 Alicia S. Arroyo
Alicia S. Arroyo David López-Escardó
David López-Escardó Eunsoo Kim
Eunsoo Kim Iñaki Ruiz-Trillo
Iñaki Ruiz-Trillo Sebastián R. Najle
Sebastián R. Najle- 1Institut de Biologia Evolutiva, CSIC-Universitat Pompeu Fabra, Barcelona, Spain
- 2Division of Invertebrate Zoology and Sackler, Institute for Comparative Genomics, American Museum of Natural History, New York, NY, United States
- 3Departament de Genètica, Microbiologia i Estadística, Institut de Recerca de la Biodiversitat, Universitat de Barcelona, Barcelona, Spain
- 4ICREA, Barcelona, Spain
- 5Instituto de Biología Molecular y Celular de Rosario (IBR-CONICET), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Rosario, Argentina
Opisthokonta represents a major lineage of eukaryotes and includes fungi and metazoans, as well as other less known unicellular groups. The latter are paraphyletic assemblages that branch in between the former two groups, and thus are important for understanding the origin and early diversification of opisthokonts. The full range of their diversity, however, has not yet been explored from diverse ecological habitats. Freshwater environments are crucial sources for new diversity; they are considered even more heterogeneous than marine ecosystems. This heterogeneity implies more ecological niches where local eukaryotic communities are located. However, knowledge of the unicellular opisthokont diversity is scarce from freshwater environments. Here, we performed an 18S rDNA metabarcoding study in the Middle Paraná River, Argentina, to characterize the molecular diversity of microbial eukaryotes, in particular unicellular members of Opisthokonta. We identified a potential novel clade branching as a sister-group to Fungi. We also detected in our data that more than 60% operational taxonomic units classified as unicellular holozoans (animals and relatives) represent new taxa at the species level. Of the remaining, the majority was assigned to the newly described holozoan species, Syssomonas multiformis. Together, our results show that a large hidden diversity of unicellular members of opisthokonts still remain to be uncovered. We also found that the geographical and ecological distribution of several taxa considered exclusive to marine environments is wider than previously thought.
Introduction
Understanding eukaryotic diversity is an essential need for humankind. It does not only provide more knowledge about evolutionary processes, but it also helps to fully describe complex ecological networks, which can lead to better conservation policies and new sources of food and medicines (Pawlowski et al., 2016). From all eukaryotes, historically, animals, fungi, and plants have stood up, hence they account for the vast majority of described species (Pawlowski et al., 2012; del Campo et al., 2014). Nevertheless, molecular surveys of biodiversity have redrawn a completely different paradigm, in which unicellular eukaryotes represent the majority of the total eukaryotic richness, while remaining mostly undescribed (Pawlowski et al., 2012; del Campo et al., 2014). Many studies have overcome this limitation using molecular data to define new clades (López-García et al., 2001; Massana et al., 2004, 2014; Šlapeta et al., 2005; Guillou et al., 2008; Marande et al., 2009; Jones et al., 2011; del Campo and Ruiz-Trillo, 2013). However, the complete view of the molecular eukaryotic diversity remains unsolved.
This lack of knowledge of eukaryotic diversity also occurs in Opisthokonta, the superclade that comprises Metazoa, Fungi and several unicellular lineages (Ruiz-Trillo et al., 2008; Paps and Ruiz-Trillo, 2010; Torruella et al., 2012). These unicellular opisthokonts are understudied compared to their multicellular relatives, even though they are essential for better understanding key evolutionary transitions, including those that took place at the origins of both animals and fungi (Ruiz-Trillo et al., 2007; Richter and King, 2013; Brunet and King, 2017; Richards et al., 2017; Sebé-Pedrós et al., 2017).
Opisthokonts are divided into two major clades: Holozoa and Holomycota. Holozoa contains animals and their unicellular relatives, namely Choanoflagellata, Filasterea and Teretosporea (or Ichthyosporea and Plurimorfea, depending on the authors) (Lang et al., 2002; Torruella et al., 2012, 2015; Hehenberger et al., 2017). Holomycota contains fungi and their closest unicellular lineages (Brown et al., 2009; Liu et al., 2009; Lara et al., 2010; López-Escardó et al., 2018). Previous studies on unicellular opisthokonts had already found new molecular lineages within or even between clades, such as marine opisthokonts (MAOP), marine fonticulids (MAFO), marine choanoflagellates (MACHO) or Clade L (Weber et al., 2012; del Campo and Ruiz-Trillo, 2013; del Campo et al., 2015). The majority of these environmental lineages of opisthokonts were described in marine environments. However, a few new species have been recently described in fresh waters; these include the genera Syssomonas and Pigoraptor (Hehenberger et al., 2017) within Holozoa and Parvularia as an early-branching Holomycota (López-Escardó et al., 2018). Therefore, there is need for exploring the diversity of unicellular opisthokonts, particularly from freshwater environments where a wide range of ecological niches exist (Logares et al., 2009a; Simon et al., 2015). This heterogeneous nature can be a potential reservoir of a diverse array of new eukaryotic lineages (Šlapeta et al., 2005; Downing, 2010).
Among freshwater habitats, lakes are the most studied environments (Charvet et al., 2012; Eiler et al., 2013; Lepère et al., 2016; Boenigk et al., 2018). For example, mountain lakes have been shown to harbor an extensive diversity within large eukaryotic lineages and a high proportion of unclassified sequences (Triadó-Margarit and Casamayor, 2012; Kammerlander et al., 2015; Filker et al., 2016; Boenigk et al., 2018). This is remarkable given the extreme oligotrophic condition of some of these lakes. There has also been research on ponds, brooks and other seasonal currents (Šlapeta et al., 2005; Simon et al., 2014, 2015). Some of these studies found novel eukaryotic clades, and showed the presence of some putatively marine-exclusive lineages in those freshwater environments. However, fewer molecular biodiversity surveys have been conducted in rivers (Berney et al., 2004; Thomas et al., 2012; Debroas et al., 2017).
In the present study, we performed a metabarcoding survey in the Middle Paraná River, Argentina. The Paraná River is the second largest river in South America, after the Amazon. It springs in southern Brazil and flows into the Atlantic Ocean, going through a great variety of ecosystems, from deserts to humid forests. It covers 2.6 million km2, making the Paraná River basin the sixth biggest river basin around the world (Iriondo and Paira, 2007). Its large volume flow allows the formation of heterogeneous wetlands and channels and drags tons of fine sediments toward the ocean (Iriondo and Paira, 2007). We chose the Middle Paraná for several reasons. First, it is less studied than the Upper Paraná River (Train and Rodrigues, 1997; Agostinho et al., 2004a,b; Lansac-Tôha et al., 2009; Roberto et al., 2009). Second, the Bermejo River, one of the main tributaries to the Middle Paraná in its origin, supplies 50–70% of the dissolved solids (Devercelli, 2006), which can be a decisive factor for determining the composition of the protist community. These sediments, together with the vegetation, create different natural barriers, in which some locations of the river are not permanently flooded, favoring a high heterogeneity between closely located sites. Finally, previous studies about protist diversity in this river focused largely on morphological analyses (Zalocar de Domitrovic et al., 2007). To our knowledge, there is no molecular diversity study done along the Paraná River.
We performed a metabarcoding survey in the Middle Paraná River with one main objective: to find potential molecular novelties within the Opisthokonta clade. We sequenced the V4 and V8-9 regions of the 18S rRNA gene in samples from five different places of the Middle Paraná River. We carried out phylogenetic analyses to look for molecular novelties both within Holozoa and Holomycota. We found that the majority of new diversity within the Holomycota identified in our study represent members of “early-branching” groups such as Chytridiomycota and Cryptomycota. We also identified two potential new clades (tentatively named as FRESHOL1 and FRESHOL1-related) that diverge as sister-groups to the classical “true” Fungi. Regarding unicellular Holozoa, more than 60% OTUs of Paraná diversity matches with uncultured sequences, showing that the hidden diversity is still very high, especially within choanoflagellates. We also identified OTUs closely related to the newly described species Syssomonas multiformis (Hehenberger et al., 2017), which strikingly, appears to be the most abundant holozoan taxon in our dataset. Finally, we detected in our freshwater samples the presence of groups that were previously known only from marine samples; these include members of Acanthoecid choanoflagellates. Our results add to a growing body of evidence suggesting that the microbial portion of Opisthokonta is still vastly under-sampled and that our current ecological understanding of these organisms is limited.
Results
Sampling, Sequencing, and Clustering
Fresh water samples were collected from five different locations in the Middle Paraná River, nearby San Nicolás de los Arroyos, Argentina (Figure 1). We fractionated each water sample by size: picoplanktonic (2–8 μm) and nanoplanktonic (8–15 μm) portions because the majority of unicellular opisthokonts have these dimensions. We performed DNA extraction and amplified the V4 and V8-9 regions of the 18S rRNA gene. Sequencing was carried out on the Illumina MiSeq platform (see details in Materials and Methods). We retrieved 3,328,000 reads that were subsequently clustered at 97% threshold following the UPARSE workflow (Edgar, 2013). A total of 8,061 OTUs (4,195 of V4 region; 3,866 of V8-9 region) were obtained. We assigned them taxonomically using our curated PR2 database (see Materials and Methods).
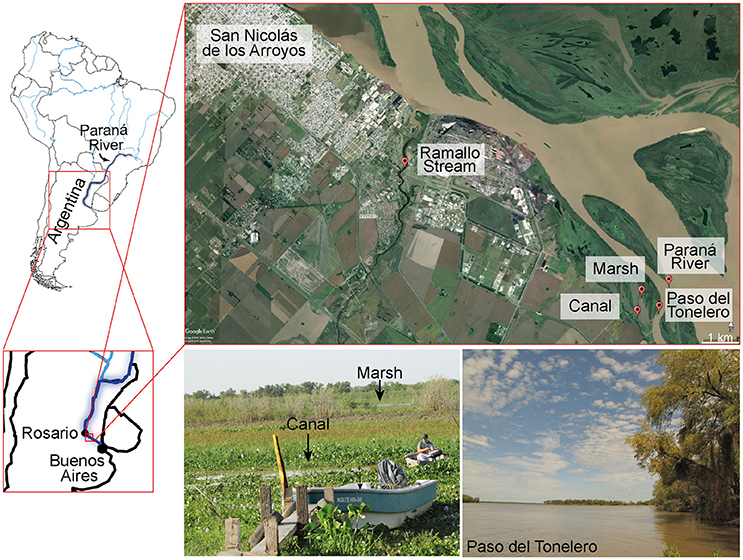
Figure 1. Localization of the sampling sites in the Middle Paraná River, Argentina. The five sampling sites are located near San Nicolás de los Arroyos, in the north of Buenos Aires Province, Argentina. Out of five sites, 4 were located in the Paraná River (Paraná River, Paso del Tonelero, Marsh and Canal), while Ramallo Stream is a small tributary of it. Specific cartographic data for each site can be found in Supplementary Table.
Diversity Patterns of Freshwater Eukaryotes
We first analyzed the overall perspective on the eukaryotic diversity present in the data. We used the Bray-Curtis dissimilarity matrix, which was plotted in a dendrogram (see Materials and Methods). Our data appeared to cluster by size fraction, rather than sampling site (Supplementary Figures 1A, 2A). The only exception is Ramallo Stream, whose two size fractions clustered together far from the rest of the sampling sites. This topology of the cluster is the same in both V4 and V8-9 regions. Moreover, the distribution of unique eukaryotic OTUs in each sampling site showed that the composition of eukaryotes at Ramallo Stream differed the most from the other sites. In fact, 41.81% of eukaryotic OTUsV4 and 28.40% of OTUsV8−9 are exclusive to Ramallo Stream (Supplementary Figure 3A). Despite these differences among sampling sites, the rarefaction curves of the whole eukaryotic diversity in both regions of the 18S rRNA gene reached saturation (Supplementary Figure 3B). This implies that most of the unicellular eukaryotic diversity in the sampling locations was recovered.
