 Yizhuo Zhang
Yizhuo Zhang Luqin Lv
Luqin Lv Su Xu
Su Xu Yijian Chen
Yijian Chen- Institute of Antibiotics, Huashan Hospital, Fudan University & Key Laboratory of Clinical Pharmacology of Antibiotics, National Health Commission, Shanghai, China
Introduction: Clostridioides difficile (C. difficile), a human pathogen that causes diarrhea and colon lesions, has garnered widespread attention. Rapid and accurate detection of bacterial virulence factors is essential for the diagnosis of C. difficile infection (CDI). To date, numerous laboratory tests have been developed; however, none fully meet the combined requirements of speed, cost-effectiveness, portability, sensitivity, and specificity. Molecular diagnostic technologies based on CRISPR-Cas systems have provided a promising solution to this challenge. Nonetheless, the limited compatibility between pre-amplification and CRISPR cleavage, coupled with the inherent selectivity of CRISPR systems for protospacer adjacent motif (PAM) sequences near the target site, poses additional constraints on the broader adoption of this approach.
Methods: Here, we developed PAM-unconventional, One-step LAMP/CRISPR-Cas12b (POLC) detection platforms for the toxin-encoding genes tcdA and tcdB of C. difficile.
Results: The POLC platforms operated at 60 °C, enabling result interpretation either through fluorescence intensity measurements or direct visualization under UV light. The limits of detection (LoDs) ranged from 3 to 14 copies/μL using a fluorescence reader and from 6 to 18 copies/μL via direct observation. Compared to qPCR, which typically requires over an hour, the POLC platforms reduced the detection time to approximately 40 minutes. Each reaction cost approximately USD 6.5, offering a substantial cost saving compared to qPCR-based commercial kits (over USD 10 per test). In clinical validation with 55 fecal samples, the tcdA POLC assay achieved 86.4% sensitivity and 84.8% specificity, while the tcdB POLC assay demonstrated 96.6% sensitivity and 100% specificity, using qPCR as the reference standard.
Discussion: Our research presents innovative CRISPR-based one-step nucleic acid detection platforms that eliminate canonical PAM sequence requirements. These platforms exhibit high sensitivity and specificity while achieving rapid detection under simple conditions, making them promising candidates for clinical diagnostics and point-of-care testing (POCT).
1 Introduction
Clostridioides difficile (C. difficile) is a Gram-positive, anaerobic bacterium responsible for both nosocomial and community-associated gastrointestinal infections (Smits et al., 2016; Guh and Kutty, 2018). C. difficile infection (CDI) typically presents with diarrhea and may progress to severe complications, such as ileus, pseudomembranous colitis, and colon necrosis (Smits et al., 2016; McDonald et al., 2018). In 2019, the Centers for Disease Control and Prevention classified CDI as one of the most serious “urgent threats” to public health (Centers for Disease Control and Prevention, 2019). It was estimated that CDI caused approximately half a million cases and up to 29,300 deaths annually in the United States (Lessa et al., 2015). Rapid and accurate diagnosis of CDI is essential to ensure timely and effective patient management.
Laboratory testing remains important for the diagnosis of CDI (Gateau et al., 2018). Current confirmatory methods include toxigenic culture (TC), C. difficile cytotoxin neutralization assay (CCNA), enzyme immunoassay (EIA) for glutamate dehydrogenase (GDH) or toxins, and nucleic acid amplification tests (NAATs) (O’Horo et al., 2012; Arimoto et al., 2016; Crobach et al., 2016, 2018; Kraft et al., 2019). TC and CCNA are time-consuming and labor-intensive (Crobach et al., 2016). GDH EIAs only serve as screening tools without diagnostic capacity (Arimoto et al., 2016), and toxin EIAs suffer from limited sensitivity in detecting toxins A (TcdA) and B (TcdB) (Crobach et al., 2016). The advent of NAATs has significantly advanced CDI diagnostics, offering shorter turnaround times, simplified procedures, and improved sensitivity and specificity (Barbut et al., 2011; Deshpande et al., 2011; Norén et al., 2011; O’Horo et al., 2012; Surawicz et al., 2013; Crobach et al., 2016). Currently, polymerase chain reaction (PCR) and loop-mediated isothermal amplification (LAMP) are widely adopted (McDonald et al., 2018). Although PCR-based methods, such as the Cepheid Xpert C. difficile assay, are highly accurate and routinely employed in clinical laboratories, their dependence on complex instrumentation limits the suitability for point-of-care settings (Mahony et al., 2009). LAMP is an isothermal amplification technique that functions at a constant temperature using basic equipment such as a water or metal bath. However, its specificity is compromised by non-specific amplification (Wang et al., 2015). These limitations highlight the urgent need for novel methods that combine sensitivity, specificity, and operational simplicity for CDI diagnosis.
CRISPR-Cas (clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins) systems have revolutionized modern biology, initially through gene editing due to their efficiency, precision, and simplicity (Cong et al., 2013; Mali et al., 2013). The discovery of trans-cleavage activity in Cas12 (Chen et al., 2018), Cas13 (East-Seletsky et al., 2016), and Cas14a (Harrington et al., 2018) has expanded their application to molecular diagnostics. Upon formation of a ternary complex (Cas protein–crRNA/sgRNA–target), Cas proteins mediate both site-specific (cis-) cleavage of the target and nonspecific collateral (trans-) cleavage of surrounding single-stranded nucleic acids (Li et al., 2018a). In diagnostic assays, cleavage of fluorescent probes releases a fluorophore, generating a detectable signal indicative of target presence (Kellner et al., 2019). Coupling CRISPR-Cas systems with nucleic acid amplification significantly enhances detection sensitivity, as demonstrated by platforms such as SHERLOCK (specific high-sensitivity enzymatic reporter unlocking; RPA-Cas13a) (Gootenberg et al., 2017), HOLMES (one-hour low-cost multipurpose highly efficient system; PCR/RT-PCR-Cas12a) (Li et al., 2018a, b), and DETECTR (DNA endonuclease-targeted CRISPR trans-reporter; RPA-Cas12a) (Chen et al., 2018), each achieving attomolar sensitivity. Moreover, the requirements for protospacer adjacent motif (PAM) recognition and precise base pairing enable CRISPR-based assays to attain single-base resolution (Chen et al., 2025). However, incompatibility between amplification and CRISPR detection remains challenging, primarily due to mismatched reaction temperatures and premature cleavage of amplification products (Lu et al., 2022). Consequently, amplification and detection are typically performed sequentially, complicating the workflow and increasing the risk of aerosol contamination. To address this, platforms such as HOLMESv2 (Li et al., 2019), STOPCovid (SHERLOCK Testing in One Pot) (Joung et al., 2020), CRISPR-SPADE (CRISPR Single Pot Assay for Detecting Emerging Variants of Concern) (Nguyen et al., 2022), and SPLENDID (Single-Pot LAMP-mediated Engineered BrCas12b for Nucleic Acid Detection of Infectious Diseases) (Nguyen et al., 2023) have leveraged thermostable Cas12b variants to enable single-tube detection. Beyond viral targets, LAMP-CRISPR platforms have also been applied to the detection of bacteria such as Mycobacterium tuberculosis (Sam et al., 2021), Klebsiella pneumoniae (Qiu et al., 2022), Pseudomonas aeruginosa (Qiu et al., 2023), and Salmonella Typhimurium (Gong et al., 2024). Notably, recent studies have demonstrated that employing non-canonical PAMs can further enhance the performance of one-pot CRISPR assays by reducing cis-cleavage activity and minimizing amplification product consumption (Lu et al., 2022). A suboptimal PAM-based Cas12a detection method (sPAMC) achieved 94.2% sensitivity and 100% specificity for SARS-CoV-2 detection within 20 minutes (Lu et al., 2022). Additionally, a one-step RPA-CRISPR detection (ORCD) platform bypassing classical PAM constraints enabled the detection of as few as 0.2 copies/μL of DNA and 0.4 copies/μL of RNA (Lin et al., 2023).
In this study, we developed the POLC (PAM-unconventional, One-step LAMP/CRISPR-Cas12b) detection platform, which integrates LAMP with a CRISPR-Cas12b system. Unlike traditional CRISPR-based assays, POLC functions independently of canonical PAM sequences, enhancing target flexibility and simultaneously promoting robust fluorescence signal generation (Figure 1). Based on this platform, we established two separate one-step, single-tube assays for the detection of tcdA and tcdB, respectively. Each assay requires only the addition of sample DNA to a premixed reagent, with results obtainable using a fluorescence plate reader or through direct visualization under UV light, demonstrating applicability in both laboratory and point-of-care settings (Figure 2).
