Mycobacterium avium subsp. paratuberculosis infection revealed by changes in gene expression and levels of MicroRNA. PloS ONE. (2016) 11:e0164461. doi: 10.1371/journal.pone.0164461
14. Raszek MM, Guan LL, Plastow GS. Use of genomic tools to improve cattle health in the context of infectious diseases. Front Genet. (2016) 7:30. doi: 10.3389/fgene.2016.00030
15. Braud M, Magee DA, Park SD, Sonstegard TS, Waters SM, MacHugh DE, et al. Genome-wide microRNA binding site variation between extinct wild aurochs and modern cattle identifies candidate microrna-regulated domestication genes. Front Genet. (2017) 8:3. doi: 10.3389/fgene.2017.00003
16. Skobel OI. Evolutionary conserved synteny of 12 genes associated with the central nervous system. In: Proceedings of the International Youth Scientific Forum “LOMONOSOV-2021”. (2021). Chesapeake, VA: MAKS Press. Available online at: https://lomonosov-msu.ru/archive/Lomonosov_2021/data/21878/126134_uid143833_report.pdf (accessed September 7, 2024).
17. Hirata Y, Zai CC, Souza RP, Lieberman JA, Meltzer HY, Kennedy JL. Association study of GRIK1 gene polymorphisms in schizophrenia: case-control and family-based studies. Hum Psychopharmacol. (2012) 27:345–51. doi: 10.1002/hup.2233
18. Le-Niculescu H, Patel SD, Bhat M, Kuczenski R, Faraone SV, Tsuang MT, et al. Convergent functional genomics of genome-wide association data for bipolar disorder: comprehensive identification of candidate genes, pathways and mechanisms. American J Med Genet Part B, Neuropsychiat Genetics. (2009) 150:155–81. doi: 10.1002/ajmg.b.30887
19. Chu J, Hong NA, Masuda CA, Jenkins BV, Nelms KA, Goodnow CC, et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci U S A. (2009) 106:2097–103. doi: 10.1073/pnas.0812819106
20. Khondoker M, Newhouse S, Westman E, Muehlboeck JS, Mecocci P, Vellas B, et al. Linking genetics of brain changes to alzheimer's disease: sparse whole genome association scan of regional MRI volumes in the ADNI and AddNeuroMed cohorts. J Alzheimer's Dis. (2015) 45:851–64. doi: 10.3233/JAD-142214
21. Vallino Costassa E, Fiorini M, Zanusso G, Peletto S, Acutis P, Baioni E, et al. Characterization of amyloid-β deposits in bovine brains. J Alzheimer's Dis. (2016) 51:875–87. doi: 10.3233/JAD-151007
22. Vilardell M, Rasche A, Thormann A, Maschke-Dutz E, Pérez-Jurado LA, Lehrach H, et al. Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics. (2011) 12:229. doi: 10.1186/1471-2164-12-229
23. Marzano F, Caratozzolo MF, Consiglio A, Licciulli F, Liuni S, Sbisà E, et al. Plant miRNAs reduce cancer cell proliferation by targeting MALAT1 and NEAT1: a beneficial cross-kingdom interaction. Front Genet. (2020) 11:552490. doi: 10.3389/fgene.2020.552490
24. Huang S, Zhou J, Gao L, et al. Plant miR397 and its functions. Functional Plant Biol. (2021) 48:361–70. doi: 10.1071/FP20342
25. Banigan MG, Kao PF, Kozubek JA, et al. Differential Expression of Exosomal microRNAs in Prefrontal Cortices of Schizophrenia and Bipolar Disorder Patients. PLoS ONE. (2013) 8:e48814. doi: 10.1371/journal.pone.0048814
26. Li H, Wang J, Xu F, Wang L, Sun G, Wang J, et al. By downregulating PBX3, miR-526b suppresses the epithelial-mesenchymal transition process in cervical cancer cells. Future Oncol. (2019) 15:1577–91. doi: 10.2217/fon-2018-0575
27. Liu YQ, Cong YZ, Jiang J, Sheng JZ Li XH, Zhao M, et al. MiR-526b suppresses cell proliferation, cell invasion and epithelial-mesenchymal transition in breast cancer by targeting Twist1. Eur Rev Med Pharmacol Sci. (2020) 24:3113–21. doi: 10.26355/eurrev_202003_20678
28. Pandey RK, Sundar S, Prajapati VK. Differential Expression of miRNA Regulates T Cell Differentiation and Plasticity During Visceral Leishmaniasis Infection. Front Microbiol. (2016) 7:206. doi: 10.3389/fmicb.2016.00206
29. Zarski LM, Weber PSD, Lee Y, Soboll Hussey G. Transcriptomic profiling of equine and viral genes in peripheral blood mononuclear cells in horses during equine herpesvirus 1 infection. Pathogens. (2021) 10:43. doi: 10.3390/pathogens10010043
