 Shiyu Liu1Peter Szatmary2
Shiyu Liu1Peter Szatmary2 Jing-wen Lin3Qiqi Wang1Robert Sutton2
Jing-wen Lin3Qiqi Wang1Robert Sutton2 Lu Chen3
Lu Chen3 Tingting Liu1*
Tingting Liu1* Wei Huang1,4*Qing Xia1*
Wei Huang1,4*Qing Xia1*- 1West China Centre of Excellence for Pancreatitis, Institute of Integrated Traditional Chinese and Western Medicine, West China-Liverpool Biomedical Research Centre, West China Hospital, Sichuan University, Chengdu, China
- 2Liverpool Pancreatitis Research Group, Liverpool University Hospitals NHS Foundation Trust and Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, United Kingdom
- 3State Key Laboratory of Biotherapy, West China Hospital, Sichuan University and Collaborative Innovation Center for Biotherapy, Chengdu, China
- 4Institutes for Systems Genetics & Immunology and Inflammation, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu, China
Acute pancreatitis is a common gastrointestinal disease characterized by inflammation of the exocrine pancreas and manifesting itself through acute onset of abdominal pain. It is frequently associated with organ failure, pancreatic necrosis, and death. Mounting evidence describes monocytes - phagocytic, antigen presenting, and regulatory cells of the innate immune system - as key contributors and regulators of the inflammatory response and subsequent organ failure in acute pancreatitis. This review highlights the recent advances of dynamic change of numbers, phenotypes, and functions of circulating monocytes as well as their underling regulatory mechanisms with a special focus on the role of lipid modulation during acute pancreatitis.
1. Introduction
Acute pancreatitis (AP) is a common gastrointestinal disease with a globally rising incidence (1). Patients present with very acute onset of severe abdominal pain often necessitating urgent hospital admission (2), an event which is thought to coincide with the time of initial pancreatic injury. After the initial insult by pancreatitis toxins, injured pancreatic acinar cells release cytokines, chemokines, cellular components, and neuropeptides to promote inflammation (3–5). Immune cells are closely related with the systemic response to pancreatic injury, thereby contributing substantially to disease severity (6–9). Neutrophils and monocytes are recruited to the pancreas during early stages of AP, followed by dendritic cells (DCs), mast cells, and T cells (8, 10). Neutrophils promote inflammation and tissue damage in AP through further secretion of cytokines and chemokines, as well as through generation of reactive oxygen species (ROS) (11), enhancement of nicotinamide adenine dinucleotide phosphate oxidase (12) and granule enzymes activity (13, 14), release of neutrophil extracellular traps (15), and promotion of trypsinogen activation (16). The critical role of neutrophils in the pathogenesis of AP has been recently reviewed (17, 18). There is some limited evidence to suggest that DCs possess both protective (19) and pro-fibroinflammatory roles (20), while mast cells are often reported to contribute to deleterious outcomes of AP (21, 22). The role of T cells in AP is complex, with AP severity correlating with T cell infiltration and being regulated by the balance of Th1/Th2 and Treg/Th17 cells (23, 24).
Monocytes/macrophages are also widely accepted as the key contributors and main inflammatory cell population during initiation and progression of systemic inflammation in AP (7, 25). Monocyte-derived macrophages form the bulk of the macrophage population in inflamed pancreatic tissue (26) and while tissue macrophages in AP have been extensively reviewed (25, 27), the role of circulating monocytes remains largely neglected. This review aims to detail the current understanding of circulating monocytes in AP.
2. Monocytes
Monocytes are short-lived mononuclear phagocytes characterized by kidney-shaped nuclei, and are the largest leucocyte subtype in blood, making up about 5-10% of circulating blood leucocytes (28, 29). They are a crucial component of the innate immune system and serve as phagocytes and antigen-presenting cells as well as having a critical role in orchestrating the immune response to infection and inflammation (30–34). Traditionally, monocytes were regarded as transitional cells – non-terminally differentiated, circulating forms of macrophages or DCs. Increasingly, distinct functions of heterogeneous subsets of monocytes with their own transcriptional profiles and phenotypes are being described in inflammatory and autoimmune diseases (30, 35).
