Abstract
Superantigens (SAgs) are a family of potent immunostimulatory exotoxins known to be produced by only a few bacterial pathogens, including Staphylococcus aureus. More than 20 distinct SAgs have been characterized from different S. aureus strains and at least 80% of clinical strains harbor at least one SAg gene, although most strains encode many. SAgs have been classically associated with food poisoning and toxic shock syndrome (TSS), for which these toxins are the causative agent. TSS is a potentially fatal disease whereby SAg-mediated activation of T cells results in overproduction of cytokines and results in systemic inflammation and shock. Numerous studies have also shown a possible role for SAgs in other diseases such as Kawasaki disease (KD), atopic dermatitis (AD), and chronic rhinosinusitis (CRS). There is also now a rich understanding of the mechanisms of action of SAgs, as well as their structures and function. However, we have yet to discover what purpose SAgs play in the life cycle of S. aureus, and why such a wide array of these toxins exists. This review will focus on recent developments within the SAg field in terms of the molecular biology of these toxins and their role in both colonization and disease.
Introduction
Bacterial superantigens (SAgs) represent a unique class of exotoxins which all function to activate enormous numbers of T lymphocytes (McCormick et al., 2001; Llewelyn and Cohen, 2002; Proft and Fraser, 2003). Although only a few SAgs have clear associations with specific human diseases, bacterial genome sequencing projects over the last decade have led to the characterization of a large and expanding family of exotoxins that includes many genetically and antigenically distinct proteins. These are found primarily in Staphylococcus aureus and Streptococcus pyogenes, but also found in a few other species of β-hemolytic streptococci, coagulase negative staphylococci, Mycoplasma arthritidis, and Yersinia pseudotuberculosis. In the following sections, we will update recent findings in the biochemistry of staphylococcal SAgs, and explore the role of SAgs in different lifestyles of S. aureus in the context of both infection and nasal colonization.
The superantigen superfamily
The staphylococcal SAgs include the staphylococcal enterotoxins (SEs), the staphylococcal enterotoxin-like (SEls) proteins, and toxic shock syndrome toxin-1 (TSST-1) (Lina et al., 2004). The SEs were originally defined by their ability to cause staphylococcal food poisoning (SFP) including emesis, and currently include the SEs A, B, C, D, E, G, H, I, R, and T. The SEl toxins, although both homologous and structurally similar to the SEs, either do not induce emesis, or have not been formally demonstrated to induce emesis, and include the SEls J, K, L, M, N, O, P, Q, S, U, V, and X. It is important to note that although designated as a “SEl” toxin, some of these may possess undemonstrated emetic activity and be reclassified in the future as bona fide enterotoxins.
An updated phylogenetic classification scheme of the SAg exotoxins (McCormick et al., 2001) is shown in Figure 1 where SAgs from staphylococci and streptococci are placed into five evolutionary groups. TSST-1 sits as an evolutionarily distinct SAg that does not induce emesis (Schlievert et al., 2000) and is the only member of the Group I SAgs. TSST-1 is believed to be the major, if not sole cause of the menstrual form of toxic shock syndrome (TSS) (Bergdoll et al., 1981; Schlievert et al., 1981). The Group II SAgs contain both staphylococcal and streptococcal SAgs including SEB, SEC, and streptococcal pyrogenic exotoxin A (SpeA). After TSST-1, SEB has been historically most commonly linked with non-menstrual-associated cases of staphylococcal TSS (Schlievert, 1986), while SpeA has been historically most commonly linked with streptococcal TSS (Stevens et al., 1989). The Group III SAgs include only staphylococcal SAgs, and in general terms, this Group contains SAgs most commonly associated with SFP such as SEA, SED, and SEE, although the Group II SAgs SEB and SEC are often implicated as well (Argudin et al., 2010). Both Group II and III SAgs contain a unique “cysteine-loop structure” that is important for emetic activity (Hovde et al., 1994). The Group IV SAgs are only populated by streptococcal SAgs and will not be discussed here. The Group V SAgs, contain mostly staphylococcal SAgs (except SpeI and related orthologues), and other than SEI which has weak emetic activity, consists of only SEl toxins. In fact, SEI is the only SAg outside of the Group II and III SAgs demonstrated to have emetic activity, although this only occurred in one of four animals tested (Munson et al., 1998). Very recently, SEl-X was described as a novel SAg that does not align well within the currently classification system, but is encoded within the core chromosome of most S. aureus strains (Wilson et al., 2011). Also of note are the staphylococcal superantigen-like proteins (SSLs) (Langley et al., 2010), and although these are also structurally similar to the staphylococcal superantigens (Baker et al., 2007; Chung et al., 2007; Ramsland et al., 2007) they do not possess SAg activity and will not be discussed within this review.
Figure 1

Phylogenetic tree of known bacterial SAgs. The unrooted tree was based on the alignment of amino acid sequences constructed with the unweighted pair group method using arithmetic averages (UPGMA) in MacVector 7.2.3. The SAg abbreviations are indicated followed by the relevant accession number. As previously proposed (McCormick et al., 2001), the five main groups of SAgs belonging to the pyrogenic toxin class are indicated. MAM, YPM, and non-Group A streptococcal SAgs are also included in the analysis. The number of times each branch was supported from 1000 bootstraps is shown as a percentage.
Conventional versus superantigen-mediated T cell activation
Normal T cell-mediated immunity is initiated through the interaction of an αβ T cell receptor (TCR) and a processed peptide antigen presented within self-major histocompatibility (pMHC) complexes (Figure 2A) (Garcia et al., 1999; Garcia and Adams, 2005). If the TCR specifically recognizes the antigen as foreign, these interactions will activate the tyrosine kinase Lck (associated with co-receptors CD4 and CD8), which in turn will activate downstream cell signaling resulting in activation of transcription factors to induce T cell proliferation and differentiation (Smith-Garvin et al., 2009). As TCRs are extraordinarily diverse molecules, only ∼0.01% of naïve T cells will recognize a given antigen (Givan et al., 1999).
Figure 2
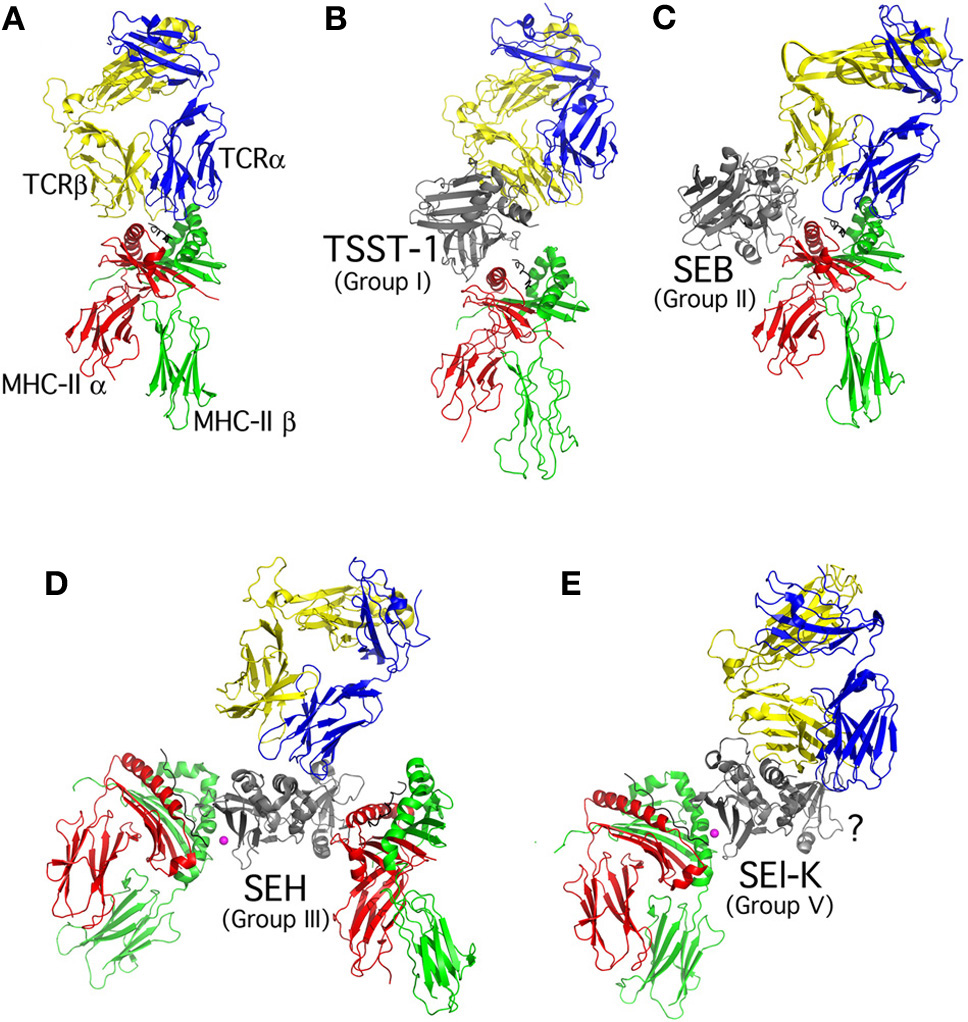
Structural overview of the SAg-mediated T cell activation complexes. Ribbon diagram models show (A) conventional T cell activation (Hennecke et al., 2000), and SAg-mediated T cell activation complexes for (B) Group I (e.g., TSST-1) (C) Group II (e.g., SEB) (D) Group III (e.g., SEH) and (E) Group V (e.g., SEl-K). Colors for TCR and MHC class II chains are labeled in Panel (A). The SAg activation complex models were generated by superposition of the TSST-Vβ (Moza et al., 2007) and TSST-MHC class II (Kim et al., 1994) structures, the SEC-Vβ (Fields et al., 1996) and SEB-MHC class II (Jardetzky et al., 1994) structures, the SEH-VαVβ (Saline et al., 2010), SEH-MHC class II β-chain (Petersson et al., 2001), and the SEA-MHC class II α-chain (Petersson et al., 2002) structures, and the SEK-Vβ (Gunther et al., 2007) and SEI-MHC II (Fernandez et al., 2006) structures. The TCR α-chain was modeled for clarity in each case from the conventional complex (Hennecke et al., 2000). The “?” in Panel (E) indicates that there is no current information regarding the presence, or absence, of the generic low-affinity MHC class II binding domain for Group V SAgs.
