 Fan Mao
Fan Mao Xiaoyang Jin1,2†
Xiaoyang Jin1,2† Yang Zhang
Yang Zhang- 1Nansha Islands Coral Reef Ecosystem National Observation and Research Station, Ministry of Natural Resources of the People’s Republic of China (MNR), Guangzhou, China
- 2State Key Laboratory of Breeding Biotechnology and Sustainable Aquaculture, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 3South China Sea Ecological Center, Ministry of Natural Resources of the People's Republic of China (MNR), Guangzhou, China
Introduction: Coral reef ecosystems are increasingly threatened by climate change-induced thermal stress, leading to widespread bleaching events. Giant clams (Tridacna spp.) are key photosymbiotic reef inhabitants that harbor extracellular dinoflagellate symbionts (Symbiodiniaceae) and contribute to reef structure and nutrient cycling. However, the molecular mechanisms underlying their response to heat stress remain poorly understood.
Methods: A non-lethal plasma sampling technique was employeed to characterize the proteomic profile of Tridacna crocea under controlled thermal stress. iTRAQ-based quantitative proteomics profiled host plasma proteins, and integrated transcriptomic analyses across five tissues assessed tissue-specific expression and the contribution of secretory factors to host–symbiont interactions.
Results: We quantified 554 host plasma proteins that differentially expressed, with significant enrichment in immune response pathways, lectin-mediated recognition, and complement system components. Integrated transcriptomic analysis of five tissues revealed tissue-specific expression patterns and underscored the role of secretory proteins in host-symbiont interactions. Key biomarkers, including C1q domain-containing proteins and lectin family members, exhibited consistent dysregulation under stress, reflecting a shift from symbiosis maintenance to immune defense.
Discussion: These data delineate a plasma proteomic signature of bleaching in T. crocea and implicate innate immune pathways in restructuring host–symbiont dynamics under heat stress. The non-lethal plasma assay, coupled with proteogenomic readouts, offers a scalable framework for monitoring giant clam health and, by extension, reef condition under ongoing climate change.
Introduction
Coral reefs are among the most diverse marine ecosystems on Earth, providing resources and services that support millions of people worldwide (van Woesik and Kratochwill, 2022). Among the reef-associated organisms, giant clams (Tridacna spp.) are unique reef builders that, like corals, establish symbiotic relationships with dinoflagellates (Symbiodiniaceae). Unlike corals, however, their association is extracellular rather than intracellular. Giant clams play vital ecological roles within reef ecosystems: their shells provide substrata for epibionts and enhance reef structural complexity, while the mantle cavity hosts diverse symbiotic and epizoic communities (Neo et al., 2015; Van Wynsberge et al., 2017). In addition, clams release symbionts, fecal matter, and gametes into the reef environment, serving as food sources for other organisms (Neo and Todd, 2012; Zhou et al., 2019). Under extreme thermal stress, corals expel their symbionts, leading to bleaching, which is often followed by mass mortality (McClanahan et al., 2019). Similarly, giant clams bleach when elevated temperatures cause loss of Symbiodiniaceae from the outer mantle, resulting in mantle paling, reduced photosynthetic capacity, impaired physiology, and eventual mortality (Savitski et al., 2015; Sayco et al., 2023, 2024; Maboloc et al., 2025).
The earliest biological responses to environmental stress manifest at the molecular level, forming the foundation of the cellular stress response. In corals, accumulation of reactive oxygen species (ROS) induces oxidative stress and triggers a cascade of gene responses, including members of the heat shock protein (HSP) family such as HSP70 and HSP90, which act as early responders to thermal stress (Louis et al., 2017). Key transcription factors such as NF-κB (NF-κB1, NF-κB2) are up-regulated under heat stress. In addition, complement component C3-like proteins (C3-Am) are activated following physical damage, and several Ca2+-signaling-related genes also display differential expression (DeSalvo et al., 2008; Rodriguez-Lanetty et al., 2009; DeSalvo et al., 2010; Kenkel et al., 2011). Based on these findings, qPCR-based gene expression biomarkers—such as HSP16 (αB-crystallin), HSP60, HSP90, actin, and spondin 2—have been applied to monitor coral health (Kenkel et al., 2014). In giant clams, both the symbionts and the host exhibit physiological and molecular responses under heat stress, including alterations in genes related to photosynthesis and nitrogen metabolism in Symbiodiniaceae (Alves Monteiro et al., 2020), as well as host antioxidant and apoptosis-related pathways (Zhou et al., 2019). Specifically, caspase-3, SOD, and CAT have been identified as key effectors in the giant clam T. crocea under heat stress. Similar to corals, molecular chaperones such as heat shock proteins also undergo significant changes during thermal stress (Zhou et al., 2019). Our recent study further revealed that, following bleaching, clam tissues adopt metabolic shifts regulated by AMP-activated protein kinase (AMPK) signaling and the FoxO–atrogin pathway as a survival strategy (Mao et al., 2025). Nevertheless, molecular biomarker studies of thermal stress in giant clams remain limited, despite emerging evidence of altered physiological and transcriptional responses (Dubousquet et al., 2016; Mao et al., 2025; Teaniniuraitemoana et al., 2025).
As bivalves with open circulatory systems, giant clams circulate plasma as their primary extracellular fluid, integrating signals between organism and environment (Gosling, 2021). Under pathological conditions, plasma composition can change substantially, reflecting both metabolic activity and environmental stress. Plasma markers are widely used for diagnostic purposes in vertebrates (Ahmad et al., 2023), and in bivalves, plasma biomarkers have likewise been applied as indicators of health status (Joyner-Matos et al., 2009; Cotou et al., 2013). Importantly, due to the unique anatomical structure of giant clams, hemolymph plasma can be obtained non-lethally through the byssal opening, allowing for repeated sampling without damaging the animal. In this study, we employed minimally invasive plasma sampling, followed by comprehensive proteomic profiling, to identify candidate biomarkers of thermal stress and bleaching. By focusing on molecular signatures detectable in plasma, we aim to extent the current knowledge gap in giant clam biomarker research (Uchida et al., 2025), elucidate the molecular mechanisms underlying clam responses to heat stress, and establish a foundation for the conservation and management of coral reef ecosystems in a changing climate.
Materials and methods
Animals and heat stress treatment
The thermal stress protocol followed our previously published methodology (Mao et al., 2025). Briefly, visibly healthy Tridacna crocea (average wet weight ~200 g) were collected from the Xisha Islands, South China Sea, and transported to the Tropical Marine Biological Research Station in Hainan (TMBRS). Upon arrival, individuals were acclimated in outdoor flow-through aquaria (33 psu, 28–29°C, natural light) prior to experimentation. Eighty individual giant clams (N = 40/group) were involved for thermal stress experiment. Clams were randomly allocated to either a control group (maintained at 28°C) or a thermal stress group (31–32°C) (Figure 1A). The experimental design comprised a 7-day acclimatization period, a 30-day heat exposure period, and a 40-day recovery period. For the heat treatment, water temperature was adjusted gradually (± 1°C/day) at the onset of both exposure and recovery to avoid thermal shock, with mean values of 32.00 ± 0.29°C during exposure and 28.51 ± 0.31°C during recovery. Control conditions were maintained at ~28.5°C throughout. In the experimental design, giant clams were first acclimated in a large glass bath (133 L) and subsequently distributed into four smaller containers (8.6 L) to reduce temperature fluctuations. Each container was supplied with flowing seawater pumped directly from the reef in front of TMBRS, with temperature regulated by a thermostatic heater and continuous aeration provided by an air stone. All experiments were carried out in glass chambers under natural sunlight exposure. Seawater temperature was monitored every 4 h with a WMZ-200LCD digital thermometer and adjusted with thermostatic heaters to ensure the target conditions were maintained. After 30-days exposure period, severely bleached individuals from temperature treatment group and healthy individuals from control group were applied for subsequent omics analysis. All sampling and experimental procedures involving giant clams were conducted in accordance with institutional and national guidelines, and were approved by Animal Ethics Committee of the South China Sea institute of Oceanology, Chinese Academy of Science (Approval No. SCSIO-2025027).