Monocytes develop primarily in the bone marrow (BM) in a highly regulated manner (33). Spleen is a primary site of extramedullary monocytopoiesis and contains a monocyte reservoir with over one million monocytes (36, 37). The common monocyte progenitor population is identified to be present in BM and spleen, which generates monocytes and monocyte-derived macrophages (38).
There are two main populations of monocytes in human and other species, termed as classical and non-classical monocytes. The proportion of these two subsets are analogous in blood and spleen (37, 39). Classical monocyte fate is regulated by the sequential action of transcription factors including PU.1 (Sfpi1), Irf8, and Klf4 (33, 40). Non-classical monocytes develop from classical monocytes under regulation of Nr4a1 (TR3/Nur77), but the possibility of directly deriving from progenitors in BM cannot be ruled out (33, 40). Intermediate monocytes are thought to be transitional populations between classical and non-classical monocytes (30). The specific markers and distinct functions of the different populations are briefly summarized in Table 1.

Table 1 Specific immune markers and functions of monocyte subsets.
3. Monocytes in AP
During AP, pancreatic acinar and ductal cells are injured in the initial aseptic environment through a series of pathological cellular and molecular events including premature trypsinogen activation, calcium overload, loss of mitochondrial membrane potential, endoplasmic reticulum stress, and impaired autophagy after exposure to pernicious factors such as alcohol metabolites, free fatty acids, bile acids, high intraductal pressure or intraductal acidification (3, 46). Injured cells liberate pro-inflammatory cytokines, chemokines (47), and other inflammatory meditators such as damage-associated molecular patterns (48, 49) locally and into circulation, promoting leucocytes infiltration and activation which exacerbates pancreatic injury, systemic inflammation, and organ failure (4).
Among inflammatory mediators, the released CC-chemokines including CC-motif chemokine ligand 2 (CCL2; or monocyte chemoattractant protein-1, MCP-1), CCL3 (macrophage inflammatory protein-1α, MIP-1α), CCL5 (RANTES), and CCL7 (MCP-3) give rise to recruitment of circulating monocytes to the inflamed pancreas (25, 50). CCL2 is a canonical chemokine produced by stromal cells or immune cells in acute inflammation, dimerizes, and binds to extracellular matrix glycosaminoglycans forming a stable gradient to attract CCR2+ inflammatory monocytes (7, 50). CCL2 levels were increased in pancreatic, systemic, lung, intestinal, and urinary samples and correlated with severity in experimental and clinical AP (51–57). The infiltrating monocytes become activated and gain distinct immune phenotype in response to the environmental cues, which afterwards either serve as monocyte reservoirs with proliferative activity or further differentiate into macrophages or DCs (33) (Figure 1). The key findings of major studies on monocytes in experimental and clinical AP are summarized in Supplementary Table 1 and Supplementary Table 2 respectively.