We later performed a general analysis to detect patterns of eukaryotic diversity. We recovered reads from all major eukaryotic groups (Adl et al., 2012; Figure 2). Regarding richness (number of OTUs), Holomycota is the richest eukaryotic group (22.8% in V4 region, 21% in V8-9 region), followed by Cercozoa (15.1%, 12.7%) and Ciliophora (10.1%, 9.1%, respectively). However, in terms of abundance (number of reads), Bacillariophyta (Stramenopiles) is the most abundant group (33.85% in V4 region, 30.3% in V8-9 region), together with Ciliophora (15.9% and 18.9%, respectively) (Figure 2).
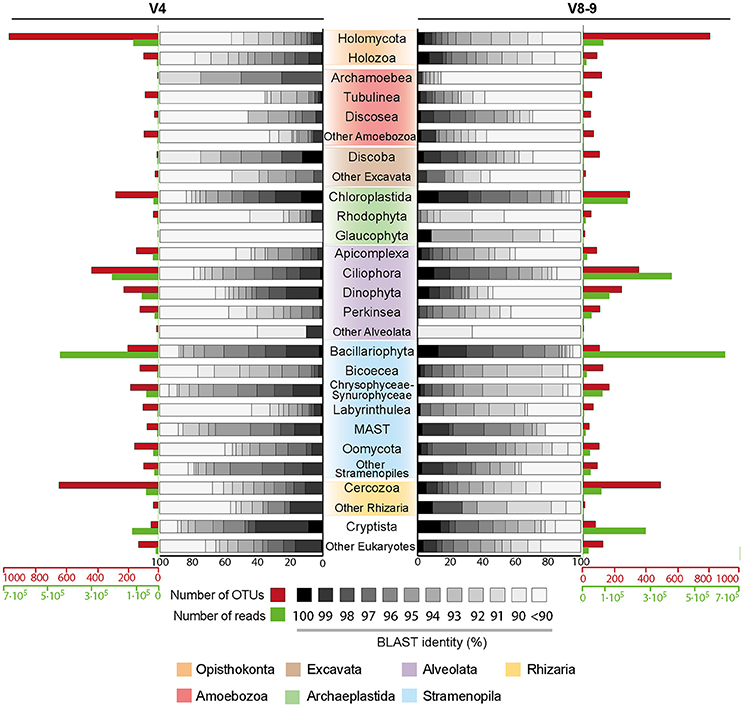
Figure 2. Eukaryotic diversity in Paraná River represented by V4 and V8-9 regions of 18S rRNA gene. Eukaryotic classification follows Adl et al. (2012). External barplots show the richness (number of OTUs) in red and the abundance (number of reads) in green for each eukaryotic group. Inner histograms show the distribution of percentages of blast identity against our reference database adapted from PR2 (see Materials and Methods) in every group.
A more specific analysis of the eukaryotic diversity across sampling sites or size fraction showed that these abundant groups are not equally distributed in our samples. For example, Holomycota accounts for = 22.94% and = 21.81% of the total eukaryotic abundance in Ramallo Stream while in the rest of samples its abundance remains below 6.5% and 2.5% in V4 and V8-9 regions, respectively (Supplementary Figures 1B, 2B). Bacillariophyta is mostly found in the 8–15 μm fraction in all samples ( = 47.98% and = 46%), while Cryptista is mostly found in the 2-8 μm fraction ( = 16.74% and = 21.51%) (Supplementary Figures 1B, 2B). Another exception is the Dinophyta, whose abundance in Marsh 8-15 μm is much higher than in the rest of the samples (32.13% in V4 and 33.03% in V8-9, compared to 2.35% and 1.79%) (Supplementary Figures 1B, 2B). In contrast, the rest of the eukaryotic groups have a more homogeneous distribution across the samples.
We then questioned whether there were differences in the level of potential novel diversity among different eukaryotic lineages by looking at the blast identities against the PR2-Opistho database (see Materials and Methods). Our analyses showed that Amoebozoa stands out in terms of potential novelty; with around half of its diversity having less than 90% of blast identity against our reference database ( = 46.55% and = 57.96%) (Figure 2 and Supplementary Table). It is worth mentioning the case of the Archamoebae, with 85.71% of OTUsV8−9 below 90% of blast identity. Alveolata also holds a high amount of diversity that could be considered unknown. If the well-known ciliates (within the Ciliophora group) are not considered, then 45.81% and 51% of the Alveolata diversity is below 90% blast similarity in V4 and V8-9 region, respectively. The groups Rhodophyta ( = 55.17% and = 47.06%) and Glaucophyta ( = 100% and = 16.67%) also have a high amount of potential novel diversity (Figure 2 and Supplementary Table). Finally, Opisthokonta contains a moderate proportion of unknown diversity ( = 32.72% and = 19.89%) (Figure 2). This group also shows a steep slope of the accumulation curve, indicating the large amount of diversity still to be discovered (Supplementary Figure 3C). This steep slope, together with the low blast identity values and the curated dataset available, offered us the possibility to further explore the putative unknown diversity retrieved within Opisthokonta.
Novel Opisthokonta Diversity
Diversity and Novelty of Holomycota
Holomycota (syn. Nucletmycea) is the group that contains Fungi and their unicellular relatives, namely Nucleariidae and Opisthosporidia (Brown et al., 2009; Liu et al., 2009; Karpov et al., 2014a). In our dataset, Holomycota is the richest eukaryotic clade (Figure 2). In particular, we obtained 956 and 813 OTUs of Holomycota in the V4 and the V8-9 region, which represents 22.8 and 21% of the total eukaryotes, respectively. The distribution of this diversity is considerably uniform across the sampling sites (Supplementary Figure 4). However, there is a singular difference between V4 and V8-9 regions. While Chytridiomycota and Cryptomycota are the most abundant groups in the V4 region, Dikarya (the group that comprises Ascomycota and Basidiomycota) is the most abundant one in the V8-9 region.
A good proportion of the total Holomycota richness (specifically, 43.93% in V4 and 23.7% in V8-9) has less than 90% of blast identity against PR2-Opistho database and, therefore, could be potential novel diversity (Figure 2 and Supplementary Table). Additionally, the majority of the Holomycota richness (63.08% of the taxonomical assignments in V4 and 51.78% in V8-9) does not match with actual described species, but with environmental sequences (Supplementary Table). Given these numbers, we went further to explore the putative new Holomycota diversity.
To look for Holomycota novelties, we performed a phylogenetic placement of all the OTUs into a curated Holomycota reference tree (see Materials and Methods) (Figure 3A, Supplementary Figures 5, 6, and Supplementary Datasheets 1, 2). In both regions, we did not recover many OTUs among the classical “true” Fungi, such as Dikarya, Glomeromycota or Zygomycota (Karpov et al., 2014a; Spatafora et al., 2016). Instead, we recovered a large number of Opisthosporidia, which is also the group with a high number of OTUs placed in both terminal and internal nodes: the latter probably indicating the presence of novel groups (see details of the phylogenetic placement approach in Materials and Methods).
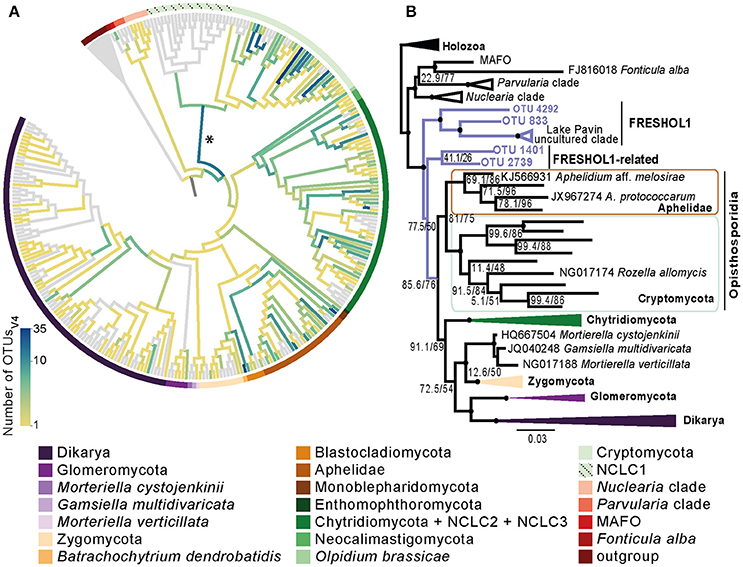
Figure 3. Diversity and novelty of Holomycota OTUs of the V4 region. (A) Mapping of all Holomycota OTUsV4 into our reference tree. The darker the branch, the higher amount of OTUsV4 it contains, as it is depicted in the reference bar. Taxonomic assignment is shown by color code indicated in the panel below. Early-diverging fungal groups, such as Cryptomycota and Chytridiomycota hold the majority of the Paraná Holomycota diversity. An asterisk indicates an assemblage of putatively novel OTUsV4, whose phylogeny is shown in (B). (B) Maximum-likelihood tree showing the phylogenetic placement of novel OTUsV4 found in our Paraná dataset in relation to Holomycota and other opisthokont lineages including the environmental clade identified from Lake Pavin (Monchy et al., 2011). We term this group FRESHOL1 (for freshwater Holomycota). We also named the closest group as FRESHOL1-related. The tree was obtained using IQ-tree software under GTR evolutionary model with nodal supports (SH-aLRT and UFBoot support) obtained through 1,000 bootstrap replicates (Nguyen et al., 2015). Black dots are depicted when SH-aLRT > 80% and UFBoot > 95%. Full alignment and tree can be found in Supplementary Datasheet 3.
In this respect, an interesting branch is the one indicated by an asterisk in Figure 3A, which branches in a very early position in the phylogeny of Holomycota. This branch, despite being quite deep, contains the highest amount of OTUsV4 placed within Holomycota. With the objective to determine the phylogenetic position of these sequences, we retained the OTUsV4 that were placed in this branch by two different software packages for phylogenetic placement (see Materials and Methods). We also searched for related full-length 18S sequences in GenBank that could help to improve the phylogenetic signal (Supplementary Datasheet 3). The final tree is shown in Figure 3B. The 4 OTUsV4 retrieved (OTU IDs 4292, 833, 1401 and 2739) plus the long GenBank 18S sequences from Lake Pavin (Monchy et al., 2011) are positioned as the sister-group of the clade formed by Opisthosporidia + Fungi with good statistical support (97.7% and 99% for both ML IQ-tree 1,000 bootstrap replicates: SH-aLRT and UFBoot, respectively) (full tree and alignments can be found in Supplementary Datasheet 3). The group formed by OTU 4292, OTU 833 and the Lake Pavin clade is well supported (SH-aLRT = 92, UFBoot = 94); thus, we name it as FRESHOL1 (for freshwater Holomycota). The specific phylogenetic position of the group formed by OTUs 1401 and 2739 varies depending on the maximum-likelihood software used, but they are always positioned early-branching to the clade that includes Fungi and Opisthosporidia. We, then, tentatively propose FRESHOL1-related to this group.