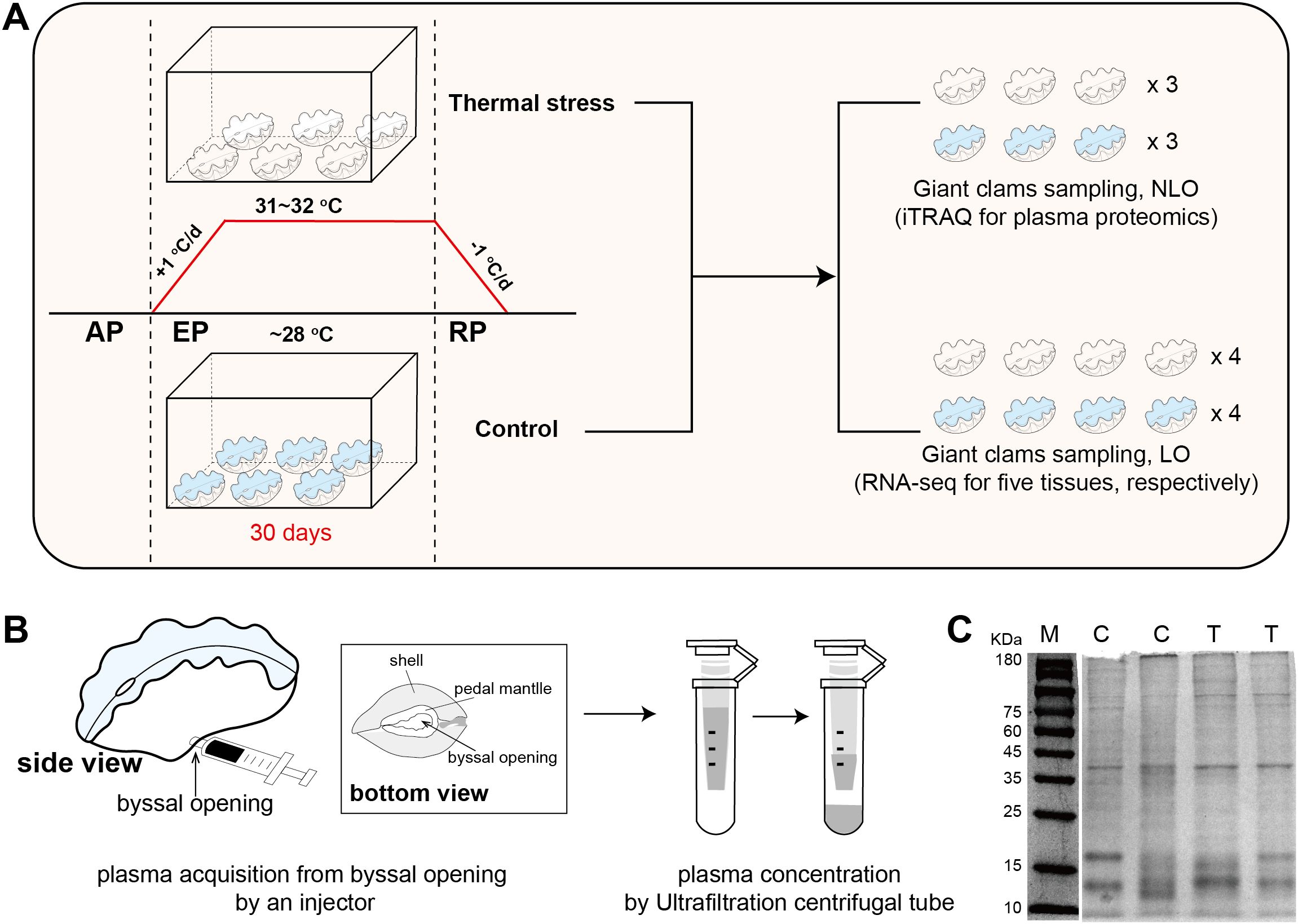
Figure 1. Experimental design of thermal stress and plasma protein sampling. (A) Workflow of the thermal stress experiment, including temperature regulation, treatment duration, and sampling schemes for proteomic and transcriptomic analyses. AP, acclimatization period; EP, exposure period; RP, recovery period; NLO, non-lethal operation; LO, lethal operation. (B) Method for non-lethal plasma sampling, with detailed anatomical locations highlighted, particularly the byssal opening. (C) SDS-PAGE analysis of plasma proteins from the control (C) and thermal stress (T) groups; M represents the molecular weight marker.
Plasma sampling and protein preparation
Plasma samples were obtained from live T. crocea individuals using a non- lethal sampling protocol. Briefly, a pre-chilled 1 mL sterile syringe was gently inserted through the byssal opening (Figure 1B) to access the extrapallial cavity, and approximately 2–3 mL of hemolymph was slowly withdrawn. This approach avoided tissue incision or structural damage, thereby minimizing physiological stress to the animal. Hemolymph was collected at 12:00 a.m. after 30 days of heat exposure. For both the thermal stress and control groups, three samples were obtained, with hemolymph from two individuals pooled in equal volumes to constitute one sample. To eliminate potential seawater contamination, the external surface of each individual and the area around the byssal opening were carefully blotted dry prior to hemolymph collection. The collected hemolymph was immediately placed on ice and processed within minutes. Cellular components were removed by centrifugation at 3,000 g for 15 min at 4°C. The resulting supernatant was then subjected to a second centrifugation at 10,000 g for 10 min under the same temperature conditions to eliminate residual cell debris and particulates. The clarified plasma fraction was concentrated using 3 kDa molecular weight cut-off ultrafiltration units (Amicon Ultra, Millipore) to obtain protein-enriched plasma samples for downstream proteomic analysis.
iTRAQ-based quantitative proteomic analysis
Protein digestion and iTRAQ labeling
For each sample, 100 μg of total protein was measured into a 1.5 mL microcentrifuge tube. The concentration of urea and SDS was adjusted to <2 M and <0.1%, respectively, using 0.5 M triethylammonium bicarbonate (TEAB) buffer. Trypsin (Promega) was added at a 1:20 (w/w) enzyme-to-protein ratio, and the mixture was vortexed and incubated at 37°C for 4 h to complete enzymatic digestion. The resulting peptides were desalted using a C18 solid-phase extraction column (Waters) and lyophilized in a vacuum centrifuge.
For labeling, lyophilized peptides were reconstituted in 0.5 M TEAB and reacted with isobaric tags from an 8-plex iTRAQ reagent kit (AB Sciex; tags 113, 114, 115 for control group; tags 117, 118, 119 for bleaching group), which had been pre-activated with isopropanol. Labeling was performed at room temperature for 2 h, to enable specific tagging of peptides from different samples.
LC–MS/MS analysis
Labeled peptides were first fractionated in the first dimension using high-pH reverse-phase chromatography on a Shimadzu LC-20AD system equipped with a Gemini C18 column (5 μm, 20 cm × 180 μm). Peptides were eluted with an acetonitrile gradient (5–80%) in pH 9.8 buffer, and 20 fractions were collected at 3.15 min intervals under UV detection at 214 nm.
The second-dimension analysis was performed using a Thermo UltiMate 3000 UHPLC system coupled to a self-packed C18 capillary column (3 μm, 75 μm × 25 cm). Peptides were separated at a flow rate of 300 nL/min using a nano-flow gradient of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B).
Mass spectrometry was carried out on a Q-Exactive HF-X instrument (Thermo Scientific) equipped with a nano-electrospray ionization (nanoESI) source, operated at 1.9 kV in data-dependent acquisition (DDA) mode. MS1 scans were acquired at a resolution of 60,000 (m/z 350–1500), and MS/MS scans were acquired at a resolution of 15,000 with higher-energy collisional dissociation (HCD). Dynamic exclusion was set to 30 s to improve identification coverage. Raw MS data were converted to Mascot generic format (MGF) files. Searches were performed against the appropriate protein database (T. crocea reference genome, CRA016517, https://ngdc.cncb.ac.cn/gsa/; Zooxanthellae reference genome, GCA_905221615.1_Stri_CCMP2592, NCBI) using Mascot (Matrix Science). Quality control was performed to ensure dataset reliability. Proteins were assembled from peptide identifications based on the parsimony principle, and filtered at the protein level using a picked protein FDR strategy with a false discovery rate threshold of ≤1% (Protein-level FDR ≤ 0.01) (Savitski et al., 2015).
Differential protein analysis and functional enrichment
Quantitative analysis of iTRAQ data was performed using IQuant (BGI, Shenzhen, China) (Wen et al., 2014), which integrates the Mascot Percolator algorithm (Brosch et al., 2009) to improve identification accuracy. Proteins were assembled according to the parsimony principle, grouping peptides into minimal non-redundant protein sets. To further control false positives at the protein level, an additional filtering step was applied at a 1% FDR threshold (Protein-level FDR ≤ 0.01) using the Picked Protein FDR strategy (Savitski et al., 2015). For comparisons with biological replicates (unpaired design), proteins were considered differentially expressed if they met both of the following criteria: average fold change (FC) > 1.2 across all pairwise comparisons, and Qvalue < 0.05 based on a two-tailed Student’s t-test.