SAg-mediated T cell activation is both quantitatively and qualitatively distinct from conventional T cell activation (Bueno et al., 2007). As the defining feature of the SAg toxin is the ability to activate T lymphocytes in a TCR β-chain variable domain (Vβ)-dependent manner (Marrack and Kappler, 1990), very large numbers of T cells can be activated upon SAg exposure. Although TCRs are extraordinarily diverse molecules, this diversity is concentrated within the CDR3 loops due to V(D)J (somatic) recombination during T cell development. However, there are a relatively limited number of possible TCR Vβ regions (∼50 are functionally expressed in humans), and thus SAgs can activate T cells in orders of magnitude above conventional processes. SAgs also do this in an extremely potent manner, and in general, most SAgs can induce measurable activation of T cells in the picogram (10−12 g) concentration range. It is often assumed that SAg-mediated T cell activation follows the normal signaling rules for conventional pMHC-mediated T cell activation and indeed this is the case with at least one major distinction. As predicted, TCR ligation by SAg will induce signals through Lck (Morgan et al., 2001), although Lck signaling is not actually required (Yamasaki et al., 1997; Bueno et al., 2006). However, signaling can proceed in the absence of Lck through a Gα11/PLCβ-dependent pathway that converges with the canonical Lck-dependent pathway at the level of ERK1/2 (Bueno et al., 2006). Since one function of the CD4/CD8 co-receptors is the recruitment of Lck, the ability of SAgs to bypass Lck is also likely related to the capability of SAgs to activate both CD4+ and CD8+ T cells, despite cross-linking with MHC class II molecules (Herrmann et al., 1992; Fuller and Braciale, 1998).
The human immune system has evolved to be able to recognize and eliminate pathogens and their antigens. However, SAgs represent the only known microbial virulence factor whose primary role is to deliberately force the activation of the adaptive immune system. This is counter-intuitive given the numerous staphylococcal virulence factors apparently designed for immune subversion and evasion (Nizet, 2007). This leads to the question as to why S. aureus produce SAgs. Given the wide variety and high prevalence of SAg genes, it is likely that these genes would be lost, especially since they are primarily encoded on mobile genetic elements, without a contribution to the fitness of S. aureus. The influence and architecture of SAg-encoding mobile genetic elements has been the subject of a comprehensive review in this issue (McGavin et al., 2012).
T cell anergy, a phenomenon where T cells become unresponsive to stimulation, has long been proposed to be an immune subversion tactic of S. aureus. Several studies have shown this ex vivo following in vivo stimulation (Kawabe and Ochi, 1990; Rellahan et al., 1990; Lee and Vitetta, 1992; Miller et al., 1999). However, SAg-induced anergy produced ex vivo does not necessarily translate into in vivo anergy (Heeg et al., 1995). In addition, high levels of purified toxin are often used in experimental mouse models that may not reflect physiological conditions. Also, there is no evidence to suggest that T cells are exhausted in nasal carriers of toxigenic S. aureus strains. Recently, a case study of TSS (likely induced by TSST-1) showed deletion followed by an expansion in the Vβ2+ subset that normalized 70 days post-convalescence. In this important study, re-stimulation of peripheral blood mononuclear cells (PBMCs) taken during the acute phase of disease with exogenous SAg resulted in proliferation of Vβ2+ cells suggesting that T cells were not rendered anergic (Rasigade et al., 2011). Recurrent cases of TSS occur, usually as a combined result of insufficient eradication of S. aureus and the inability to form anti-SAg antibodies. The proliferative response of T cells was assessed from a patient with recurrent TSS and there was no reduction in the ability of the patients T cells to respond in vitro (Arvand and Hahn, 1996). Thus, at least in some patients, true anergy of Vβ specific T cell subsets may not occur and suggests that this activity is not the purpose of SAg activity for S. aureus. Recurrent TSS also implies that T cells are not rendered anergic as they are able to react to SAg stimulation during multiple episodes.
Recently, Taylor and Llewelyn (2010) demonstrated that human PBMCs exposed to SAgs resulted in a dose-dependent, Vβ specific increase in CD25+ FoxP3+ cells, indicative of a regulatory T cell (Treg) phenotype. The immunosuppressive qualities of these SAg-induced Tregs have been attributed to the expression of IL-10 and may have a role in prolonging commensalism (Taylor and Llewelyn, 2010).
Staphylococcal superantigens and their host receptors
SAg pro-toxins include a secretion signal that is cleaved from the N-terminus upon export via the general Sec-dependent secretion pathway. SAgs are released as non-enzymatic, relatively small proteins, with the final toxin product ranging in size from ∼22 to 29 kDa. All SAgs are made of two structurally similar domains, linked through a central α-helix. The larger N-terminal domain contains a β-barrel motif similar to an OB-fold, while the smaller C-terminal domain contains the β-grasp motif, which is similar to immunoglobulin-binding domains (Mitchell et al., 2000).
Pioneering crystallographic studies with staphylococcal enterotoxins B in complex with human leukocyte antigen (HLA)-DR1 (Jardetzky et al., 1994), and SEC3 in complex with the mouse TCR Vβ8.2-chain (Fields et al., 1996), established a molecular framework by how SAgs can activate so many T cells (Li et al., 1999). These studies demonstrated that SAgs bind to lateral surfaces of both TCRs, and pMHC class II molecules, to “distort” the normal TCR-pMHC II interaction, such that the CDR3 loops of both TCR α- and β-chains (which are key for antigen recognition) are wedged away from the antigenic peptide (Figures 2B–E). Through this mechanism, activation of the T cell is no longer antigen specific, but now dependent upon which Vβs can be bound by that particular SAg explaining how SAgs are Vβ-specific (Li et al., 1999). Large numbers of SAg-activated T cells can then release a multitude of proinflammatory cytokines which in severe cases may lead to the “cytokine storm” phenomenon characteristic of TSS (McCormick et al., 2001). Activation of antigen presenting cells (APCs) by SAgs also contributes to cytokine release due to the involvement of MyD88, which upregulates NF-κB, leading to production of proinflammatory molecules (Kissner et al., 2011).
Recent years have seen a number of further advances in the structural characterization of the staphylococcal SAgs, and there is now a broader picture as to how SAgs from the different evolutionary groups (Figure 1) function to distort the normal process of T cell activation (Figure 2). For example, the Group I SAg TSST-1 (Figure 2B), which is extremely specific for the human Vβ2+ T cells (Choi et al., 1989), forms a unique T cell activation complex by binding the MHC II α-chain through a relatively low-affinity interface that is highly influenced by different antigenic peptides within MHC II (Kim et al., 1994; Wen et al., 1996). Also, TSST-1 recognizes unique amino acid insertions from Vβ2 within both CDR2 and framework region (FR) 3, explaining the extreme Vβ-specificity of this SAg (Moza et al., 2006, 2007; Rahman et al., 2011). There are no direct TCR-MHC II contacts in this T cell activation complex. Group II SAgs (Figure 2C) such as SEB, SEC3, and SpeA, are more “promiscuous” in their Vβ-targets, and engage TCR Vβ through “conformation-dependent” mechanisms that are thought to be less dependent on specific Vβ amino acid side-chains (Fields et al., 1996; Li et al., 1998; Sundberg et al., 2002). These SAgs bind the MHC II α-chain through an N-terminal, low-affinity binding domain, yet in contrast to TSST-1, this binding is antigenic peptide-independent (Jardetzky et al., 1994). Group III SAgs (Figure 2D) consist of only staphylococcal SAgs, and these toxins are thought to be able to cross-link MHC II molecules (Abrahmsen et al., 1995; Hudson et al., 1995) through both a low-affinity site similar to Group II, (Petersson et al., 2002) as well as a high-affinity, zinc-dependent MHC II β-chain interface located within the β-grasp domain of the SAg (Petersson et al., 2001). The only structural information for how Group III SAgs engage TCR is for SEH (Saline et al., 2010), which represents somewhat of an outlier within Group III, and is the only known Vα-specific SAg (Petersson et al., 2003; Pumphrey et al., 2007). Group IV SAgs are restricted to only streptococcal members, and these toxins bind Vβ similar to the Group II SAgs, although with a larger footprint (Sundberg et al., 2002), and contain a high-affinity MHC II β-chain binding domain similar to Group III (Li et al., 2001). Considerable evidence indicates the presence of a low-affinity MHC II α-chain interaction, likely similar to Group II (Swietnicki et al., 2003; Tripp et al., 2003; Kasper et al., 2008), although this interaction has not been characterized structurally. The Group V SAgs contain a high-affinity MHC II β-chain binding domain (Fernandez et al., 2006) similar to Group III, and bind the TCR Vβ with a more “lateral” position extending into FR4 (Figure 2E) (Gunther et al., 2007). There is currently no information relating to the presence, or absence, of the generic low-affinity MHC II interface with Group V SAgs.
