 Paul Hodor1*
Paul Hodor1* Christopher E. Pope2Kathryn B. Whitlock3Lucas R. Hoffman1,2David L. Limbrick4
Christopher E. Pope2Kathryn B. Whitlock3Lucas R. Hoffman1,2David L. Limbrick4 Patrick J. McDonald5
Patrick J. McDonald5 Jason S. Hauptman1,2Jeffrey G. Ojemann1,2
Jason S. Hauptman1,2Jeffrey G. Ojemann1,2 Tamara D. Simon6,7 on behalf of Cerebrospinal FLuId MicroBiota in Shunts Study (CLIMB) Group
Tamara D. Simon6,7 on behalf of Cerebrospinal FLuId MicroBiota in Shunts Study (CLIMB) Group- 1Seattle Children’s Hospital, Seattle, WA, United States
- 2Department of Pediatrics, University of Washington, Seattle, WA, United States
- 3New Harmony Statistical Consulting LLC, Clinton, WA, United States
- 4Department of Neurosurgery, Washington University in St. Louis, St. Louis, MO, United States
- 5Division of Neurosurgery, University of British Columbia, Vancouver, BC, Canada
- 6Children’s Hospital Los Angeles, Los Angeles, CA, United States
- 7Department of Pediatrics, Keck School of Medicine at the University of Southern California, Los Angeles, CA, United States
Understanding the etiology of cerebrospinal fluid (CSF) shunt infections and reinfections requires detailed characterization of associated microorganisms. Traditionally, identification of bacteria present in the CSF has relied on culture methods, but recent studies have used high throughput sequencing of 16S rRNA genes. Here we evaluated the method of shotgun DNA sequencing for its potential to provide additional genomic information. CSF samples were collected from 3 patients near the beginning and end of each of 2 infection episodes. Extracted total DNA was sequenced by: (1) whole genome amplification followed by shotgun sequencing (WGA) and (2) high-throughput sequencing of the 16S rRNA V4 region (16S). Taxonomic assignments of sequences from WGA and 16S were compared with one another and with conventional microbiological cultures. While classification of bacteria was consistent among the 3 approaches, WGA provided additional insights into sample microbiological composition, such as showing relative abundances of microbial versus human DNA, identifying samples of questionable quality, and detecting significant viral load in some samples. One sample yielded sufficient non-human reads to allow assembly of a high-quality Staphylococcus epidermidis genome, denoted CLIMB1, which we characterized in terms of its MLST profile, gene complement (including putative antimicrobial resistance genes), and similarity to other annotated S. epidermidis genomes. Our results demonstrate that WGA directly applied to CSF is a valuable tool for the identification and genomic characterization of dominant microorganisms in CSF shunt infections, which can facilitate molecular approaches for the development of better diagnostic and treatment methods.
Introduction
Hydrocephalus is a common cause of neurological disability in children (Williams et al., 2007). Cerebrospinal fluid (CSF) shunt placement allows children with hydrocephalus to survive and avoid ongoing brain injury. However, CSF shunts cause new chronic surgical problems, including the need for surgical revisions from catheter obstruction and infections (Kestle, 2003; Simon et al., 2009; Simon et al., 2012). CSF shunt infection treatment usually requires surgical removal of the CSF shunt, two weeks of intravenous antibiotics tailored to organisms recovered from conventional culture, and a second surgery to place a new CSF shunt (Kestle et al., 2006; Simon et al., 2010). Despite this aggressive treatment, reinfection rates range from 20 to 25% (Kulkarni et al., 2001; Kestle et al., 2006; Tuan et al., 2011). Reinfection rates are higher still for children with their second CSF shunt infection (Tuan et al., 2011). An improved understanding of the mechanisms of infection is critical to effectively treat more than 2,000 CSF shunt infections diagnosed each year (Simon et al., 2008).
Among the 20 to 25% of patients with treated CSF shunt infection who develop reinfection, it is unclear whether reinfections are caused by an organism that persists from one infection to the next, or are independent infection events. In the majority of reinfections (70%), the organism(s) recovered by culture differ between the first and second infection (Tuan et al., 2011). Detailed genomic characterization of microorganisms from CSF can help answer questions about the microbial determinants of infection and reinfection.
Several methods exist for characterizing microorganisms in CSF and other clinical specimens. Microbiological culture methods, typically bacterial cultures, are standard in the routine clinical laboratory practice (Baron et al., 2013; Pittman et al., 2014). While sensitive, culture methods are limited to detecting specific human pathogens and often do not provide strain information without additional investigation. Multilocus sequence typing (MLST) can identify strain subtypes for certain species, based on characteristic gene sequence markers (Belén et al., 2009). 16S rRNA sequencing can identify a broader range of species than either culture or MLST, including some not detectable by culture, but is generally less sensitive and often cannot discriminate microbes to the species level (Church et al., 2020). Next-generation sequencing applications are increasingly being used to address the limitations of these traditional methods (Mitchell and Simner, 2019), potentially combined with mass spectrometry (Hrabak et al., 2020). Whole genome sequencing offers advantages such as detailed genomic characterization, strain discrimination, identification of putative antibiotic resistance genes, and characterization of metabolic capacity, all of which are relevant in studying the dynamics of infection and treatment response. However, in the case of CSF shunt infection, samples have low microbial loads, and therefore a DNA enrichment method must be applied prior to sequencing. Whole genome amplification and shotgun sequencing (WGA) is one such method, which was recently applied to a cohort of hospitalized patients with infectious meningitis and encephalitis (Wilson et al., 2019), identifying potential pathogens that were previously underappreciated.
Here we describe a proof of concept study to assess the usefulness of WGA for characterizing the microbiota of CSF shunt infections, comparing results of WGA with those of conventional microbiological culture and 16S rRNA high throughput sequencing (16S).
Materials and Methods
Study Subjects
The cohort considered in this study was previously described (Whitlock et al., 2021). Eligible subjects were children ≤18 years old undergoing treatment for conventional culture-confirmed CSF shunt infection at either Seattle Children’s Hospital (SCH) or Primary Children’s Hospital (PCH). Enrollment occurred from 2010 to present at SCH and from 2008 to 2015 at PCH. In this study we considered a subset of children who failed treatment for CSF shunt infection (i.e. had CSF shunt reinfection) and had CSF collected both near the beginning and end of both infection episodes (n = 3).
CSF Specimen Collection
Collection and storage of CSF was done under standard sterile conditions. The initial CSF sample for diagnosis of infection was obtained by needle aspiration of the shunt reservoir outside the operating room in a bedside “shunt tap”. The CSF sample near the beginning of the infection, which was analyzed in this study, was either left over from the initial diagnostic sample or was obtained in the operating room under sterile conditions from the system being removed during the first surgery to treat infection. Samples near the end of the infection were generally obtained under sterile bedside conditions through a sampling port within sterile extension tubing attached to the external ventricular drain.
CSF samples were stored at 4°C upon collection, aliquoted into vials of ~100 µl, and stored at -70°C. PCH samples were shipped overnight to Seattle on dry ice for analysis.
Conventional Culture Identification of Bacteria
All samples were tested by routine CSF aerobic culture in hospital-certified laboratories at both SCH and PCH. The methodology followed guidelines of the Clinical and Laboratory Standards Institute guidelines (https://clsi.org).