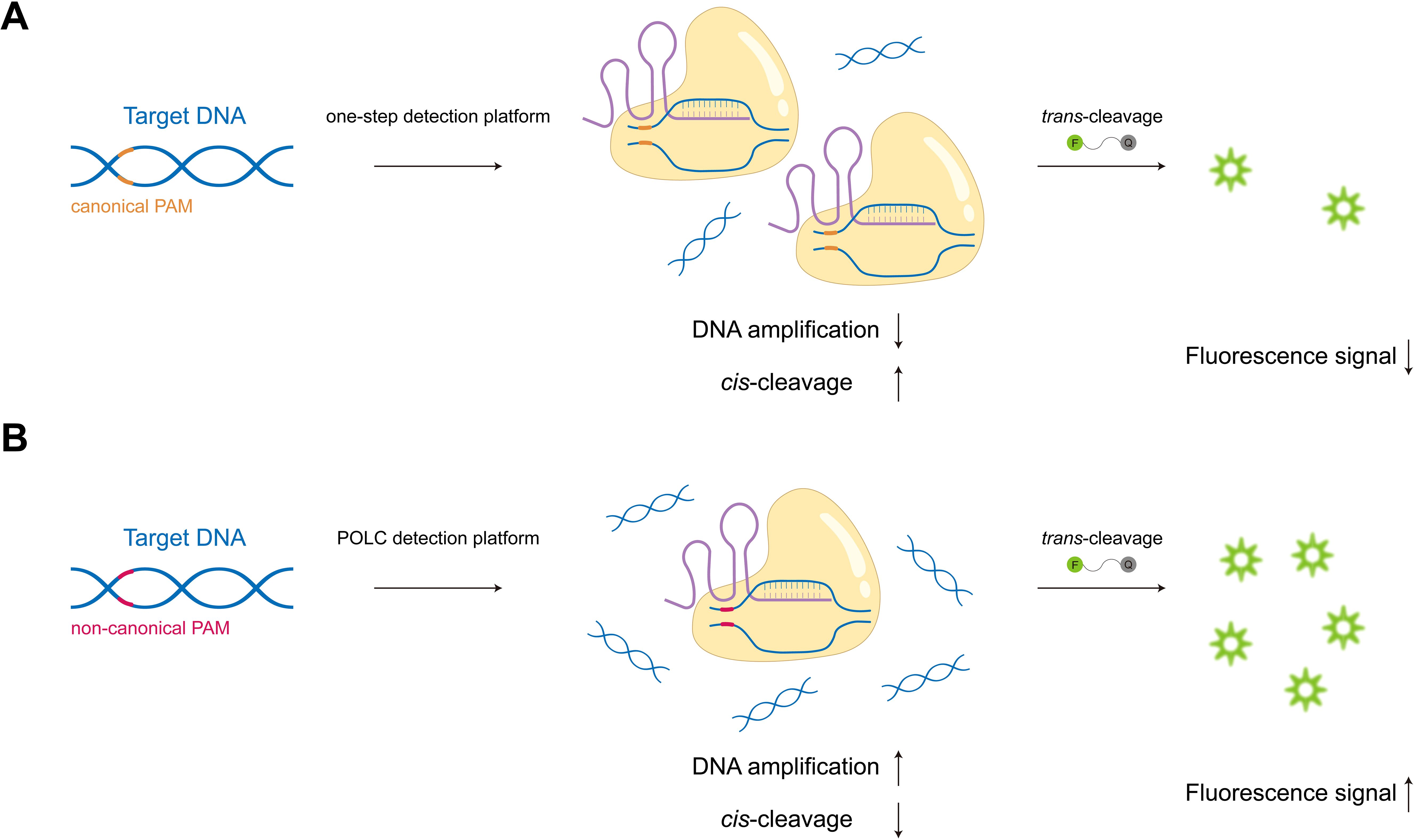
Figure 1. Schematic illustration of one-step CRISPR-based detection platforms under canonical and non-canonical PAM conditions. (A) When recognizing canonical PAMs, Cas effectors exhibit high cis-cleavage activity towards target nucleic acids, impairing target enrichment and resulting in weak fluorescence signals. (B) In the POLC platforms, the use of non-canonical PAMs weakens Cas binding, allowing amplification to dominate the early phase of the reaction. This leads to effective target accumulation and enhances fluorescence signal production.
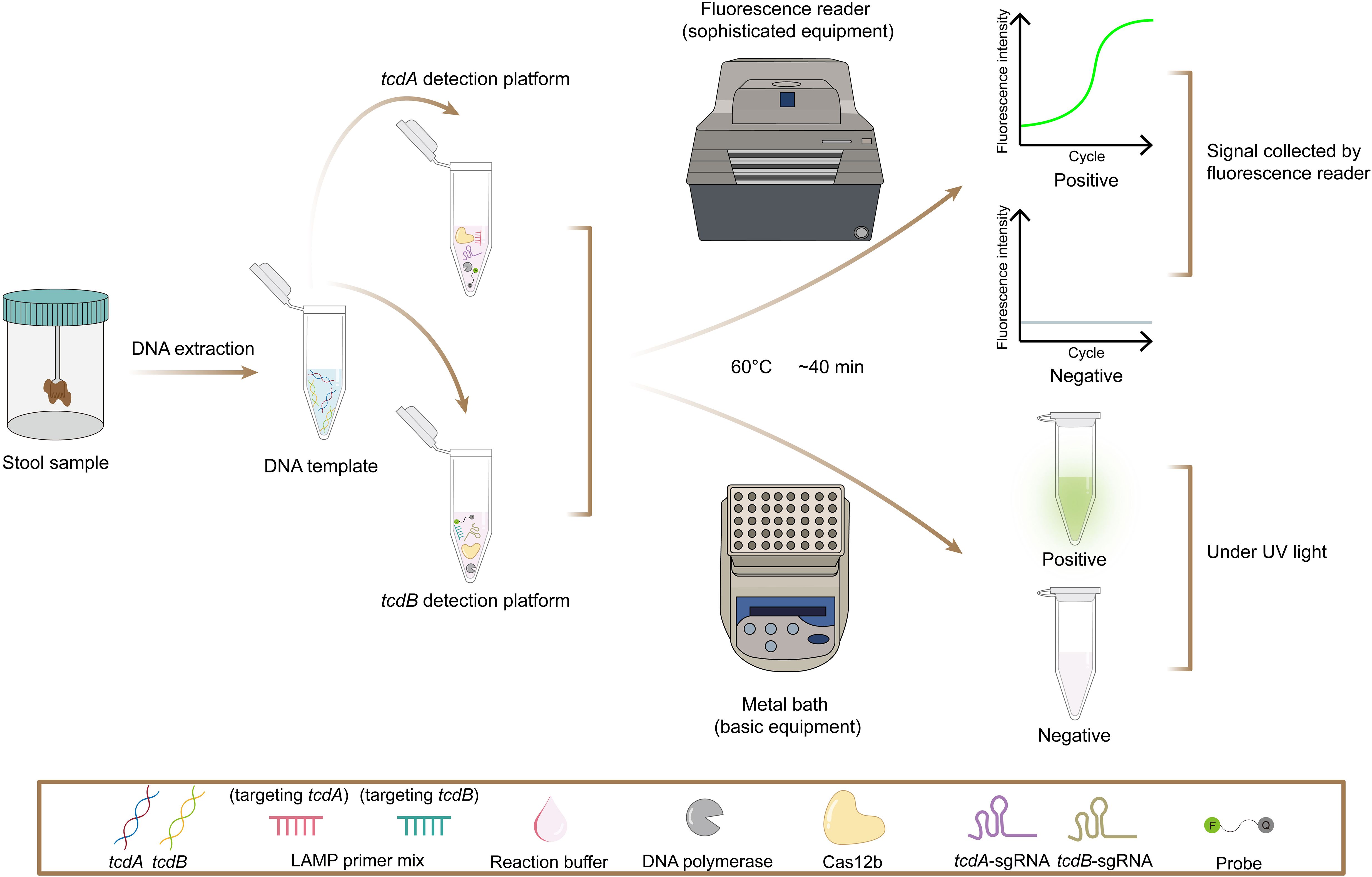
Figure 2. Workflow of the POLC detection platforms targeting tcdA and tcdB.
2 Materials and methods
2.1 Reagents and instrumentation
The 10× Isothermal Amplification Buffer, 100 mM MgSO4 Solution, and Bst 2.0 Warmstart® DNA Polymerase, were purchased from New England Biolabs Inc. (MA, USA). The dNTP mix (10 mM) was obtained from Hongene Biotech Corp. (Shanghai, China). All nucleotides and additives, including glycine, formamide, and tetramethyl ammonium chloride (TMAC), were sourced from Sangon Biotech Co., Ltd. (Shanghai, China), while betaine and dimethyl sulfoxide (DMSO) were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China) and Sigma-Aldrich Co. (MO, USA), respectively. The 10× EasyTaq® Buffer was provided by Transgen Biotech. Co., Ltd. (Beijing, China). Salmon sperm DNA solution, SYTO™ 9 dye, and nuclease-free water were purchased from Thermo Fisher Scientific Inc. (MA, USA). Alicyclobacillus acidiphilus Cas12b (AapCas12b) and the Cas12b High Yield sgRNA Synthesis and Purification Kit were obtained from TOLO Biotech. Co., Ltd. (Shanghai, China). The 10× SYBR Green DigitalAmp® PCR Kit was purchased from Zhenzhun Bio-Tech. Co., Ltd. (Shanghai, China). The QIAamp® Fast DNA Stool Mini Kit was obtained from Qiagen Co., Ltd. (Shanghai, China), and the C. difficile toxin A/B gene detection kit was sourced from Hongweitest Biotechnology Co., Ltd. (Jiangsu, China).
Instruments for in vitro transcription included a GE series thermal cycler (Bio-Gener Technology Co., Ltd, Hangzhou, China) for annealing and incubation, and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., MA, USA) for sgRNA quantification. The AccuMini series digital PCR system was purchased from Zhenzhun Bio-Tech. Co., Ltd. (Shanghai, China). LAMP and POLC assays were performed on the LineGene 9600 Plus (Bioer Technology Co., Ltd, Hangzhou, China). The qPCR for clinical validation was conducted on the QuantStudio™ 5 Real-Time PCR Instrument (Applied Biosystems Inc., CA, USA).
2.2 LAMP primer design and template preparation
LAMP primers targeting the reference sequences of tcdA (Gene ID: 66353160) and tcdB (Gene ID: 66353157) were designed using the NEB® LAMP Primer Design Tool (https://lamp.neb.com/) and PrimerExplorer version 5 software (http://primerexplorer.jp/lampv5e/index.html). To check sequence conservation within the primer design regions, ClustalW multiple sequence alignment (Bioedit Sequence Alignment Editor software, v.7.2.5) was performed on tcdA and tcdB sequences retrieved from the National Center for Biotechnology Information (NCBI) nucleotide database. The analysis revealed that the targeted regions of tcdA were highly conserved, whereas those of tcdB exhibited relatively lower conservation. BLAST (Basic Local Alignment Search Tool) analysis further confirmed that the amplification regions and adjacent zones of the tcdA and tcdB reference sequences shared high homology with C. difficile 630, designated as tcdA and tcdB1, respectively. Two additional tcdB variants homologous to ribotype 027 (RT027) and 078 (RT078) strains were identified and named tcdB2 and tcdB3, respectively. The sequences of tcdA, tcdB1, tcdB2, and tcdB3 were synthesized and cloned into pUC57 vectors to generate four types of standard plasmids, which served as template DNA. Detailed information on the template sequences was shown in Supplementary Figure 1.