Diversity and Novelty of Holozoa
Holozoa is the group that contains Metazoa and their closest unicellular relatives, namely Choanoflagellata, Filasterea, Ichthyosporea, and Plurimorfea, the latter with an uncertain phylogenetic position in relation to Ichthyosporea (Paps and Ruiz-Trillo, 2010; Torruella et al., 2015; Hehenberger et al., 2017). Holozoa also contains some exclusively environmental clades, mainly within choanoflagellates and ichthyosporeans (del Campo and Ruiz-Trillo, 2013). We detected 180 unicellular Holozoa OTUs in both V4 and V8-9 regions. The majority of these OTUs were assigned to Choanoflagellata ( = 64.45%, both V4 and V8-9 regions), followed by Ichthyosporea ( = 22.81%) and other holozoan lineages (namely, MAOP1 and Syssomonas multiformis; = 6.6%). We did not identify any Filasterea reads in our samples, neither in V4 nor in V8-9 region. Finally, around 6% of all unicellular holozoans could not be assigned to any defined clade, so they were classified as Holozoa-unclassified (Figure 4A and Supplementary Table).
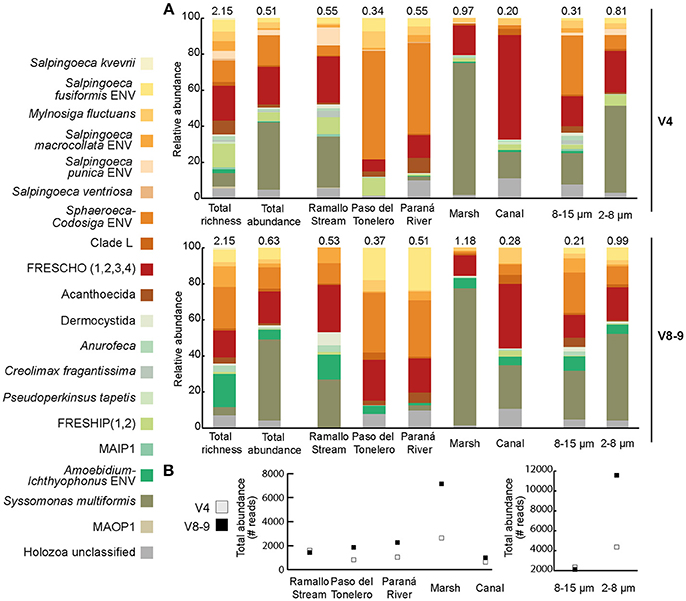
Figure 4. (A) Distribution and relative abundance of the unicellular Holozoa as a whole, across sampling sites and by size fraction. Note that Syssomonas multiformis, Sphaeroeca-Codosiga environmental clade and the environmental group freshwater choanoflagellates (FRESCHO) account for most of the detected diversity. Numbers on top of each histogram shows the relative proportion of the unicellular Holozoa with respect to all eukaryotes. ENV stands for environmental, those sequences in our dataset that expand the diversity of a specific clade or species. (B) Distribution of reads across sampling sites (left) and size fraction (right) depending on the 18S region considered. Even though the diversity patters are similar, the V8-9 region detects more abundance in each sampling site, especially in the 2–8 μm fraction.
Only 33.79% of the Holozoa OTUs that could be properly assigned in both regions were associated to a defined genus or species level. To the contrary, the majority of them (59.14% in V4 region; 62.07% in V8-9) were associated with environmental uncultured sequences (Figures 5, 6). This implies that most of the unicellular Holozoa diversity found in Paraná River is still undescribed.
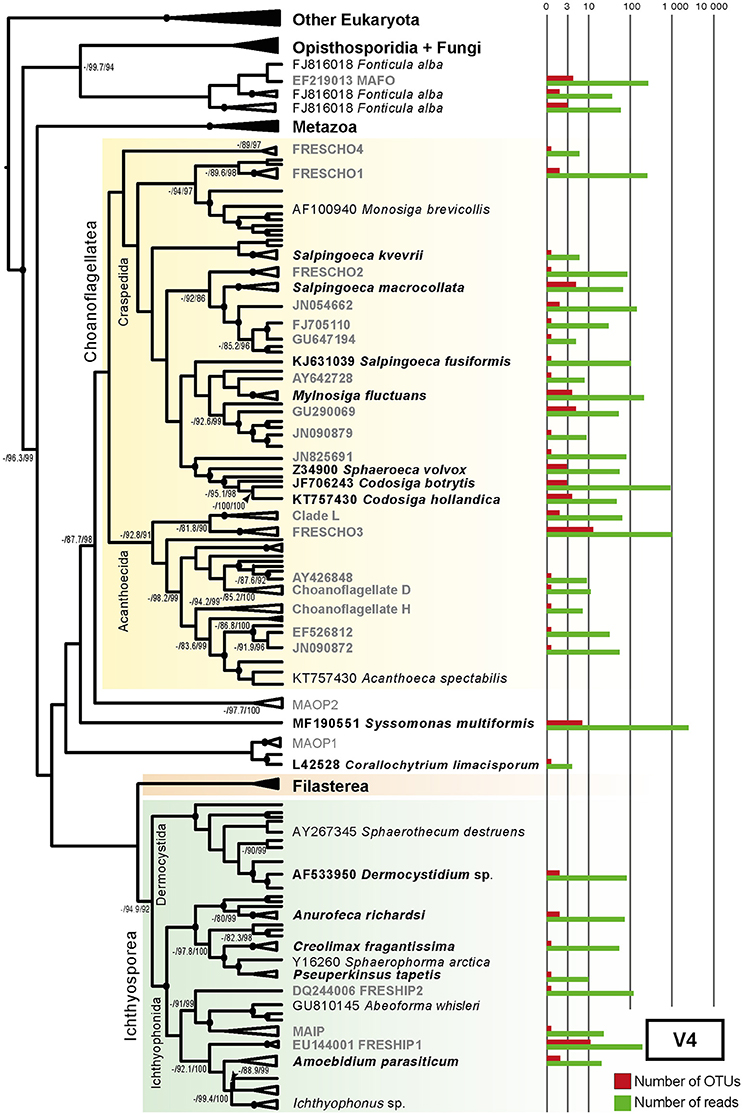
Figure 5. Maximum-likelihood consensus tree of all GenBank sequences showing the diversity and novelty of unicellular Holozoa in V4 region. The tree was constructed using RAxML v.8.2.8 under GTRCAT evolutionary model with a nodal support obtained through 1,000 bootstrap replicates (Stamatakis, 2014). Three nodal supports are shown: ML bootstrap/SH-aLRT/UFBoot. Nodal support is depicted as black dots when all three supports are above 90%, as a numerical value when ML bootstrap is below 90% and absent if at least two of the supports were below 90%. Barplots show the number of OTUs (red) and the number of reads (green) associated with their closest GenBank full-length representatives, either environmental (gray) or morphologically described (black) (see Materials and Methods). The more abundant groups are S. multiformis, the recently described species from a lake in Vietnam (Hehenberger et al., 2017) and several environmental clades, such as FRESCHO3 or FRESHIP1. Alignments and phylogenetic trees can be found in Supplementary Datasheet 5.
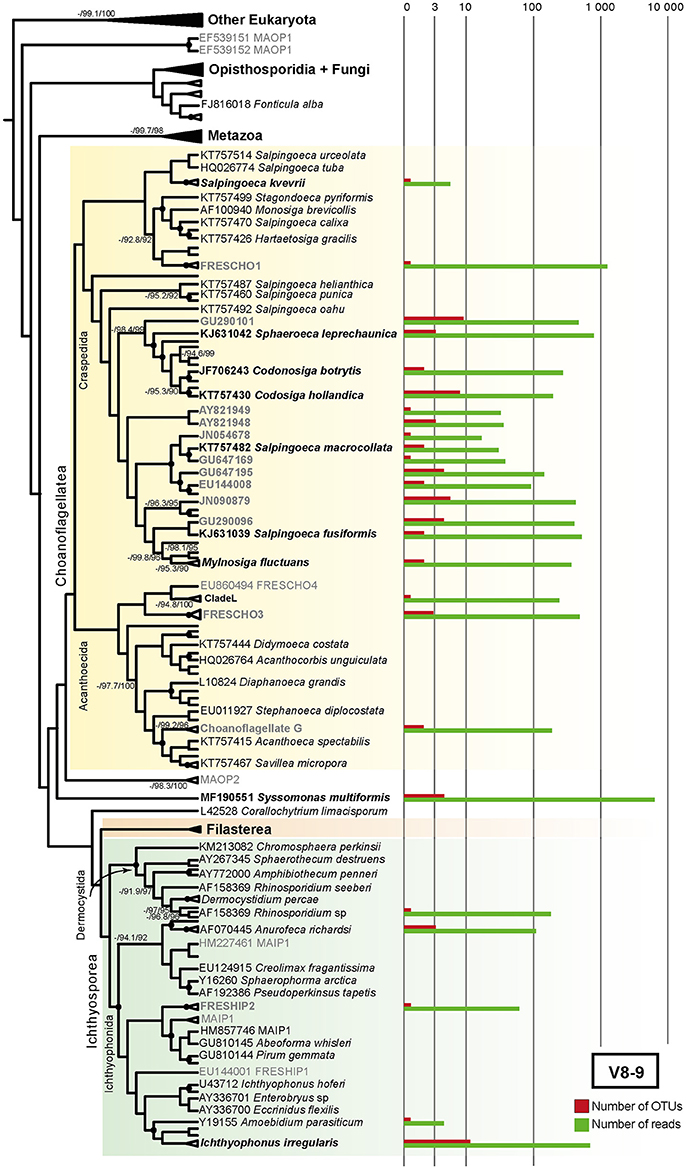
Figure 6. Maximum-likelihood (ML) consensus tree of all GenBank reference sequences showing the diversity and novelty of unicellular Holozoa in V8-9 region. The tree was constructed using RAxML v.8.2.8 under GTRCAT evolutionary model with a nodal support obtained through 100 bootstrap replicates (Stamatakis, 2014). Three nodal supports are shown: ML bootstrap/SH-aLRT/UFBoot. Nodal support is depicted as black dots when all three supports are above 90%, as a numerical value when ML bootstrap is below 90% and absent if at least two of the supports were below 90%. Barplots show the number of OTUs (in red) and the number of reads (green) associated with their closest GenBank full-length representatives, either environmental (gray) or morphologically described (black) (see Materials and Methods). The more abundant groups are S. multiformis, the recently described species from a Vietnamese lake (Hehenberger et al., 2017), the environmental clade FRESCHO1 and several uncultured choanoflagellate sequences closely related to Salpingoeca macrocollata. Alignments and phylogenetic trees can be found in Supplementary Datasheet 5.
We then investigated the putative novelty within Holozoa. In this case, and given the smaller number of OTUs compared to Holomycota, we performed a deeper and phylogenetically-based analysis to classify this diversity (see Materials and Methods for more details). We first grouped our OTUs into smaller clades based on the same BLAST hit. For each group, we constructed a tree with the reference Holozoa sequences, our OTUs and GenBank full sequences with high identity to our OTUs. Then, we built a tree with only the references and the GenBank full sequences. Finally, we performed a phylogenetic placement of the OTUs into those trees to check the phylogenetic position obtained previously. With this procedure, we succeed in classifying more accurately the holozoan OTUs because it was based on a phylogenetic method rather than only on the blast-taxonomical classification.
In our study, the majority of the Choanoflagellata diversity falls into uncultured GenBank sequences (Figures 5, 6). However, we also recovered a few described species. Specifically, in both V4 and V8-9 regions we detected the recently described Salpingoeca kvevrii, S. macrocollata and Codosiga hollandica. Among environmental choanoflagellates, 19.35% of unicellular Holozoa V4 richness and 14.94% of V8-9 richness are FRESCHO groups (freshwater choanoflagellates) (del Campo and Ruiz-Trillo, 2013; Figures 4A, 5, 6), being FRESCHO3 the richest and most abundant group in our dataset. We could also assign 10 OTUs of both regions with a total of 303 reads to Acanthoecida, a traditionally marine group of choanoflagellates (Carr et al., 2008; Leadbeater, 2008; Figures 4A, 5, 6).