DNA Extraction, 16S rRNA Gene Amplification and Sequencing
A diagram of the experimental procedures, beginning with DNA extraction and ending with taxonomic assignment of sequences, is shown in Supplementary Figure 1.
DNA was extracted and purified from CSF samples using the AGOWA mag Mini DNA isolation kit (AGOWA, LGC Genomics, Berlin, Germany) and CSF microbiota amplicon library construction was carried out using a one-step PCR amplification targeting the V4 region of the bacterial 16S rRNA gene as described (Whitlock et al., 2021).
Sequencing of the pooled libraries was carried out for 600 cycles on an Illumina MiSeq desktop sequencer using the Miseq Reagent Kit v3.
Whole Genome Amplification and Shotgun Sequencing
Whole genome amplification of DNA purified from CSF samples and two mock community samples was carried out using the REPLI-g Mini Kit (Qiagen, CN: 150023) in accordance with the manufacturer’s recommendations. For each sample, including mock communities and no-template controls, a random-fragment library was constructed using the Nextera DNA Sample Preparation Kit (Illumina) with dual indexing and sequenced on the HiSeq 2500 platform to produce 96-bp paired-end reads.
Taxonomic Assignment of 16S Sequences With DADA2
Sequencing data were analyzed using the denoising program DADA2 (Callahan et al., 2016) (version1.6.0) as described (Nelson et al., 2019), and aligned to the SILVA 16S reference database (v. 132) (Pruesse et al., 2007) to produce a 16S amplicon taxa table for downstream computational analysis.
Taxonomic Assignment of Shotgun Sequences With Kraken
Shotgun sequence data were analyzed using the program Kraken (Wood and Salzberg, 2014) against a custom database that included full genomic sequence of human, bacteria, virus, and archaea obtained from GenBank.
De Novo Assembly and Annotation
Non-human DNA reads were assembled and annotated with the Comprehensive Genome Analysis service on the PATRIC website (Wattam et al., 2017). This analysis included assembly using SPAdes (Bankevich et al., 2012), a standard pipeline for annotating sequences with open reading frames and hypothetical function, and prediction of antibiotic resistance genes using the comprehensive antibiotic resistance database (CARD) (McArthur et al., 2013).
Multilocus sequence typing (MLST) analysis was performed on the PubMLST.org website (Jolley et al., 2018) for S. epidermidis (Thomas et al., 2007) and on Institute Pasteur MLST website (http://bigsdb.pasteur.fr) for K. pneumoniae (Diancourt et al., 2005).
The PATRIC Similar Genome Finder was searched with the assembled S. epidermidis CLIMB1 genome, and the closest 100 matching genomes were identified. A rooted phylogenetic tree was built using the Codon Tree method, with Staphylococcus caprae C87 as an outgroup.
Results
Sequencing-Defined Taxonomic Composition of CSF DNA
CSF samples were expected to contain substantial amounts of human DNA, along with DNA from other types of organisms, both pathogenic and opportunistic. We estimated the relative DNA abundance of organisms from the major domains of life in CSF samples by the distribution of reads in WGA (Figure 1). Human DNA constituted the overwhelming majority of reads in 10 out of 12 samples (range: 76.5 to 99.9%, mean: 95.4%, median: 98.7%). Two replicates of one sample (P3 I2 B) yielded 60.8% and 87.2% human reads, respectively, and another (P2 I1 E) yielded fewer than 0.5% human reads in both replicates. The low abundance of human DNA in these latter 2 samples, together with the detection of typical bacterial false positives (e.g. Delftia spp., Bradyrhizobium spp.), indicated potential quality issues with the latter 2 samples. These samples were therefore excluded from further analysis. The proportion of sequencing reads contributed by bacteria in the remaining 10 samples ranged from 0.1 to 22.8% (mean: 3.1%, median: 0.6%). In 2 samples collected near the beginning of an infection episode, the proportion of bacterial reads was particularly high: Sample P1 I1 B – 15.7% and 22.8%, and P2 I2 B – 6.4% and 7.1%. Viral sequences were observed in 2 samples at low frequency (< 0.2%). No archaeal sequences were detected in any of the CSF samples.
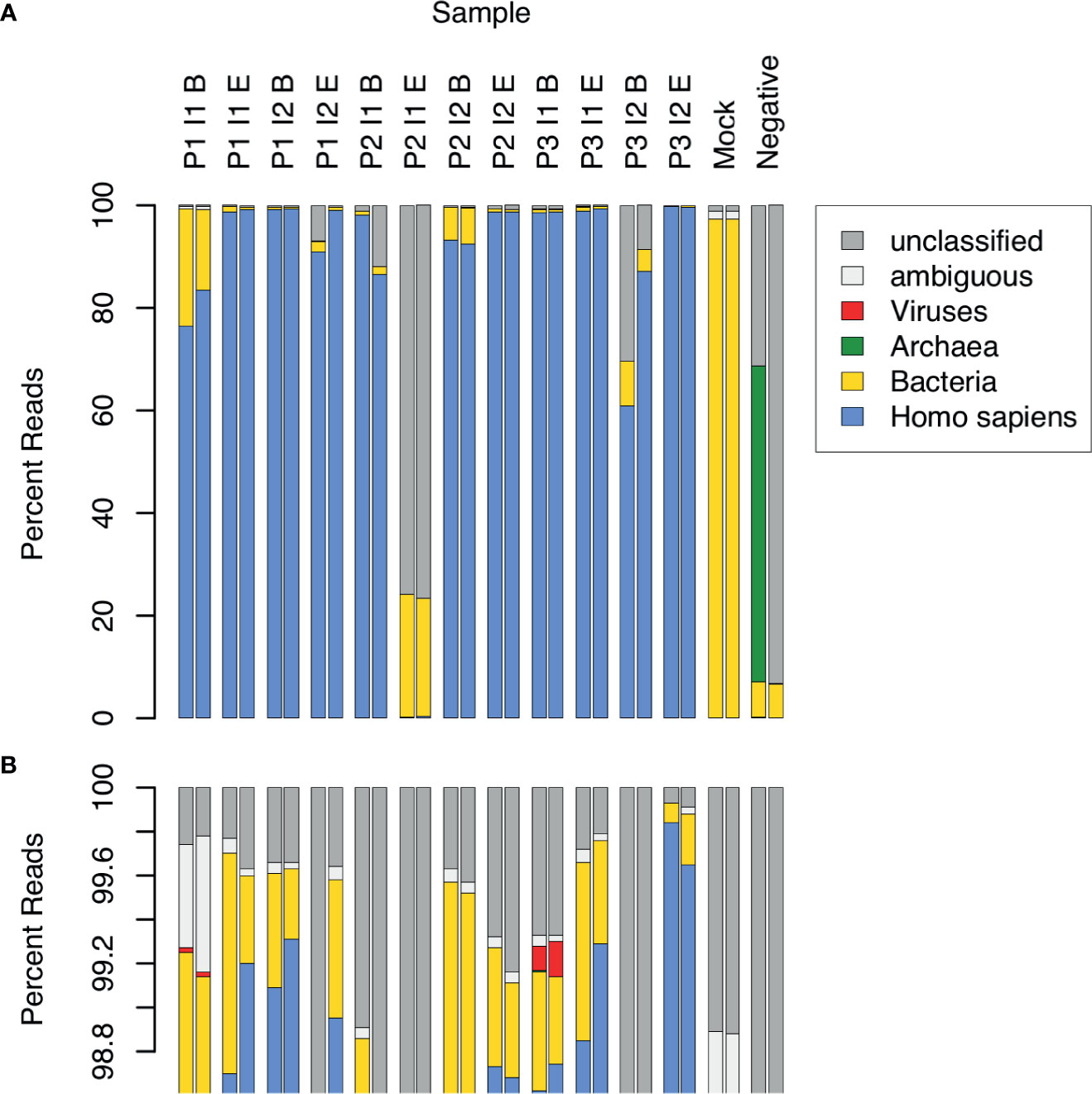
Figure 1 Relative abundance of sequence reads in CSF samples across major domains of life. 12 CSF samples were analyzed in duplicate by WGA. The samples came from 3 patients (P1, P2, P3), each having 2 infection episodes (I1, I2), with one sample collected near the beginning (B) and the other near the end (E) of the episode. Control samples consisted of a mock bacterial community (Mock) and a negative, no-template sample (Negative). Assignment of sequence reads to major domains of life was performed with Kraken: human (blue), bacteria (yellow), archaea (green), viruses (red), ambiguous, i.e. assignment to more than 1 domain (white), and unclassified, i.e. not assigned to any domain (grey). The lower panel (B) is an expanded view of (A), showing the top percentile range and revealing 2 samples in which viral sequences were detected.
