 Yao Tian1,2
Yao Tian1,2 Yun Zhao2*
Yun Zhao2* Yuqiong Kang2Junru Wu2Yuefeng Meng2Xia Hu2
Yuqiong Kang2Junru Wu2Yuefeng Meng2Xia Hu2 Ming Huang3
Ming Huang3 Bo Lan3*Feiyu Kang2
Bo Lan3*Feiyu Kang2 Baohua Li2*
Baohua Li2*- 1Safety and Quality Technology Research Center, China Waterborne Transport Research Institute, Beijing, China
- 2Institute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, China
- 3Department of Mechanical Engineering, Imperial College London, London, United Kingdom
Understanding the behavior of lithium-ion battery electrolytes during thermal runaway is essential for designing safer batteries. However, current reports on electrolyte decomposition behaviors often focus on reactions with electrode materials. Herein we use quantum chemical calculations to develop a model for the thermal decomposition mechanism of electrolytes under both electrolyte and ambient atmosphere conditions. The thermal stability is found to be associated with the dielectric constants of electrolyte constituents. Within the electrolyte, the solvation effects between molecules increase electrolyte stability, making thermal decomposition a more difficult process. Furthermore, Li+ is observed to facilitate electrolyte thermal decomposition, as the energy required for the thermal decomposition reactions of molecules decreases when they are bonded with Li+. It is hoped that this study will offer a theoretical basis for understanding the complex reactions occurring during thermal runaway events.
1 Introduction
Lithium-ion batteries (LIBs) have become an indispensable technology in the realms of personal electronics, electric vehicles, and energy storage systems (Dunn et al., 2011). The global demand for LIBs surged to 948.1 GW h in 2022, marking a significant annual rise of approximately 66.7% (Zhao et al., 2023). However, this increase has been accompanied by a series of fire incidents (Chen et al., 2021). Throughout the decade of 2011–2021, there were 32 reported instances of fires and explosions globally in energy storage systems, with some occurrences still reported as recently as November 2022. In 2022 alone, China witnessed roughly 20,000 fires related to electric bikes and approximately 2,300 associated with electric vehicles. The frequent incidents related to LIBs have raised safety concerns due to potential ramifications towards human life and property.
These accidents predominantly originate from the thermal runaway of LIBs, a phenomenon representing uncontrolled, exothermic reactions within the battery induced by thermal, mechanical, or electrical stresses (Liu et al., 2018). These lead to an abrupt increase in temperature, alongside intensive heat release and flammable gas production. The complex process of thermal runaway includes the degradation of the solid electrolyte interface (SEI) film, electrolyte decomposition, reactions between the anode, cathode and the electrolyte, oxygen liberation from the cathode materials, separator melting, among other processes (Liu et al., 2020). The intricate interrelations among these reactions further fuel additional combustion processes via the generated radicals and heat (Kalhoff et al., 2015). Notably, the electrolyte, as a critical component, affects the evolution of thermal runaway in LIBs, transitioning from a state of mild combustion to a complete loss of temperature control. Moreover, the electrolyte is a major source of the flammable and toxic gases released during thermal runaway, with its thermal decomposition and combustion reactions considerably influencing the safety profile and potential hazards associated with battery fires (Wang et al., 2019).
Commercially available LIBs generally utilize electrolytes comprising lithium hexafluorophosphate (LiPF6) dissolved in a blend of various carbonate solvents (Xu, 2014). Previous reports have demonstrated the decomposition of LiPF6 and its reactions with carbonate solvents under the condition of LiCx presence. However, a comprehensive understanding of the thermal decomposition characteristics of the electrolyte itself remains insufficiently explored (Choi et al., 2008; Gachot et al., 2008; Haik et al., 2011; Gachot et al., 2012; Zheng et al., 2022). The electrolyte can be transformed into substantial volumes of gases such as CH4 (methane), C2H4 (ethylene), C2H6 (ethane), as well as carbon monoxide (CO) and carbon dioxide (CO2) (Campion et al., 2005; Heiskanen et al., 2019; Henschel et al., 2020; Rinkel et al., 2020). During thermal runaway, the battery temperature sharply increases, intensifying the reactions related to the electrolyte. Hence, understanding the breakdown and oxidation pathways of the electrolyte is crucial, imparting valuable theoretical insight into the thermal runaway process within LIBs.
Herein we use quantum chemical calculations to conceptualize a thermal decomposition mechanism model for the electrolyte in both its liquid and gas environments. Specifically, the study focuses on exploring the catalytic influence of Li+ on the thermal decomposition sequences of electrolyte constituents. By elucidating these mechanisms, which constitute the main contribution and novelty of this work, we aim to provide a robust theoretical foundation for the scientific investigation and numerical simulation of LIBs during uncontrolled states and their corresponding fire mechanisms.
2 Computational details
In the initial stage of thermal runaway, the battery temperature remains relatively modest and experiences a gradual increase. However, if the heat generation within the LIBs becomes uncontrollable, the battery temperature could undergo a remarkably sharp increase, potentially exceeding 600°C within a few seconds. Consequently, electrolyte decomposition during the thermal runaway process may occur via two distinctive pathways. At lower battery temperatures, the electrolyte transitions into gaseous molecules that undergo further decomposition to generate smaller molecular entities. On the contrary, at elevated temperatures, the electrolyte directly disintegrates into smaller molecules while still in its liquid environment, which subsequently vaporize and combust. Hence, the primary objective of this research is to investigate the different breakdown mechanisms of the electrolyte in liquid and gas environments.