30. Yu J, He K, Ren T, Lou Y, Zhao A. High-throughput sequencing reveals differential expression of miRNAs in prehierarchal follicles of laying and brooding geese. Physiol Genomics. (2016) 48:455–63. doi: 10.1152/physiolgenomics.00011.2016
31. Lee J, Lee S, Son J, Lim H, Kim E, Kim D, et al. Analysis of circulating-microRNA expression in lactating Holstein cows under summer heat stress. PLoS ONE (2020) 15:e0231125. doi: 10.1371/journal.pone.0231125
32. Zheng Y, Chen KL, Zheng XM Li HX, Wang GL. Identification and bioinformatics analysis of microRNAs associated with stress and immune response in serum of heat-stressed and normal Holstein cows. Cell Stress Chaperones. (2014) 19:973–81. doi: 10.1007/s12192-014-0521-8
33. Zhang BW, Cai HF, Wei XF, Sun JJ, Lan XY, Lei CZ, et al. miR-30-5p Regulates Muscle Differentiation and Alternative Splicing of Muscle-Related Genes by Targeting MBNL. Int J Mol Sci. (2016) 17:E182. doi: 10.3390/ijms17020182
34. Stowe HM, Calcatera SM, Dimmick MA, Andrae JG, Duckett SK, Pratt SL. The bull sperm microRNAome and the effect of fescue toxicosis on sperm microRNA expression. PLoS ONE. (2014) 9:e113163. doi: 10.1371/journal.pone.0113163
35. Lai CY, Wu YT Yu SL, Yu YH, Lee SY, Liu CM, et al. Modulated expression of human peripheral blood microRNAs from infancy to adulthood and its role in aging. Aging Cell. (2014) 13:679–89. doi: 10.1111/acel.12225
36. Mishra R, Krishnamoorthy P, Kumar H. MicroRNA-30e-5p Regulates SOCS1 and SOCS3 During Bacterial Infection. Front Cell Infect Microbiol. (2021) 10:604016. doi: 10.3389/fcimb.2020.604016
37. He M, Zhang H, Tang Z, et al. Diagnostic and therapeutic potential of exosomal MicroRNAs for neurodegenerative diseases. Neural Plast. (2021) 2021:8884642. doi: 10.1155/2021/8884642
38. Cheng T, Ding S, Liu S, Li X, Tang X, Sun L.. Resolvin D1 improves the Treg/Th17 imbalance in systemic lupus erythematosus through miR-30e-5p. Front Immunol. (2021) 12:668760. doi: 10.3389/fimmu.2021.668760
39. Dieter C, Assmann TS, Costa AR, Canani LH, de Souza BM, Bauer AC, et al. MiR-30e-5p and MiR-15a-5p expressions in plasma and urine of type 1 diabetic patients with diabetic kidney disease. Front Genet. (2019) 10:563. doi: 10.3389/fgene.2019.00563
40. Zhang S, Li G, Liu C, Lu S, Jing Q, Chen X, et al. miR-30e-5p represses angiogenesis and metastasis by directly targeting AEG-1 in squamous cell carcinoma of the head and neck. Cancer Sci. (2020) 111:356–68. doi: 10.1111/cas.14259
41. Glazko V, Zybailov B, Glazko T. Asking the right question about the genetic basis of domestication: what is the source of genetic diversity of domesticated species? Adv Genetic Eng. (2015) 04:1000125. doi: 10.4172/2169-0111.1000125
42. Chen S, Liu S, Shi S, Jiang Y, Cao M, Tang Y, et al. Comparative epigenomics reveals the impact of ruminant-specific regulatory elements on complex traits. BMC Biol. (2022) 20:273. doi: 10.1186/s12915-022-01459-0
43. Low WY, Rosen BD, Ren Y, Bickhart DM, To TH, Martin FJ, et al. Gaur genome reveals expansion of sperm odorant receptors in domesticated cattle. BMC Genom. (2022) 23:1. doi: 10.1186/s12864-022-08561-1
44. Stroupe S, Forgacs D, Harris A, Derr JN, Davis BW. Genomic evaluation of hybridization in historic and modern North American Bison (Bison bison). Sci Rep. (2022) 12:6397. doi: 10.1038/s41598-022-09828-z
45. Bovine Genome Sequencing and Analysis Consortium Elsik CG Tellam RL Worley KC Gibbs RA Muzny DM . The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science . (2009) 324:522–28. doi: 10.1126/science.1169588
46. Bovine HapMap Consortium; Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, et al. Genome-Wide Survey of SNP Variation Uncovers the Genetic Structure of Cattle Breeds. Science. (2009) 324:528–532. doi: 10.1126/science.1167936
47. Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaSci. (2020) 9:giaa021. doi: 10.1093/gigascience/giaa021