Taxonomic assignment of WGA DNA reads identified 16 bacterial species that comprised 5% or more of bacterial reads in at least 1 CSF sample (Figure 2). Species with less than 5% of reads were assumed to be mostly artifactual and were excluded from the analysis. Although the Kraken software has excellent specificity (Lindgreen et al., 2016), it is difficult to distinguish true positives from contaminants. Consistency between replicates was limited even among species above the 5% threshold. Only the following species were found in both replicates of the same CSF sample: S. epidermidis in 2 samples (P1 I1 B and P2 I1 B), and K. pneumoniae (P2 I2 B) and S. pyogenes (P3 I1 B) in one sample each. S. aureus sequences were present in all samples, including the no template negative controls.
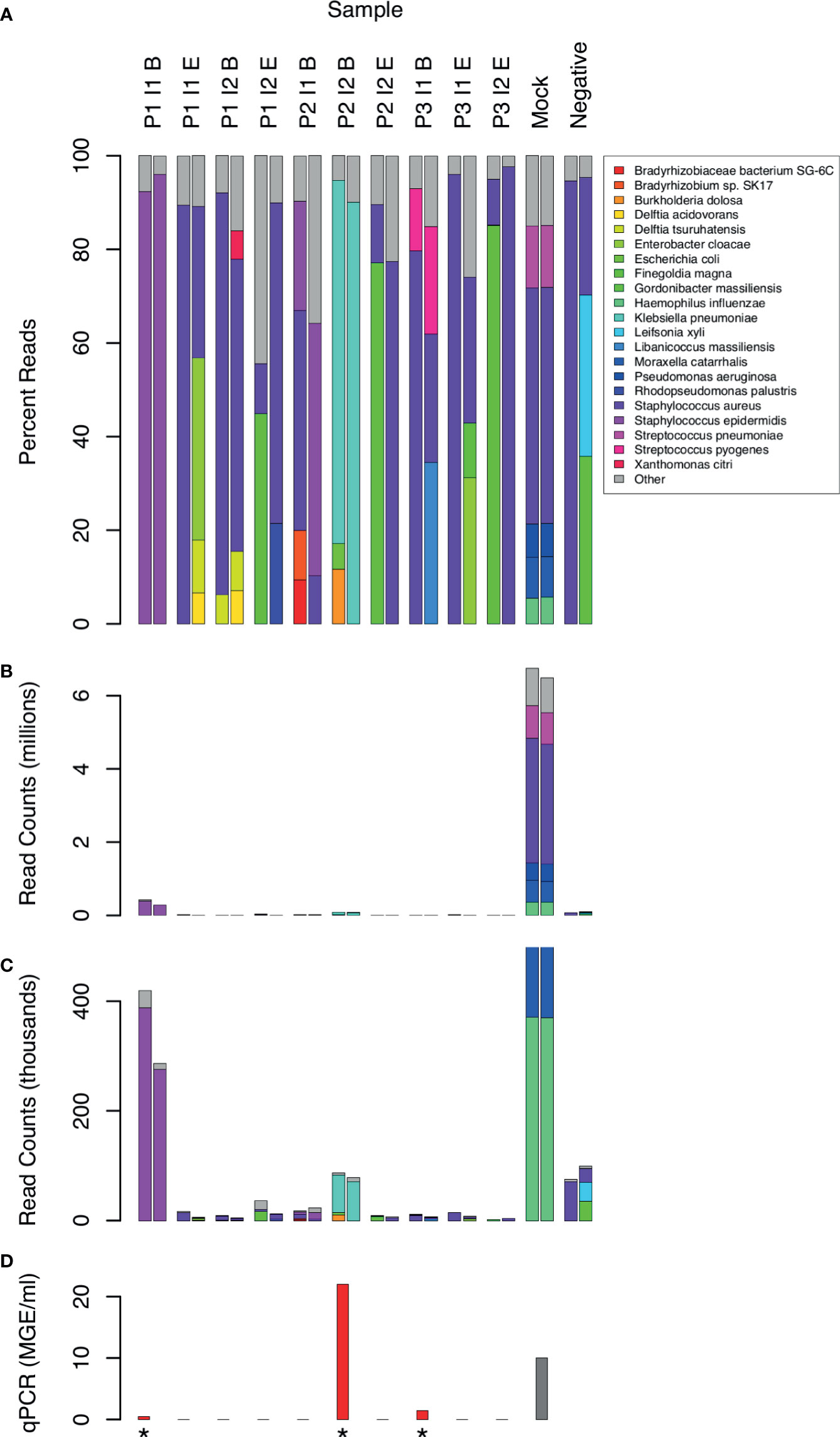
Figure 2 Distribution of bacterial species in sequence reads from 10 CSF samples. Sample labels are as in Figure 1. 2 samples with low human DNA content were excluded. 21 species that yielded 5% or more bacterial reads in at least one sample are shown by name. Less frequent species assignments are grouped into the “Other” category. Reads are plotted as percentages of bacterial reads (A) and counts (B, C). Panel C shows the same data as B, but scaled to highlight the experimental samples, which had much lower counts than the mock positive control. qPCR measurements of 16S RNA are shown for comparison (D) in units of genome equivalents per ml. The gray bar (Mock) represents a computed, non-experimental value. Measurements above the limit of detection are marked with an asterisk.
The absolute number of bacterial reads, although not a rigorous measure of abundance, was comparatively high in 2 samples (Figure 2, samples P1 I1 B and P2 I2 B). In both cases, the non-human reads were assigned to 1 predominant bacterial species (Staphylococcus epidermis in sample P1 I1 B and Klebsiella pneumoniae in P2 I2 B). These were also 2 of the 3 clinical samples yielding 16S rRNA above the level of detection by qPCR (Figure 2).
In addition to S. epidermidis, sample P1 I1 B had about 300 (0.02%) reads assigned to a Staphylococcus phage StB20-like genome. StB20 was previously described as a lysogenic bacteriophage isolated from a coagulase-negative Staphylococcus capitis (Deghorain et al., 2012). Sample P3 I1 B had 2000-3000 (0.1-0.2%) sequences mapped to multiple torque teno virus (TTV) genomes. TTV has been previously identified in CSF (Simner et al., 2018; Manso et al., 2020), but its clinical significance is unclear. Our preliminary findings from assembling the TTV reads indicate that the sample contained potentially 5 or more strains of TTV and torque teno mini virus.
