 Jinyan Li1,2
Jinyan Li1,2 Haibo Feng
Haibo Feng Dechun Chen
Dechun Chen Huanrong Zhang
Huanrong Zhang Yi Liao
Yi Liao- 1College of Animal Husbandry and Veterinary Medicine, Southwest Minzu University, Chengdu, China
- 2Institute of Qinghai-Tibetan Plateau, Southwest Minzu University, Chengdu, China
Mycobacteria pose significant global health burdens, with Mycobacterium tuberculosis complex causing tuberculosis-a leading infectious killer claiming over 1.25 million lives annually-and NTM driving pulmonary and ulcerative infections, particularly in immunocompromised populations. Autophagy, a conserved cellular degradation pathway, serves as a critical mechanism of host defense against mycobacteria by delivering bacteria to the lysosome. As a response, mycobacteria have evolved intricate strategies to subvert or exploit autophagy for survival. Consequently, autophagy exhibits a dichotomous role in mycobacterial infection: functioning as a protective mechanism of host while simultaneously serving as a virulence determinant hijacked by bacteria for their survival. This review synthesizes current insights into the molecular mechanisms mediating host-initiated autophagy during mycobacterial infection, as well as the bacterial strategies for subverting or hijacking autophagic pathways. While autophagy may be hijacked by mycobacteria, substantial evidence from numerous studies demonstrates that autophagy-activating agents may be beneficial in restricting mycobacteria infection, even with multidrug-resistant strains. This review also systematizes promising agents that enhance autophagy to improve bacterial clearance. By synthesizing the latest research findings, this article aims to enhance our understanding of the intricate relationship between autophagy and mycobacteria, paving the way for efficient host-directed therapies (HDTs) against this severely harmful pathogen.
1 Introduction
Mycobacteria include the Mycobacterium tuberculosis complex and nontuberculous mycobacteria (NTM). The Mycobacterium tuberculosis complex can cause tuberculosis, which is a grave worldwide public health peril, claiming over 1.25 million lives each year. NTM can act as causal pathogens causing pulmonary and ulcerative human diseases (Crilly et al., 2020; Johansen et al., 2020; Kilinç et al., 2021). As successful intracellular bacteria, mycobacteria primarily resides within macrophages and phagocytes. Macrophages and phagocytes, which are the host cells of mycobacteria, activate multiple immune pathways to eliminate the invading bacteria. However, mycobacteria have developed diverse tactics to circumvent host immune elimination. The emergence of drug-resistant strains has reduced the cure rate of treating mycobacterial infections with antibiotics alone (Falkinham, 2018). Progress in the design of next generation antimycobacterial vaccines and agents demands a systematic understanding of the crosstalk between mycobacteria and host immune signaling pathways, which is complex and many aspects still need to be explored in more detail.
Autophagy represents an evolutionarily conserved biological process in eukaryotes, spanning from yeast to humans. Autophagy is employed to sustain cellular homeostasis by degrading organelles, proteins, nucleic acids, and lipids and recycling their components when the cell is subjected to nutrient deficiencies or invasion by pathogenic microorganisms (Lam et al., 2017). As a pivotal mechanism for sustaining cellular homeostasis, autophagy is involved in the prevention of multiple diseases. Besides, autophagy plays a vital role in the regulation of multiple immune responses, encompassing inflammation, innate and adaptive immunity, and antibacterial defenses. Therefore, it is not a wonder that under many pathological conditions perturbed autophagy can be implicated in a variety of diseases, including neurodegeneration, infection, inflammation, metabolic derangement, neoplasia and aging-related pathologies (Dikic and Elazar, 2018).
Within the context of host resistance to multiple intracellular bacterial pathogens, including mycobacteria, autophagy exerts a critical function by delivering endogenous and exogenous cargo to lysosome (Gutierrez et al., 2004; Yoshimori and Amano, 2009; Yuk et al., 2012; Manzanillo et al., 2013; Gomes and Dikic, 2014; Siqueira et al., 2018). Accumulating research show that autophagy affects both innate and adaptive immune responses (Yang et al., 2021; Nieto Ramirez et al., 2024). As a hedge, mycobacteria have also evolved various strategies to modulate autophagy and other immune pathways to evade host clearance (Chai et al., 2020). However, there is also study showing that autophagy is not relevant to the outcomes of mycobacterial infections. Autophagy genes protect the host against mycobacteria by reducing immune damage after infection rather than by enhancing autophagy (Kimmey et al., 2015). Although there is still debate about the specific role of autophagy in the eradication of mycobacteria, many autophagy-inducing drugs/agents can help better eliminate bacteria and reduce inflammatory damage, making autophagy induction a promising target for host-directed therapy (HDT) synergizing with existing therapies against mycobacteria (Yang, 2017; Kilinç et al., 2021; Zhao et al., 2023; Liu et al., 2024). In this review, we summarize the mechanistic insights into autophagy during mycobacterial infection and briefly discuss recent discoveries of autophagy-modulating agents that facilitate mycobacterial restriction.
2 Classification and processes of autophagy
Based on the different ways of transporting unwanted or harmful cytoplasmic cargo to lysosomes, autophagy in eukaryotic cells can be subdivided into three major types: microautophagy, molecular chaperone-mediated autophagy (CMA), and macroautophagy. During microautophagy, the lysosomal membrane invagination directly wraps intracytoplasmic cargo (Mijaljica et al., 2011) or these substances directly enter multivesicular bodies (MVBs) (Sahu et al., 2011). During CMA, intracytoplasmic unfolded proteins, recognized by molecular chaperones, enter the lysosome for degradation in a lysosomal-associated membrane protein 2A (LAMP2A)-dependent manner (Orenstein and Cuervo, 2010; Wang Y. et al., 2025). Macroautophagy is characterized by the appearance of a special double-membrane vesicle (DMV) called autophagosome that envelops the cargo to facilitate its delivery to the lysosome for degradation. Macroautophagy is the most extensively studied type and will be denoted as “autophagy” for brevity in the following part of this article. Autophagic degradation can be either non-selective or selective. Autophagic receptors serve as key determinants for selective autophagy, as they specifically recognize intracellular cargoes and mediate autophagosome formation. Cargoes include intracellular pathogens, mitochondria, endoplasmic reticula, peroxisomes, protein aggregates and lipid droplets, with their degradation via selective autophagy termed xenophagy, mitophagy, ER-phagy, pexophagy, aggrephagy and lipophagy, respectively.
The autophagy process is molecularly orchestrated by a series of proteins known as autophagy-associated proteins (ATGs). ATGs, first identified in yeast research, function through their mammalian homologs by forming five distinct complexes to mediate autophagy (Nakatogawa et al., 2009; Harnett et al., 2017).
ULK1 complex, which is composed of serine-threonine kinase, Unc-51 like kinase-1(ULK1), focal adhesion kinase family-interacting protein of 200 kDa (FIP200), ATG13 and ATG101 (Bento et al., 2015; Dikic and Elazar, 2018).
Class III phosphatidylinositol 3-kinase (PI3KC3) complex, which is composed of vacuolar protein sorting 34 (VPS34), autophagy and beclin 1 regulator 1 (AMBRA), ATG6, ATG14, phosphoinositide-3-kinase-regulatory subunit 4 (PIK3R4), and UV radiation resistance-associated gene protein (UVRAG) (Kametaka et al., 1998; Liang et al., 2006; Matsunaga et al., 2009; Bento et al., 2015; Dikic and Elazar, 2018).
WD repeat domain phosphoinositide-interacting (WIPI) proteins (Proikas-Cezanne et al., 2004; Polson et al., 2010; Harnett et al., 2017).
ATG12-ATG5-ATG16L complex (Mizushima et al., 1998, 1999; Bento et al., 2015; Dikic and Elazar, 2018).
Microtubule-associated 1 light chain 3 (LC3), a core autophagic protein, exists in two isoforms: LC3-I and LC3-II. LC3-II formed via conjugation of phosphatidylethanolamine (PE) to LC3-I by ATG3 and ATG7 (Kirisako et al., 1999; Weidberg et al., 2010; Bento et al., 2015; Dikic and Elazar, 2018; Frudd et al., 2018).
The above five complexes are sequentially involved in five stages of autophagy, including signal induction, nucleation, elongation, fusion and degradation (Figure 1).
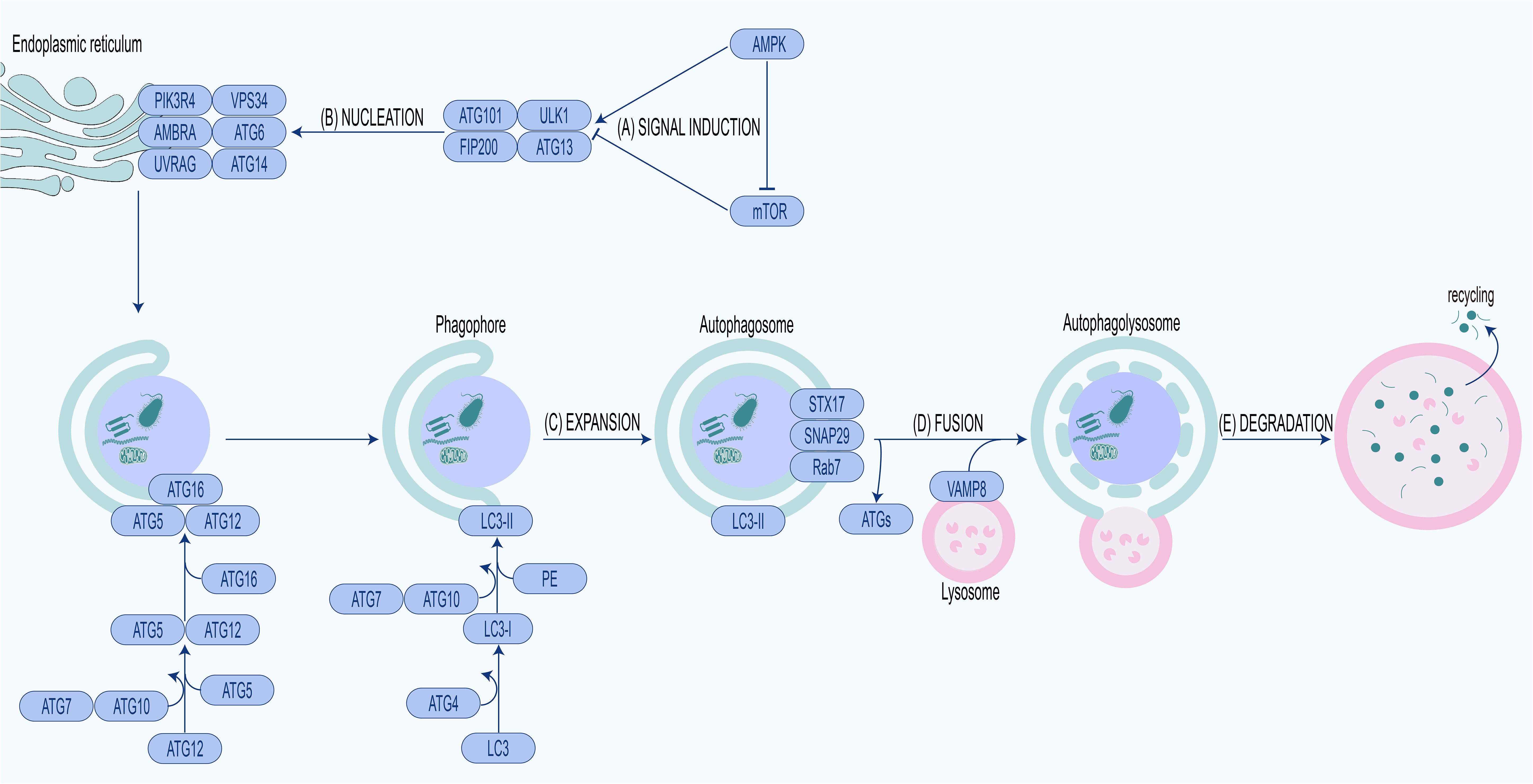
Figure 1. Molecular mechanism of autophagy.(A) Signal induction. Activation of AMPK or inhibition of mTOR leads to ULK1 complex activation and autophagy initiation. (B) Nucleation. Activated ULK1 complex phosphorylates PI3KC3 complex, producing PI3P, recruiting WIPIs and ATG12-ATG5-ATG16L complex. (C) Expansion. Recruited ATG12-ATG5-ATG16L complex conjugates PE to LC3-I to form LC3-II, expanding phagophore to form the autophagosome. (D) Fusion. Autophagosome fuses with lysosome to form autophagolysosome under the mediation of fusion proteins. (E) Degradation. The cargo in the autophagolysosome is degraded for recycling.
2.1 Signal induction
The pivotal modulators of autophagy are two proteins with antagonistic effects, mechanistic/mammalian target of rapamycin (mTOR) and AMP activated protein kinase (AMPK) (Kim et al., 2011; Shang and Wang, 2011; Suzuki et al., 2025). mTOR is a serine/threonine protein kinase that regulates autophagy in response to hormones, nutrients, energy levels and oxygen content (Shang and Wang, 2011; Singh and Subbian, 2018). When nutrients are abundant, AMPK switches to an inactive state while mTOR undergoes activation. Activated mTOR binds to ULK1 and phosphorylates particular amino acid residues to inactivate it, thereby blocking ULK1-mediated autophagy initiation. Under nutrient deprivation, AMPK is activated, activating ULK1, ATG6, and VPS34 while inactivating mTOR, thereby enabling autophagy initiation (Hosokawa et al., 2009; Langer et al., 2024).
2.2 Nucleation
The activated ULK1 complex phosphorylates PI3KC3 complex and recruits it to phagophore assembly site (PAS). PAS is generally located in the endoplasmic reticulum, especially at the endoplasmic reticulum–mitochondria contact sites. The PI3KC3 complex recruited to the PAS catalyzes the production of phosphatidylinositol-3-phosphate (PI3P) (Nascimbeni et al., 2017). With the increase of PI3P in the PAS, WIPIs, as PI3P effector proteins, are also recruited to the PAS (Nair et al., 2010; Zhao et al., 2022). WIPIs can interact with ATG16L to facilitate recruitment of the ATG12-ATG5-ATG16L complex, which is indispensable for LC3 lipidation and autophagosome assembly (Strong et al., 2021).
2.3 Expansion
The ATG12-ATG5-ATG16L complex is an E3-like enzyme that plays a scaffolding role in LC3-I lipidation at sites where phagosomal membranes are to be formed (Fujita et al., 2008; Zhao et al., 2022). Upon autophagic induction, LC3-I is connected to PE through the catalysis of the E1-like enzyme ATG7 and the E2-like enzyme ATG3, forming LC3-II. Then LC3-II attaches to the inner and outer membranes of the phagosome and is removed from the autophagosome membrane by ATG4 prior to fusion with the late endosome/lysosome (Hussey et al., 2009; Carneiro and Travassos, 2013). Lipolysis of LC3 is in association with the ATG16L complex assembly as ATG12-ATG5 conjugation decreased significantly with the loss of LC3-II in ATG3-deficient cells (Sou et al., 2008; Lystad et al., 2019).
2.4 Fusion
Preceding the fusion of autophagosomes and lysosomes, ATGs are removed while fusion-associated molecules are recruited to autophagosomes. The autophagosomal membrane-anchored Syntaxin 17 (STX17), synaptosomal-associated protein 29 (SNAP29) and Rab7 interact with lysosomal membrane-anchored vesicle-associated membrane protein 8 (VAMP8), enabling fusion of autophagosomes with lysosomes and subsequent autophagolysosome formation (Furuta et al., 2010; Itakura et al., 2012; Hyttinen et al., 2013; Morelli et al., 2014; Zhao et al., 2021).
2.5 Degradation
When the autophagolysosome is formed, degradation of cargo inside autophagosome begins (Yang and Klionsky, 2010).
3 Autophagy against mycobacteria
3.1 Autophagy against M. tuberculosis
Host cells generate autophagy primarily by recognizing bacterial damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) (Deretic et al., 2015; Shibutani et al., 2015). When DAMPs and PAMPs are recognized, the cell generates signaling cascades that rapidly lead to the co-localization of autophagy machinery and cargo (van der Vaart et al., 2014a). The calcium signal is a DAMP. A recent study identified the tumor necrosis factor-like weak inducer of apoptosis (TWEAK) as pivotal mediator of calcium-associated autophagy. TWEAK binding to fibroblast growth factor-inducible 14 (Fn14) promotes calcium channel activation, leading to calcium influx and downstream activation of AMPK signaling to induce autophagy. Persistent TWEAK-Fn14 signaling also triggers mitochondrial ROS accumulation and cell death in late infection. Genetic depletion of Fn14 or TWEAK blockade suppresses autophagy and cell death, significantly enhancing mycobacterial survival in macrophages (Chen et al., 2022). The transcription of autophagic genes is further enhanced by the engagement of transcription factors, including nuclear factor-κB (NF-κB) and transcription factor EB (TFEB), which in turn promotes autophagy (van der Vaart et al., 2014b; Pastore et al., 2016). Notably, additional evidence from a separate study indicates that in non-immune epithelial cells, mycobacterial infection upregulates the expression of TLR2/4/7 to inhibit ROS, autophagy, and apoptosis in a MyD88-dependent manner, thereby promoting bacterial survival. This suggests that the TLR pathway exhibits dual regulatory roles in host defense across different cellular contexts (Singh et al., 2025).
In addition to DAMPs and PAMPs, cytokines also regulate host cell autophagy during M. tuberculosis infection. Stimulation of autophagic pathways by IFN-γ in mycobacteria infected macrophages causes colocalization of autophagy factors LC3 and mycobacterial autophagosome, indicating that intracellular mycobacteria may be target of host autophagy. IFN-γ also induces maturation of mycobacterial autophagosome, reducing intracellular viability of mycobacteria (Gutierrez et al., 2004; Songane et al., 2012; Dey et al., 2015; Watson et al., 2015; Ning et al., 2021; Yang et al., 2021). Subsequent study confirmed this finding and further revealed that although starvation and IFN-γ can trigger autophagy in host macrophage during M. tuberculosis infection, T helper (Th) 2 cytokines can reverse this anti-mycobacteria mechanism through AKT signaling and transcription 6 (STAT6) pathways respectively (Harris et al., 2007). It is seen that different cytokines may exhibit distinct autophagy-inducing effects. Furthermore, autophagy plays a role in additional anti-tuberculosis mechanisms such as increasing lysosomal bactericidal activity (Alonso et al., 2007; Purdy, 2011), modulating expression of scavenger receptors (SRs) (Bonilla et al., 2013) and increasing mycobacterial antigen presentation (Jagannath et al., 2009; Khan et al., 2021; Wu et al., 2022). In conclusion, these studies confirm the contribution of autophagy to host innate and adaptive immune defense against mycobacterial pathogens.
Furthermore, miRNAs may additionally modulate autophagy in mycobacteria-infected host cells. MiR-155 exhibits the opposite regulation of autophagy in different cells. In M. tuberculosis-infected dendritic cells, miR-155 impedes the formation of autophagosome and autophagolysosome (Etna et al., 2018), whereas in infected macrophages, miR-155 augments autophagic flux by interacting with Ras homologue enriched in brain (Rheb), which negatively regulates autophagy (Wang et al., 2013). The autophagy induced in M. tuberculosis infected macrophage is greatly repressed by miR-142-3p overexpression, which also prevents phagolysosome formation and enhances M. tuberculosis viability in macrophages. Additionally, by specifically targeting the 3’-UTR, miR-142-3p negatively regulates ATG16L1 and ATG4 expression, resulting substantial abatement of autophagy (Qu et al., 2021). Analogous to miR-142-3p, miR-874-3p and miR-129-3p also reduce macrophage autophagy through inhibiting ATG16L1 and ATG4 expression respectively (Qu et al., 2019; Luo et al., 2021). Apart from ATG16L1, ULK and ATG7 are also targets of miRNA to regulate autophagy in mycobacterial host cells. MiR-106a and miR-20a both downregulate ATG7 and ATG16L1 expression, whereas miR-106a also downregulates ULK expression to repress autophagy and facilitate M. tuberculosis survival (Guo et al., 2016; Liu et al., 2020). MiR-1958 exhibits similar inhibitory effect on autophagy and promotional effect on M. tuberculosis survival in macrophages. The mechanism is that miR-1958 directly targets the 3’UTR of ATG5, downregulates ATG5 expression, blocks autophagosome-lysosome fusion, impairs autophagic flux, and thus facilitates intracellular M. tuberculosis survival (Ding et al., 2019). ATGs are not the only target that miRNAs regulate to influence autophagy during mycobacteria infection. The flow of calcium ions from the endoplasmic reticulum toward the cytoplasmic matrix can cause autophagy. MiR-27a, which is increased in M. tuberculosis infection context, targets the ER-located Ca2+ transporter channel auxiliary subunit, downregulates Ca2+ signaling, thus inhibits autophagosome maturation, promotes M. tuberculosis survival (Liu et al., 2018). MiRNA possesses 3p and 5p arms. different arm of miR-30a plays contrary role on autophagy and anti-M. tuberculosis activity. MiR-30a-3p inhibits autophagosome and autophagolysosome formation and favors M. tuberculosis survival, whereas miR-30a-5p has the exact opposite effect (Behura et al., 2019, 2021). Similar to miR-30a, the role that the 3p and 5p arm of miR-125a plays in modulating is also opposite. MiR-125a-3p inhibits autophagy activation and anti-mycobacteria effect by targeting UVRAG, miR-125a-5p enhances autophagy activation and antimicrobial effects by inhibiting STAT3 (Kim et al., 2015; Wang Y. et al., 2020). MiR-17, another miRNA targeting STAT3, increases autophagy against M. tuberculosis by inhibiting upstream and downstream signaling of STAT3 (Kumar et al., 2016). Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), a factor that plays a role in promoting autophagy, is inhibited on expression by miR-889, resulting survival of M. tuberculosis (Chen et al., 2020). It has been reported that increased miR-23a-5p triggered by M. tuberculosis infection dramatically prevented autophagy in macrophages. Mechanistically, miR-23a-5p can inhibit TLR2/MyD88/NF-κB signaling by decreasing TLR2 expression (Gu et al., 2017). MiR-18a, belonging to the miR-17 family, demonstrates increased expression during M. tuberculosis infection and inhibits autophagy by downregulating ATM-AMPK signaling, resulting in increased bacterial survival (Yuan et al., 2020). A study has confirmed that miR-207 inhibited macrophage autophagy by directly binding to lysosome-associated membrane protein 2 (LAMP2), thus enhancing the survival of M. tuberculosis (Du et al., 2025). Damage-regulated autophagy modulator 2 (DRAM2), which interacts with UVRAG, is relevant to the promotion of autophagy. MiR144* can decrease DRAM2 expression and formation of autophagosomes, facilitate M. tuberculosis survival, by targeting 3’-untranslated region of DRAM2 (Kim et al., 2017). Additionally, a recent study revealed that miR-25-3p promotes macrophage autophagy by targeting dual specificity phosphatase 10 (DUSP10) to activate ERK1/2 phosphorylation, significantly increasing the expression of autophagy-related proteins like LC3-II and Beclin1, thereby reducing intracellular M. bovis survival (Yuan et al., 2023).