Within the SAg family of toxins, each member is able to efficiently activate large numbers of T cells, regardless of subtle, or dramatic, differences within the different SAg-mediated T cell activation complexes. However, the one common structural feature of all characterized SAgs, with the exception of the Vα-specific SEH, is the engagement of the Vβ CDR2 loop, and this loop appears to be the critical determinant for Vβ-specificity (Rahman et al., 2011).
Recently, it was demonstrated that SEB can bind to the costimulatory molecule CD28, which is constitutively expressed on naïve T cells and binds B7 ligands on APCs. The CD28 binding site is divergent from both the TCR and MHC II binding domains of SEB, and is relatively conserved amongst the SAg family. Disruption of CD28 binding by peptide antagonists reduced mortality rates in mice administered with D-galactosamine and SEB by downregulating Th1, but not Th2 cytokines (Arad et al., 2011). These lines of evidence support the proposal that CD28 binding by SAgs is important to the function of SAgs. Further research elucidating downstream mechanisms will clarify the exact role of CD28 during T cell activation by SAgs.
Staphylococcal superantigens and disease
Staphylococcal food poisoning
The first disease linked to the staphylococcal SAgs was SFP, and evidence that a staphylococcal toxin caused the illness dates back to 1930, where filterable supernatants from a “yellow staphylococcus” was able to induce SFP in human volunteers (Dack et al., 1930; Jordan, 1930). The role of the staphylococcal SAgs in foodborne disease has been reviewed recently in detail (Argudin et al., 2010; Hennekinne et al., 2011), and will not be discussed here.
Toxic shock syndrome
The other human disease clearly caused by the staphylococcal SAgs is TSS. This disease was described in 1927 by Franklin Stevens as staphylococcal scarlet fever (Stevens, 1927), and was named “toxic shock syndrome” by Todd and colleagues in 1978 to describe a systemic illness in seven children caused by non-invasive S. aureus (Todd et al., 1978). The pathogenesis of TSS is due to a SAg-induced cytokine storm owing to the massive activation of T cells in individuals lacking neutralizing antibodies to the particular SAg [reviewed in (McCormick et al., 2001)]. The disease is a capillary leak syndrome where patients develop fever, rash, hypotension, multiorgan involvement and convalescent desquamation (McCormick et al., 2001). S. aureus can cause the menstrual form of TSS, which historically occurred in young women in association with high absorbency tampons, and non-menstrual TSS, which can occur from virtually any S. aureus infection, although infrequently from bacteremia (McCormick et al., 2001). Although most staphylococcal SAgs are functionally capable of inducing TSS in experimental animals, only a few select SAgs have historically been associated with the disease. This is somewhat surprising given the large “collection” of these extremely potent toxins. The TSST-1 SAg was linked to the menstrual form of TSS in 1981 (Bergdoll et al., 1981; Schlievert et al., 1981), although it is also clear that other SAgs, primarily TSST-1, SEB and SEC, are capable of causing the non-menstrual form (Bohach et al., 1990; McCormick et al., 2001).
During the early 1980s, there were a high number of menstrual TSS cases in young women associated with the use of high absorbency tampons (Shands et al., 1980) and the estimated incidence of all forms of TSS at this time was 13.7/100,000 (Osterholm and Forfang, 1982). By the mid-1980s, following the removal of these products from the marker, and public awareness campaigns as well as product labeling, the overall incidence dropped to 0.53/100,000 with a case-fatality rate of ∼4% (Gaventa et al., 1989). A recent population based surveillance for TSS in Minnesota between 2000 and 2006 demonstrates that this rate has been relatively stable and that TSST-1 was still the major cause in most cases (DeVries et al., 2011). Of note, although the community acquired MRSA clonal strain USA300 has dramatically increased in prevalence in the U.S., this strain does not appear to cause many cases of TSS (DeVries et al., 2011). Although the overall incidence of TSS appears low, it has been suggested that severe SAg-mediated disease remains underreported (DeVries et al., 2011), due to both the strict CDC case definition (Centers for Disease Control and Prevention, 2011) as well as prompt and appropriate medical attention that would prevent the most severe forms of SAg intoxication. Indeed, TSS is still a major problem, and cases of non-menstrual TSS pediatric burn patients can be extremely dangerous if not recognized early (White et al., 2005).
Apart from the more overt forms of SAg-mediated diseases, there is significant evidence that SAgs also can play a role in a number of other diseases and these will be discussed below.
Kawasaki disease
Kawasaki Disease (KD) was first described by Tomisaku Kawasaki in 1967 (Van Crombruggen et al., 2011) and is now the leading cause of acquired heart disease in children from developed nations. KD is an acute, self-limiting vasculitis, typically affecting the coronary arteries, and thought to be triggered by an infectious agent in genetically susceptible individuals (Yeung, 2010). Although the etiology of KD is not known, there is compelling evidence that bacterial SAgs are involved, and could be causal in association with host genetic factors (Matsubara and Fukaya, 2007). First, the clinical presentation of KD has features reminiscent of TSS, including fever, a desquamating rash and erythema of the mucous membranes. SAg producing S. aureus and S. pyogenes have been isolated from KD patients, and seroconversion with anti-SAg antibodies has also been demonstrated. Perhaps the strongest evidence of SAg involvement however, is the demonstration of Vβ skewing in KD patients (Abe et al., 1992). A number of studies have found primarily Vβ2 expansion (Leung et al., 1995b) providing a link to either TSST-1 or SpeC which are both Vβ2 specific (Rahman et al., 2011). Others, however, have found expansion of various Vβ families (Nomura et al., 1998; Yoshioka et al., 1999), potentially implicating other SAgs with different Vβ profiles. Treatment of KD involves the use of intravenous immunoglobulin (IVIG) (Newburger et al., 1986), and IVIG is well known to contain SAg neutralizing antibodies (Darenberg et al., 2004; Schrage et al., 2006). Although there is no direct evidence to suggest SAg involvement, there also exists the Kawasaki-like syndrome, which in contrast to KD occurs primarily in adults with severe immunosuppression including HIV/AIDS (Stankovic et al., 2007).
Chronic rhinosinusitis
Chronic rhinosinusitis (CRS) is a group of disorders characterized by inflammation of the nose and paranasal sinuses for at least 3 months duration (Van Crombruggen et al., 2011). CRS can occur with or without nasal polyps, and accumulated evidence is now convincing that S. aureus SAgs can contribute to, at least in some cases, CRS with nasal polyposis (Van Zele et al., 2004). In this disease, SAgs are thought to skew the cytokine response towards a TH2 phenotype inducing both eosinophilia and the production of polyclonal IgE, which in turn could be further linked to asthma (Bachert et al., 2010). There is no single SAg associated specifically to this disease (Van Zele et al., 2008; Heymans et al., 2010), and as noted (Van Crombruggen et al., 2011), a causal relationship with S. aureus has not been established.
Atopic dermatitis
Atopic dermatitis (AD) represents a chronic and relapsing T cell-mediated inflammatory skin disorder with IgE-mediated sensitization to allergens. AD most often affects infants and young children, but may persist into adulthood, or may first develop in adults as late-onset AD. AD has both genetic and environmental contributions but nearly all AD patients are colonized by S. aureus. This is likely due to both the damaged skin barrier and impaired host immune responses. Significant evidence also indicates an important role for the staphylococcal SAgs in exacerbating the disease [reviewed in (Schlievert et al., 2010)]. SAgs have long been known to induce the skin homing receptor cutaneous lymphocyte-associated antigen (CLA) on T cells to recruit these cells to the skin (Leung et al., 1995a). Very recent evidence indicates that skin homing, phenotypically Treg (CD4+ FoxP3+) cells from AD patients may actually display a TH2 phenotype in response to SEB stimulation (Lin et al., 2011). AD patients may also develop anti-SAg IgE antibodies that can further worsen the condition (Leung et al., 1993; Bunikowski et al., 1999; Lin et al., 2000). AD is often treated with glucocorticoids and SAgs have been shown to induce glucocorticoid resistance in PBMCs (Hauk et al., 2000). A recent study that examined essentially the entire staphylococcal SAg family found that isolates from steroid resistant AD patients contained significantly more SAgs genes than isolates from non-steroid resistant patients or menstrual isolates provoking the idea that steroid treatment may actually select for SAgs in these strains (Schlievert et al., 2008).
Guttate psoriasis
Guttate psoriasis is an acute form of psoriasis mediated by autoreactive T cells that typically develops in young adults and children. This inflammatory skin disease is typically preceded by streptococcal pharyngitis, and the streptococcal SAgs, in particular SpeC, and Vβ2+ T cells have been implicated (Leung et al., 1995c). Some associations have also been made with S. aureus and chronic plaque psoriasis (Sayama et al., 1998; Balci et al., 2009).
Nasal colonization and staphylococcal superantigens
Staphylococcal colonization can be defined by the presence and multiplication of S. aureus in the absence of infection or disease. In humans, the anterior nares are the most common area colonized by S. aureus and the prevalence of nasal colonization is particularly high within the general population. Individuals have been typically classified into three separate groups based on their nasal carriage status: persistent, intermediate, or non-carriers. Approximately 20% of the general population are persistent carriers of S. aureus, ∼30% are intermittent carriers, and ∼50% are non-carriers (Wertheim et al., 2005). In the event of an infection, carriers have a better prognosis than non-carriers (von Eiff et al., 2001; Wertheim et al., 2004); however, nasal colonization increases the risk of infection by fourfold (Safdar and Bradley, 2008). Furthermore, it is believed that ∼80% of S. aureus bloodstream infections come from an endogenous source (von Eiff et al., 2001), and this can be particularly dangerous in a hospital setting if a nasal carrier is immunocompromised and the colonizing strain is resistant to antibiotics.