Comparison of Identified Microorganisms by Cultivation, 16S, and WGA
Identification of bacterial species by culture remains the gold standard for the microbiological characterization of CSF. Of the samples in this study, 6 were culture positive. We examined whether WGA and 16S data were consistent with culture results (Table 1). Culture made 7 bacterial species identifications in the 6 samples. Of these, WGA identified the same species as the most or second-most abundant in 5 cases. In the remaining 2 cases, the same species were present in the WGA data, but at an abundance too low to be called out as positives. 16S results resembled those from WGA, albeit at the genus and family levels. For example, S. epidermidis and S. aureus could not be distinguished by 16S.
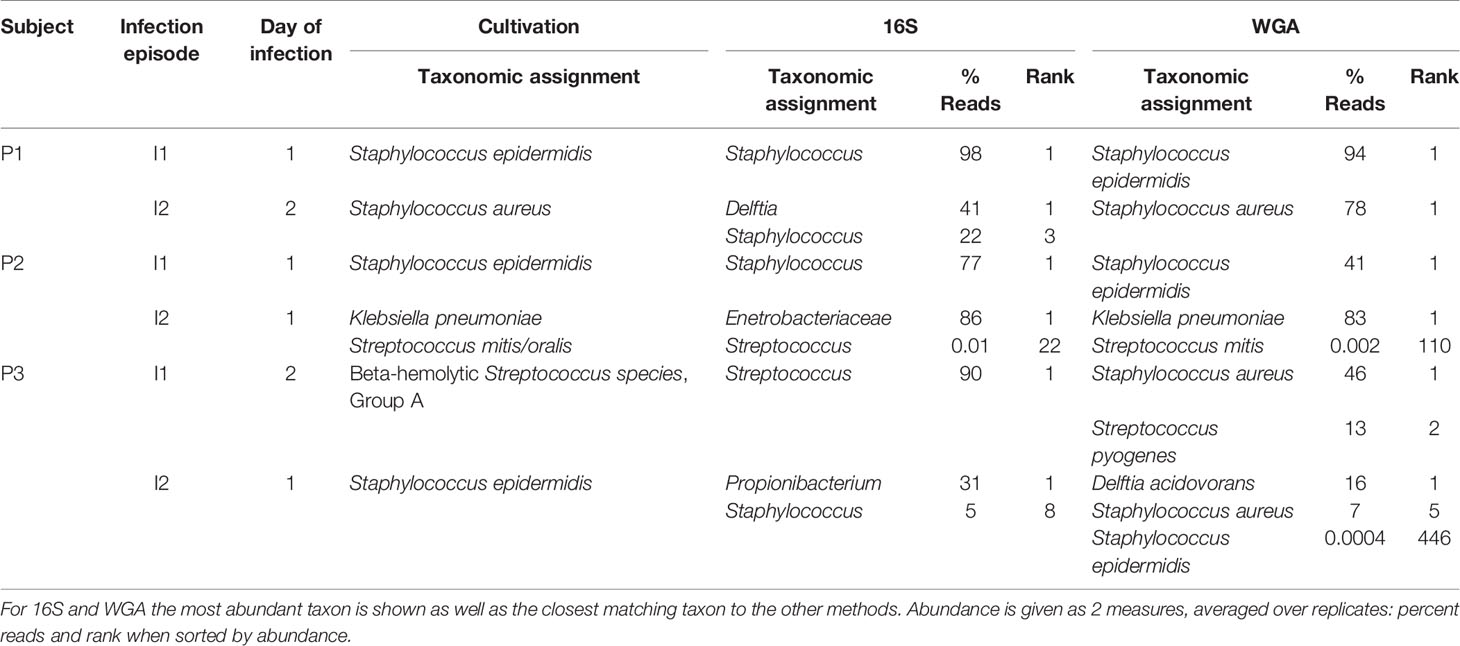
Table 1 Comparison of taxonomic assignments of bacteria by microbiology lab culture, 16S, and WGA for 6 samples that were culture-positive.
De Novo Genome Assembly
A set of contigs of sufficient quality was assembled de novo from one sample, P1 I1 B, yielding a draft S. epidermidis genome, which we refer to hereafter as CLIMB1 (see below). Assembly of the non-human reads from sample P2 I2 B yielded 55 contigs spanning 610 Kbp of DNA. 410 Kbp (68%) of this sequence showed over 99% identity to multiple K. pneumoniae chromosomal sequences from GenBank. 136 Kbp (22%) of the assembled sequence had a 100% match to an unnamed plasmid of 239 Kbp total length from K. pneumoniae KP14003. None of the 7 K. pneumoniae MLST genes were present in the assembled sequence. A possible explanation is that the contigs represent only a partial assembly of a putative K. pneumoniae genome, which missed the MLST genes by chance due to low coverage (< 10%) of the chromosomal sequence.
Characterization of the S. epidermidis CLIMB1 Genome
The S. epidermidis CLIMB1 assembly consisted of 76 contigs comprising a total of 2.42 Mbp (Figure 3), in the range of typical S. epidermidis genomes (Conlan et al., 2012). The contig N50 value of 67.5 Kbp and L50 value of 12 reflects an acceptable quality of assembly. There were a total of 2,337 predicted protein coding sequences. Of these, 13 were annotated as putative antibiotic resistance genes based on a CARD database search (Table 2).