In contrast, our Paraná diversity of ichthyosporeans mainly falls in described cultured species (Figures 5, 6). Both regions of 18S rRNA gene recover freshwater species, such as Amoebodium parasiticum or Anurofeca richardsi, even though they are parasites (Mendoza et al., 2002; Glockling et al., 2013). We also detected environmental ichthyosporeans. For instance, the environmental clade formed around A. parasiticum and Ichthyophonus irregularis species accounts for up to 18.39% of the total Holozoa richness in the V8-9 region (named as Amoebidium-Ichthyophonus ENV in Figure 4A). The environmental clade FRESHIP (freshwater ichthyosporeans) (del Campo and Ruiz-Trillo, 2013) was also identified from our samples. In fact, it is the richest and most abundant ichthyosporean group in the V4 region (12.9% of the total Holozoa abundance) (Figure 4A).
Surprisingly, the most abundant OTUs of all unicellular holozoans in our dataset is associated to the recently described Syssomonas multiformis, putatively the sister-group to Corallochytrium limacisporum (Hehenberger et al., 2017; Figures 5, 6). These OTUs account for 37.36% of the total unicellular Holozoa abundance in the V4 region and 45% in the V8-9 region (Figure 4A). It is significantly more abundant in Ramallo Stream and Marsh sampling sites, and completely absent in Paso del Tonelero (Figure 4A).
Discussion
V4 and V8-9 Regions of the 18S rRNA Gene Recover Different Eukaryotic Diversity
We decided to sequence both V4 and V8-9 regions of the 18S rRNA gene. Both V4 and V9 are considered suitable regions for biodiversity studies given their great variability and the combination of high and low entropy in these sections, which allows forgood primer binding (Behnke et al., 2011; Hadziavdic et al., 2014; Tanabe et al., 2016). They also have been widely used in molecular diversity studies, so the research community knows the advantages and limitations of both regions (Amaral-Zettler et al., 2009; Stoeck et al., 2010; Massana et al., 2015). In fact, we overcame one of the main limitations of the V9 region, which is its short length (Amaral-Zettler et al., 2009; Pawlowski et al., 2011), by sequencing the V8 region as well. This strategy had already been proven to be reliable as demonstrated by Taib et al. (2013). In addition, a combination of V4 and V8-9 regions compensate the existing primer bias (Amaral-Zettler et al., 2009; Pawlowski et al., 2011), leading to a more realistic picture of the eukaryotic diversity.
The diversity and distribution patterns are similar between both regions. However, there are some examples of a biased recovery of the eukaryotic diversity (Figures 2, 4–6 and Supplementary Figure 4). We found out that only the V4 region could detect some eukaryotic groups such as Picobiliphyta, Dictyostelea and Malawimonadidae (Supplementary Table). V4 region also included unaffiliated OTUs: 9 OTUsV4 did not match to any group, and were therefore labeled as Eukaryota-unclassified. In contrast, all OTUsV8−9 were properly assigned to an extant eukaryotic group. Moreover, the V8-9 region seems to detect less novelty for each lineage than V4 (on average, 8.29% less). We also found a different proportion of groups depending on each 18S rRNA gene region. This can be seen in the distribution of Holomycota diversity across sampling sites (Supplementary Figure 4). V4 shows that Chytridiomycota and Cryptomycota are the most abundant groups in each sample, while V8-9 detects Dikarya as the most abundant group. The environmental group of freshwater ichthyosporeans (FRESHIP) is another example (del Campo and Ruiz-Trillo, 2013). FRESHIP is definitely better represented in V4 (12.9% of the total Holozoa richness, 4.67% of the abundance) than in V8-9 samples (1.15% and 0.4%, respectively) (Figure 4A and Supplementary Table).
The lower level of novelty in the V8-9 region compared to the V4 may at least be explained by the higher variability of the V4 region (Stoeck et al., 2010). However, even though in our PR2-Opistho database the number of sequences that contain the V8-9 region was only 11.17% of the total, BLAST identities were higher for OTUsV8−9. The differences in the proportion of richness and abundance between both regions in some eukaryotic groups are likely to be due to primer bias effect. In addition to that, indels or variation among sequences can also change the properties of primers favoring the amplification of some groups above others, explaining the unequal recovery of diversity and novelty in eukaryotes (Pawlowski et al., 2011; Giner et al., 2016).
Eukaryotic Diversity in Paraná River is Similar to Other Freshwater Environments
According to the rarefaction curves, our sampling effort was enough to recover most of the unicellular eukaryotic diversity (Supplementary Figure 3B). The sampling was limited, however, to a two-month period (Supplementary Table), so we may have missed seasonal changes in community composition and structure throughout the year. Our study shows, however, that in general eukaryotic diversity is similar across sampling sites, with the exception of some groups such as Holomycota or Dinophyta. It also shows that Ramallo Stream sampling site differs the most from the other sites in its eukaryotic composition (Supplementary Figures 1A, 2A) and contains the largest propotion of unique OTUs (Supplementary Figure 3A). One explanation could be that Ramallo Stream, unlike the other sampling sites, is a tributary of the Paraná River and the sample was taken almost 2 km away from the river's mouth (Figure 1). Thus, Ramallo Stream does not belong to the same hydrological system as the rest of sites.
The richest and most abundant groups in our Paraná samples are Stramenopiles, Alveolates and Rhizarians, together with Holomycota (Figure 2). This is in agreement with previous freshwater studies (Taib et al., 2013; Simon et al., 2014, 2015). The main reason of this large number might not be the abundance of the organisms themselves but the greater amount of rRNA gene copies in their genomes (Medinger et al., 2010; Torres-Machorro et al., 2010; Pawlowski et al., 2011). Another important eukaryotic group in fresh waters are the small unicellular algae cryptophytes, which belong to Cryptista clade. Cryptista appear to be moderately abundant in our samples, reaching up to 25% of the total eukaryotic abundance in the 2–8 μm size fraction in almost all samples (Figure 2, Supplementary Figures 1B, 2B). The abundance of this group is, however, not as large as in other surveys (Šlapeta et al., 2005; Taib et al., 2013; Simon et al., 2014). This pattern might be explained by the fluctuations of its abundance over time, variations in flow rate of the river or the presence of oxygenated waters (Šlapeta et al., 2005; Devercelli, 2006; Simon et al., 2015).
Regarding the molecular novelty in eukaryotes, Amoebozoa is the group with more putative novel diversity (Figure 2). This is also in agreement with previous studies that found a large undetected diversity within this lineage, and highlights the importance of unraveling it to understand its evolution (Romeralo et al., 2011; Fiz-Palacios et al., 2013; Tekle et al., 2016; Kang et al., 2017).
Large Amount of Novel Diversity of Early-Diverging Holomycota
During the past decade, an increasingly large number of undescribed taxa within or related to Fungi have been detected, leading to the “dark matter fungi” concept (Hibbett et al., 2011; Jones et al., 2011; Kagami et al., 2014; Grossart et al., 2016; Yahr et al., 2016). This dark matter comprises a huge fungal diversity, widespread across different ecosystems, which have never been cultured, taxonomically classified or morphologically studied. Among the genetic markers to study Fungi, the internal transcribed spacer (ITS) has been proposed as the best one because of its high resolution for species identification (Schoch et al., 2012). However, we decided to work with 18S rDNA because the aim of our study was not to define fungal diversity at species level, but to search for more distinctly related lineages in a broader scope.
In our Paraná samples, a significant proportion of all Holomycota have less than 90% blast identity against our PR2-Opistho database ( = 43.93% and = 23.7%) (Supplementary Table). Most of this putative novel diversity is placed within either Chytridiomycota or Cryptomycota (Figure 3A and Supplementary Figures 5, 6). Nonetheless, we found 4 OTUsV4 that branch close to but clearly outside the Opisthosporidia and Fungi clade and do not correspond to any known group (Figure 3B). Although this result should be taken with caution, it certainly suggests the existence of novel species that do not belong to any of the currently known and defined Holomycota lineages. Little can be said about these OTUs besides that they appear only in two sampling sites, Ramallo Stream and Marsh, and that the OTU_833 seems to correspond to a bigger organism as it was only found in the 8–15 μm fraction. On the other hand, the other OTUs were found in the smaller fraction. It has been hypothesized that the presence of novel early-branching groups in different size fractions could be related to a multi-stage life cycle (in which there would be two different types of cells: a small one, spores or cysts, and a big one, in form of a multi-cellular sporangia) or to symbiotic interactions (Richards et al., 2015). We cannot claim whether these hypotheses explain our observations. What we can assert is that we have found a new group of 18S rDNA sequences at the base of Holomycota, which we named FRESHOL1 and FRESHOL1-related. Their position between nucleariids and Opisthosporidia suggests they are unrelated to any previously described early-branching holomycotan lineage. Further studies using single-cell genomics or isolating the organisms would be necessary to formally describe these new taxa and to more precisely place them in phylogenies. Thus, our study highlights the importance of metabarcoding analyses. Using short fragments of the 18S rRNA gene is an important first step toward detecting novel eukaryotic diversity, paving the way for further isolation and description of new taxa (del Campo and Massana, 2011; Gómez et al., 2011; Cavalier-Smith and Scoble, 2013; Shiratori et al., 2017; López-Escardó et al., 2018).
New Geographical Distribution and Evolutionary Novel Diversity Within Unicellular Holozoa
The uncovered diversity of unicellular holozoans has been increasing in recent years, proving that the closest unicellular relatives of animals are more abundant than previously thought, their genomes are more complex and their ecological traits are wider and different, even within the same lineage (del Campo and Ruiz-Trillo, 2013; Torruella et al., 2015; Grau-Bové et al., 2017; Sebé-Pedrós et al., 2017). Even though our Paraná samples recovered a moderate amount of putative novelty (21.51% in V4 region; 16.1% in V8-9 region), these samples expand the geographic distribution of some holozoan species and also expand the diversity within some small groups, specially the environmental ones.
First, within choanoflagellates, we detected OTUs related to C. hollandica (11 OTUs with 230 reads in both 18S regions) (Figures 5, 6). C. hollandica is a recently described species isolated from a freshwater pond in Madeira Island, Portugal (Carr et al., 2017) and was also detected from 18S rDNA survey work in freshwater systems conducted in the Netherlands (Valster et al., 2010). The presence of C. hollandica OTUs in Paraná River shows that this species has a wider distribution across different freshwater habitats around the globe. The Codosiga clade, together with Sphaeroeca OTUs, dominates the unicellular Holozoa community in Paso del Tonelero and Paraná River sampling sites ( = 55.22%, = 32.18%) (shown as Sphaeroeca-Codosiga ENV in Figure 4A). Among the environmental choanoflagellates, FRESCHO3 is the most abundant group in the V4 data, representing 15% of the total unicellular Holozoa richness and abundance (Figure 4A, Supplementary Table). This result agrees with a previous survey based on GenBank data, where FRESCHO3 was defined as an exclusively freshwater clade holding the largest proportion of environmental choanoflagellate sequences (del Campo and Ruiz-Trillo, 2013).
Second, we found that the most abundant OTUs in our samples were related to the newly described species Syssomonas multiformis (Figures 4A, 5, 6). This flagellate was isolated from a freshwater lake in Vietnam and it was shown to be sister to Corallochytrium limacisporum (Hehenberger et al., 2017). Even though S. multiformis OTUs were present in all our sampling sites, they were more abundant in Marsh (Figure 4A). Marsh sampling site is the only spot of this marshland that never gets dried, remaining as a pond even when the water level of the Paraná River remains low during the dry season (S. R. Najle, personal observation). Perhaps S. multiformis fitness is higher in still waters, rather than in running waters.