Interestingly, different mycobacteria differ in their ability to induce autophagy in host cells. proteomics revealed that M. bovis significantly increased the expression of 51 autophagy- and inflammation-related genes and activated the NF-κB pathway while M. tuberculosis only upregulated 8 energy metabolism-related proteins with weaker autophagy activation (Cai et al., 2023). Variations in the capacity to activate autophagy may not only partially dictate the survival outcomes of distinct mycobacterial strains but also underlie differences in their pathogenic potential.
3.2 Autophagy against NTM
As the mechanism of host resistance against pathogens, autophagy is also seen in nontuberculous mycobacterial infection (Daley and Winthrop, 2020). M. avium complex (MAC) is one of the most prevailing NTM species. Similar to M. tuberculosis-infected host cells, autophagy appears in MAC-infected macrophages through miR-125a-5p induced STAT3 (Wang Y. et al., 2020). Due to evolutionary conservation (Zhang et al., 2024), M. marinum can infect both humans and ectotherms and is often used as a model microbe of M. tuberculosis (Menon et al., 2025). Similar to M. tuberculosis, M. marinum also induces autophagy in host for the removal of intracellular bacteria (Chen et al., 2018; Kjellin et al., 2019). Researches on zebrafish models reveals that mediating by STING and p62, the DRAM1 functions downstream of TLR to activate autophagy, inducing defensive autophagy against M. marinum (van der Vaart et al., 2014a, 2014b; Zhang et al., 2020). Research on M. marinum infected dictyostelium discoideum reveals that, in addition to direct phagocytosis and removal of bacteria, the autophagy mechanism can repair disruption at the mycobacteria-containing vacuole in parallel with the endosomal sorting complex required for transport (ESCRT), thereby suppresses M. marinum proliferation (López-Jiménez et al., 2018). M. smegmatis and M. fortuitum are both fast-growing non-pathogenic NTMs, and both can induce strong autophagy independent mTOR pathway in host cells (Zullo and Lee, 2012). The autophagy induced by M. smegmatis in THP-1 macrophages relies on cell surface recognition receptor TLR2 but not bacterial ubiquitination, suggesting that host cells may remove M. smegmatis through non-selective autophagy (Bah et al., 2016). M. terrae is a slow-growing NTM that can cause intractable debilitating disease due to its antibiotic resistance (Wang et al., 2019). During M. terrae infection, autophagy induced by IL-17 is indispensable for antibacterial reaction in macrophages (Orosz et al., 2016).
3.3 Xenophagy against mycobacteria
In early studies, researchers believed that autophagy was a non-selective process, which indiscriminately wrapped all organelles and macromolecular complexes within a region of the cytoplasm into autophagosome for degradation. More recently, researchers observed that autophagy could act as a selective pathway that delivers specific organelles, invading organisms, or macromolecular complexes to autophagic machinery. During selective autophagy, recognition of cargo tends to be achieved by ubiquitylation that does not occur during non-selective autophagy (Gubas and Dikic, 2022). The ubiquitinated cargo is then bound to receptors proteins. Receptors contain LC3interacting region (LIR) domains and ubiquitinbinding domains that anchor the LC3-containing phagophore to the cargo for engulfment (Kirkin and Rogov, 2019; Abdrakhmanov et al., 2020). Host selective autophagy for invading organisms is termed “xenophagy” (Galluzzi et al., 2017). Xenophagy is pivotal in orchestrating the host immune defense against mycobacteria because deletion of related genes allows the bacterium to proliferate abundantly (Shariq et al., 2023).
3.2.1 Ubiquitination of mycobacteria
The mycobacterial ubiquitylation mediated by ubiquitin-ligating enzyme is the critical step for xenophagy origination. Two E3 ubiquitin ligases, parkin and smurf1, attach ubiquitin to bacteria during mycobacteria infection.
Parkin, as a member of the RBR ubiquitin ligase family, harbors multiple conserved domains, such as RING1, RING2, UBL, RING0, REP, and IBR (Dove and Klevit, 2017). Phosphorylation at S65 of the UBL domain and Ub elicits dramatic conformational changes and activation of parkin (Gladkova et al., 2018; Sauvé et al., 2018). In addition to its role in apoptosis, lipid metabolism, and inflammatory responses, parkin is involved in xenophagy against mycobacteria (Manzanillo et al., 2013; Romagnoli et al., 2023). M. tuberculosis utilizes its ESX-1 type VII secretion system to disrupt the phagosome membrane and enter the cytosol, where parkin catalyzes the K63-linked polyubiquitination to bacteria or bacteria-related membrane structures via its E3-Ub ligase activity. Autophagy receptors bind to ubiquitinated bacteria or membrane structures via the ubiquitinbinding domain and then recruit LC3-containing phagophore via the LIR domain. Parkin knockout in macrophage reduces LC3 lipidation and increases survival of M. tuberculosis, whereas parkin overexpression reduces bacterial proliferation (Manzanillo et al., 2013). Animal experiments have yielded consistent results. Parkin knockout mice exhibit more severe symptoms and higher mortality compared to wild-type mice. Meanwhile, parkin knockout increases mycobacterial replication and proliferation in the lung, spleen, and liver of mice (Manzanillo et al., 2013). These results suggest that ubiquitination and subsequent xenophagy facilitated by parkin play an essential role in the control of mycobacteria. Intriguingly, in macrophage infected with M. tuberculosis, only a portion of the intracellular bacteria is linked to the K63-linked Ub chains, the other portion is attached to the K48-linked Ub chains. These results suggest that other E3-Ub ligase are implicated in the ubiquitination of mycobacteria and relevant structures in addition to parkin.
Smurf1, an additional E3 ubiquitin ligase, mediates ubiquitination of intracellular mycobacteria. In addition to the E3-Ub ligase domain, smurf1 has a C2 phospholipid-binding domain, and both domains are involved in xenophagy against M. tuberculosis, as mutation in either of the two domains exhibits a deficiency in recruiting polyubiquitin, the autophagy receptor, the LC3 protein and the lysosome to M. tuberculosis relevant structures and exhibits increased mycobacterial survival in host cells (Franco et al., 2017). Analogous to parkin, smurf1 activity depends on ESX-1-driven translocation of M. tuberculosis from phagosomal compartments to the cytoplasmic matrix (Chandra and Philips, 2025). Contrary to parkin, smurf1 connects K48-linked Ub chains, not K65-linked ones, to M. tuberculosis. During chronic infection, smurf1-deficient mice display increased mycobacteria proliferation in the lungs and spleens compared to wild-types, whereas during acute infection, smurf1-deficient exhibit no effect on mycobacterial proliferation. Intriguingly, during acute infection, parkin-deficient mice display more M. tuberculosis proliferation. Whether these results are related to the different ubiquitin linkages catalyzed by parkin and smurf1 needs to be further explored.
Recently, tripartite motif 32 (TRIM32), another E3-Ub ligase belonging to the TRIM proteins family, was identified to be engaged in the ubiquitination of M. tuberculosis in host cells. TRIM32 knockdown in THP1 cells induces enhanced M. tuberculosis replication, owing to blocked bacterial ubiquitination, decreased autophagy recruitment and reduced autophagosome formation (Romagnoli et al., 2023). Another study identified a TRAF-like E3 ubiquitin ligase, TrafE, which integrates ESCRT and autophagy pathways by recruiting ALIX and Vps32 to damaged membranes during M. marinum infection. TrafE deficiency leads to reduced K63-polyubiquitination, impaired xenophagy, and premature host cell death, highlighting its critical role in membrane repair and bacterial restriction (Raykov et al., 2023). An in vitro autoubiquitination investigation revealed that the E3-Ub ligase, makorin ring finger protein 1 (MKRN1), in coordination with the ubiquitin-activating enzyme E1 (UBE1) and ubiquitin conjugating enzyme E2 D3 (UBE2D3), catalyzed the ubiquitination of M. tuberculosis, but not B. subtilis suggesting that MKRN1 may be a M. tuberculosis-specific E3-Ub ligase (Subrahmanian et al., 2020). However, whether MKRN1 catalyzes intracellular ubiquitination of M. tuberculosis or other mycobacteria needs to be further investigated.
In addition to being catalyzed by E3-Ub ligase, the M. tuberculosis can directly anchor to host ubiquitin chains through the mycobacterial surface protein Rv1468c, which harbors the eukaryotic-like ubiquitin-associated (UBA) domain (Chai et al., 2019). During Salmonella typhimurium infection, host galectin-8 identifies bacterially disrupted phagosomes, recruiting the autophagy receptor and LC3, inducing xenophagy (Thurston et al., 2012). These findings suggest that host can initiate xenophagy in a ubiquitin-independent manner. Considering that M. tuberculosis disrupted phagosomes may also be recognized by galectin (Schnettger et al., 2017; Morrison et al., 2023), it is not hard to understand that the host can also initiate xenophagy during mycobacteria infection through a galectin (rather than ubiquitin)-dependent manner (Bell et al., 2021). Furthermore, given the expanding number of eukaryotic-like effectors found in M. tuberculosis (Forrellad et al., 2013; Chai et al., 2018; Krause and Dikic, 2022), it is not implausible that M. tuberculosis may directly initiate host xenophagy by utilizing bacterial structural proteins that can be recognized by autophagy receptors or LC3 family members through protein–protein interaction.
3.2.2 Autophagy receptors of xenophagy against mycobacteria
Beyond the pivotal roles of E3 ubiquitin ligases and diverse initiation mechanisms in xenophagy against mycobacteria, a set of specialized autophagy receptors further orchestrates the selective engulfment and elimination of intracellular mycobacteria, representing a critical downstream framework in this immune response. Autophagy receptors SQSTM1, CALCOCO2, NBR1, OPTN, and TAX1BP1 play crucial roles in mediating the attachment of intracellular mycobacteria to phagophores during xenophagy.
SQSTM1 was initially found to be a selective autophagy receptor for cytosolic protein aggregates (Komatsu et al., 2007; Pankiv et al., 2007; Tung et al., 2010; Chen et al., 2024). SQSTM1 contains not only a dimerization domain, a LIR domain, and a UBA domain, which are common to xenophagy receptors, but also a PB1 domain, a Ub ligase-interacting region and zinc finger. SQSTM1 self-oligomerization via the PB1 domain is indispensable for its function in selective autophagy (Itakura and Mizushima, 2011; Kim et al., 2016). To efficiently deliver cargo to the phagophore, SQSTM1 needs to interact with not only ubiquitin-modified cargo, but also other effector proteins (Clausen et al., 2010; Filimonenko et al., 2010; Chai et al., 2019; Zhang et al., 2019). The UBA domain of SQSTM1 is phosphorylated and ubiquitinated, which in turn binds to K63 linked cargo ubiquitin chains (Pilli et al., 2012; Lee et al., 2017; Lee and Weihl, 2017). The ubiquitination of SQSTM1 is catalyzed by effector proteins that bind to it. These effector proteins are primarily E3-Ub ligases like tripartite motif containing 50 (TRIM50), TNF receptor associated factor 6 (TRAF6), SMURF2, RNF166 and kelch-like ECH-associated protein 1 (KEAP1) (Komatsu et al., 2010; Lau et al., 2010; Heath et al., 2016; Lee et al., 2017). SQSTM1 participates in autophagic degradation of N-terminal arginylated proteins (Cha-Molstad et al., 2017; Zhang et al., 2018). SQSTM1 is involved in xenophagy of mycobacteria. SQSTM1 binds ubiquitinated Rv1468c protein on the surface of M. tuberculosis and brings M. tuberculosis to LC3-associated autophagosomes for xenophagy clearance (Chai et al., 2019). In M. marinum infected zebrafish model, SQSTM1 increases co-localization of LC3 with mycobacteria and inhibites bacteria multiplication (Zhang et al., 2019). Intriguingly, the antimycobacterial activity of SQSTM1 is not limited to xenophagy. SQSTM1 can deliver specific ribosomal and bulk ubiquitinated cytosolic proteins from the cytoplasm to autolysosomes for processing into molecules with antimycobacterial activity (Ponpuak et al., 2010). In addition, SQSTM1 is involved in the regulation of cyclic GMP-AMP synthase (cGAS)- Stimulator of Interferon Genes (STING) pathway. cGAS-STING pathway sensing to DNA induces phosphorylation of TBK1, which in turn activates IRF3, causing type-1 interferon expression. Phosphorylated TBK1 catalyzes the phosphorylation of SQSTM1, which induces the STING translocating to phagophores and degrading and avoids the overproduction of type-1 interferon (Prabakaran et al., 2018). TBK1 is a very specific molecule, which acts as a downstream of STING and participates in the generation of type-1 interferon (Wang et al., 2018) and also participates in mitophagy and xenophagy within mycobacteria-infected macrophages (Song et al., 2022). Given that type-I interferon, mitophagy, and xenophagy all modulate host-mediated clearance of mycobacteria, TBK1 emerges as a promising target for developing non-antibiotic therapeutics against these pathogens.
TAX1BP1 contains the LIR structural domain common to autophagy receptors for binding LC3, and unlike SQSTM1, TAX1BP1 does not contain the UBA domain. TAX1BP1 relies on the C-terminal overlapping Ub and myosin VI (MYO6) interacting domain to recognize ubiquitin (Ceregido et al., 2014; Whang et al., 2017; Fu et al., 2018). As with other autophagy receptors, TAX1BP1 targets ubiquitylated M. tuberculosis to LC3-containing phagophores, resulting clearance of bacteria (Budzik et al., 2020).
CALCOCO2 relies on the LIR and the unique CLIR to bind to LC3 and on the C2H2 zinc finger to recognize ubiquitin (Xie et al., 2015). CALCOCO2 delivers ubiquitinated M. tuberculosis to the phagosome, a process that requires the mycobacterial ESX-1type VII secretion system. BCG lacking ESX-1 cannot be delivered to the phagosome, and restoration of ESX-1 in BCG reverses this process (Watson et al., 2012).
OPTN, as an autophagy receptor, is involved in xenophagy, aggrephagy and mitophagy (Qiu et al., 2022). The LIR structural domain of OPTN is located between the two coil domains at the N-terminus, while the ubiquitin binding domains UBAN and zinc finger are located at the c-terminus. The UBAN domain preferentially recognizes linear ubiquitin chain (Li et al., 2018; Herhaus et al., 2019). The terminal coil domains of OPTN assemble into a heterotetrameric complex with the TBK1 C-terminus, thereby regulating OPTN function in selective autophagy (Morton et al., 2008; Li et al., 2016). OPTN has different phosphorylation sites and performs different functions. Phosphorylation of the S172 residue positioned close to the LIR by TBK1 leads to increased binding affinity of OPTN for LC3-family proteins. Phosphorylation of S473 in the UBAN domain by TBK1 potentiates OPTN binding to Ub, facilitating selective autophagy (Heo et al., 2015; Richter et al., 2016; Li et al., 2018). Phosphorylated OPTN exhibits co-localization with M. tuberculosis in macrophages (Budzik et al., 2020). Zebrafish model studies show that OPTN deficiency diminishes LC3-mycobacteria co-localization, thereby promoting bacterial proliferation, whereas OPTN overexpression augments LC3-bacterial co-localization, leading to reduced bacterial replication (Zhang et al., 2019). OPTN -deficient macrophages infected with high MOI mycobacteria exhibit enhanced cell death, reduced LC3-II levels, and altered Pro-IL-1β expression (Ramachandran et al., 2024). These results suggest that OPTN may play a role in xenophagy against mycobacteria.
NBR1 harbors a coiled-coil domain enabling its dimerization, a PB1 domain that interacts with the corresponding domain of SQSTM1, in addition to LIRs and a UBA domain (Lamark et al., 2003; Lange et al., 2005; Whitehouse et al., 2010; Rogov et al., 2014). Although NBR1 can act independently of and even antagonize SQSTM1 (Kirkin et al., 2009; Deosaran et al., 2013; Nishimura et al., 2024), the assembly of NBR1-SQSTM1 complex significantly enhances the efficacy of pexophagy and simaphagy (Deosaran et al., 2013; Migliano et al., 2024). Interaction with SQSTM1 may have altered the conformation of NBR1, thereby modulating its affinity for ubiquitin and LC3. Similar to SQSTM1 and TAX1BP1, NBR1 is engaged in xenophagy against M. tuberculosis, as NBR1 can recognize and bind the ubiquitinated M. tuberculosis surface protein PE_PGRS29 (Chai et al., 2019).
Exploring autophagy receptors plays a pivotal role in unraveling the molecular mechanisms of selective autophagy. With the increasing identification of these receptors, researchers have progressively turned their focus toward the interactions and regulatory networks among different autophagy receptors. Zebrafish models showed that the autophagic receptors OPTN and p62 function complementarily to independently restrict mycobacterial growth, while the autophagy modulator Dram1 can restore the association of autophagosomes with bacteria and lysosomal acidification even in the absence of both receptors, indicating that the three factors function independently yet synergistically in anti-mycobacterial immunity (Xie and Meijer, 2023). TBK1 phosphorylates autophagy receptors, enhancing their binding affinity to ubiquitinated substrates and promoting the occurrence of selective autophagy. Our study found that mitophagy competitively recruited TBK1 to mitochondria, reducing the translocation of TBK1 to mycobacteria and thereby inhibiting xenophagy (Song et al., 2022).
4 Mycobacteria regulate host autophagy for survival
While the host employs intricate autophagy mechanisms involving selective and non-selective autophagy to combat mycobacterial invasion, mycobacteria have evolved elaborate evasion strategies to subvert or exploit autophagy for their survival (Figure 2; Table 1), highlighting a dynamic interplay between host defense and pathogen evasion.