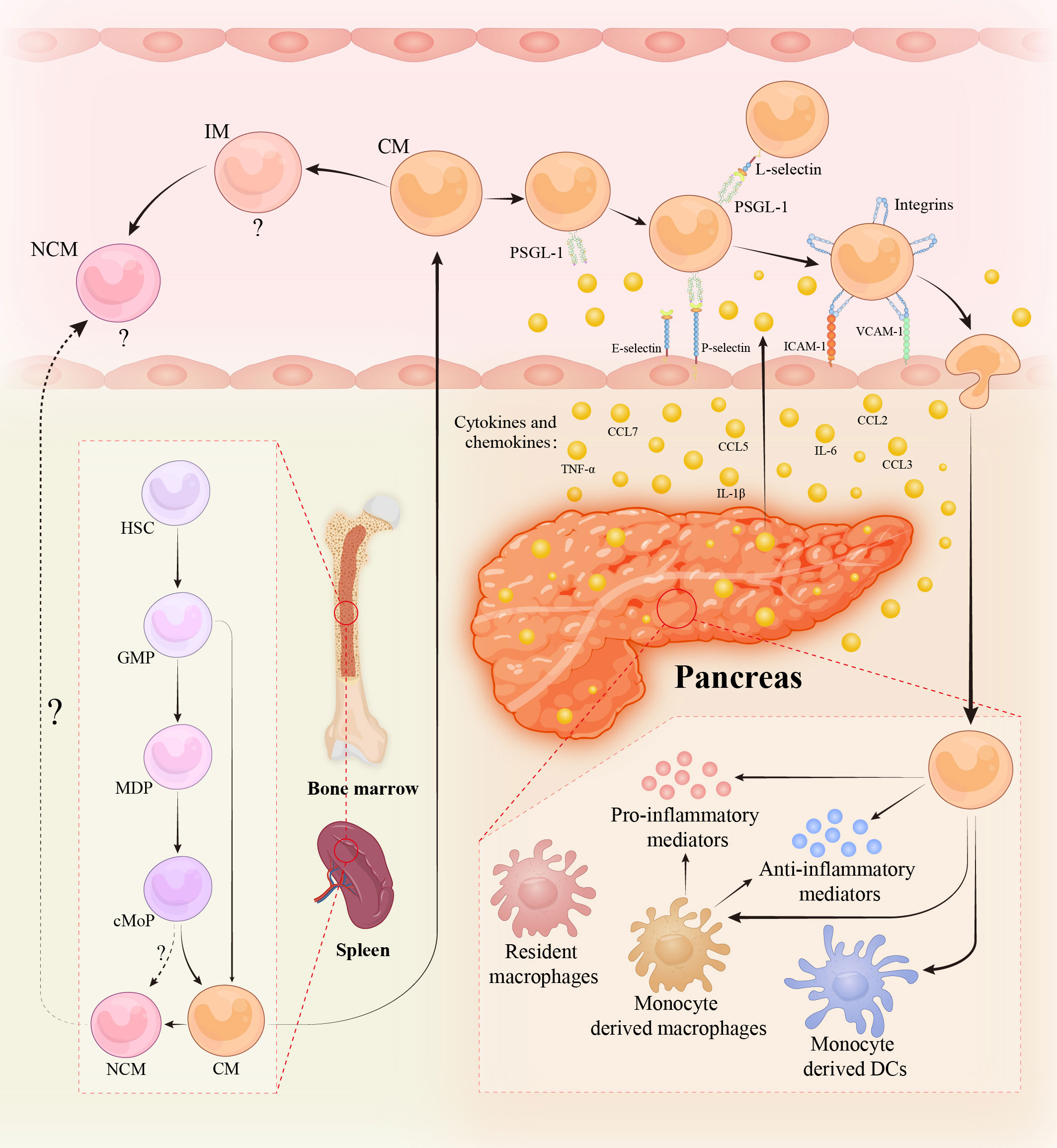
Figure 1 Monocytes in acute pancreatitis. Monocytes are continuously produced in bone marrow (BM) and spleen from hematopoietic stem cells (HSCs) via granulocyte and macrophage progenitors (GMPs), monocyte/dendritic cell (DC) progenitors (MDPs), and common monocyte progenitors (cMoPs) intermediates. During acute and severe inflammation, monocytes might be directly generated from GMPs in response to the need for rapid generation. Non-classical monocytes (NCMs) are derived from classical monocytes (CMs) and might be directly developed from cMoPs. The existence of NCMs in BM/spleen and the regulatory mechanisms of their egress into circulation are disputable and obscure. Intermediate monocytes (IMs) are the transitional cells of CMs and NCMs with distinct functions. The role of NCMs and IMs in acute pancreatitis (AP) is still vague. During AP, pancreatic acinar and ductal cells are injured and liberate damage-associated molecular patterns, cytokines, chemokines, and various pro-inflammatory mediators, leading to the recruitment of circulating monocytes to the inflamed pancreas. Monocytes migration across the endothelium requires a series of sequential adhesive interactions between monocytes and endothelial cells mediated by the indicated endothelial adhesion molecules and monocyte selectin and integrin ligands. When monocytes influx into the pancreas, they either develop into monocyte-derived macrophages/DCs or maintain monocyte-state. The infiltrated monocytes and monocyte-derived macrophages exhibit heterogeneous phenotypes such as pro-inflammatory or anti-inflammatory activities, depending on the environmental cues. Abbreviations: PSGL-1, P-selectin glycoprotein ligand 1; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cellular adhesion molecule-1.