To minimize false negatives, primer positions were slightly adjusted to target conserved regions, and degenerate bases were introduced at mutation sites. Three primer sets were designed for each tcdB plasmid (tcdB1-P1 to P3, tcdB2-P1 to P3, tcdB3-P1 to P3). Primer sets tcdB1-P3, tcdB2-P3, and tcdB3-P3 were generated by modifying the F2 region of the forward inner primer (FIP) from tcdB1-P2, tcdB2-P2, and tcdB3-P2, respectively, to further improve sequence conservation. For tcdA, three primer sets (tcdA-P1 to P3) were designed. Primer specificity was verified by BLAST analysis, and sequences were provided in Supplementary Tables 1, 2.
2.3 Accurate quantification of plasmid templates
The copy number of the serially diluted plasmids used in subsequent experiments was quantified by 10× SYBR Green DigitalAmp® PCR Kit according to the operation instructions. Each 20 μL reaction contained 2 μL of 10× SYBR Green PCR Master Mix, 1 μL of PCR primer mix, 2 μL of standard plasmid, and 15 μL of nuclease-free water. The thermal cycling procedure was as follows: 95 °C for 10 minutes, followed by 45 cycles of 95 °C for 30 seconds and 58 °C for 45 seconds. After sample loading, amplification, and chip reading, data were finally analyzed by digital PCR analysis software. The primers used in this reaction were listed in Supplementary Table 3.
2.4 LAMP primer and additive screening
To screen for the most efficient LAMP primers, tcdA-P1 to P3 were evaluated for the amplification of tcdA-plasmid. Similarly, tcdB1-P1 to P3, tcdB2-P1 to P3, and tcdB3-P1 to P3 were assessed using their corresponding tcdB1-, tcdB2-, and tcdB3-plasmid. Primer sets demonstrating superior amplification efficiency for each tcdB variant were subsequently subjected to cross-validation by amplifying all three tcdB plasmids. Sets achieving consistently high and stable amplification across all templates were selected for further development. LAMP reactions were performed following the standard protocol provided by NEB® (https://www.neb.cn/zh-cn/protocols/2014/12/29/typical-lamp-protocol-m0538) in a final volume of 25 μL. Each reaction included 5 μL of plasmid diluted in salmon sperm DNA solution (to reduce loss during gradient dilution) as the positive control, or 5 μL of salmon sperm DNA alone as the negative control. SYTO™ 9 (1 μM) was added as the fluorescent dye, and real-time amplification was monitored on the LineGene 9600 Plus via the FAM channel (470–525 nm).
To improve nucleic acid amplification performance, several additives previously recognized as PCR enhancers were tested at various final concentrations: betaine (0/200/400/600 mM), DMSO (0/2/4/6%), formamide (0/1/2/3%), TMAC (0/10/20/30 mM), and glycine (0/240/360/480 mM). Betaine and DMSO function by reducing secondary structure formation in GC-enriched regions, thereby preventing DNA polymerase dissociation from the template (Henke et al., 1997; Choi et al., 1999; Jensen et al., 2010). Formamide and TMAC help minimize non-specific priming events (Sarkar et al., 1990; Jang and Kim, 2022). Notably, recent studies have shown that glycine can enhance the sensitivity of nucleic acid amplification and promote one-pot CRISPR-based detection (Joung et al., 2020; Ooi et al., 2021). Therefore, it was also included for evaluation and comparison with the other additives. All LAMP reactions were conducted at 65°C, with fluorescence signals collected once per minute over 25 cycles.
2.5 POLC detection platforms
After the establishment of LAMP systems, 20 bp conserved regions within the amplification loci of tcdA and tcdB were selected as target sequences for sgRNA design. To develop the POLC detection platforms, non-canonical protospacer adjacent motifs (PAMs) were employed to attenuate the cis-cleavage activity of Cas12b. Transcription templates were prepared in 20 μL reaction mixtures containing 1× EasyTaq® Buffer, 2 μM T7 promoter (5’-TAATACGACTCACTATAGG-3’), 2 μM target antisense polynucleotide, and nuclease-free water. Annealing was performed on a thermal cycler (95°C for 5 minutes, cooling to 25°C at 0.1°C/s, followed by incubation at 25°C). In vitro transcription and sgRNA purification were carried out using the Cas12b High Yield sgRNA Synthesis and Purification Kit. The synthesized sgRNAs were quantified by NanoDrop and diluted to the working concentration of 10 μM. All sgRNA sequences used in this study were listed in Supplementary Table 3.
The POLC detection platforms integrated LAMP with non-canonical CRISPR-Cas12b systems. On the basis of the LAMP components described above, sgRNAs and AapCas12b were added to the mixture, while SYTO™ 9 was replaced with a fluorescence-quenched single-stranded DNA (ssDNA) probe, designated 8C-FQ (Supplementary Table 3). The initial concentrations of sgRNAs, AapCas12b, and 8C-FQ were set at 500 nM. Template volume was adjusted to 1 μL (quantified plasmids for the positive groups, and salmon sperm DNA for the negative control). The preliminary reaction temperature was maintained at 60°C. The screening of sgRNA was conducted under varying Mg2+ concentrations (8–12 mM in 1 mM increments) and dNTP concentrations (0.8-1.4 mM in 0.2 mM increments), which were systematically cross-tested.
To enhance detection efficacy, key reaction parameters were optimized, including the concentrations of AapCas12b, sgRNAs, fluorescent probes, and glycine, as well as the reaction temperature. AapCas12b was examined at concentrations of 125, 250, 500, and 750 nM, with sgRNA concentrations adjusted to achieve molar ratios of 1:1, 1:1.5, and 1:2 (AapCas12b:sgRNA). Probe concentrations were evaluated at 250, 500, 750 nM, and 1 μM. Glycine was tested at concentrations of 0, 240, 360, 480, 600, and 720 mM. Although LAMP typically operates at 60-65°C, AapCas12b exhibits reduced enzymatic activity above 60°C (Notomi et al., 2000; Joung et al., 2020). Therefore, temperature optimization was conducted near 60°C. For the tcdA platform, reaction temperatures of 57, 58.2, 59, 60, and 61 °C were screened. For the tcdB platform, the tested temperatures were 58, 59.2, 60, 61, and 62°C. The LineGene 9600 Plus instrument imposes a fixed temperature gradient, resulting in non-uniform intervals.
2.6 Sensitivity and specificity of the POLC detection platforms
Plasmids quantified by digital PCR were serially diluted and used as template DNA to assess the limit of detection (LoD) of the POLC platforms targeting tcdA and tcdB, respectively. Each dilution level was tested in eight replicates, with salmon sperm DNA serving as the negative control. To evaluate the portability and visual detection performance of the POLC platforms, the prepared reaction mixtures were incubated in a metal bath at 60°C for approximately 40 minutes, followed by exposure to UV light. Results were interpreted based on color changes visible to the naked eye, and the LoD for visual detection was determined.
The specificity of the POLC detection platforms was validated using nucleic acids extracted from several common opportunistic pathogens in the human intestinal tract, including C. difficile, Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, Staphylococcus aureus, Enterococcus faecalis, Proteus mirabilis, and Bacteroides fragilis. Strain details were provided in Supplementary Table 4. All nucleic acids were extracted by the conventional boiling method. Each pathogen was tested in triplicate, with nuclease-free water serving as the negative control.
2.7 Clinical validation
To verify the practicability of the POLC detection platforms, stool samples were collected from patients with suspected CDI at Huashan Hospital, Fudan University—a teaching hospital with multiple campuses in Shanghai, China. The enrolled patients met all of the following criteria: (1) age ≥18 years; (2) receipt of antibiotic therapy or chemotherapy within the past 60 days; (3) diarrhea occurring three or more times per day; and (4) stool classified as Bristol types 5-7. Exclusion criteria included: (1) chronic diarrhea; (2) use of laxatives within three days prior to symptom onset; and (3) diarrhea with a known cause, such as lactose intolerance. The study was approved by the Institutional Review Board of Huashan Hospital (20230737). All samples were collected after obtaining informed consent and were stored at -80°C.
Fecal DNA was extracted using the QIAamp® Fast DNA Stool Mini Kit according to the manufacturer’s instructions, and was used as the template for both the POLC detection platforms and qPCR. The qPCR was conducted on the QuantStudio™ 5 Real-Time PCR system using a C. difficile toxin A/B gene detection kit, which served as the reference method. Each qPCR reaction had a total volume of 32 μL, comprising 25 μL of nucleic acid reaction buffer, 2 μL of internal control, and 5 μL of sample DNA (or Buffer ATE for the negative control). The thermal cycling protocol was as follows: 50°C for 2 minutes, 95°C for 5 minutes, followed by 40 cycles of 95°C for 15 seconds and 55°C for 30 seconds.
2.8 Data visualization and statistical analysis
Data visualization and statistical analysis were performed by GraphPad Prism 10 (Graphpad Software Inc., CA, USA), OriginPro 2021 (OriginLab Corp., MA, USA), Adobe Illustrator 2024 (Adobe Systems Inc., CA, USA), and SPSS 29.0 (IBM Corp., NY, USA). McNemar’s two-sided test was used to evaluate statistical differences between the results obtained from the POLC detection platforms and qPCR. A P value < 0.05 was considered statistically significant. Consistency between these two methods was assessed by the Kappa statistic, interpreted according to the following scale: 0.00-0.20 (slight), 0.21-0.40 (fair), 0.41-0.60 (moderate), 0.61-0.80 (substantial), and 0.81-1.00 (almost perfect) (Landis and Koch, 1977).