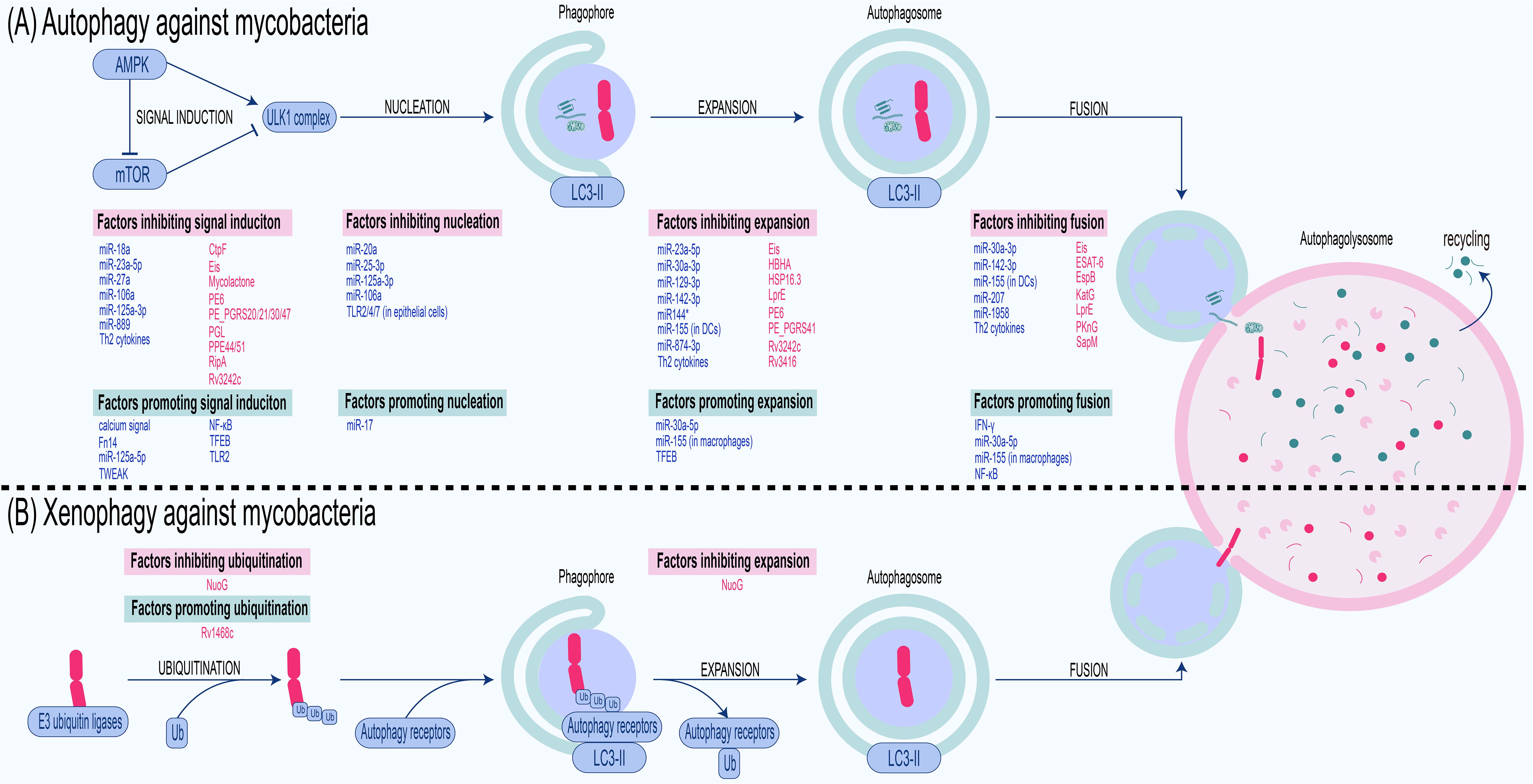
Figure 2. Antimycobacterial autophagy and xenophagy: processes and regulators. (A) Autophagy against mycobacteria. Cells sense nutritional or infection signals via the AMPK/mTOR pathway, activate the ULK1 complex, and recruit LC3-II to the phagophore with the assistance of ATGs. The phagophore embedded with LC3-II expands into an autophagosome, which engulfs components such as bacteria, cytosolic organelles, and macromolecules. This autophagosome then fuses with the lysosome to form an autophagolysosome where the cargo is degraded. Thus, autophagy against mycobacteria also involves steps including signal induction, nucleation, expansion, and fusion, each of which is inhibited or promoted by factors derived from the host and bacteria. (B) Xenophagy against mycobacteria. Upon entering the host cytoplasm, mycobacteria are ubiquitinated by host E3 ubiquitin ligases. The ubiquitinated bacteria are then recognized by autophagy receptors. These receptors bind ubiquitinated pathogens via their UBA domain while simultaneously anchoring to LC3 on the phagophore through their LIR domain . This dual binding recruits the pathogens to the phagophore, which subsequently elongates under the mediation of ATGs to envelop the mycobacteria, forming an autophagosome. The autophagosome then fuses with a lysosome to generate an autolysosome, wherein the bacteria are degraded. Host-derived factors are marked in blue, whereas bacterial factors are marked in red.
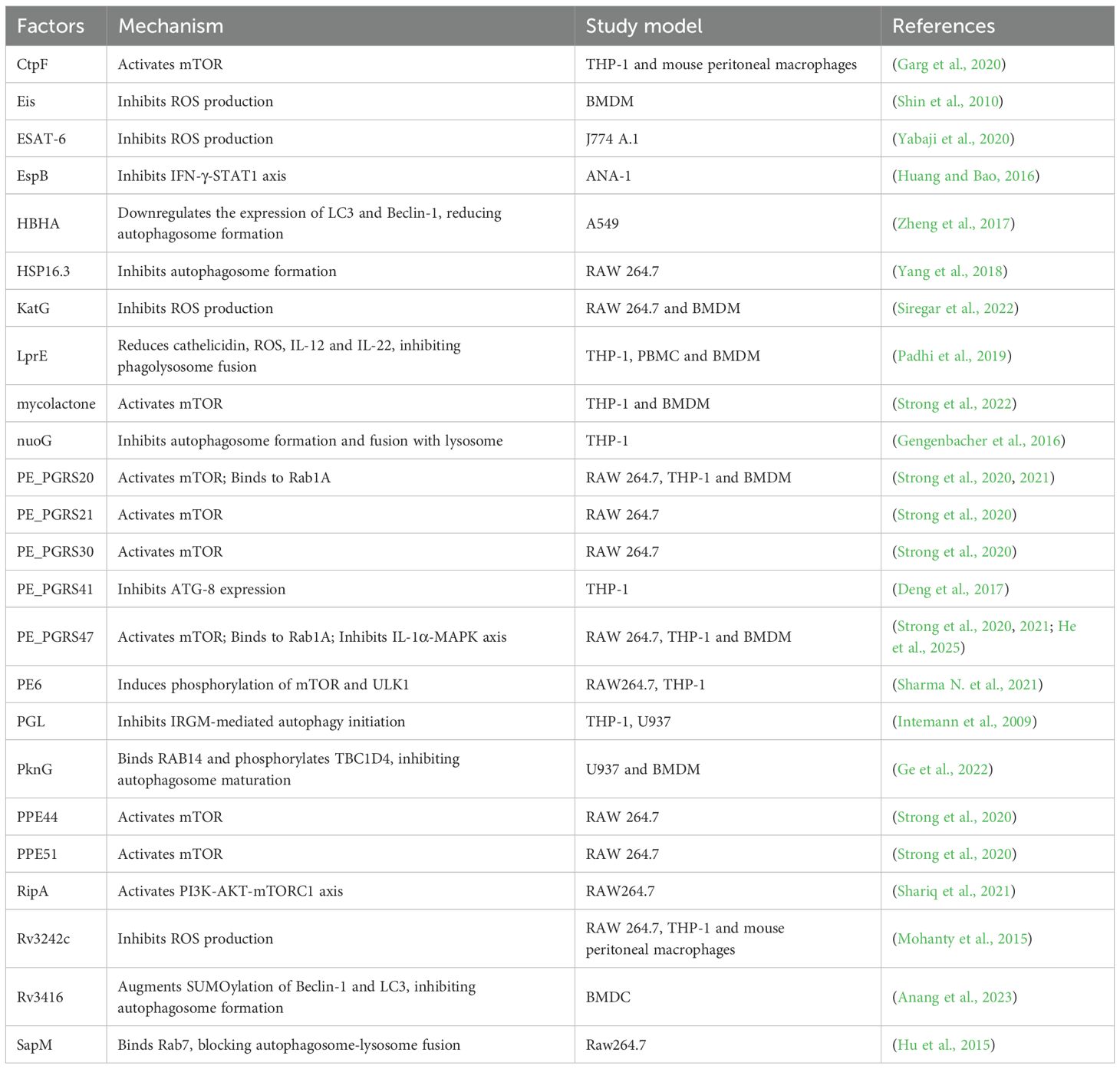
Table 1. Strategies of mycobacteria to regulate host autophagy.
Determinants that inhibit host autophagic clearance are often virulent factors of mycobacteria. As a classical virulence factor in mycobacteria, the ESX-1 secretory system is involved in the modulation of host autophagy. ESX-1 secretion-associated protein B (EspB) of M. tuberculosis reduces mRNA and protein levels of IFN-γ receptor 1 and decreases IFN-induced STAT1 phosphorylation in murine ANA-1 macrophage cells, thereby inhibiting LC3 expression and autophagosome maturation (Huang and Bao, 2016). Macrophages infected with ESAT-6-expressing mycobacteria exhibited elevated levels of SOD-2, decreased autophagosome-lysosome fusion, and increased survival of bacteria. These effects of ESAT-6 may be related to the SOD-2-mediated reduction of ROS (Yabaji et al., 2020). M. ulcerans is the etiological agent of Buruli ulcer, the third most prevalent mycobacterial infection worldwide. A study shows that M. ulcerans secretes mycolactone to induce necrotic cell death in macrophages, promoting bacterial escape while maintaining mTOR activation to suppress autophagy, as demonstrated by restored pathogenicity in a tetracycline-inducible mycolactone expression system (Strong et al., 2022). Phenolic glycolipid (PGL) is one of the virulence factors of M. africanum. Because Euro-American clades of M. tuberculosis lack the gene required for PGL biosynthesis, these clades are readily cleared by host autophagy enhanced by IRGM mutations. In contrast, this enhanced autophagic response is suppressed by M. africanum, which harbors an intact pks1/15 gene to produce PGL (Intemann et al., 2009). Since PGL reduces the synthesis of cytokines capable of inducing autophagy, like IL-6 and IL-12 (Reed et al., 2004; Cambier et al., 2017), it is hypothesized that the inhibition of autophagy by PGL may be partially mediated by the reduction of these cytokines.
The PE/PPE family, which can disrupt the host immune response, has a higher virulence compared to other mycobacterial proteins (Sharma et al., 2022). PE6 induces phosphorylation of mTOR and ULK1, thereby inhibiting the conversion of LC3I to LC3II and reducing autophagy (Sharma N. et al., 2021). Six PE/PPE family proteins with activating effect on mTOR and inhibitory effects on autophagy were screened by loss-of-function screening of an M. tuberculosis transposon mutant library. Expression of these proteins in M. smegmatis confirmed their autophagy inhibitory effect. The inhibition of autophagy conferred higher survival and replication of mycobacteria in host cells. The expression of these PE/PPE proteins varied under different stress conditions, suggesting that PE/PPE may confer adaptability of M. tuberculosis to a wide range of conditions (Strong et al., 2020). PE_PGRS20 and PE_PGRS47 inhibit autophagy initiation by binding to Rab1A of host cells, thereby increasing bacterial survival. Intriguingly, PE_PGRS20 and PE_PGRS47 also inhibit antigen presentation of host cells. This may be due to the fact that antigen presentation is dependent on phagocytic degradation of the mycobacteria by the host cell, and the inhibition of autophagy by PE_PGRS20 and PE_PGRS47 reduces the amount of antigen available for presentation from bacteria degradation (Saini et al., 2016; Strong et al., 2021). Additionally, PE_PGRS47 can synergistically inhibit macrophage autophagy and apoptosis by downregulating IL-1α secretion and inhibiting the MAPK signaling pathway (particularly p38 and ERK1/2), thereby promoting the survival of M. smegmatis within macrophages. This indicates that the multifunctional virulence factor PE_PGRS47 can evade host immunity such as autophagy through multiple pathways (He et al., 2025). PE_PGRS41 is also an autophagy inhibitor because PE_PGRS41 knock-in M. smegmatis reduced autophagy in infected macrophages (Deng et al., 2017). Under the pressure of adapting to the host, some PE/PPE family genes of M. bovis may be deleted or mutated, so that M. bovis has evolved a strategy to evade host autophagy without relying on PE/PPE family proteins. M. bovis inhibits autophagic clearance and promotes its intracellular survival by disrupting autophagosome-lysosome fusion and exploiting host energy metabolism remodeling (Cai et al., 2023).
In addition to the PE/PPE family, certain enzymes in mycobacteria can also inhibit autophagy, thereby facilitating bacterial survival. M. tuberculosis-secreted acid phosphatase SapM can inhibit autophagy by reducing phagosome-lysosome fusion and lysosomal acidification. SapM interacts with GTPase Rab7 through CT domain, thereby inhibiting autophagosome maturation and enhancing autophagosomes accumulation (Hu et al., 2015). RipA is a peptidoglycan hydrolase of M. tuberculosis. Survival of RipA-expressing M. smegmatis is increased in macrophages. Mechanistically, RipA interacts with the LIR of autophagy receptors and inhibits ULK by activating PI3K-AKT-mTORC1 signaling, thereby inhibiting antibacterial autophagy (Shariq et al., 2021). The enhanced intracellular survival (Eis) gene, which is an acetyltransferase, can increase the bacterial viability of mycobacteria, as literally indicated. Eis knockout M. tuberculosis causes infected macrophages to produce more ROS and autophagosomes, suggesting that Eis may inhibit autophagy by reducing ROS production. Genetic recombination assays showed that the inhibitory effect of Eis on ROS and autophagosomes production is dependent on its N-acetyltransferase domain (Shin et al., 2010). A calcium transporting P2A ATPase of M. tuberculosis, CtpF, inhibits autophagy by affecting calcium efflux. During the early stage of M. tuberculosis infection of macrophages, CtpF expression is elevated under conditions of macrophage stress, like hypoxia, elevated nitric oxide concentrations, and acidic environments. Elevated CtpF allows calcium efflux and activates mTOR, thus inhibiting autophagy and enhancing mycobacteria survival (Garg et al., 2020). Visfatin of host cells is associated with autophagy and ROS production during M. tuberculosis infection. Rv3242c of M. tuberculosis, encoding a phosphoribosyltransferase, can inhibit visfatin level in infected macrophages, thereby suppressing autophagy and ROS production. Rv3242c-expressing M. smegmatis activates MAPK and increases IL-10 production, suggesting that the inhibitory effect of Rv3242c on autophagy may be mediated through the MAPK pathway and IL-10 (Mohanty et al., 2015). KatG also inhibits host autophagy by reducing ROS. KatG, a catalase-peroxidase, is upregulated in the M. tuberculosis Beijing strain but not in H37Rv. The upregulated KatG neutralizes mitochondrial ROS generated during M. tuberculosis Beijing strain infection, thereby blocking autophagosome maturation and enhancing intracellular bacterial survival (Siregar et al., 2022). Protein kinase G (PKnG), the eukaryotic-like serine/threonine protein kinase, is secreted by pathogenic mycobacteria in infected macrophages, where it initiates autophagy but prevents fusion of the autophagosome and lysosome (Walburger et al., 2004; Ge et al., 2022). For autophagy initiation, PKnG binds directly to the pleckstrin homology domain of AKT, thereby reverting the inhibitory effect of AKT for autophagy. For autophagosome maturation inhibition, PKnG binds directly to host small GTPase RAB14 and inhibits the GTPase activity of RAB14. In addition, PKnG phosphorylates TBC1 domain family member 4 (TBC1D4), depriving it of the ability to activate RAB14 (Ge et al., 2022). Xenophagy against mycobacteria is associated with NADH dehydrogenase. NuoG, which encodes NADH dehydrogenase I subunit G, was previously recognized as an anti-apoptotic virulence factor of M. tuberculosis. However, knockout of NuoG allows more LC3 to be recruited to intracellular M. bovis, suggesting that NuoG is also involved in the inhibition of xenophagy (Gengenbacher et al., 2016). Regulation of NADH dehydrogenase determines the ability of different strains to induce bactericidal xenophagy.
Some other functional proteins apart from enzymes can also inhibit host autophagy. Latent mycobacteria infection is dependent on the balance between host and pathogenic bacteria, and heat shock proteins (HSPs) of M. tuberculosis play a key role in this process. Autophagosomes in HSP16.3 mutant M. tuberculosis infected macrophages are significantly more than that in wild strain infected cells, suggesting that HSP16.3 may increase bacterial survival by inhibiting host autophagy (Yang et al., 2018). Heparin-binding hemagglutinin (HBHA) of M. tuberculosis is another autophagy inhibitor. In A549 cells, HBHA avoids phagosome maturation by inhibiting LC3 expression, thereby increasing the survival of M. smegmatis expressing HBHA (Zheng et al., 2017). The inhibition of autophagy by M. bovis or Rv3416 of M. tuberculosis may be mediated through enhanced SUMOylation rather than ubiquitination. Inhibiting SUMOylation enhances the expression of autophagy markers and promotes autophagy, whereas M. bovis or Rv3416 of M. tuberculosis augment the SUMOylation of these autophagic molecules to suppress autophagy in infected bone marrow derived dendritic cells (BMDCs) (Anang et al., 2023). Mb3523c, a structural protein of M. bovis belonging to the Mce4 family, promotes bacterial evasion of clearance by inducing host CMA and ferroptosis. Mechanistically, Mb3523c protein promotes CMA by interacting with host HSP90 at Y237 and G241 sites, stabilizing LAMP2A on lysosomes to facilitate GPX4 degradation via the CMA pathway, thereby inducing ferroptosis to enhance bacterial pathogenicity and dissemination (Wang H. et al., 2025). Lipoprotein is a critical class of virulence proteins of M. tuberculosis, and its virulence is associated with the manipulation of autophagy. LprE mutant M. tuberculosis causes more autophagy-associated protein expression and more recruitment of lysosomal and phagosomal proteins in infected macrophages, suggesting that LprE inhibits autophagosome formation and fusion of autophagosomes with lysosomes. LprE inhibits phago-lysosome fusion because of downregulation of IL-12 and IL-22 (Padhi et al., 2019). As an important virulence protein of M. tuberculosis, the mechanism by which lipoproteins inhibit autophagy and increase intracellular bacterial load has been ambiguous and more investigation is needed.
Mycobacteria can also regulate autophagy by phosphorylating/dephosphorylating upstream molecules of autophagy. Phosphoproteome analysis revealed that M. tuberculosis infection induces extensive dephosphorylation of host proteins in macrophages, particularly in MAPK and PI3K signaling pathways critical for autophagy activation, whereas avirulent M. bovis infection elicits milder phosphorylation changes and preserves partial autophagic signaling (Choudhary et al., 2020). A recent study revealed the role of potassium ion in the induction of autophagy. Mycobacterial infection can upregulate the surface expression of potassium ion channel Kir2.1 in epithelial cells and macrophages. Inhibition of Kir2.1 can promote autophagy and apoptosis by enhancing oxidative burst and activating the MAPK/NF-κB pathway, significantly reducing bacterial survival, suggesting that Kir2.1 may assist mycobacterial immune escape by regulating ion homeostasis (Sinha et al., 2024). However, the specific factors by which mycobacteria regulate host protein phosphorylation status and enhance potassium channel protein expression remain to be elucidated.
Although many studies have shown that autophagy is an important part of the host’s response to clear mycobacteria, a growing amount of research has challenged this opinion. On the one hand, in addition to autophagy, autophagy genes are also involved in non-autophagic processes (Galluzzi and Green, 2019), and it is difficult to conclude that the changes in intracellular mycobacteria survival after the deletion of a certain host autophagy gene are necessarily related to autophagy. For instance, an ingenious investigation showed no change in mycobacteria proliferation after abrogating host autophagy by knocking out the autophagy genes ATG3, ATG7, ATG12, ATG14, or ATG16l1, suggesting that autophagy is dispensable for inhibiting mycobacteria proliferation. Concurrently, the deletion of ATG5 resulted in a marked increase in bacterial proliferation, ultimately leading to the demise of all infected mice. The research team put forth the hypothesis that ATG5 exerts its antimycobacterial effects through preventing neutrophil-mediated immunopathology, rather than autophagy (Kimmey et al., 2015). So, further investigation is required to elucidate the precise function of autophagy genes in combating mycobacteria. On the other hand, certain virulence factors associated with mycobacteria species have been observed to induce autophagosome formation while simultaneously inhibiting the fusion of these autophagosomes with lysosomes. In addition to the previously mentioned PKnG, ESX-1 secretory system can also be considered as a virulent factor of this kind. ESX-1 secretory system is necessary for the induction of host xenophagy during early stages of M. tuberculosis infection (Watson et al., 2012, 2015); however, it also inhibits the fusion of autophagosomes with lysosomes during the late infection stages (Romagnoli et al., 2012; Chandra et al., 2015). The dysregulation of organelles has the potential to induce the secretion of antimicrobial and inflammatory cytokines (Zhou et al., 2011; Liao et al., 2019) and autophagy may assist in the removal of dysregulated organelles, thereby reducing the secretion of these cytokines (Nakahira et al., 2011; Di Rita et al., 2021; Han et al., 2021).In the event of M. tuberculosis inhibiting the entirety of the autophagic flux, this would result in dysregulated organelles remaining incompletely cleared and an increase in cytokine secretion. To prevent the maturation of bacteriophage-containing autophagosomes, while allowing for the maturation of bacteriophage-free autophagosomes, M. tuberculosis employed a subtle strategy to expel Rab7 from M. tuberculosis-containing autophagosomes. This approach ensured the uninterrupted progression of entire autophagic fluxes (Chandra and Kumar, 2016). This evidence suggests that autophagosomes may serve as a niche for mycobacteria replication.
5 Agents targeting autophagy against mycobacteria
The ability of mycobacteria to inhibit autophagy may be negatively correlated with their pathogenicity (Gonzalez-Orozco et al., 2022). Faced with the sophisticated strategies employed by mycobacteria to regulate autophagy for survival, the development of therapeutic interventions that modulate autophagy in the host has emerged as a promising approach in combating mycobacterial infections (Table 2). This type of approach is generally called HDT.