3.1. Dynamic changes of monocyte numbers
Human pancreas is often difficult to obtain for mechanistic studies. Therefore, experimental animal models have been widely employed to investigate the pathobiology of AP for over a century and a half (58). Of the experimental animals, mice and rats are preferred as the mammalian pancreas differs markedly from non-mammals in structure and function and due to the relative ease of genetic manipulation in these species (59). Repetitive administration of caerulein, a cholecystokinin analogue, causes clinical, biochemical and histopathological changes in rodent pancreas similar to those seen in human AP (60). Caerulein-induced AP (CER-AP) is the most popular experimental model, as it is easily reproducible, simple to perform and the severity can be adjusted by different dosing regimens (58). Further, a septic condition with profound systemic inflammation and organ failure can be produced by superimposing a single injection of lipopolysaccharide (LPS) on CER-AP (CER/LPS-AP) (58). Choline-deficient ethionine-supplemented diet and intraperitoneal administration of L-arginine are other less-invasive ways to induce AP (CDE-AP and ARG-AP, respectively) (58, 61). These AP models are often associated with significant systemic inflammation, organ failure and a certain rate of mortality, thereby mirroring the pathobiology of SAP (58). Both ductal infusion of bile acid, sodium taurocholate induced-AP (NaTC-AP) and pancreatic duct ligation-induced AP (PDL-AP) are designed to mimic human biliary pancreatitis and have features of clinical SAP (62, 63).
Observational studies looking at monocyte subtypes in circulation or within the pancreas in association with different stages of AP as well as its severity, together with depletion studies in experimental models form the backbone of our understanding of the role of monocytes in AP. Most detailed studies of the timeline of monocyte response during AP come from experimental models, where the time of disease onset is carefully controlled.
In mice with CER-AP, the number of circulating monocytes peaked at 12 h and pancreatic monocytes at 24 h, both returning to baseline during the recovery phase (by 168 h) (64). Following NaTC-AP and in PDL-AP in rats, monocyte numbers in peripheral blood were significantly elevated from 30 min and 6 h respectively (65, 66). An increased frequency of monocytes was observed both in circulation and in the pancreas at 72 h in mice with CDE-AP but this was not statistically significant in circulation (64). In a similar manner, peripheral monocytes were elevated in AP patients on admission, especially those who had more severe clinical phenotype (67–69). Monocyte levels remained elevated up to one week after admission (67–69). Clinically, lymphocyte-to-monocyte ratio (LMR) could serve as one of the predictive indices for persistent organ failure and mortality (70–75). Compared with mild AP (MAP; no organ failure, no local or systemic complications) or moderately severe AP (MSAP; transient organ failure or local or systemic complications) patients, those with severe AP (SAP; persistent organ failure) as defined by the revised Atlanta classification (76) had a lower LMR (71, 73, 74, 77, 78). Paradoxically, SAP patients with uncertain time of disease onset had reduced lymphocyte and monocyte counts at the early stage of their ICU stay, perhaps indicating an immune anergy later in severe disease, which recovered to normal on day 7 (79). Depletion of monocytes/macrophages in circulation and tissue by clodronate liposomes reduced disease severity in CER-AP mice and NaTC-AP rats (80, 81) if used prior to AP induction. The method was also used to describe the dynamic phenotypes (pro-inflammatory and restorative) during CER-AP and recovery (26, 82). Conditional depletion of monocytes, achieved by administration of diphtheria toxin (DT) to mice with transgenic expression of the human diphtheria toxin receptor [DTR] coupled to the CD11b promoter (CD11b-DTR), reduced pancreatic edema and necrosis in CER-AP and NaTC-AP (83). Targeting the CCL2 axis with genetic ablation or pharmacological inhibition reduced the number of monocytes/macrophages in the pancreas and ameliorated the severity of CER-AP, ARG-AP, and NaTC-AP models (84–86).