2.1 Computational contents
All calculations in this work were performed using Gaussian 16 (Frisch et al., 2016) and GaussView 6.0 (Dennington et al., 2009) software. The default condition for the calculation process is 298.15 K and 1.01*105 Pa. This research focuses on a blended electrolyte formulated with a volumetric ratio of 1:1:1 comprising ethylene carbonate (EC), dimethyl carbonate (DMC), and diethyl carbonate (DEC), and uses lithium hexafluorophosphate (LiPF6) as the lithium salt at a molar concentration of 1 mol L−1. The study examines the breakdown reaction mechanisms of LiPF6, EC, and DMC molecules in both liquid and gas environments. Given the analogous reaction mechanisms between DMC and DEC, the analysis does not differentiate between the two in the main text to maintain conciseness. An in-depth exploration of the DEC decomposition routes within the liquid electrolyte and its gas environment is presented in Supplementary Material Supplementary Figure S1.
2.2 Computational methods of electrolyte in its liquid environment
We have explored the breakdown reaction mechanisms of the main components of the electrolyte within the liquid environment. To simulate these conditions, a solvent model that neglects specific solvent properties was employed, representing a generalized approach that overlooks the microscopic state of individual solvent molecules and treats them as a homogeneously distributed medium. The advantage of such solvent models lies in their ability to accurately capture the average solvation effect while significantly reducing computational demands.
In this work, a solvation model based on density (SMD), along with Density Functional Theory (DFT) methods, was used for quantum chemical simulations. The main molecular models of the electrolyte are displayed in Figure 1, and the corresponding model parameters can be found in Table 1. Parameters for commercial electrolytes were determined through the dielectric constants of their individual components, weighted according to their volumetric ratios.
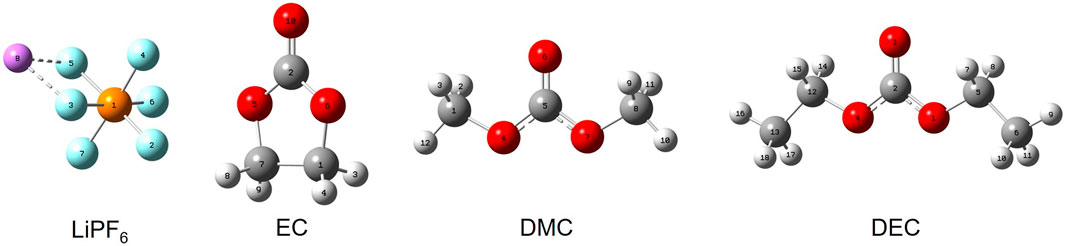
FIGURE 1. Configurations of key electrolyte constituents. Note: Purple represents lithium (Li), cyan represents fluorine (F), orange represents phosphorus (P), gray represents carbon (C), red represents oxygen (O), and white represents hydrogen (H).
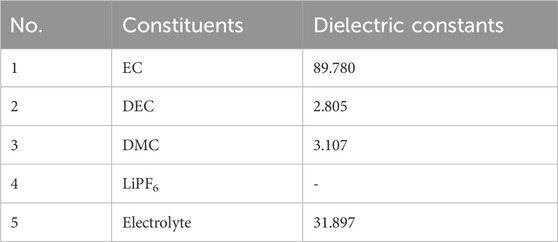
TABLE 1. Parameters of commercial electrolytes.
In the computational process, we initially perform geometric optimization of the molecules and functional groups in the electrolyte using the B3LYP density functional and the 6-311++G(d,p) basis set. Both dispersion forces and thermodynamic corrections are taken into account simultaneously. Subsequently, we conduct high-accuracy electronic energy calculations on the geometrically optimized molecules and functional groups using the M06-2X/6-31G(d) basis set. All calculations were conducted under the standard conditions of 298.15 K and 1.01 × 105 Pa. Finally, Gibbs free energy calculations are performed. The formula for calculating the Gibbs free energy of each molecule and functional group is given as follows:
In the equation, ΔG represents Gibbs free energy in kJ/mol; E represents electronic energy at the M05-2X/6-31G(d) level in kJ/mol; G (corr) represents the Gibbs free energy correction factor at the B3LYP/6-311++G(d,p) level in kJ/mol; ΔGdec.(AB) represents the reaction Gibbs free energy of the process when molecule or functional group AB decomposes into molecule or functional groups A and B, in kJ/mol; ΔGAB, ΔGA, ΔGB represent the Gibbs free energy of molecule or functional group AB, A, and B respectively, in kJ/mol.
2.3 Computational methods of electrolyte in its gas state
In examining the thermal breakdown mechanisms of the main components of the electrolyte in a gas state, we initially applied the B3LYP density functional approach with the 6-311++G(d,p) basis set for the optimization of molecular and functional group geometries. Calculations included dispersion forces and thermodynamic adjustments to improve the accuracy of the simulations. Next, we applied the M06-2X method with the def2-TZVP basis set for precise electronic energy calculations on the optimized molecular and functional group structures, with the goal of ensuring the accuracy of the energy information. Lastly, based on the results of these electronic energy calculations, we computed Gibbs free energy to assess the thermal stability of the molecules and functional groups.
2.4 Transition state search and Intrinsic Reaction Coordinate pathway analysis
In this study, we used the Transition State (TS)-Berny method to identify transition states involved in the electrolyte’s thermal decomposition reactions. We further validated these traced transition state pathways using the Intrinsic Reaction Coordinate (IRC) method to verify their links to respective reactants and products. The functional and basis set employed for the transition state search remained consistent with the aforementioned calculations.
3 Results and discussion
3.1 Electrolyte on molecular reactivity
Our analysis of the electrolyte for different constituents within commercial electrolytes revealed that these environments significantly influence the respective oxidation and reduction properties, as well as overall thermal stability of the constituents. An increase in molecular polarity within the electrolyte enhances the electrostatic interactions between molecules, thereby profoundly influencing both the equilibrium state and the rate of reaction.