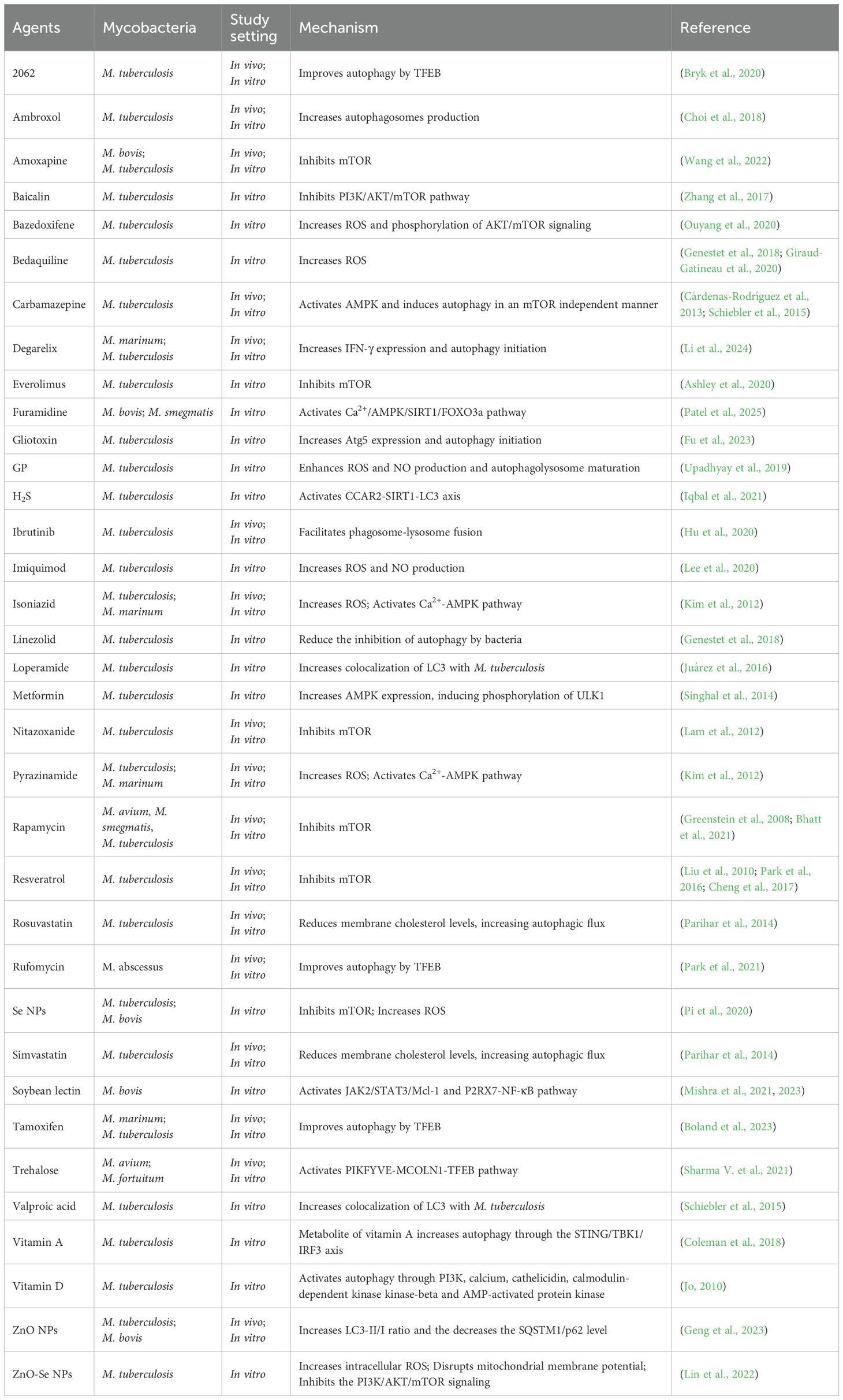
Table 2. Agents targeting autophagy against mycobacteria.
5.1 Classic autophagy inducers
Rapamycin is one of the most extensively researched autophagy inducers. Indeed, in an experimental context, rapamycin demonstrated notable inhibitory activity against mycobacterial infections (Greenstein et al., 2008; Bhatt et al., 2021). However, the absorption of rapamycin in the gastrointestinal tract exhibits considerable fluctuations, necessitating monitoring during administration and causing significant inconvenience in the clinical use of the drug. The administration of rapamycin has been observed to induce interstitial pneumonitis, which may potentially offset some of the positive antimicrobial effects observed in cases of mycobacteria pulmonale infection. Additionally, rifampicin, a standard treatment for tuberculosis, stimulates expression of the hepatic enzyme CYP3A4, which metabolizes rapamycin. These factors restrict the clinical application of rapamycin in the treatment of mycobacteria infections. Everolimus, as a rapamycin analog, also targets mTOR and can significantly reduce the intracellular M. tuberculosis burden in granulomas (Ashley et al., 2020). However, experiments in M. tuberculosis-infected THP-1 macrophages, a different picture emerged. Everolimus stimulated autophagy by increasing ROS and autophagosome formation, but it did not promote autolysosome generation or significantly inhibit intracellular M. tuberculosis replication (Bianco et al., 2023). This contradiction highlights the importance of more preclinical experiments when everolimus comes to drug repurpose. Ibrutinib, as another agent targeting the mTOR pathway, can significantly increase the colocalization of LC3 and M. tuberculosis as well as auto-lysosome fusion, significantly reducing the M. tuberculosis load in the mediastinal node and spleen of infected mice (Hu et al., 2020). If further research can confirm that everolimus and ibrutinib do not have the clinical limitations of rapamycin, then these agents could be promising HDT drugs.
Metformin, renowned for its AMPK-mediated inhibitive role in mTOR signaling pathway, is prevalent in managing type-2 diabetes, and it triggers ROS generation, phagosome maturation, autophagy in vitro (Singhal et al., 2014; Yang, 2017; Russell et al., 2019). The administration of metformin to healthy human volunteers led to a notable downregulation of genes associated with the mTOR signaling pathway and an upregulation of genes associated with phagocytosis and the generation of ROS. In vitro, the metformin treatment in mononuclear cells isolated from peripheral blood of healthy donors resulted in enhancement of cellular metabolism and suppression of mTOR downstream effectors, p70S6K and 4EBP1 (Lachmandas et al., 2019). Several clinical studies have shown that the use of metformin effectively reduces the risk of active tuberculosis and mortality in patients with diabetes mellitus and effectively promotes sputum culture conversion in cavitary pulmonary patients (Degner et al., 2018; Lee M. et al., 2018; Lee Y. et al., 2018). Consistent with the above studies, Singhal et al. showed that metformin restricted the growth of drug-resistant M. tuberculosis strains in an AMPK-dependent manner, alleviated lung lesions in infected mice, increased ROS production and phagosome maturation, and improved the efficacy of anti-tuberculosis drugs (Singhal et al., 2014). However, another experiment conducted by Dutta et al. showed that the combination of metformin did not improve the activity of first-line anti-tuberculosis drugs in mice (Dutta et al., 2017). This discrepancy cannot be explained by the dose of metformin because the same dose was used in both experiments (250 mg/kg). It is speculated that this difference may be related to the different experimental mouse strains used in the two experiments. Given that Dutta et al. used RHZE and rifampicin while Singhal et al. used isoniazid or ethambutol, another seemingly more plausible theory is that the powerful drug RHZE utilized in the Dutta’s experiment masked the effects of metformin, or that rifampicin sped up the clearance of metformin.
As classic autophagy inducers, rapamycin and metformin have shown the potential to eliminate mycobacteria, but issues such as side effects, drug interactions and consistency of therapeutic efficacy have restricted their clinical application. Future research needs to focus on analogue optimization, delivery system innovation and combined therapeutic strategies to promote the clinical transformation of host-directed therapy in the fight against mycobacterial infections.
5.2 Antibiotics
Commonly used anti-tuberculosis antibiotics isoniazid and pyrazinamide have the effect of inducing autophagy and ROS production in host cells infected by M. tuberculosis (Kim et al., 2012). Isoniazid and pyrazinamide increase cellular and mitochondrial ROS and facilitate phagosome-lysosome fusion in M. tuberculosis-infected host cells. In vivo, its antimycobacterial efficacy relies on host autophagy, as autophagy-defective models show reduced survival in the context of administering antibiotics (Kim et al., 2012). Another antibiotic that can induce host cell autophagy is nitazoxanide. Nitazoxanide can directly restrain M. tuberculosis proliferation in vitro, and it exerts a stronger inhibitory effect within host cells. Mechanistically, nitazoxanide suppresses the quinone oxidoreductase in host cells, thereby blocking the mTOR signaling pathway and enhancing autophagy (Lam et al., 2012). Bedaquiline and 2062, respectively a new antibiotic and a small molecule substance, can both increase autophagy and phagosome-lysosome fusion by activating TFEB (Bryk et al., 2020; Giraud-Gatineau et al., 2020). Given the effectiveness of autophagy in clearing intracellular bacteria, it is worth exploring whether first-line anti-tuberculosis drugs, which have abundant safety evaluation data, can be used as adjunctive medication against NTMs, even if they do not have significant activity direct against NTMs.
Rufomycin can partially restore the expression and nuclear translocation of TFEB in BMDMs infected with the M. abscessus, activate the mRNA expression of autophagy/lysosome-related genes downstream of TFEB, and increase the colocalization of M. abscessus phagosomes and lysosomes, indicating that rufomycin can enhance autophagy during M. abscessus infection (Park et al., 2021).
Antibiotic-induced autophagy is not necessarily due to direct stimulation of host cells. After macrophages are infected with M. tuberculosis pre-treated with rifampicin, linezolid or bedaquiline, autophagy activation and efficacy are enhanced, indicating that antibiotics can promote host cell autophagic clearance by altering bacterial protein synthesis or energy metabolism (Genestet et al., 2018). Exposure to isoniazid, bedaquiline, rifampicin, and O-floxacin causes M. tuberculosis to exhibit higher NADH: NAD+ ratios in infected macrophages, facilitating the production of more ROS (Bhat et al., 2016). ROS is an inducer of autophagy. The ROS produced by M. tuberculosis exposed to antibiotics may be released into the infected host cells, thereby inducing autophagy. It is speculated that this is at least part of the mechanism by which antibiotics are effective against M. tuberculosis (Piccaro et al., 2014).
Although some commonly used anti-tuberculosis antibiotics show the potential to induce autophagy, the association between their antibacterial activity and autophagy remains unclear, and the mechanism of action needs to be refined. In the future, efforts should be focused on mechanism analysis and exploration of the potential for adjuvant therapy.
5.3 Other chemical agents
Multidrug-resistant M. tuberculosis has evolved mechanisms to resist autophagy, resulting in basal autophagy being insufficient to eliminate the bacteria. The anticonvulsant drug carbamazepine can induce sufficient autophagy to clear M. tuberculosis through a pathway that does not rely on mTOR but reduces intracellular myoinositol (Schiebler et al., 2015). In addition, carbamazepine can also activate AMPK. Although this result does not come from M. tuberculosis infection experiments, it does not rule out the possibility that AMPK is involved in the process of carbamazepine-induced autophagy in infected cells (Cárdenas-Rodríguez et al., 2013). Another anticonvulsant medication valproic acid and the anti-diarrhea drug loperamide can also enhance autophagy, and this enhancement is evidenced by the increased co-localization of LC3 and M. tuberculosis (Schiebler et al., 2015; Juárez et al., 2016). Furamidine, a minor groove binder of DNA, is also an autophagy inducer that acts on AMPK. Furamidine induces autophagy in differentiated THP-1 cells via the Ca2+/AMPK/silent mating type information regulation 1 (SIRT1)/forkhead box O3 (FOXO3a) signaling pathway, reducing intracellular M. tuberculosis burden through enhanced autophagic flux, as evidenced by LC3-II conversion, autophagic vacuole accumulation, and activation of autophagic markers (Patel et al., 2025).
Degarelix, a synthetic decapeptide GnRH antagonist, inhibits luteinizing and follicle-stimulating hormone production to reduce testosterone and estrogen synthesis, and was clinically approved for prostate cancer treatment (Devos et al., 2023). A recent study shows that degarelix induces autophagy initiation in macrophages via a PI3K-dependent pathway, potentially synergizing with IFN-γ upregulation to enhance antimycobacterial activity, without altering classical autophagic flux. These findings highlight degarelix as a novel host-directed therapeutic candidate for TB by targeting early autophagic mechanisms (Li et al., 2024). Gliotoxin, a metabolite derived from marine fungi, is a bioactive compound with potential antibacterial properties. Experiments showed that gliotoxin significantly increased the LC3-II/LC3-I ratio and ATG5 expression to promote autophagy, and the autophagy inhibitor 3-MA could suppress the induced autophagy and restore gliotoxin-inhibited M. tuberculosis infection. Since 3-MA mainly inhibits the initiation of autophagy, gliotoxin might suppress M. tuberculosis infection in macrophages by promoting autophagy initiation (Fu et al., 2023). The study found that amoxapine can inhibit mTOR activation, induce autophagy in macrophages, increase the level of LC3B-II, promote autophagosome formation without affecting autophagic flux. After inhibiting autophagy with 3-MA or knocking down ATG16L1, the antibacterial effect of amoxapine against intracellular mycobacteria was significantly weakened, indicating that autophagy plays a crucial role in the process of amoxapine inhibiting the growth of intracellular mycobacteria (Wang et al., 2022). Dimethyl itaconate is another agent that can induce autophagy initiation, but it has not been approved for clinical treatment. Dimethyl itaconate can enhance autophagic flux and phagolysosomal fusion in macrophages infected with mycobacteria, as evidenced by increased autophagic LC3-II accumulation and bacterial colocalization with lysosomes (Kim et al., 2023).
The decrease of membrane cholesterol levels mediated by simvastatin and rosuvastatin promotes autophagy and phagosomal maturation, reduces bacterial load and lung burdens, and improves histopathologic changes (Parihar et al., 2014). Fluvastatin, another member of statins, possesses moderate antimycobacterial activity against M. tuberculosis (Battah et al., 2019). It is plausible to hypothesize that fluvastatin reduces membrane cholesterol synthesis by inhibiting hydroxy-methyl-glutaryl-CoA (HMG-CoA) reductase, thereby promoting phagosomal maturation and autophagy to reduce intracellular bacterial burdens in infected host cells.
Selective estrogen receptor modulators, such as tamoxifen and bazedoxifene, have been shown to exhibit the effect of inhibiting the growth of M. tuberculosis within macrophages. In vitro and in vivo, tamoxifen increases autophagy related vesicles, enhances mycobacterial localization in lysosomes, and its antimycobacterial effect is associated with autophagy -lysosomal pathway modulation, as inhibition of lysosomal activity reduces its efficacy (Boland et al., 2023). Treatment with bazedoxifene increases autophagosome formation and the expression of LC3B-II protein in infected macrophages. This indicates that the anti-mycobacterial activity of bazedoxifene might be related to autophagy. Autophagy indeced by bazedoxifene is associated with an increase in ROS and the phosphorylation of the AKT/mTOR signaling pathway (Ouyang et al., 2020).
Imiquimod, a drug for treating superficial basal cell carcinoma, induces BNIP3-mediated mitophagy through stimulating TLR7 of macrophage. On the other hand, imiquimod induces NO production through the GSK-3β-mediated signaling pathway, which leads to autophagy in the late stage. The autophagy triggered by imiquimod can effectively eliminate M. tuberculosis within macrophages (Lee et al., 2020).
Ambroxol, a lead compound identified from screens for autophagy-inducing drugs, represents a potential host-directed therapy adjunct to conventional antibiotic chemotherapy against M. tuberculosis. At clinically relevant doses, ambroxol elicited autophagy both in vitro and in vivo, thereby promoting the elimination of mycobacteria by host. Moreover, ambroxol additionally potentiated the antimicrobial activity of rifampin in vivo, demonstrating synergistic effects in combating mycobacterial infection (Choi et al., 2018).
A study found that H2S can sulfhydrate GAPDH and cause its translocation to the nucleus, where it interacts with cell cycle and apoptosis regulator 2 (CCAR2), disrupts the CCAR2-SIRT1 complex, activating SIRT1. Subsequently, SIRT1 deacetylates LC3B, enabling its translocation to the cytoplasm and inducing autophagy. Moreover, H2S-induced autophagy can promote the trafficking of M. tuberculosis to lysosomes and restrict its intracellular growth. This process depends on the sulfhydration of GAPDH, and SIRT1 is crucial for H2S-induced autophagy and the inhibition of M. tuberculosis growth (Iqbal et al., 2021). Since GAPDH is widely present in various types of cells, autophagy dependent on the sulfhydration of GAPDH may occur in various cells, enabling H2S to serve as a broad-spectrum autophagy inducer. However, considering the toxicity of H2S, developing low-toxicity analogs might be a better strategy.
Clinically approved chemical drugs with validated safety profiles, particularly those indicated for patients with comorbidities involving their primary approved indications and mycobacterial infections, warrant mechanistic exploration of their synergistic therapeutic potential and precise druggable targets to advance rational combination therapies.
5.4 Natural products
The catalytic product of vitamin D-1-hydroxylase, calcitriol, exhibits antibacterial effects. By activating TLR to upregulate vitamin D-1-hydroxylase, cathelicidin can be induced, indicating that calcitriol may exert its antibacterial effect through cathelicidin (Liu et al., 2006; Periyasamy et al., 2020). Cathelicidin may induce autophagy through pathways such as TFEB, AMPK, ULK1 and MAPK (Ikutama et al., 2023; Xi et al., 2024; Yang et al., 2024).
Although studies have shown that vitamin D can induce autophagy through multiple pathways (Jo, 2010), based on meta-analyses from multiple randomized controlled trials, vitamin D does not have consistent efficacy in the HDT against tuberculosis (Wu et al., 2018; Jolliffe et al., 2019; Tang et al., 2020). Although a meta-analysis shows that vitamin D deficiency may be a risk factor for tuberculosis (Huang et al., 2017), high-dose use of vitamin D cannot effectively accelerate the sputum culture conversion process in the entire trial population, but it is only effective in MDR-TB cases or patients with a specific genotype, such as polymorphisms in the vitamin D receptor gene (Tukvadze et al., 2015; Ganmaa et al., 2017). The combined use of vitamin D and phenylbutyrate (PBA) can induce the expression of cathelicidin and cathelicidin-induced autophagy in macrophages, reducing M. tuberculosis proliferation (Coussens et al., 2015; Rekha et al., 2015). However, the effect of this combined therapy on increasing cathelicidin expression is only observed at very specific doses of PBA (Mily et al., 2013). It is possible that due to the specific doses of PBA, multiple randomized controlled trials of the combined use of vitamin D and PBA cannot yield consistent results (Mily et al., 2015; Bekele et al., 2018), leading to vitamin D not being able to become an effective HDT for treating mycobacterial infections.
Similar to vitamin D, vitamin A may be involved in host resistance to M. tuberculosis. Vitamin A deficiency was significantly associated with elevated tuberculosis incidence among HIV-infected individuals (Tenforde et al., 2017). Mechanistically, the metabolite of vitamin A, all-trans retinoic acid, increases autophagy through the STING/TBK1/IRF3 axis, enhancing the colocalization of M. tuberculosis autophagic vesicles and acidified lysosomes (Coleman et al., 2018). Although vitamin A can reduce the mycobacterial load in mice, there is a shortage of consistent evidence regarding its benefits in tuberculosis patients (Karyadi et al., 2002; Lawson et al., 2010; Visser et al., 2011; Wang J. et al., 2020). As a result, whether vitamin A can be used as an adjunct therapy for tuberculosis remains indeterminate.
Many studies have confirmed the protective effects of autophagy in alleviating the excessive inflammatory response caused by M. tuberculosis (Castillo et al., 2012). Excessive activation of inflammasomes can lead to the excessive secretion of pro-inflammatory cytokines. The excessive secretion of these cytokines triggered by M. tuberculosis infection can lead to lung damage, which is detrimental to recovery. Excessive inflammasome activation can be mitigated by the autophagic clearance of endogenous stimuli and inflammasome components (Nakahira et al., 2011; Zhou et al., 2011; Shi et al., 2012). In this way, autophagy exerts protective effects in mycobacterial infections from two aspects: on one hand, autophagy can engulf and eliminate pathogens; on the other hand, autophagy can reduce inflammatory damage caused by the infection. Baicalin induces autophagy in M. tuberculosis-infected macrophages through the PI3K/AKT/mTOR pathway while simultaneously reducing inflammasome activation by inhibiting the PI3K/AKT/NF-κB pathway. Induced autophagy can also clear inflammasomes, thereby reducing the production of inflammatory cytokines. Therefore, baicalin can be considered a candidate drug for eliminating M. tuberculosis and reducing inflammatory lung damage (Zhang et al., 2017).