It was first demonstrated in the 1970s that monocytes increased proliferative activity in BM during inflammation, resulting in monocytosis (87). Intriguingly, cholesterol metabolism plays a crucial role in regulating proliferation of hematopoietic stem and progenitor cells (88). And hypercholesterolemia is reported to increase circulating monocytes via promoting hematopoietic stem and progenitor cell proliferation in BM and seed in spleen (88). However rapid changes in the number of circulating leucocytes are a function of release of cells from storage pools, rather than de novo synthesis of cells. Classical monocytes have been shown to egress from BM in a CCR2-dependent manner. CCL2 and CCL7 are ligands for CCR2 and help maintain the stable levels of circulating monocytes (89). Whether non-classical monocytes exist in the BM and the mechanism of them exiting the BM are as yet unclear. The spleen contains a mobilizable monocyte reservoir in the subcapsular red pulp, with morphology and transcriptomes of Ly6Chigh monocytes indistinguishable from those in blood (39, 90). Angiotensin II or CCR2 signaling is reported to induce monocytes egress from the spleen to the sites of injury and tumor, such as myocardial infarction and lung adenocarcinoma, resulting in increased circulating monocytes mirrored with reduced splenic monocytes (39, 91). Splenectomy related experiments demonstrate that spleen contributed almost 50% of the monocytes to the infarcted heart and 30% of the monocytes, all the Ly6Chi monocytes to the growing atheroma (92, 93).
In mild CER-AP, increased numbers of peripheral and pancreatic Ly6Chi monocytes were observed concomitant with decreased Ly6Chi monocytes in BM (94). However, the percentage of splenic Ly6Chi monocytes was no different to that of control mice, indicating monocytes were principally mobilized from the BM instead of spleen in this setting (94). In sodium deoxycholate-induced AP rats, splenocytes were recruited to the systemic circulation and resulted in splenic atrophy (95). Splenectomy after AP induction in this model reduced serum amylase, inflammatory cytokine levels and pancreatic histological injury (96). The precise role of splenic monocytes in AP remains unclear.
3.2. Phenotypic changes of monocyte immune markers
Heterogeneity of monocyte subsets is increasingly becoming apparent (33). However, studies addressing the role of monocytes in experimental and clinical AP principally pertain to classical monocytes. The involvement of non-classical, or indeed intermediate monocytes in the pathogenesis of AP is still unclear.
3.2.1. Classical monocytes
Classical monocytes are described by the expression of the LPS-binding receptor CD14 and the absence of the FcγRIII (CD16) receptor. In AP patients, the proportion and absolute numbers of circulating classical monocytes (CD14++CD16-) increase compared to healthy controls and correlate with escalating severity (MAP < MSAP < SAP) (64, 97–99). Numbers of classical monocytes remain high in SAP patients, whereas in MSAP patients, numbers decrease within one week after AP onset (98, 99). This finding correlates with experimental models of AP, where elevated numbers of equivalent monocytes [CD11bhighCD11c-Gr-1low monocytes/macrophages (86)] in circulation and in the pancreas were shown to correlate with disease severity in CER-AP and ARG-AP in mice (6, 86, 94). In CD11b-DTR mice, administration of DT which conditionally and specifically depletes monocytes/macrophages, abolished the increase of pancreatic Ly6Chi monocytes and was associated with reduced edema and necrosis of the pancreas in CER-AP (83). This process was reversed by adoptive transfer of Ly6Chi monocytes from mice of non-DT-treated CD11b-DTR or from tumor necrosis factor-alpha (Tnfα)+/+ donors, but not Tnfα-/- donors, demonstrating that the pathological role of Ly6Chi monocytes in this setting depends on the TNF-α signaling pathway (83). Ly6C+ inflammatory monocytes have also been shown to exhibit significant phenotypic heterogeneity using novel technologies. Seven distinct groups of monocytes could be identified by cell surface markers and cytokine profiles using CyTOF analysis in CER-AP and six in CDE-AP and AP from patient blood samples (64). While these novel subsets of monocytes require validation and further characterization, they offer a significant potential for targeted interventions based on precision cytometry.