3 Results
3.1 Design of the LAMP systems
To develop a highly sensitive detection platform based on CRISPR-Cas systems, it was essential to integrate nucleic acid amplification methods. Among these, LAMP is a well-established technique capable of rapidly generating large amounts of target DNA through self-cycling mechanisms (Notomi et al., 2000). We conducted the reactions using specifically designed primers targeting the tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid. The quantitative results for each plasmid type obtained from digital PCR were shown in Supplementary Figure 2. The optimal LAMP primers were selected based on time-to-detection, fluorescence intensity, and signal reproducibility. In this context, time-to-detection refers to the point at which the fluorescence signal exceeds the baseline threshold, marking the initiation of exponential amplification. As shown in Figure 3A, the system utilizing the primer tcdA-P2 could detect the tcdA-plasmid within 15 cycles (18 minutes) and generate the highest fluorescence intensity during the detection process. As shown in Figure 3B, the primers tcdB1-P3, tcdB2-P1, tcdB2-P3, and tcdB3-P3, which targeted highly conserved regions of the template sequences, exhibited a shorter time-to-detection and higher fluorescence intensity during the amplification of their corresponding plasmids. To minimize false negatives resulting from incomplete primer-template matching, the aforementioned efficient primers (i.e. tcdB1-P3, tcdB2-P1, tcdB2-P3, and tcdB3-P3) were used to amplify all three tcdB plasmids for versatility testing. As shown in Figure 3C, the primer tcdB1-P3 exhibited low efficiency in amplifying the tcdB2-plasmid, producing only a weak fluorescence signal at 25 cycles. Likewise, the primer tcdB2-P1 significantly delayed time-to-detection when used for tcdB1- and tcdB3-plasmid amplification. Both systems employing the primers tcdB2-P3 and tcdB3-P3 successfully detected the tcdB1-, tcdB2-, and tcdB3-plasmid within 15 cycles (18 minutes). However, amplification of the tcdB1-plasmid with the primer tcdB2-P3 showed poor reproducibility, whereas the primer tcdB3-P3 exhibited high stability across all three plasmids. Finally, the primers tcdA-P2 (Figure 3A) and tcdB3-P3 (Figure 3C) were chosen to develop POLC detection platforms targeting tcdA and tcdB, respectively. The location of selected primers was displayed in Supplementary Figure 1.
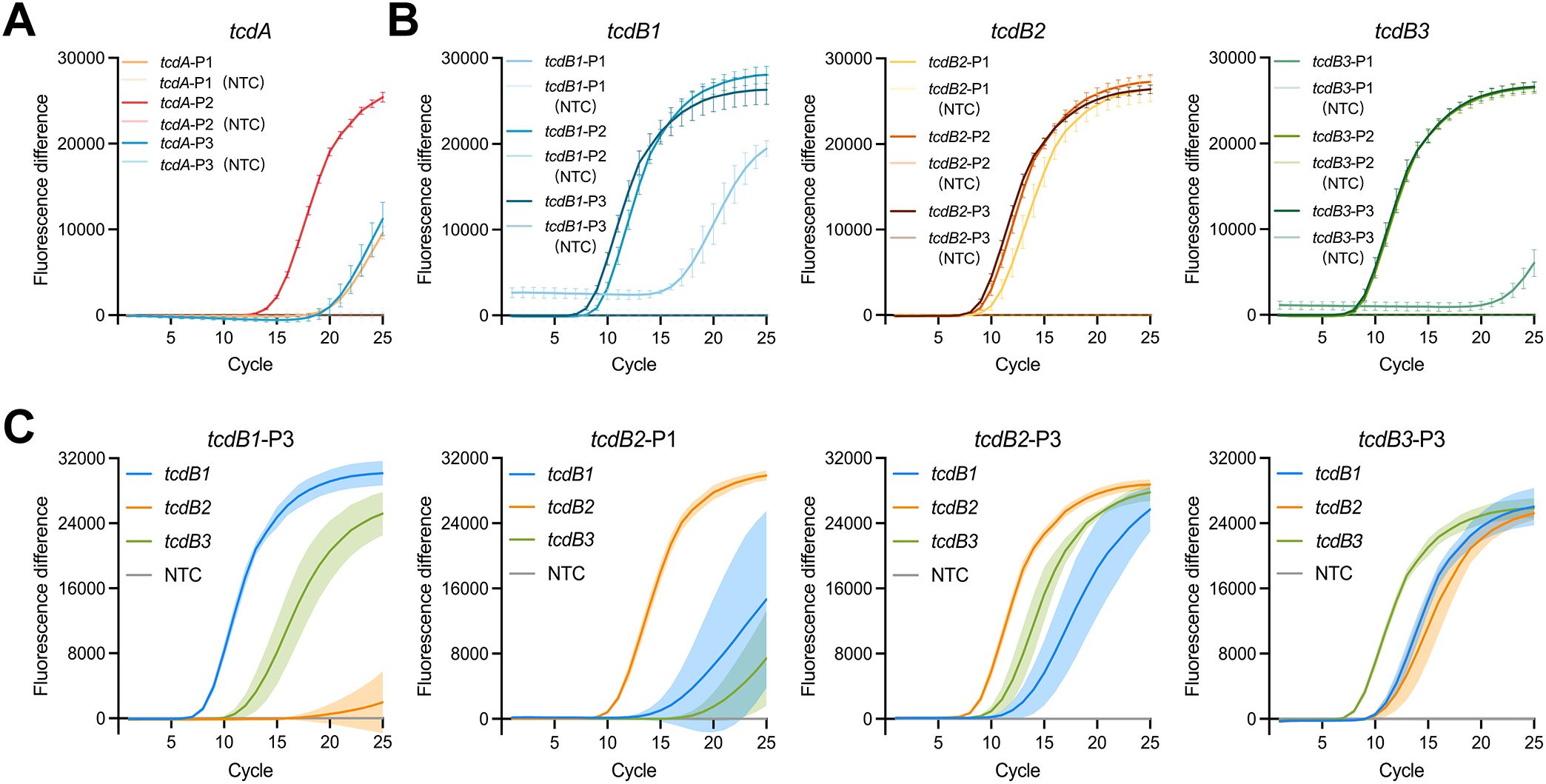
Figure 3. LAMP primer screening. (A) Screening of primers targeting tcdA (n = 6). Positive control, tcdA-plasmid. Negative control, salmon sperm DNA. (B) Initial screening of primers targeting tcdB (n = 6). Positive control, tcdB1-, tcdB2-, and tcdB3-plasmid. Negative control, salmon sperm DNA. (C) Final screening of primers targeting tcdB (n = 4). Positive control, tcdB1-, tcdB2-, and tcdB3-plasmid. Negative control, salmon sperm DNA. Fluorescence difference was calculated by subtracting the fluorescence intensity of the negative control from that of the positive control at each cycle. Error bars and shaded areas represent mean ± SD. NTC, no template control.
To further improve the efficiency of LAMP, several additives were evaluated. As shown in Supplementary Figure 3, glycine was more effective in reducing time-to-detection and enhancing fluorescence intensity. Among the tested concentrations, the system supplemented with 480 mM glycine exhibited the highest fluorescence intensity at the end of the reaction, indicating its potential to optimize the amplification yield under this condition. However, most other additives showed no significant improvement in reaction efficiency and, in some cases, exhibited even inhibitory effects. Therefore, 480 mM glycine was chosen for subsequent experiments.
3.2 Establishment of the POLC detection platforms
In CRISPR-Cas12b-based nucleic acid testing, the sgRNA not only ensures specificity by strictly base-pairing with target sequences, but also guides Cas12b in locating, recognizing, and binding to the targets (Weng et al., 2023). Cas12b is an endonuclease-capable effector that cleaves both target DNA and non-specific ssDNA via its RuvC domain (Yan et al., 2019). Notably, Mg2+ is essential for activating the RuvC domain (Zetsche et al., 2015; Swarts et al., 2017; Dai et al., 2019). Furthermore, it influences DNA polymerase activity during nucleic acid amplification by interacting with dNTPs and nucleic acid backbones (Batra et al., 2006; Xia et al., 2011). Consequently, this study established the POLC detection platforms by screening sgRNAs and the concentrations of Mg2+ and dNTP mix. As shown in Figure 4, the optimal reaction efficiency was achieved with tcdA-sgRNA-1 and tcdB-sgRNA-3 in the construction of tcdA and tcdB POLC detection platforms, respectively. This improvement was evidenced by a lower Ct value and a greater fold change in fluorescence intensity. The distribution of the preferential sgRNAs was shown in Supplementary Figure 1. Additionally, the tcdA POLC detection platform incorporating tcdA-sgRNA-1 exhibited superior performance at Mg2+ and dNTP mix concentrations of 8 mM/0.8 mM, 8 mM/1 mM, and 9 mM/1 mM. Similarly, the tcdB POLC detection platform with tcdB-sgRNA-3 exhibited improved efficiency at Mg2+ and dNTP mix concentrations of 8 mM/1 mM, 8 mM/1.2 mM, 9 mM/1 mM, 9 mM/1.2 mM, and 10 mM/1.4 mM.