Resveratrol induces autophagy by directly binding to the ATP-binding pocket of mTOR or promoting the interaction between mTOR and its inhibitor DEPTOR, thereby suppressing mTOR signaling (Liu et al., 2010; Park et al., 2016). Resveratrol, a SIRT1 activator, reduces intracellular growth of drug-susceptible and drug-resistant M. tuberculosis strains by inducing phagosome-lysosome fusion and autophagy in a SIRT1-dependent manner while dampening M. tuberculosis-mediated inflammatory responses via deacetylation of RelA/p65. In M. tuberculosis-infected mice, Resveratrol ameliorates lung pathology, reduces chronic inflammation, and enhances the efficacy of anti-TB drugs, highlighting its potential as a host-directed therapy for tuberculosis (Cheng et al., 2017).
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is an agent that can reduce the M. tuberculosis and Mav burden. In hosts infected with either M. tuberculosis or Mav, GM-CSF enhanced phagosome maturation and inhibited bacterial growth. Autophagy might play a significant role in GM-CSF activated immunity against M. tuberculosis and NTM (Kedzierska et al., 2000; Nannini et al., 2002; de Silva et al., 2007; Hariadi and Blackwood, 2017).
Soybean lectin, a glycoprotein isolated from soybean seeds with immunomodulatory activity, triggers IL-6 secretion through the P2RX7-dependent PI3K/AKT/CREB pathway, which activates the JAK2/STAT3/Mcl-1 pathway in an autocrine manner to induce autophagy and eliminate intracellular mycobacteria in differentiated THP-1 macrophages (Mishra et al., 2023). Furthermore, there is also a study showing that soybean lectin induces autophagy in differentiated THP-1 cells via a P2RX7-NF-κB-dependent pathway, increasing ROS generation and enhancing autophagic flux, thereby restricting intracellular M. tuberculosis growth in infected cells (Mishra et al., 2021).
Trehalose is a naturally occurring disaccharide with mTOR-independent autophagy-inducing properties. HIV infection was shown to inhibit autophagy flux in macrophages, promoting the survival of M. tuberculosis and NTM. Conversely, trehalose induced autophagy via a phosphoinositide kinase-FYVE finger containing (PIKFYVE)- Transient receptor potential channel mucolipin-1 (TRPML1)-dependent pathway, promoting TFEB nuclear translocation, upregulating autophagy and lysosomal genes, enhancing phagosome-lysosome colocalization, and thus restricting intracellular mycobacterial survival during both single and HIV co-infection (Sharma V. et al., 2021).
Exploring efficacy variability from the perspective of genetic polymorphisms and developing genotype-matched optimized protocols may facilitate the promotion of natural products in mycobacterial disease treatment.
5.5 Nanoparticles
ZnO nanoparticles (ZnO NPs) exhibit antibacterial effects against various M. tuberculosis strains, including multidrug-resistant strains. Moreover, ZnO NPs can dose-dependently induce autophagy and reduce mycobacteria load within macrophages. However, high-dose ZnO NPs can cause ferroptosis. Studies have shown that the combination of ferroptosis inhibitor and ZnO NPs can induce sufficient levels of autophagy to eliminate M. tuberculosis while avoiding acute lung injury that may be caused by ferroptosis in vivo (Geng et al., 2023).
Se nanoparticles (Se NPs) are another type of nanoparticles that not only have antibacterial activity (Huang et al., 2019; Estevez et al., 2020) but also activate host cell immune responses, such as autophagy (Pi et al., 2020). The autophagy activated by Se NPs is related to the alteration of ROS production, mitochondrial membrane potential, and the PI3K/AKT/mTOR signaling pathway, playing a crucial role in clearing intracellular M. tuberculosis.
The novel zinc oxide selenium nanoparticles (ZnO-Se NPs) made by combining ZnO NPs and Se NPs, like ZnO NPs or Se NPs, have the ability to directly suppress extracellular M. tuberculosis and stimulate the host cell immune response for intracellular M. tuberculosis elimination. ZnO-Se NPs induce host cell autophagy by increasing intracellular ROS, disturbing mitochondrial membrane potential, and blocking the PI3K/AKT/mTOR signaling pathway (Lin et al., 2022). Thus, nanoparticles like ZnO NPs, Se NPs and ZnO-Se NPs, which combine direct bactericidal and autophagy-activating effects, have the potential to develop into new therapeutic agents against mycobacteria.
Similar to ZnO-Se NPs, β-Glucan particles (GP) can trigger strong immune responses, including autophagy, within M. tuberculosis-infected macrophages. Unlike ZnO-Se NPs, GP cannot directly eliminate bacteria, but it can serve as a delivery system to transport Rifabutin (RB) nanoparticles into macrophages, enhancing the bactericidal efficacy of RB (Upadhyay et al., 2019).
Graphene oxide (GO) nanoparticles cannot directly kill bacteria extracellularly, nor can they induce autophagy or other immune responses within M. tuberculosis-infected macrophages. However, they can serve as drug delivery vehicles to transport drugs with autophagy-inducing or bactericidal activities into host cells, thereby achieving and maintaining high intracellular drug concentrations, which facilitates the clearance of M. tuberculosis (Saifullah et al., 2017; Pi et al., 2019; De Maio et al., 2020). Curcumin is an effective autophagy inducer, but poor bioavailability limits its application prospects (Lopresti, 2018). Encapsulating curcumin in a polylactic acid-glycolic acid shell to obtain polymerized in situ curcumin nanoparticles can effectively improve the bioavailability of curcumin and significantly increase autophagy in M. tuberculosis-infected macrophages (Gupta et al., 2023).
In addition to chemical drugs, nanoparticles can also deliver nucleic acids to induce autophagy. Nucleic acids are widely involved in the regulation of autophagy in host cells infected by M. tuberculosis. Liposomal nanoparticles can simultaneously deliver chemical drugs and nucleic acids into cells, combining the advantages of chemotherapy, nanotechnology, and nucleic acid therapy for autophagy induction. Delivering a certain concentration of siRNA and anti-tuberculosis drugs into THP-1 cells infected with M. tuberculosis through liposomal nanoparticles can significantly increase the proportion of autophagic cells (Niu et al., 2015).
Nanomedicines demonstrate remarkable advantages in therapeutic, preventive, and diagnostic applications for diseases. Numerous nanomaterials exhibit promising potential in enhancing mycobacterial clearance through direct or indirect modulation of autophagy. Continued exploration of their anti-tuberculosis mechanisms, in vivo metabolic profiles, biosafety, and degradation patterns will facilitate the development of novel therapeutic strategies against mycobacteria.
6 Conclusion and prospects
Autophagy, a conserved cellular mechanism, plays a pivotal role in the host’s defense against mycobacterial infections, encompassing both non-selective and selective pathways. However, mycobacteria, including M. tuberculosis and NTM, have evolved sophisticated strategies to subvert autophagy, such as deploying PE/PPE family proteins, SapM, and ESX-1 secretion systems to inhibit autophagosome-lysosome fusion or activate mTOR signaling.
HDT targeting autophagy has emerged as a promising approach. Classic autophagy inducers, antibiotics, natural products and nanomaterials have shown efficacy in preclinical models by enhancing autophagic flux or bacterial clearance. Notably, nanomedicines offer dual advantages of direct bactericidal activity and autophagy induction, overcoming limitations like poor bioavailability.
Key challenges and future directions include:
Mechanistic clarification. Disentangling the specific roles of autophagy genes from their non-autophagic functions and understanding how mycobacterial virulence factors differentially modulate autophagy at distinct infection stages.
Multi-omics integrated modeling. Integrating transcriptome, proteome, and metabolome data to construct a predictive biomarker model for autophagy regulation therapy.
Therapeutic optimization. Developing autophagy-inducing agents with minimal side effects and addressing drug interactions. Combination therapies and nanocarrier-based delivery systems may enhance efficacy and reduce toxicity.
Translational research. Advancing preclinical findings to clinical trials, particularly for NTM and drug-resistant tuberculosis. Genotype-specific therapies and personalized medicine approaches warrant exploration.
In summary, autophagy represents a critical nexus of host-pathogen interaction in mycobacterial diseases. Future research integrating molecular mechanisms, innovative drug delivery, and clinical translation will unlock its full potential for developing effective, host-centric therapies against these persistent pathogens.
Author contributions
JL: Writing – original draft, Software, Methodology. HF: Writing – review & editing, Validation. DC: Writing – review & editing, Investigation, Software. HZ: Supervision, Writing – review & editing. YL: Investigation, Supervision, Funding acquisition, Validation, Writing – review & editing, Writing – original draft, Project administration, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Natural Science Foundation of Sichuan (Project Nos. 2023NSFSC0176, 2024NSFJQ0005) and Southwest Minzu University Research Start-Up Fund for Introduced Talents (Project Nos. RQD2021071).
Acknowledgments
We acknowledge the Scientific and Technological Innovation Team for Qinghai-Tibetan Plateau Research in Southwest Minzu University (Grant No. 2024CXTD15) for generous support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdrakhmanov, A., Gogvadze, V., and Zhivotovsky, B. (2020). To eat or to die: Deciphering selective forms of autophagy. Trends Biochem. Sci. 45, 347–364. doi: 10.1016/j.tibs.2019.11.006
Alonso, S., Pethe, K., Russell, D. G., and Purdy, G. E. (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U S A. 104, 6031–6036. doi: 10.1073/pnas.0700036104
Anang, V., Singh, A., Kumar Rana, A., Saraswati, S. S. K., Bandyopadhyay, U., Verma, C., et al. (2023). Mycobacteria modulate SUMOylation to suppresses protective responses in dendritic cells. PloS One 18, e0283448. doi: 10.1371/journal.pone.0283448
Ashley, D., Hernandez, J., Cao, R., To, K., Yegiazaryan, A., Abrahem, R., et al. (2020). Antimycobacterial effects of everolimus in a human granuloma model. J. Clin. Med. 9, 2043. doi: 10.3390/jcm9072043
Bah, A., Lacarrière, C., and Vergne, I. (2016). Autophagy-related proteins target ubiquitin-free mycobacterial compartment to promote killing in macrophages. Front. Cell Infect. Microbiol. 6. doi: 10.3389/fcimb.2016.00053
Battah, B., Chemi, G., Butini, S., Campiani, G., Brogi, S., Delogu, G., et al. (2019). A repurposing approach for uncovering the anti-tubercular activity of fda-approved drugs with potential multi-targeting profiles. Molecules 24, 4373. doi: 10.3390/molecules24234373
Behura, A., Das, M., Kumar, A., Naik, L., Mishra, A., Manna, D., et al. (2021). ESAT-6 impedes IL-18 mediated phagosome lysosome fusion via microRNA-30a upon calcimycin treatment in mycobacteria infected macrophages. Int. Immunopharmacol. 101, 108319. doi: 10.1016/j.intimp.2021.108319
Behura, A., Mishra, A., Chugh, S., Mawatwal, S., Kumar, A., Manna, D., et al. (2019). ESAT-6 modulates calcimycin-induced autophagy through microRNA-30a in mycobacteria infected macrophages. J. Infect. 79, 139–152. doi: 10.1016/j.jinf.2019.06.001
Bekele, A., Gebreselassie, N., Ashenafi, S., Kassa, E., Aseffa, G., Amogne, W., et al. (2018). Daily adjunctive therapy with vitamin d(3) and phenylbutyrate supports clinical recovery from pulmonary tuberculosis: A randomized controlled trial in Ethiopia. J. Intern. Med. 284, 292–306. doi: 10.1111/joim.12767
Bell, S. L., Lopez, K. L., Cox, J. S., Patrick, K. L., and Watson, R. O. (2021). Galectin-8 senses phagosomal damage and recruits selective autophagy adapter TAX1BP1 to control Mycobacterium tuberculosis infection in macrophages. mBio 12, e0187120. doi: 10.1128/mBio.01871-20
Bento, C. F., Empadinhas, N., and Mendes, V. (2015). Autophagy in the fight against tuberculosis. DNA Cell Biol. 34, 228–242. doi: 10.1089/dna.2014.2745
Bhat, S. A., Iqbal, I. K., and Kumar, A. (2016). Imaging the NADH: Nad(+) homeostasis for understanding the metabolic response of Mycobacterium to physiologically relevant stresses. Front. Cell Infect. Microbiol. 6. doi: 10.3389/fcimb.2016.00145
Bhatt, K., Bhagavathula, M., Verma, S., Timmins, G. S., Deretic, V. P., Ellner, J. J., et al. (2021). Rapamycin modulates pulmonary pathology in a murine model of Mycobacterium tuberculosis infection. Dis. Model. Mech. 14, dmm049018. doi: 10.1242/dmm.049018
Bianco, D. M., De Maio, F., Santarelli, G., Palucci, I., Salustri, A., Bianchetti, G., et al. (2023). Evaluation of everolimus activity against Mycobacterium tuberculosis using in vitro models of infection. Antibiotics (Basel) 12, 171. doi: 10.3390/antibiotics12010171
Boland, R., Heemskerk, M. T., Forn-Cuní, G., Korbee, C. J., Walburg, K. V., Esselink, J. J., et al. (2023). Repurposing tamoxifen as potential host-directed therapeutic for tuberculosis. mBio 14, e0302422. doi: 10.1128/mbio.03024-22
Bonilla, D. L., Bhattacharya, A., Sha, Y., Xu, Y., Xiang, Q., Kan, A., et al. (2013). Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39, 537–547. doi: 10.1016/j.immuni.2013.08.026
Bryk, R., Mundhra, S., Jiang, X., Wood, M., Pfau, D., Weber, E., et al. (2020). Potentiation of rifampin activity in a mouse model of tuberculosis by activation of host transcription factor eb. PloS Pathog. 16, e1008567. doi: 10.1371/journal.ppat.1008567
Budzik, J. M., Swaney, D. L., Jimenez-Morales, D., Johnson, J. R., Garelis, N. E., Repasy, T., et al. (2020). Dynamic post-translational modification profiling of Mycobacterium tuberculosis-infected primary macrophages. Elife 9, e51461. doi: 10.7554/eLife.51461
Cai, Y., Gao, W., Wang, P., Zhang, G., Wang, X., Jiang, L., et al. (2023). Comparative proteome analysis revealed the differences in response to both Mycobacterium tuberculosis and Mycobacterium bovis infection of bovine alveolar macrophages. Front. Cell Infect. Microbiol. 13. doi: 10.3389/fcimb.2023.1266884
Cambier, C. J., O’Leary, S. M., O’Sullivan, M. P., Keane, J., and Ramakrishnan, L. (2017). Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity 47, 552–565.e554. doi: 10.1016/j.immuni.2017.08.003
Cárdenas-Rodríguez, N., Coballase-Urrutia, E., Rivera-Espinosa, L., Romero-Toledo, A., Sampieri, A., Ortega-Cuellar, D., et al. (2013). Modulation of antioxidant enzymatic activities by certain antiepileptic drugs (valproic acid, oxcarbazepine, and topiramate): Evidence in humans and experimental models. Oxid. Med. Cell Longev. 2013, 598493. doi: 10.1155/2013/598493
Carneiro, L. A. and Travassos, L. H. (2013). The interplay between NLRs and autophagy in immunity and inflammation. Front. Immunol. 4. doi: 10.3389/fimmu.2013.00361
Castillo, E. F., Dekonenko, A., Arko-Mensah, J., Mandell, M. A., Dupont, N., Jiang, S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. U S A. 109, E3168–E3176. doi: 10.1073/pnas.1210500109
Ceregido, M. A., Spínola Amilibia, M., Buts, L., Rivera-Torres, J., Garcia-Pino, A., Bravo, J., et al. (2014). The structure of TAX1BP1 ubz1 + 2 provides insight into target specificity and adaptability. J. Mol. Biol. 426, 674–690. doi: 10.1016/j.jmb.2013.11.006
Chai, Q., Wang, L., Liu, C. H., and Ge, B. (2020). New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 17, 901–913. doi: 10.1038/s41423-020-0502-z
Chai, Q., Wang, X., Qiang, L., Zhang, Y., Ge, P., Lu, Z., et al. (2019). A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 10, 1973. doi: 10.1038/s41467-019-09955-8
Chai, Q., Zhang, Y., and Liu, C. H. (2018). Mycobacterium tuberculosis: An adaptable pathogen associated with multiple human diseases. Front. Cell Infect. Microbiol. 8. doi: 10.3389/fcimb.2018.00158
Cha-Molstad, H., Yu, J. E., Feng, Z., Lee, S. H., Kim, J. G., Yang, P., et al. (2017). P62/SQSTM1/sequestosome-1 is an n-recognin of the n-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 8, 102. doi: 10.1038/s41467-017-00085-7
Chandra, P., Ghanwat, S., Matta, S. K., Yadav, S. S., Mehta, M., Siddiqui, Z., et al. (2015). Mycobacterium tuberculosis inhibits rab7 recruitment to selectively modulate autophagy flux in macrophages. Sci. Rep. 5, 16320. doi: 10.1038/srep16320
Chandra, P. and Kumar, D. (2016). Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy 12, 608–609. doi: 10.1080/15548627.2016.1139263
Chandra, P. and Philips, J. A. (2025). Usp8 promotes intracellular infection by enhancing escrt-mediated membrane repair, limiting xenophagy, and reducing oxidative stress. Autophagy 21, 298–314. doi: 10.1080/15548627.2024.2395134
Chen, D. Y., Chen, Y. M., Lin, C. F., Lo, C. M., Liu, H. J., and Liao, T. L. (2020). MicroRNA-889 inhibits autophagy to maintain mycobacterial survival in patients with latent tuberculosis infection by targeting tweak. mBio 11, e03045-19. doi: 10.1128/mBio.03045-19
Chen, Y. D., Lin, X. P., Ruan, Z. L., Li, M., Yi, X. M., Zhang, X., et al. (2024). Plk2-mediated phosphorylation of SQSTM1 s349 promotes aggregation of polyubiquitinated proteins upon proteasomal dysfunction. Autophagy 20, 2221–2237. doi: 10.1080/15548627.2024.2361574
Chen, Y. M., Liu, P. Y., Tang, K. T., Liu, H. J., and Liao, T. L. (2022). Tweak-fn14 axis induces calcium-associated autophagy and cell death to control mycobacterial survival in macrophages. Microbiol. Spectr. 10, e0317222. doi: 10.1128/spectrum.03172-22
Chen, Z., Shao, X. Y., Wang, C., Hua, M. H., Wang, C. N., Wang, X., et al. (2018). Mycobacterium marinum infection in zebrafish and microglia imitates the early stage of tuberculous meningitis. J. Mol. Neurosci. 64, 321–330. doi: 10.1007/s12031-018-1026-1
Cheng, C. Y., Gutierrez, N. M., Marzuki, M. B., Lu, X., Foreman, T. W., Paleja, B., et al. (2017). Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2, eaaj1789. doi: 10.1126/sciimmunol.aaj1789
Choi, S. W., Gu, Y., Peters, R. S., Salgame, P., Ellner, J. J., Timmins, G. S., et al. (2018). Ambroxol induces autophagy and potentiates rifampin antimycobacterial activity. Antimicrob. Agents Chemother. 62, e01019-18. doi: 10.1128/aac.01019-18
Choudhary, E., Bullen, C. K., Goel, R., Singh, A. K., Praharaj, M., Thakur, P., et al. (2020). Relative and quantitative phosphoproteome analysis of macrophages in response to infection by virulent and avirulent mycobacteria reveals a distinct role of the cytosolic RNA sensor RIG-I in Mycobacterium tuberculosis pathogenesis. J. Proteome Res. 19, 2316–2336. doi: 10.1021/acs.jproteome.9b00895
Clausen, T. H., Lamark, T., Isakson, P., Finley, K., Larsen, K. B., Brech, A., et al. (2010). P62/SQSTM1 and alfy interact to facilitate the formation of p62 bodies/alis and their degradation by autophagy. Autophagy 6, 330–344. doi: 10.4161/auto.6.3.11226
Coleman, M. M., Basdeo, S. A., Coleman, A. M., Cheallaigh, C. N., Peral de Castro, C., McLaughlin, A. M., et al. (2018). All-trans retinoic acid augments autophagy during intracellular bacterial infection. Am. J. Respir. Cell Mol. Biol. 59, 548–556. doi: 10.1165/rcmb.2017-0382OC
Coussens, A. K., Wilkinson, R. J., and Martineau, A. R. (2015). Phenylbutyrate is bacteriostatic against Mycobacterium tuberculosis and regulates the macrophage response to infection, synergistically with 25-hydroxy-Vitamin D3. PloS Pathog. 11, e1005007. doi: 10.1371/journal.ppat.1005007
Crilly, N. P., Ayeh, S. K., and Karakousis, P. C. (2020). The new frontier of host-directed therapies for Mycobacterium avium complex. Front. Immunol. 11. doi: 10.3389/fimmu.2020.623119
Daley, C. L. and Winthrop, K. L. (2020). Mycobacterium avium complex: Addressing gaps in diagnosis and management. J. Infect. Dis. 222, S199–s211. doi: 10.1093/infdis/jiaa354
Degner, N. R., Wang, J. Y., Golub, J. E., and Karakousis, P. C. (2018). Metformin use reverses the increased mortality associated with diabetes mellitus during tuberculosis treatment. Clin. Infect. Dis. 66, 198–205. doi: 10.1093/cid/cix819
De Maio, F., Palmieri, V., Santarelli, G., Perini, G., Salustri, A., Palucci, I., et al. (2020). Graphene oxide-linezolid combination as potential new anti-tuberculosis treatment. Nanomaterials (Basel) 10, 1431. doi: 10.3390/nano10081431
Deng, W., Long, Q., Zeng, J., Li, P., Yang, W., Chen, X., et al. (2017). Mycobacterium tuberculosis PE_pgrs41 enhances the intracellular survival of m. Smegmatis within macrophages via blocking innate immunity and inhibition of host defense. Sci. Rep. 7, 46716. doi: 10.1038/srep46716
Deosaran, E., Larsen, K. B., Hua, R., Sargent, G., Wang, Y., Kim, S., et al. (2013). NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 126, 939–952. doi: 10.1242/jcs.114819
Deretic, V., Kimura, T., Timmins, G., Moseley, P., Chauhan, S., and Mandell, M. (2015). Immunologic manifestations of autophagy. J. Clin. Invest. 125, 75–84. doi: 10.1172/jci73945
de Silva, T. I., Cope, A., Goepel, J., and Greig, J. M. (2007). The use of adjuvant granulocyte-macrophage colony-stimulating factor in hiv-related disseminated atypical mycobacterial infection. J. Infect. 54, e207–e210. doi: 10.1016/j.jinf.2006.11.005
Devos, G., Tosco, L., Baldewijns, M., Gevaert, T., Goffin, K., Petit, V., et al. (2023). Arneo: A randomized phase ii trial of neoadjuvant degarelix with or without apalutamide prior to radical prostatectomy for high-risk prostate cancer. Eur. Urol. 83, 508–518. doi: 10.1016/j.eururo.2022.09.009
Dey, B., Dey, R. J., Cheung, L. S., Pokkali, S., Guo, H., Lee, J. H., et al. (2015). A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat. Med. 21, 401–406. doi: 10.1038/nm.3813
Dikic, I. and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Ding, S., Qu, Y., Yang, S., Zhao, Y., and Xu, G. (2019). Novel miR-1958 promotes Mycobacterium tuberculosis survival in RAW264.7 cells by inhibiting autophagy via Atg5. J. Microbiol. Biotechnol. 29, 989–998. doi: 10.4014/jmb.1811.11062
Di Rita, A., Angelini, D. F., Maiorino, T., Caputo, V., Cascella, R., Kumar, M., et al. (2021). Characterization of a natural variant of human NDP52 and its functional consequences on mitophagy. Cell Death Differ. 28, 2499–2516. doi: 10.1038/s41418-021-00766-3
Dove, K. K. and Klevit, R. E. (2017). Ring-between-ring E3 ligases: Emerging themes amid the variations. J. Mol. Biol. 429, 3363–3375. doi: 10.1016/j.jmb.2017.08.008
Du, W., Dai, Y., Yue, L., Ma, T., and Wu, L. (2025). MiR-207 targets autophagy-associated protein LAMP2 to regulate the mechanism of macrophage-Mycobacterium tuberculosis interaction (Chinese). Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 41, 97–104.