3.2.2. Non-classical monocytes
Non-classical monocytes are described as expressing both CD14 and CD16. Studies investigating the role of non-classical monocytes in AP are absent from the published records. In sterile injury including wounds, liver injury and myocardial infarction, a biphasic response was observed with an initial influx of Ly6Chi monocytes producing high levels of interleukin-1 beta (IL-1β) and TNF-α followed by a delayed response from Ly6Clo monocytes/macrophages secreting transforming growth factor-beta (TGF-β) and vascular endothelial growth factor for tissue repair (100–103). Non-classical monocytes also express human leukocyte antigen (HLA)-DR, which is a valuable biomarker of the immune status, whereby a percentage of HLA-DR+ monocytes of less than 80% characterizes immunosuppression and below 30% marks immunoparalysis (104). Several studies have demonstrated a downregulation of HLA-DR on monocytes is associated with an increase in AP severity (79, 99, 105–122), and serves as a predictor for SAP, infectious complications, and mortality in AP patients (105, 108–111, 113, 119, 120, 122–125).
Reprograming of Ly6Chi monocytes at the site of inflammation or injury is thought to be more efficient than recruitment of Ly6Clo monocytes to restore homeostasis (82). Cytokines such as IL-4 and IL-10 in the local environment drive the switch of monocytes phenotype from pro-inflammatory Ly6ChiCCR2+ monocytes to reparative Ly6CloCX3CR1+Nr4a1hi monocytes (102). Non-classical monocytes also appeared to be atheroprotective in mice and were associated with vascular repair and improved organ function in kidney and heart models of ischemia reperfusion-induced injury (35). Studies investigating the role of non-classical monocytes in AP are clearly needed.
3.2.3. Intermediate monocytes
Though intermediate monocytes are normally considered to be a transitional state between classical monocytes and non-classical monocytes, they are able to generate reactive oxygen species (ROS) and pro-inflammatory cytokines (TNF-α and IL-1β) in response to LPS, have high proangiogenic capacity and are effective at antigen processing and presentation with strong expression of HLA-DR and Toll-like receptors (TLRs) (126, 127). While a previous study reported that intermediate monocyte (CD14+CD16+) counts were comparable amongst AP patients and healthy controls within 24 or 72 h of disease onset (97), a more recent study demonstrated that the percentage of intermediate monocytes was reduced in MAP patients compared with healthy controls within 24 h of admission (mean, 2.4% vs. 3.1%, respectively) (127). The reduction of intermediate monocyte numbers was reported to be possibly mediated by a disintegrin and metalloproteinase 17 (ADAM17) and contribute to increased susceptibility to infection, which needs further validation (127). A recent study demonstrated that genetic (Adam17ex/ex mice, which displayed marked reduction in global ADAM17 expression) or pharmacological blockade of ADAM17 ameliorated the severity of CER-AP mice (17). Considering the ubiquitous expression and multifunctional role of ADAM17 in inflammation (16), it is necessary to figure out the specific pathogenetic effect of ADAM17 on different cells including monocytes so as to exploit a precise therapeutic approach of cell- or tissue-specific ADAM17 inhibition. Overall, the kinetics of circulating intermediate monocytes, their roles in the pathogenesis of AP as well as the underlying mechanisms of alterations in numbers and functions remain to be elusive due to limited studies.
3.2.4. M1 and M2 polarization
M1 and M2 polarization, initially proposed for macrophages and defined by the expression of several cellular markers of polarization, can also be seen in peripheral monocytes in a number of diseases (128–130). M1 monocytes are classically activated and function as pro-inflammatory cells producing inflammatory mediators including TNF-α, IL-1β, IL-6, and IL-12; M2 monocytes are alternatively activated and release anti-inflammatory or regulatory molecules including IL-10 and TGF-β (68, 131). Different subsets of polarization usually dominate at different stages of a disease (68), with the pro-inflammatory M1 polarization state dominating early, followed by a late M2 polarization predominating, dampening inflammation and promoting tissue repair and reorganization.