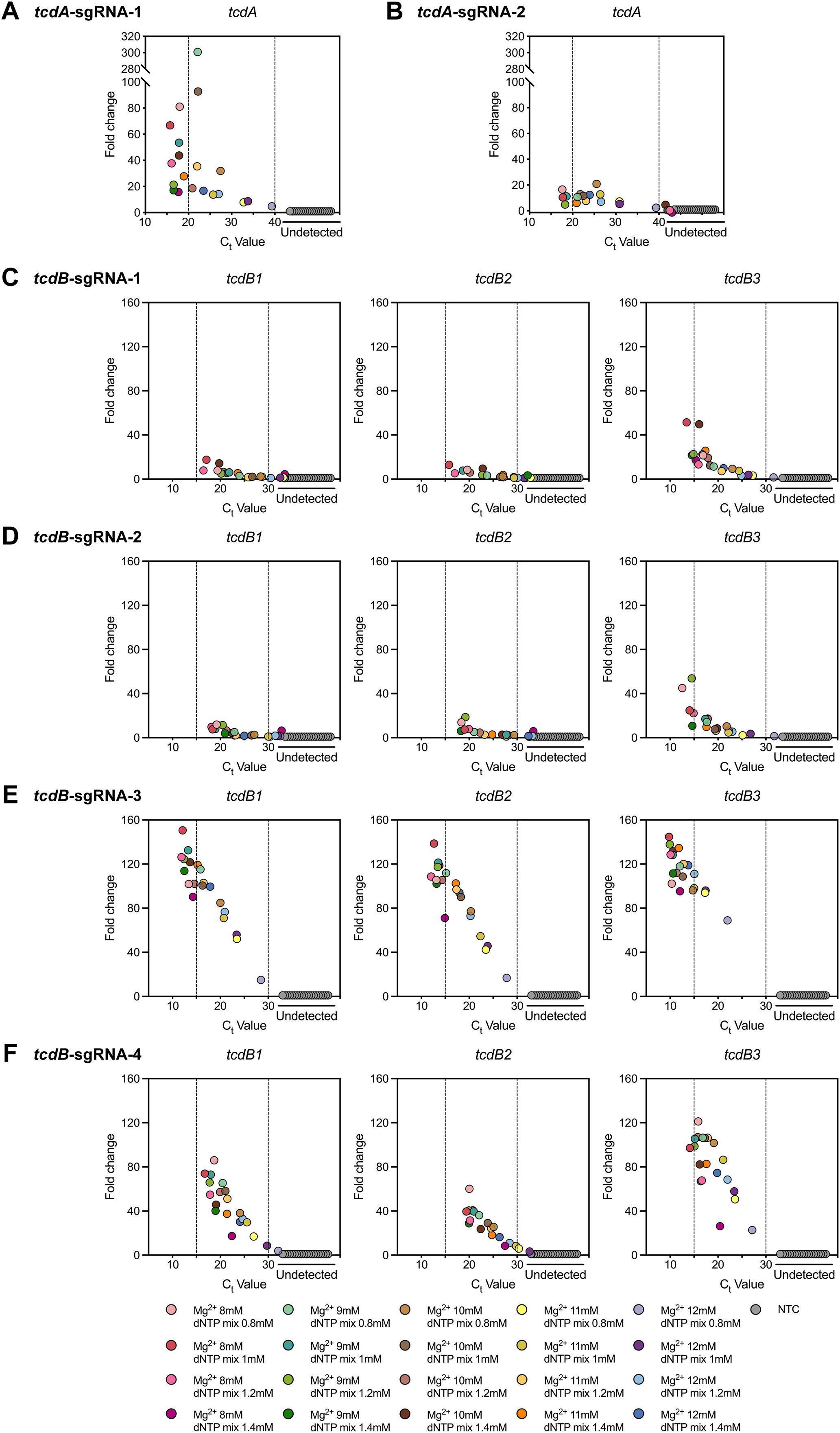
Figure 4. Establishment of the POLC detection platforms using different sgRNAs and various concentrations of Mg2+ and dNTP mix. (A, B) Responses to the usage of tcdA-sgRNA-1 and tcdA-sgRNA-2, respectively. Positive control, tcdA-plasmid. Negative control, salmon sperm DNA. (C-F) Responses to the usage of tcdB-sgRNA-1, tcdB-sgRNA-2, tcdB-sgRNA-3, and tcdB-sgRNA-4 in order. Positive control, tcdB1-, tcdB2-, and tcdB3-plasmid. Negative control, salmon sperm DNA. Fold change was defined as the ratio of the fluorescence intensity difference between the last and first cycles in the positive control to that in the negative control. NTC, no template control.
3.3 Optimization of the POLC detection platforms
To optimize the POLC detection platforms, firstly, the concentrations of Mg2+ and dNTP mix, corresponding to the points near the upper left corner with better performance (Figures 4A, E), were selected for repetition. As shown in Supplementary Figure 4A, the fluorescence change of the tcdA POLC detection platform was relatively low. Considering its potential impact on the platform’s sensitivity, the configuration with a higher ordinate value on the left was preferred, given the similar Ct values. As a result, the concentrations of Mg2+ and dNTP mix were set to 8 and 0.8 mM, respectively. The tcdB POLC detection platform, targeting all three plasmids (i.e. tcdB1-, tcdB2-, and tcdB3-plasmid), performed well under conditions of 8 mM Mg2+ and 1 mM dNTP mix, with particularly high efficiency in detecting the most common sequence tcdB1 (Supplementary Figure 4A).
Secondly, the amounts of sgRNAs and AapCas12b were determined. The results showed that the fluorescence curves for AapCas12b at concentrations of 125 and 250 nM closely mirrored those of the negative control, with all curves clustering near the abscissa axis. Considering time-to-detection, fluorescence intensity, and cost, the optimal conditions were achieved with AapCas12b and sgRNA concentrations of 500 nM for the tcdA POLC detection platform and 750 nM for the tcdB POLC detection platform (Supplementary Figure 4B).
Thirdly, the optimal reaction temperature was ascertained. For the tcdA POLC detection platform, reactions conducted at 57 and 61°C failed to generate detectable signals across all replicates and were therefore excluded from further analysis. Among the remaining conditions (58.2, 59, and 60°C), statistical analysis indicated that reactions at 60°C yielded the lowest Ct values, meaning that the tcdA assay produced positive results most rapidly at this temperature (Supplementary Figure 4C). For the tcdB POLC platform, although the Ct values at 60 and 61°C were not significantly different (Supplementary Figure 4C), the endpoint fluorescence intensity was higher at 60°C (Supplementary Figure 4D), supporting its selection as the preferred temperature.
Next, the ssDNA probe concentration needed to be decided. As shown in Supplementary Figure 4E, the efficiency of fluorescence signal generation was improved at higher probe concentrations. However, further increasing the concentration might lead to an elevated fluorescence background in the reaction system lacking target nucleic acids, as well as incur unnecessary cost increases. Thus, the probe of 1 μM was chosen for both tcdA and tcdB POLC detection platforms.
Finally, glycine concentrations were tested. For the tcdA POLC detection platform, the reaction without glycine failed to produce positive results within 40 cycles (Supplementary Figure 4F). As the glycine concentration increased, both the time-to-detection was shortened and the fluorescence intensity was enhanced (Supplementary Figure 4F). Hence, 720 mM glycine was selected for the tcdA POLC detection platform. Regarding the tcdB POLC detection platform, only weak signals were generated in the absence of glycine, while the reaction efficiency markedly improved with glycine addition (Supplementary Figure 4F). Due to the similar efficiencies observed with 480, 600, and 720 mM glycine, the concentration of 480 mM was preferred to save costs (Supplementary Figure 4F).
3.4 LoD and specificity of the POLC detection platforms
Serial dilutions of tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid were used to evaluate the LoD of the POLC platforms. As shown in Figures 5A, B, the lowest template copy numbers at which all eight replicates yielded positive fluorescence signals were approximately 14, 9, 14, and 3 copies/μL for tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid, respectively. In the visual detection assay (Figure 5C), color changes were observed when template copy numbers reached or exceeded 18, 11, 17, and 6 copies/μL for tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid, respectively. The LoD values obtained from fluorescence-based and visual readouts were comparable.
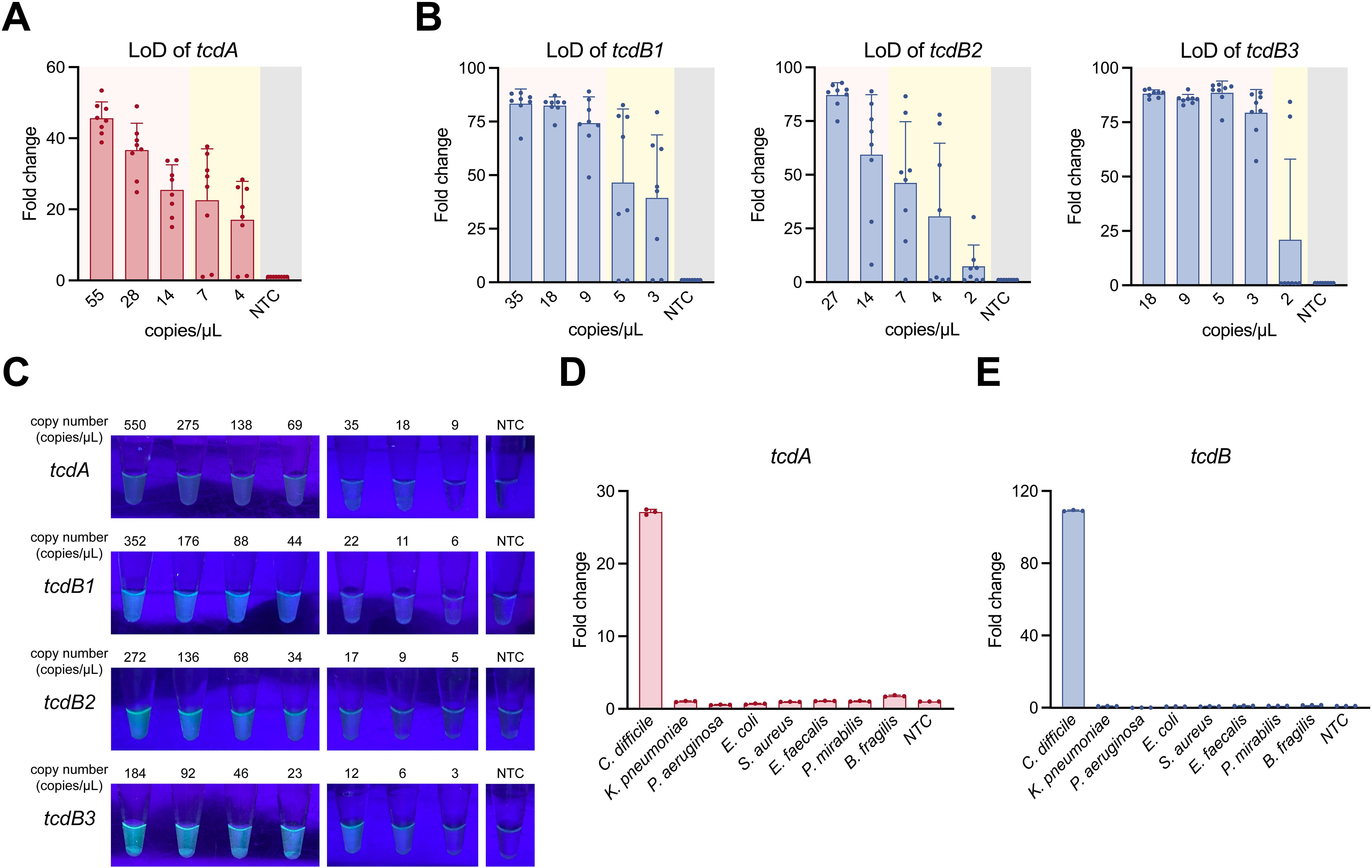
Figure 5. LoD and specificity of the POLC detection platforms. (A, B) LoD of the POLC detection platforms targeting the tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid was determined using a qPCR instrument (n = 8). The pink area indicates consistent positive detection results, whereas the yellow area indicates inconsistent results (both positive and negative) at the same plasmid copy number. The gray area represents the negative control (salmon sperm DNA solution). LoD was defined as the lowest template copy number at which the fluorescence intensity of all positive tests reached the detection threshold. Error bars represent mean ± SD. NTC, no template control. (C) LoD visualization of the POLC detection platforms targeting the tcdA-, tcdB1-, tcdB2-, and tcdB3-plasmid under UV illumination. (D, E) The specificity of the POLC detection platforms targeting tcdA and tcdB (n = 3). Error bars represent mean ± SD. NTC, no template control.
