 Prabodh Kumar Bajpai
Prabodh Kumar Bajpai Arye Harel1
Arye Harel1 Oz Barazani
Oz Barazani- 1Institute of Plant Science, Agricultural Research Organization, Rishon LeZion, Israel
- 2Department of Entomology, Institute of Environmental Sciences, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel
Populations of Eruca sativa (Brassicaceae) derived from arid and Mediterranean habitats exhibit ecotypic differentiation. Here, pooled DNA sequencing was used to assess adaptive genome differentiation in the two ecotypes. Differentiated SNP loci were scanned with the empirical FST outlier method and by correlating allele frequencies with environmental parameters. Genetic diversity values were relatively higher in the pooled arid genome, whereas the pooled Mediterranean genome exhibited stronger directional selection, indicating the impact of climatic conditions on genetic diversity. GO enrichment analysis categorized the annotated differentiated loci according to biological processes, revealing a large set of candidate genes related to abiotic and biotic stress responses. Allelic variation was detected in regulatory elements and coding regions (synonymous and non-synonymous mutations) of genes belonging to different transcription factors and phytohormone signaling, suggesting adaptation to both abiotic and biotic conditions. Furthermore, SNP mutations were also found in genic regions belonging to the synthesis of secondary metabolites, including aliphatic glucosinolates and their hydrolyzed bioactive compounds, among others. The results of this eco-genomic study demonstrate the role of divergent abiotic and biotic selection factors in evolutionary processes leading to adaptive ecotypic differentiation.
Introduction
The divergence of traits and ecotypic differentiation is considered to be an adaptive response to environmental conditions (Nosil, 2012; Zaidem et al., 2019). Thus, understanding the factors and processes that contribute to genetic differentiation is a fundamental step in evolutionary studies. Additionally, knowledge of genes and loci associated with intraspecific phenotypic differentiation is essential to understand the basis of adaptive divergence and variation in functional traits. In recent years, increased access and ease of genomic sequencing has revolutionized the ability to link genetic variation with local adaptation (Ellegren, 2014; Tiffin and Ross-Ibarra, 2014). Thus, ecological genomic studies in plants used inter-species differentiation to associate genomic regions with geographic and climatic conditions, mainly in Arabidopsis sp. and other closely related species (Fischer et al., 2013; Kubota et al., 2015; Honjo and Kudoh, 2019; Takou et al., 2019). Complementary studies with non-model classical species, such as Arabis alpina for example, detected genomic regions that are linked to environmental gradients and thus could be associated with adaptation to cold and pathogen resistance (Lobreaux and Miquel, 2020). Therefore, studies that aim to understand local adaptations utilize species whose distribution covers pronounced environmental gradients (e.g., Hancock et al., 2011; Fischer et al., 2013; Kubota et al., 2015; Rellstab et al., 2016). However, the ecological relevance of the association between single nucleotide polymorphism (SNP) in plant populations and environmental conditions remained speculative in most cases.
The topographic and aridity gradients that exists in the southeast Mediterranean region, ranging from relatively humid Mediterranean to arid desert environments over relatively short distances, mark this region as a prime live laboratory for studies aiming to understand evolutionary processes. Several studies conducted in this region demonstrated intra-specific phenological and morphological variation (Aronson et al., 1990; Petrů et al., 2006; Liancourt and Tielborger, 2009; Kigel et al., 2011; Waitz et al., 2021), emphasizing the possible role of abiotic selective factors on phenotypic and ecotypic divergence (Metz et al., 2020). Nevertheless, to date, no study in this region has yet attempted to explore the links between environmental heterogeneity, genome-wide variation, and phenotypic divergence.
Populations of the self-incompatible annual Eruca sativa (Brassicaceae) in the region are thriving in diverse habitats along the short and narrow strip of the Jordan Valley (Table 1; Westberg et al., 2013). Previous studies have shown that populations from arid and Mediterranean habitats show spatial co-variation within several ecologically important functional traits, including seed dormancy and longevity (Barazani et al., 2012; Hanin et al., 2013), and flowering time (Westberg et al., 2013). Natural variation in populations of E. sativa also includes differences in trichome density (Westberg et al., 2013), floral traits (Barazani et al., 2019), and genetic differentiation of induced defense against insects (Ogran et al., 2016, 2019), traits that possibly evolved as a response to biotic selection factors (Ogran et al., 2020). Moreover, it was also recently shown that despite a significant gene flow among populations, the phenotypic differentiation in E. sativa evolved due to diversifying selection (Bajpai et al., 2022). Thus, overall, our previous results indicate that the inter-population differentiation in E. sativa can be efficiently utilized to understand the effect of natural selection on genomic variation. For this purpose, we used a pooled genome sequencing (Pool-Seq) approach and identified more than 300,000 single nucleotide sites whose frequency significantly differed across the genome of two contrasting arid and Mediterranean populations of E. sativa. Our analyses aimed to identify genomic regions showing footprints of selection in association with environmental parameters. To do this, we used FST outlier analysis to identify differentiated genes. Further analysis of environmental association using Bayesian statistics, was applied to understand their putative adaptive roles. This study is among few that address ecotypic differentiation in non-model plant species, offering a unique opportunity to understand genomic changes associated with the responses of organisms to their biotic and abiotic environment.
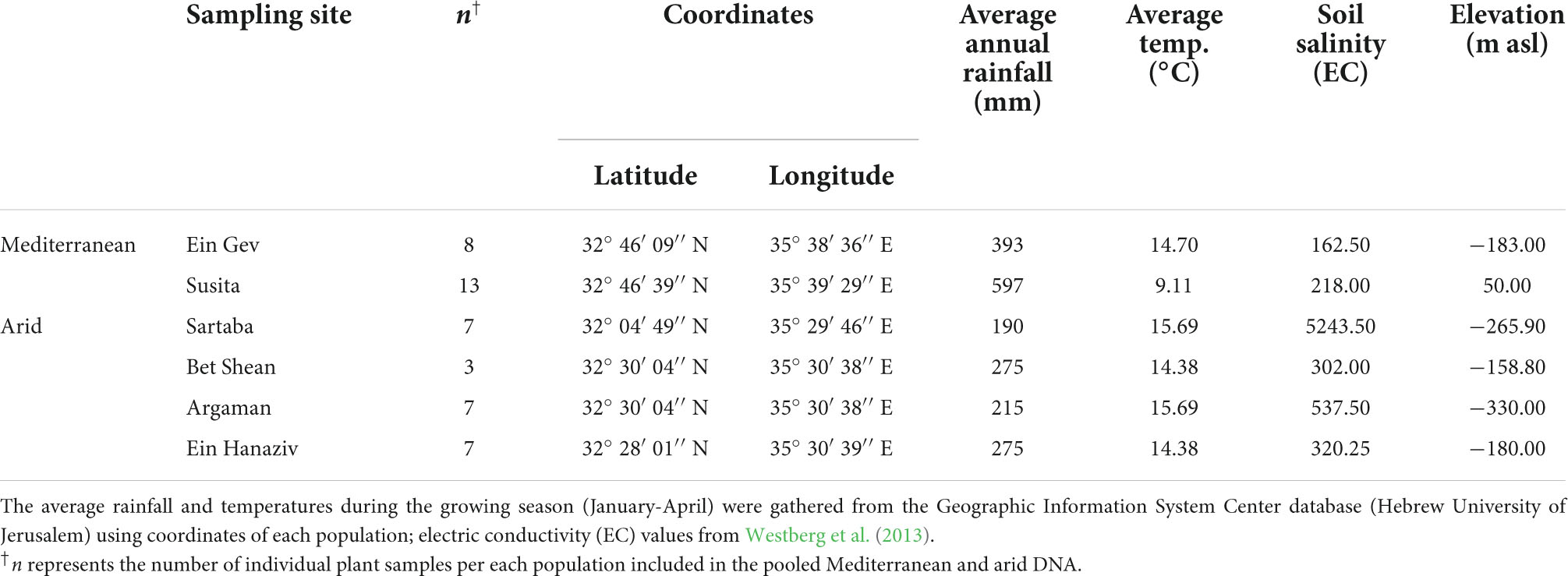
Table 1. The investigated populations of E. sativa, their location, and environmental characteristics.
Materials and methods
The studied populations, plant material, and DNA extraction
Populations of E. sativa in the east Mediterranean are distributed along a steep climatic gradient in the Jordan Valley, from arid (<200 mm rainfall year–1) to Mediterranean (>450 mm rainfall year–1) habitats (Table 1). Several environmental characteristics differentiate the arid and the Mediterranean sites, including elevation, average temperatures during the growing season, annual rainfall, and soil salinity (Table 1). In addition, a recent entomological study revealed differences in herbivory pressure at the sites, showing higher frequencies of the specialist moth Plutella xylostella at arid sites than at Mediterranean sites, and vice versa for generalist aphids (Ogran et al., 2020).
Significant correlations were found between all abiotic environmental factors, i.e., average annual rainfall, average temperatures during the growing season, and soil salinity, as well as between these and their geographical location (latitude) (Supplementary Data Sheet 1, Table S1). In addition, the Pearson correlation test revealed significant correlations between all the environmental characteristics and herbivory pressure, i.e., the frequency of P. xylostella and aphids in the two regions. Thus, to avoid multi-collinearity, in the analyses for detecting environmental-specific outlier loci (below), we used rainfall as the environmental variable, the most important environmental parameter, which limits plant survival in this region (Aronson et al., 1990, 1992; Petrů et al., 2006; Volis, 2007; Kigel et al., 2011; Westberg et al., 2013).