The solvation effects within the electrolyte primarily arise from electrostatic interactions between the solvent and the molecules or ions. Solvation-induced bond breakage within a solvent environment promotes charge separation, thereby impacting the potential energy distribution of electrons within the molecules. Under certain circumstances, the energy barriers of the transition states in chemical reactions may be lowered, thus decreasing the requisite activation energy and accelerating the reaction rate. Solvation effects not only change the binding interactions between solvent and solute molecules but also influence interactions involving polar solvents, molecular dipole moments, and electrostatic forces.
As depicted in Figure 2, there is a significant disparity in the Highest Occupied Molecular Orbital—Lowest Unoccupied Molecular Orbital (HOMO-LUMO) energy levels of the electrolyte constituent molecules between the liquid and gas environments. The diminished LUMO energy levels of Li+ in the electrolyte suggest a reduction in reducing ability. Furthermore, PF6− and LiPF6 present lower oxidation potential and increased reduction potential within the electrolyte, thereby enhancing their chemical stability.
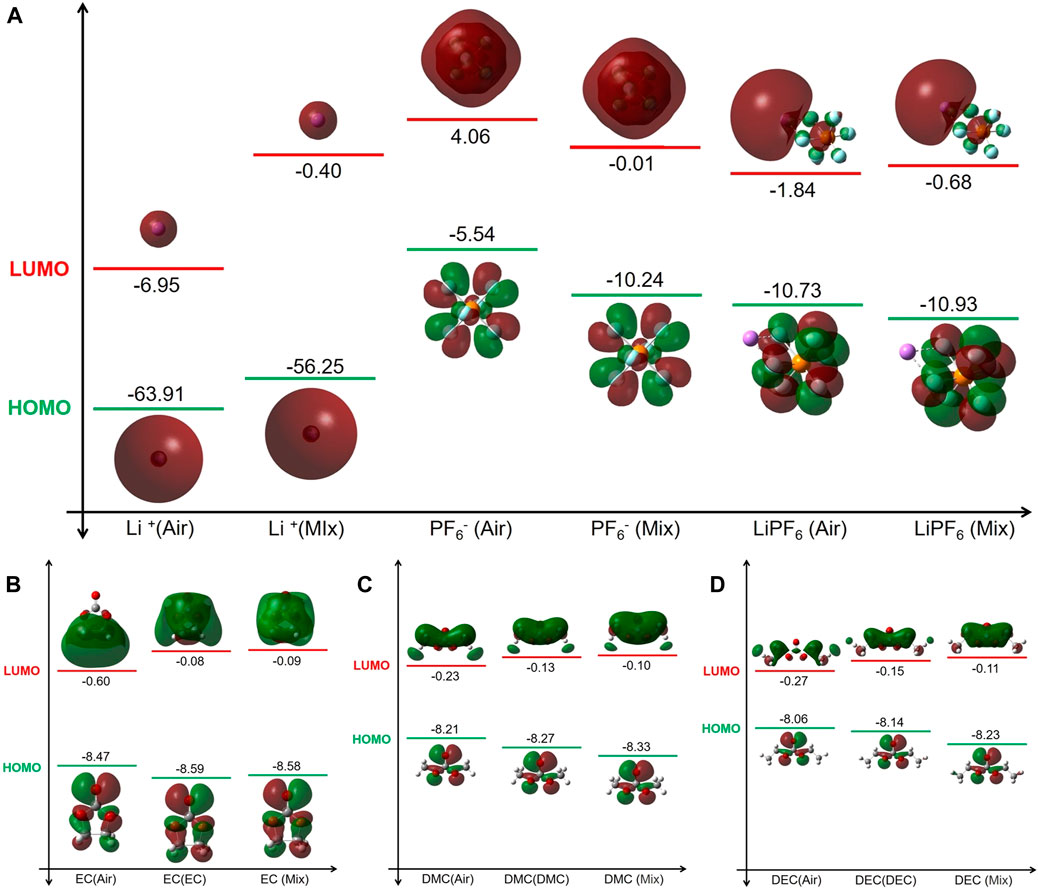
FIGURE 2. HOMO and LUMO energy levels of key electrolyte constituents under different conditions. (A) LiPF6. (B) EC. (C) DMC. (D) DEC. “Gas” denotes the molecular or ionic species in a gas state while ‘Mix’ indicates the species in the electrolyte (liquid environment).
The HOMO energy levels of EC in both the electrolyte and EC solution environments are lower than in the gas environment, whilst the LUMO levels are higher. This suggests a decrease in both oxidative and reducing tendencies within the electrolyte, resulting in increased stability. Given the inherently high polarity of EC molecules, their dielectric constant in an EC solution considerably surpasses that in the electrolyte, implying that the electrolyte exerts a minimal influence on EC molecules. Consequently, EC demonstrates similar stability in both these electrolyte. Conversely, in the gas situation, due to the absence of a uniform medium, EC molecules are more prone to redox reactions, leading to decreased stability.
The energy level calculations for HOMO-LUMO conducted within DMC and DEC solutions, as well as commercial electrolyte environments, reveal consistent patterns. Both DMC and DEC molecules, when in their respective solutions or within the electrolyte, show a decrease in HOMO levels and an increase in LUMO levels compared to those in normal gas. This signifies a decrease in the oxidation and reduction abilities of these molecules within these liquid media, consequently enhancing molecule stability. Notably, this stability increase is more evident in the electrolyte than in pure DMC or DEC solutions. This increase in stability can be attributed to the pronounced solvent effect inherent within the electrolyte. In a milieu characterized by a high dielectric constant (such as the electrolyte’s dielectric constant ε = 31.897 which exceeds DMC solution’s ε = 3.107 and DEC solution’s ε = 2.805), molecules like DMC and DEC are predisposed to transfer their electrostatic energy to molecules possessing higher dielectric constants, resulting in their own electrostatic reduction and amplification of the solvent effect. As such, a high-dielectric-constant medium substantially enhances the stability of DMC and DEC molecules.