Dutta, N. K., Pinn, M. L., and Karakousis, P. C. (2017). Metformin adjunctive therapy does not improve the sterilizing activity of the first-line antitubercular regimen in mice. Antimicrob. Agents Chemother. 61, e00652-17. doi: 10.1128/aac.00652-17
Estevez, H., Palacios, A., Gil, D., Anguita, J., Vallet-Regi, M., González, B., et al. (2020). Antimycobacterial effect of selenium nanoparticles on Mycobacterium tuberculosis. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00800
Etna, M. P., Sinigaglia, A., Grassi, A., Giacomini, E., Romagnoli, A., Pardini, M., et al. (2018). Mycobacterium tuberculosis-induced miR-155 subverts autophagy by targeting Atg3 in human dendritic cells. PloS Pathog. 14, e1006790. doi: 10.1371/journal.ppat.1006790
Falkinham, J. O. (2018). Challenges of NTM drug development. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01613
Filimonenko, M., Isakson, P., Finley, K. D., Anderson, M., Jeong, H., Melia, T. J., et al. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein alfy. Mol. Cell. 38, 265–279. doi: 10.1016/j.molcel.2010.04.007
Forrellad, M. A., Klepp, L. I., Gioffré, A., Sabio y García, J., Morbidoni, H. R., de la Paz Santangelo, M., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Franco, L. H., Nair, V. R., Scharn, C. R., Xavier, R. J., Torrealba, J. R., Shiloh, M. U., et al. (2017). The ubiquitin ligase smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe 22, 421–423. doi: 10.1016/j.chom.2017.08.005
Frudd, K., Burgoyne, T., and Burgoyne, J. R. (2018). Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 9, 95. doi: 10.1038/s41467-017-02352-z
Fu, T., Liu, J., Wang, Y., Xie, X., Hu, S., and Pan, L. (2018). Mechanistic insights into the interactions of nap1 with the skich domains of NDP52 and TAX1BP1. Proc. Natl. Acad. Sci. U S A. 115, E11651–e11660. doi: 10.1073/pnas.1811421115
Fu, J., Luo, X., Lin, M., Xiao, Z., Huang, L., Wang, J., et al. (2023). Marine-fungi-derived gliotoxin promotes autophagy to suppress mycobacteria tuberculosis infection in macrophage. Mar. Drugs 21, 616. doi: 10.3390/md21120616
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 19, 2092–2100. doi: 10.1091/mbc.e07-12-1257
Furuta, N., Fujita, N., Noda, T., Yoshimori, T., and Amano, A. (2010). Combinational soluble n-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell. 21, 1001–1010. doi: 10.1091/mbc.e09-08-0693
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Galluzzi, L. and Green, D. R. (2019). Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699. doi: 10.1016/j.cell.2019.05.026
Ganmaa, D., Munkhzul, B., Fawzi, W., Spiegelman, D., Willett, W. C., Bayasgalan, P., et al. (2017). High-dose Vitamin D(3) during tuberculosis treatment in Mongolia. A randomized controlled trial. Am. J. Respir. Crit. Care Med. 196, 628–637. doi: 10.1164/rccm.201705-0936OC
Garg, R., Borbora, S. M., Bansia, H., Rao, S., Singh, P., Verma, R., et al. (2020). Mycobacterium tuberculosis calcium pump ctpf modulates the autophagosome in an mTOR-dependent manner. Front. Cell Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.00461
Ge, P., Lei, Z., Yu, Y., Lu, Z., Qiang, L., Chai, Q., et al. (2022). Tuberculosis pkng manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy 18, 576–594. doi: 10.1080/15548627.2021.1938912
Genestet, C., Bernard-Barret, F., Hodille, E., Ginevra, C., Ader, F., Goutelle, S., et al. (2018). Antituberculous drugs modulate bacterial phagolysosome avoidance and autophagy in Mycobacterium tuberculosis-infected macrophages. Tuberculosis (Edinb). 111, 67–70. doi: 10.1016/j.tube.2018.05.014
Geng, S., Hao, P., Wang, D., Zhong, P., Tian, F., Zhang, R., et al. (2023). Zinc oxide nanoparticles have biphasic roles on Mycobacterium-induced inflammation by activating autophagy and ferroptosis mechanisms in infected macrophages. Microb. Pathog. 180, 106132. doi: 10.1016/j.micpath.2023.106132
Gengenbacher, M., Nieuwenhuizen, N., Vogelzang, A., Liu, H., Kaiser, P., Schuerer, S., et al. (2016). Deletion of nuog from the vaccine candidate Mycobacterium bovis BCG δurec::Hly improves protection against tuberculosis. mBio 7, e00679-16. doi: 10.1128/mBio.00679-16
Giraud-Gatineau, A., Coya, J. M., Maure, A., Biton, A., Thomson, M., Bernard, E. M., et al. (2020). The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. Elife 9, e55692. doi: 10.7554/eLife.55692
Gladkova, C., Maslen, S. L., Skehel, J. M., and Komander, D. (2018). Mechanism of parkin activation by pink1. Nature 559, 410–414. doi: 10.1038/s41586-018-0224-x
Gomes, L. C. and Dikic, I. (2014). Autophagy in antimicrobial immunity. Mol. Cell. 54, 224–233. doi: 10.1016/j.molcel.2014.03.009
Gonzalez-Orozco, M., Strong, E. J., Paroha, R., and Lee, S. (2022). Reversing BCG-mediated autophagy inhibition and mycobacterial survival to improve vaccine efficacy. BMC Immunol. 23, 43. doi: 10.1186/s12865-022-00518-z
Greenstein, R. J., Su, L., Juste, R. A., and Brown, S. T. (2008). On the action of cyclosporine a, rapamycin and tacrolimus on m. Avium including subspecies paratuberculosis. PloS One 3, e2496. doi: 10.1371/journal.pone.0002496
Gu, X., Gao, Y., Mu, D. G., and Fu, E. Q. (2017). MiR-23a-5p modulates mycobacterial survival and autophagy during Mycobacterium tuberculosis infection through TLR2/myd88/NF-κB pathway by targeting TLR2. Exp. Cell Res. 354, 71–77. doi: 10.1016/j.yexcr.2017.03.039
Gubas, A. and Dikic, I. (2022). A guide to the regulation of selective autophagy receptors. FEBS J. 289, 75–89. doi: 10.1111/febs.15824
Guo, L., Zhao, J., Qu, Y., Yin, R., Gao, Q., Ding, S., et al. (2016). Microrna-20a inhibits autophagic process by targeting Atg7 and Atg16L1 and favors mycobacterial survival in macrophage cells. Front. Cell Infect. Microbiol. 6. doi: 10.3389/fcimb.2016.00134
Gupta, P. K., Jahagirdar, P., Tripathi, D., Devarajan, P. V., and Kulkarni, S. (2023). Macrophage targeted polymeric curcumin nanoparticles limit intracellular survival of Mycobacterium tuberculosis through induction of autophagy and augment anti-TB activity of isoniazid in RAW 264.7 macrophages. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1233630
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. doi: 10.1016/j.cell.2004.11.038
Han, B., Jiang, W., Cui, P., Zheng, K., Dang, C., Wang, J., et al. (2021). Microglial pgc-1α protects against ischemic brain injury by suppressing neuroinflammation. Genome Med. 13, 47. doi: 10.1186/s13073-021-00863-5
Hariadi, N. I. and Blackwood, R. A. (2017). Disseminated Mycobacterium avium complex in an adolescent with perinatally-acquired hiv infection. Infect. Dis. Rep. 9, 6884. doi: 10.4081/idr.2017.6884
Harnett, M. M., Pineda, M. A., Latré de Laté, P., Eason, R. J., Besteiro, S., Harnett, W., et al. (2017). From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. BioMed. J. 40, 9–22. doi: 10.1016/j.bj.2016.12.004
Harris, J., De Haro, S. A., Master, S. S., Keane, J., Roberts, E. A., Delgado, M., et al. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27, 505–517. doi: 10.1016/j.immuni.2007.07.022
He, X., He, Y., Deng, X., Lu, N., Li, A., Gao, S., et al. (2025). Rv2741 promotes Mycobacterium survival by modulating macrophage function via the IL-1α-MAPK axis. ACS Infect. Dis. 11, 676–688. doi: 10.1021/acsinfecdis.4c00790
Heath, R. J., Goel, G., Baxt, L. A., Rush, J. S., Mohanan, V., Paulus, G. L. C., et al. (2016). Rnf166 determines recruitment of adaptor proteins during antibacterial autophagy. Cell Rep. 17, 2183–2194. doi: 10.1016/j.celrep.2016.11.005
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The pink1-parkin mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and tbk1 activation to promote mitophagy. Mol. Cell. 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Herhaus, L., van den Bedem, H., Tang, S., Maslennikov, I., Wakatsuki, S., Dikic, I., et al. (2019). Molecular recognition of m1-linked ubiquitin chains by native and phosphorylated UBAN domains. J. Mol. Biol. 431, 3146–3156. doi: 10.1016/j.jmb.2019.06.012
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. 20, 1981–1991. doi: 10.1091/mbc.e08-12-1248
Hu, Y., Wen, Z., Liu, S., Cai, Y., Guo, J., Xu, Y., et al. (2020). Ibrutinib suppresses intracellular Mycobacterium tuberculosis growth by inducing macrophage autophagy. J. Infect. 80, e19–e26. doi: 10.1016/j.jinf.2020.03.003
Hu, D., Wu, J., Wang, W., Mu, M., Zhao, R., Xu, X., et al. (2015). Autophagy regulation revealed by sapm-induced block of autophagosome-lysosome fusion via binding rab7. Biochem. Biophys. Res. Commun. 461, 401–407. doi: 10.1016/j.bbrc.2015.04.051
Huang, D. and Bao, L. (2016). Mycobacterium tuberculosis espb protein suppresses interferon-γ-induced autophagy in murine macrophages. J. Microbiol. Immunol. Infect. 49, 859–865. doi: 10.1016/j.jmii.2014.11.008
Huang, T., Holden, J. A., Heath, D. E., O’Brien-Simpson, N. M., and O’Connor, A. J. (2019). Engineering highly effective antimicrobial selenium nanoparticles through control of particle size. Nanoscale 11, 14937–14951. doi: 10.1039/c9nr04424h
Huang, S. J., Wang, X. H., Liu, Z. D., Cao, W. L., Han, Y., Ma, A. G., et al. (2017). Vitamin D deficiency and the risk of tuberculosis: A meta-analysis. Drug Des. Devel Ther. 11, 91–102. doi: 10.2147/dddt.S79870
Hussey, S., Travassos, L. H., and Jones, N. L. (2009). Autophagy as an emerging dimension to adaptive and innate immunity. Semin. Immunol. 21, 233–241. doi: 10.1016/j.smim.2009.05.004
Hyttinen, J. M., Niittykoski, M., Salminen, A., and Kaarniranta, K. (2013). Maturation of autophagosomes and endosomes: A key role for rab7. Biochim. Biophys. Acta 1833, 503–510. doi: 10.1016/j.bbamcr.2012.11.018
Ikutama, R., Peng, G., Tsukamoto, S., Umehara, Y., Trujillo-Paez, J. V., Yue, H., et al. (2023). Cathelicidin LL-37 activates human keratinocyte autophagy through the P2X7, mechanistic target of rapamycin, and MAPK pathways. J. Invest. Dermatol. 143, 751–761.e757. doi: 10.1016/j.jid.2022.10.020
Intemann, C. D., Thye, T., Niemann, S., Browne, E. N., Amanua Chinbuah, M., Enimil, A., et al. (2009). Autophagy gene variant IRGM -261t contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by m. Africanum strains. PloS Pathog. 5, e1000577. doi: 10.1371/journal.ppat.1000577
Iqbal, I. K., Bajeli, S., Sahu, S., Bhat, S. A., and Kumar, A. (2021). Hydrogen sulfide-induced GAPDH sulfhydration disrupts the CCAR2-SIRT1 interaction to initiate autophagy. Autophagy 17, 3511–3529. doi: 10.1080/15548627.2021.1876342
Itakura, E., Kishi-Itakura, C., and Mizushima, N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. doi: 10.1016/j.cell.2012.11.001
Itakura, E. and Mizushima, N. (2011). P62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 192, 17–27. doi: 10.1083/jcb.201009067
Jagannath, C., Lindsey, D. R., Dhandayuthapani, S., Xu, Y., Hunter, R. L., Jr., and Eissa, N. T. (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 15, 267–276. doi: 10.1038/nm.1928
Jo, E. K. (2010). Innate immunity to mycobacteria: Vitamin d and autophagy. Cell Microbiol. 12, 1026–1035. doi: 10.1111/j.1462-5822.2010.01491.x
Johansen, M. D., Herrmann, J. L., and Kremer, L. (2020). Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. N. at Rev. Microbiol. 18, 392–407. doi: 10.1038/s41579-020-0331-1
Jolliffe, D. A., Ganmaa, D., Wejse, C., Raqib, R., Haq, M. A., Salahuddin, N., et al. (2019). Adjunctive Vitamin D in tuberculosis treatment: Meta-analysis of individual participant data. Eur. Respir. J. 53, 1802003. doi: 10.1183/13993003.02003-2018
Juárez, E., Carranza, C., Sánchez, G., González, M., Chávez, J., Sarabia, C., et al. (2016). Loperamide restricts intracellular growth of Mycobacterium tuberculosis in lung macrophages. Am. J. Respir. Cell Mol. Biol. 55, 837–847. doi: 10.1165/rcmb.2015-0383OC
Kametaka, S., Okano, T., Ohsumi, M., and Ohsumi, Y. (1998). Apg14p and apg6/vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 273, 22284–22291. doi: 10.1074/jbc.273.35.22284
Karyadi, E., West, C. E., Schultink, W., Nelwan, R. H., Gross, R., Amin, Z., et al. (2002). A double-blind, placebo-controlled study of vitamin a and zinc supplementation in persons with tuberculosis in Indonesia: Effects on clinical response and nutritional status. Am. J. Clin. Nutr. 75, 720–727. doi: 10.1093/ajcn/75.4.720
Kedzierska, K., Mak, J., Mijch, A., Cooke, I., Rainbird, M., Roberts, S., et al. (2000). Granulocyte-macrophage colony-stimulating factor augments phagocytosis of Mycobacterium avium complex by human immunodeficiency virus type 1-infected monocytes/macrophages in vitro and in vivo. J. Infect. Dis. 181, 390–394. doi: 10.1086/315191
Khan, A., Sayedahmed, E. E., Singh, V. K., Mishra, A., Dorta-Estremera, S., Nookala, S., et al. (2021). A recombinant bovine adenoviral mucosal vaccine expressing mycobacterial antigen-85b generates robust protection against tuberculosis in mice. Cell Rep. Med. 2, 100372. doi: 10.1016/j.xcrm.2021.100372
Kilinç, G., Saris, A., Ottenhoff, T. H. M., and Haks, M. C. (2021). Host-directed therapy to combat mycobacterial infections. Immunol. Rev. 301, 62–83. doi: 10.1111/imr.12951
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, B. W., Kwon, D. H., and Song, H. K. (2016). Structure biology of selective autophagy receptors. BMB Rep. 49, 73–80. doi: 10.5483/bmbrep.2016.49.2.265
Kim, J. K., Lee, H. M., Park, K. S., Shin, D. M., Kim, T. S., Kim, Y. S., et al. (2017). MiR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein dram2. Autophagy 13, 423–441. doi: 10.1080/15548627.2016.1241922
Kim, J. J., Lee, H. M., Shin, D. M., Kim, W., Yuk, J. M., Jin, H. S., et al. (2012). Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11, 457–468. doi: 10.1016/j.chom.2012.03.008
Kim, Y. J., Park, E. J., Lee, S. H., Silwal, P., Kim, J. K., Yang, J. S., et al. (2023). Dimethyl itaconate is effective in host-directed antimicrobial responses against mycobacterial infections through multifaceted innate immune pathways. Cell Biosci. 13, 49. doi: 10.1186/s13578-023-00992-x
Kim, J. K., Yuk, J. M., Kim, S. Y., Kim, T. S., Jin, H. S., Yang, C. S., et al. (2015). MicroRNA-125a inhibits autophagy activation and antimicrobial responses during mycobacterial infection. J. Immunol. 194, 5355–5365. doi: 10.4049/jimmunol.1402557
Kimmey, J. M., Huynh, J. P., Weiss, L. A., Park, S., Kambal, A., Debnath, J., et al. (2015). Unique role for Atg5 in neutrophil-mediated immunopathology during m. Tuberculosis infection. Nature. 528, 565–569. doi: 10.1038/nature16451
Kirisako, T., Baba, M., Ishihara, N., Miyazawa, K., Ohsumi, M., Yoshimori, T., et al. (1999). Formation process of autophagosome is traced with apg8/aut7p in yeast. J. Cell Biol. 147, 435–446. doi: 10.1083/jcb.147.2.435
Kirkin, V., Lamark, T., Sou, Y. S., Bjørkøy, G., Nunn, J. L., Bruun, J. A., et al. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 33, 505–516. doi: 10.1016/j.molcel.2009.01.020
Kirkin, V. and Rogov, V. V. (2019). A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell. 76, 268–285. doi: 10.1016/j.molcel.2019.09.005
Kjellin, J., Pränting, M., Bach, F., Vaid, R., Edelbroek, B., Li, Z., et al. (2019). Investigation of the host transcriptional response to intracellular bacterial infection using dictyostelium discoideum as a host model. BMC Genomics 20, 961. doi: 10.1186/s12864-019-6269-x
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223. doi: 10.1038/ncb2021
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163. doi: 10.1016/j.cell.2007.10.035
Krause, D. S. and Dikic, I. (2022). Mycobacterium tuberculosis hijacks ubiquitin to inhibit pyroptosis. Mol. Cell. 82, 4588–4590. doi: 10.1016/j.molcel.2022.11.020
Kumar, R., Sahu, S. K., Kumar, M., Jana, K., Gupta, P., Gupta, U. D., et al. (2016). MicroRNA 17-5p regulates autophagy in Mycobacterium tuberculosis-infected macrophages by targeting Mcl-1 and STAT3. Cell Microbiol. 18, 679–691. doi: 10.1111/cmi.12540
Lachmandas, E., Eckold, C., Böhme, J., Koeken, V., Marzuki, M. B., Blok, B., et al. (2019). Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J. Infect. Dis. 220, 139–150. doi: 10.1093/infdis/jiz064
Lam, A., Prabhu, R., Gross, C. M., Riesenberg, L. A., Singh, V., and Aggarwal, S. (2017). Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L218–l229. doi: 10.1152/ajplung.00162.2017
Lam, K. K., Zheng, X., Forestieri, R., Balgi, A. D., Nodwell, M., Vollett, S., et al. (2012). Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PloS Pathog. 8, e1002691. doi: 10.1371/journal.ppat.1002691
Lamark, T., Perander, M., Outzen, H., Kristiansen, K., Øvervatn, A., Michaelsen, E., et al. (2003). Interaction codes within the family of mammalian phox and bem1p domain-containing proteins. J. Biol. Chem. 278, 34568–34581. doi: 10.1074/jbc.M303221200
Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E., et al. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science 308, 1599–1603. doi: 10.1126/science.1110463
Langer, H. T., Rohm, M., Goncalves, M. D., and Sylow, L. (2024). AMPK as a mediator of tissue preservation: Time for a shift in dogma? Nat. Rev. Endocrinol. 20, 526–540. doi: 10.1038/s41574-024-00992-y
Lau, A., Wang, X. J., Zhao, F., Villeneuve, N. F., Wu, T., Jiang, T., et al. (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 30, 3275–3285. doi: 10.1128/mcb.00248-10
Lawson, L., Thacher, T. D., Yassin, M. A., Onuoha, N. A., Usman, A., Emenyonu, N. E., et al. (2010). Randomized controlled trial of zinc and vitamin a as co-adjuvants for the treatment of pulmonary tuberculosis. Trop. Med. Int. Health 15, 1481–1490. doi: 10.1111/j.1365-3156.2010.02638.x
Lee, M. C., Chiang, C. Y., Lee, C. H., Ho, C. M., Chang, C. H., Wang, J. Y., et al. (2018). Metformin use is associated with a low risk of tuberculosis among newly diagnosed diabetes mellitus patients with normal renal function: A nationwide cohort study with validated diagnostic criteria. PloS One 13, e0205807. doi: 10.1371/journal.pone.0205807