Gene Ontology (GO) enrichment analysis of differentially expressed proteins (DEPs) was performed by comparing the DEP set to the background of all identified proteins, using a hypergeometric test to identify significantly enriched GO terms. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment was conducted using the same statistical principle, comparing observed pathway memberships in DEPs against the expected distribution in the background set.
RNA extraction, transcriptome sequencing and processing
Healthy clams from the control group and bleached clams from the thermal stress group were sampled for five tissues (siphonal mantle, inner mantle, hemocytes, ctenidium, and digestive gland). Similar to plasma collection, tissues were sampled at 12:00 a.m. after 30 days of heat exposure. Four healthy giant clams from the control group and four bleached individuals from the thermal stress group were collected for tissue sampling. Total RNA was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. RNA quality was assessed using an Agilent 2100 Bioanalyzer and RNase-free agarose gel electrophoresis. mRNA was enriched with oligo(dT) beads, fragmented, and reverse transcribed with the NEBNext Ultra RNA Library Prep Kit (NEB, USA). Libraries were end-repaired, adenylated, adapter-ligated, and sequenced on an Illumina NovaSeq 6000 platform (Gene Denovo, Guangzhou, China).
Raw reads were filtered with fastp v0.18.0 (Chen et al., 2018), and rRNA reads were removed using Bowtie2 v2.2.8 (Langmead and Salzberg, 2012). Clean reads were mapped to the T. crocea reference genome (CRA016517, https://ngdc.cncb.ac.cn/gsa/) using HISAT2 v2.2.1 (Kim et al., 2015) (“-rna-strandness RF” option and default parameters). Gene expression levels were quantified and differential expression was analyzed with DESeq2 v1.45.0 (Love et al., 2014). Genes with FDR < 0.05 and |fold change| ≥ 2 were defined as differentially expressed. KEGG pathway enrichment was performed to identify significantly enriched metabolic and signaling pathways relative to the genomic background.
Integrated transcriptomic and proteomic analysis
Integrated analysis combined RNA-seq and quantitative proteomic datasets. Protein identifiers were mapped to gene symbols based on Ensembl and UniProt annotations; unmatched entries were denoted as “–”. A nine-quadrant plot was generated with transcriptomic log2FC values on the x-axis and proteomic log2FC values on the y-axis, with the defined thresholds as boundaries to partition the plot into nine regions. Thresholds for differential expression were set at |log2FC| ≥ 1 for transcriptomic data and |log2FC| ≥ log2 (1.2) ≈ 0.263 for proteomic data. Genes were further classified by statistical significance (FDR < 0.05), and significant genes were highlighted with their gene symbols. Visualization was performed in R (RStudio) using the tidyverse, ggplot2, and ggrepel packages.
Result
Non-lethal collection of giant clam plasma proteins
Giant clams were subjected to thermal stress treatment following the protocol described in our previous study (Mao et al., 2025). At the end of the heat exposure (day 30), multiple tissues were collected for multi-tissue transcriptome sequencing (RNA-seq). Meanwhile, plasma samples were obtained for iTRAQ-based proteomic analysis (Figure 1A). Transcriptome and plasma protein samples were collected from separate individuals. Plasma samples were acquired using a non-lethal sampling technique developed in this study, designed for potential future application in non-lethal monitoring of giant clam physiological and molecular status. The key procedure involved inserting a pre-chilled sterile syringe through the byssal opening to access the mantle cavity and gently aspirating the hemolymph without causing tissue damage (Figure 1B). The collected hemolymph was centrifuged, and the supernatant was concentrated to obtain plasma protein samples.
To assess sample quality, four representative plasma protein samples (two from controls, two from bleaching group) were subjected to SDS–PAGE. Clear protein bands were revealed within the 10–180 kDa range (Figure 1C), with highly consistent banding patterns between groups, indicating high reproducibility and reliability of sample preparation. Notably, distinct dominant bands were observed at approximately 40 kDa, 17 kDa, and 13 kDa, which likely correspond to major storage proteins in giant clam plasma.
Quantitative plasma proteomics reveals host-specific signatures of thermal stress response
To investigate thermal stress responses, plasma protein identification was performed against both the Tridacna crocea and symbiotic zooxanthellae protein databases (Table 1). In the T. crocea database, a total of 1,837 host-derived proteins were identified, corresponding to 21,970 unique spectra and 8,422 unique peptides, with an average protein sequence coverage of 28.7%. In contrast, only 11 proteins were matched to the zooxanthellae database, indicating that the host plasma proteome predominantly reflects host-derived protein expression. Despite the large number of spectra acquired, many remained unmatched likely due to incomplete reference coverage for both the Symbiodiniaceae symbionts, sequence divergence among clades, contributions from associated microbiota and environmental proteins, unexpected post−translational modifications, co−fragmentation, and stringent FDR filtering. For the host proteome, the peptide length distribution (Figure 2A) showed that most identified peptides ranged from 7 to 16 amino acids, accounting for the majority of the dataset. The protein coverage distribution (Figure 2B) revealed that a large proportion of proteins exhibited coverage within the 0–20% range.
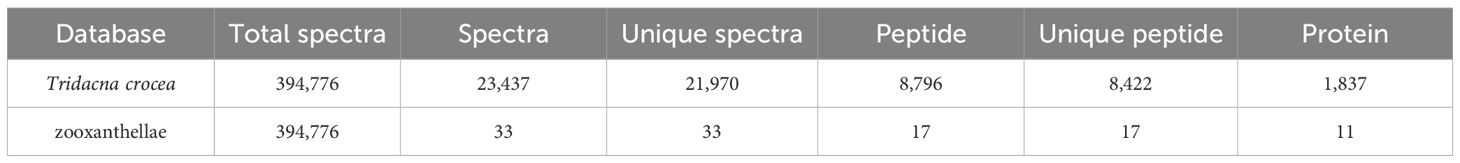
Table 1. Plasma proteins identified with matching entries in the Tridacna crocea and zooxanthellae databases.
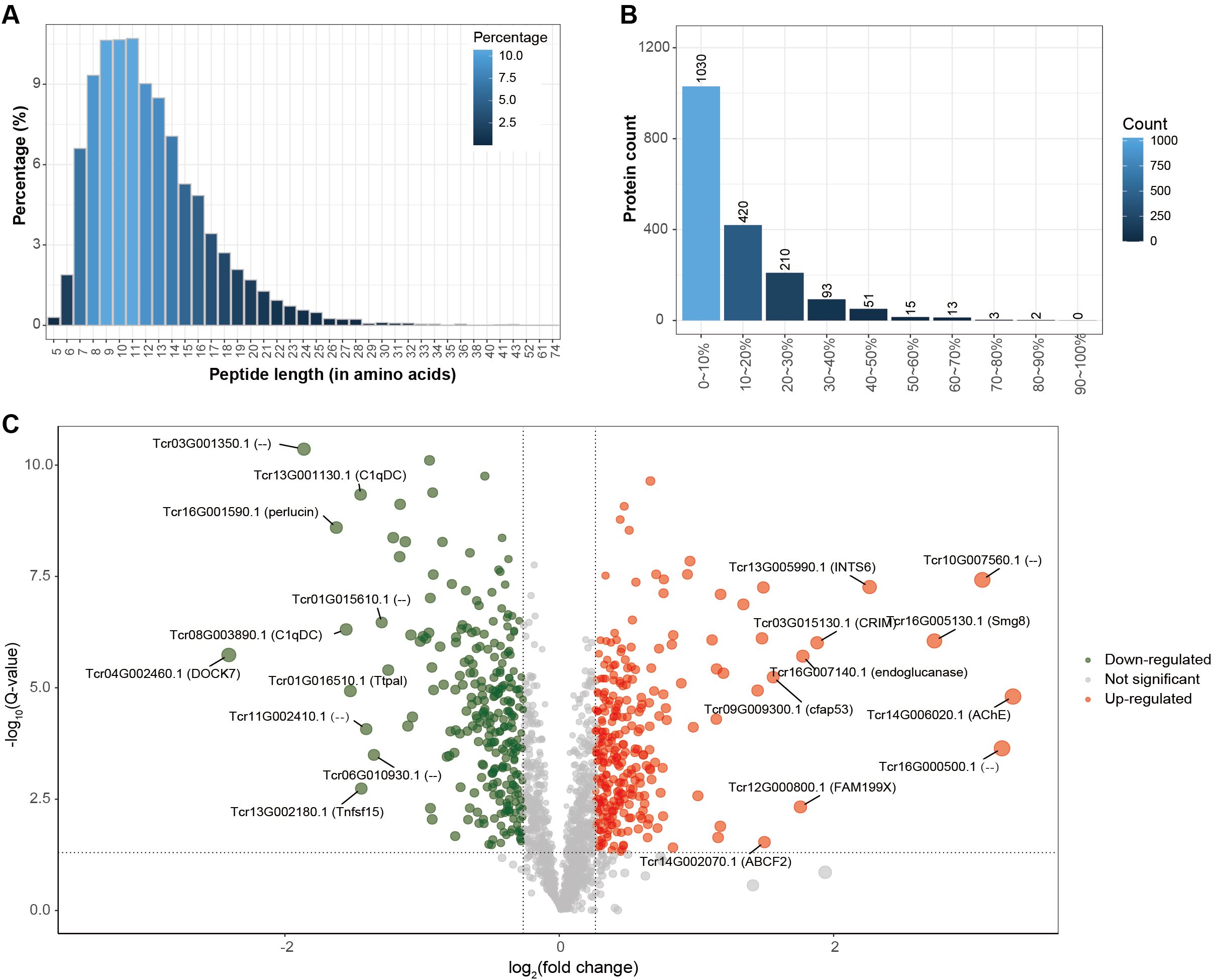
Figure 2. Proteomic identification of host proteins. (A) Peptide length distribution, showing the proportion of peptides of different lengths (X-axis: peptide length; Y-axis: percentage of peptides). (B) Protein coverage distribution, where the X-axis represents protein coverage ranges and the Y-axis indicates the corresponding number of proteins. (C) Volcano plot of differentially expressed proteins, displaying log2 fold-change (X-axis) versus –log10 Q-value (Y-axis). Differential expression was defined by thresholds of Q-value < 0.05 and fold-change > 1.2. Ten significantly upregulated or downregulated proteins in bleached clams compared with controls are highlighted with gene IDs and gene symbols or names; proteins without annotation are labeled as “–”.