We previously described the genetic diversity between nine populations of E. sativa along this climatic gradient (Westberg et al., 2013). Our previous results indicated that genetic diversity values were similar in all populations. In addition, Bayesian clustering divided populations from Mediterranean and arid environments to two main phylogeographic clusters, with a population with a mixed ancestry of the clusters at the border of the two regions (Westberg et al., 2013). Accordingly, leaf samples were collected from plants in each of six selected populations representing the two main genetic clusters of arid (Sartaba, Argaman, Bet Shean, and Ein Hanaziv) and Mediterranean (Ein Gev, Susita) habitats [Table 1 and see Westberg et al. (2013)]; the population with a mixed ancestry was not included. DNA was extracted with the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany), and DNA quality and quantity were measured on UV-Vis Spectrometer (NanoDrop 8000, Thermo Scientific) as well as on 1% agarose gel.
Sequencing and read cleaning
The Pool-Seq approach provides an alternative, cost-effective way, to sequence DNA (Schlotterer et al., 2014). Several studies used this approach to quantify allele frequency differences between populations to infer about selection processes (Boitard et al., 2012; Ferretti et al., 2013; Fischer et al., 2013). Thus, this approach can provide insight into the evolutionary and demographic history of populations, to identify regions under selection, and alleles whose frequencies differ consistently between populations.
Equal quantities of 21 and 24 individual DNA samples, representing four arid and two Mediterranean populations, respectively (Table 1), were pooled together to make two samples of 3 μg DNA each, at a concentration of 40 ng/μl. Using the True-seq DNA PCR Free kit (Illumina), each pooled DNA sample was converted separately into a paired-end 151 bp genomic sequencing library. The two libraries were then sequenced on an Illumina HiSeq X Ten platform (Macrogen, South Korea). Whole genome sequences (WGS) of the arid and Mediterranean regions (AR and MR, respectively) DNA pools were analyzed at 135X coverage, considering genome size of 851 Mb (Bell et al., 2020). Data quality was then checked using FastQC1 and was cleaned by trimming low quality reads by a perl script (trim-fastq.pl) provided in the PoPoolation program (Kofler et al., 2011a). To ensure the sequences’ accuracy, a Phred quality score was kept at Q30, including a threshold of 50 bp minimum read length.
Reads mapping and SNP calling
Trimmed paired-end reads of each AR and MR genomes were mapped to the Eruca vesicaria (L.) Cav. (syn. E. sativa Miller) genome.2 The index command in the Burrows-Wheeler aligner (BWA) program was used for sequence alignment and to prepare a reference genome (Li and Durbin, 2009, 2010). Reads were mapped using the BWA mem command. The mapped reads were further sorted with Picard tools3 and tagged duplicates were removed. Samtools ver. 1.7 (Li et al., 2009) was then used for variant calling, removing ambiguously mapped reads (q score < 20), and to create a mpileup file combining outputs from both samples. Genomic indels were identified and removed using PoPoolation2 program using perl scripts identify-genomic-indels-regions.pl and filter-pileup-by-gtf.pl, respectively. The PoPoolation2 script mpileup2sync.jar was then used to create a sync file from the indel filtered data by keeping base quality threshold of 30. The PoPoolation2 program (script: subsample-synchronised.pl) was further used to submsample the data for removing potential sequencing errors. The subsampled data was then used to estimate SNPs’ allele frequency (script: snp-frequency-diff.pl) by maintaining a minimum minor allele count of 4, and minimum and maximum coverage of 20 and 200, respectively.
Genome-wide diversity analysis
Genome-wide patterns of diversity were characterized by estimation of three genetic diversity parameters: (i) nucleotide diversity (π); (ii) Watterson’s θ (θw); and (iii) Tajima’s D (TD). All genetic diversity parameters were estimated from individual sample pool files, i.e., the AR and MR pool files. Each pool file was sub-sampled to a uniform coverage of 20 before the analysis. Using PoPoolation2, the genetic diversity parameters were estimated in non-overlapping 10-kb windows across the genome (Kofler et al., 2011a), by maintaining a minimum allele count of 2. All genetic diversity parameters were checked for normality and homogeneity of variance using the Komolgorov Smirnov and Leven’s tests, respectively. As none of the genetic diversity parameters followed the assumption of normal distribution, the non-parametric independent sample Mann–Whitney U test/Wilcoxon rank-sum test (Guo et al., 2016) was conducted to test for significant differences in genetic diversity parameters between the two AR and MR genomes.
Screening for differentiated SNPs–candidate loci analysis
Candidate loci analyses were performed in two steps. First, using Fisher’s exact (FE) test, we screened genomic regions that exhibited significant genetic differentiation between the two genomes by comparing allelic frequencies (Raymond and Rousset, 1995; Goudet et al., 1996; Ryman and Jorde, 2001); the FE test was performed using the perl script fisher-test.pl in PoPoolation2 program. The Benjamini-Hochberg false discovery rate (FDR) correction was further applied to the obtained P-values using R (version 4.0.2) (R Core Team., 2020). SNPs in specific genomic comparison with FDR values lower than 0.01 were defined as significantly differentiated. Second, we further screened significantly differentiated SNPs based on the genetic differentiation (FST) values. The FST value for each SNP was determined using the perl script fst-sliding.pl in PoPoolation2 (Kofler et al., 2011b), after which SNPs falling in the upper 5% tail of the FST distribution range were selected. The allele frequency difference of most variable alleles (AFDMVA) of the AR and MR pooled genomes was estimated by subtracting the MR allele frequency from that of the AR.
Gene assignment and functional annotation
Genomic annotations and sequences for E. vesicaria genome version 1.1 were downloaded from the JGI genome portal (Nordberg et al., 2013). The gene annotations were extracted from the general feature format (GFF) using Python scripts. Using Python scripts, candidate SNPs (above) were annotated as being part of coding region sequences (CDS), potential gene promoters [2000 bp upstream to the 5′ of the untranslated region (UTR)], or as not in the range of gene coding region. For SNPs located within a CDS, the potential non-synonymous effect caused by change in nucleotides was computed from the reference sequence of the E. vesicaria genome. Functional annotation of SNPs associated with CDS or promoters was carried out using the annotation file of the E. vesicaria genome by looking at GO IDs and best hits of the associated gene in Arabidopsis thaliana, Oryza sativa, and Chlamydomonas reinhardtii. Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs and their related pathways were also identified with the web-based KEGG Automatic Annotation Server (KASS) (Moriya et al., 2007) against a selected list of genomes (Supplementary Data Sheet 1, Table S2) using the SBH (Sequencing by hybridization) method and default parameters and thresholds.
Environmental association analysis
A Bayesian approach implemented in the Bayenv2 program was used to assess the association between environmental parameters and allele frequencies (Gunther and Coop, 2013). Environmental specific outlier loci were detected by controlling sampling errors due to the sample pools. This approach also accounts for covariance due to population size history (Gunther and Coop, 2013). To perform this analysis, we first removed 258 monomorphic SNPs and 468 SNPs with insufficient allele count information from the total of 153,560 annotated SNPs (above). The 152,834 significant polymorphic SNPs obtained were used to build a reference covariance matrix of allele frequencies using 400000 MCMC iterations. The allele frequencies matrix was correlated with standardized rainfall, to obtain correlation statistics, called Z statistics (Bayenv2). Z statistic ranges from 0 to 0.5, with values close to the upper range showing a strong correlation with the environmental parameter. The allele states of the pooled AR and MR genomes were further estimated as described above.
Gene set enrichment analysis
Gene ontology (GO) enrichment analysis (Ge et al., 2020) was used for examining enriched biological processes in two lists of significantly differentiated SNPs: (1) based on allele frequency and FST (AF-FST); and (2) based on environmental parameter association. The AF-FST and environmental parameter association analyses yielded a total of 2,417 and 517 SNPs, respectively, that were annotated to distinct Arabidopsis genes (see Section “Results”). Both gene lists were further subjected to GO analysis using the ShinyGO website.4 Fisher’s exact test with FDR correction was used to identify significantly enriched GO identifier; GO terms with FDR less than 0.05 were considered significantly enriched. In the case of SNPs based on AF-FST analysis, the REVIGO program,5 which uses a simple clustering algorithm to find a representative subset of the terms, was applied with default parameters to shorten and remove redundant GO terms (Supek et al., 2011). The KEGG Mapper6 and the AmiGO gene ontology resource7 were also used to categorize the orthologous A. thaliana gene IDs into biological processes.
The ShinyGO tool was further applied to the list of 2,417 AF-FST SNPs for the identification of enriched cis regulatory elements (CREs) or transcription factor (TF) binding motifs in the promoter regions (300 bp upstream of genes). The ShinyGO platform used the stress responsive transcription factors database (STIFDB8). Fisher’s exact test with FDR correction was applied to select significantly (P < 0.05) enriched CREs and transcription factors.
Results
Sequencing and mapping statistics
The whole genome Illumina sequencing produced more than 381 × 106 and 374 × 106 raw reads in the arid and Mediterranean pooled DNA samples, respectively (Supplementary Data Sheet 1, Table S3). After quality check and trimming, 375 × 106 arid and 367 × 106 Mediterranean cleaned pair-end reads were obtained. The Q20 of the arid and Mediterranean genome libraries were 97.69 and 97.17%, respectively, and the respective Q30 values were 94.58 and 93.68% (Supplementary Data Sheet 1, Table S3). Mapping against the E. vesicaria genome yielded more than 98% of mapped reads in both the arid and Mediterranean genome pools (Supplementary Data Sheet 1, Table S4).
Genome-wide variation
Nucleotide diversity values (π) ranged from 2.3 × 10–5 to 6.1 × 10–2 in the pooled genome of the Mediterranean region (MR), while it ranged from 0.0 to 5.6 × 10–2 in the pooled genome of the arid region (AR). When averaged for 10 kb windows, genome-wide estimates were 1.10 × 10–2 and 1.15 × 10–2 for the MR and AR pooled genomes, respectively (Table 2). Estimates of θw ranged from 4.11 × 10–5 to 5.40 × 10–2 and 0.0 to 5.05 × 10–2 for the MR and AR pooled genomes, respectively. The θw average 10 kb windows values were relatively similar for the MR and AR pooled genomes (1.12 × 10–2 and 1.16 × 10–2, respectively). Nevertheless, the mean rank values of both π and θw were significantly higher in the AR pooled genome than the MR pooled genome (Mann–Whitney U test P < 0.001). In addition, the negative TD values, indicating deviation from neutrality, were significantly lower in the MR pooled genome than in the AR pooled genome (Mann–Whitney U test P < 0.001) (Table 2).