Several bacteria were used to examine the specificity of the POLC detection platforms. As shown in Figures 5D, E, only the reaction with the nucleic acids extracted from toxigenic C. difficile yielded positive results, whereas the reactions involving nucleic acids extracted from the remaining bacteria as well as nuclease-free water corresponded to negative results.
3.5 Clinical validation of the POLC detection platforms
To assess the feasibility of the POLC platforms for detecting tcdA and tcdB in fecal specimens, 55 clinical stool samples were collected between August 2023 and February 2024. Nucleic acids extracted from these samples served as templates for both the POLC platforms and qPCR. The fluorescence signals generated by the POLC platforms and qPCR were shown in Figures 6A, B, respectively. To facilitate direct comparison of tcdA and tcdB detection by the two methods, the qualitative results were visualized in a heatmap with color-coded blocks: yellow indicates samples positive by both methods; white indicates samples negative by both; red indicates samples positive by the POLC platforms but negative by qPCR; and green indicates samples negative by the POLC platforms but positive by qPCR (Figure 6C). In addition, the Ct values obtained using the POLC platforms and qPCR for the same samples were presented in Supplementary Tables 5, 6, respectively. Notably, for most positive samples, the Ct values derived from the POLC platforms were lower than those from qPCR. Furthermore, by eliminating the need for thermal cycling, the POLC platforms significantly reduced the reaction time to approximately 40 minutes, compared to 73 minutes required for qPCR.
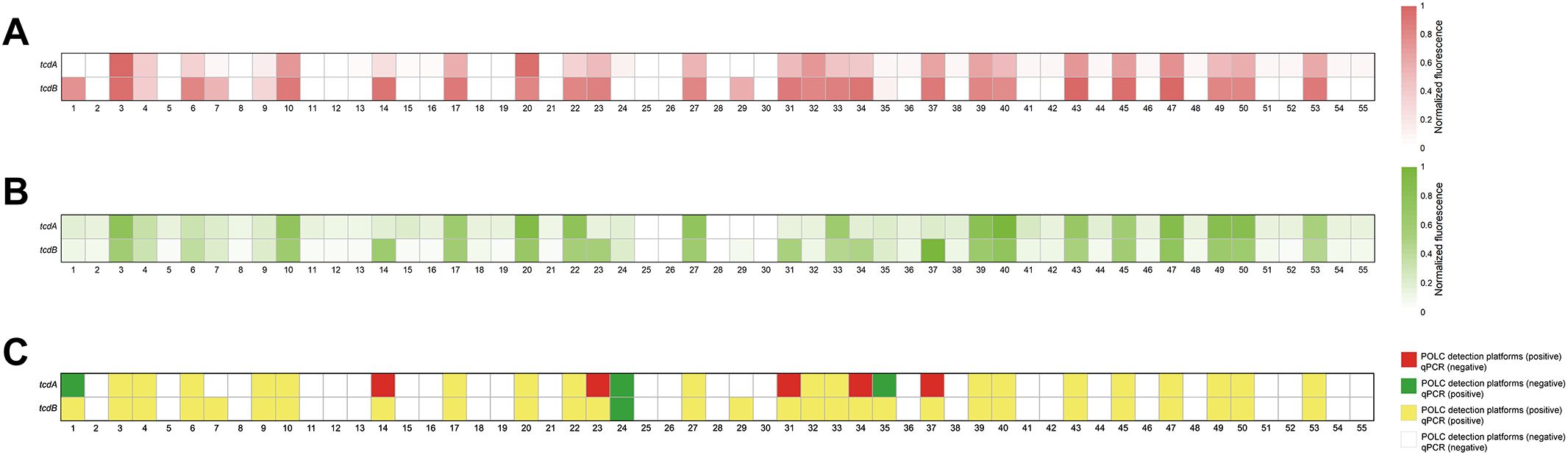
Figure 6. Detection of clinical specimens from patients with suspected CDI by the POLC detection platforms and qPCR. (A) Results from clinical samples using the POLC detection platforms. (B) Results from clinical samples using qPCR. (C) Comparison of qualitative results from clinical samples using the POLC detection platforms and qPCR.
The relationship between the results obtained by the POLC platforms and qPCR was summarized through cross-tabulations (Tables 1, 2). Using qPCR as the reference standard, the sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) of the POLC platform for tcdA detection were 86.4% (19/22), 84.8% (28/33), 79.2% (19/24), and 90.3% (28/31), respectively. For tcdB, the corresponding values were 96.6% (28/29), 100% (26/26), 100% (28/28), and 96.3% (26/27), respectively (Table 3). Additionally, the Kappa statistic was applied to assess the agreement between the two approaches. The Kappa coefficients for tcdA and tcdB detection were 0.701 (substantial agreement) and 0.964 (almost perfect agreement), respectively (Table 3). Although results interpreted by the two methods were not entirely identical in a few cases, McNemar’s test yielded exact P values greater than 0.05, indicating no statistically significant difference between the POLC platforms and qPCR (Table 3).
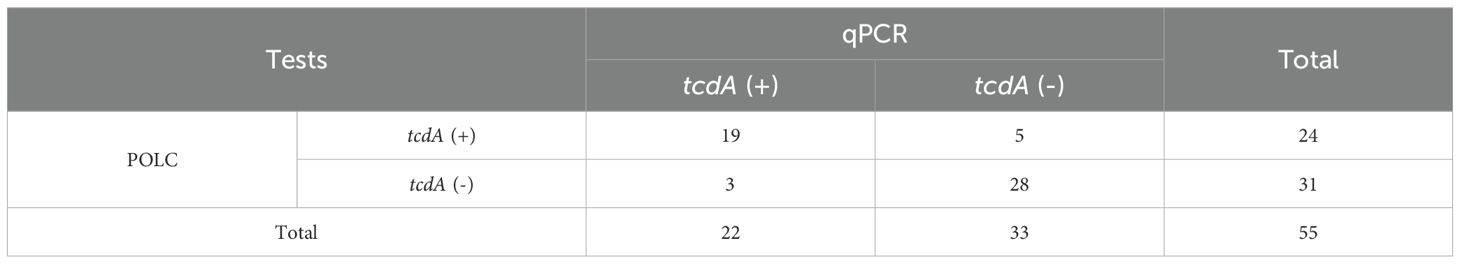
Table 1. Detection of tcdA in clinical samples using POLC and qPCR.
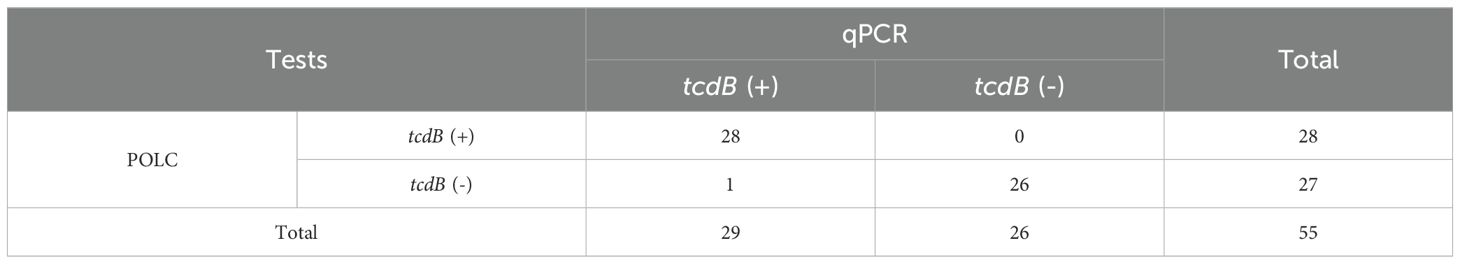
Table 2. Detection of tcdB in clinical samples using POLC and qPCR.
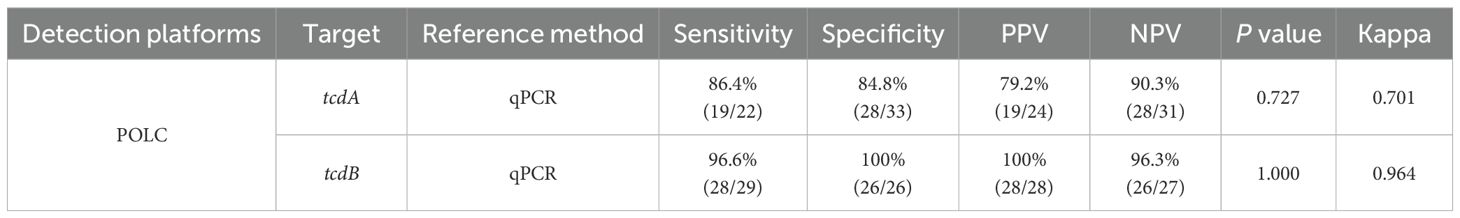
Table 3. Statistical analysis of clinical sample verification.
4 Discussion
CDI has caught much attention from public health due to its high levels of morbidity and mortality, as well as the large medical expenditure involved (Smits et al., 2016; Zhang et al., 2016; Guh et al., 2020). In clinical settings, the diagnosis of CDI primarily employs PCR-based methods targeting toxin-encoding genes. Despite their excellent sensitivity and specificity, these methods hinge on thermal cycling process, which leads to a strong dependence on sophisticated temperature control equipment and subsequently drives up the detection costs. As a result, the application of such methods in on-site detection is markedly limited. In light of this, there is an urgent need to develop a detection method suitable for POCT or easily implementable in resource-limited areas.
CRISPR-Cas systems have found widespread applications in the detection of nucleic acids for disease diagnosis. When CRISPR-Cas systems are coupled with nucleic acid amplification techniques, a notable enhancement in detection sensitivity can be achieved (Gootenberg et al., 2017; Myhrvold et al., 2018; Teng et al., 2019). However, the limited compatibility between nucleic acid amplification and CRISPR cleavage often necessitates a two-step detection procedure (Lin et al., 2022). In two-pot assays, amplification and CRISPR cleavage are performed in separate tubes, and the transfer of amplification products increases the risk of contamination (Jiang et al., 2023a). In one-pot assays, the two systems are often physically isolated within a single tube and require centrifugation to initiate the CRISPR reaction (Jiang et al., 2023b; Liu et al., 2023). Although this reduces the risk of contamination, it still compromises operational simplicity.