Further analysis of the host-derived plasma proteome revealed that 554 proteins were significantly differentially expressed under thermal stress (fold change |FC| ≥ 1.2 and Q-value < 0.05), representing 30.2% of the total identified proteins (Table 2). The differentially expressed proteins (DEPs) exhibited distinct functional patterns (Figure 2C). Among the 286 significantly down-regulated proteins in bleached clams compared with controls notable representatives included SMG8 (protein SMG8), AChE (acetylcholinesterase), ABCF2 (ATP-binding cassette sub-family F member 2-like), CFAP53 (cilia- and flagella-associated protein 53-like), and endoglucanase. In contrast, 268 up-regulated proteins in bleached clams compared with controls were mainly enriched in immune response–related pathways, with key examples including DOCK7 (dedicator of cytokinesis protein), perlucin, and C1qDC (C1q domain–containing protein).
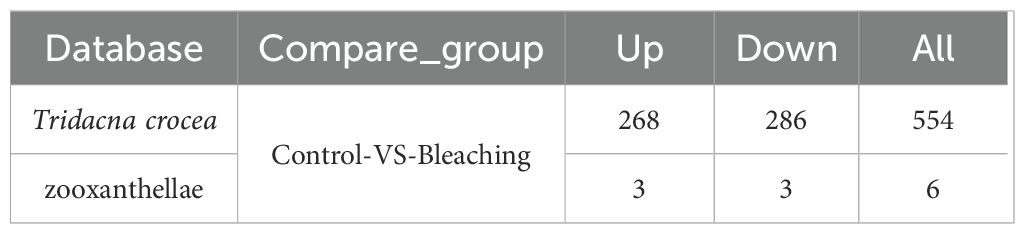
Table 2. Differentially expressed plasma proteins identified in the Tridacna crocea and zooxanthellae databases.
Notably, only six differentially expressed proteins were derived from zooxanthellae, including glyceraldehyde-3-phosphate dehydrogenase, probable serine/threonine-protein kinase, HSP70, probable polyketide synthase, and ubiquitin-40S ribosomal protein, with three upregulated and three downregulated.
Database used for protein identification; Total spectra, the total number of MS/MS spectra acquired; Spectra, the number of spectra matched to peptide sequences; Unique spectra, the number of distinct spectra matched; Peptide, the total number of peptide sequences identified; Unique peptide, the number of non-redundant peptides identified; and Protein, the total number of proteins identified.
The numbers indicate up/down-regulated genes in bleached clams relative to controls.
Plasma differential protein functional enrichment reveals key pathways
To further explore the biological functions of the differentially expressed proteins (DEPs), we performed KEGG pathway enrichment analysis on the 554 DEPs. As shown in Figure 3A, 153 proteins (27.6% of all DEPs) were significantly enriched in major KEGG categories (p < 0.05, FDR-adjusted), with immune system-related pathways being particularly prominent, encompassing 68 proteins (44.4% of the enriched proteins, representing 30.96% of the total proteins annotated to these pathways).
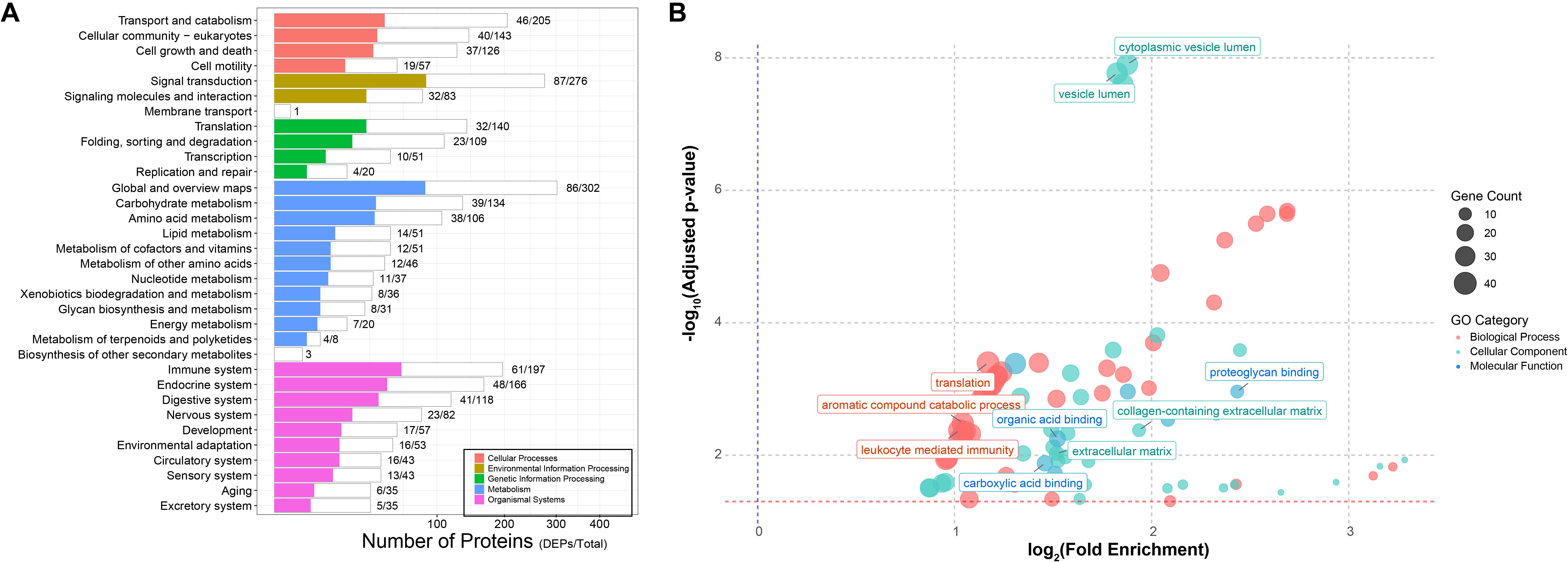
Figure 3. Functional enrichment of host DEPs. (A) KEGG pathway enrichment. White bars represent the total number of identified proteins mapped to each pathway, while colored bars indicate the number of differentially expressed proteins enriched in the corresponding pathways. (B) GO enrichment analysis. Different colors denote the three major GO categories (red, biological process; green, cellular components; blue, molecular function), with bubble size representing the number of enriched proteins. Highly enriched pathways are highlighted in the plot.