Numbers of peripheral M1 monocytes (defined by an absence of CD163 and presence of IL-12 or MAC387) and M2 monocytes (defined by CD206 and CD163 positivity) were both significantly elevated in SAP compared to MAP patients or healthy controls (131, 132); and the numbers correlated with Acute Physiology and Chronic Health Evaluation II score (131, 132), one of commonly used composite clinical indices to predict severity of AP (133). By day 3, a higher positive M1:M2 ratio can be observed in the blood of SAP compared with MAP or healthy controls (68). Unfortunately, peripheral monocyte polarization does not appear to be useful in early differentiation of disease severity (i.e., before 48 hours after disease onset) and it remains unclear whether the relationship of disease severity and monocyte polarization is in any way causal.
3.3. Function alterations in monocytes
Numerous human and rodent studies have demonstrated the functional variation of monocytes in AP, including alterations in activation, adhesion and phagocytosis. Distinct expression of surface markers and intracellular molecules, and differential change of elaborative regulatory pathways including gaseous signaling pathways in monocytes are associated with specific functional dysregulation such as hyperfunction or anergy, which may together serve as useful tools to assess the immune status during AP (Figure 2).
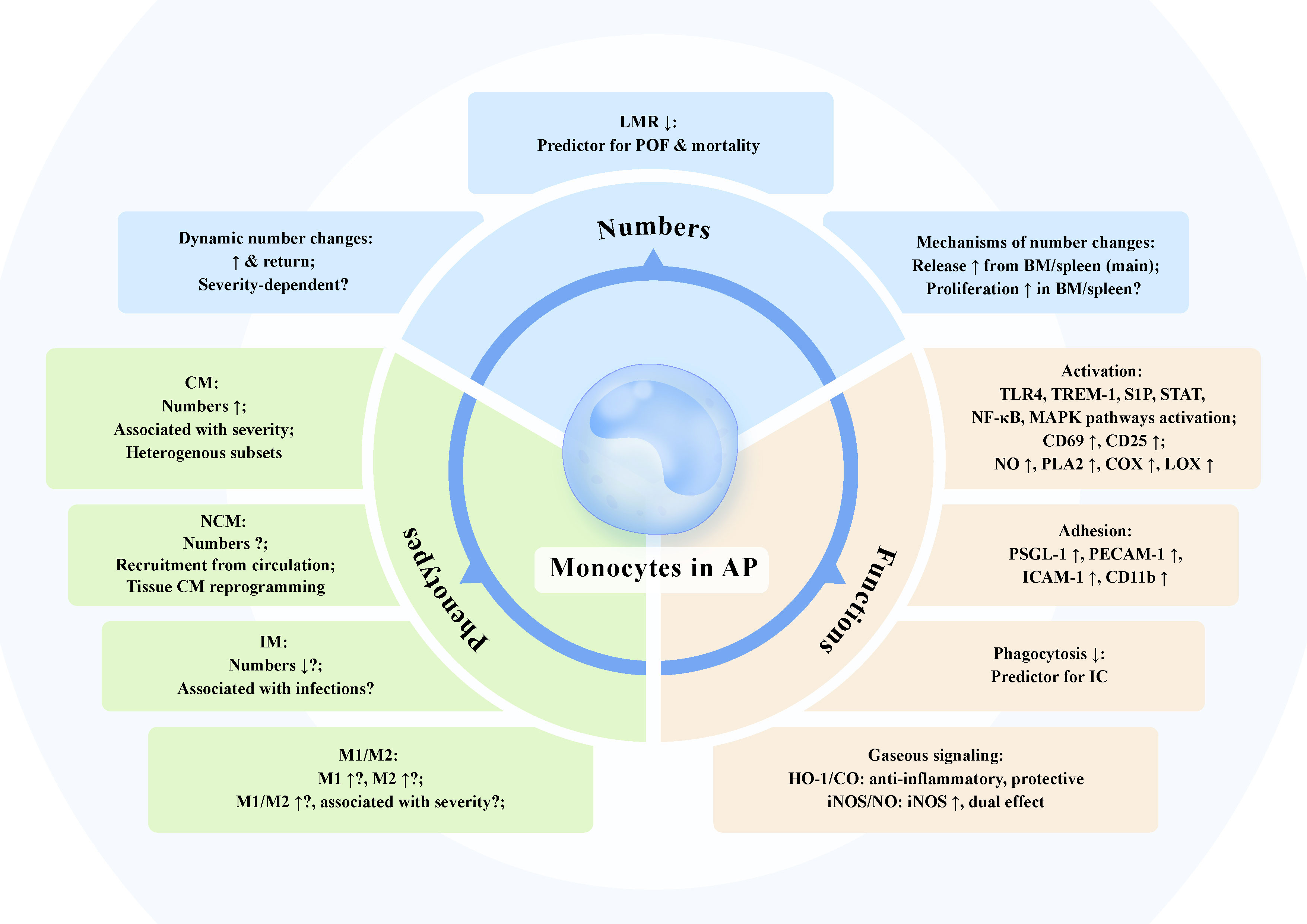
Figure 2 The dynamic changes of monocyte numbers, phenotypes, and functions in acute pancreatitis. Numbers: The number of monocytes in circulation and pancreas is dynamically elevated and might return at various timepoints of experimental and clinical acute pancreatitis (AP). The association of the increased number of monocytes with severity of AP has not been fully verified, despite that the increase in monocytes is more pronounced in more severe phenotype in some cases. Lymphocyte-to-monocyte ratio (LMR) is negatively correlated with AP severity and serves as a predictive index for persistent organ failure and mortality. The augmented number of monocytes within a short time is mainly the function of the release from storage pools. The increased proliferation of monocytes and associated progenitors in bone marrow (BM) and spleen may also contribute