Table 2. Genome-wide diversity parameters of the Mediterranean and arid DNA pools (mean ± SE): nucleotide diversity (π), Watterson θ (θw), and Tajima’s D (TD) in 10-kb non-overlapping windows.
Candidate loci analysis and the identification of differentiated SNPs
A total of 11,917,267 SNPs were obtained by mapping the pair-end reads of each of the two genome pools against the E. vesicaria genome. Following a comparison of allelic frequencies by FE test, 333,631 significant differentiated SNPs were found. The average FST value for the total (11,917,267) and significant (333,355; as assessed with the PoPooplation2 program) SNPs were 0.08 ± 0.10 and 0.46 ± 0.11, respectively.
Out of the total 333,631 significant differentiated SNPs, 327,196 were annotated (Supplementary Data Sheet 1, Figure S1). Further grouping of the annotated SNPs according to gene model predictions (extracted from the GFF file), revealed that most (53.0%) were categorized to non-genic regions. Among the 153,560 SNPs that were detected in genic regions, 17.9% were categorized to promoters, 15.6% to CDS regions, 11.2% to mRNA, and 2.3% to 3′ and 5′ UTR; 53.0% of the annotated SNPs were not within a genic region (NIG) (Supplementary Data Sheet 1, Figure S1). In addition, among the 51,082 SNPs that were categorized to CDS, 41.5% were defined as non-synonymous (Supplementary Data Sheet 1, Figure S1 inset).
Enrichment analysis of significantly differentiated SNPs based on allele frequency-FST
Out of the 333,355 significant SNPs obtained (above), 16,668 were detected in the upper 5% tail of the FST distribution. Among the latter, only 2,417 were annotated to distinct A. thaliana gene IDs (Supplementary Data Sheet 2). Gene-set enrichment analysis categorized the 2,417 gene IDs to 191 enriched biological processes (see “Significant GO terms” in Supplementary Data Sheet 2). The 20 important biological processes that were specified by REVIGO (“Enriched GO terms by REVIGO” in Supplementary Data Sheet 2), included 1,403 genes. SNPs on the list of genes derived from the REVIGO AF-FST enrichment analysis were mostly (53.0%) in CDS regions, while 46.9% of other SNPs belonged to regulatory regions (promoter and 5′ and 3′ UTR) (Figure 1A). In addition, among the loci that were associated with CDS region, 267 (38.5%) possessed non-synonymous mutations (“Gene IDs and allelic state” in Supplementary Data Sheet 2). The values of AFDMVA of the respective loci in the AF-FST were ≥ 0.5 (see SNPs IDs in Supplementary Data Sheet 2). Among the distinct gene IDs, 408 genes were listed in “response to stress” (Figure 2A), which was directly linked with 187 genes that were enriched in “response to external stimulus” biological processes.
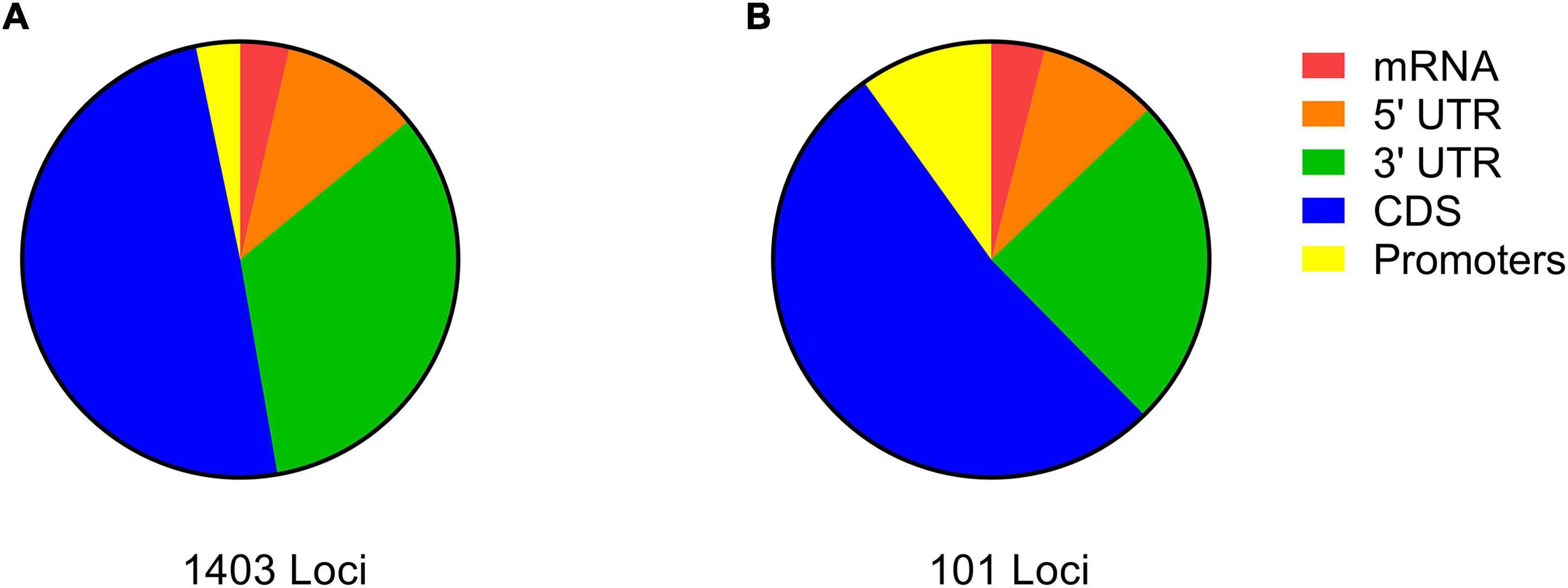
Figure 1. The categorization of differentiated loci detected by the AF-FST (A) and environmental parameters (B) to different genic regions.
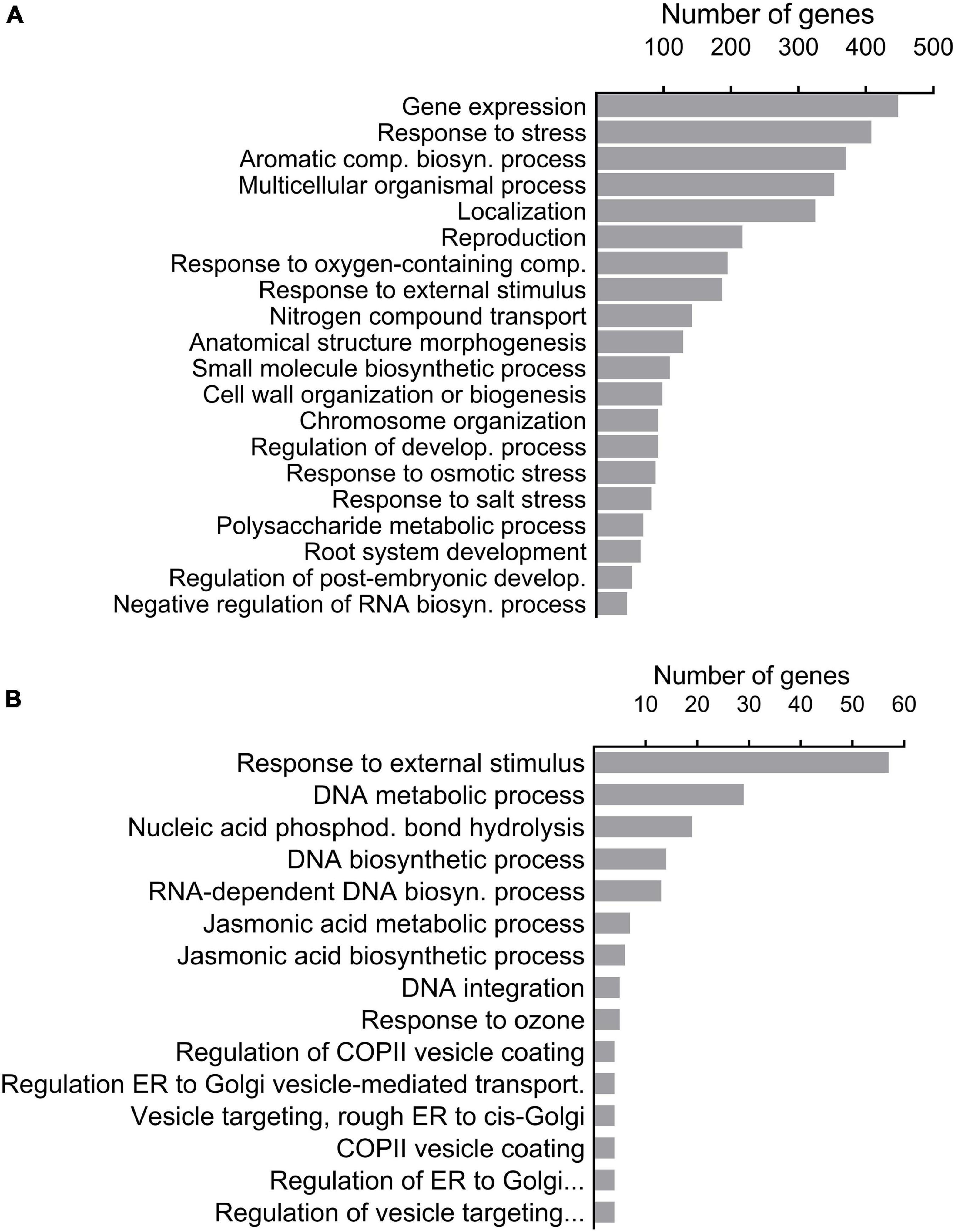
Figure 2. GO enriched biological processes of significantly differentiated SNPs (P < 0.05, Fisher’s exact test with FDR correction) that were screened on the basis of AF-FST (A) and environmental parameters (B).