In this study, we developed one-step detection platforms for tcdA and tcdB in Clostridioides difficile by integrating LAMP with CRISPR-Cas12b, which we termed POLC. To address the limited compatibility mentioned above, we optimized the reaction buffer and designed sgRNAs targeting sequences adjacent to non-canonical PAMs. Although isothermal amplification buffers were available for both LAMP and CRISPR cleavage, ion concentrations still needed optimization to meet the requirements of both systems. In one-step CRISPR-based assays, recognition of canonical PAMs by Cas proteins can trigger strong cis-cleavage activity, leading to premature degradation of amplification products and delayed fluorescence signal generation. In contrast, non-canonical PAMs reduce such cis-cleavage activity, allowing amplification to proceed more efficiently. This facilitates target enrichment and timely activation of Cas-mediated trans-cleavage, producing more robust fluorescence signals (Lu et al., 2022). Moreover, the LAMP reaction may generate single-stranded target DNA, enabling PAM-independent Cas activation. As a result, all components could be premixed, supporting the formulation of a POLC reaction buffer suitable for future commercial diagnostic kits.
Through meticulous optimization of components and reaction temperatures, the POLC platforms achieved efficient isothermal operation, with LoDs ranging from 3 to 14 copies/μL when measured using a qPCR instrument, and from 6 to 18 copies/μL when interpreted visually under UV light. Specificity testing revealed no cross-reactivity with other common gut bacteria. In addition to excellent analytical performance, the POLC platforms demonstrated notable cost-effectiveness, with an estimated cost of approximately USD 6.5 per reaction, compared to around USD 10 for commercial qPCR kits and USD 26 for the Cepheid Xpert C. difficile assay. Moreover, the isothermal workflow and direct UV-based detection eliminate the need for specialized thermal cyclers and fluorescence monitoring systems, further reducing operational costs. These features make POLC particularly well-suited for POCT.
To assess the clinical applicability of the POLC platforms, 55 stool samples from patients with suspected CDI were analyzed. Including approximately 25 minutes for fecal DNA extraction, the total turnaround time for POLC was about one hour—roughly 30 minutes shorter than that required for qPCR. Notably, positive signals were often detectable much earlier during the reaction process: some appeared as early as 15 minutes, and most were observed within 30 minutes (Ct values listed in Supplementary Table 5), further demonstrating the platform’s rapidity. In terms of diagnostic performance, the POLC platforms exhibited high sensitivity and specificity, with results comparable to those obtained by qPCR. Although minor discrepancies were observed, statistical analysis revealed no significant differences. These discrepancies may be attributed to sequence variations in the tcdA and tcdB target regions among different C. difficile strains. The diagnostic capability of the tcdA POLC assay was slightly lower than that of the tcdB assay. This may be due to the initial assumption that the tcdA target region was relatively conserved, which led to less rigorous screening of its LAMP primers and sgRNAs. These factors may have adversely affected downstream detection performance.
In addition, several limitations remain to be addressed. First, the tcdA and tcdB assays were conducted independently, meaning that simultaneous detection of both genes requires two separate reactions, which somewhat compromises workflow simplicity. Second, C. difficile also harbors other virulence-associated genes, including tcdC, cdtA, and cdtB (Awad et al., 2014). At present, the detection scope of the POLC platforms remains limited, and future efforts should focus on developing a multiplex assay capable of identifying multiple targets. Finally, incorporating a nucleic acid-free extraction approach may further enhance operational efficiency and reduce overall detection time.
5 Conclusion
Our study devised one-step CRISPR-based diagnostic platforms, termed POLC, for the rapid detection of the tcdA and tcdB genes in C. difficile. By integrating LAMP with non-canonical CRISPR-Cas12b systems, the platforms allow direct sample addition to premixed reagents, followed by simple isothermal incubation. Clinical validation demonstrated that the POLC assays achieve high sensitivity and specificity comparable to qPCR. Moreover, by eliminating the need for thermal cycling and real-time fluorescence monitoring systems, the POLC platforms not only shorten detection time but also simplify equipment requirements and reduce costs, highlighting their strong potential for POCT.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Huashan Hospital, Fudan University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YZ: Writing – original draft, Methodology, Investigation, Conceptualization. LL: Investigation, Methodology, Writing – original draft. SX: Funding acquisition, Resources, Writing – review & editing. YC: Supervision, Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by Shanghai Pujiang Program (2021PJD008) (SX) and Shanghai Municipal Science and Technology Major Project (2023SHZDZX02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1594271/full#supplementary-material
References
Arimoto, J., Horita, N., Kato, S., Fuyuki, A., Higurashi, T., Ohkubo, H., et al. (2016). Diagnostic test accuracy of glutamate dehydrogenase for Clostridium difficile: Systematic review and meta-analysis. Sci. Rep. 6, 29754. doi: 10.1038/srep29754
Awad, M. M., Johanesen, P. A., Carter, G. P., Rose, E., and Lyras, D. (2014). Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microbes 5, 579–593. doi: 10.4161/19490976.2014.969632
Barbut, F., Monot, M., Rousseau, A., Cavelot, S., Simon, T., Burghoffer, B., et al. (2011). Rapid diagnosis of Clostridium difficile infection by multiplex real-time PCR. Eur. J. Clin. Microbiol. Infect. Dis. 30, 1279–1285. doi: 10.1007/s10096-011-1224-z
Batra, V. K., Beard, W. A., Shock, D. D., Krahn, J. M., Pedersen, L. C., and Wilson, S. H. (2006). Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure (London England: 1993) 14, 757–766. doi: 10.1016/j.str.2006.01.011
Centers for Disease Control and Prevention (2019). Antibiotic Resistance Threats in the UNITED STATES 2019. Available online at: http://www.cdc.gov/antimicrobial-resistance/data-research/threats (Accessed November 3, 2024).
Chen, L., Hu, M., and Zhou, X. (2025). Trends in developing one-pot CRISPR diagnostics strategies. Trends In Biotechnol. 43, 98–110. doi: 10.1016/j.tibtech.2024.07.007
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Sci. (New York N.Y.) 360, 436–439. doi: 10.1126/science.aar6245
Choi, J. S., Kim, J. S., Joe, C. O., Kim, S., Ha, K. S., and Park, Y. M. (1999). Improved cycle sequencing of GC-rich DNA template. Exp. Mol. Med. 31, 20–24. doi: 10.1038/emm.1999.3
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Sci. (New York N.Y.) 339, 819–823. doi: 10.1126/science.1231143
Crobach, M. J. T., Planche, T., Eckert, C., Barbut, F., Terveer, E. M., Dekkers, O. M., et al. (2016). European Society of Clinical Microbiology and Infectious Diseases: update of the diagnostic guidance document for Clostridium difficile infection. Clin. Microbiol. Infection 22 Suppl 4, S63–S81. doi: 10.1016/j.cmi.2016.03.010
Crobach, M. J. T., Vernon, J. J., Loo, V. G., Kong, L. Y., Péchiné, S., Wilcox, M. H., et al. (2018). Understanding clostridium difficile colonization. Clin. Microbiol. Rev. 31. doi: 10.1128/CMR.00021-17
Dai, Y., Somoza, R. A., Wang, L., Welter, J. F., Li, Y., Caplan, A. I., et al. (2019). Exploring the trans-cleavage activity of CRISPR-Cas12a (cpf1) for the development of a universal electrochemical biosensor. Angewandte Chemie (International Ed. In English) 58, 17399–17405. doi: 10.1002/anie.201910772
Deshpande, A., Pasupuleti, V., Rolston, D. D. K., Jain, A., Deshpande, N., Pant, C., et al. (2011). Diagnostic accuracy of real-time polymerase chain reaction in detection of Clostridium difficile in the stool samples of patients with suspected Clostridium difficile Infection: a meta-analysis. Clin. Infect. Dis. 53, e81–e90. doi: 10.1093/cid/cir505
East-Seletsky, A., O’Connell, M. R., Knight, S. C., Burstein, D., Cate, J. H. D., Tjian, R., et al. (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. doi: 10.1038/nature19802
Gateau, C., Couturier, J., Coia, J., and Barbut, F. (2018). How to: diagnose infection caused by Clostridium difficile. Clin. Microbiol. Infection 24, 463–468. doi: 10.1016/j.cmi.2017.12.005
Gong, J., Jiang, Y., Zhang, D., Li, T., Fu, L., and Dou, X. (2024). One-tube detection of Salmonella Typhimurium using LAMP and CRISPR-Cas12b. Microbiol. Spectr. 12, e0127124. doi: 10.1128/spectrum.01271-24
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Sci. (New York N.Y.) 356, 438–442. doi: 10.1126/science.aam9321
Guh, A. Y. and Kutty, P. K. (2018). Clostridioides difficile infection. Ann. Internal Med. 169, ITC49–ITC64. doi: 10.7326/AITC201810020
Guh, A. Y., Mu, Y., Winston, L. G., Johnston, H., Olson, D., Farley, M. M., et al. (2020). Trends in U.S. Burden of clostridioides difficile infection and outcomes. New Engl. J. Med. 382, 1320–1330. doi: 10.1056/NEJMoa1910215
Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., et al. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Sci. (New York N.Y.) 362, 839–842. doi: 10.1126/science.aav4294