Finally, we did not recover any Filasterea in our dataset (Figures 5, 6, Supplementary Table). This result is similar to other metabarcoding studies, which failed to retrieve any filasterean in marine or fresh-water samples (del Campo and Ruiz-Trillo, 2013; del Campo et al., 2015). This failure is puzzling because, after aligning the primers against known filastereans in our PR2-Opistho database, we checked that primer mismatch should not be the reason. Even though half of the filasterean species were isolated in fresh waters (Stibbs et al., 1979; Hehenberger et al., 2017), the most likely explanation is that they were in very low abundance, not found due to seasonal fluctuations, or simply not present in this environment.
Detection of Marine Groups in Paraná Freshwater Samples
In the microbial world, transitions from marine to fresh waters are thought to be rare mainly due to the sharp change in salinity and the harsh competition against better adapted freshwater organisms (Logares et al., 2009b). However, there is an increasing number of studies that show that some “marine” lineages are also present in freshwater ecosystems, although in lower abundances (Alverson et al., 2007; Bråte et al., 2010; Simon et al., 2013, 2014; Debroas et al., 2017).
In this context, MAST (for marine Stramenopila), a group of mostly uncultured bacterivorous flagellates, is a good example. They were initially thought to be exclusively marine, although some clades have also been detected in a wide range of ecosystems, including fresh waters (Massana et al., 2004, 2014; Simon et al., 2014, 2015). In our Paraná dataset, we detected several MAST groups and thus, confirmed its wider spatial distribution. MAST-12 was the second largest group of all MAST in the V4 region (35.94%) and the most abundant in the V8-9 region (71.04%) (Supplementary Table). The majority of these OTUs belong to MAST-12C, a versatile group found in freshwaters, marine habitats or even soils (Massana et al., 2014). However, we also detected 4 OTUs from MAST-12A in both regions, which so far had only been recovered from the Artic and Atlantic oceans (Massana et al., 2014). However, 58.56% of the total MAST abundance in the V4 region and 24.83% in the V8-9 region had a blast identity lower than 97%, so these OTUs could not be assigned properly to any specific MAST subgroups (we thus classified these as MAST-Unk, for unkown; see Supplementary Table). Thus, our data has uncovered a high proportion of MAST diversity with the potential to be novel. However, our MAST clades only represent 0.34 and 0.51% of the total eukaryotic diversity in V4 and V8-9 region, respectively (Supplementary Table). This proportion is minor compared to the >10% typically found in marine ecosystems (Massana and Pedrós-Alió, 2008). In any case, the MAST richness found in our samples is comparable to that found in other freshwater studies (Simon et al., 2014, 2015).
On the contrary, we did not recover any MALV (marine Alveolata), a group of uncultured parasites that accounts for a significant proportion (16.1%) of the microeukaryotic global diversity in the oceans (Guillou et al., 2008; Massana and Pedrós-Alió, 2008; de Vargas et al., 2015). So far, MALV has not been detected in freshwater environments or in continental habitats. Thus, it seems that MALV, as opposed to MAST, is truly an exclusively marine group (Guillou et al., 2008; Logares et al., 2009b).
Within Holozoa, we also detected traditional marine species in our Paraná freshwater samples. For example, considering both 18S rDNA regions, we detected a total of 18 OTUs with 379 reads of both Salpingoeca kvevrii and S. macrocollata, which are newly described choanoflagellate species isolated from a saltmarsh (Carr et al., 2017). Thus, this is the first record of them in a freshwater sub-tropical environment. We could also assign 10 OTUs with a total of 303 reads, in both 18S rDNA regions, to Acanthoecida, a traditionally considered marine group of choanoflagellates (Carr et al., 2008; Leadbeater, 2008; Figures 4A, 5, 6). These acanthoecids were identified mainly from Paraná River sampling site and the 8–15 μm size fraction (Figure 4A). There are only two described freshwater acanthoecid species so far: Acanthocorbis mongolica (Paul, 2012) and Stephanoeca arndtii (Nitsche, 2014), although the taxonomic identification of only the latter has been confirmed by 18S rDNA sequencing. In any case, our OTUs matched with environmental sequences that originated mainly from marine samples. Only the GenBank sequence JN090872 had been detected in a Greek freshwater lake as well (Oikonomou et al., 2012). Our finding of several freshwater OTUs within acanthoecids demonstrates that there is also an unknown freshwater diversity of this choanoflagellate group, although the OTUs are not as abundant as in marine environments (del Campo et al., 2015).
Our observations provide more evidence that, although rare, transitions from marine to freshwater ecosystems are present in different eukaryotic lineages, as it has already been seen for dinoflagellates, haptophytes or stramenopiles (Logares et al., 2009b). These findings demonstrate a more complex picture of the eukaryotic diversity, in which several lineages have adapted to completely different habitats, while some present strong biogeographic patterns.
Materials and Methods
Characteristics of the Sampling Sites
The five sampling sites were located in the North of Buenos Aires Province, Argentina (Figure 1; Supplementary Table). The hydrochemistry characterization of the area has been previously reported (Depetris, 1976; Devercelli, 2006; Roberto et al., 2009). One of the main characteristics of this river is the fluctuation of the water volume (Roberto et al., 2009). For example, pH and conductivity have an indirect correlation to the water discharge over time (Devercelli, 2006). According to this author, Middle Paraná has a pH of 7.55, a conductivity of 100 μS cm−1 and an oxygen concentration near saturation. The salinity of the river has different origins, such as sedimentary rocks, saline soils, groundwater seepage and precipitations (Depetris, 1976). It was measured as 276 mg L−1, which is notably low compared to the salinity of marine waters, generally established as 35,000 mg L−1 (Culkin and Smith, 1980; Millero et al., 2008). Regarding vegetation, the water surface of the Middle Paraná River tends to be occupied by macrophytes, such as water hyacinths (Eichornia crassipes, E. azurea) and water lettuce (Pistia stratiotes) (Figure 1; Paira and Drago, 2007).
Sampling
Samples were collected during October and November, 2012. Half to one liter of surface water (20 cm depth) was pre-filtered through a 15 μm nylon mesh strainer to remove debris. A water sample of up to 250 ml was first filtered onto an 8 μm polycarbonate membrane filter by gravity (Millipore) and consequently filtered onto a 2 μm filter under gentle vacuum using a Millipore Sterifil Aseptic system (Millipore). Each filter was folded and placed in a 2 ml cryovial, and stored at −80°C until DNA extraction.
DNA Extraction and Next-Generation Sequencing
A DNeasy Blood & Tissue kit (Qiagen, Valencia, CA) was used for DNA extraction. Purification was done following the manufacturer instructions. PCR amplification of either the V4 or V8-9 regions of 18S rDNA were performed as previously described (Kim et al., 2016). Both V4 and V8-9 regions of the 18S rRNA gene were sequenced because of their extensive use for eukaryotic diversity (Nickrent and Sargent, 1991; Amaral-Zettler et al., 2009; de Vargas et al., 2015; Massana et al., 2015) and to compensate the existing primer bias of each region. Amplicon libraries were prepared as in Kim et al. (2016) (further details about library preparation can be found in Supplementary Table). Sequencing was performed on Illumina MiSeq platform (2 × 300 bp) at the Cornell University Sequencing Core (New York; USA). Raw reads generated in this study are available at ENA with the following accession identifications: PRJEB23471 (V4 region) and PRJEB23493 (V8-9 region).
Clustering and Taxonomical Annotation
The clustering process was carried out following the UPARSE workflow included in USEARCH v8.1.1861 (Edgar, 2013). Firstly, raw reads were checked for sequencing errors using BayesHammer (Nikolenko et al., 2013). Then, sequences from forward and reverse primers were assembled using PEAR (Zhang et al., 2014). After quality filtering, there were a total amount of 1,420,587 assembled reads corresponding to the V4 region of the 18S rRNA gene and 2,286,416 reads corresponding to the V8-9 region. These clean reads were then clustered into Operational Taxonomic Units (OTUs) using UPARSE with a 97% similarity threshold, resulting in a total of 4,598 OTUs for V4 and 4,176 OTUs for V8-9 region. Even though there is no clear consensus on the optimal threshold to use in clustering algorithms (Nebel et al., 2011; Brown et al., 2015), we decided to use 97% because it has been widely used (Stoeck et al., 2010; Kammerlander et al., 2015; Massana et al., 2015; Giner et al., 2016; Kim et al., 2016). We are aware that this threshold might group species or even genus into the same OTU. However, in this way, it reduces the amount of singletons or artifactual OTUs and the consequent overestimation of rare diversity (Kunin et al., 2010; Behnke et al., 2011). To achieve our objective on finding unknown diversity, we opted to forgo fine taxonomical resolution.
These OTUs were filtered against chimeras using both PR2 (Guillou et al., 2013) and SILVA119 databases (Quast et al., 2013). We then obtained a total of 4,316 OTUs for V4 and 4,051 for V8-9. The taxonomic assignment of the OTUs was done through a BLASTn (Camacho et al., 2009) against our modified version of PR2 database (hereinafter PR2-Opistho) using as thresholds 75% BLAST identity and 0.0001 e-value. PR2-Opistho contains exactly the same sequences from PR2 database for all eukaryotes except for Opisthokonta. Given that the main purpose of this study is to find molecular novelties within opisthokonts, we added our well-curated and most-updated sequences of this clade (based mostly in del Campo and Ruiz-Trillo, 2013). Finally, we refined the OTU table removing those OTUs from animals (Metazoa) and land plants (Embryophyta). As these groups are multicellular, they could interfere with our estimations of diversity and richness of microbial eukaryotes. Consequently, our final dataset contained a total of 4,195 OTUs for V4 region and 3,866 for V8-9 region. The final OTU tables for both 18S regions can be found in the Supplementary Table.
Data Analysis
General patterns of eukaryotic diversity (richness and abundance) were calculated selecting the first two taxonomical ranks of all eukaryotic groups (Adl et al., 2012). In order to know how much putative novelty existed in eukaryotes, we used the blast identity score as proxy. For that purpose, we computed the proportion of every eukaryotic group in relation to each category of blast identity using an in-home script. To know the distribution of eukaryotic diversity in all samples, we computed Bray-Curtis dissimilarity matrix using the “vegan” package of R Studio v.1.0.136 (https://CRAN.R-project.org/package=vegan). We also performed Venn diagrams to show the distribution of unique and shared OTUs among sampling sites. Venn diagrams were computed using “VennDiagram” package in R Studio v.1.0.136 (https://CRAN.R-project.org/package=VennDiagram). Rarefaction and accumulation curves of each eukaryotic group were calculated to estimate if our sampling effort was enough to recover the eukaryotic community in this part of the Paraná River. Rarefaction curves were computed using the “vegan” package in R Studio v.1.0.136 (https://CRAN.R-project.org/package=vegan) while accumulation curves were calculated with EstimateS v.9.1.0 and plotted with R Studio v.1.0.136 (Colwell and Elsensohn, 2014; https://www.rstudio.com).
To address the novelty within Opisthokonta, we used a phylogenetic placement approach, which is suitable for comparing short amplicons (like our OTUs) with full-length sequences (in our case, 18S reference sequences from GenBank) (Matsen et al., 2010; Berger et al., 2011). Basically, the approach consists of placing each OTU, individually and independently, into a fixed reference tree using a maximum-likelihood model. In this approach, an OTU may have different locations within different branches of the tree, each of them with their corresponding likelihood ratio. The more internally an OTU is located, the more different it is compared to the known sequences at the tips of the reference tree.