Although a myriad of bacterial factors play a role in determining nasal colonization, it has not yet been established whether or not SAgs are involved. Epidemiological studies evaluating S. aureus SAg gene distribution in nasal swabs compared with blood isolates concluded that there were no differences between blood and nasal isolates in the number of toxins, or a correlation to a particular toxin and that toxin gene distribution was widespread and highly varied (Holtfreter et al., 2007). S. aureus strains encoding the same SAg genes can produce different amounts of toxin (Varshney et al., 2009) and this may make correlations difficult in epidemiological studies, which often rely on genomic typing instead of protein quantification.
The particular molecular switch by how colonized bacteria become pathogenic is likely a mixture of host-pathogen and environmental factors that leads to a breach in the mucosa and subsequent infection. The role of the two-component regulatory system agr has been classically associated with dissemination and the release of secretory proteins and downregulation of surface associated proteins (Recsei et al., 1986). Many SAgs such as TSST-1 are regulated by agr (Recsei et al., 1986), which appears to be dampened during colonization, suggesting that agr-controlled SAgs may not be involved in colonization. This has been further supported where the constitutive expression of RNAIII, the effector molecule of the agr system, reduces nasal colonization in rats (Pynnonen et al., 2011). Thus, it is likely that agr is downregulated during colonization, which has been demonstrated in human studies (Burian et al., 2010). It has however, been suggested that certain SAgs such as staphylococcal enterotoxin A (SEA), which is not regulated by agr, may play a role early on in colonization (Bohach and Schlievert, 2007). Although many persistent carriers contain the bacteriophage that carries SEA, this genetic element does not appear to play a role early on during colonization (Verkaik et al., 2011). Furthermore, the sea gene has been correlated with sepsis (Ferry et al., 2005), although the presence of SEA has yet to be confirmed in blood during sepsis. This work also demonstrated a correlation between the egc operon of SAgs and colonization (Ferry et al., 2005). A follow-up study using recombinant SAgs found that both types (egc and non-egc) of SAgs induced similar proliferative activity on PBMCs (Grumann et al., 2008). However, the proliferative potential of supernatants taken from patients with strains containing egc genes demonstrated that strains encoding egc SAgs do not have as high proliferative activity as strains encoding non-egc SAgs, suggesting that egc SAgs are not made in quantities as high as non-egc SAgs. A lack of neutralizing antibodies against egc-encoded SAgs was also found in serum from healthy humans indicating either a lack of egc toxins being produced by S. aureus or an inability to produce neutralizing antibodies by the host (Holtfreter et al., 2004). It is interesting that only non-egc encoded SAgs have been implicated in toxin-mediated diseases. Thus, the role of egc-encoded SAgs in colonization requires further investigation.
It is difficult to directly ascertain whether or not SAgs are produced in vivo during colonization mainly due to the presence of S. aureus protein A, which binds the Fc portion of antibodies, thereby causing background levels of assays to be quite high. However, analysis of the immunological response can provide important information. In particular, both Vβ-specific T cell activation and SAg-neutralizing antibodies are indirect ways of determining if SAgs have encountered the immune system. While Vβ-skewing has been studied in the context of severe disease (Ferry et al., 2008b), it has long been known that the general population develops anti-SAg antibodies capable of neutralizing these toxins (Vergeront et al., 1983). Also, persistent nasal carriers of S. aureus have been found to have neutralizing antibodies against the SAgs produced by the colonizing strain (Holtfreter et al., 2006; Kolata et al., 2011). Levels of neutralizing antibodies against TSST-1 and SEA were significantly higher in persistent nasal carriers than non-carriers (Verkaik et al., 2009) suggesting that these SAgs are actively produced during nasal colonization.
To what extent, if any, do SAgs play during colonization has not yet been experimentally addressed. Intranasal vaccination in rodents with deactivated TSST-1 was able to decrease mortality rates from TSST-1 producing S. aureus septic challenge and significantly decreased the bacterial load in organs (Narita et al., 2008). This was a TSST-1 specific response, as challenge with non-TSST-1 producing S. aureus did not result in a significant reduction in bacterial load when compared to non-vaccinated mice. The same vaccination strategy protected against nasal challenge only during the initial colonization phase (days 1 and 3). Since the model only evaluated colonization up to day 7, it is difficult to assess whether or not this is able to have a lasting effect against S. aureus nasal persistence, since there were not significant effects at day 5 (Narita et al., 2008).
Staphylococcal peptidoglycan-embedded molecules have been found to downregulate the immune response stimulated by SAgs (Chau et al., 2009). This effect was most effective at high cell densities suggesting that it is important in a state of colonization or a biofilm as opposed to free-living planktonic cells. Thus, if a colonized population of S. aureus is producing SAgs, any invading “rogue” cells that are not a part of the main colony may be killed by an activated immune system, while the dense colony is able to downregulate this response in the local area to prevent clearance. This suggests a role for SAgs as checkpoints of dissemination. Evidence suggests that when SAgs are systemic in the case of TSS (Ferry et al., 2008b), S. aureus is able to prevent dissemination, which may be partly why bacteremia is rarely associated with staphylococcal TSS. This is also supported by the observation that sepsis patients lack Vβ-skewing unlike TSS patients (Ferry et al., 2008a), suggesting that bacterial dissemination prevents toxin production.
Future directions for research
The collective SAg research community has contributed enormously to an advanced understanding of SAg biology. Yet, there remain a number of important avenues for further study and consideration.
Although SAgs are defined by Vβ-specificity, different human MHC II molecules are also clearly important for the response to SAgs (Yeung et al., 1996; Medina et al., 2001; Kotb et al., 2002; Llewelyn et al., 2004; Goldmann et al., 2005; Nooh et al., 2007). Mouse models (such as C57BL/6 and BALBc) have been hampered by the fact that mouse MHC II do not respond in the same way, and are not as sensitive to SAgs, as human MHC (Yeung et al., 1996). Alternative models include rabbits that respond more appropriately (Parsonnet et al., 1987; Dinges and Schlievert, 2001; Buonpane et al., 2007), as well as transgenic mouse strains that express human MHC class II molecules (Yeung et al., 1996; DaSilva et al., 2002). Models of TSS also often utilize a liver-damaging reagent such as D-galactosamine in conjunction with high levels of SAgs. Liver and gut pathology has recently been implicated in the course of TSS in a humanized transgenic HLA-DR3 mouse model without the use of D-galactosamine (Tilahun et al., 2011a,b) and thus, D-galactosamine may mask pathologies normally induced via TSS. Lastly, although many studies using purified recombinant SAgs have yielded many insights, SAg function is still rarely studied in the context of live infections using genetically defined knockout strains. More work using live infections with appropriate SAg-responsive models is needed to be able to coordinate SAg production with other virulence factors.
Although a number of studies have evaluated the presence of S. aureus, and correlations of particular SAg genes with particular clinical syndromes, the presence of the gene does not equate to expression and function of the actual toxin. Indeed, the original discovery of TSST-1 as the causal agent of menstrual TSS was made due to the high level production of this toxin from menstrual TSS strains (Bergdoll et al., 1981; Schlievert et al., 1981). For many human diseases where SAgs may contribute to, or drive the pathology, there is likely not being a single toxin responsible given that they can all activate numerous T cells. As we now know the Vβ skewing patterns of virtually all the known staphylococcal SAgs (Thomas et al., 2009; Seo et al., 2010; Wilson et al., 2011), further systematic evaluations focused on SAg expression coupled with function in relation to particular clinical syndromes (Ferry et al., 2008b), are warranted.
The large family of SAgs continue to grow, and the YPM and MAM SAgs seem to have developed their SAg-activity through convergent evolution as these toxins are not orthologous to the pyrogenic toxin SAgs, or to each other. Also, the animal model of KD utilizes an uncharacterized SAg from the cell wall preparation of Lactobacillus casei to induce the disease in mice (Yeung, 2007). L. casei is found commonly in the intestinal tract, is widely used by the dairy industry, and is clearly not a pathogen. It is easy to speculate that uncharacterized SAgs could be produced by other microorganisms associated with human immune mediated sequelae.
Arguably the most interesting question that remains in this field is why do S. aureus possess such a large, genetically and antigenically distinct, extremely potent, and seemingly redundant group of these toxins? SAgs skew responses toward TH1 during severe disease, but toward TH2 responses during atopic disease in genetically predisposed individuals. TH1 skewing can result in delayed development of neutralizing antibody and perhaps this is an important in vivo survival strategy. Many patients following menstrual TSS fail to develop anti-TSST-1 antibodies (Stolz et al., 1985) so this can occur from TSS. Conversely, humans clearly develop anti-TSST-1 antibodies such that by age 1, ∼50% have antibody titers considered to be protective (Vergeront et al., 1983). An interesting hypothesis has been proposed where excessive T cell expansion may act as a sponge to titrate IL-2 necessary for further T cell expansion, essentially causing immunosuppression (Llewelyn and Cohen, 2002). Similarly, massive expansion of Vβ-specific T cells may induce a loss of overall receptor diversity filling up the “space,” providing an alternative method of immune escape. Continued efforts into understanding the complex biology of SAgs will undoubtedly answer many of these questions. It is clear that these remarkable toxins represent a highly unique and well adapted virulence factor, although the evolutionary function of these toxin in the life cycle of S. aureus still remains unclear.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Acknowledgments
Research on bacterial superantigens in the McCormick laboratory is supported by operating funds from the Canadian Institutes of Health Research. Stacey X. Xu is supported by a Queen Elizabeth II Graduate Scholarship in Science and Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abe J. Kotzin B. L. Jujo K. Melish M. E. Glode M. P. Kohsaka T. Leung D. Y. (1992). Selective expansion of T cells expressing T-cell receptor variable regions V beta 2 and V beta 8 in Kawasaki disease. Proc. Natl. Acad. Sci. U.S.A. 89, 4066–4070.