In summary, an electrolyte environment significantly increases the solvent effect for molecules such as LiPF6, EC, DMC, and DEC, significantly reducing their oxidation and reduction abilities as seen in normal gas and further enhancing their stability. This solvation effect is especially significant when the solvent’s dielectric constant exceeds that of the molecule itself.
3.2 LiPF6 decomposition in electrolyte
Within LIBs, LiPF6 typically exists in various forms, including Li+, PF6− ions, and LiPF6 molecules. In the event of overheating, LiPF6 undergoes thermally induced degradation reactions within the electrolyte. Detailed pathways for the thermally induced breakdown of LiPF6 in the electrolyte are presented in Figure 3 and Supplementary Table S1.
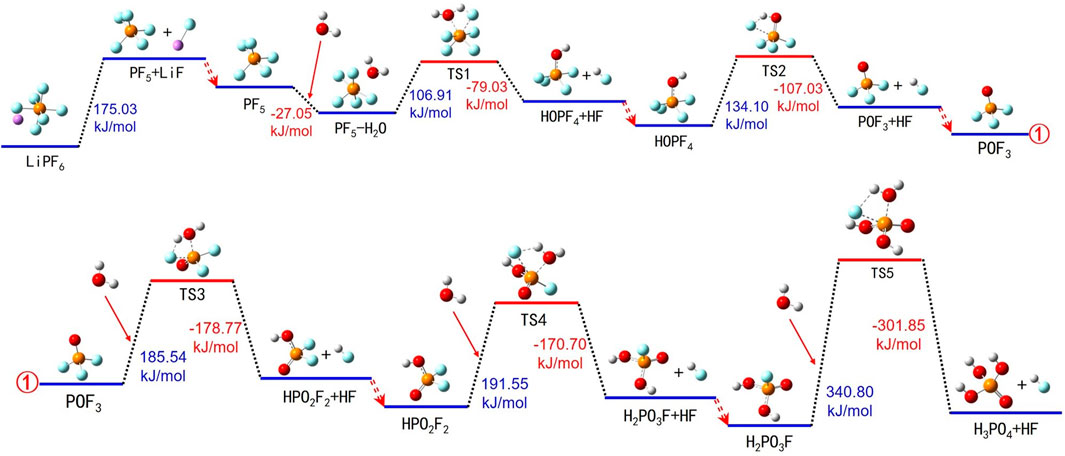
FIGURE 3. Thermal decomposition reaction pathways of LiPF6 in the electrolyte. ①represents the connection between the two images.
During the thermal degradation of LiPF6, the process begins with LiPF6 absorbing energy and breaking down into solid LiF and gaseous PF5. Subsequently, PF5 interacts with H2O, forming a PF5-H2O compound and releasing energy. The PF5-H2O compound then absorbs energy, transitions into an intermediate species TS1, and decomposes into PF4-OH and HF. PF4-OH continues to absorb energy, forms another intermediate species TS2, and eventually breaks down into POF3 and HF, accompanied by energy release. The binding process of POF3 with H2O involves energy absorption, resulting in the formation of intermediate species TS3, which ultimately decomposes into HPO2F2 and HF. HPO2F2 reacts with H2O, absorbing energy to form intermediate species TS4, and subsequently splitting into H2PO3F and HF. Finally, H2PO3F interacts with H2O, forms transitional species TS5 through energy absorption, and breaks down into H3PO4 and HF. The thermal decomposition process of LiPF6 includes many reaction steps, along with various intermediate products and transitional substances, thus involving multiple adsorption and desorption processes.
The thermally induced breakdown process of LiPF6 in the electrolyte solution can be divided into two main stages: the first stage involves the decomposition of LiPF6 into LiF and PF5, wherein LiF precipitates as an insoluble solid and PF5 remains dissolved in the electrolyte for subsequent reactions; the second stage entails the reaction between H2O and PF5, which produces gaseous HF and a variety of phosphate compounds. Within this reaction, water primarily functions as a hydrogen donor (H or H+), and free hydrogen atoms and hydroxyl groups (OH−) within the electrolyte behave similarly. Their reaction process mirrors that of H2O.
In summary, throughout the thermally induced degradation process of LiPF6, entities such as H2O, free hydrogen atoms, H+, and OH− play pivotal roles as reducing agents and proton donors, facilitating the conversion of F atoms in LiPF6 into HF. Concurrently, water and OH− also serve as oxygen donors, aiding the transformation of P atoms in LiPF6 into phosphate compounds. During the thermal degradation cycles of LiPF6, the produced HF continues to dissolve in the electrolyte and partially dissociates into H+ and F−. The generated H+ ions then interact with reaction intermediates such as PF5, POF3, HPO2F2, and H2PO3F, further promoting their decomposition.
3.3 EC decomposition in liquid and gas environments
Figure 4A and Supplementary Table S2 show the thermally induced decomposition pathways of EC molecules in the electrolyte. As temperatures rise, EC molecules absorb energy and undergo structural transformations with the elongation of the carbon-oxygen bond between carbons 2 and 7 and oxygen atoms 5 and 6, transitioning into an intermediate stage TS1. Subsequently, TS1 degrades into acetaldehyde (CH3CHO) and CO2. The thermal decomposition of acetaldehyde then proceeds via two distinct pathways. In one pathway, acetaldehyde dissociates into free methyl radicals (-CH3) and formyl radicals (-CHO). The formyl radical further decomposes into free hydrogen atoms and CO gas. In a concurrent pathway, acetaldehyde reacts with a free -CH3 to form a different intermediate specie TS2. This intermediate then decomposes into a methyloxyl anion (CH3CO-) and CH4. The sequence concludes with CH3CO- transitioning into an additional intermediate TS3, which subsequently breaks down into a -CH3 and CO.