We divided the dataset into Holozoa and Holomycota. With regards Holomycota, we built a reference tree including the main lineages of Fungi, Opisthosporidia and unicellular clades of Holomycota, together with environmental sequences (Karpov et al., 2014b; Richards et al., 2015) (Supplementary Datasheet 1). We then performed a phylogenetic placement of all Holomycota OTUs into this curated reference tree using pplacer package (Matsen et al., 2010). The –keep-factor option was set to 0.1 to avoid noisy signal due to low likelihood placements. Genesis package v 0.12.1 (Czech and Stamatakis, 2016) was used to calculate the amount of OTUs placed in each branch. These alignments and phylogenetic placements of Holomycota in jplace format of both V4 and V8-9 regions can be found in Supplementary Datasheet 2.
To check the putative new clade at the base of Opisthosporidia and Fungi, we only kept those OTUs that were placed in this branch by using two softwares for phylogenetic placement: pplacer and RAxML-EPA (Berger et al., 2011). We also searched for related 18S long sequences in GenBank in order to get better phylogenetic signal and avoid possible artifacts. In this search, we detected full 18S sequences from a metabarcoding study in Lake Pavin, France (Monchy et al., 2011). The final tree was built using the Lake Pavin sequences plus our putative novel OTUs using IQ-tree software (Nguyen et al., 2015) under a GTR model of evolution with 1,000 bootstrap replicates for SH-aLRT (Guindon et al., 2010) and 1000 ultrafast bootstrap replicates (Minh et al., 2013). The alignments and phylogeny can be found in Supplementary Datasheet 3.
With regards to Holozoa, we proceeded in a different way than with Holomycota. As the number of OTUs were smaller and thus, easier to manage, we could perform a more accurate phylogenetic method to classify this diversity. First, we built a reference dataset with well-curated sequences from Choanoflagellata, Filasterea, Ichthyosporea, Plurimorfea, and several environmental clades (del Campo and Ruiz-Trillo, 2013; del Campo et al., 2015; Carr et al., 2017) (Supplementary Datasheet 4). From our Paraná dataset, we trusted the taxonomical assignment of the OTUs with a blast identity higher than 95%. If not, we considered that these OTUs could be putative novelties or prone to be incorrectly assigned, so they were chosen for further phylogenetic classification. Then, we grouped our OTUs into 69 groups based on the same BLAST hit retrieved. We constructed 69 phylogenetic trees, each of them with different small groups of these OTUs plus the first blast hit of these OTUs against GenBank. These GenBank representatives may help to correctly place the Holozoa diversity since they are usually longer 18S sequences. As most of our OTUs formed a monophyletic group with these representatives, we only kept the GenBank representatives in addition to the original reference tree. In this way, we finally created a reference tree with long GenBank sequences, which represented the unicellular diversity of holozoans found in Paraná River. All trees were built by using RAxML v8.2.8 (Stamatakis, 2014), considering a GTRCAT substitution model and 100 best trees and a nodal support obtained through 100 bootstrap replicates. Using the same dataset, we also constructed ML trees using IQ-tree (Nguyen et al., 2015) to have SH-aLRT and UFBoot through 1,000 replicates. Alignments, RAxML and IQ-tree trees of both V4 and V8-9 regions are included in the Supplementary Datasheet 5. It is worth mentioning that the reference tree of V4 and V8-9 region were different because of the different GenBank representatives included and the differences in the phylogenetic estimates inferred by each 18S rRNA gene region. We finally carried out a phylogenetic placement of the OTUs with less than 95% blast identity to confirm their phylogenetic position. For that purpose, we used both pplacer software and RAxML-EPA algorithm. Although both are useful to place short environmental sequences into a reference tree, there are subtle differences between them, which might affect the final phylogeny. For this reason, we compared their placement outputs using Genesis package v0.12.1 (Czech and Stamatakis, 2016). We only kept for downstream analysis those OTUs whose first placement (the one with highest likelihood weight ratio) was identical. The final taxonomical assignment of the OTUs was done associating each OTU to a representative in the reference tree. Holozoan OTUs that could not be properly assigned to a specific group were classified as Holozoa-unclassified.
Data Availability
Raw reads generated in this study are available at ENA (https://www.ebi.ac.uk/ena) with the following accession identifiers: PRJEB23471 (V4 region) and PRJEB23493 (V8-9 region).
Author Contributions
SN and EK conceived and designed the project. SN collected the samples. SN and EK produced the DNA libraries. DL-E and AA carried out the analysis. AA designed the figures and wrote the manuscript. SN and IR-T supervised the project and corrected the manuscript. All authors corrected and approved the final version of the manuscript.
Funding
This work was supported by an American Museum of Natural History's Lerner-Gray Fund for Marine Research grant to SN, a European Research Council Consolidator Grant (ERC-2012-Co-616960), and grants (BFU2014-57779-P and BFU2017-90114-P) from Ministerio de Economía y Competitividad (MINECO), Agencia Estatal de Investigación (AEI), and Fondo Europeo de Desarrollo Regional (FEDER) to IR-T.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mr. Edgardo P. Guglielmetti for technical assistance in the field and Dr. Ramón Massana for his comments on MAST diversity. SN acknowledges the Lerner Gray Memorial Fund of the American Museum of Natural History for aid received. We greatly acknowledge the reviewers' comments, which helped to improve the original manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2018.00099/full#supplementary-material
References
Adl, S. M., Simpson, A. G., Lane, C. E., Lukeš, J., Bass, D., Bowser, S. S., et al. (2012). The revised classification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–493. doi: 10.1111/j.1550-7408.2012.00644.x
Agostinho, A. A., Gomes, L. C., Veríssimo, S.K., and Okada, E. (2004a). Flood regime, dam regulation and fish in the Upper Paraná River: effects on assemblage attributes, reproduction and recruitment. Rev. Fish Biol. Fish. 14, 11–19. doi: 10.1007/s11160-004-3551-y
Agostinho, A. A., Thomaz, S. M., and Gomes, L. C. (2004b). Threats for biodiversity in the floodplain of the Upper Paraná River: effects of hydrological regulation by dams. Ecohydrol. Hydrobiol. 4, 255–256.
Alverson, A. J., Jansen, R. K., and Theriot, E. C. (2007). Bridging the rubicon: phylogenetic analysis reveals repeated colonizations of marine and fresh waters by thalassiosiroid diatoms. Mol. Phylogenet. Evol. 45, 193–210. doi: 10.1016/j.ympev.2007.03.024
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W., and Huse, S. M. (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA Genes. PLoS ONE 4:e6372. doi: 10.1371/annotation/50c43133-0df5-4b8b-8975-8cc37d4f2f26
Behnke, A., Engel, M., Christen, R., Nebel, M., Klein, R. R., and Stoeck, T. (2011). Depicting more accurate pictures of protistan community complexity using pyrosequencing of hypervariable SSU rRNA gene regions. Environ. Microbiol. 13, 340–349. doi: 10.1111/j.1462-2920.2010.02332.x
Berger, S. A., Krompass, D., and Stamatakis, A. (2011). Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol. 60, 291–302. doi: 10.1093/sysbio/syr010
Berney, C., Fahrni, J., and Pawlowski, J. (2004). How many novel eukaryotic “kingdoms?” Pitfalls and limitations of environmental DNA surveys. BMC Biol. 2:13. doi: 10.1186/1741-7007-2-13
Boenigk, J., Wodniok, S., Bock, C., Beisser, D., Hempel, C., Grossmann, L., et al. (2018). Geographic distance and mountain ranges structure freshwater protist communities on a European scal? Metab. Metagen. 2:e21519. doi: 10.3897/mbmg.2.21519
Bråte, J., Logares, R., Berney, C., Ree, D. K., Klaveness, D., Jakobsen, K. S., et al. (2010). Freshwater Perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA. ISME J. 4, 1144–1153. doi: 10.1038/ismej.2010.39
Brown, E. A., Chain, F. J., Crease, T. J., Macisaac, H. J., and Cristescu, M. E. (2015). Divergence thresholds and divergent biodiversity estimates: can metabarcoding reliably describe zooplankton communities? Ecol. Evol. 5, 2234–2251. doi: 10.1002/ece3.1485
Brown, M. W., Spiegel, F. W., and Silberman, J. D. (2009). Phylogeny of the “forgotten” cellular slime mold, Fonticula alba, reveals a key evolutionary branch within Opisthokonta. Mol. Biol. Evol. 26, 2699–2709. doi: 10.1093/molbev/msp185
Brunet, T., and King, N. (2017). The origin of animal multicellularity and cell differentiation. Dev. Cell 43, 124–140. doi: 10.1016/j.devcel.2017.09.016
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST plus: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Carr, M., Leadbeater, B. S., Hassan, R., Nelson, M., and Baldauf, S. L. (2008). Molecular phylogeny of choanoflagellates, the sister group to Metazoa. Proc. Natl. Acad. Sci. U.S.A. 105, 16641–16646. doi: 10.1073/pnas.0801667105
Carr, M., Richter, D. J., Fozouni, P., Smith, T. J., Jeuck, A., Leadbeater, B. S. C., et al. (2017). A six-gene phylogeny provides new insights into choanoflagellate evolution. Mol. Phylogenet. Evol. 107, 166–178. doi: 10.1016/j.ympev.2016.10.011
Cavalier-Smith, T., and Scoble, J. M. (2013). Phylogeny of Heterokonta: incisomonas marina, a uniciliate gliding opalozoan related to Solenicola (Nanomonadea), and evidence that Actinophryida evolved from raphidophytes. Eur. J. Protistol. 49, 328–353. doi: 10.1016/j.ejop.2012.09.002
Charvet, S., Vincent, W. F., Comeau, A., and Lovejoy, C. (2012). Pyrosequencing analysis of the protist communities in a High Arctic meromictic lake: DNA preservation and change. Front. Microbiol. 3:422. doi: 10.3389/fmicb.2012.00422
Colwell, R. K., and Elsensohn, J. E. (2014). EstimateS turns 20: statistical estimation of species richness and shared species from samples, with non-parametric extrapolation. Ecography 37, 609–613. doi: 10.1111/ecog.00814
Culkin, F., and Smith, N. (1980). Determination of the concentration of potassium chloride solution having the same electrical conductivity, at 15°C and infinite frequency, as standard seawater of salinity 35.0000%0 (Chlorinity 19.37394%0). IEEE J. Ocean. Eng. 5, 22–23. doi: 10.1109/JOE.1980.1145443
Czech, L., and Stamatakis, A. (2016). Genesis. A Toolkit for Working with Phylogenetic Data. GitHub. Available online at: https://github.com/lczech/genesis
Debroas, D., Domaizon, I., Humbert, J., Jardillier, L., and Lepère C Oudart, A. (2017). Overview of freshwater microbial eukaryotes diversity : a first analysis of publicly available metabarcoding data. FEMS Microb. Ecol. 93, 1–14. doi: 10.1093/femsec/fix023
del Campo, J., Mallo, D., Massana, R., de Vargas, C., Richards, T. A., and Ruiz-trillo, I. (2015). Diversity and distribution of unicellular opisthokonts along the European coast analysed using high-throughput sequencing. Environ. Microbiol. 17, 3195–207. doi: 10.1111/1462-2920.12759
del Campo, J., and Massana, R. (2011). Emerging diversity within chrysophytes, choanoflagellates and bicosoecids based on molecular surveys. Protist 162, 435–448. doi: 10.1016/j.protis.2010.10.003
del Campo, J., and Ruiz-Trillo, I. (2013). Environmental survey meta-analysis reveals hidden diversity among unicellular opisthokonts. Mol. Biol. Evol. 30, 802–805. doi: 10.1093/molbev/mst006
del Campo, J., Sieracki, M. E., Molestina, R., Keeling, P., Massana, R., and Ruiz-Trillo, I. (2014). The others: our biased perspective of eukaryotic genomes. Trends Ecol. Evol. 29, 252–259. doi: 10.1016/j.tree.2014.03.006