48. Kim J, Lee C, Ko BJ, Yoo DA, Won S, Phillippy AM, et al. False gene and chromosome losses in genome assemblies caused by GC content variation and repeats. Genome Biol. (2022) 23:204. doi: 10.1186/s13059-022-02765-0
49. Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. (2021) 592:737–46. doi: 10.1038/s41586-021-03451-0
50. Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N, et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Current Biol: CB. (2008) 18:R157–158. doi: 10.1016/j.cub.2008.01.019
51. Oppenheimer J, Rosen BD, Heaton MP, Vander Ley BL, Shafer WR, Schuetze FT, et al. A reference genome assembly of American Bison, Bison bison bison. J Heredity Koepfli K-P. (2021) 112:174–83. doi: 10.1093/jhered/esab003
52. Seidl F, Levis NA, Jones CD, Monroy-Eklund A, Ehrenreich IM, Pfennig KS. Variation in hybrid gene expression: implications for the evolution of genetic incompatibilities in interbreeding species. Mol Ecol. (2019) 28:4667–79. doi: 10.1111/mec.15246
53. Sloan DB, Warren JM, Williams AM, Kuster SA, Forsythe ES. Incompatibility and interchangeability in molecular evolution. Genome Biol Evol. (2022) 15:evac184. doi: 10.1093/gbe/evac184
54. Heng J, Heng HH. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems. (2021) 208:104476. doi: 10.1016/j.biosystems.2021.104476
55. Ghiurcuta CG, Moret BME. Evaluating synteny for improved comparative studies. Bioinformatics. (2014) 30:i9–i18. doi: 10.1093/bioinformatics/btu259
56. Liu H, Shi J, Cai Z, Huang Y, Lv M, Du H, et al. Evolution and Domestication Footprints Uncovered from the Genomes of Coix. Mol Plant. (2020) 13:295–308. doi: 10.1016/j.molp.2019.11.009
57. Restrepo-Montoya D, McClean PE, Osorno JOrthology M. and synteny analysis of receptor-like kinases “RLK” and receptor-like proteins “RLP” in legumes. BMC Genomics. (2021) 22:113. doi: 10.1186/s12864-021-07384-w
58. Schnable JC. Genome evolution in maize: from genomes back to genes. Annu Rev Plant Biol. (2015) 66:329–43. doi: 10.1146/annurev-arplant-043014-115604
59. Simakov O, Marlétaz F, Yue JX, O'Connell B, Jenkins J, Brandt A, et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat Ecol Evol. (2020) 4:820–30. doi: 10.1038/s41559-020-1156-z
60. Zeder MA. Core questions in domestication research. Proc Natl Acad Sci U S A. (2015) 112:3191–8. doi: 10.1073/pnas.1501711112
61. Damas J, Corbo M, Kim J, Turner-Maier J, Farré M, Larkin DM, et al. Evolution of the ancestral mammalian karyotype and syntenic regions. Proc Natl Acad Sci U S A. (2022) 119:e2209139119
62. Snyder-Mackler N, Burger JR, Gaydosh L, Belsky DW, Noppert GA, Campos FA, et al. Social determinants of health and survival in humans and other animals. Science. (2020) 368:eaax. doi: 10.1126/science.aax9553
63. Wilkins AS. A striking example of developmental bias in an evolutionary process: The ‘domestication syndrome'. Evol Dev. (2020) 22:143–53. doi: 10.1111/ede.12319
64. Duarte INH, Bessa Ade F O, Rola LD, et al. Cross-population selection signatures in Canchim composite beef cattle. PLoS ONE. (2022) 17:e0264279. doi: 10.1371/journal.pone.0264279
65. Ghoreishifar SM, Eriksson S, Johansson AM, Khansefid M, Moghaddaszadeh-Ahrabi S, Parna N, et al. Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genetics Select Evol. (2020) 52:52. doi: 10.1186/s12711-020-00571-5
66. Pitt D, Bruford MW, Barbato M. Orozco-terWengel P, Martínez R, Sevane N. Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics. Evol Appl. (2019) 12:105–22. doi: 10.1111/eva.12641
67. Wang GD, Shao XJ, Bai B, Wang J, Wang X, Cao X, et al. Structural variation during dog domestication: insights from gray wolf and dhole genomes. Nat Sci Rev. (2019) 6:110–22. doi: 10.1093/nsr/nwy076
68. Ivancevic AM, Kortschak RD, Bertozzi T, Adelson DL. Horizontal transfer of BovB and L1 retrotransposons in eukaryotes. Genome Biol. (2018) 19:85. doi: 10.1186/s13059-018-1456-7
69. Choudhary MN, Friedman RZ, Wang JT, Jang HS, Zhuo X, Wang T. Co-opted transposons help perpetuate conserved higher-order chromosomal structures. Genome Biol. (2020) 21:28. doi: 10.1186/s13059-019-1916-8