Enrichment analysis of significantly differentiated SNPs based on environmental parameters
The 152,834 SNPs belonging to the coding and promoters region (Supplementary Data Sheet 1, Figure S1, above) were further subjected to environmental association analysis (EVA). Among the analyzed SNPs, 763 fell in the upper 0.5% tail of Z statistics (ranging from 0.438 to 0.423). The association between EVA and allele frequencies (using Bayenv2.0) revealed 517 SNPs with distinct 102 A. thaliana gene IDs (Supplementary Data Sheet 3). The majority of the SNPs detected in the EVA-GO enrichment analysis (53 loci) belonged to the CDS of their orthologous A. thaliana gene IDs, among them 50.9% possessed non-synonymous mutations (Figure 1B; “Gene IDs and allelic state” in Supplementary Data Sheet 3). Similar to the AF-FST analysis, the AFDMVA values of SNPs associated with gene IDs were ≥ 0.5.
The GO enrichment analysis displayed 16 enriched biological processes, among these responses to external stimuli (57 genes) and JA metabolic and biosynthesis processes (7 and 6 genes, respectively) (Figure 2B).
Genomic regions associated with ecological adaptations
The KEGG mapper and AmiGO tools were used to detect genomic regions with ecological relevance, i.e., water deprivation and defense against herbivores. The AmiGO tool categorized 24 genes of the 1,403 gene IDs derived from the REVIGO enrichment analysis (above), as belonging to water deprivation response, eight of them were in the CDS region (Table 3 and see “Gene IDs and allelic state” in Supplementary Data Sheet 2). The majority of candidate genes belonged to the phytohormone signaling pathways, mainly ABA (ABI5, ABA1, ABA2, NCED5, CBL1), but also auxin (ARF3 and AVP1) and cytokinin (ARR10). Others involved TF activity (ANAC059 and MYBs), protein kinases (CIPK6, ABR), and genes involved in defense as PAL1, involved in the first steps of the phenylpropanoid pathway, and RD19 encoding a cysteine proteinase (Table 3).
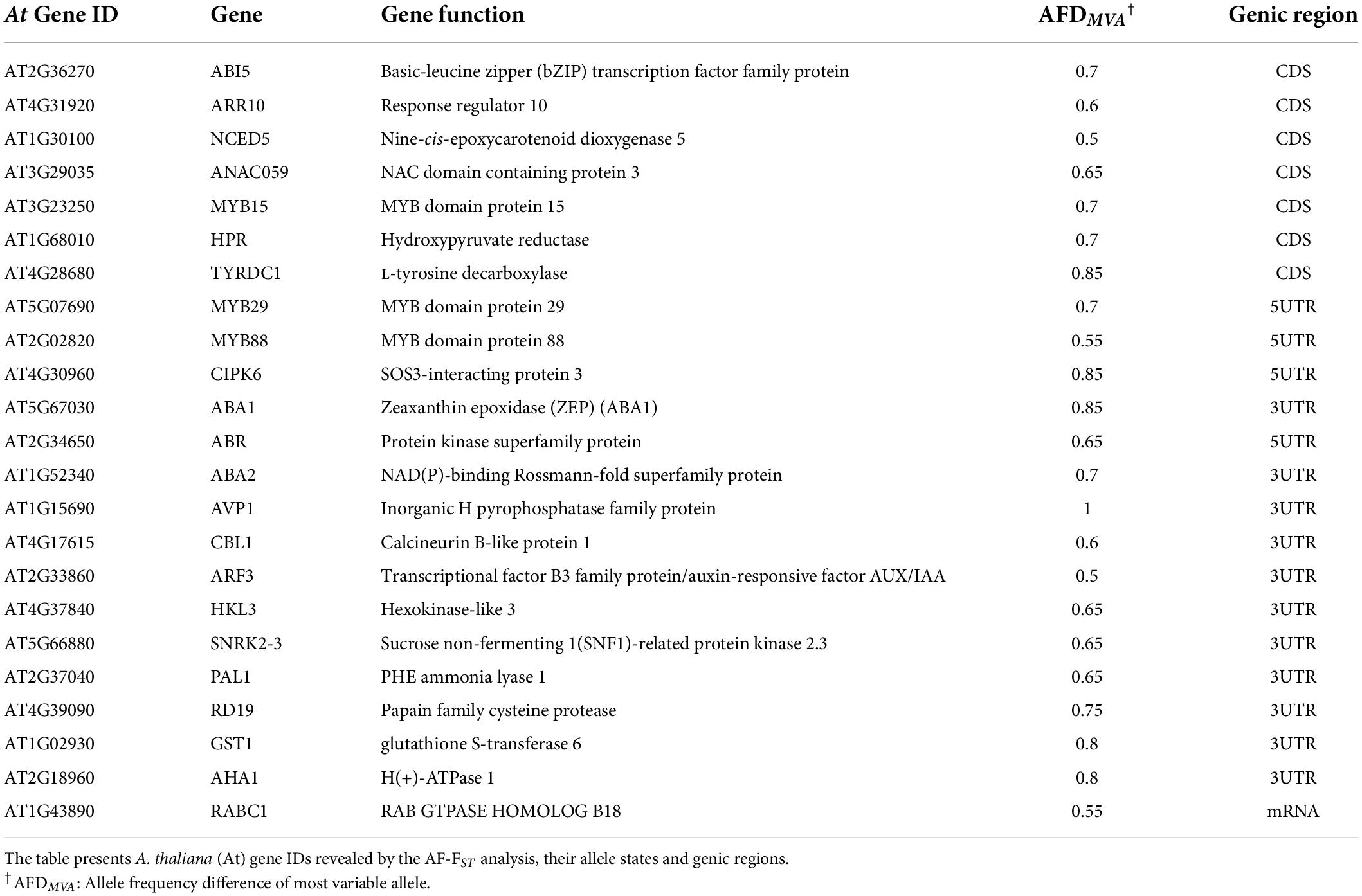
Table 3. Loci associated with response of plants to water deprivation.
Further analysis with the KEGG mapper categorized the detected 1,403 gene IDs into 114 pathways, among which 112 genes belonged to the biosynthesis of secondary metabolites. These included genes involved in synthesizing terpenoids, carotenoids, flavonoids, glucosinolates, sesquiterpenoids, and triterpenoids, among others (“KEGG pathways” in Supplementary Data Sheet 2). Loci possessing non-synonymous mutations included genes involved in synthesizing secondary metabolites, including genes associated with phytohormone signaling (below), cutin synthesis and proteinase inhibitors (Table 4). NSP1 and ESM, encoding nitrile specifier and epithiospecifier modifier proteins, play a role in the hydrolysis of glucosinolates to nitriles and isothiocyanates (ITCs), respectively, also possessed non-synonymous mutations in their coding regions (Table 4).
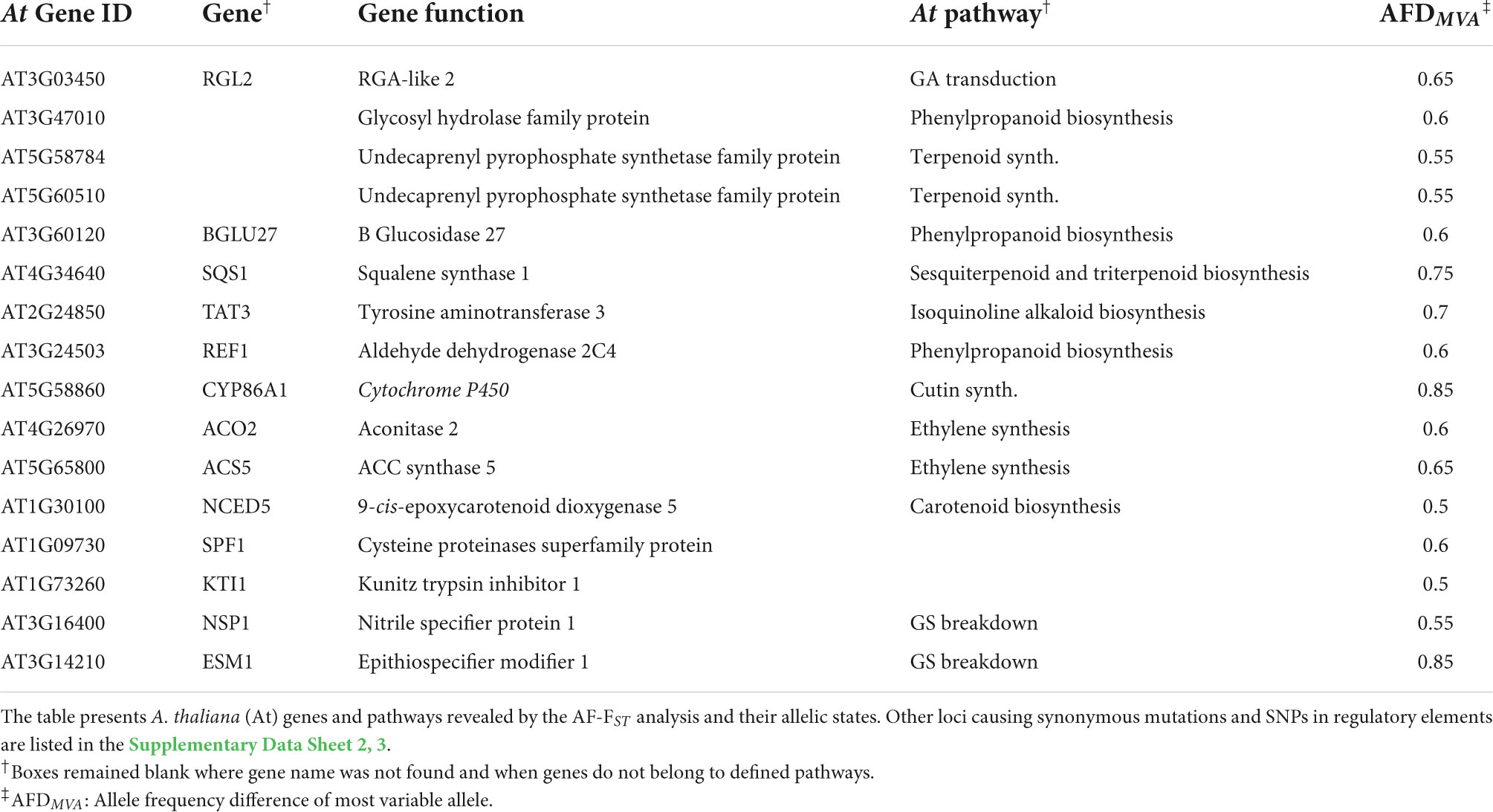
Table 4. SNPs with non-synonymous mutations in the CDS regions of genes associated with induced defense against herbivores.