FIGURE 4. Thermal decomposition reaction pathways of EC in both (A) electrolyte and (B) gas states.
The degradation reactions of EC molecules in the gas state are illustrated in Figure 4B and Supplementary Table S3. Although these reactions share similarities with those in the electrolyte, the absorption or release of energy in each reaction is different. Comparatively, the energy absorbed by EC molecules during the initial ring-opening transition to the intermediate state TS1 in the gas state is analogous to that within the battery solution. However, the subsequent decomposition of TS1 into acetaldehyde and CO2 is an exothermic process, releasing more energy in the gas state than in the electrolyte. Moreover, the further degradation of acetaldehyde into other entities requires less energy in the gas state compared to the reaction in the electrolyte. Conversely, the energy required to facilitate the reaction of acetaldehyde with the free -CH3 is higher in the gas state.
3.4 DMC decomposition in liquid and gas environments
Figure 5A and Supplementary Table S4 illustrate the decomposition pathways of DMC molecules in the electrolyte. The structure of DMC exhibits a high degree of symmetry. This symmetry allows for a primarily linear sequence of bond ruptures upon absorbing heat. Specifically, the hydrogen atoms at positions 2, 3, 9, 10, 11, and 12 in the molecule are interchangeable, and the oxygen atoms at positions 3 and 7 exhibit identical characteristics.
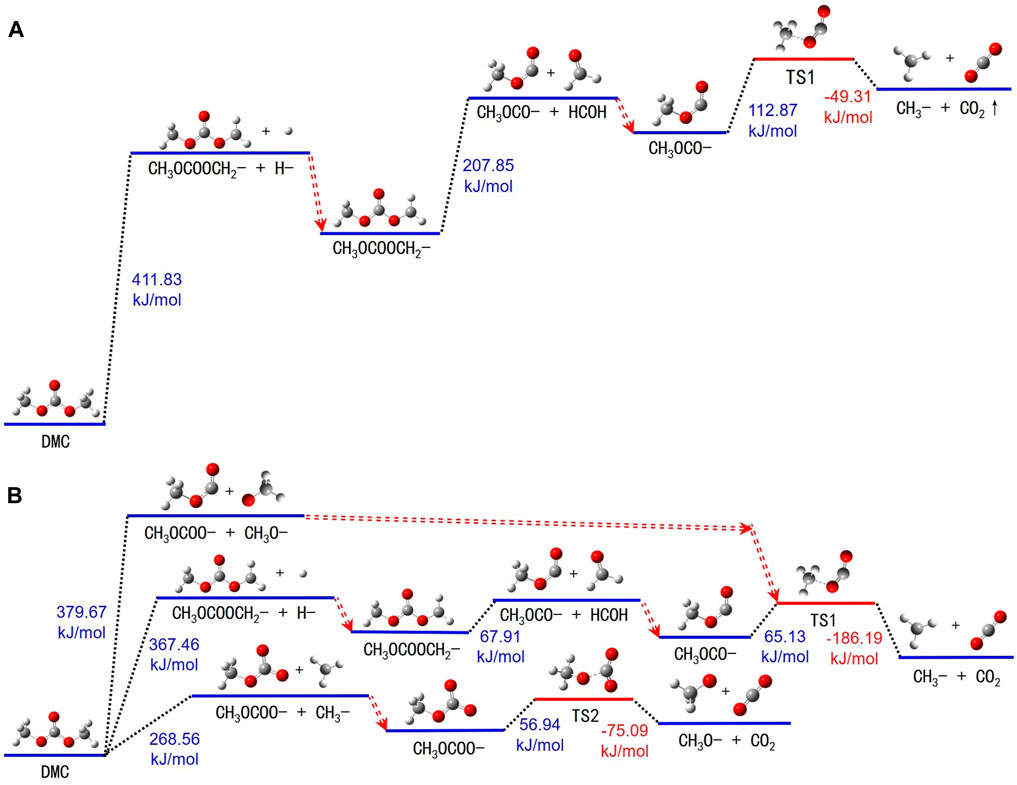
FIGURE 5. Thermal decomposition reaction pathways of DMC in both (A) electrolyte and (B) gas states.
Upon thermal excitation, DMC molecules absorb energy which triggers the breakage of the bond between carbon atom 1 and any one of its associated hydrogen atoms. This results in the formation of methyl hydrogen carbonate (-CH2OCOOCH3) and yields a free hydrogen atom. Next, CH3OCOOCH2- undergoes further energy absorption, leading to the rupture of the bond between carbon atom 5 and oxygen atom 4, thereby generating methylene (-OCH2-) and -OCOCH3. This process of bond breakage requires an energy input of approximately 207.85 kJ/mol. Subsequently, the -OCOCH3 intermediate absorbs additional energy, transitioning into an intermediate state TS1. Ultimately, TS1 decomposes into a -CH3 and CO2.
An extensive review of the decomposition reactions of DMC in the gas state is illustrated in Figure 5B and Supplementary Table S5. These reactions can be categorized into three predominant pathways. Initially, DMC molecules absorb energy, prompting the carbon-oxygen bond between carbon atom 5 and oxygen atom 7 to rupture. This step results in the formation of a -OCOCH3 and a free methoxyl (-OCH3) molecule. Secondly, energy is absorbed by DMC molecules to trigger the breakage of the carbon-hydrogen bond linking carbon atom 8 and hydrogen atom 10, yielding -CH2OCOOCH3 and a detached hydrogen atom. Further decomposition of -CH3OCOOCH2- produces -OCOCH3 and formaldehyde (-CH2O). Methyl carbonate generated in the first two stages absorbs energy to transition into an intermediate state TS1. This state then decomposes into a -CH3 and CO2. Lastly, DMC molecules break down into a formate (-CH3OCOO-) and a free -CH3 upon absorbing energy. The -CH3OCOO- subsequently absorbs additional energy and transits into another intermediate TS2, which further disintegrates into a methoxyl radical and carbon dioxide.