2
Abrahmsen L. Dohlsten M. Segren S. Bjork P. Jonsson E. Kalland T. (1995). Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO J. 14, 2978–2986.
3
Arad G. Levy R. Nasie I. Hillman D. Rotfogel Z. Barash U. Supper E. Shpilka T. Minis A. Kaempfer R. (2011). Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol.9:e100-1149. 10.1371/journal.pbio.1001149
4
Argudin M. A. Mendoza M. C. Rodicio M. R. (2010). Food poisoning and Staphylococcus aureus enterotoxins. Toxins2, 1751–1773. 10.3390/toxins2071751
5
Arvand M. Hahn H. (1996). T-cell activation and proliferation in a case of recurrent menstrual toxic shock syndrome. Zentralbl. Bakteriol. 284, 164–169.
6
Bachert C. Zhang N. Holtappels G. De Lobel L. Van Cauwenberge P. Liu S. Lin P. Bousquet J. Van Steen K. (2010). Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J. Allergy Clin. Immunol. 126, 962–968, 968 e961–966. 10.1016/j.jaci.2010.07.007
7
Baker H. M. Basu I. Chung M. C. Caradoc-Davies T. Fraser J. D. Baker E. N. (2007). Crystal structures of the staphylococcal toxin SSL5 in complex with sialyl Lewis X reveal a conserved binding site that shares common features with viral and bacterial sialic acid binding proteins. J. Mol. Biol. 374, 1298–1308. 10.1016/j.jmb.2007.09.091
8
Balci D. D. Duran N. Ozer B. Gunesacar R. Onlen Y. Yenin J. Z. (2009). High prevalence of Staphylococcus aureus cultivation and superantigen production in patients with psoriasis. Eur. J. Dermatol. 19, 238–242. 10.1684/ejd.2009.0663
9
Bergdoll M. S. Crass B. A. Reiser R. F. Robbins R. N. Davis J. P. (1981). A new staphylococcal enterotoxin, enterotoxin F, associated with toxic- shock-syndrome Staphylococcus aureus isolates. Lancet1, 1017–1021.
10
Bohach G. A. Fast D. J. Nelson R. D. Schlievert P. M. (1990). Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit. Rev. Microbiol. 17, 251–272. 10.3109/10408419009105728
11
Bohach G. Schlievert P. M. (2007). “Staphylococcal and streptococcal superantigens: an update,” in Superantigens: Molecular Basis for the Role in Human Diseases, eds FraserJ. D.KotbM.(Washington, DC: ASM Press), 21–36.
12
Bueno C. Criado G. McCormick J. K. Madrenas J. (2007). T cell signalling induced by bacterial superantigens. Chem. Immunol. Allergy93, 161–180. 10.1159/0000100894
13
Bueno C. Lemke C. D. Criado G. Baroja M. L. Ferguson S. S. Rahman A. K. Tsoukas C. D. McCormick J. K. Madrenas J. (2006). Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity25, 67–78. 10.1016/j.immuni.2006.04.012
14
Bunikowski R. Mielke M. Skarabis H. Herz U. Bergmann R. L. Wahn U. Renz H. (1999). Prevalence and role of serum IgE antibodies to the Staphylococcus aureus-derived superantigens SEA and SEB in children with atopic dermatitis. J. Allergy Clin. Immunol. 103, 119–124.
15
Buonpane R. A. Churchill H. R. Moza B. Sundberg E. J. Peterson M. L. Schlievert P. M. Kranz D. M. (2007). Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat. Med. 13, 725–729. 10.1038/nm1584
16
Burian M. Rautenberg M. Kohler T. Fritz M. Krismer B. Unger C. Hoffmann W. H. Peschel A. Wolz C. Goerke C. (2010). Temporal expression of adhesion factors and activity of global regulators during establishment of Staphylococcus aureus nasal colonization. J. Infect. Dis. 201, 1414–1421. 10.1086/651619
17
Centers for Disease Control Prevention. (2011). Toxic Shock Syndrome; 2011 Case Definition.
18
Chau T. A. McCully M. L. Brintnell W. An G. Kasper K. J. Vines E. D. Kubes P. Haeryfar S. M. McCormick J. K. Cairns E. Heinrichs D. E. Madrenas J. (2009). Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat. Med. 15, 641–648. 10.1038/nm.1965
19
Choi Y. W. Kotzin B. Herron L. Callahan J. Marrack P. Kappler J. (1989). Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc. Natl. Acad. Sci. U.S.A. 86, 8941–8945.
20
Chung M. C. Wines B. D. Baker H. Langley R. J. Baker E. N. Fraser J. D. (2007). The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol. Microbiol. 66, 1342–1355. 10.1111/j.1365-2958.2007.05989.x
21
Dack G. M. Cary W. E. Woolpert O. Wiggers H. (1930). An outbreak of food poisoning proved to be due to a yellow hemolytic staphylococcus. J. Prev. Med. 4, 167–175.
22
Darenberg J. Soderquist B. Normark B. H. Norrby-Teglund A. (2004). Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: implications for therapy of toxic shock syndrome. Clin. Infect. Dis. 38, 836–842. 10.1086/381979
23
DaSilva L. Welcher B. C. Ulrich R. G. Aman M. J. David C. S. Bavari S. (2002). Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J. Infect. Dis. 185, 1754–1760.
24
DeVries A. S. Lesher L. Schlievert P. M. Rogers T. Villaume L. G. Danila R. Lynfield R. (2011). Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One6:e22997. 10.1371/journal.pone.0022997
25
Dinges M. M. Schlievert P. M. (2001). Comparative analysis of lipopolysaccharide-induced tumor necrosis factor alpha activity in serum and lethality in mice and rabbits pretreated with the staphylococcal superantigen toxic shock syndrome toxin 1. Infect. Immun. 69, 7169–7172. 10.1128/IAI.69.11.7169-7172.2001
26
Fernandez M. M. Guan R. Swaminathan C. P. Malchiodi E. L. Mariuzza R. A. (2006). Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J. Biol. Chem. 281, 25356–25364. 10.1074/jbc.M603969200
27
Ferry T. Thomas D. Bouchut J. C. Lina G. Vasselon-Raina M. Dauwalder O. Gillet Y. Vandenesch F. Floret D. Etienne J. (2008a). Early diagnosis of staphylococcal toxic shock syndrome by detection of the TSST-1 Vbeta signature in peripheral blood of a 12-year-old boy. Pediatr. Infect. Dis. J. 27, 274–277. 10.1097/INF.0b013e31815c93a3
28
Ferry T. Thomas D. Genestier A. L. Bes M. Lina G. Vandenesch F. Etienne J. (2005). Comparative prevalence of superantigen genes in Staphylococcus aureus isolates causing sepsis with and without septic shock. Clin. Infect. Dis. 41, 771–777. 10.1086/432798
29
Ferry T. Thomas D. Perpoint T. Lina G. Monneret G. Mohammedi I. Chidiac C. Peyramond D. Vandenesch F. Etienne J. (2008b). Analysis of superantigenic toxin Vbeta T-cell signatures produced during cases of staphylococcal toxic shock syndrome and septic shock. Clin. Microbiol. Infect. 14, 546–554. 10.1111/j.1469-0691.2008.01975.x
30
Fields B. A. Malchiodi E. L. Li H. Ysern X. Stauffacher C. V. Schlievert P. M. Karjalainen K. Mariuzza R. A. (1996). Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature384, 188–192. 10.1038/384188a0
31
Fuller C. L. Braciale V. L. (1998). Selective induction of CD8+ cytotoxic T lymphocyte effector function by staphylococcus enterotoxin B. J. Immunol. 161, 5179–5186.
32
Garcia K. C. Adams E. J. (2005). How the T cell receptor sees antigen–a structural view. Cell122, 333–336. 10.1016/j.cell.2005.07.015
33
Garcia K. C. Teyton L. Wilson I. A. (1999). Structural basis of T cell recognition. Annu. Rev. Immunol. 17, 369–397. 10.1146/annurev.immunol.17.1.369
34
Gaventa S. Reingold A. L. Hightower A. W. Broome C. V. Schwartz B. Hoppe C. Harwell J. Lefkowitz L. K. Makintubee S. Cundiff D. R. Sitze S. Toxic Shock Syndrome Study Group. (1989). Active surveillance for toxic shock syndrome in the United States, 1986.Rev. Infect. Dis.11 (Suppl. 1), S28–S34.
35
Givan A. L. Fisher J. L. Waugh M. Ernstoff M. S. Wallace P. K. (1999). A flow cytometric method to estimate the precursor frequencies of cells proliferating in response to specific antigens. J. Immunol. Methods230, 99–112. 10.1016/S0022-1759(99)00136-2
36
Goldmann O. Lengeling A. Bose J. Bloecker H. Geffers R. Chhatwal G. S. Medina E. (2005). The role of the MHC on resistance to group a streptococci in mice. J. Immunol. 175, 3862–3872.
37
Grumann D. Scharf S. S. Holtfreter S. Kohler C. Steil L. Engelmann S. Hecker M. Volker U. Broker B. M. (2008). Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J. Immunol. 181, 5054–5061.