Depetris, P. J. (1976). Hydrochemistry of the Paraná River. Limnol. Oceanogr. 21, 736–739. doi: 10.4319/lo.1976.21.5.0736
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1261605–1261605. doi: 10.1126/science.1261605
Devercelli, M. (2006). Phytoplankton of the middle Paraná river during an anomalous hydrological period: a morphological and functional approach. Hydrobiologia 563, 465–478. doi: 10.1007/s10750-006-0036-0
Downing, J. A. (2010). Emerging global role of small lakes and ponds: little things mean a lot. Limnetica 29, 9–24.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Eiler, A., Drakare, S., Bertilsson, S., Pernthaler, J., Peura, S., Rofner, C., et al. (2013). Unveiling distribution patterns of freshwater phytoplankton by a next generation sequencing based approach. PLoS ONE 8:e53516. doi: 10.1371/journal.pone.0053516
Filker, S., Sommaruga, R., Vila, I., and Stoeck, T. (2016). Microbial eukaryote plankton communities of high-mountain lakes from three continents exhibit strong biogeographic patterns. Mol. Ecol. 25, 2286–2301. doi: 10.1111/mec.13633
Fiz-Palacios, O., Romeralo, M., Ahmadzadeh, A., Weststrand, S., Ahlberg, P. E., and Baldauf, S. (2013). Did terrestrial diversification of amoebas (amoebozoa) occur in synchrony with land plants? PLoS ONE 8:e74374. doi: 10.1371/journal.pone.0074374
Giner, C. R., Forn, I., Romac, S., Logares, R., de Vargas, C., and Massana, R. (2016). Environmental sequencing provides reasonable estimates of the relative abundance of specific picoeukaryotes. Appl. Environ. Microbiol. 82, 4757–4766. doi: 10.1128/AEM.00560-16
Glockling, S. L., Marshall, W. L., and Gleason, F. H. (2013). Phylogenetic interpretations and ecological potentials of the Mesomycetozoea (Ichthyosporea). Fungal Ecol. 6, 237–247. doi: 10.1016/j.funeco.2013.03.005
Gómez, F., Moreira, D., Benzerara, K., and López-García, P. (2011). Solenicola setigera is the first characterized member of the abundant and cosmopolitan uncultured marine stramenopile group MAST-3. Environ. Microbiol. 13, 193–202. doi: 10.1111/j.1462-2920.2010.02320.x
Grau-Bové, X., Torruella, G., Donachie, S., Suga, H., Leonard, G., Richards, T. A., et al. (2017). Dynamics of genomic innovation in the unicellular ancestry of animals. Elife 6, 1–35. doi: 10.7554/eLife.26036
Grossart, H. P., Wurzbacher, C., James, T. Y., and Kagami, M. (2016). Discovery of dark matter fungi in aquatic ecosystems demands a reappraisal of the phylogeny and ecology of zoosporic fungi. Fungal Ecol. 19, 28–38. doi: 10.1016/j.funeco.2015.06.004
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The protist ribosomal reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Guillou, L., Viprey, M., Chambouvet, A., Welsh, R. M., Kirkham, A. R., Massana, R., et al. (2008). Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environ. Microbiol. 10, 3349–3365. doi: 10.1111/j.1462-2920.2008.01731.x
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hadziavdic, K., Lekang, K., Lanzen, A., Jonassen, I., Thompson, E. M., and Troedsson, C. (2014). Characterization of the 18s rRNA gene for designing universal eukaryote specific primers. PLoS ONE 9:e87624. doi: 10.1371/journal.pone.0087624
Hehenberger, E., Tikhonenkov, D. V., Kolisko, M., del Campo, J., Esaulov, A. S., Mylnikov, A. P., et al. (2017). Novel predators reshape holozoan phylogeny and reveal the presence of a two-component signaling system in the ancestor of animals. Curr. Biol. 27, 2043–2050.e6. doi: 10.1016/j.cub.2017.06.006
Hibbett, D. S., Ohman, A., Glotzer, D., Nuhn, M., Kirk, P., and Nilsson, R. H. (2011). Progress in molecular and morphological taxon discovery in Fungi and options for formal classification of environmental sequences. Fungal Biol. Rev. 25, 38–47. doi: 10.1016/j.fbr.2011.01.001
Iriondo, M. H., and Paira, A. R. (2007). “Physical Geography of the Basin,” in The Middle Paraná River: Limnology of a Subtropical Wetland, eds M. H. Irondo, J. C. Paggi, and M. J. Parma (Heidelberg: Spring-Verlag), 7–31.
Jones, M. D., Forn, I., Gadelha, C., Egan, M. J., Bass, D., Massana, R., et al. (2011). Discovery of novel intermediate forms redefines the fungal tree of life. Nature 474, 200–203. doi: 10.1038/nature09984
Kagami, M., Miki, T., and Takimoto, G. (2014). Mycoloop: chytrids in aquatic food webs. Front. Microbiol. 5:166. doi: 10.3389/fmicb.2014.00166
Kammerlander, B., Breiner, H. W., Filker, S., Sommaruga, R., Sonntag, B., and Stoeck, T. (2015). High diversity of protistan plankton communities in remote high mountain lakes in the European Alps and the Himalayan mountains. FEMS Microbiol. Ecol. 91, 1–10. doi: 10.1093/femsec/fiv010
Kang, S., Tice, A. K., Spiegel, F. W., Silberman, J. D., Pánek, T., Cepicka, I., et al. (2017). Between a pod and a hard test: the deep evolution of amoebae. Mol. Biol. Evol. 34, 2258–2270. doi: 10.1093/molbev/msx162
Karpov, S. A., Mamkaeva, M. A., Aleoshin, V. V., Nassonova, E., Lilje, O., and Gleason, F. H. (2014a). Morphology, phylogeny, and ecology of the aphelids (Aphelidea, Opisthokonta) and proposal for the new superphylum Opisthosporidia. Front. Microbiol. 5:112. doi: 10.3389/fmicb.2014.00112
Karpov, S. A., Mamkaeva, M. A., Benzerara, K., Moreira, D., and López-García, P. (2014b). Molecular phylogeny and ultrastructure of aphelidium aff. melosirae (Aphelida, Opisthosporidia). Protist 165, 512–526. doi: 10.1016/j.protis.2014.05.003
Kim, E., Sprung, B., Duhamel, S., Filardi, C., and Kyoon Shin, M. (2016). Oligotrophic lagoons of the South Pacific Ocean are home to a surprising number of novel eukaryotic microorganisms. Environ. Microbiol. 18, 4549–4563. doi: 10.1111/1462-2920.13523
Kunin, V., Engelbrektson, A., Ochman, H., and Hugenholtz, P. (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123. doi: 10.1111/j.1462-2920.2009.02051.x
Lang, B. F., O'Kelly, C., Nerad, T., Gray, M. W., and Burger, G. (2002). The closest unicellular relatives of animals. Curr. Biol. 12, 1773–1778. doi: 10.1016/S0960-9822(02)01187-9
Lansac-Tôha, F. A., Bonecker, C. C., Velho, L. F., Simões, N. R., Dias, J. D., Alves, G. M., et al. (2009). Biodiversity of zooplankton communities in the Upper Paraná River floodplain: interannual variation from long-term studies. Brazilian J. Biol. 69, 539–549. doi: 10.1590/S1519-69842009000300009
Lara, E., Moreira, D., and López-García, P. (2010). The environmental clade LKM11 and rozella form the deepest branching clade of fungi. Protist 161, 116–121. doi: 10.1016/j.protis.2009.06.005
Leadbeater, B. S. C. (2008). Choanoflagellate evolution : the morphological perspective. Protistology 5, 256–267.
Lepère, C., Domaizon, I., Hugoni, M., Vellet, A., and Debroas, D. (2016). Diversity and dynamics of active small microbial eukaryotes in the anoxic zone of a freshwater meromictic lake (Pavin, France). Front. Microbiol. 7:130. doi: 10.3389/fmicb.2016.00130
Liu, Y., Steenkamp, E. T., Brinkmann, H., Forget, L., Philippe, H., and Lang, B. F. (2009). Phylogenomic analyses predict sistergroup relationship of nucleariids and fungi and paraphyly of zygomycetes with significant support. BMC Evol. Biol. 9:272. doi: 10.1186/1471-2148-9-272
Logares, R., Boltovskoy, A., Bensch, S., Laybourn-Parry, J., and Rengefors, K. (2009a). Genetic diversity patterns in five protist species occurring in lakes. Protist 160, 301–317. doi: 10.1016/j.protis.2008.10.004
Logares, R., Bråte, J., Bertilsson, S., Clasen, J. L., Shalchian-Tabrizi, K., and Rengefors, K. (2009b). Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol. 17, 414–422. doi: 10.1016/j.tim.2009.05.010
López-Escardó, D., López-García, P., Moreira, D., Ruiz-Trillo, I., and Torruella, G. (2018). Parvularia atlantis gen. et sp. nov., a Nucleariid Filose Amoeba (Holomycota, Opisthokonta). J. Eukaryot. Microbiol. 65, 170–179. doi: 10.1111/jeu.12450
López-García, P., Rodríguez-Valera F, Pedrós-Alió, C., and Moreira, D. (2001). Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature 409, 603–607. doi: 10.1038/35054537
Marande, W., López-García, P., and Moreira, D. (2009). Eukaryotic diversity and phylogeny using small- and large-subunit ribosomal RNA genes from environmental samples. Environ. Microbiol. 11, 3179–3188. doi: 10.1111/j.1462-2920.2009.02023.x
Massana, R., Castresana, J., Balagué, V., Guillou, L., Romari, K., Groisillier, A., et al. (2004). Phylogenetic and ecological analysis of novel marine stramenopiles. Appl. Environ. Microbiol. 70, 3528–3534. doi: 10.1128/AEM.70.6.3528-3534.2004
Massana, R., del Campo, J., Sieracki, M. E., Audic, S., and Logares, R. (2014). Exploring the uncultured microeukaryote majority in the oceans: reevaluation of ribogroups within stramenopiles. ISME J. 8, 854–866. doi: 10.1038/ismej.2013.204
Massana, R., Gobet, A., Audic, S., Bass, D., Bittner, L., Boutte, C., et al. (2015). Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ. Microbiol. 17, 4035–4049. doi: 10.1111/1462-2920.12955
Massana, R., and Pedrós-Alió, C. (2008). Unveiling new microbial eukaryotes in the surface ocean. Curr. Opin. Microbiol. 11, 213–218. doi: 10.1016/j.mib.2008.04.004
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11:538. doi: 10.1186/1471-2105-11-538
Medinger, R., Nolte, V., Pandey, R. V., Jost, S., Ottenwälder, B., Schlötterer, C., et al. (2010). Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol. Ecol. 19, 32–40. doi: 10.1111/j.1365-294X.2009.04478.x
Mendoza, L., Taylor, J. W., and Ajello, L. (2002). The class Mesomycetozoea: a heterogeneous group of microorganisms at the animal-fungal boundary. Annu. Rev. Microbiol. 56, 315–344. doi: 10.1146/annurev.micro.56.012302.160950
Millero, F. J., Feistel, R., Wright, D. G., and McDougall, T. J. (2008). The composition of standard seawater and the definition of the reference-composition salinity scale. Deep Sea Res. I Oceanogr. Res. Pap. 55, 50–72. doi: 10.1016/j.dsr.2007.10.001
Minh, B. Q., Nguyen, M. A., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Monchy, S., Sanciu, G., Jobard, M., Rasconi, S., Gerphagnon, M., Chabé, M., et al. (2011). Exploring and quantifying fungal diversity in freshwater lake ecosystems using rDNA cloning/sequencing and SSU tag pyrosequencing. Environ. Microbiol. 13, 1433–1453. doi: 10.1111/j.1462-2920.2011.02444.x
Nebel, M., Pfabel, C., Stock, A., Dunthorn, M., and Stoeck, T. (2011). Delimiting operational taxonomic units for assessing ciliate environmental diversity using small-subunit rRNA gene sequences. Environ. Microbiol. Rep. 3, 154–158. doi: 10.1111/j.1758-2229.2010.00200.x
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nickrent, D. L., and Sargent, M. L. (1991). An overview of the secondary structure of the V4 region of eukaryotic small-subunit ribosomal RNA. Nucleic Acids Res. 19, 227–235. doi: 10.1093/nar/19.2.227
Nikolenko, S. I., Korobeynikov, A. I., and Alekseyev, M. A. (2013). BayesHammer: bayesian clustering for error correction in single-cell sequencing. BMC Genomics 14:S7. doi: 10.1186/1471-2164-14-S1-S7
Nitsche, F. (2014). Stephanoeca arndti spec. nov.-First cultivation success including molecular and autecological data from a freshwater acanthoecid choanoflagellate from Samoa. Eur. J. Protistol. 50, 412–421. doi: 10.1016/j.ejop.2014.03.004
Oikonomou, A., Katsiapi, M., Karayanni, H., Moustaka-Gouni, M., and Kormas, K. A. (2012). Plankton microorganisms coinciding with two consecutive mass fish kills in a newly reconstructed lake. Sci. World J. 2012, 1–14. doi: 10.1100/2012/504135
Paira, A. R., and Drago, E. C. (2007). “Origin, Evolution and Types of Floodplain Water Bodies,” in The Middle Paraná River: Limnology of a Subtropical Wetland, eds M. H. Iriondo, J. C. Paggi, and M. J. Parma (Heidelberg: Springer-Verlag), 53–81.