70. Hanif Q, Farooq M, Amin I, Mansoor S, Zhang Y, Khan QM. In silico identification of conserved miRNAs and their selective target gene prediction in indicine (Bos indicus) cattle. PLoS ONE. 13:e0206154. doi: 10.1371/journal.pone.0206154
71. Manku HK, Dhanoa JK, Kaur S, Arora JS, Mukhopadhyay CS. Biocomputational identification and validation of novel microRNAs predicted from bubaline whole genome shotgun sequences. Comput Biol Chem. (2017) 70:96–106. doi: 10.1016/j.compbiolchem.2017.08.005
72. Qin S, Jin P, Zhou X, Chen L, Ma F. The role of transposable elements in the origin and evolution of microRNAs in human. PLoS ONE. (2015) 10:e0131365. doi: 10.1371/journal.pone.0131365
73. Li Z, Xu R, Li N. MicroRNAs from plants to animals, do they define a new messenger for communication? Nutr Metab. (2018) 15:68. doi: 10.1186/s12986-018-0305-8
74. Chen Z, Qiu H, Ma L, Luo J, Sun S, Kang K, et al. miR-30e-5p and miR-15a synergistically regulate fatty acid metabolism in goat mammary epithelial cells via LRP6 and YAP1. Int J Mol Sci. (2016) 17:1909. doi: 10.3390/ijms17111909
75. Eisenberg I, Eran A, Nishino I, et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Nat Acad Sci. (2007) 104:17016–21. doi: 10.1073/pnas.0708115104
76. Ballouz S, Dobin A, Gillis JA. Is it time to change the reference genome? Genome Biol. (2019) 20:159. doi: 10.1186/s13059-019-1774-4
77. Matthews CA, Watson-Haigh NS, Burton RA, Sheppard AE. A gentle introduction to pangenomics. Brief Bioinformat . (2024) 25: bbae588. doi: 10.1093/bib/bbae588
78. Cow [Bos taurus] Genomic Dataset. (2011). Available online at: http://www.repeatmasker.org/genomes/bosTau7/RepeatMasker-rm405-db20140131/bosTau7.fa.out.gz (accessed April 18, 2017).
79. Smit A, Hubley R, Green P. RepeatMasker Open-4.0. (2013). Available online at: http://www.repeatmasker.org (accessed April 18, 2017).
80. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. (2014). 42:D68–73. doi: 10.1093/nar/gkt1181
81. Billa PA, Faulconnier Y, Ye T, et al. Deep RNA-Seq reveals miRNome differences in mammary tissue of lactating Holstein and Montbéliarde cows. BMC Genomics. (2019) 20:621. doi: 10.1186/s12864-019-5987-4
82. Chen Z, Xie Y, Luo J, Chen T, Xi Q, Zhang Y, et al. Milk exosome-derived miRNAs from water buffalo are implicated in immune response and metabolism process. BMC Vet Res. (2020) 16:123. doi: 10.1186/s12917-020-02339-x
83. Khudiakov AA, Panshin DD, Fomicheva YV, Knyazeva AA, Simonova KA, Lebedev DS, et al. Different expressions of pericardial fluid microRNAs in patients with arrhythmogenic right ventricular cardiomyopathy and ischemic heart disease undergoing ventricular tachycardia ablation. Front Cardiovas Med. (2021) 8:647812. doi: 10.3389/fcvm.2021.647812
84. Chair SY, Chan JYW, Waye MMY, Liu T, Law BMH, Chien WT. Exploration of potential genetic biomarkers for heart failure: a systematic review. Int J Environ Res Public Health. (2021) 18:5904. doi: 10.3390/ijerph18115904
85. Le Guillou S, Leduc A, Laubier J, Barbey S, Rossignol MN, Lefebvre R, et al. Characterization of Holstein and normande whole milk mirnomes highlights breed specificities. Sci Rep. (2019) 9:20345. doi: 10.1038/s41598-019-56690-7
Keywords: cattle, bison, retrotransposons, microRNA, miR-30, artificial selection, domestication, RTE-BovB
Citation: Kosovsky GY, Glazko GV and Skobel OI (2025) Bos taurus and Bison bison conservative retrotransposon recombination products. Front. Vet. Sci. 12:1516731. doi: 10.3389/fvets.2025.1516731
Received: 24 October 2024; Accepted: 07 April 2025;
Published: 30 April 2025.
Edited by:
Mario Barbato, University of Messina, ItalyReviewed by:
Malgorzata Tokarska, Polish Academy of Sciences, PolandSarika Sahu, Indian Council of Agricultural Research (ICAR), India
Copyright © 2025 Kosovsky, Glazko and Skobel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Galina V. Glazko, Z3ZnbGF6a29AdWFtcy5lZHU=