38
Gunther S. Varma A. K. Moza B. Kasper K. J. Wyatt A. W. Zhu P. Rahman A. K. Li Y. Mariuzza R. A. McCormick J. K. Sundberg E. J. (2007). A novel loop domain in superantigens extends their T cell receptor recognition site. J. Mol. Biol. 371, 210–221. 10.1016/j.jmb.2007.05.038
39
Hauk P. J. Hamid Q. A. Chrousos G. P. Leung D. Y. (2000). Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. J. Allergy Clin. Immunol. 105, 782–787. 10.1067/mai.2000.105807
40
Heeg K. Gaus H. Griese D. Bendigs S. Miethke T. Wagner H. (1995). Superantigen-reactive T cells that display an anergic phenotype in vitro appear functional in vivo. Int. Immunol. 7, 105–114.
41
Hennecke J. Carfi A. Wiley D. C. (2000). Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 19, 5611–5624. 10.1093/emboj/19.21.5611
42
Hennekinne J. A. De Buyser M. L. Dragacci S. (2011). Staphylococcus aureus and its food poisoning toxins: characterization and outbreak investigation.FEMS Microbiol. Rev. . [Epub ahead of print]. 10.1111/j.1574-6976.2011.00311.x
43
Herrmann T. Baschieri S. Lees R. K. Macdonald H. R. (1992). In vivo responses of CD4+ and CD8+ cells to bacterial superantigens.Eur. J. Immunol.22, 1935–1938. 10.1002/eji.1830220739
44
Heymans F. Fischer A. Stow N. W. Girard M. Vourexakis Z. Des Courtis A. Renzi G. Huggler E. Vlaminck S. Bonfils P. Mladina R. Lund V. Schrenzel J. Francois P. Lacroix J. S. (2010). Screening for staphylococcal superantigen genes shows no correlation with the presence or the severity of chronic rhinosinusitis and nasal polyposis.PLoS One5:e9525. 10.1371/journal.pone.0009525
45
Holtfreter S. Bauer K. Thomas D. Feig C. Lorenz V. Roschack K. Friebe E. Selleng K. Lovenich S. Greve T. Greinacher A. Panzig B. Engelmann S. Lina G. Broker B. M. (2004). egc-Encoded superantigens from Staphylococcus aureus are neutralized by human sera much less efficiently than are classical staphylococcal enterotoxins or toxic shock syndrome toxin.Infect. Immun.72, 4061–4071. 10.1128/IAI.72.7.4061-4071.2004
46
Holtfreter S. Grumann D. Schmudde M. Nguyen H. T. Eichler P. Strommenger B. Kopron K. Kolata J. Giedrys-Kalemba S. Steinmetz I. Witte W. Broker B. M. (2007). Clonal distribution of superantigen genes in clinical Staphylococcus aureus isolates. J. Clin. Microbiol. 45, 2669–2680. 10.1128/JCM.00204-07
47
Holtfreter S. Roschack K. Eichler P. Eske K. Holtfreter B. Kohler C. Engelmann S. Hecker M. Greinacher A. Broker B. M. (2006). Staphylococcus aureus carriers neutralize superantigens by antibodies specific for their colonizing strain: a potential explanation for their improved prognosis in severe sepsis.J. Infect. Dis.193, 1275–1278. 10.1086/503048
48
Hovde C. J. Marr J. C. Hoffmann M. L. Hackett S. P. Chi Y. I. Crum K. K. Stevens D. L. Stauffacher C. V. Bohach G. A. (1994). Investigation of the role of the disulphide bond in the activity and structure of staphylococcal enterotoxin C1. Mol. Microbiol. 13, 897–909.
49
Hudson K. R. Tiedemann R. E. Urban R. G. Lowe S. C. Strominger J. L. Fraser J. D. (1995). Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J. Exp. Med. 182, 711–720.
50
Jardetzky T. S. Brown J. H. Gorga J. C. Stern L. J. Urban R. G. Chi Y. I. Stauffacher C. Strominger J. L. Wiley D. C. (1994). Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature368, 711–718. 10.1038/368711a0
51
Jordan E. O. (1930). The production by staphylococci of a substance causing food poisoning. JAMA94, 1648–1650.
52
Kasper K. J. Xi W. Rahman A. K. Nooh M. M. Kotb M. Sundberg E. J. Madrenas J. McCormick J. K. (2008). Molecular requirements for MHC class II alpha-chain engagement and allelic discrimination by the bacterial superantigen streptococcal pyrogenic exotoxin C. J. Immunol. 181, 3384–3392.
53
Kawabe Y. Ochi A. (1990). Selective anergy of V beta 8+, CD4+ T cells in Staphylococcus enterotoxin B-primed mice. J. Exp. Med. 172, 1065–1070.
54
Kim J. Urban R. G. Strominger J. L. Wiley D. C. (1994). Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science266, 1870–1874. 10.1126/science.7997880
55
Kissner T. L. Ruthel G. Alam S. Ulrich R. G. Fernandez S. Saikh K. U. (2011). Activation of MyD88 signaling upon staphylococcal enterotoxin binding to MHC class II molecules.PLoS One6:e15985. 10.1371/journal.pone.0015985
56
Kolata J. Bode L. G. Holtfreter S. Steil L. Kusch H. Holtfreter B. Albrecht D. Hecker M. Engelmann S. Van Belkum A. Volker U. Broker B. M. (2011). Distinctive patterns in the human antibody response to Staphylococcus aureus bacteremia in carriers and non-carriers. Proteomics11, 3914–3927. 10.1002/pmic.201000760
57
Kotb M. Norrby-Teglund A. McGeer A. El-Sherbini H. Dorak M. T. Khurshid A. Green K. Peeples J. Wade J. Thomson G. Schwartz B. Low D. E. (2002). An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat. Med. 8, 1398–1404. 10.1038/nm800
58
Langley R. Patel D. Jackson N. Clow F. Fraser J. D. (2010). Staphylococcal superantigen super-domains in immune evasion. Crit. Rev. Immunol. 30, 149–165.
59
Lee W. T. Vitetta E. S. (1992). Memory T cells are anergic to the superantigen staphylococcal enterotoxin B. J. Exp. Med. 176, 575–579.
60
Leung D. Y. Gately M. Trumble A. Ferguson-Darnell B. Schlievert P. M. Picker L. J. (1995a). Bacterial superantigens induce T cell expression of the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen, via stimulation of interleukin 12 production. J. Exp. Med. 181, 747–753.
61
Leung D. Y. Harbeck R. Bina P. Reiser R. F. Yang E. Norris D. A. Hanifin J. M. Sampson H. A. (1993). Presence of IgE antibodies to staphylococcal exotoxins on the skin of patients with atopic dermatitis. Evidence for a new group of allergens. J. Clin. Invest.92, 1374–1380. 10.1172/JCI116711
62
Leung D. Y. Meissner C. Fulton D. Schlievert P. M. (1995b). The potential role of bacterial superantigens in the pathogenesis of Kawasaki syndrome.J. Clin. Immunol.15, 11S–17S.
63
Leung D. Y. Travers J. B. Giorno R. Norris D. A. Skinner R. Aelion J. Kazemi L. V. Kim M. H. Trumble A. E. Kotb M. (1995c). Evidence for a streptococcal superantigen-driven process in acute guttate psoriasis. J. Clin. Invest. 96, 2106–2112. 10.1172/JCI118263
64
Li H. Llera A. Malchiodi E. L. Mariuzza R. A. (1999). The structural basis of T cell activation by superantigens. Annu. Rev. Immunol. 17, 435–466. 10.1146/annurev.immunol.17.1.435
65
Li H. Llera A. Tsuchiya D. Leder L. Ysern X. Schlievert P. M. Karjalainen K. Mariuzza R. A. (1998). Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity9, 807–816.
66
Li Y. Li H. Dimasi N. McCormick J. K. Martin R. Schuck P. Schlievert P. M. Mariuzza R. A. (2001). Crystal structure of a superantigen bound to the high-affinity, zinc- dependent site on MHC class II. Immunity14, 93–104.
67
Lin Y. T. Shau W. Y. Wang L. F. Yang Y. H. Hwang Y. W. Tsai M. J. Tsao P. N. Chiang B. L. (2000). Comparison of serum specific IgE antibodies to staphylococcal enterotoxins between atopic children with and without atopic dermatitis. Allergy55, 641–646.
68
Lin Y. T. Wang C. T. Chao P. S. Lee J. H. Wang L. C. Yu H. H. Yang Y. H. Chiang B. L. (2011). Skin-homing CD4+ Foxp3+ T cells exert Th2-like function after staphylococcal superantigen stimulation in atopic dermatitis patients. Clin. Exp. Allergy41, 516–525. 10.1111/j.1365-2222.2010.03681.x
69
Lina G. Bohach G. A. Nair S. P. Hiramatsu K. Jouvin-Marche E. Mariuzza R. (2004). Standard nomenclature for the superantigens expressed by Staphylococcus. J. Infect. Dis. 189, 2334–2336. 10.1086/420852
70
Llewelyn M. Cohen J. (2002). Superantigens: microbial agents that corrupt immunity. Lancet Infect. Dis. 2, 156–162.
71
Llewelyn M. Sriskandan S. Peakman M. Ambrozak D. R. Douek D. C. Kwok W. W. Cohen J. Altmann D. M. (2004). HLA class II polymorphisms determine responses to bacterial superantigens. J. Immunol. 172, 1719–1726.