Given that KEGG coverage was less than 50%, we further conducted Gene Ontology (GO) annotation analysis. A total of 313 DEPs were successfully mapped to 82 GO terms, including 39 biological processes (BP), 36 cellular components (CC), and 7 molecular functions (MF) (Supplementary Table 1, Figure 3B). In the BP category, protein translation (GO: 0006412), aromatic compound catabolic process (GO: 0009439), and leukocyte-mediated immune response (GO: 0002443) were the most significantly enriched, containing 40, 37, and 37 proteins, respectively. Within the CC category, proteins were predominantly associated with cytoplasmic vesicle lumen (GO: 0060205) and vesicle lumen (GO: 0031983), each including 34 proteins, indicating that many identified proteins are vesicle-related, although their precise origin (exosomal versus intracellular release due to cell lysis) requires further verification. Notably, extracellular matrix (ECM)-related terms, such as extracellular matrix (GO: 0031012) and collagen-containing extracellular matrix (GO: 0062023), were also significantly enriched. In the MF category, structural molecule activity (GO: 0005198) was the most enriched term. Other notable functions included proteoglycan binding (GO: 0043398), carboxylic acid binding (GO: 0031406), and organic acid binding (GO: 0043177). Collectively, these functional enrichment analyses indicate that bleaching induces significant alterations in the clam plasma proteome.
Tissue origin and secretory features of plasma proteins
Given the open circulatory system of giant clams, plasma proteins are likely derived from multiple tissues. To systematically investigate the tissue origin and functional relevance of plasma proteins under thermal stress, we integrated plasma proteome data with transcriptomes from five key tissues (Figures 4A-E). Specifically, in the siphonal mantle, 3 proteins were upregulated and 9 downregulated in sync with plasma; in the inner mantle, 7 were upregulated and 8 downregulated; hemocytes showed 5 upregulated and 6 downregulated proteins; gills exhibited 5 upregulated and 7 downregulated proteins; and the digestive gland displayed 9 upregulated and 7 downregulated proteins. Several proteins exhibited consistent differential expression across multiple tissues (Figure 4F). For example, Zwei Ig domain protein ZIG-3 (ZIG3) and proteolysis-inducing factor (PIF) were highly expressed in the digestive gland, gills, and inner mantle, while transglutaminase 1 (TGM1) was mainly expressed in the gills, inner mantle, and siphonal mantle. Complement C1q-like 4 (C1QL4) was significantly downregulated in the gills, inner mantle, and siphonal mantle. C-type lectin domain family 4 member M (CL4M) showed high expression in the digestive gland, gills, and hemocytes, and aminoacylase 1 (ACY1) displayed elevated expression in the gills, hemocytes, and inner mantle. Xylanase promoter-binding protein 1 (XPP1) was enriched in the digestive gland, hemocytes, inner mantle, and siphonal mantle, whereas major facilitator superfamily domain-containing 9 (MFSD9) was highly expressed in the gills, hemocytes, inner mantle, and siphonal mantle.
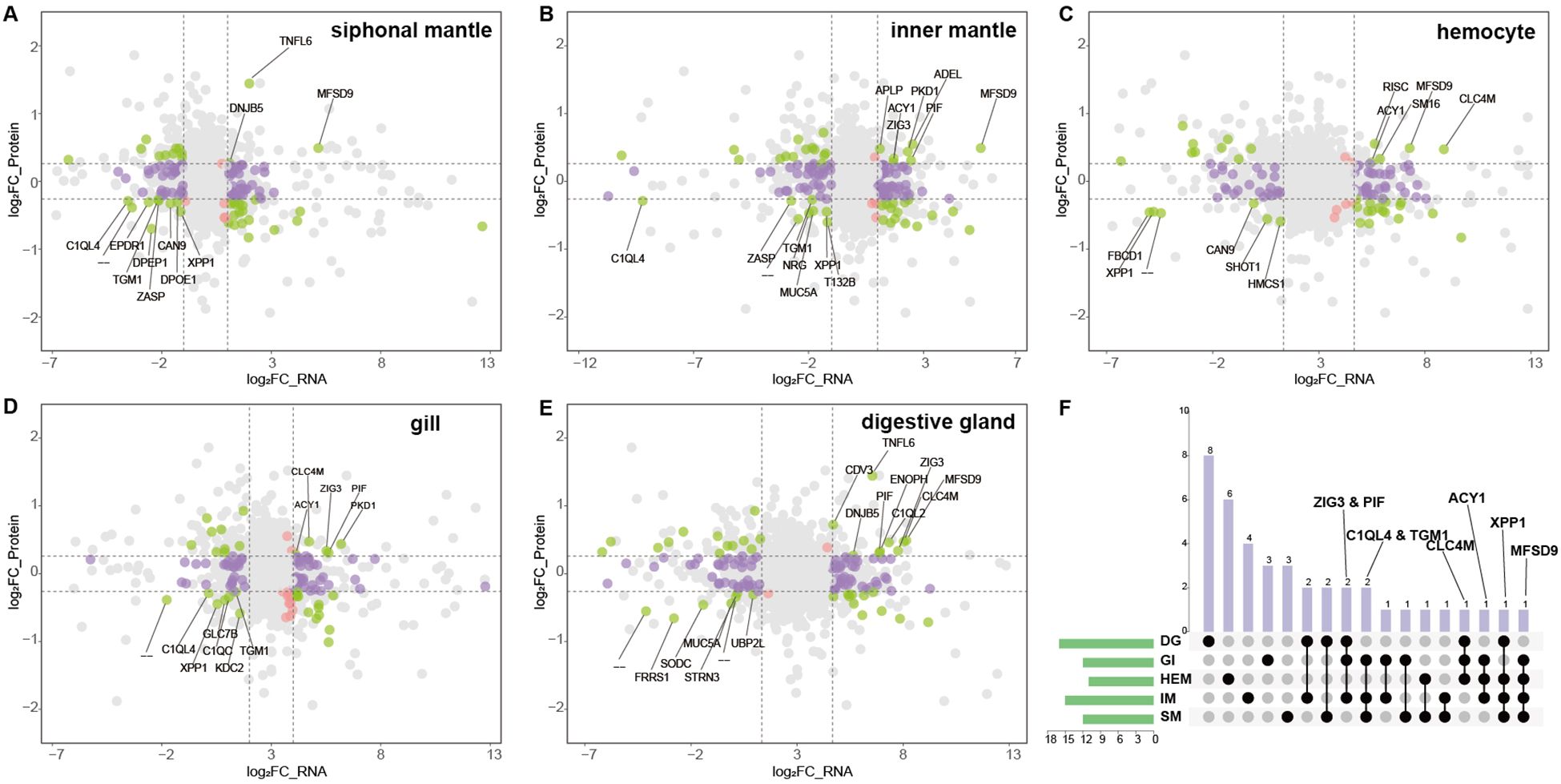
Figure 4. Integrated transcriptomic and proteomic analysis across tissues. (A–E) Nine-quadrant plots of transcriptome–proteome correlation in five tissues. The X-axis represents log2 fold-change in transcript expression, and the Y-axis represents log2 fold-change in protein expression. Colored points indicate proteins identified in the proteome. Proteins differentially expressed in both the transcriptome and proteome are shown in green; those differentially expressed only in the transcriptome in purple; and those differentially expressed only in the proteome in red. Genes exhibiting concordant significant up- or down-regulation in both datasets (quadrants 3 and 7) are labeled with their gene symbols. All other points not meeting these criteria are shown in gray. (F) Overlap between upregulated transcripts and significantly upregulated proteins across the five tissues. Rows represent tissues and columns represent their pairwise intersections. Black solid circles indicate the presence of overlap, whereas light gray circles indicate no overlap. Tissue abbreviations: DG, digestive gland; GI, gill; HEM, hemocytes; IM, inner mantle; SM, siphonal mantle.
To exclude potential contamination from intracellular proteins released by lysed cells, secretory proteins were further analyzed using SignalP 5.0 for signal peptide prediction (Supplementary Table 2). Among the DEPs, a total of 164 classical secretory proteins were identified, of which 129 were annotated (73 upregulated, 56 downregulated). The remaining proteins are predominantly intracellular, which may reflect physiological release into the circulation under bleaching−induced stress or, to a lesser extent, hemocyte lysis during sample collection and preparation. Functional annotation revealed three major categories of these secretory proteins: complement system proteins (15 C1q domain-containing proteins, 11 upregulated, 4 downregulated), pattern recognition receptors (14 lectin family proteins, 9 upregulated, 5 downregulated), and biomineralization-related proteins (3, including PIF, Galaxin, and Perlucin). Notably, expression changes of lectins, C1q, and PIF closely correlated with the bleaching process, indicating that these characteristic secretory proteins represent candidate molecular markers of host thermal stress response in giant clams, although further validation in natural reef populations is warranted.