Henke, W., Herdel, K., Jung, K., Schnorr, D., and Loening, S. A. (1997). Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 25, 3957–3958. doi: 10.1093/nar/25.19.3957
Jang, M. and Kim, S. (2022). Inhibition of non-specific amplification in loop-mediated isothermal amplification via tetramethylammonium chloride. Biochip J. 16, 326–333. doi: 10.1007/s13206-022-00070-3
Jensen, M. A., Fukushima, M., and Davis, R. W. (2010). DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis. PLoS One 5, e11024. doi: 10.1371/journal.pone.0011024
Jiang, T., Hu, X., Lin, C., Xia, Z., Yang, W., Zhu, Y., et al. (2023a). Rapid visualization of Clostridioides difficile toxins A and B by multiplex RPA combined with CRISPR-Cas12a. Front. In Microbiol. 14. doi: 10.3389/fmicb.2023.1119395
Jiang, T., Hu, X., and Shen, J. (2023b). Establishment of a novel detection platform for clostridioides difficile toxin genes based on orthogonal CRISPR. Microbiol. Spectr. 11, e0188623. doi: 10.1128/spectrum.01886-23
Joung, J., Ladha, A., Saito, M., Kim, N.-G., Woolley, A. E., Segel, M., et al. (2020). Detection of SARS-CoV-2 with SHERLOCK one-pot testing. New Engl. J. Med. 383, 1492–1494. doi: 10.1056/NEJMc2026172
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O., and Zhang, F. (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. doi: 10.1038/s41596-019-0210-2
Kraft, C. S., Parrott, J. S., Cornish, N. E., Rubinstein, M. L., Weissfeld, A. S., McNult, P., et al. (2019). A laboratory medicine best practices systematic review and meta-analysis of nucleic acid amplification tests (NAATs) and algorithms including NAATs for the diagnosis of clostridioides (Clostridium) difficile in adults. Clin. Microbiol. Rev. 32. doi: 10.1128/CMR.00032-18
Landis, J. R. and Koch, G. G. (1977). The measurement of observer agreement for categorical data. Biometrics 33, 159–174. doi: 10.2307/2529310
Lessa, F. C., Mu, Y., Bamberg, W. M., Beldavs, Z. G., Dumyati, G. K., Dunn, J. R., et al. (2015). Burden of Clostridium difficile infection in the United States. New Engl. J. Med. 372, 825–834. doi: 10.1056/NEJMoa1408913
Li, S.-Y., Cheng, Q.-X., Liu, J.-K., Nie, X.-Q., Zhao, G.-P., and Wang, J. (2018a). CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 28, 491–493. doi: 10.1038/s41422-018-0022-x
Li, S.-Y., Cheng, Q.-X., Wang, J.-M., Li, X.-Y., Zhang, Z.-L., Gao, S., et al. (2018b). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery 4, 20. doi: 10.1038/s41421-018-0028-z
Li, L., Li, S., Wu, N., Wu, J., Wang, G., Zhao, G., et al. (2019). HOLMESv2: A CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synthetic Biol. 8, 2228–2237. doi: 10.1021/acssynbio.9b00209
Lin, K., Guo, J., Guo, X., Li, Q., Li, X., Sun, Z., et al. (2023). Fast and visual detection of nucleic acids using a one-step RPA-CRISPR detection (ORCD) system unrestricted by the PAM. Analytica Chimica Acta 1248, 340938. doi: 10.1016/j.aca.2023.340938
Lin, M., Yue, H., Tian, T., Xiong, E., Zhu, D., Jiang, Y., et al. (2022). Glycerol additive boosts 100-fold sensitivity enhancement for one-pot RPA-CRISPR/Cas12a assay. Analytical Chem. 94, 8277–8284. doi: 10.1021/acs.analchem.2c00616
Liu, Y., Liu, H., Yu, G., Sun, W., Aizaz, M., Yang, G., et al. (2023). One-tube RPA-CRISPR Cas12a/Cas13a rapid detection of methicillin-resistant Staphylococcus aureus. Analytica Chimica Acta 1278, 341757. doi: 10.1016/j.aca.2023.341757
Lu, S., Tong, X., Han, Y., Zhang, K., Zhang, Y., Chen, Q., et al. (2022). Fast and sensitive detection of SARS-CoV-2 RNA using suboptimal protospacer adjacent motifs for Cas12a. Nat. Biomed. Eng. 6, 286–297. doi: 10.1038/s41551-022-00861-x
Mahony, J. B., Blackhouse, G., Babwah, J., Smieja, M., Buracond, S., Chong, S., et al. (2009). Cost analysis of multiplex PCR testing for diagnosing respiratory virus infections. J. Clin. Microbiol. 47, 2812–2817. doi: 10.1128/JCM.00556-09
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Sci. (New York N.Y.) 339, 823–826. doi: 10.1126/science.1232033
McDonald, L. C., Gerding, D. N., Johnson, S., Bakken, J. S., Carroll, K. C., Coffin, S. E., et al. (2018). Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and society for healthcare epidemiology of America (SHEA). Clin. Infect. Dis. 66, e1–e48. doi: 10.1093/cid/cix1085
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Sci. (New York N.Y.) 360, 444–448. doi: 10.1126/science.aas8836
Nguyen, L. T., Macaluso, N. C., Pizzano, B. L. M., Cash, M. N., Spacek, J., Karasek, J., et al. (2022). A thermostable Cas12b from Brevibacillus leverages one-pot discrimination of SARS-CoV-2 variants of concern. EBioMedicine 77, 103926. doi: 10.1016/j.ebiom.2022.103926
Nguyen, L. T., Rananaware, S. R., Yang, L. G., Macaluso, N. C., Ocana-Ortiz, J. E., Meister, K. S., et al. (2023). Engineering highly thermostable Cas12b via de novo structural analyses for one-pot detection of nucleic acids. Cell Rep. Med. 4, 101037. doi: 10.1016/j.xcrm.2023.101037
Norén, T., Alriksson, I., Andersson, J., Akerlund, T., and Unemo, M. (2011). Rapid and sensitive loop-mediated isothermal amplification test for Clostridium difficile detection challenges cytotoxin B cell test and culture as gold standard. J. Clin. Microbiol. 49, 710–711. doi: 10.1128/JCM.01824-10
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, E63. doi: 10.1093/nar/28.12.e63
O’Horo, J. C., Jones, A., Sternke, M., Harper, C., and Safdar, N. (2012). Molecular techniques for diagnosis of Clostridium difficile infection: systematic review and meta-analysis. Mayo Clinic Proc. 87, 643–651. doi: 10.1016/j.mayocp.2012.02.024
Ooi, K. H., Liu, M. M., Tay, J. W. D., Teo, S. Y., Kaewsapsak, P., Jin, S., et al. (2021). An engineered CRISPR-Cas12a variant and DNA-RNA hybrid guides enable robust and rapid COVID-19 testing. Nat. Commun. 12, 1739. doi: 10.1038/s41467-021-21996-6
Qiu, X., Liu, X., Ma, X., Wang, R., Chen, S., Li, F., et al. (2022). One-pot isothermal LAMP-CRISPR-based assay for Klebsiella pneumoniae detection. Microbiol. Spectr. 10, e0154522. doi: 10.1128/spectrum.01545-22
Qiu, X., Liu, X., Wang, R., Ma, X., Han, L., Yao, J., et al. (2023). Accurate, sensitive, and rapid detection of Pseudomonas aeruginosa based on CRISPR/Cas12b with one fluid-handling step. Microbiol. Spectr. 11, e0352322. doi: 10.1128/spectrum.03523-22
Sam, I. K., Chen, Y.-Y., Ma, J., Li, S.-Y., Ying, R.-Y., Li, L.-X., et al. (2021). TB-QUICK: CRISPR-Cas12b-assisted rapid and sensitive detection of Mycobacterium tuberculosis. J. Infection 83, 54–60. doi: 10.1016/j.jinf.2021.04.032
Sarkar, G., Kapelner, S., and Sommer, S. S. (1990). Formamide can dramatically improve the specificity of PCR. Nucleic Acids Res. 18, 7465. doi: 10.1093/nar/18.24.7465
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H., and Kuijper, E. J. (2016). Clostridium difficile infection. Nat. Rev. Dis. Primers 2, 16020. doi: 10.1038/nrdp.2016.20
Surawicz, C. M., Brandt, L. J., Binion, D. G., Ananthakrishnan, A. N., Curry, S. R., Gilligan, P. H., et al. (2013). Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 108, 478–498. doi: 10.1038/ajg.2013.4
Swarts, D. C., van der Oost, J., and Jinek, M. (2017). Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233. doi: 10.1016/j.molcel.2017.03.016
Teng, F., Guo, L., Cui, T., Wang, X.-G., Xu, K., Gao, Q., et al. (2019). CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 20, 132. doi: 10.1186/s13059-019-1742-z
Wang, D.-G., Brewster, J. D., Paul, M., and Tomasula, P. M. (2015). Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules (Basel Switzerland) 20, 6048–6059. doi: 10.3390/molecules20046048
Weng, Z., You, Z., Yang, J., Mohammad, N., Lin, M., Wei, Q., et al. (2023). CRISPR-Cas biochemistry and CRISPR-based molecular diagnostics. Angewandte Chemie (International Ed. In English) 62, e202214987. doi: 10.1002/anie.202214987
Xia, S., Wang, M., Blaha, G., Konigsberg, W. H., and Wang, J. (2011). Structural insights into complete metal ion coordination from ternary complexes of B family RB69 DNA polymerase. Biochemistry 50, 9114–9124. doi: 10.1021/bi201260h
Yan, W. X., Hunnewell, P., Alfonse, L. E., Carte, J. M., Keston-Smith, E., Sothiselvam, S., et al. (2019). Functionally diverse type V CRISPR-Cas systems. Sci. (New York N.Y.) 363, 88–91. doi: 10.1126/science.aav7271
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. doi: 10.1016/j.cell.2015.09.038
Keywords: Clostridioides difficile, LAMP, CRISPR-Cas12b, nucleic acid detection, point-of-care testing
Citation: Zhang Y, Lv L, Xu S and Chen Y (2025) Innovative nucleic acid detection of Clostridioides difficile utilizing the PAM-unconventional, one-step LAMP/CRISPR-Cas12b detection platforms. Front. Cell. Infect. Microbiol. 15:1594271. doi: 10.3389/fcimb.2025.1594271
Received: 15 March 2025; Accepted: 12 May 2025;
Published: 29 May 2025.
Edited by:
Yuetian Yu, Shanghai Jiao Tong University, ChinaReviewed by:
Hans Christiaan Van Leeuwen, Netherlands Organisation for Applied Scientific Research, NetherlandsAtif Khurshid Wani, Lovely Professional University, India
Reza Mohammadhassan, University of Otago, New Zealand
Ambreen Zahra, University of Agriculture, Faisalabad, Pakistan
Tsz Chun Ng, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2025 Zhang, Lv, Xu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Su Xu, eHVzdV9oc0BmdWRhbi5lZHUuY24=; Yijian Chen, Y2hlbnlpamlhbkBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work and share first authorship