Loci associated with phytohormone signaling
In addition to differentiated loci belonging to auxin and cytokinin pathways (Table 3), the AF-FST analysis detected SNPs possessing non-synonymous mutations in the CDS region of the ethylene synthesis genes (ACO2 and ACS5) and the gibberellic acid (GA) transduction pathway (Table 4 and see Supplementary Data Sheet 2). Interestingly, our AF-FST and EVA analyses detected 14 differentiated SNPs linked to α-linolenic metabolism and JA signaling pathway (Table 5), among which non-synonymous SNP mutations were identified in the CDS regions of OPR1 and LOX3, AOC2 and AOC3, all involved in JA metabolism. Additional SNPs included a synonymous mutation in the CDS regions of LOX2, and other differentiated loci in genic regions were detected in regulatory elements of JA metabolism and its transduction pathway (Table 5).
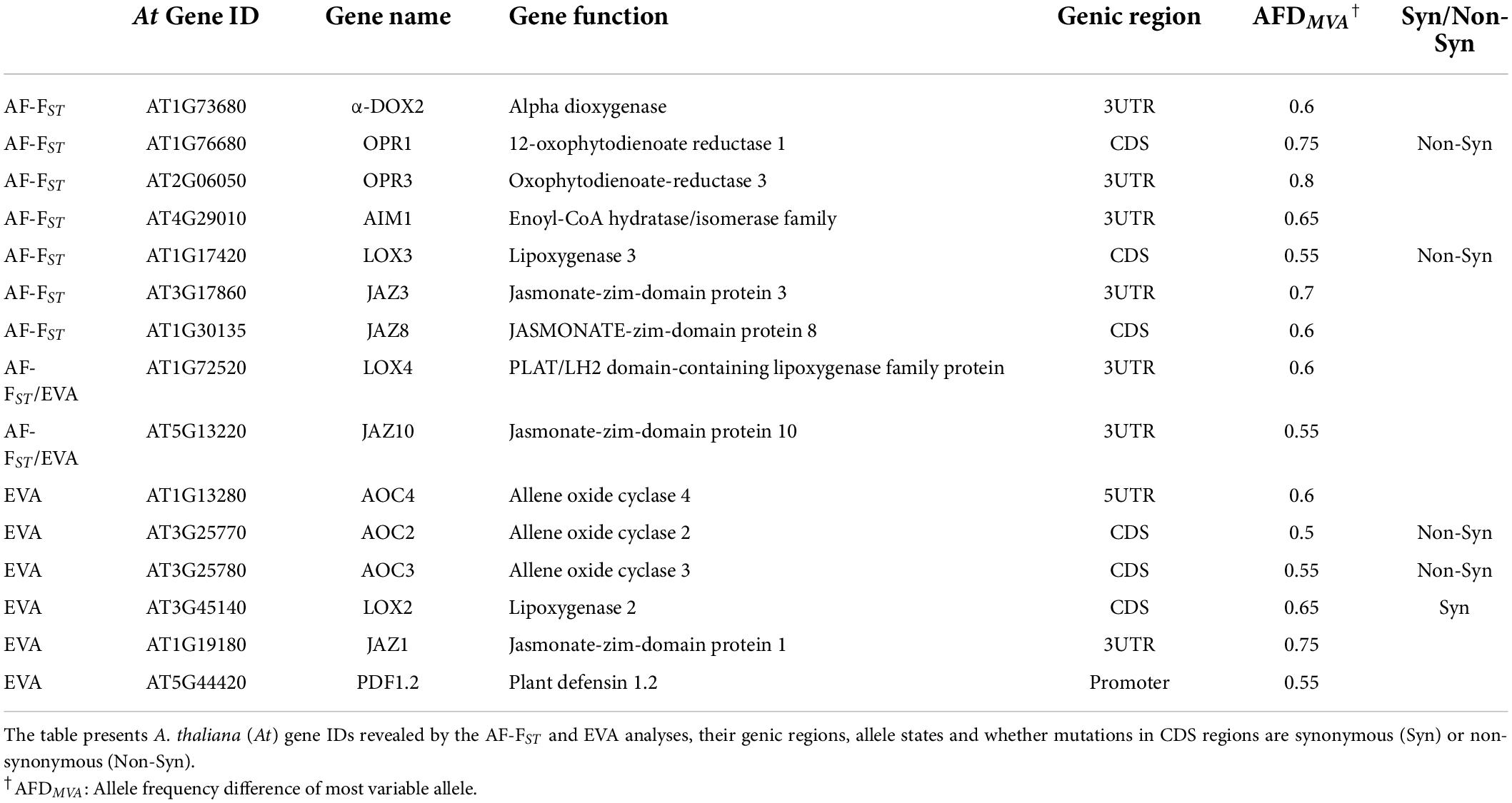
Table 5. Loci associated with jasmonic acid metabolism and transduction pathway.
Analysis of enrichment of cis regulatory elements and stress responsive transcription factors
The majority of the highly enriched CREs and TF binding motifs detected by ShinyGO included members of the HD-ZIP TF family, with eight belonging to the HD-ZIP I subfamily (Supplementary Data Sheet 1, Table S5). Other highly enriched CRE included TFs belonging to MADS box and AT hook TF families, among others. ShinyGO also detected 12 highly enriched TFs which belong to 1,849 genes of the STIFDB database (Table 6). TFs involved in hormone synthesis, hormone transduction pathways and secondary metabolite synthesis included 588 MYB gene family members, which bind to four different CRE, as well as bHLH/MYC, NAC and bZIP TFs. Other enriched stress responsive TFs included 505 HFS, bHLH/MYC and AP2/DREB family members involved in the regulation of plant response to abiotic stresses (Table 6), among them MYB29 and MYB88 (Table 3 and see “Gene IDs and allelic state” in Supplementary Data Sheet 2).
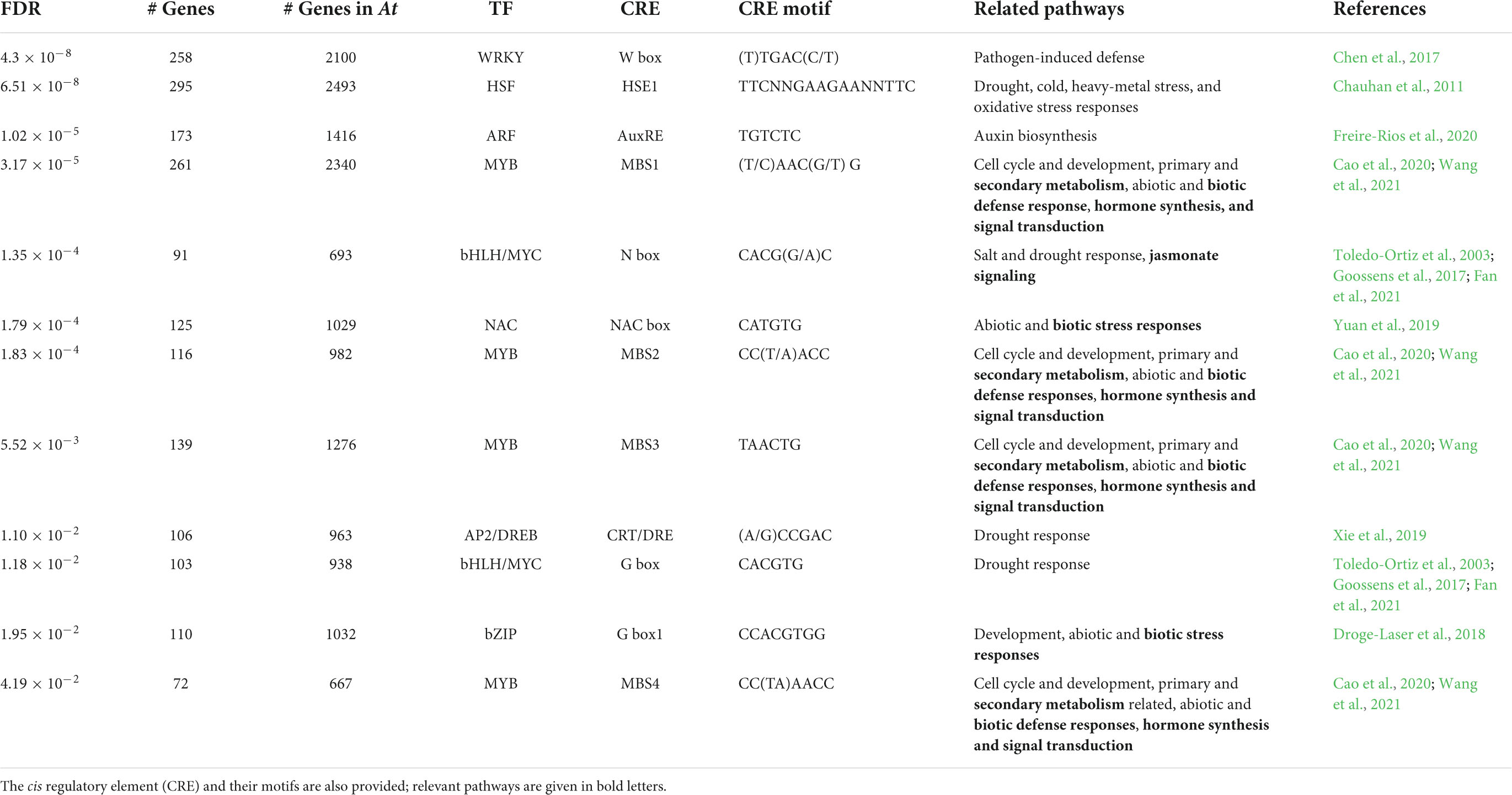
Table 6. List of enriched stress responsive transcription factors (TF), their obtained FDR values and the number of genes found for each TF group among those found in A. thaliana (At).