In comparing the thermal resistance of DMC molecules in the liquid and gas environments, DMC is more stable against decomposition in the electrolyte. This enhanced stability may stem from the larger dielectric constant (ε = 31.897) of the electrolyte compared to DMC’s (ε = 3.107). Also, DMC shows enhanced thermal decomposition resistance compared to EC and LiPF6 in the electrolyte. This can be attributed to DMC’s inherently lower dielectric constant, which mitigates its static electric potential, thereby prompting an easier transfer of static charge from DMC to molecules with a higher dielectric constant, and consequently enhancing its thermal stability.
3.5 The catalytic effect of Li+ on the thermal decomposition of EC and DMC
A comprehensive examination of the chemical alterations experienced by EC molecules in conjunction with Li+ in the electrolyte is depicted in Figure 6A and Supplementary Table S6. In the electrolyte, EC and DMC form solvated structures with Li+, ensuring optimal battery performance. Li+ is generally solvated with the 10th oxygen atom of an EC molecule, leading to an energy release and the formation of an EC + Li+ cluster. Subsequently, the structure of EC + Li undergoes a transformation wherein the Li atom occupies the fifth and 10th positions of the oxygen atoms. The thermal decomposition of the balanced EC + Li can be categorized into three predominant pathways.
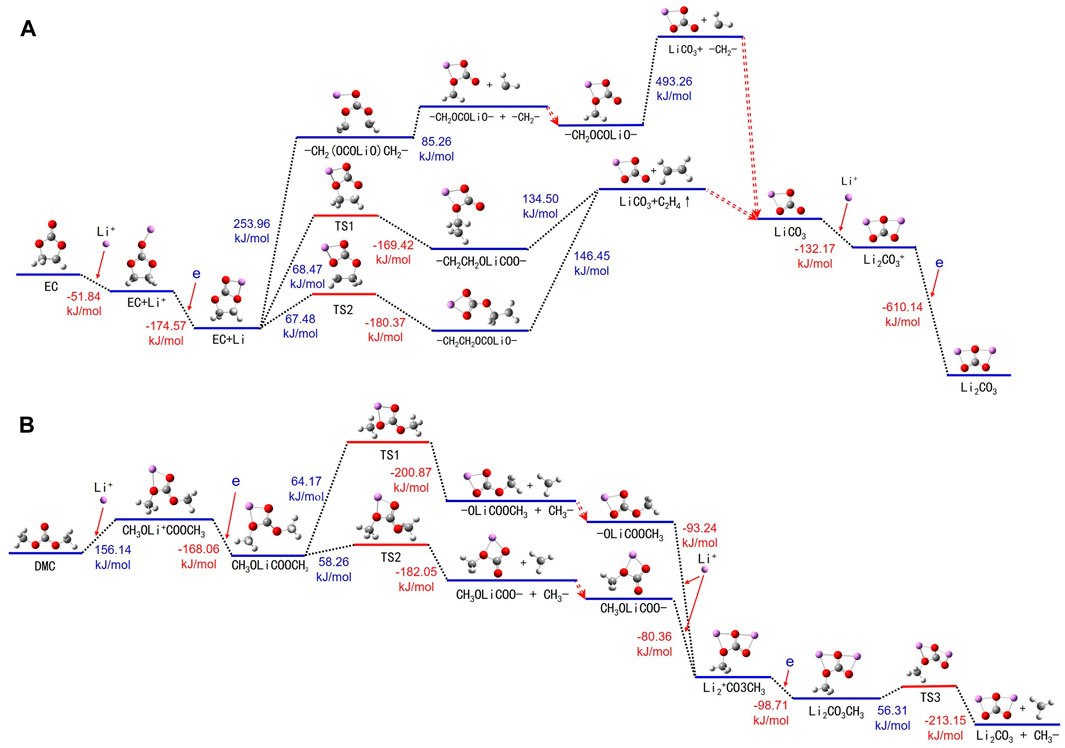
FIGURE 6. Thermal decomposition reaction pathways of (A) EC + Li+ and (B) DMC + Li+ in both liquid and gas environments.
Firstly, the energy adsorbed EC + Li leads to the rupture of the bond shared by carbon atoms 1 and 7, resulting in an elongated segment -CH2(OCOLiO)CH2-. On acquiring additional energy, this chain further decomposes into -CH2(OCOLiO) and a -CH2- fragment, with the former continuing to break down into lithium carbonate (LiCO3) and yet another -CH2- segment. Second, the energy adsorbed EC + Li breaks the bond between the oxygen atom 5 and carbon atom 7, yielding a transition state -CH2CH2OLiCOO-, which further decomposes into LiCO3 and ethylene gas (C2H4). The third pathway involves the cleavage between oxygen atom 6 and carbon atom 1 of the EC + Li, inducing a transition state CH2CH2OCOLiO-, which further decomposes to produce LiCO3 and C2H4. The lithium carbonate generated throughout these stages continues to react with additional Li+, leading to energy release and the formation of Li2CO3+. Then, Li2CO3+ attains an electron, becoming Li2CO3.
Figure 6B and Supplementary Table S7 show the routes of thermal decomposition for DMC molecules in the presence Li+ in the electrolyte. The initiating mechanism includes DMC molecules absorbing energy, followed by the formation of DMC + Li+. During this phase, Li+ notably affiliates with the fourth and sixth (or the sixth and seventh) oxygen atoms of the DMC molecule. These DMC + Li+ complexes then migrate to the anode, where they acquire an electron, thereby transitioning into the DMC + Li complex and concurrently liberating energy.