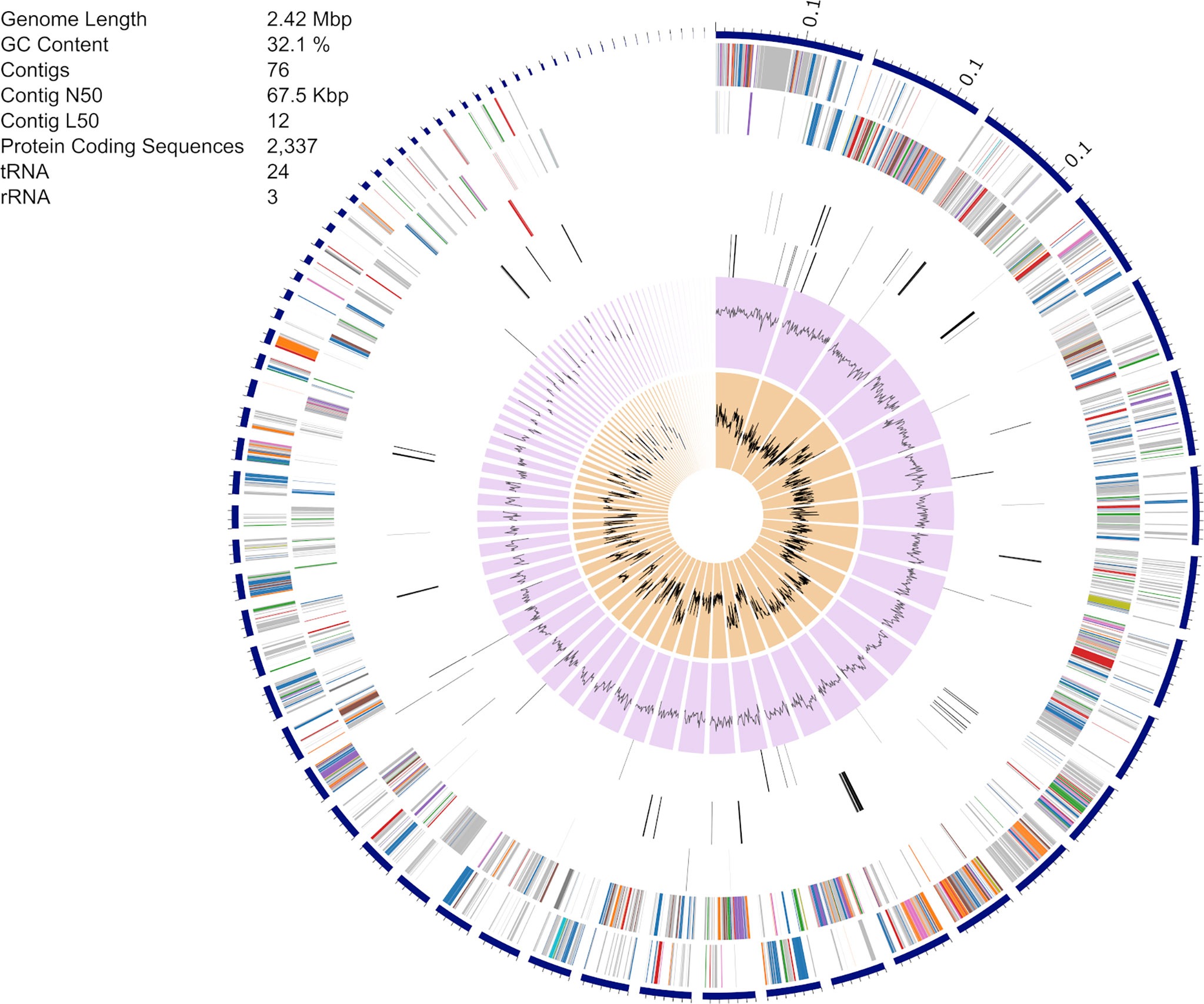
Figure 3 Assembly of the S. epidermidis CLIMB1 genome. The circular display of the assembly shows, from outer to inner rings, the contigs, coding sequences on the forward strand, coding sequences on the reverse strand, RNA genes, coding sequences of putative antimicrobial resistance genes, coding sequences of putative virulence genes, GC content, and GC skew. The embedded table includes summary statistics of the assembly.
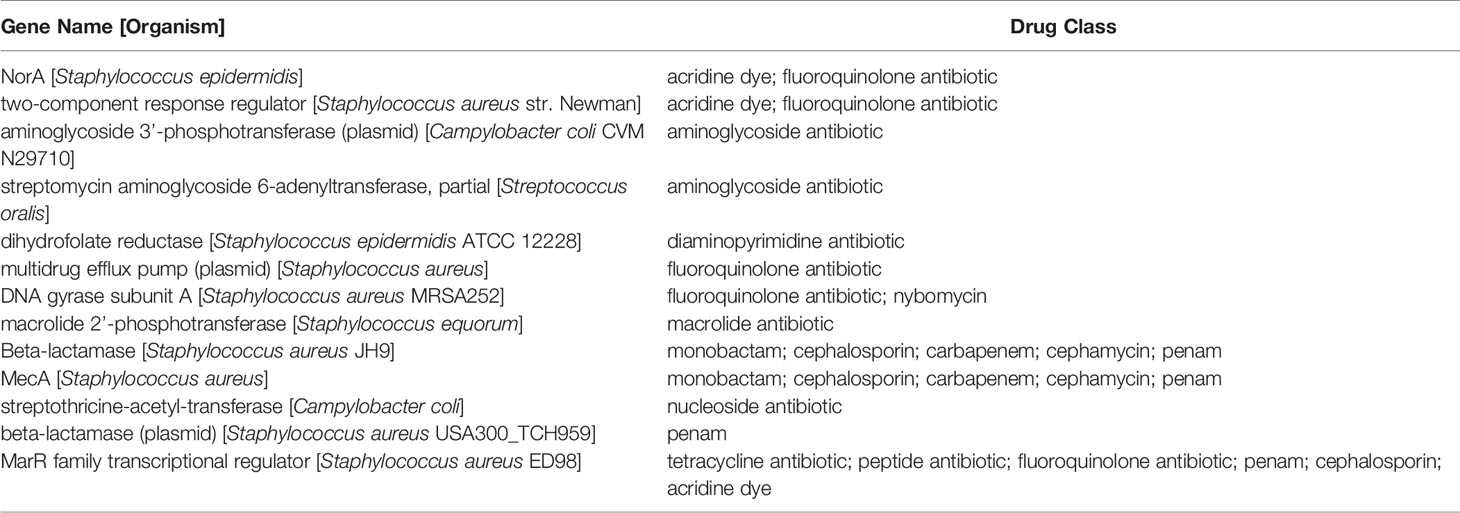
Table 2 Predicted antibiotic resistance genes in the S. epidermidis CLIMB1 genome ascertained from CARD.
The S. epidermidis CLIMB1 genome assembly contained all seven MLST genes of S. epidermidis (arcC, aroE, gtr, mutS, pyrR, tpiA, yqiL). MLST sequences of CLIMB1 assign this strain to profile 16-2-1-2-2-15-1-1 or ST-16. The PubMLST database contains 3 other previously described isolates with profile ST-16, 2 from human(s) in the USA in 2001 and Russia in 2010 and 1 from an environmental source in Poland in 2007.
A phylogenetic tree was constructed with the S. epidermidis CLIMB1 genome and the 100 most similar genomes from the PATRIC database (Figure 4). Most of the included genomes were annotated as S. epidermidis strains. 6 genomes were described only as Staphylococcus strains. CLIMB1 belonged to a compact clade of 26 S. epidermidis genomes, 9 of which represented all strains with the ST-16 MLST profile. The clade included a variety of clinical and environmental isolates. One strain, NIHLM049, was described as a commensal strain that clustered with nosocomial isolates in a pan-genome study of 28 genomes (Conlan et al., 2012). A group of 7 genomes (DE prefix) were from environmental samples collected from the Duke University campus (NCBI BioProject PRJNA543692). Another 6 genomes (SEPI) were ICU isolates from neonatal or critical care units at the University of Washington Medical Center (Roach et al., 2015). 4 genomes (APC) were from isolates from human milk obtained in Ireland (Angelopoulou et al., 2020).