Paps, J., and Ruiz-Trillo, I. (2010). “Animals and their unicellular ancestors,” in Encyclopedia of Life Sciences (Chichester: John Wiley and Sons, Ltd). doi: 10.1002/9780470015902.a0022853
Paul, M. (2012). Acanthocorbis mongolica nov. spec.-description of the first freshwater loricate choanoflagellate (Acanthoecida) from a Mongolian lake. Eur. J. Protistol. 48, 1–8. doi: 10.1016/j.ejop.2011.07.001
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C., et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 10:e1001419. doi: 10.1371/journal.pbio.1001419
Pawlowski, J., Christen, R., Lecroq, B., Bachar, D., Shahbazkia, H. R., Amaral-Zettler, L., et al. (2011). Eukaryotic richness in the abyss: insights from pyrotag sequencing. PLoS ONE 6:e18169. doi: 10.1371/journal.pone.0018169
Pawlowski, J., Lejzerowicz, F., Apotheloz-Perret-Gentil, L., Visco, J., and Esling, P. (2016). Protist metabarcoding and environmental biomonitoring: time for change. Eur. J. Protistol. 55, 12–25. doi: 10.1016/j.ejop.2016.02.003
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Richards, T. A., Leonard, G., Mahé, F., del Campo, J., Romac, S., Jones, M. D., et al. (2015). Molecular diversity and distribution of marine fungi across 130 European environmental samples. Proc. R. Soc. B Biol. Sci. 282, 1–10. doi: 10.1098/rspb.2015.2243
Richards T. A., Leonard G., and Wideman J. G. (2017). What defines the “kingdom” fungi? Microbiol. Spectrum 5. doi: 10.1128/microbiolspec.FUNK-0044-2017
Richter, D. J., and King, N. (2013). The genomic and cellular foundations of animal origins. Annu. Rev. Genet 47, 509–537. doi: 10.1146/annurev-genet-111212-133456
Roberto, M. C., Santana, N. N., and Thomaz, S. M. (2009). Limnology in the Upper Paraná River floodplain: large-scale spatial and temporal patterns, and the influence of reservoirs. Brazilian J. Biol. 69, 717–725. doi: 10.1590/S1519-69842009000300025
Romeralo, M., Cavender, J. C., Landolt, J. C., Stephenson, S. L., and Baldauf, S. L. (2011). An expanded phylogeny of social amoebas (Dictyostelia) shows increasing diversity and new morphological patterns. BMC Evol. Biol. 11:84. doi: 10.1186/1471-2148-11-84
Ruiz-Trillo, I., Burger, G., Holland, P. W., King, N., Lang, B. F., Roger, A. J., et al. (2007). The origins of multicellularity: a multi-taxon genome initiative. Trends Genet. 23, 113–118. doi: 10.1016/j.tig.2007.01.005
Ruiz-Trillo, I., Roger, A. J., Burger, G., Gray, M. W., and Lang, B. F. (2008). A phylogenomic investigation into the origin of metazoa. Mol. Biol. Evol. 25, 664–672. doi: 10.1093/molbev/msn006
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Sebé-Pedrós, A., Degnan, B. M., and Ruiz-Trillo, I. (2017). The origin of Metazoa, a unicellular perspective. Nat. Rev. Genet. 18, 498–512. doi: 10.1038/nrg.2017.21
Shiratori, T., Thakur, R., Ishida, K., and ichiro (2017). Pseudophyllomitus vesiculosus (Larsen and Patterson 1990) Lee, 2002, a poorly studied phagotrophic biflagellate is the first characterized member of stramenopile environmental clade MAST-6. Protist 168, 439–451. doi: 10.1016/j.protis.2017.06.004
Simon, M., Jardillier, L., Deschamps, P., Moreira, D., Restoux, G., Bertolino, P., et al. (2014). Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems. Environ. Microbiol. 17, 3610–3627. doi: 10.1111/1462-2920.12591
Simon, M., López-García, P., Deschamps, P., Moreira, D., Restoux, G., Bertolino, P., et al. (2015). Marked seasonality and high spatial variability of protist communities in shallow freshwater systems. ISME J. 9, 1–13. doi: 10.1038/ismej.2015.6
Simon, M., López-García, P., Moreira, D., and Jardillier, L. (2013). New haptophyte lineages and multiple independent colonizations of freshwater ecosystems. Environ. Microbiol. Rep. 5, 322–332. doi: 10.1111/1758-2229.12023
Slapeta, J., Moreira, D., and López-García, P. (2005). The extent of protist diversity: insights from molecular ecology of freshwater eukaryotes. Proc. R. Soc. B Biol. Sci. 272, 2073–2081. doi: 10.1098/rspb.2005.3195
Spatafora, J. W., Chang, Y., Benny, G. L., Lazarus, K., Smith, M. E., Berbee, M. L., et al. (2016). A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108, 1028–1046. doi: 10.3852/16-042
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stibbs, H. H., Owczarzak, A., Bayne, C. J., and DeWan, P. (1979). Schistosome sporocyst-killing amoebae isolated from Biomphalaria glabrata. J. Invertebr. Pathol. 33, 159–170. doi: 10.1016/0022-2011(79)90149-6
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D., Breiner, H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Taib, N., Mangot, J. F., Domaizon, I., Bronner, G., and Debroas, D. (2013). Phylogenetic affiliation of SSU rRNA genes generated by massively parallel sequencing: new insights into the freshwater protist diversity. PLoS ONE 8:e58950. doi: 10.1371/journal.pone.0058950
Tanabe, A. S., Nagai, S., Hida, K., Yasuike, M., Fujiwara, A., Nakamura, Y., et al. (2016). Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Mol. Ecol. Resour. 16, 402–414. doi: 10.1111/1755-0998.12459
Tekle, Y. I., Anderson, O. R., Katz, L. A., Maurer-Alcalá XX, Romero, M. A. C., and Molestina, R. (2016). Phylogenomics of “Discosea”: a new molecular phylogenetic perspective on Amoebozoa with flat body forms. Mol. Phylogenet. Evol. 99, 144–154. doi: 10.1016/j.ympev.2016.03.029
Thomas, M. C., Selinger, L. B., and Inglis, G. D. (2012). Seasonal diversity of planktonic protists in Southwestern Alberta rivers over a 1-year period as revealed by terminal restriction fragment length polymorphism and 18S rRNA gene library analyses. Appl. Environ. Microbiol. 78, 5653–5660. doi: 10.1128/AEM.00237-12
Torres-Machorro, A. L., Hernández, R., Cevallos, A. M., and López-Villaseñor, I. (2010). Ribosomal RNA genes in eukaryotic microorganisms: Witnesses of phylogeny? FEMS Microbiol. Rev. 34, 59–86. doi: 10.1111/j.1574-6976.2009.00196.x
Torruella, G., de Mendoza, A., Grau-Bové, X., Antó, M., Chaplin, M. A., del Campo, J., et al. (2015). Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr. Biol. 25, 2404–2410. doi: 10.1016/j.cub.2015.07.053
Torruella, G., Derelle, R., Paps, J., Lang, B. F., Roger, A. J., Shalchian-Tabrizi, K., et al. (2012). Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 29, 531–544. doi: 10.1093/molbev/msr185
Train, S., and Rodrigues, L. C. (1997). Temporal fluctuations of the phytoplankton community of the Baía River, in the upper Paraná River floodplain, Mato Grosso do Sul, Brazil. Hydrobiologia 361, 125–134. doi: 10.1023/A:1003118200157
Triadó-Margarit, X., and Casamayor, E. O. (2012). Genetic diversity of planktonic eukaryotes in high mountain lakes (Central Pyrenees, Spain). Environ. Microbiol. 14, 2445–2456. doi: 10.1111/j.1462-2920.2012.02797.x
Valster, R. M., Wullings, B. A., and Van Der Kooij, D. (2010). Detection of protozoan hosts for legionella pneumophila in engineered water systems by using a biofilm batch test. Appl. Environ. Microbiol. 76, 7144–7153. doi: 10.1128/AEM.00926-10
Weber, F., del Campo, J., Wylezich, C., Massana, R., and Jürgens, K. (2012). Unveiling trophic functions of uncultured protist taxa by incubation experiments in the Brackish Baltic sea. PLoS ONE 7:e41970. doi: 10.1371/journal.pone.0041970
Yahr, R., Schoch, C. L., and Dentinger, B. T. (2016). Scaling up discovery of hidden diversity in fungi: impacts of barcoding approaches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 1–11. doi: 10.1098/rstb.2015.0336
Zalocar de Domitrovic, Y., Poi de Neiff, A. S., and Casco, S. L. (2007). Abundance and diversity of phytoplankton in the Paraná River (Argentina) 220 km downstream of the Yacyretá reservoir. Brazilian J. Biol. 67, 53–63. doi: 10.1590/S1519-69842007000100008
Keywords: unicellular Holozoa, early-branching Holomycota, metabarcoding, Paraná river, molecular diversity, freshwater
Citation: Arroyo AS, López-Escardó D, Kim E, Ruiz-Trillo I and Najle SR (2018) Novel Diversity of Deeply Branching Holomycota and Unicellular Holozoans Revealed by Metabarcoding in Middle Paraná River, Argentina. Front. Ecol. Evol. 6:99. doi: 10.3389/fevo.2018.00099
Received: 27 March 2018; Accepted: 25 June 2018;
Published: 12 July 2018.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Jeffrey D. Silberman, University of Arkansas, United StatesYonas Isaak Tekle, Spelman College, United States
Copyright © 2018 Arroyo, López-Escardó, Kim, Ruiz-Trillo and Najle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastián R. Najle, bmFqbGVAaWJyLWNvbmljZXQuZ292LmFy; c2ViYXN0aWFuLm5hamxlQHVwZi5lZHU=