Lee, Y., Chou, T. F., Pittman, S. K., Keith, A. L., Razani, B., and Weihl, C. C. (2017). Keap1/cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 20, 1994. doi: 10.1016/j.celrep.2017.08.019
Lee, Y. J., Han, S. K., Park, J. H., Lee, J. K., Kim, D. K., Chung, H. S., et al. (2018). The effect of metformin on culture conversion in tuberculosis patients with diabetes mellitus. Korean J. Intern. Med. 33, 933–940. doi: 10.3904/kjim.2017.249
Lee, H. J., Kang, S. J., Woo, Y., Hahn, T. W., Ko, H. J., and Jung, Y. J. (2020). TLR7 stimulation with imiquimod induces selective autophagy and controls Mycobacterium tuberculosis growth in mouse macrophages. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01684
Lee, Y. and Weihl, C. C. (2017). Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy 13, 1615–1616. doi: 10.1080/15548627.2017.1339845
Li, J., Gao, J., Gao, Y., Shi, C., Guo, X., Huang, H., et al. (2024). Degarelix limits the survival of mycobacteria and granuloma formation. Microb. Pathog. 197, 107046. doi: 10.1016/j.micpath.2024.107046
Li, F., Xie, X., Wang, Y., Liu, J., Cheng, X., Guo, Y., et al. (2016). Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and tbk1 proteins. Nat. Commun. 7, 12708. doi: 10.1038/ncomms12708
Li, F., Xu, D., Wang, Y., Zhou, Z., Liu, J., Hu, S., et al. (2018). Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by tbk1-mediated phosphorylation. Autophagy 14, 66–79. doi: 10.1080/15548627.2017.1391970
Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., et al. (2006). Autophagic and tumour suppressor activity of a novel beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699. doi: 10.1038/ncb1426
Liao, Y., Hussain, T., Liu, C., Cui, Y., Wang, J., Yao, J., et al. (2019). Endoplasmic reticulum stress induces macrophages to produce IL-1β during Mycobacterium bovis infection via a positive feedback loop between mitochondrial damage and inflammasome activation. Front. Immunol. 10. doi: 10.3389/fimmu.2019.00268
Lin, W., Fan, S., Liao, K., Huang, Y., Cong, Y., Zhang, J., et al. (2022). Engineering zinc oxide hybrid selenium nanoparticles for synergetic anti-tuberculosis treatment by combining Mycobacterium tuberculosis killings and host cell immunological inhibition. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.1074533
Liu, F., Chen, J., Wang, P., Li, H., Zhou, Y., Liu, H., et al. (2018). MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat. Commun. 9, 4295. doi: 10.1038/s41467-018-06836-4
Liu, K., Hong, D., Zhang, F., Li, X., He, M., Han, X., et al. (2020). MicroRNA-106a inhibits autophagy process and antimicrobial responses by targeting ULK1, Atg7, and Atg16L1 during mycobacterial infection. Front. Immunol. 11. doi: 10.3389/fimmu.2020.610021
Liu, P. T., Stenger, S., Li, H., Wenzel, L., Tan, B. H., Krutzik, S. R., et al. (2006). Toll-like receptor triggering of a vitamin d-mediated human antimicrobial response. Science 311, 1770–1773. doi: 10.1126/science.1123933
Liu, Y., Wang, J., Yang, J., Xia, J., Yu, J., Chen, D., et al. (2024). Nanomaterial-mediated host directed therapy of tuberculosis by manipulating macrophage autophagy. J. Nanobiotechnology. 22, 608. doi: 10.1186/s12951-024-02875-w
Liu, M., Wilk, S. A., Wang, A., Zhou, L., Wang, R. H., Ogawa, W., et al. (2010). Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and deptor. J. Biol. Chem. 285, 36387–36394. doi: 10.1074/jbc.M110.169284
López-Jiménez, A. T., Cardenal-Muñoz, E., Leuba, F., Gerstenmaier, L., Barisch, C., Hagedorn, M., et al. (2018). The escrt and autophagy machineries cooperate to repair esx-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PloS Pathog. 14, e1007501. doi: 10.1371/journal.ppat.1007501
Lopresti, A. L. (2018). The problem of curcumin and its bioavailability: Could its gastrointestinal influence contribute to its overall health-enhancing effects? Adv. Nutr. 9, 41–50. doi: 10.1093/advances/nmx011
Luo, H. L., Pi, J., Zhang, J. A., Yang, E. Z., Xu, H., Luo, H., et al. (2021). Circular RNA trappc6b inhibits intracellular Mycobacterium tuberculosis growth while inducing autophagy in macrophages by targeting microRNA-874-3p. Clin. Transl. Immunol. 10, e1254. doi: 10.1002/cti2.1254
Lystad, A. H., Carlsson, S. R., and Simonsen, A. (2019). Toward the function of mammalian Atg12-Atg5-Atg16L1 complex in autophagy and related processes. Autophagy 15, 1485–1486. doi: 10.1080/15548627.2019.1618100
Manzanillo, P. S., Ayres, J. S., Watson, R. O., Collins, A. C., Souza, G., Rae, C. S., et al. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501, 512–516. doi: 10.1038/nature12566
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., et al. (2009). Two beclin 1-binding proteins, Atg14l and rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396. doi: 10.1038/ncb1846
Menon, A. R., Prest, R. J., Tobin, D. M., and Champion, P. A. (2025). Mycobacterium marinum as a model for understanding principles of mycobacterial pathogenesis. J. Bacteriol. 207, e0004725. doi: 10.1128/jb.00047-25
Migliano, S. M., Schultz, S. W., Wenzel, E. M., Takáts, S., Liu, D., Mørk, S., et al. (2024). Removal of hypersignaling endosomes by simaphagy. Autophagy 20, 769–791. doi: 10.1080/15548627.2023.2267958
Mijaljica, D., Prescott, M., and Devenish, R. J. (2011). Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 7, 673–682. doi: 10.4161/auto.7.7.14733
Mily, A., Rekha, R. S., Kamal, S. M., Akhtar, E., Sarker, P., Rahim, Z., et al. (2013). Oral intake of phenylbutyrate with or without vitamin d3 upregulates the cathelicidin LL-37 in human macrophages: A dose finding study for treatment of tuberculosis. BMC Pulm Med. 13, 23. doi: 10.1186/1471-2466-13-23
Mily, A., Rekha, R. S., Kamal, S. M., Arifuzzaman, A. S., Rahim, Z., Khan, L., et al. (2015). Significant effects of oral phenylbutyrate and Vitamin D3 adjunctive therapy in pulmonary tuberculosis: A randomized controlled trial. PloS One 10, e0138340. doi: 10.1371/journal.pone.0138340
Mishra, A., Behura, A., Kumar, A., Ghosh, A., Naik, L., Mawatwal, S., et al. (2021). Soybean lectin induces autophagy through P2RX7 dependent activation of NF-κB-ROS pathway to kill intracellular mycobacteria. Biochim. Biophys. Acta Gen. Subj. 1865, 129806. doi: 10.1016/j.bbagen.2020.129806
Mishra, A., Kumar, A., Naik, L., Patel, S., Das, M., Behura, A., et al. (2023). Soybean lectin-triggered IL-6 secretion induces autophagy to kill intracellular mycobacteria through P2RX7 dependent activation of the JAK2/STAT3/Mcl-1 pathway. Cytokine 171, 156366. doi: 10.1016/j.cyto.2023.156366
Mizushima, N., Noda, T., and Ohsumi, Y. (1999). Apg16p is required for the function of the apg12p-apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888–3896. doi: 10.1093/emboj/18.14.3888
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., et al. (1998). A protein conjugation system essential for autophagy. Nature 395, 395–398. doi: 10.1038/26506
Mohanty, S., Jagannathan, L., Ganguli, G., Padhi, A., Roy, D., Alaridah, N., et al. (2015). A mycobacterial phosphoribosyltransferase promotes bacillary survival by inhibiting oxidative stress and autophagy pathways in macrophages and zebrafish. J. Biol. Chem. 290, 13321–13343. doi: 10.1074/jbc.M114.598482
Morelli, E., Ginefra, P., Mastrodonato, V., Beznoussenko, G. V., Rusten, T. E., Bilder, D., et al. (2014). Multiple functions of the SNARE protein snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in drosophila. Autophagy 10, 2251–2268. doi: 10.4161/15548627.2014.981913
Morrison, H. M., Craft, J., Rivera-Lugo, R., Johnson, J. R., Golovkine, G. R., Bell, S. L., et al. (2023). Deficiency in galectin-3, -8, and -9 impairs immunity to chronic Mycobacterium tuberculosis infection but not acute infection with multiple intracellular pathogens. PloS Pathog. 19, e1011088. doi: 10.1371/journal.ppat.1011088
Morton, S., Hesson, L., Peggie, M., and Cohen, P. (2008). Enhanced binding of tbk1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 582, 997–1002. doi: 10.1016/j.febslet.2008.02.047
Nair, U., Cao, Y., Xie, Z., and Klionsky, D. J. (2010). Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J. Biol. Chem. 285, 11476–11488. doi: 10.1074/jbc.M109.080374
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the nalp3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi: 10.1038/nrm2708
Nannini, E. C., Keating, M., Binstock, P., Samonis, G., and Kontoyiannis, D. P. (2002). Successful treatment of refractory disseminated Mycobacterium avium complex infection with the addition of linezolid and mefloquine. J. Infect. 44, 201–203. doi: 10.1053/jinf.2002.0970
Nascimbeni, A. C., Giordano, F., Dupont, N., Grasso, D., Vaccaro, M. I., Codogno, P., et al. (2017). Er-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 36, 2018–2033. doi: 10.15252/embj.201797006
Nieto Ramirez, L. M., Mehaffy, C., and Dobos, K. M. (2024). Systematic review of innate immune responses against Mycobacterium tuberculosis complex infection in animal models. Front. Immunol. 15. doi: 10.3389/fimmu.2024.1467016
Ning, H., Zhang, W., Kang, J., Ding, T., Liang, X., Lu, Y., et al. (2021). Subunit vaccine ESAT-6:c-di-amp delivered by intranasal route elicits immune responses and protects against Mycobacterium tuberculosis infection. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.647220
Nishimura, S., Linares, J. F., L’Hermitte, A., Duran, A., Cid-Diaz, T., Martinez-Ordoñez, A., et al. (2024). Opposing regulation of the STING pathway in hepatic stellate cells by NBR1 and p62 determines the progression of hepatocellular carcinoma. Mol. Cell. 84, 4660–4676.e4610. doi: 10.1016/j.molcel.2024.09.026
Niu, N. K., Yin, J. J., Yang, Y. X., Wang, Z. L., Zhou, Z. W., He, Z. X., et al. (2015). Novel targeting of pegylated liposomes for codelivery of TGF-β1 sirna and four antitubercular drugs to human macrophages for the treatment of mycobacterial infection: A quantitative proteomic study. Drug Des. Devel Ther. 9, 4441–4470. doi: 10.2147/dddt.S79369
Orenstein, S. J. and Cuervo, A. M. (2010). Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 21, 719–726. doi: 10.1016/j.semcdb.2010.02.005
Orosz, L., Papanicolaou, E. G., Seprényi, G., and Megyeri, K. (2016). IL-17A and IL-17F induce autophagy in RAW 264.7 macrophages. BioMed. Pharmacother. 77, 129–134. doi: 10.1016/j.biopha.2015.12.020
Ouyang, Q., Zhang, K., Lin, D., Feng, C. G., Cai, Y., and Chen, X. (2020). Bazedoxifene suppresses intracellular Mycobacterium tuberculosis growth by enhancing autophagy. mSphere 5, e00124-20. doi: 10.1128/mSphere.00124-20
Padhi, A., Pattnaik, K., Biswas, M., Jagadeb, M., Behera, A., and Sonawane, A. (2019). Mycobacterium tuberculosis LprE suppresses TLR2-dependent cathelicidin and autophagy expression to enhance bacterial survival in macrophages. J. Immunol. 203, 2665–2678. doi: 10.4049/jimmunol.1801301
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Parihar, S. P., Guler, R., Khutlang, R., Lang, D. M., Hurdayal, R., Mhlanga, M. M., et al. (2014). Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 209, 754–763. doi: 10.1093/infdis/jit550
Park, D., Jeong, H., Lee, M. N., Koh, A., Kwon, O., Yang, Y. R., et al. (2016). Resveratrol induces autophagy by directly inhibiting mTOR through atp competition. Sci. Rep. 6, 21772. doi: 10.1038/srep21772
Park, C. R., Paik, S., Kim, Y. J., Kim, J. K., Jeon, S. M., Lee, S. H., et al. (2021). Rufomycin exhibits dual effects against Mycobacterium abscessus infection by inducing host defense and antimicrobial activities. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.695024
Pastore, N., Brady, O. A., Diab, H. I., Martina, J. A., Sun, L., Huynh, T., et al. (2016). TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258. doi: 10.1080/15548627.2016.1179405
Patel, S., Naik, L., Das, M., Nayak, D. K., Dandsena, P. K., Mishra, A., et al. (2025). Furamidine-induced autophagy exerts an anti-mycobacterial effect in a SIRT1-pampk-FOXO3a-dependent manner by elevation of intracellular ca(2+) level expression. Microbiol. Res. 290, 127976. doi: 10.1016/j.micres.2024.127976
Periyasamy, K. M., Ranganathan, U. D., Tripathy, S. P., and Bethunaickan, R. (2020). Vitamin D - a host directed autophagy mediated therapy for tuberculosis. Mol. Immunol. 127, 238–244. doi: 10.1016/j.molimm.2020.08.007
Pi, J., Shen, L., Shen, H., Yang, E., Wang, W., Wang, R., et al. (2019). Mannosylated graphene oxide as macrophage-targeted delivery system for enhanced intracellular m.Tuberculosis killing efficiency. Mater Sci. Eng. C Mater Biol. Appl. 103, 109777. doi: 10.1016/j.msec.2019.109777
Pi, J., Shen, L., Yang, E., Shen, H., Huang, D., Wang, R., et al. (2020). Macrophage-targeted isoniazid-selenium nanoparticles promote antimicrobial immunity and synergize bactericidal destruction of tuberculosis bacilli. Angew Chem. Int. Ed Engl. 59, 3226–3234. doi: 10.1002/anie.201912122
Piccaro, G., Pietraforte, D., Giannoni, F., Mustazzolu, A., and Fattorini, L. (2014). Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 7527–7533. doi: 10.1128/aac.03169-14
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M. A., et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. doi: 10.1016/j.immuni.2012.04.015
Polson, H. E., de Lartigue, J., Rigden, D. J., Reedijk, M., Urbé, S., Clague, M. J., et al. (2010). Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6, 506–522. doi: 10.4161/auto.6.4.11863
Ponpuak, M., Davis, A. S., Roberts, E. A., Delgado, M. A., Dinkins, C., Zhao, Z., et al. (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341. doi: 10.1016/j.immuni.2010.02.009
Prabakaran, T., Bodda, C., Krapp, C., Zhang, B. C., Christensen, M. H., Sun, C., et al. (2018). Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by tbk1. EMBO J. 37, e97858. doi: 10.15252/embj.201797858
Proikas-Cezanne, T., Waddell, S., Gaugel, A., Frickey, T., Lupas, A., and Nordheim, A. (2004). Wipi-1alpha (wipi49), a member of the novel 7-bladed wipi protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23, 9314–9325. doi: 10.1038/sj.onc.1208331
Purdy, G. E. (2011). Taking out TB-lysosomal trafficking and mycobactericidal ubiquitin-derived peptides. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00007
Qiu, Y., Wang, J., Li, H., Yang, B., Wang, J., He, Q., et al. (2022). Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy 18, 73–85. doi: 10.1080/15548627.2021.1908722
Qu, Y., Ding, S., Ma, Z., Jiang, D., Xu, X., Zhang, Y., et al. (2019). MiR-129-3p favors intracellular BCG survival in RAW264.7 cells by inhibiting autophagy via Atg4b. Cell Immunol. 337, 22–32. doi: 10.1016/j.cellimm.2019.01.004
Qu, Y., Gao, Q., Wu, S., Xu, T., Jiang, D., and Xu, G. (2021). MicroRNA-142-3p inhibits autophagy and promotes intracellular survival of Mycobacterium tuberculosis by targeting Atg16L1 and Atg4c. Int. Immunopharmacol. 101, 108202. doi: 10.1016/j.intimp.2021.108202
Ramachandran, G., Yeruva, C. V., Swarup, G., and Raghunand, T. R. (2024). A cytoprotective role for optineurin during mycobacterial infection of macrophages. Biochem. Biophys. Rep. 38, 101672. doi: 10.1016/j.bbrep.2024.101672
Raykov, L., Mottet, M., Nitschke, J., and Soldati, T. (2023). A traf-like E3 ubiquitin ligase trafe coordinates escrt and autophagy in endolysosomal damage response and cell-autonomous immunity to Mycobacterium marinum. Elife 12, e85727. doi: 10.7554/eLife.85727
Reed, M. B., Domenech, P., Manca, C., Su, H., Barczak, A. K., Kreiswirth, B. N., et al. (2004). A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87. doi: 10.1038/nature02837
Rekha, R. S., Rao Muvva, S. S., Wan, M., Raqib, R., Bergman, P., Brighenti, S., et al. (2015). Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy 11, 1688–1699. doi: 10.1080/15548627.2015.1075110
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by tbk1 enhances its binding to ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U S A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Rogov, V., Dötsch, V., Johansen, T., and Kirkin, V. (2014). Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell. 53, 167–178. doi: 10.1016/j.molcel.2013.12.014
Romagnoli, A., Di Rienzo, M., Petruccioli, E., Fusco, C., Palucci, I., Micale, L., et al. (2023). The ubiquitin ligase trim32 promotes the autophagic response to Mycobacterium tuberculosis infection in macrophages. Cell Death Dis. 14, 505. doi: 10.1038/s41419-023-06026-1
Romagnoli, A., Etna, M. P., Giacomini, E., Pardini, M., Remoli, M. E., Corazzari, M., et al. (2012). Esx-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370. doi: 10.4161/auto.20881
Russell, S. L., Lamprecht, D. A., Mandizvo, T., Jones, T. T., Naidoo, V., Addicott, K. W., et al. (2019). Compromised metabolic reprogramming is an early indicator of cd8(+) t cell dysfunction during chronic Mycobacterium tuberculosis infection. Cell Rep. 29, 3564–3579.e3565. doi: 10.1016/j.celrep.2019.11.034
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell. 20, 131–139. doi: 10.1016/j.devcel.2010.12.003
Saifullah, B., Chrzastek, A., Maitra, A., Naeemullah, B., Fakurazi, S., Bhakta, S., et al. (2017). Novel anti-tuberculosis nanodelivery formulation of ethambutol with graphene oxide. Molecules 22, 1560. doi: 10.3390/molecules22101560
Saini, N. K., Baena, A., Ng, T. W., Venkataswamy, M. M., Kennedy, S. C., Kunnath-Velayudhan, S., et al. (2016). Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_pgrs47. Nat. Microbiol. 1, 16133. doi: 10.1038/nmicrobiol.2016.133
Sauvé, V., Sung, G., Soya, N., Kozlov, G., Blaimschein, N., Miotto, L. S., et al. (2018). Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 25, 623–630. doi: 10.1038/s41594-018-0088-7