72
Marrack P. Kappler J. (1990). The staphylococcal enterotoxins and their relatives. Science248, 705–711. 10.1126/science.2185544
73
Matsubara K. Fukaya T. (2007). The role of superantigens of group A Streptococcus and Staphylococcus aureus in Kawasaki disease. Curr. Opin. Infect. Dis. 20, 298–303. 10.1097/QCO.0b013e3280964d8c
74
McCormick J. K. Yarwood J. M. Schlievert P. M. (2001). Toxic shock syndrome and bacterial superantigens: an update. Annu. Rev. Microbiol. 55, 77–104. 10.1146/annurev.micro.55.1.77
75
McGavin M. J. Arsic B. Nickerson N. N. (2012). Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30.Front. Cell. Inf. Microbio.2: 48. 10.3389/fcimb.2012.00048
76
Medina E. Goldmann O. Rohde M. Lengeling A. Chhatwal G. S. (2001). Genetic control of susceptibility to group A streptococcal infection in mice. J. Infect. Dis. 184, 846–852. 10.1086/323292
77
Miller C. Ragheb J. A. Schwartz R. H. (1999). Anergy and cytokine-mediated suppression as distinct superantigen-induced tolerance mechanisms in vivo. J. Exp. Med. 190, 53–64. 10.1084/jem.190.1.53
78
Mitchell D. T. Levitt D. G. Schlievert P. M. Ohlendorf D. H. (2000). Structural evidence for the evolution of pyrogenic toxin superantigens. J. Mol. Evol. 51, 520–531.
79
Morgan M. M. Labno C. M. Van Seventer G. A. Denny M. F. Straus D. B. Burkhardt J. K. (2001). Superantigen-induced T cell:B cell conjugation is mediated by LFA-1 and requires signaling through Lck, but not ZAP-70. J. Immunol. 167, 5708–5718.
80
Moza B. Buonpane R. A. Zhu P. Herfst C. A. Rahman A. K. McCormick J. K. Kranz D. M. Sundberg E. J. (2006). Long-range cooperative binding effects in a T cell receptor variable domain. Proc. Natl. Acad. Sci. U.S.A. 103, 9867–9872. 10.1073/pnas.0600220103
81
Moza B. Varma A. K. Buonpane R. A. Zhu P. Herfst C. A. Nicholson M. J. Wilbuer A. -K. Seth N. P. Wucherpfennig K. W. McCormick J. K. Kranz D. M. Sundberg E. J. (2007). Structural basis of T-cell specificity and activation by the bacterial superantigen TSST-1. EMBO J. 26, 1187–1197. 10.1038/sj.emboj.7601531
82
Munson S. H. Tremaine M. T. Betley M. J. Welch R. A. (1998). Identification and characterization of staphylococcal enterotoxin types G and I from Staphylococcus aureus. Infect. Immun. 66, 3337–3348.
83
Narita K. Hu D. L. Tsuji T. Nakane A. (2008). Intranasal immunization of mutant toxic shock syndrome toxin 1 elicits systemic and mucosal immune response against Staphylococcus aureus infection. FEMS Immunol. Med. Microbiol. 52, 389–396. 10.1111/j.1574-695X.2008.00384.x
84
Newburger J. W. Takahashi M. Burns J. C. Beiser A. S. Chung K. J. Duffy C. E. Glode M. P. Mason W. H. Reddy V. Sanders S. P. Shulman S. T. Wiggins J. W. Hicks R. V. Fulton D. R. Lewis A. B. Leung D. Y. M. Colton T. Rosen F. S. Melish M. E. (1986). The treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 315, 341–347. 10.1056/NEJM198608073150601
85
Nizet V. (2007). Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 120, 13–22. 10.1016/j.jaci.2007.06.005
86
Nomura Y. Masuda K. Shinkoda Y. Sameshima K. Oku S. Yoshinaga M. Miyata K. (1998). Twenty-five types of T-cell receptor Vbeta family repertoire in patients with Kawasaki syndrome. Eur. J. Pediatr. 157, 981–986. 10.1007/s004310050982
87
Nooh M. M. El-Gengehi N. Kansal R. David C. S. Kotb M. (2007). HLA transgenic mice provide evidence for a direct and dominant role of HLA class II variation in modulating the severity of streptococcal sepsis. J. Immunol. 178, 3076–3083.
88
Osterholm M. T. Forfang J. C. (1982). Toxic-shock syndrome in Minnesota: results of an active-passive surveillance system. J. Infect. Dis. 145, 458–464.
89
Parsonnet J. Gillis Z. A. Richter A. G. Pier G. B. (1987). A rabbit model of toxic shock syndrome that uses a constant, subcutaneous infusion of toxic shock syndrome toxin 1. Infect. Immun. 55, 1070–1076.
90
Petersson K. Hakansson M. Nilsson H. Forsberg G. Svensson L. A. Liljas A. Walse B. (2001). Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 20, 3306–3312. 10.1093/emboj/20.13.3306
91
Petersson K. Pettersson H. Skartved N. J. Walse B. Forsberg G. (2003). Staphylococcal enterotoxin H induces V alpha-specific expansion of T cells. J. Immunol. 170, 4148–4154.
92
Petersson K. Thunnissen M. Forsberg G. Walse B. (2002). Crystal structure of a SEA variant in complex with MHC class II reveals the ability of SEA to crosslink MHC molecules. Structure10, 1619–1626.
93
Proft T. Fraser J. D. (2003). Bacterial superantigens. Clin. Exp. Immunol. 133, 299–306.
94
Pumphrey N. Vuidepot A. Jakobsen B. Forsberg G. Walse B. Lindkvist-Petersson K. (2007). Cutting edge: evidence of direct TCR alpha-chain interaction with superantigen. J. Immunol. 179, 2700–2704.
95
Pynnonen M. Stephenson R. E. Schwartz K. Hernandez M. Boles B. R. (2011). Hemoglobin promotes Staphylococcus aureus nasal colonization.PLoS Pathog.7: e1002104. 10.1371/journal.ppat.1002104
96
Rahman A. K. Bonsor D. A. Herfst C. A. Pollard F. Peirce M. Wyatt A. W. Kasper K. J. Madrenas J. Sundberg E. J. McCormick J. K. (2011). The T cell receptor beta-chain second complementarity determining region loop (CDR2beta) governs T cell activation and Vbeta specificity by bacterial superantigens. J. Biol. Chem. 286, 4871–4881. 10.1074/jbc.M110.189068
97
Ramsland P. A. Willoughby N. Trist H. M. Farrugia W. Hogarth P. M. Fraser J. D. Wines B. D. (2007). Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc. Natl. Acad. Sci. U.S.A. 104, 15051–15056. 10.1073/pnas.0706028104
98
Rasigade J. P. Thomas D. Perpoint T. Peyramond D. Chidiac C. Etienne J. Vandenesch F. Lina G. Ferry T. (2011). T-cell response to superantigen restimulation during menstrual toxic shock syndrome. FEMS Immunol. Med. Microbiol. 62, 368–371. 10.1111/j.1574-695X.2011.00808.x
99
Recsei P. Kreiswirth B. O'Reilly M. Schlievert P. Gruss A. Novick R. P. (1986). Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol. Gen. Genet. 202, 58–61.
100
Rellahan B. L. Jones L. A. Kruisbeek A. M. Fry A. M. Matis L. A. (1990). In vivo induction of anergy in peripheral V beta 8+ T cells by staphylococcal enterotoxin B.J. Exp. Med.172, 1091–1100.
101
Safdar N. Bradley E. A. (2008). The risk of infection after nasal colonization with Staphylococcus aureus. Am. J. Med. 121, 310–315. 10.1016/j.amjmed.2007.07.034
102
Saline M. Rodstrom K. E. Fischer G. Orekhov V. Y. Karlsson B. G. Lindkvist-Petersson K. (2010). The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat. Commun. 1, 119. 10.1038/ncomms1117
103
Sayama K. Midorikawa K. Hanakawa Y. Sugai M. Hashimoto K. (1998). Superantigen production by Staphylococcus aureus in psoriasis. Dermatology196, 194–198.
104
Schlievert P. M. (1986). Staphylococcal enterotoxin B and toxic-shock syndrome toxin-1 are significantly associated with non-menstrual TSS [letter]. Lancet1, 1149–1150.
105
Schlievert P. M. Case L. C. Strandberg K. L. Abrams B. B. Leung D. Y. (2008). Superantigen profile of Staphylococcus aureus isolates from patients with steroid-resistant atopic dermatitis. Clin. Infect. Dis. 46, 1562–1567. 10.1086/586746
106
Schlievert P. M. Jablonski L. M. Roggiani M. Sadler I. Callantine S. Mitchell D. T. Ohlendorf D. H. Bohach G. A. (2000). Pyrogenic toxin superantigen site specificity in toxic shock syndrome and food poisoning in animals. Infect. Immun. 68, 3630–3634. 10.1128/IAI.68.6.3630-3634.2000
107
Schlievert P. M. Shands K. N. Dan B. B. Schmid G. P. Nishimura R. D. (1981). Identification and characterization of an exotoxin from Staphylococcus aureus associated with toxic-shock syndrome. J. Infect. Dis. 143, 509–516.