Comparative analysis of bleaching-responsive proteins conserved between corals and Tridacna
Based on the list of coral heat-stress response genes (Louis et al., 2017), we characterized homologous proteins in Tridacna plasma based on semantic protein annotation to identify potential common biomarkers of reef organisms (Figure 5). Most of the coral heat-stress response genes were detected in giant clam plasma, including GST, SOD, calmodulin, lectins, and several HSPs, while others such as GAPDH, cytochrome P450, and catalase were not identified. All identified immune-related proteins belonged to the lectin family, among which 14 were significantly upregulated (p < 0.05) and 8 were downregulated (p < 0.05). Heat stress–related proteins were mainly represented by three heat shock protein family members, HSP90, HSP70, and HSP75. Notably, HSP90 exhibited a significantly downregulated expression pattern, whereas both HSP70 and HSP75 were upregulated. Two key antioxidant enzymes, glutathione S-transferase (GST) and superoxide dismutase (SOD), were differentially expressed. Among them, only one GST isoform (Tcr06G005840.1) was significantly upregulated (fold change = 1.40), while all other isoforms showed a downregulation trend. In addition, calmodulin, a key regulator of calcium signaling, also showed significant differential expression following bleaching. Collectively, these findings support the conservation of stress-response mechanisms between Tridacna and corals.
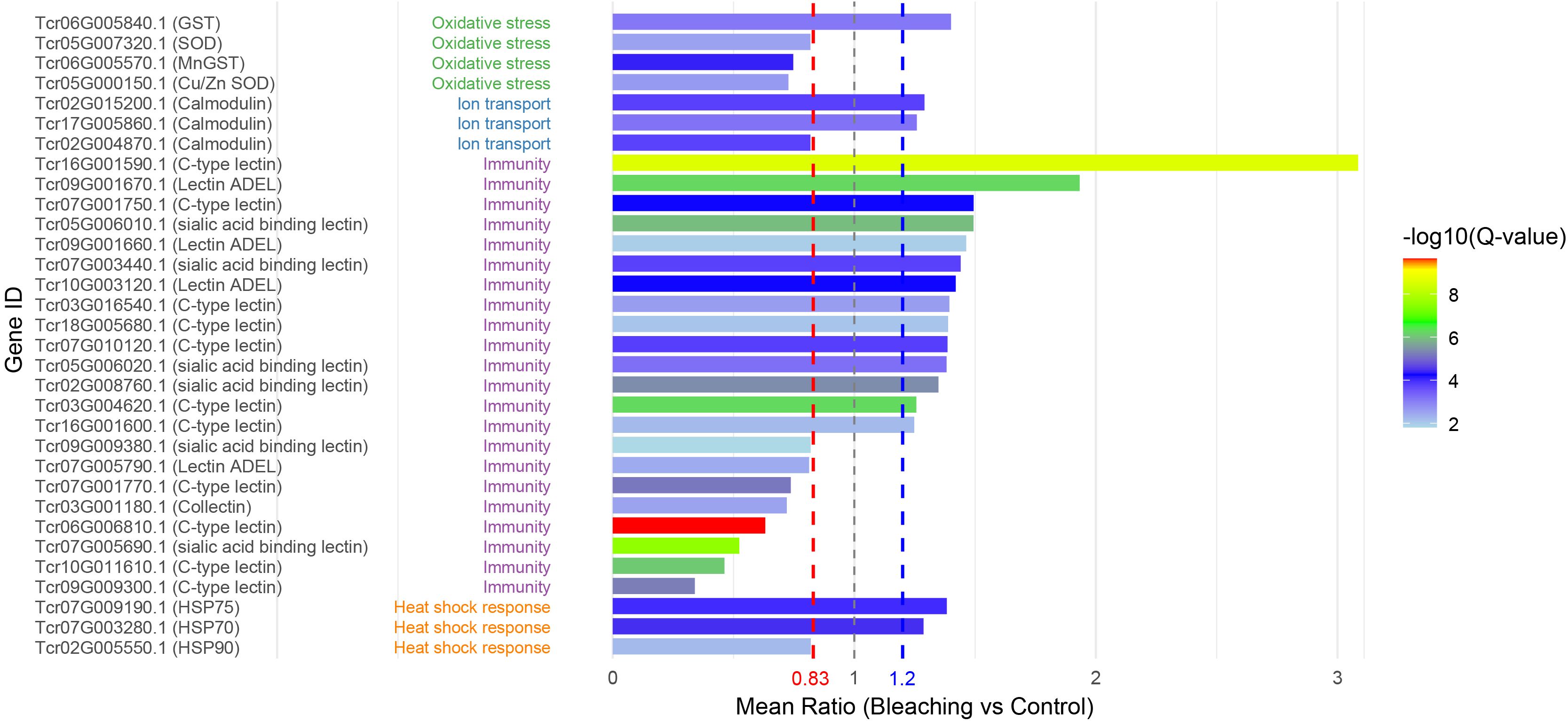
Figure 5. Homologous proteins in Tridacna plasma responsive to thermal stress. Gene IDs are derived from annotations of the T. crocea genome, with corresponding gene symbols or names shown after each ID. Proteins are functionally classified into four major categories: oxidative stress, ion transport, immunity, and heat shock response. Their expression changes in plasma under thermal stress are illustrated in bar plots, with red and blue dashed lines indicating the thresholds for differential expression.
Discussion
Against the backdrop of accelerating global climate change, bleaching in reef-associated organisms has emerged as one of the most pressing challenges for marine ecosystems. As a key functional group within coral reef, bleaching of giant clams reflects a direct stress response and indicates potential community-level imbalance. In this study, we investigated molecular markers associated with the disruption of the Tridacna–Symbiodiniaceae symbiosis under heat stress in Tridacna crocea, the boring giant clam. By combining a novel non-lethal plasma sampling strategy with high-resolution proteomic profiling, we provide the first proteomic characterization of giant clam plasma before and after bleaching. Given that the type and composition of Symbiodiniaceae harbored by the host can strongly influence thermal resilience—with some clades exhibiting greater tolerance to elevated temperatures than others (Ladner et al., 2012; Barshis et al., 2014)—the proteomic signatures observed here likely reflect both host responses and symbiont community dynamics. Building on this context, our results show that the bleaching-associated stress response in Tridacna involves not only immune activation but also modulation of symbiotic interactions, with characteristic molecular signatures enriched in lectin family proteins and complement component C1q.
Among these, changes in the lectin family were particularly prominent in the giant clam heat-stress response. Lectin-mediated glycan–lectin signaling pathways are a hallmark of the innate immune microbe-associated molecular patterns–pattern recognition receptors (MAMP–PRR) mechanism (Stegmann and Lepenies, 2024) and play a central role in cnidarian–dinoflagellate symbiosis recognition (Davy et al., 2012; Maruyama et al., 2022; Parkinson et al., 2018). In higher animals, host lectins typically bind to glycans on invading organisms to trigger immune responses, whereas in cnidarians, glycoconjugates secreted by Symbiodiniaceae are specifically recognized by host lectins (Jimbo et al., 2000, 2005; Kvennefors et al., 2008; Vidal-Dupiol et al., 2009; Jimbo et al., 2010; Kvennefors et al., 2010), maintaining a stable symbiosis. Lectins display remarkable diversity across taxa—for instance, 67 C-type lectins have been predicted in the genome of Nematostella vectensis (Wood-Charlson and Weis, 2009), reflecting the structural and functional complexity of lectin-mediated signaling in cnidarian–dinoflagellate interactions. Bivalves, in contrast, harbor an even broader lectin repertoire (Regan et al., 2021). Here, we identified 22 distinct lectins in giant clam plasma, with some upregulated under heat stress (potentially linked to immune defense) and others downregulated. The latter pattern is consistent with the reduced expression of PdC lectins observed in cnidarians co-localized with Symbiodiniaceae under elevated temperatures (Vidal-Dupiol et al., 2009), suggesting that heat stress may impair the “symbiosis-maintaining” lectin expression profile. Such molecular changes hold promise as potential biomarkers for heat stress and bleaching in giant clams.