The thermal decomposition of the DMC + Li complex continues along two possible paths: The first path involves a bond breaking between carbon atom 1 and oxygen atom 4, resulting in the formation of the CH3OOCLiO- and -CH3. CH3OOCLiO- further interacts with Li+, with interaction happening at oxygen atoms 6 and 7, thereby forming (CH3OLiOLiO)+. The second pathway involves the DMC + Li breaking the bond between carbon atom 8 and oxygen atom 7, creating CH3OLiOCO- and -CH3. CH3OLiOCO- then also interacts with Li+ at oxygen atoms 4 and 6, forming (CH3OLiOLiO)+. Subsequently (CH3OLiOLiO)+ acquires an electron to form CH3OLiOLiO-. Following this, CH3OLiOLiO- consumes energy, transitions to a TS1 state, and ultimately breaks up into Li2CO3 and -CH3.
The thermal decomposition process of DMC molecules is similar to that of EC, consisting of three principal stages. Initially, DMC molecules interact with Li+, undergoing an electron transfer to form DMC + Li complexes. In the subsequent phase, -CH3 groups are cleaved from the DMC + Li complexes, resulting in intermediate species such as CH3OOCLiO- or CH3OLiOCO-. These intermediates engage in further reactions with Li+, again involving electron transfer to ultimately produce (CH3OLiOLiO)+ complexes. The final stage sees the decomposition of these complexes, yielding Li2CO3 and CH3 radicals. The generated CH3 radicals have the potential to combine, forming ethane (C2H6). Analogous to the behavior of EC molecules, the presence of Li+ facilitates a more energy-efficient thermal decomposition of DMC in the electrolyte.
The aforementioned study reveals the thermal decomposition mechanisms of EC + Li+ and DMC + Li+ in the electrolyte, clarifying that the energy requirement for these reactions is significantly reduced in the presence of Li+. This phenomenon may be caused by two reasons. Firstly, the complexes formed through the interaction of electrolyte molecules with Li+ acquire a positive charge, thereby enhancing their potential to attract electrons. Upon electron uptake, the electric potential energy of these positively charged complexes increases, facilitating their transition into the thermal decomposition phase. Secondly, Li+ and Li atoms exhibit greater polarity compared to electrolyte molecules, suggesting that their integration into these complexes introduces significant changes to the electric potential energy profile of the complex, alongside modifications to atomic distances and configurations, as demonstrated in Figure 7. These changes lead to the reconfiguration of bond structures and bond energies, which subsequently makes thermal decomposition reactions more feasible. The thermal decomposition mechanism of DEC molecules follows this pattern, with supporting data provided in Supplementary Figure S3 and Supplementary Tables S8–S10. In summary, the participation of Li+ or Li atoms in reactions with battery electrolyte constituents, such as EC, DMC, and DEC, can augment the thermal decomposition propensity of these molecules, thereby escalating the risk of thermal runaway and subsequent fire hazards.

FIGURE 7. Changes in electrostatic potential distribution and molecular structure before and after binding (A) EC, (B) DMC, and (C) DEC with Li+.
4 Conclusion
In this study, we used quantum chemistry calculation methods to explore the thermal decomposition and oxidation processes of main constituents of LiPF6, EC, DMC, and DEC in both liquid and gas environments. This enabled us to build complete chemical reaction models, with the main findings being: Thermal decomposition reactions in the gas state are quite different from those in the electrolyte. In the electrolyte, the solvation effect between molecules enhances the stability of EC, DMC, and DEC, making thermal decomposition more difficult. Among these constituents, EC molecules, due to their higher dielectric constant, exhibit lower thermal stability and higher tendency to decompose, compared to DMC and DEC, which have lower dielectric constants. During the thermal runaway of LIBs, Li+ assists the thermal decomposition of the electrolyte. The energy required for the thermal decomposition reactions of molecules like EC, DMC, and DEC decreases upon bonding with Li+, with the breakdown producing hydrocarbon gases and Li2CO3. It should be noted that the thermal decomposition during the thermal runaway of LIBs is extremely complicated, resulting from the interactions and buildup of multiple chemical reactions. The quantum calculations in this study could provide a theoretical basis for understanding the complex reactions during thermal runaway process.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
YT: Conceptualization, Formal Analysis, Methodology, Software, Validation, Visualization, Writing–original draft, Writing–review and editing, Investigation. YZ: Conceptualization, Software, Writing–original draft, Formal Analysis, Methodology, Validation, Visualization, Writing–review and editing. YK: Data curation, Formal Analysis, Investigation, Software, Writing–review and editing. JW: Data curation, Methodology, Validation, Visualization, Writing–review and editing. YM: Formal Analysis, Methodology, Resources, Writing–review and editing. XH: Formal Analysis, Methodology, Software, Validation, Writing–review and editing. HD: Formal Analysis, Methodology, Resources, Validation, Visualization, Writing–review and editing. MH: Formal Analysis, Validation, Visualization, Writing–review and editing. BL: Data curation, Formal Analysis, Funding acquisition, Methodology, Resources, Validation, Visualization, Writing–review and editing. FK: Formal Analysis, Funding acquisition, Resources, Supervision, Writing–review and editing. BL: Conceptualization, Funding acquisition, Project administration, Supervision, Formal Analysis, Investigation, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was financially supported by the Shenzhen Science and Technology Program (KCXFZ20211020163810015), Shenzhen Science and Technology Program (KCXST20221021111201003), National Natural Science Foundation of China (No. 52261160384 and 52072208), Fundamental Research Project of Shenzhen (No. JCYJ20220818101004009), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01N111), Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515110531), and China Postdoctoral Science Foundation (No. 2022M721800).