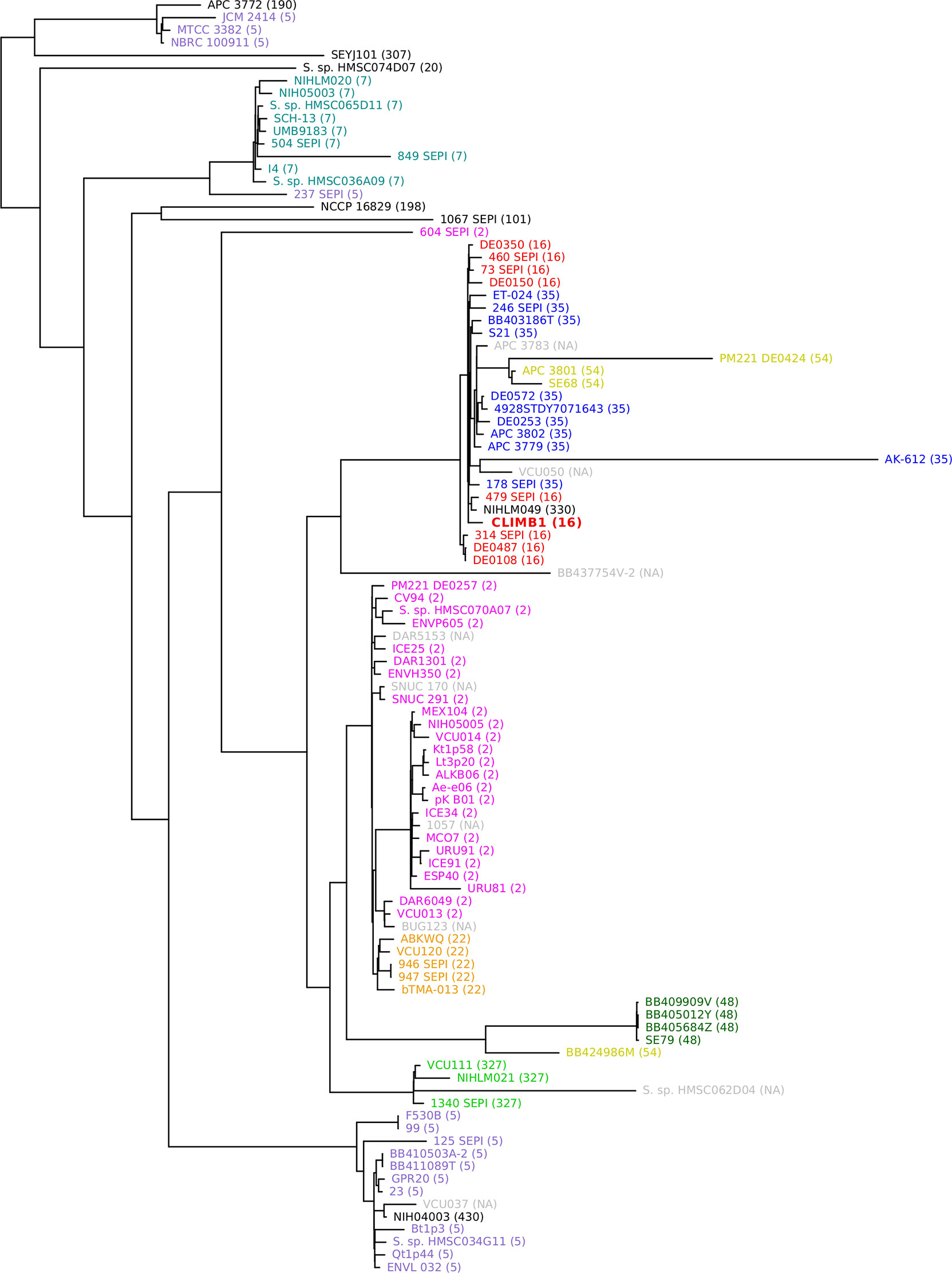
Figure 4 Phylogenetic tree of S. epidermidis, including CLIMB1 (red, center) and the most similar strains with known genomes, based on genomic sequence comparison. MLST profiles are indicated in parentheses and by color coding.
Discussion
This proof of concept study showed the promise of WGA when applied to CSF shunt infection samples. WGA results were consistent with conventional microbiological culture and 16S. Dominant bacterial species observed in this study have been previously reported in CSF shunt infections (Simon et al., 2014a; Simon et al., 2014b; Whitlock et al., 2021). S. epidermidis and S. aureus are common occurrences, hypothesized to originate from the normal skin flora. K. pneumoniae was previously found in 1 CSF shunt infection patient (Simon et al., 2014b).
Compared to 16S, WGA provided resolution at the species or strain level for both bacteria and viruses. Compared to bacterial culture, both 16S and WGA identified additional organisms of potential clinical relevance. In some cases, the species recovered by bacterial culture were identified in 16S and WGA at only low relative levels. WGA data allowed us to assemble the entire genomic sequence of an S. epidermidis strain from a CSF shunt sample. Finally, WGA identified the presence of TTV viral genomes in CSF shunt infection samples, which neither bacterial culture nor 16S could detect.
WGA holds the promise of detailed molecular characterization of genomic DNA, which implies precise strain identification, evaluation of putative antibiotic resistance genes, and characterization of metabolic capacity. For example, the S. epidermidis genome we assembled with WGA/SS data yielded information about molecular strain type (MLST), antimicrobial resistance gene content, and phylogenetic relationships to previously identified strains. These results were achieved without physical removal of the highly abundant human DNA from CSF shunt infection specimens before processing for sequencing. However, this analysis was only possible for a sample with one predominant bacterial species, which yielded a sufficient number of sequencing reads. Therefore, the utility of WGA may be optimal at the beginning of CSF shunt infection, before the bacterial infection is cleared by antibiotic treatment.
This work builds on an earlier report of the application of WGA to a cohort of hospitalized patients with infectious meningitis and encephalitis (Wilson et al., 2019). In contrast to the earlier study, in which a minority (13%) of participants had hardware present, all children in the current cohort had CSF shunts. In addition, our study cohort had CSF shunt infection documented by bacterial culture, while WGA both confirmed culture diagnosis and identified potential additional bacterial and/or viral organisms present.
Because reinfection occurs at high rates among patients with CSF shunt infection, detailed genomic characterization of microorganisms in CSF can help answer questions about the etiology of infection and reinfection. We previously explored the relationship between infection and reinfection using 16S (Whitlock et al., 2021). In that study we showed that 16S results were generally consistent with culture-based methods and that 16S may detect organisms missed by culture at the end of infection treatment but detected by culture at reinfection. However, the CSF microbiota identified by 16S only weakly correlated within patients at the end of infection and beginning of reinfection. For this study, we explored whether WGA might provide critical information about the microbiota present at the end of infection. Instead, our results indicate that this method only yields useful results when sufficient microbial DNA is present, which occurs only at the beginning rather than the end of infection.
We therefore suggest a more targeted approach to address the question of microorganism persistence from one infection to another, which relies on the high resolution information provided by WGA and the enhanced sensitivity and specificity of PCR. The approach would apply WGA to CSF samples at the beginning of a reinfection to obtain the genomic sequence of the principal infecting organism. The sequence information would be used to develop strain-specific PCR primers, which would be used to detect the strain in late samples of the previous infection. A positive result would provide strong evidence for the hypothesis that microorganisms present at low levels in CSF during treatment of an infection can lead to reinfection. Such an approach would be based on existing methodology. For example, methods for designing specific primers based on bacterial genomic sequence have been described in the literature (Kechin et al., 2020). Nested PCR is a common technique for increasing sensitivity and specificity of DNA sequence detection (Green and Sambrook, 2019).
We acknowledge several limitations for this proof of concept study. Extraction and sequencing of DNA from CSF samples is challenging due to low DNA abundance. In addition, analysis of microbial sequences is limited by the high proportion of human DNA, even during an active infection. As a result, the sensitivity of WGA is not better than that of 16S and worse than culture. Only a small fraction of the CSF samples we analyzed produced a sufficient number of bacterial reads to allow partial genomic assembly of predominant species. An expanded study would be needed to better understand the applicability of the approach. WGA may only be useful for CSF infections where one or a small number of microorganisms dominate. Alternately, as has been shown in aseptic meningitis (Wilson et al., 2019), WGA may indicate the presence of other microorganisms, such as viruses present in the setting of CSF shunt infection. An additional consideration when choosing WGA is cost. Processing by 16S is estimated at $80/sample, while WGA can cost twice as much or more, depending on the depth of sequencing. Other factors that contribute to an increased cost of WGA are a potentially longer turn-around time for sequencing (3-6 days compared to 3 days for 16S) and a more complex computational pipeline.