Complement component C1q also exhibited characteristic expression changes. Traditionally, C1q is considered a pattern-recognition molecule that detects pathogen-associated molecular patterns (PAMPs) (Bredt et al., 1977; Santoro et al., 1980) and damage-associated molecular patterns (DAMPs) (Bohlson et al., 2014), thereby activating the complement cascade—a mechanism well established in mammals and bivalves such as oysters and mussels (Kojouharova et al., 2010; Gerdol et al., 2011; Zhang et al., 2015). As a bivalve lineage with a long-standing symbiosis with Symbiodiniaceae, giant clams must finely tune their immune recognition of “self”, symbionts, and pathogens. Recent study has shown that C1q in giant clams not only retains its ancestral antimicrobial role but also recognizes multiple symbiosis-related surface signals from Symbiodiniaceae (Yi et al., 2025). Here, we observed a significant downregulation of this C1q protein under heat stress, consistent with the downregulation of pattern-recognition molecules reported in corals and other symbiotic organisms under environmental stress (Vidal-Dupiol et al., 2009). This finding suggests that C1q suppression may represent an early molecular warning signal of symbiosis breakdown. Notably, several lectins and C1q proteins were up-regulated in the plasma of bleached clams. While these proteins have been discussed in the context of symbiosis maintenance, their classical roles in innate immune defense cannot be overlooked (Sun et al., 2023; Jeyachandran et al., 2024). This suggests that bleaching and heat stress not only perturb symbiotic interactions but also trigger broader changes in host immune function.
Additionally, heat shock proteins (HSPs) and antioxidant enzymes were markedly differentially expressed in plasma, reinforcing the notion of a shared molecular response between giant clams and corals under thermal stress (Louis et al., 2017; Howells et al., 2021). Overall, our study provides a comprehensive view of the multilayered molecular responses of Tridacna during heat stress and bleaching, including pattern-recognition proteins (lectins and C1q), stress-defense proteins (HSPs), regulators of antioxidant capacity, and material transport. Importantly, the detection of these molecules directly in plasma underscores their potential utility as candidate biomarkers for liquid biopsy–based monitoring of heat stress and symbiotic state. More broadly, this molecular marker suite offers insights that may inform future monitoring of reef ecosystem health under climate change.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal studies were approved by South China Sea Institute of Oceanology, Chinese Academy of Sciense. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
FM: Formal Analysis, Funding acquisition, Methodology, Validation, Writing – original draft, Writing – review & editing. XJ: Formal Analysis, Visualization, Writing – original draft. CC: Formal Analysis, Writing – original draft. WY: Formal Analysis, Resources, Writing – original draft. YZ: Formal Analysis, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Open Fund of Nansha Islands Coral Reef Ecosystem National Observation and Research Station (NO. NSICR23204), the National Key R&D Program of China (Grant number 2022-20), National Science Foundation of China (42494882, 42449303, U22A20533), the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001), and the Science and Technology Program of Guangzhou, China (2024A04J6278).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2025.1695021/full#supplementary-material
References
Ahmad A., Imran M., and Ahsan H. (2023). Biomarkers as biomedical bioindicators: approaches and techniques for the detection, analysis, and validation of novel Biomarkers of diseases. Pharmaceutics 15, 1630. doi: 10.3390/pharmaceutics15061630
Alves Monteiro H. J., Brahmi C., Mayfield A. B., Vidal-Dupiol J., Lapeyre B., and Le Luyer J. (2020). Molecular mechanisms of acclimation to long-term elevated temperature exposure in marine symbioses. Global Change Biol. 26, 1271–1284. doi: 10.1111/gcb.14907
Barshis D. J., Ladner J. T., Oliver T. A., and Palumbi S. R. (2014). Lineage-specific transcriptional profiles of Symbiodinium spp. unaltered by heat stress in a coral host. Mol. Biol. Evol. 31, 1343–1352. doi: 10.1093/molbev/msu107
Bohlson S. S., O’Conner S. D., Hulsebus H. J., Ho M.-M., and Fraser D. A. (2014). Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front. Immunol. 5, 402. doi: 10.3389/fimmu.2014.00402
Bredt W., Wellek B., Brunner H., and Loos M. (1977). Interactions between mycoplasma pneumoniae and the first components of complement. Infection Immun. 15, 7–12. doi: 10.1128/iai.15.1.7-12.1977
Brosch M., Yu L., Hubbard T., and Choudhary J. (2009). Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res. 8, 3176–3181. doi: 10.1021/pr800982s
Chen S., Zhou Y., Chen Y., and Gu J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cotou E., Tsangaris C., and Henry M. (2013). Comparative study of biochemical and immunological biomarkers in three marine bivalves exposed at a polluted site. Environ. Sci. pollut. Res. 20, 1812–1822. doi: 10.1007/s11356-012-1150-3
Davy S. K., Allemand D., and Weis V. M. (2012). Cell biology of cnidarian-dinoflagellate symbiosis. Microbiol. Mol. Biol. Rev. 76, 229–261. doi: 10.1128/MMBR.05014-11
DeSalvo M. K., Sunagawa S., Voolstra C. R., and Medina M. (2010). Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Mar. Ecol. Prog. Ser. 402, 97–113. doi: 10.3354/meps08372
DeSalvo M. K., Voolstra C. R., Sunagawa S., Schwarz J. A., Stillman J. H., Coffroth M. A., et al. (2008). Differential gene expression during thermal stress and bleaching in the Caribbean coral Montastraea faveolata. Mol. Ecol. 17, 3952–3971. doi: 10.1111/j.1365-294X.2008.03879.x
Dubousquet V., Gros E., Berteaux-Lecellier V., Viguier B., Raharivelomanana P., Bertrand C., et al. (2016). Changes in fatty acid composition in the giant clam Tridacna maxima in response to thermal stress. Biol. Open 5, 1400–1407. doi: 10.1242/bio.017921
Gerdol M., Manfrin C., De Moro G., Figueras A., Novoa B., Venier P., et al. (2011). The C1q domain containing proteins of the Mediterranean mussel Mytilus galloprovincialis: a widespread and diverse family of immune-related molecules. Dev. Comp. Immunol. 35, 635–643. doi: 10.1016/j.dci.2011.01.018
Gosling E. (2021). “Physiology of the circulatory, respiratory and excretory systems,” in Marine Mussels: Ecology, Physiology, Genetics and Culture. (Chichester, UK: John Wiley & Sons Ltd), 357–413. doi: 10.1002/9781119293927.ch7
Howells E. J., Abrego D., Liew Y. J., Burt J. A., Meyer E., and Aranda M. (2021). Enhancing the heat tolerance of reef-building corals to future warming. Sci. Adv. 7, eabg6070. doi: 10.1126/sciadv.abg6070
Jeyachandran S., Radhakrishnan A., and Ragavendran C. (2024). Harnessing the power of mollusc lectins as immuno-protective biomolecules. Mol. Biol. Rep. 51, 182. doi: 10.1007/s11033-023-09018-8
Jimbo M., Koike K., Sakai R., Muramoto K., and Kamiya H. (2005). Cloning and characterization of a lectin from the octocoral Sinularia lochmodes. Biochem. Biophys. Res. Commun. 330, 157–162. doi: 10.1016/j.bbrc.2005.02.137
Jimbo M., Yamashita H., Koike K., Sakai R., and Kamiya H. (2010). Effects of lectin in the scleractinian coral Ctenactis eChinata on symbiotic zooxanthellae. Fisheries Sci. 76, 355–363. doi: 10.1007/s12562-009-0204-z
Jimbo M., Yanohara T., Koike K., Koike K., Sakai R., Muramoto K., et al. (2000). The D-galactose-binding lectin of the octocoral Sinularia lochmodes: characterization and possible relationship to the symbiotic dinoflagellates. Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 125, 227–236. doi: 10.1016/s0305-0491(99)00173-x
Joyner-Matos J., Andrzejewski J., Briggs L., Baker S. M., Downs C. A., and Julian D. (2009). Assessment of cellular and functional biomarkers in bivalves exposed to ecologically relevant abiotic stressors. J. Aquat. Anim. Health 21, 104–116. doi: 10.1577/H08-066.1