Discussion
This study describes signatures of adaptive genomic differentiation in populations of E. sativa thriving in contrasting Mediterranean and arid habitats. By employing various investigative approaches to a large SNP dataset, our study links genomic differentiation, phenotypic divergence, and selection drivers to reveal footprints of selection in specific genes that fulfill ecological requirements in distinct arid and Mediterranean habitats of E. sativa.
Genetic divergence and signatures of selection
The genome screen (Supplementary Data Sheet 1, Table S3) and FE test of allele frequencies yielded 333,631 differentiated SNPs, out of which 98.1% were annotated to the genome of E. vesicaria, thus indicating a relatively high coverage of the de novo assembled genome in relation to the reference. Overall, at the genome-wide level, low average pairwise FST values for all the detected SNPs indicated relatively low genetic differentiation between AR and MR populations, supporting the results of a previous study (Westberg et al., 2013). These results are primarily expected in self-incompatible species with continuous distribution along a relatively short and narrow distribution range [Table 1 and see Westberg et al. (2013)]. However, despite the potential gene flow between populations, nucleotide diversity values and the Watterson θw parameter, as an estimator of mutation rate, as well as the allele frequency AFDMVA values of the significantly differentiated loci (below), indicated significant higher genetic diversity in the arid genome pool than in the Mediterranean (Table 2; Supplementary Data Sheet 2, 3). Similarly, microsatellite polymorphism among populations of wild emmer wheat (Triticum dicoccoides) in Israel showed higher diversity values in peripheral sites exhibiting harsh conditions as compared to populations from more favorable habitats (Fahima et al., 2002). Thus, our results provide further evidence for the link between genetic distribution and ecological heterogeneity in the southeast Mediterranean, and in turn, selection.
Despite the generally low level of genome-wide genetic differentiation (above), we discovered high FST values in differentiated candidate loci, categorizing them as having a higher probability of being actively selected. Furthermore, significantly lower TD value in the MR pooled genome than in the AR (Table 2) indicated that pooled MR genome evolved under stronger directional selection pressure (Weedall and Conway, 2010). Results of our recent study indicated that arid populations of E. sativa possess higher phenological traits’ variation than a Mediterranean population (Bajpai et al., 2022). Accordingly, we speculate that conditions in the Mediterranean habitats led to the fixation of alleles and decreased levels of genetic diversity, in comparison to populations in arid, harsh and unpredictable conditions (Table 2). In addition, the negative TD values in both regions (Table 2) suggest that E. sativa experienced either selective sweeps in the recent past, or recent expansion which resulted in decreased genetic variation, something also shown in A. thaliana (Vasseur et al., 2018).
Further FST-based analysis of the differentiated loci, and correlation of their allele frequency with environmental variables, enabled the inference, detection and selection of outlier loci that undergo natural selection (Kubota et al., 2015). The AF-FST and EVA analyses indicated that differentiation between the two genomes include high amounts of outlier loci with associations to functional genes (Figure 1). Moreover, the GO enrichment analysis linked a large number of the differentiated SNPs to pathways associated with response to abiotic stress, such as osmotic and salt stress (Figure 2A). In addition, both the AF-FST and EVA analyses categorized the annotated genomic regions with biotic interaction (Figure 2). Similar studies that assessed genomic divergence between populations of A. hallerii (Fischer et al., 2013; Kubota et al., 2015) and Arabis alpina (Lobreaux and Miquel, 2020) along topographical and climatic gradients, detected GO-enriched pathways and candidate genes associated with various stress responses. In E. sativa, signatures of genomic adaptations could be assessed regarding abiotic (Westberg et al., 2013; Ogran et al., 2021) and biotic conditions (Ogran et al., 2020).
Evidence of local adaptation
Among the 1,403 differential loci detected by the AF-FST analysis, 24 loci were annotated to A. thaliana orthologues involved in response to water deprivation (Table 3). Furthermore, the majority of the candidate loci were situated in CDS and regulatory regions of functional genes (Figure 1), suggesting an impact of differential conditions on signatures of local adaptation at the genomic level.
ABA is the major key phytohormone playing a role in the regulation of plant response to abiotic stresses (Nakashima et al., 2014; Clauw et al., 2015), mainly by inducing stomatal closure under water stress and regulating stress-related responses (Sreenivasulu et al., 2012). Transcriptional changes in plants of E. sativa that were exposed to water deficiency included significant induction in the expression of ABA1 in AR plants, as compared to control and drought-exposed MR plants (Ogran et al., 2021). Here, we show that genomic differentiation between the two regions includes SNP mutations in regulatory elements of ABA metabolism and signaling pathways (Table 3). Furthermore, differentiated genomic regions also included SNPs in regulatory elements of genes of the auxin and cytokinin signaling pathways (Table 3), which possibly point to their role in the regulation and modification of plant development under stress conditions (Bielach et al., 2017). Signatures of local adaption to abiotic conditions in candidate genes included those involved in the response of plants to oxidative stress (GST1, PAL1), as well as in important transcription factors (MYB, NAC, bHLH/MYC, and ARF) (Tables 3, 6). Functional analysis studies have shown that members of these TF families are involved in the regulation of genes connected to phytohormone synthesis and signaling transduction, consequently influencing plant responses to both abiotic and biotic stress (see references cited in Table 5). Thus, genomic differentiation can be associated with abiotic selection factors linked to adaptive resilience to water stress.
Genomic differentiation in loci linked to induced defense against herbivores
One of the striking results of this study is the relatively high number of differentiated loci detected in genes associated with JA signaling and the biosynthesis of secondary metabolites (Tables 4, 5; Supplementary Data Sheet 2, 3). In Brassicaceae, glucosinolates are the main chemical metabolites produced for defense against herbivores. The prominent glucosinolate compound in leaves of E. sativa is the aliphatic 4-mercaptobutyl glucosinolate (Bennett et al., 2002), and aliphatic glucosinolates are more effective than indole glucosinolates against generalist herbivores (e.g., Arany et al., 2008). Thus, the allelic differences detected in the regulatory element BCAT-1 (Supplementary Data Sheet 2), involved in chain elongation of aliphatic methionine-derived glucosinolates (Schuster et al., 2006), can be linked to the role of glucosinolate in defense against herbivores, especially in MR plants (Ogran et al., 2016). However, the defensive role of glucosinolates is attributed to their hydrolysis products, mainly the toxic ITCs. The enzyme myrosinase, and the associated nitrile-specifiers (NSP) and epithiospecifier proteins (i.e., ESP and ESM), mediate the hydrolysis of glucosinolates. In A. thaliana, allelic mutation in the epithiospecifier protein (ESP) directs glucosinolate hydrolysis to simple nitriles at the expense of ITCs (Lambrix et al., 2001), thus increasing the susceptibility of the Ler ecotype to generalist herbivores (Lambrix et al., 2001) but decreasing oviposition by the specialist Pieris rapae (Mumm et al., 2008). Similarly, non-synonymous mutations were detected in the CDS loci of the epithiospecifier modifier 1 (ESM1) and nitrile specifier protein (NSP1) (Table 4). Accordingly, it can be concluded that the results of this study support the assumption that the release of simple nitriles in AR defends the plants from the specialist P. xylostella (Ogran et al., 2016). Moreover, we have previously shown that differences between populations in response to herbivory include higher expression of trypsin and KUNITZ proteinase inhibitors in AR than MR plants (Ogran et al., 2016, 2021). Thus, the non-synonymous allelic differentiation in KTI1 (Table 4) suggest a role of proteinase inhibitors as counter adaption to the specialist herbivore (Travers-Martin and Muller, 2008) in the AR plants. In addition, non-synonymous mutations in the cutin synthesis gene (CYP86A1, Table 4), responsible for the thickening of the epidermis cell walls, and close association between induced activity of proteinase inhibitors and trichomes in plants of E. sativa, suggest a potential role of mechanical defenses against herbivores. However, it remains to be investigated whether these defense mechanisms are effective against larvae of P. xylostella, and whether differential expression of nitrile specifier genes make the AR plants less apparent for oviposition by specialist moth females.
Induced defenses against herbivores are regulated mainly by the phytohormone JA and its interactions with ethylene and salicylic acid. The crosstalk between JA with ABA is also important in mediating plant response to specialist herbivores (Vos et al., 2019) and in combined drought and herbivory stress (Nguyen et al., 2016a,b). Regulation of phytohormone synthesis mainly depends on different TFs, and transcriptional regulators govern the crosstalk between them. The genomic scan of populations across different elevations of Arabis alpina by Lobreaux and Miquel (2020) emphasized the involvement of the NAC062 TF in adaptation to climatic and biotic conditions. Thus, allelic differences in SNP loci associated with various TFs (Tables 3, 6), and transcriptional regulators such as JAZ and PDF1.2 (Tables 4, 5) suggest that they are involved in governing different defense responses in the two populations (Ogran et al., 2016, 2019). Plants of E. sativa in both arid and semi-arid habitats are often exposed to simultaneous abiotic and biotic stress. In addition, an on-going investigation suggests that differences between floral attraction traits, i.e., petal color and scent (Barazani et al., 2019), might have evolved as a response to different pollinators. Thus, our results support the assumption that local adaptions mirror signatures of combined abiotic and biotic conditions (Ogran et al., 2021).