Despite these limitations, our findings demonstrate the promise of WGA for research of CSF shunt infection. WGA results: (1) were consistent with culture-based methods, (2) identified all bacteria detected in culture to the species level, while taxa were classified by 16S rRNA to only genus or family level, and (3) provided additional insights regarding viruses present and strain identity of predominant bacteria. Further work is needed to better understand the utility of WGA in the setting of CSF shunts and CSF shunt infection, including a comparison between the effectiveness of the WGA with other approaches to characterizing the microbiota such as strain-specific PCR primers.
Data Availability Statement
DNA sequence data presented in this study can be found online at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov) under BioProject PRJNA707514.
Ethics Statement
The study received initial Institutional Review Board approvals from the Seattle Children’s Research Institute on Febryary 8, 2011 and the University of Utah, as well as approval from the Primary Children’s Hospital Privacy Board on January 14, 2009. For all study subjects, except those from Primary Children’s Hospital prior to March 18, 2010, consent was obtained for additional CSF to be collected on each occasion that regular CSF samples were obtained during treatment for CSF shunt infection. Prior to March 18, 2010 at Primary Children’s Hospital, we used CSF remaining for after routine processing and testing in the Primary Children’s Hospital Microbiology Laboratory.
CLIMB Group Members
Current membership of the CLIMB group includes: Alexander Cheong, Gabriel Haller, Julie McGalliard, Diego Morales, Amanda Morgan, Alexander Rangel-Humphrey, and Lisa Wick. Past membership includes: Dan Berger, Whitney Bond, Haley Botteron, Courtney Dethlefs, Jessica Foster, Mohammed Gabir, Robert Johnson, Deanna Mercer, and Linda Shih.
Author Contributions
TS, PH, CP, KW, and LH conceived and designed the study. DL, PM, JH, JO, and TS provided oversight over study enrollment and material collection. CP performed the experiments. PH, CP, and KW analyzed the data. PH, TS, and CP wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by R01 NS095979. DL receives research funding and equipment for unrelated projects through Microbot Medical, Inc. and Medtronic, Inc. LH receives additional support from the National Institutes of Health via K24 HL141669.
Conflict of Interest
Author KW was employed by company New Harmony Statistical Consulting LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the children and families who participated in the study at SCH and PCH. We thank those who made CSF sample collection at SCH and PCH possible, including: Anne J. Blaschke and Jay Riva-Cambrin who led enrollment at PCH; the pediatric neurosurgeons and neurosurgical staff at SCH and PCH; Hydrocephalus Clinical Research Network coordinators Amy Anderson and Tracey Habrock-Bach who identified eligible children to our study staff; and study staff who enrolled patients and ensured appropriate collection and storage of CSF specimens including Marshal Werfelman at SCH and Chris Stockmann (deceased), Priscilla Cowan, Trenda Barney, and Abby Phillips at PCH.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.699506/full#supplementary-material
References
Angelopoulou, A., Warda, A. K., O'connor, P. M., Stockdale, S. R., Shkoporov, A. N., Field, D., et al. (2020). Diverse Bacteriocins Produced by Strains From the Human Milk Microbiota. Front. Microbiol. 11, 788. doi: 10.3389/fmicb.2020.00788
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A New Genome Assembly Algorithm and its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baron, E. J., Miller, J. M., Weinstein, M. P., Richter, S. S., Gilligan, P. H., Thomson, R. B., Jr., et al. (2013). A Guide to Utilization of the Microbiology Laboratory for Diagnosis of Infectious Diseases: 2013 Recommendations by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM)(a). Clin. Infect. Dis. 57, e22–e121. doi: 10.1093/cid/cit278
Belén, A., Pavón, I., Maiden, M. C. J. (2009). Multilocus Sequence Typing. Methods Mol. Biol. 551, 129–140. doi: 10.1007/978-1-60327-999-4_11
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P. (2016). DADA2: High-Resolution Sample Inference From Illumina Amplicon Data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Church, D. L., Cerutti, L., Gurtler, A., Griener, T., Zelazny, A., Emler, S. (2020). Performance and Application of 16S rRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 33, e00053–19. doi: 10.1128/CMR.00053-19
Conlan, S., Mijares, L. A., Program, N. C. S., Becker, J., Blakesley, R. W., Bouffard, G. G., et al. (2012). Staphylococcus Epidermidis Pan-Genome Sequence Analysis Reveals Diversity of Skin Commensal and Hospital Infection-Associated Isolates. Genome Biol. 13, R64. doi: 10.1186/gb-2012-13-7-r64
Deghorain, M., Bobay, L. M., Smeesters, P. R., Bousbata, S., Vermeersch, M., Perez-Morga, D., et al. (2012). Characterization of Novel Phages Isolated in Coagulase-Negative Staphylococci Reveals Evolutionary Relationships With Staphylococcus Aureus Phages. J. Bacteriol. 194, 5829–5839. doi: 10.1128/JB.01085-12
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A., Brisse, S. (2005). Multilocus Sequence Typing of Klebsiella Pneumoniae Nosocomial Isolates. J. Clin. Microbiol. 43, 4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005
Green, M. R., Sambrook, J. (2019). Nested Polymerase Chain Reaction (PCR). Cold Spring Harb. Protoc. 2019, 175–178. doi: 10.1101/pdb.prot095182
Hrabak, J., Bitar, I., Papagiannitsis, C. C. (2020). Combination of Mass Spectrometry and DNA Sequencing for Detection of Antibiotic Resistance in Diagnostic Laboratories. Folia Microbiol. (Praha) 65, 233–243. doi: 10.1007/s12223-019-00757-5
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.org Website and Their Applications. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Kechin, A., Borobova, V., Boyarskikh, U., Khrapov, E., Subbotin, S., Filipenko, M. (2020). NGS-PrimerPlex: High-Throughput Primer Design for Multiplex Polymerase Chain Reactions. PloS Comput. Biol. 16, e1008468. doi: 10.1371/journal.pcbi.1008468
Kestle, J. R. (2003). Pediatric Hydrocephalus: Current Management. Neurol. Clin. 21, 883–895, vii. doi: 10.1016/S0733-8619(03)00016-1