Kenkel C. D., Aglyamova G., Alamaru A., Bhagooli R., Capper R., Cunning R., et al. (2011). Development of gene expression markers of acute heat-light stress in reef-building corals of the genus Porites. PloS One 6, e26914. doi: 10.1371/journal.pone.0026914
Kenkel C. D., Sheridan C., Leal M. C., Bhagooli R., Castillo K. D., Kurata N., et al. (2014). Diagnostic gene expression biomarkers of coral thermal stress. Mol. Ecol. Resour. 14, 667–678. doi: 10.1111/1755-0998.12218
Kim D., Langmead B., and Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kojouharova M., Reid K., and Gadjeva M. (2010). New insights into the molecular mechanisms of classical complement activation. Mol. Immunol. 47, 2154–2160. doi: 10.1016/j.molimm.2010.05.011
Kvennefors E. C. E., Leggat W., Hoegh-Guldberg O., Degnan B. M., and Barnes A. C. (2008). An ancient and variable mannose-binding lectin from the coral Acropora millepora binds both pathogens and symbionts. Dev. Comp. Immunol. 32, 1582–1592. doi: 10.1016/j.dci.2008.05.010
Kvennefors E. C. E., Leggat W., Kerr C. C., Ainsworth T. D., Hoegh-Guldberg O., and Barnes A. C. (2010). Analysis of evolutionarily conserved innate immune components in coral links immunity and symbiosis. Dev. Comp. Immunol. 34, 1219–1229. doi: 10.1016/j.dci.2010.06.016
Ladner J. T., Barshis D. J., and Palumbi S. R. (2012). Protein evolution in two co-occurring types of Symbiodinium: an exploration into the genetic basis of thermal tolerance in Symbiodinium clade D. BMC Evol. Biol. 12, 217. doi: 10.1186/1471-2148-12-217
Langmead B. and Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Louis Y. D., Bhagooli R., Kenkel C. D., Baker A. C., and Dyall S. D. (2017). Gene expression biomarkers of heat stress in scleractinian corals: promises and limitations. Comp. Biochem. Physiol. Part C: Toxicol. Pharmacol. 191, 63–77. doi: 10.1016/j.cbpc.2016.08.007
Love M. I., Huber W., and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Maboloc E. A., Cabaitan P. C., and Mingoa-Licuanan S. S. (2025). Bleaching in the giant clams after typhoon Megi. Mar. Biodivers. 55, 64. doi: 10.1007/S12526-025-01550-Z
Mao F., Xiao S., Dang X., Cui G., Gaitán-Espitia J. D., Thiyagarajan V., et al. (2025). Metabolic shifts and muscle remodeling as pro-survival and energy compensation strategies in photosymbiotic giant clams after bleaching. Environ. Sci. Technol. 59, 10239–10252. doi: 10.1021/acs.est.5c00474
Maruyama S., Mandelare-Ruiz P. E., McCauley M., Peng W., Cho B. G., Wang J., et al. (2022). Heat stress of algal partner hinders colonization success and alters the algal cell surface glycome in a cnidarian-algal symbiosis. Microbiol. Spectr. 10, e0156722. doi: 10.1128/spectrum.01567-22
McClanahan T. R., Darling E. S., Maina J. M., Muthiga N. A., Jupiter S. D., Arthur R., et al. (2019). Temperature patterns and mechanisms influencing coral bleaching during the 2016 El Niño. Nat. Climate Change 9, 845–851. doi: 10.1038/s41558-019-0576-8
Neo M. L., Eckman W., Vicentuan K., Teo S. L. M., and Todd P. A. (2015). The ecological significance of giant clams in coral reef ecosystems. Biol. Conserv. 181, 111–123. doi: 10.1016/j.biocon.2014.11.004
Neo M. L. and Todd P. A. (2012). Population density and genetic structure of the giant clams Tridacna crocea and T. squamosa on Singapore’s reefs. Aquat. Biol. 14, 265–275. doi: 10.3354/ab00400
Parkinson J. E., Tivey T. R., Mandelare P. E., Adpressa D. A., Loesgen S., and Weis V. M. (2018). Subtle differences in symbiont cell surface glycan profiles do not explain species-specific colonization rates in a model cnidarian-algal symbiosis. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00842
Regan T., Stevens L., Penaloza C., Houston R. D., Robledo D., and Bean T. P. (2021). Ancestral physical stress and later immune gene family expansions shaped bivalve mollusc evolution. Genome Biol. Evol. 13, evab177. doi: 10.1093/gbe/evab177
Rodriguez-Lanetty M., Harii S., and Hoegh-Guldberg O. V. E. (2009). Early molecular responses of coral larvae to hyperthermal stress. Mol. Ecol. 18, 5101–5114. doi: 10.1111/j.1365-294X.2009.04419.x
Santoro F., Ouaissi M. A., Pestel J., and Capron A. (1980). Interaction between Schistosoma mansoni and the complement system: binding of C1q to schistosomula. J. Immunol. 124, 2886–2891. doi: 10.4049/jimmunol.124.6.2886
Savitski M. M., Wilhelm M., Hahne H., Kuster B., and Bantscheff M. (2015). A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell Proteomics 14, 2394–2404. doi: 10.1074/mcp.M114.046995
Sayco S. L. G., Alabort Pomares A., Cabaitan P. C., and Kurihara H. (2024). Reproductive consequences of thermal stress-induced bleaching in the giant clam Tridacna crocea. Mar. Environ. Res. 193, 106280. doi: 10.1016/j.marenvres.2023.106280
Sayco S. L. G., Cabaitan P. C., and Kurihara H. (2023). Bleaching reduces reproduction in the giant clam Tridacna gigas. Mar. Ecol. Prog. Ser. 706, 47–56. doi: 10.3354/meps14251
Stegmann F. and Lepenies B. (2024). Myeloid C-type lectin receptors in host–pathogen interactions and glycan-based targeting. Curr. Opin. Chem. Biol. 82, 102521. doi: 10.1016/j.cbpa.2024.102521
Sun J., Wang L., and Song L. (2023). The primitive complement system in molluscs. Dev. Comp. Immunol. 139, 104565. doi: 10.1016/j.dci.2022.104565
Teaniniuraitemoana V., Monaco C. J., Celariès M., Jauffrais T., and Van Wynsberge S. (2025). Light intensity modulates the effect of thermal stress on giant clams and their symbiotic zooxanthellae. Coral Reefs. 44, 1449–1465. doi: 10.1007/s00338-025-02708-8
Uchida T., Li Y., Yamashita H., Shimada G., and Shinzato C. (2025). Microbiome of the boring giant clam provides insights into holobiont resilience under coral reef environmental stress. Environ. Microbiol. 27, e70161. doi: 10.1111/1462-2920.70161
van Woesik R. and Kratochwill C. (2022). A global coral-bleaching database 1980–2020. Sci. Data 9, 20. doi: 10.1038/s41597-022-01121-y
Van Wynsberge S., Andréfouët S., Gaertner-Mazouni N., Wabnitz C. C. C., Menoud M., Le Moullac G., et al. (2017). Growth, survival and reproduction of the giant clam Tridacna maxima (Röding 1798, Bivalvia) in two contrasting lagoons in French Polynesia. PLoS One 12, e0170565. doi: 10.1371/journal.pone.0170565
Vidal-Dupiol J., Adjeroud M., Roger E., Foure L., Duval D., Mone Y., et al. (2009). Coral bleaching under thermal stress: putative involvement of host/symbiont recognition mechanisms. BMC Physiol. 9, 14. doi: 10.1186/1472-6793-9-14
Wen B. O., Zhou R., Feng Q., Wang Q., Wang J., and Liu S. (2014). IQuant: an automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 14, 2280–2285. doi: 10.1002/pmic.201300361
Wood-Charlson E. M. and Weis V. M. (2009). The diversity of C-type lectins in the genome of a basal metazoan, Nematostella vectensis. Dev. Comp. Immunol. 33, 881–889. doi: 10.1016/j.dci.2009.01.008
Yi W., Tang Y., Kawsar M. A., Huang K., Jin X., Yu Z., et al. (2025). A novel C1q domain-containing protein from Tridacna crocea exhibits dual functionality in symbiont recognition and immune defense. Fish Shellfish Immunol. 165, 110509. doi: 10.1016/j.fsi.2025.110509
Zhang L., Li L., Guo X., Litman G. W., Dishaw L. J., and Zhang G. (2015). Massive expansion and functional divergence of innate immune genes in a protostome. Sci. Rep. 5, 8693. doi: 10.1038/srep08693
Keywords: plasma proteomics, thermal stress, Tridacna crocea, photosymbiosis, bleaching
Citation: Mao F, Jin X, Chen C, Yi W and Zhang Y (2025) Plasma proteomic profiling reveals molecular signatures of thermal stress and bleaching in the photosymbiotic giant clam Tridacna crocea. Front. Ecol. Evol. 13:1695021. doi: 10.3389/fevo.2025.1695021
Received: 29 August 2025; Accepted: 28 October 2025;
Published: 17 November 2025.
Edited by:
Ruiqi Li, University of Colorado Boulder, United StatesReviewed by:
Lisa Ann Kirkendale, Western Australian Museum, AustraliaCecilia Conaco, University of the Philippines Diliman, Philippines
Copyright © 2025 Mao, Jin, Chen, Yi and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zhang, eXpoYW5nQHNjc2lvLmFjLmNu
†These authors have contributed equally to this work