Acknowledgments
We also acknowledge the support of the Testing Technology Center of Materials and Devices of Tsinghua Shenzhen International Graduate School (SIGS), and the Major Science and Technology Infrastructure Project of Material Genome Big-science Facilities Platform supported by Municipal Development and Reform Commission of Shenzhen. BL would like to acknowledge support from the Imperial College Research Fellowship scheme.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2024.1356672/full#supplementary-material
References
Campion, C. L., Li, W., and Lucht, B. L. (2005). Thermal decomposition of LiPF6-based electrolytes for lithium-ion batteries. J. Electrochem. Soc. 152 (12), A2327. doi:10.1149/1.2083267
Chen, Y., Kang, Y., Zhao, Y., Wang, L., Liu, J., Li, Y., et al. (2021). A review of lithium-ion battery safety concerns: the issues, strategies, and testing standards. J. Energy Chem. 59, 83–99. doi:10.1016/j.jechem.2020.10.017
Choi, N.-S., Profatilova, I. A., Kim, S.-S., and Song, E. H. (2008). Thermal reactions of lithiated graphite anode in LiPF6-based electrolyte. Thermochim. Acta 480, 10–14. doi:10.1016/j.tca.2008.09.017
Dunn, B., Kamath, H., and Tarascon, J.-M. (2011). Electrical energy storage for the grid: a battery of choices. Science 334 (6058), 928–935. doi:10.1126/science.1212741
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, revision C.01[CP/OL].
Gachot, G., Grugeon, S., Armand, M., Pilard, S., Guenot, P., Tarascon, J. M., et al. (2008). Deciphering the multi-step degradation mechanisms of carbonate-based electrolyte in Li batteries. J. Power Sources 178, 409–421. doi:10.1016/j.jpowsour.2007.11.110
Gachot, G., Grugeon, S., Eshetu, G. G., Mathiron, D., Ribière, P., Armand, M., et al. (2012). Thermal behaviour of the lithiated-graphite/electrolyte interface through GC/MS analysis. Electrochimica Acta 83, 402–409. doi:10.1016/j.electacta.2012.08.016
Haik, O., Ganin, S., Gershinsky, G., Zinigrad, E., Markovsky, B., Aurbach, D., et al. (2011). On the thermal behavior of lithium intercalated graphites. J. Electrochem. Soc. 158, A913–A923. doi:10.1149/1.3598173
Heiskanen, S. K., Kim, J., and Lucht, B. L. (2019). Generation and evolution of the solid electrolyte interphase of lithium-ion batteries. Joule 3 (10), 2322–2333. doi:10.1016/j.joule.2019.08.018
Henschel, J., Peschel, C., Klein, S., Horsthemke, F., Winter, M., and Nowak, S. (2020). Clarification of decomposition pathways in a state-of-the-art lithium ion battery electrolyte through 13C-labeling of electrolyte components. Angew. Chem. Int. Ed. 59 (15), 6128–6137. doi:10.1002/anie.202000727
Kalhoff, J., Eshetu, G. G., Bresser, D., and Passerini, S. (2015). Safer electrolytes for lithium-ion batteries: state of the art and perspectives. ChemSusChem 8 (13), 2154–2175. doi:10.1002/cssc.201500284
Liu, B., Jia, Y., Yuan, C., Wang, L., Gao, X., Yin, S., et al. (2020). Safety issues and mechanisms of lithium-ion battery cell upon mechanical abusive loading: a review. Energy Storage Mater. 24, 85–112. doi:10.1016/j.ensm.2019.06.036
Liu, K., Liu, Y., Lin, D., Pei, A., and Cui, Y. (2018). Materials for lithium-ion battery safety. Sci. Adv. 4 (6), eaas9820. doi:10.1126/sciadv.aas9820
Rinkel, B. L., Hall, D. S., Temprano, I., and Grey, C. P. (2020). Electrolyte oxidation pathways in lithium-ion batteries. J. Am. Chem. Soc. 142 (35), 15058–15074. doi:10.1021/jacs.0c06363
Wang, Q., Jiang, L., Yu, Y., and Sun, J. (2019). Progress of enhancing the safety of lithium ion battery from the electrolyte aspect. Nano Energy 55, 93–114. doi:10.1016/j.nanoen.2018.10.035
Xu, K. (2014). Electrolytes and interphases in Li-ion batteries and beyond. Chem. Rev. 114 (23), 11503–11618. doi:10.1021/cr500003w
Zhao, Y., Kang, Y., Wozny, J., Lu, J., and Du, H. (2023). Recycling of sodium-ion batteries. Nat. Rev. Mater. 8 (9), 623–634. doi:10.1038/s41578-023-00574-w
Keywords: lithium-ion batteries, electrolyte decomposition, thermal runaway, quantum chemical calculation, solvation
Citation: Tian Y, Zhao Y, Kang Y, Wu J, Meng Y, Hu X, Huang M, Lan B, Kang F and Li B (2024) Quantum chemical calculation study on the thermal decomposition of electrolyte during lithium-ion battery thermal runaway. Front. Energy Res. 12:1356672. doi: 10.3389/fenrg.2024.1356672
Received: 16 December 2023; Accepted: 29 February 2024;
Published: 08 March 2024.
Edited by:
Burak Ulgut, Bilkent University, TürkiyeCopyright © 2024 Tian, Zhao, Kang, Wu, Meng, Hu, Huang, Lan, Kang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Zhao, eXpoYW8uemp1dEBob3RtYWlsLmNvbQ==; Bo Lan, Ym8ubGFuQGltcGVyaWFsLmFjLnVr; Baohua Li, bGliaEBtYWlsLnN6LnRzaW5naHVhLmVkdS5jbg==