Schiebler, M., Brown, K., Hegyi, K., Newton, S. M., Renna, M., Hepburn, L., et al. (2015). Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 7, 127–139. doi: 10.15252/emmm.201404137
Schnettger, L., Rodgers, A., Repnik, U., Lai, R. P., Pei, G., Verdoes, M., et al. (2017). A rab20-dependent membrane trafficking pathway controls m. tuberculosis replication by regulating phagosome spaciousness and integrity. Cell Host Microbe 21, 619–628.e615. doi: 10.1016/j.chom.2017.04.004
Shang, L. and Wang, X. (2011). AMPK and mTOR coordinate the regulation of ULK1 and mammalian autophagy initiation. Autophagy 7, 924–926. doi: 10.4161/auto.7.8.15860
Shariq, M., Quadir, N., Alam, A., Zarin, S., Sheikh, J. A., Sharma, N., et al. (2023). The exploitation of host autophagy and ubiquitin machinery by Mycobacterium tuberculosis in shaping immune responses and host defense during infection. Autophagy 19, 3–23. doi: 10.1080/15548627.2021.2021495
Shariq, M., Quadir, N., Sharma, N., Singh, J., Sheikh, J. A., Khubaib, M., et al. (2021). Mycobacterium tuberculosis ripa dampens tlr4-mediated host protective response using a multi-pronged approach involving autophagy, apoptosis, metabolic repurposing, and immune modulation. Front. Immunol. 12. doi: 10.3389/fimmu.2021.636644
Sharma, T., Alam, A., Ehtram, A., Rani, A., Grover, S., Ehtesham, N. Z., et al. (2022). The Mycobacterium tuberculosis PE_PGRS protein family acts as an immunological decoy to subvert host immune response. Int. J. Mol. Sci. 23, 525. doi: 10.3390/ijms23010525
Sharma, V., Makhdoomi, M., Singh, L., Kumar, P., Khan, N., Singh, S., et al. (2021). Trehalose limits opportunistic mycobacterial survival during hiv co-infection by reversing hiv-mediated autophagy block. Autophagy 17, 476–495. doi: 10.1080/15548627.2020.1725374
Sharma, N., Shariq, M., Quadir, N., Singh, J., Sheikh, J. A., Hasnain, S. E., et al. (2021). Mycobacterium tuberculosis protein pe6 (rv0335c), a novel tlr4 agonist, evokes an inflammatory response and modulates the cell death pathways in macrophages to enhance intracellular survival. Front. Immunol. 12. doi: 10.3389/fimmu.2021.696491
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Shibutani, S. T., Saitoh, T., Nowag, H., Münz, C., and Yoshimori, T. (2015). Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16, 1014–1024. doi: 10.1038/ni.3273
Shin, D. M., Jeon, B. Y., Lee, H. M., Jin, H. S., Yuk, J. M., Song, C. H., et al. (2010). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PloS Pathog. 6, e1001230. doi: 10.1371/journal.ppat.1001230
Singh, A., Singh, A., Saraswati, S. S. K., Rana, A. K., Singh, A., Verma, C., et al. (2025). Suppressive effects of toll-like receptor 2, toll-like receptor 4, and toll-like receptor 7 on protective responses to Mycobacterium bovis BCG from epithelial cells. Microbes Infect. 27, 105428. doi: 10.1016/j.micinf.2024.105428
Singh, P. and Subbian, S. (2018). Harnessing the mTOR pathway for tuberculosis treatment. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00070
Singhal, A., Jie, L., Kumar, P., Hong, G. S., Leow, M. K., Paleja, B., et al. (2014). Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 6, 263ra159. doi: 10.1126/scitranslmed.3009885
Sinha, V., Singh, A., Singh, A., Saraswati, S. S. K., Rana, A. K., Kalra, K., et al. (2024). Potassium ion channel kir2.1 negatively regulates protective responses to Mycobacterium bovis BCG. J. Leukoc. Biol. 116, 644–656. doi: 10.1093/jleuko/qiae068
Siqueira, M. D. S., Ribeiro, R. M., and Travassos, L. H. (2018). Autophagy and its interaction with intracellular bacterial pathogens. Front. Immunol. 9. doi: 10.3389/fimmu.2018.00935
Siregar, T. A. P., Prombutara, P., Kanjanasirirat, P., Kunkaew, N., Tubsuwan, A., Boonmee, A., et al. (2022). The autophagy-resistant Mycobacterium tuberculosis beijing strain upregulates katg to evade starvation-induced autophagic restriction. Pathog. Dis. 80, ftac004. doi: 10.1093/femspd/ftac004
Song, Y., Ge, X., Chen, Y., Hussain, T., Liang, Z., Dong, Y., et al. (2022). Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy 18, 1401–1415. doi: 10.1080/15548627.2021.1987671
Songane, M., Kleinnijenhuis, J., Netea, M. G., and van Crevel, R. (2012). The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb). 92, 388–396. doi: 10.1016/j.tube.2012.05.004
Sou, Y. S., Waguri, S., Iwata, J., Ueno, T., Fujimura, T., Hara, T., et al. (2008). The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell. 19, 4762–4775. doi: 10.1091/mbc.e08-03-0309
Strong, L. M., Chang, C., Riley, J. F., Boecker, C. A., Flower, T. G., Buffalo, C. Z., et al. (2021). Structural basis for membrane recruitment of Atg16l1 by WIPI2 in autophagy. Elife 10, e70372. doi: 10.7554/eLife.70372
Strong, E., Hart, B., Wang, J., Orozco, M. G., and Lee, S. (2022). Induced synthesis of mycolactone restores the pathogenesis of Mycobacterium ulcerans in vitro and in vivo. Front. Immunol. 13, e00269-20. doi: 10.3389/fimmu.2022.750643
Strong, E. J., Jurcic Smith, K. L., Saini, N. K., Ng, T. W., Porcelli, S. A., and Lee, S. (2020). Identification of autophagy-inhibiting factors of Mycobacterium tuberculosis by high-throughput loss-of-function screening. Infect. Immun. 88. doi: 10.1128/iai.00269-20
Strong, E. J., Ng, T. W., Porcelli, S. A., and Lee, S. (2021). Mycobacterium tuberculosis PE_pgrs20 and PE_pgrs47 proteins inhibit autophagy by interaction with rab1a. mSphere 6, e0054921. doi: 10.1128/mSphere.00549-21
Subrahmanian, M., Marimuthu, J., Sairam, T., and Sankaran, R. (2020). In vitro ubiquitination of Mycobacterium tuberculosis by E3 ubiquitin ligase, mkrn1. Biotechnol. Lett. 42, 1527–1534. doi: 10.1007/s10529-020-02873-6
Suzuki, H., Hasegawa, S., Fushimi, S., Tagami, K., Nishikawa, M., Kondo, Y., et al. (2025). Metformin prevents diabetes development in type 1 diabetes models via suppression of mTOR and STAT3 signaling in immune cells. Sci. Rep. 15, 10641. doi: 10.1038/s41598-025-93647-5
Tang, X. Z., Huang, T., Ma, Y., Fu, X. Y., Qian, K., Yi, Y. L., et al. (2020). Meta-analysis of efficacy and safety of vitamin d supplementation in the treatment of pulmonary tuberculosis. Zhonghua Yi Xue Za Zhi. 100, 2525–2531. doi: 10.3760/cma.j.cn112137-20200121-00146
Tenforde, M. W., Yadav, A., Dowdy, D. W., Gupte, N., Shivakoti, R., Yang, W. T., et al. (2017). Vitamin A and d deficiencies associated with incident tuberculosis in hiv-infected patients initiating antiretroviral therapy in multinational case-cohort study. J. Acquir. Immune Defic. Syndr. 75, e71–e79. doi: 10.1097/qai.0000000000001308
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Tukvadze, N., Sanikidze, E., Kipiani, M., Hebbar, G., Easley, K. A., Shenvi, N., et al. (2015). High-dose vitamin d3 in adults with pulmonary tuberculosis: A double-blind randomized controlled trial. Am. J. Clin. Nutr. 102, 1059–1069. doi: 10.3945/ajcn.115.113886
Tung, Y. T., Hsu, W. M., Lee, H., Huang, W. P., and Liao, Y. F. (2010). The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell Mol. Neurobiol. 30, 795–806. doi: 10.1007/s10571-010-9507-y
Upadhyay, T. K., Fatima, N., Sharma, A., Sharma, D., and Sharma, R. (2019). Nano-rifabutin entrapment within glucan microparticles enhances protection against intracellular Mycobacterium tuberculosis. Artif. Cells Nanomed Biotechnol. 47, 427–435. doi: 10.1080/21691401.2018.1559180
van der Vaart, M., Korbee, C. J., Lamers, G. E., Tengeler, A. C., Hosseini, R., Haks, M. C., et al. (2014a). The DNA damage-regulated autophagy modulator dram1 links mycobacterial recognition via TLR-myd88 to autophagic defense. Cell Host Microbe 15, 753–767. doi: 10.1016/j.chom.2014.05.005
van der Vaart, M., Korbee, C. J., Lamers, G. E., Tengeler, A. C., Hosseini, R., Haks, M. C., et al. (2014b). The DNA damage-regulated autophagy modulator dram1 links mycobacterial recognition via TLR-myd88 to autophagic defense [corrected. Cell Host Microbe 15, 753–767. doi: 10.1016/j.chom.2014.05.005
Visser, M. E., Grewal, H. M., Swart, E. C., Dhansay, M. A., Walzl, G., Swanevelder, S., et al. (2011). The effect of vitamin a and zinc supplementation on treatment outcomes in pulmonary tuberculosis: A randomized controlled trial. Am. J. Clin. Nutr. 93, 93–100. doi: 10.3945/ajcn.110.001784
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., et al. (2004). Protein kinase g from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804. doi: 10.1126/science.1099384
Wang, Y., Chen, C., Xu, X. D., Li, H., Cheng, M. H., Liu, J., et al. (2020). Levels of miR-125a-5p are altered in Mycobacterium avium-infected macrophages and associate with the triggering of an autophagic response. Microbes Infect. 22, 31–39. doi: 10.1016/j.micinf.2019.07.002
Wang, J., Hussain, T., Yue, R., Liao, Y., Li, Q., Yao, J., et al. (2018). MicroRNA-199a inhibits cellular autophagy and downregulates IFN-β expression by targeting tbk1 in Mycobacterium bovis infected cells. Front. Cell Infect. Microbiol. 8. doi: 10.3389/fcimb.2018.00238
Wang, H., Liu, D., Ge, X., Wang, Y., and Zhou, X. (2025). Mycobacterium bovis mb3523c protein regulates host ferroptosis via chaperone-mediated autophagy. Autophagy 21, 1335–1352. doi: 10.1080/15548627.2025.2468139
Wang, Y., Liu, J., Wang, H., Jiang, P., Cao, L., Lu, S., et al. (2025). Multiple regulatory mechanisms, functions and therapeutic potential of chaperone-mediated autophagy. Theranostics 15, 2778–2793. doi: 10.7150/thno.107761
Wang, J., Sha, J., Strong, E., Chopra, A. K., and Lee, S. (2022). Fda-approved amoxapine effectively promotes macrophage control of mycobacteria by inducing autophagy. Microbiol. Spectr. 10, e0250922. doi: 10.1128/spectrum.02509-22
Wang, J., Xiong, K., Wang, Q., Zhao, S., Liu, Y., and Ma, A. (2020). Adjunctive Vitamin A and d during pulmonary tuberculosis treatment: A randomized controlled trial with a 2 × 2 factorial design. Food Funct. 11, 4672–4681. doi: 10.1039/c9fo02751c
Wang, J., Yang, K., Zhou, L., Minhaowu, Wu, Y., Zhu, M., et al. (2013). MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting rheb. PloS Pathog. 9, e1003697. doi: 10.1371/journal.ppat.1003697
Wang, R., Yao, X., and Li, R. (2019). Mycetoma in China: A case report and review of the literature. Mycopathologia 184, 327–334. doi: 10.1007/s11046-019-00324-z
Watson, R. O., Bell, S. L., MacDuff, D. A., Kimmey, J. M., Diner, E. J., Olivas, J., et al. (2015). The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type i interferons and activate autophagy. Cell Host Microbe 17, 811–819. doi: 10.1016/j.chom.2015.05.004
Watson, R. O., Manzanillo, P. S., and Cox, J. S. (2012). Extracellular m. Tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. doi: 10.1016/j.cell.2012.06.040
Weidberg, H., Shvets, E., Shpilka, T., Shimron, F., Shinder, V., and Elazar, Z. (2010). LC3 and gate-16/gabarap subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802. doi: 10.1038/emboj.2010.74
Whang, M. I., Tavares, R. M., Benjamin, D. I., Kattah, M. G., Advincula, R., Nomura, D. K., et al. (2017). The ubiquitin binding protein TAX1BP1 mediates autophagasome induction and the metabolic transition of activated t cells. Immunity 46, 405–420. doi: 10.1016/j.immuni.2017.02.018
Whitehouse, C. A., Waters, S., Marchbank, K., Horner, A., McGowan, N. W., Jovanovic, J. V., et al. (2010). Neighbor of brca1 gene (nbr1) functions as a negative regulator of postnatal osteoblastic bone formation and p38 MAPK activity. Proc. Natl. Acad. Sci. U S A. 107, 12913–12918. doi: 10.1073/pnas.0913058107
Wu, Y., Tian, M., Zhang, Y., Peng, H., Lei, Q., Yuan, X., et al. (2022). Deletion of BCG_2432c from the bacillus calmette-guérin vaccine enhances autophagy-mediated immunity against tuberculosis. Allergy 77, 619–632. doi: 10.1111/all.15158
Wu, H. X., Xiong, X. F., Zhu, M., Wei, J., Zhuo, K. Q., and Cheng, D. Y. (2018). Effects of vitamin d supplementation on the outcomes of patients with pulmonary tuberculosis: A systematic review and meta-analysis. BMC Pulm Med. 18, 108. doi: 10.1186/s12890-018-0677-6
Xi, L., Du, J., Xue, W., Shao, K., Jiang, X., Peng, W., et al. (2024). Cathelicidin LL-37 promotes wound healing in diabetic mice by regulating TFEB-dependent autophagy. Peptides 175, 171183. doi: 10.1016/j.peptides.2024.171183
Xie, X., Li, F., Wang, Y., Wang, Y., Lin, Z., Cheng, X., et al. (2015). Molecular basis of ubiquitin recognition by the autophagy receptor calcoco2. Autophagy 11, 1775–1789. doi: 10.1080/15548627.2015.1082025
Xie, J. and Meijer, A. H. (2023). Xenophagy receptors optn and p62 and autophagy modulator dram1 independently promote the zebrafish host defense against Mycobacterium marinum. Front. Cell Infect. Microbiol. 13. doi: 10.3389/fcimb.2023.1331818
Yabaji, S. M., Dhamija, E., Mishra, A. K., and Srivastava, K. K. (2020). ESAT-6 regulates autophagous response through SOD-2 and as a result induces intracellular survival of Mycobacterium bovis BCG. Biochim. Biophys. Acta Proteins Proteom. 1868, 140470. doi: 10.1016/j.bbapap.2020.140470
Yang, C. S. (2017). Advancing host-directed therapy for tuberculosis. Microb. Cell. 4, 105–107. doi: 10.15698/mic2017.03.565
Yang, Z. and Klionsky, D. J. (2010). Eaten alive: A history of macroautophagy. Nat. Cell Biol. 12, 814–822. doi: 10.1038/ncb0910-814
Yang, R., Peng, Y., Pi, J., Liu, Y., Yang, E., Shen, X., et al. (2021). A CD4+CD161+ t-cell subset present in unexposed humans, not tb patients, are fast acting cells that inhibit the growth of intracellular mycobacteria involving CD161 pathway, perforin, and IFN-γ/autophagy. Front. Immunol. 12. doi: 10.3389/fimmu.2021.599641
Yang, N., Yu, G., Lai, Y., Zhao, J., Chen, Z., Chen, L., et al. (2024). A snake cathelicidin enhances transcription factor eb-mediated autophagy and alleviates ROS-induced pyroptosis after ischaemia-reperfusion injury of island skin flaps. Br. J. Pharmacol. 181, 1068–1090. doi: 10.1111/bph.16268
Yang, L., Zhang, C., Zhao, Y., Zhao, N., Wu, P., Zhang, H., et al. (2018). Effects of Mycobacterium tuberculosis mutant strain hsp16.3 gene on murine RAW 264.7 macrophage autophagy. DNA Cell Biol. 37, 7–14. doi: 10.1089/dna.2016.3599
Yoshimori, T. and Amano, A. (2009). Group a streptococcus: A loser in the battle with autophagy. Curr. Top. Microbiol. Immunol. 335, 217–226. doi: 10.1007/978-3-642-00302-8_10
Yuan, Q., Chen, H., Yang, Y., Fu, Y., and Yi, Z. (2020). MiR-18a promotes mycobacterial survival in macrophages via inhibiting autophagy by down-regulation of atm. J. Cell Mol. Med. 24, 2004–2012. doi: 10.1111/jcmm.14899
Yuan, W., Zhan, X., Liu, W., Ma, R., Zhou, Y., Xu, G., et al. (2023). Mmu-miR-25-3p promotes macrophage autophagy by targeting dusp10 to reduce mycobacteria survival. Front. Cell Infect. Microbiol. 13. doi: 10.3389/fcimb.2023.1120570
Yuk, J. M., Yoshimori, T., and Jo, E. K. (2012). Autophagy and bacterial infectious diseases. Exp. Mol. Med. 44, 99–108. doi: 10.3858/emm.2012.44.2.032
Zhang, M., Adroub, S., Ummels, R., Asaad, M., Song, L., Pain, A., et al. (2024). Comprehensive pan-genome analysis of Mycobacterium marinum: Insights into genomic diversity, evolution, and pathogenicity. Sci. Rep. 14, 27723. doi: 10.1038/s41598-024-75228-0
Zhang, Y., Mun, S. R., Linares, J. F., Ahn, J., Towers, C. G., Ji, C. H., et al. (2018). Zz-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 9, 4373. doi: 10.1038/s41467-018-06878-8
Zhang, Q., Sun, J., Wang, Y., He, W., Wang, L., Zheng, Y., et al. (2017). Antimycobacterial and anti-inflammatory mechanisms of baicalin via induced autophagy in macrophages infected with Mycobacterium tuberculosis. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02142
Zhang, R., Varela, M., Forn-Cuní, G., Torraca, V., van der Vaart, M., and Meijer, A. H. (2020). Deficiency in the autophagy modulator dram1 exacerbates pyroptotic cell death of mycobacteria-infected macrophages. Cell Death Dis. 11, 277. doi: 10.1038/s41419-020-2477-1
Zhang, R., Varela, M., Vallentgoed, W., Forn-Cuni, G., van der Vaart, M., and Meijer, A. H. (2019). The selective autophagy receptors optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PloS Pathog. 15, e1007329. doi: 10.1371/journal.ppat.1007329
Zhao, Y. G., Codogno, P., and Zhang, H. (2021). Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 22, 733–750. doi: 10.1038/s41580-021-00392-4
Zhao, L., Fan, K., Sun, X., Li, W., Qin, F., Shi, L., et al. (2023). Host-directed therapy against Mycobacterium tuberculosis infections with diabetes mellitus. Front. Immunol. 14, 9550. doi: 10.3389/fimmu.2023.1305325
Zhao, L., You, W., Sun, D., Xu, H., You, X., Xu, H., et al. (2022). Vps21 directs the PI3K-PI(3)P-Atg21-Atg16 module to phagophores via VPS8 for autophagy. Int. J. Mol. Sci. 23. doi: 10.3390/ijms23179550
Zheng, Q., Li, Z., Zhou, S., Zhang, Q., Zhou, L., Fu, X., et al. (2017). Heparin-binding hemagglutinin of Mycobacterium tuberculosis is an inhibitor of autophagy. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00033
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in nlrp3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Keywords: mycobacterium, autophagy, molecular mechanism, host-pathogen interaction, host-directed therapy
Citation: Li J, Feng H, Chen D, Zhang H and Liao Y (2025) Autophagy in mycobacterial infections: molecular mechanisms, host-pathogen interactions, and therapeutic opportunities. Front. Cell. Infect. Microbiol. 15:1640647. doi: 10.3389/fcimb.2025.1640647
Received: 04 June 2025; Accepted: 21 July 2025;
Published: 07 August 2025.
Edited by:
Percy Schröttner, Technische Universität Dresden, GermanyReviewed by:
Alessandra Vitaliti, University of Rome Tor Vergata, ItalyAndressa Francielli Bonjorno, Sao Paulo State Universty, Brazil
Copyright © 2025 Li, Feng, Chen, Zhang and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Liao, MjczODA2MTQ2QHFxLmNvbQ==