108
Schlievert P. M. Strandberg K. L. Lin Y. C. Peterson M. L. Leung D. Y. (2010). Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J. Allergy Clin. Immunol. 125, 39–49. 10.1016/j.jaci.2009.10.039
109
Schrage B. Duan G. Yang L. P. Fraser J. D. Proft T. (2006). Different preparations of intravenous immunoglobulin vary in their efficacy to neutralize streptococcal superantigens: implications for treatment of streptococcal toxic shock syndrome. Clin. Infect. Dis. 43, 743–746. 10.1086/507037
110
Seo K. S. Park J. Y. Terman D. S. Bohach G. A. (2010). A quantitative real time PCR method to analyze T cell receptor Vbeta subgroup expansion by staphylococcal superantigens. J. Transl. Med. 8, 2. 10.1186/1479-5876-8-2
111
Shands K. N. Schmid G. P. Dan B. B. Blum D. Guidotti R. J. Hargrett N. T. Anderson R. L. Hill D. L. Broome C. V. Band J. D. Fraser D. W. (1980). Toxic-shock syndrome in menstruating women: association with tampon use and Staphylococcus aureus and clinical features in 52 cases. N. Engl. J. Med. 303, 1436–1442. 10.1056/NEJM198012183032502
112
Smith-Garvin J. E. Koretzky G. A. Jordan M. S. (2009). T cell activation. Annu. Rev. Immunol. 27, 591–619. 10.1146/annurev.immunol.021908.132706
113
Stankovic K. Miailhes P. Bessis D. Ferry T. Broussolle C. Seve P. (2007). Kawasaki-like syndromes in HIV-infected adults. J. Infect. 55, 488–494. 10.1016/j.jinf.2007.09.005
114
Stevens D. L. Tanner M. H. Winship J. Swarts R. Ries K. M. Schlievert P. M. Kaplan E. (1989). Severe group A streptococcal infections associated with a toxic shock- like syndrome and scarlet fever toxin A. N. Engl. J. Med. 321, 1–7. 10.1056/NEJM198907063210101
115
Stevens F. A. (1927). The occurence of Staphylococcus aureus infection with a scarlitiniform rash. JAMA88, 1957–1958.
116
Stolz S. J. Davis J. P. Vergeront J. M. Crass B. A. Chesney P. J. Wand P. J. Bergdoll M. S. (1985). Development of serum antibody to toxic shock toxin among individuals with toxic shock syndrome in Wisconsin. J. Infect. Dis. 151, 883–889.
117
Sundberg E. J. Li H. Llera A. S. McCormick J. K. Tormo J. Schlievert P. M. Karjalainen K. Mariuzza R. A. (2002). Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes.Structure10, 687–699.
118
Swietnicki W. Barnie A. M. Dyas B. K. Ulrich R. G. (2003). Zinc binding and dimerization of Streptococcus pyogenes pyrogenic exotoxin C are not essential for T-cell stimulation. J. Biol. Chem. 278, 9885–9895. 10.1074/jbc.M206957200
119
Taylor A. L. Llewelyn M. J. (2010). Superantigen-induced proliferation of human CD4+CD25- T cells is followed by a switch to a functional regulatory phenotype. J. Immunol. 185, 6591–6598. 10.4049/jimmunol.1002416
120
Thomas D. Dauwalder O. Brun V. Badiou C. Ferry T. Etienne J. Vandenesch F. Lina G. (2009). Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect. Immun.77, 2043–2050. 10.1128/IAI.01388-08
121
Tilahun A. Y. Holz M. Wu T. T. David C. S. Rajagopalan G. (2011a). Interferon gamma-dependent intestinal pathology contributes to the lethality in bacterial superantigen-induced toxic shock syndrome.PLoS One6:e16764. 10.1371/journal.pone.0016764
122
Tilahun A. Y. Marietta E. V. Wu T. T. Patel R. David C. S. Rajagopalan G. (2011b). Human leukocyte antigen class II transgenic mouse model unmasks the significant extrahepatic pathology in toxic shock syndrome. Am. J. Pathol. 178, 2760–2773. 10.1016/j.ajpath.2011.02.033
123
Todd J. K. Kapral F. A. Fishaut M. Welch T. R. (1978). Toxic shock syndrome associated with phage group 1 staphylococci. Lancet2, 1116–1118.
124
Tripp T. J. McCormick J. K. Webb J. M. Schlievert P. M. (2003). The zinc-dependent major histocompatibility complex class II binding site of streptococcal pyrogenic exotoxin C is critical for maximal superantigen function and toxic activity. Infect. Immun. 71, 1548–1550. 10.1128/IAI.71.3.1548-1550.2003
125
Van Crombruggen K. Zhang N. Gevaert P. Tomassen P. Bachert C. (2011). Pathogenesis of chronic rhinosinusitis: inflammation. J. Allergy Clin. Immunol. 128, 728–732. 10.1016/j.jaci.2011.07.049
126
Van Zele T. Gevaert P. Watelet J. B. Claeys G. Holtappels G. Claeys C. Van Cauwenberge P. Bachert C. (2004). Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis.J. Allergy Clin. Immunol.114, 981–983. 10.1016/j.jaci.2004.07.013
127
Van Zele T. Vaneechoutte M. Holtappels G. Gevaert P. Van Cauwenberge P. Bachert C. (2008). Detection of enterotoxin DNA in Staphylococcus aureus strains obtained from the middle meatus in controls and nasal polyp patients. Am. J. Rhinol. 22, 223–227. 10.2500/ajr.2008.22.3161
128
Varshney A. K. Mediavilla J. R. Robiou N. Guh A. Wang X. Gialanella P. Levi M. H. Kreiswirth B. N. Fries B. C. (2009). Diverse enterotoxin gene profiles among clonal complexes of Staphylococcus aureus isolates from the Bronx, New York. Appl. Environ. Microbiol. 75, 6839–6849. 10.1128/AEM.00272-09
129
Vergeront J. M. Stolz S. J. Crass B. A. Nelson D. B. Davis J. P. Bergdoll M. S. (1983). Prevalence of serum antibody to staphylococcal enterotoxin F among Wisconsin residents: implications for toxic-shock syndrome. J. Infect. Dis. 148, 692–698.
130
Verkaik N. J. Benard M. Boelens H. A. De Vogel C. P. Nouwen J. L. Verbrugh H. A. Melles D. C. Van Belkum A. Van Wamel W. J. (2011). Immune evasion cluster-positive bacteriophages are highly prevalent among human Staphylococcus aureus strains, but they are not essential in the first stages of nasal colonization. Clin. Microbiol. Infect. 17, 343–348. 10.1111/j.1469-0691.2010.03227.x
131
Verkaik N. J. De Vogel C. P. Boelens H. A. Grumann D. Hoogenboezem T. Vink C. Hooijkaas H. Foster T. J. Verbrugh H. A. Van Belkum A. Van Wamel W. J. (2009). Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J. Infect. Dis. 199, 625–632. 10.1086/596743
132
von Eiff C. Becker K. Machka K. Stammer H. Peters G. (2001). Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group.N. Engl. J. Med.344, 11–16. 10.1056/NEJM200101043440102
133
Wen R. Cole G. A. Surman S. Blackman M. A. Woodland D. L. (1996). Major histocompatibility complex class II-associated peptides control the presentation of bacterial superantigens to T cells. J. Exp. Med. 183, 1083–1092.
134
Wertheim H. F. Melles D. C. Vos M. C. Van Leeuwen W. Van Belkum A. Verbrugh H. A. Nouwen J. L. (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762. 10.1016/S1473-3099(05)70295-4
135
Wertheim H. F. Vos M. C. Ott A. Van Belkum A. Voss A. Kluytmans J. A. Van Keulen P. H. Vandenbroucke-Grauls C. M. Meester M. H. Verbrugh H. A. (2004). Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet364, 703–705. 10.1016/S0140-6736(04)16897-9
136
White M. C. Thornton K. Young A. E. (2005). Early diagnosis and treatment of toxic shock syndrome in paediatric burns. Burns31, 193–197. 10.1016/j.burns.2004.09.017
137
Wilson G. J. Seo K. S. Cartwright R. A. Connelley T. Chuang-Smith O. N. Merriman J. A. Guinane C. M. Park J. Y. Bohach G. A. Schlievert P. M. Morrison W. I. Fitzgerald J. R. (2011). A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia.PLoS Pathog.7:e1002271. 10.1371/journal.ppat.1002271
138
Yamasaki S. Tachibana M. Shinohara N. Iwashima M. (1997). Lck-independent triggering of T-cell antigen receptor signal transduction by staphylococcal enterotoxins. J. Biol. Chem. 272, 14787–14791. 10.1074/jbc.272.23.14787
139
Yeung R. S. (2007). Lessons learned from an animal model of Kawasaki disease. Clin. Exp. Rheumatol. 25, S69–S71.
140
Yeung R. S. (2010). Kawasaki disease: update on pathogenesis. Curr. Opin. Rheumatol. 22, 551–560. 10.1097/BOR.0b013e32833cf051
141
Yeung R. S. Penninger J. M. Kundig T. Khoo W. Ohashi P. S. Kroemer G. Mak T. W. (1996). Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: supersensitivity to superantigen-induced septic shock. Eur. J. Immunol. 26, 1074–1082. 10.1002/eji.1830260518
142
Yoshioka T. Matsutani T. Iwagami S. Toyosaki-Maeda T. Yutsudo T. Tsuruta Y. Suzuki H. Uemura S. Takeuchi T. Koike M. Suzuki R. (1999). Polyclonal expansion of TCRBV2- and TCRBV6-bearing T cells in patients with Kawasaki disease. Immunology96, 465–472.
Summary
Keywords
superantigen, staphylococcal enterotoxin, Staphylococcus aureus , colonization
Citation
Xu SX and McCormick JK (2012) Staphylococcal superantigens in colonization and disease. Front. Cell. Inf. Microbio. 2:52. doi: 10.3389/fcimb.2012.00052
Received
01 February 2012
Accepted
29 March 2012
Published
17 April 2012
Volume
2 - 2012
Edited by
Martin J. McGavin, University of Western Ontario, Canada
Reviewed by
Thomas Proft, University of Auckland, New Zealand; Victor J. Torres, New York University School of Medicine, USA
Copyright
© 2012 Xu and McCormick.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John K. McCormick, Department of Microbiology and Immunology, University of Western Ontario, 1151 Richmond St., London, ON N6A 5C1, Canada. e-mail: john.mccormick@schulich.uwo.ca
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.