Summary
The results of this study successfully linked genomic differentiation in functional genes with environmental conditions and known phenotypic divergence in induced defenses, thus linking genomic differentiation with selection factors. Genomic differentiation in the two contrasting habitats of E. sativa, despite possible gene flow between sites, addresses fundamental evolutionary processes of ecotypic differentiation. These processes highlight the role of ecological interactions in the alteration of CDS and regulatory genic regions of functional genes involved in abiotic and biotic plant interactions. However, whether the genomic scan can be linked to signatures of selection of various other important phenological or morphological life history traits that differ between the AR and the MR populations (Barazani et al., 2012, 2019; Hanin et al., 2013; Westberg et al., 2013) or plant adaptations to different pollinators, remains to be investigated.
Data availability statement
The datasets presented in this study can be found in online repositories. The raw sequencing data are deposited in the NCBI Sequence Read Archive (SRA) database as BioProject PRJNA810733.
Author contributions
OB and SS conceived the study and obtained relevant funding. PB conducted the study and together with AH designed the bioinformatic work and performed the computational analyses. All authors contributed to the writing and finalizing of the manuscript.
Funding
This study was supported by the Israel Science Foundation (grant no. 2037/17).
Acknowledgments
We thank Dr. Zach Dunseth for his valuable assistance in editing the original manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.938981/full#supplementary-material
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://bmap.jgi.doe.gov
- ^ https://broadinstitute.github.io/picard/
- ^ http://bioinformatics.sdstate.edu/go/
- ^ http://revigo.irb.hr/
- ^ https://www.genome.jp/kegg/tool/map_pathway2.html
- ^ http://geneontology.org/
- ^ http://caps.ncbs.res.in/stifdb
References
Arany, A. M., de Jong, T. J., Kim, H. K., van Dam, N. M., Choi, Y. H., Verpoorte, R., et al. (2008). Glucosinolates and other metabolites in the leaves of Arabidopsis thaliana from natural populations and their effects on a generalist and a specialist herbivore. Chemoecology 18, 65–71. doi: 10.1007/s00049-007-0394-8
Aronson, J., Kigel, J., Shmida, A., and Klein, J. (1992). Adaptive phenology of desert and Mediterranean populations of annual plants grown with and without water-stress. Oecologia 89, 17–26. doi: 10.1007/Bf00319010
Aronson, J. A., Kigel, J., and Shmida, A. (1990). Comparative plant sizes and reproductive strategies in desert and Mediterranean populations of ephemeral plants. Isr. J. Bot. 39, 413–430.
Bajpai, P. K., Weiss, H., Dvir, G., Hanin, N., Wasserstrom, H., and Barazani, O. (2022). Phenotypic differentiation and diversifying selection in populations of Eruca sativa along an aridity gradient. BMC Ecol. Evol. 22:40. doi: 10.1186/s12862-022-01996-w
Barazani, O., Erez, T., Ogran, A., Hanin, N., Barzilai, M., Dag, A., et al. (2019). Natural variation in flower color and scent in populations of Eruca sativa (Brassicaceae) affects pollination behavior of honey bees. J. Insect Sci. 19:6. doi: 10.1093/jisesa/iez038
Barazani, O., Quaye, M., Ohali, S., Barzilai, M., and Kigel, J. (2012). Photo-thermal regulation of seed germination in natural populations of Eruca sativa Miller (Brassicaceae). J. Arid Environ. 85, 93–96.
Bell, L., Chadwick, M., Puranik, M., Tudor, R., Methven, L., Kennedy, S., et al. (2020). The Eruca sativa genome and transcriptome: a targeted analysis of sulfur metabolism and glucosinolate biosynthesis pre and postharvest. Front. Plant Sci. 11:525102. doi: 10.3389/Fpls.2020.525102
Bennett, R. N., Mellon, F. A., Botting, N. P., Eagles, J., Rosa, E. A. S., and Williamson, G. (2002). Identification of the major glucosinolate (4-mercaptobutyl glucosinolate) in leaves of Eruca sativa L. (salad rocket). Phytochemistry 61, 25–30. doi: 10.1016/S0031-9422(02)00203-0
Bielach, A., Hrtyan, M., and Tognetti, V. B. (2017). Plants under stress: involvement of auxin and cytokinin. Int. J. Mol. Sci. 18:1427. doi: 10.3390/Ijms18071427
Boitard, S., Schlotterer, C., Nolte, V., Pandey, R. V., and Futschik, A. (2012). Detecting selective sweeps from pooled next-generation sequencing samples. Mol. Biol. Evol. 29, 2177–2186. doi: 10.1093/molbev/mss090
Cao, Y. P., Li, K., Li, Y. L., Zhao, X. P., and Wang, L. H. (2020). MYB transcription factors as regulators of secondary metabolism in plants. Biol. Basel 9:61. doi: 10.3390/Biology9030061
Chauhan, H., Khurana, N., Agarwal, P., and Khurana, P. (2011). Heat shock factors in rice (Oryza sativa L.): genome-wide expression analysis during reproductive development and abiotic stress. Mol. Genet. Genom. 286, 171–187. doi: 10.1007/s00438-011-0638-8
Chen, F., Hu, Y., Vannozzi, A., Wu, K. C., Cai, H. Y., Qin, Y., et al. (2017). The WRKY transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 36, 311–335. doi: 10.1080/07352689.2018.1441103
Clauw, P., Coppens, F., De Beuf, K., Dhondt, S., Van Daele, T., Maleux, K., et al. (2015). Leaf responses to mild drought stress in natural variants of Arabidopsis. Plant Physiol. 167, 800–816. doi: 10.1104/pp.114.254284
Droge-Laser, W., Snoek, B. L., Snel, B., and Weiste, C. (2018). The Arabidopsis bZIP transcription factor family - an update. Curr. Opin. Plant Biol. 45, 36–49. doi: 10.1016/j.pbi.2018.05.001
Ellegren, H. (2014). Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 29, 51–63. doi: 10.1016/j.tree.2013.09.008
Fahima, T., Röder, M., Wendehake, K., Kirzhner, V., and Nevo, E. (2002). Microsatellite polymorphism in natural populations of wild emmer wheat, Triticum dicoccoides, in Israel. Theor. Appl. Genet. 104, 17–29. doi: 10.1007/s001220200002
Fan, Y., Yang, H., Lai, D. L., He, A. L., Xue, G. X., Feng, L., et al. (2021). Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in sorghum [Sorghum bicolor (L.) Moench]. BMC Genom. 22:778. doi: 10.1186/s12864-021-07652-9
Ferretti, L., Ramos-Onsins, S. E., and Perez-Enciso, M. (2013). Population genomics from pool sequencing. Mol. Ecol. 22, 5561–5576. doi: 10.1111/mec.12522
Fischer, M. C., Rellstab, C., Tedder, A., Zoller, S., Gugerli, F., Shimizu, K. K., et al. (2013). Population genomic footprints of selection and associations with climate in natural populations of Arabidopsis halleri from the Alps. Mol. Ecol. 22, 5594–5607. doi: 10.1111/mec.12521
Freire-Rios, A., Tanaka, K., Crespo, I., van der, W. E., Sizentsova, Y., Levitsky, V., et al. (2020). Architecture of DNA elements mediating ARF transcription factor binding and auxin-responsive gene expression in Arabidopsis. Proc. Natl. Acad. Sci.U.S.A. 117, 24557–24566. doi: 10.1073/pnas.2009554117
Ge, S. X., Jung, D. M., and Yao, R. A. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Goossens, J., Mertens, J., and Goossens, A. (2017). Role and functioning of bHLH transcription factors in jasmonate signalling. J. Exp. Bot. 68, 1333–1347. doi: 10.1093/jxb/erw440
Goudet, J., Raymond, M., deMeeus, T., and Rousset, F. (1996). Testing differentiation in diploid populations. Genetics 144, 1933–1940.
Gunther, T., and Coop, G. (2013). Robust identification of local adaptation from allele frequencies. Genetics 195, 205–220. doi: 10.1534/genetics.113.152462
Guo, B. C., Li, Z. T., and Merila, J. (2016). Population genomic evidence for adaptive differentiation in the Baltic Sea herring. Mol. Ecol. 25, 2833–2852. doi: 10.1111/mec.13657
Hancock, A. M., Brachi, B., Faure, N., Horton, M. W., Jarymowycz, L. B., Sperone, F. G., et al. (2011). Adaptation to climate across the Arabidopsis thaliana Genome. Science 334, 83–86. doi: 10.1126/science.1209244
Hanin, N., Quaye, M., Westberg, E., and Barazani, O. (2013). Soil seed bank and among-years genetic diversity in arid populations of Eruca sativa Miller (Brassicaceae). J. Arid Environ. 91, 151–154. doi: 10.1016/j.jaridenv.2013.01.004
Honjo, M. N., and Kudoh, H. (2019). Arabidopsis halleri: a perennial model system for studying population differentiation and local adaptation. AoB Plants 11:lz076. doi: 10.1093/aobpla/plz076
Kigel, J., Konsens, I., Rosen, N., Rotem, G., Kon, A., and Fragman-Sapir, O. (2011). Relationships between flowering time and rainfall gradients across Mediterranean-desert transects. Isr. J. Ecol. Evol. 57, 91–109.