Kestle, J. R., Garton, H. J., Whitehead, W. E., Drake, J. M., Kulkarni, A. V., Cochrane, D. D., et al. (2006). Management of Shunt Infections: A Multicenter Pilot Study. J. Neurosurg. 105, 177–181. doi: 10.3171/ped.2006.105.3.177
Kulkarni, A. V., Rabin, D., Lamberti-Pasculli, M., Drake, J. M. (2001). Repeat Cerebrospinal Fluid Shunt Infection in Children. Pediatr. Neurosurg. 35, 66–71. doi: 10.1159/000050393
Lindgreen, S., Adair, K. L., Gardner, P. P. (2016). An Evaluation of the Accuracy and Speed of Metagenome Analysis Tools. Sci. Rep. 6, 19233. doi: 10.1038/srep19233
Manso, C. F., Bibby, D. F., Mohamed, H., Brown, D. W. G., Zuckerman, M., Mbisa, J. L. (2020). Enhanced Detection of DNA Viruses in the Cerebrospinal Fluid of Encephalitis Patients Using Metagenomic Next-Generation Sequencing. Front. Microbiol. 11, 1879. doi: 10.3389/fmicb.2020.01879
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mitchell, S. L., Simner, P. J. (2019). Next-Generation Sequencing in Clinical Microbiology: Are We There Yet? Clin. Lab. Med. 39, 405–418. doi: 10.1016/j.cll.2019.05.003
Nelson, M. T., Pope, C. E., Marsh, R. L., Wolter, D. J., Weiss, E. J., Hager, K. R., et al. (2019). Human and Extracellular DNA Depletion for Metagenomic Analysis of Complex Clinical Infection Samples Yields Optimized Viable Microbiome Profiles. Cell Rep. 26, 2227–2240.e2225. doi: 10.1016/j.celrep.2019.01.091
Pittman, M. E., Thomas, B. S., Wallace, M. A., Weber, C. J., Burnham, C. A. (2014). Routine Testing for Anaerobic Bacteria in Cerebrospinal Fluid Cultures Improves Recovery of Clinically Significant Pathogens. J. Clin. Microbiol. 52, 1824–1829. doi: 10.1128/JCM.00193-14
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: A Comprehensive Online Resource for Quality Checked and Aligned Ribosomal RNA Sequence Data Compatible With ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Roach, D. J., Burton, J. N., Lee, C., Stackhouse, B., Butler-Wu, S. M., Cookson, B. T., et al. (2015). A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PloS Genet. 11, e1005413. doi: 10.1371/journal.pgen.1005413
Simner, P. J., Miller, H. B., Breitwieser, F. P., Pinilla Monsalve, G., Pardo, C. A., Salzberg, S. L., et al. (2018). Development and Optimization of Metagenomic Next-Generation Sequencing Methods for Cerebrospinal Fluid Diagnostics. J. Clin. Microbiol. 56, e00472–18. doi: 10.1128/JCM.00472-18
Simon, T. D., Hall, M., Dean, J. M., Kestle, J. R., Riva-Cambrin, J. (2010). Reinfection Following Initial Cerebrospinal Fluid Shunt Infection. J. Neurosurg. Pediatr. 6, 277–285. doi: 10.3171/2010.5.PEDS09457
Simon, T. D., Hall, M., Riva-Cambrin, J., Albert, J. E., Jeffries, H. E., Lafleur, B., et al. (2009). Infection Rates Following Initial Cerebrospinal Fluid Shunt Placement Across Pediatric Hospitals in the United States. Clinical Article. J. Neurosurg. Pediatr. 4, 156–165. doi: 10.3171/2009.3.PEDS08215
Simon, T. D., Pope, C. E., Browd, S. R., Ojemann, J. G., Riva-Cambrin, J., Mayer-Hamblett, N., et al. (2014a). Evaluation of Microbial Bacterial and Fungal Diversity in Cerebrospinal Fluid Shunt Infection. PloS One 9, e83229. doi: 10.1371/journal.pone.0083229
Simon, T. D., Riva-Cambrin, J., Srivastava, R., Bratton, S. L., Dean, J. M., Kestle, J. R. (2008). Hospital Care for Children With Hydrocephalus in the United States: Utilization, Charges, Comorbidities, and Deaths. J. Neurosurg. Pediatr. 1, 131–137. doi: 10.3171/PED/2008/1/2/131
Simon, T. D., Van Yserloo, B., Nelson, K., Gillespie, D., Jensen, R., Mcallister, J. P., 2nd, et al. (2014b). Use of Quantitative 16S rRNA PCR to Determine Bacterial Load Does Not Augment Conventional Cerebrospinal Fluid (CSF) Cultures Among Children Undergoing Treatment for CSF Shunt Infection. Diagn. Microbiol. Infect. Dis. 78, 188–195. doi: 10.1016/j.diagmicrobio.2013.06.027
Simon, T. D., Whitlock, K. B., Riva-Cambrin, J., Kestle, J. R., Rosenfeld, M., Dean, J. M., et al. (2012). Revision Surgeries are Associated With Significant Increased Risk of Subsequent Cerebrospinal Fluid Shunt Infection. Pediatr. Infect. Dis. J. 31, 551–556. doi: 10.1097/INF.0b013e31824da5bd
Thomas, J. C., Vargas, M. R., Miragaia, M., Peacock, S. J., Archer, G. L., Enright, M. C. (2007). Improved Multilocus Sequence Typing Scheme for Staphylococcus Epidermidis. J. Clin. Microbiol. 45, 616–619. doi: 10.1128/JCM.01934-06
Tuan, T. J., Thorell, E. A., Hamblett, N. M., Kestle, J. R., Rosenfeld, M., Simon, T. D. (2011). Treatment and Microbiology of Repeated Cerebrospinal Fluid Shunt Infections in Children. Pediatr. Infect. Dis. J. 30, 731–735. doi: 10.1097/INF.0b013e318218ac0e
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the All-Bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Whitlock, K. B., Pope, C. E., Hodor, P., Hoffman, L. R., Limbrick, D. L., Jr., Mcdonald, P. J., et al. (2021). Characterization of Cerebrospinal Fluid (CSF) Microbiota From Patients With CSF Shunt Infection and Reinfection Using High Throughput Sequencing of 16S Ribosomal RNAgenes. PloS One 16, e0244643. doi: 10.1371/journal.pone.0244643
Williams, M. A., Mcallister, J. P., Walker, M. L., Kranz, D. A., Bergsneider, M., Del Bigio, M. R., et al. (2007). Priorities for Hydrocephalus Research: Report From a National Institutes of Health-Sponsored Workshop. J. Neurosurg. 5 Suppl Pediatrics, 345–357. doi: 10.3171/PED-07/11/345
Wilson, M. R., Sample, H. A., Zorn, K. C., Arevalo, S., Yu, G., Neuhaus, J., et al. (2019). Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N Engl. J. Med. 380, 2327–2340. doi: 10.1056/NEJMoa1803396
Keywords: cerebrospinal fluid, CSF shunt infection, microbiota, Staphylococcus epidermidis CLIMB1, high throughput DNA sequencing
Citation: Hodor P, Pope CE, Whitlock KB, Hoffman LR, Limbrick DL, McDonald PJ, Hauptman JS, Ojemann JG and Simon TD (2021) Molecular Characterization of Microbiota in Cerebrospinal Fluid From Patients With CSF Shunt Infections Using Whole Genome Amplification Followed by Shotgun Sequencing. Front. Cell. Infect. Microbiol. 11:699506. doi: 10.3389/fcimb.2021.699506
Received: 03 May 2021; Accepted: 29 July 2021;
Published: 20 August 2021.
Edited by:
Costas C. Papagiannitsis, University of Thessaly, GreeceReviewed by:
Jaroslav Hrabak, Charles University, CzechiaAlberto Antonelli, University of Florence, Italy
Efthymia Petinaki, University of Thessaly, Greece
Copyright © 2021 Hodor, Pope, Whitlock, Hoffman, Limbrick, McDonald, Hauptman, Ojemann and Simon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Hodor, cGF1bC5ob2RvckBzZWF0dGxlY2hpbGRyZW5zLm9yZw==