Kofler, R., Orozco-terWengel, P., De Maio, N., Pandey, R. V., Nolte, V., Futschik, A., et al. (2011a). PoPoolation: a Toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One 6:e15925. doi: 10.1371/journal.pone.0015925
Kofler, R., Pandey, R. V., and Schlotterer, C. (2011b). PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436. doi: 10.1093/bioinformatics/btr589
Kubota, S., Iwasaki, T., Hanada, K., Nagano, A. J., Fujiyama, A., Toyoda, A., et al. (2015). A genome scan for genes underlying microgeographic-scale local adaptation in a wild Arabidopsis species. PLoS Genet. 11:e1005361. doi: 10.1371/journal.pgen.1005361
Lambrix, V., Reichelt, M., Mitchell-Olds, T., Kliebenstein, D. J., and Gershenzon, J. (2001). The Arabidopsis epithiospecifier protein promotes the hydrolysis of glucosinolates to nitriles and influences Trichoplusia ni herbivory. Plant Cell 13, 2793–2807. doi: 10.1105/tpc.13.12.2793
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liancourt, P., and Tielborger, K. (2009). Competition and a short growing season lead to ecotypic differentiation at the two extremes of the ecological range. Funct. Ecol. 23, 397–404. doi: 10.1111/j.1365-2435.2008.01497.x
Lobreaux, S., and Miquel, C. (2020). Identification of Arabis alpina genomic regions associated with climatic variables along an elevation gradient through whole genome scan. Genomics 112, 729–735. doi: 10.1016/j.ygeno.2019.05.008
Metz, J., Lampei, C., Baumler, L., Bocherens, H., Dittberner, H., Henneberg, L., et al. (2020). Rapid adaptive evolution to drought in a subset of plant traits in a large-scale climate change experiment. Ecol. Lett. 23, 1643–1653. doi: 10.1111/ele.13596
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucl. Acids Res. 35:W182–W185. doi: 10.1093/nar/gkm321
Mumm, R., Burow, M., Bukovinszkine’Kiss, G., Kazantzidou, E., Wittstock, U., Dicke, M., et al. (2008). Formation of simple nitriles upon glucosinolate hydrolysis affects direct and indirect defense against the specialist herbivore, Pieris rapae. J. Chem. Ecol. 34, 1311–1321. doi: 10.1007/s10886-008-9534-z
Nakashima, K., Yamaguchi-Shinozaki, K., and Shinozaki, K. (2014). The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 5:170. doi: 10.3389/Fpls.2014.00170
Nguyen, D., D’Agostino, N., Tytgat, T. O., Sun, P., Lortzing, T., Visser, E. J., et al. (2016a). Drought and flooding have distinct effects on herbivore-induced responses and resistance in Solanum dulcamara. Plant Cell Environ. 39, 1485–1499. doi: 10.1111/pce.12708
Nguyen, D., Rieu, I., Mariani, C., and van Dam, N. M. (2016b). How plants handle multiple stresses: hormonal interactions underlying responses to abiotic stress and insect herbivory. Plant Mol. Biol. 91, 727–740. doi: 10.1007/s11103-016-0481-8
Nordberg, H., Cantor, M., Dusheyko, S., Hua, S., Poliakov, A., Shabalov, I., et al. (2013). The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucl. Acids Res. 42:D26–D32. doi: 10.1093/nar/gkt1069
Ogran, A., Conner, J., Agrawal, A. A., and Barazani, O. (2020). Evolution of phenotypic plasticity: genetic differentiation and additive genetic variation for induced plant defence in wild arugula Eruca sativa. J. Evol. Biol. 33, 237–246. doi: 10.1111/jeb.13558
Ogran, A., Faigenboim, A., and Barazani, O. (2019). Transcriptome responses to different herbivores reveal differences in defense strategies between populations of Eruca sativa. BMC Genom. 20:843. doi: 10.1186/S12864-019-6217-9
Ogran, A., Landau, N., Hanin, N., Levy, M., Gafni, Y., and Barazani, O. (2016). Intraspecific variation in defense against a generalist lepidopteran herbivore in populations of Eruca sativa (Mill.). Ecol. Evol. 6, 363–374. doi: 10.1002/ece3.1805
Ogran, A., Wasserstrom, H., Barzilai, M., Faraj, T., Dai, N., Carmi, N., et al. (2021). Water deficiency and induced defense against a generalist insect herbivore in desert and Mediterranean populations of Eruca sativa. J. Chem. Ecol. 47, 768–776. doi: 10.1007/s10886-021-01292-9
Petrů, M., Tielbörger, K., Belkin, R., Sternberg, M., and Jeltsch, F. (2006). Life history variation in an annual plant under two opposing environmental constraints along an aridity gradient. Ecography 29, 66–74.
R Core Team. (2020). R: A language And Environment for Statistical Computing. Version 4.0. 2 (Taking Off Again). Vienna: R Foundation for Statistical Computing.
Raymond, M., and Rousset, F. (1995). An exact test for population differentiation. Evolution 49, 1280–1283. doi: 10.2307/2410454
Rellstab, C., Zoller, S., Walthert, L., Lesur, I., Pluess, A. R., Graf, R. E., et al. (2016). Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol. Ecol. 25, 5907–5924. doi: 10.1111/mec.13889
Ryman, N., and Jorde, P. E. (2001). Statistical power when testing for genetic differentiation. Mol. Ecol. 10, 2361–2373. doi: 10.1046/j.0962-1083.2001.01345.x
Schlotterer, C., Tobler, R., Kofler, R., and Nolte, V. (2014). Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 749–763. doi: 10.1038/nrg3803
Schuster, J., Knill, T., Reichelt, M., Gershenzon, J., and Binder, S. (2006). BRANCHED-CHAIN AMINOTRANSFERASE4 is part of the chain elongation pathway in the biosynthesis of methionine-derived glucosinolates in Arabidopsis. Plant Cell 18, 2664–2679. doi: 10.1105/tpc.105.039339
Sreenivasulu, N., Harshavardhan, V. T., Govind, G., Seiler, C., and Kohli, A. (2012). Contrapuntal role of ABA: does it mediate stress tolerance or plant growth retardation under long-term drought stress? Gene 506, 265–273. doi: 10.1016/j.gene.2012.06.076
Supek, F., Bosnjak, M., Skunca, N., and Smuc, T. (2011). REVIGO Summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Takou, M., Wieters, B., Kopriva, S., Coupland, G., Linstadter, A., and de Meaux, J. (2019). Linking genes with ecological strategies in Arabidopsis thaliana. J. Exp. Bot. 70, 1141–1151. doi: 10.1093/jxb/ery447
Tiffin, P., and Ross-Ibarra, J. (2014). Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 29, 673–680. doi: 10.1016/j.tree.2014.10.004
Toledo-Ortiz, G., Huq, E., and Quail, P. H. (2003). The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 15, 1749–1770. doi: 10.1105/tpc.013839
Travers-Martin, N., and Muller, C. (2008). Matching plant defence syndromes with performance and preference of a specialist herbivore. Funct. Ecol. 22, 1033–1043. doi: 10.1111/j.1365-2435.2008.01487
Vasseur, F., Exposito-Alonso, M., Ayala-Garay, O. J., Wang, G., Enquist, B. J., Vile, D., et al. (2018). Adaptive diversification of growth allometry in the plant Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 115, 3416–3421. doi: 10.1073/pnas.1709141115
Volis, S. (2007). Correlated patterns of variation in phenology and seed production in populations of two annual grasses along an aridity gradient. Evol. Ecol. 21, 381–393.
Vos, I. A., Verhage, A., Watt, L. G., Vlaardingerbroek, I., Schuurink, R. C., Pieterse, C. M., et al. (2019). Abscisic acid is essential for rewiring of jasmonic acid-dependent defenses during herbivory. bioRxiv [Preprint]. doi: 10.1101/747345
Waitz, Y., Wasserstrom, H., Hanin, N., Landau, N., Faraj, T., Barzilai, M., et al. (2021). Close association between flowering time and aridity gradient for Sarcopoterium spinosum in Israel. J. Arid Environ. 188:104468.
Wang, X. P., Niu, Y. L., and Zheng, Y. (2021). Multiple functions of MYB transcription factors in abiotic stress responses. Int. J. Mol. Sci. 22:6125. doi: 10.3390/Ijms22116125
Weedall, G. D., and Conway, D. J. (2010). Detecting signatures of balancing selection to identify targets of anti-parasite immunity. Trends Parasitol. 26, 363–369. doi: 10.1016/j.pt.2010.04.002
Westberg, E., Ohali, S., Shevelevich, A., Fine, P., and Barazani, O. (2013). Environmental effects on molecular and phenotypic variation in populations of Eruca sativa across a steep climatic gradient. Ecol. Evol. 3, 2471–2484. doi: 10.1002/ece3.646
Xie, Z., Nolan, T. M., Jiang, H., and Yin, Y. (2019). AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front. Plant Sci. 10:228. doi: 10.3389/fpls.2019.00228
Yuan, X., Wang, H., Cai, J., Li, D., and Song, F. (2019). NAC transcription factors in plant immunity. Phytopathol. Res. 1:3.
Keywords: ecotypes, eco-genomics, induced defense, selection, adaptation
Citation: Bajpai PK, Harel A, Shafir S and Barazani O (2022) Whole genome sequencing reveals footprints of adaptive genetic variation in populations of Eruca sativa. Front. Ecol. Evol. 10:938981. doi: 10.3389/fevo.2022.938981
Received: 08 May 2022; Accepted: 11 July 2022;
Published: 22 August 2022.
Edited by:
Antonio Carvajal-Rodríguez, University of Vigo, SpainReviewed by:
Maria Saura, Instituto Nacional de Investigación y Tecnología Agroalimentaria (INIA), SpainIvo Chelo, University of Lisbon, Portugal
Copyright © 2022 Bajpai, Harel, Shafir and Barazani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oz Barazani, YmFyYXphbmlAYWdyaS5nb3YuaWw=