to the increased circulating monocytes in AP. Phenotypes: Circulating classical monocytes (CM) levels are associated with escalating AP severity. Novel heterogeneous subsets of monocytes in AP have been identified. The role of non-classical monocytes (NCM) and intermediate monocytes (IM) in AP is still not known. Decreased number of IM in AP might associate with increased susceptibility to infections. Increased M1 and M2 monocytes together with higher M1/M2 ratio can be observed in AP, which possibly correlate with disease severity. Functions: Monocyte activation along with disturbed intracellular multiple signaling profiles and elevated activation markers including CD69 and CD25, leads to massive production of nitric oxide (NO) together with induction of phospholipase A2 (PLA2), cyclooxygenase (COX), and lipoxygenase (LOX). Monocytes in AP exhibit increased adhesion molecules including P-selectin glycoprotein ligand 1 (PSGL-1), platelet endothelial cell adhesion molecule-1 (PECAM-1), and intercellular adhesion molecule 1 (ICAM-1), promoting monocyte adhesion to endothelium and migration to the inflamed pancreas. Impaired phagocytosis of monocytes in AP might be a useful predictor for infectious complication. Heme oxygenase-1 (HO-1)/carbon monoxide (CO) and inducible NO synthase (iNOS)/NO are pivotal gaseous signaling pathways in monocytes of AP. HO-1/CO pathway in monocytes exerts anti-inflammatory effect to dampen AP severity. Increased expression of iNOS in monocytes is observed in AP and the generated NO has dual effect on the production of pro-inflammatory mediators depending on concentrations.
3.3.1. Activation
Monocyte activation takes place in response to activation of TLR4, triggering receptor expressed on myeloid cells-1 (TREM-1) or sphingosine-1-phosphate (S1P) signaling, transduced via the signal transducer and activator of transcription (STAT), nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways (134). Disorders of these signaling profiles in monocytes in AP are briefly summarized in Table 2. Evidence for the enhanced activation of monocytes in AP can be seen in both experimental and clinical AP. CD69, the early activation inducer molecule, is a C-type lectin binding protein with a transmembrane and intracellular signaling domain expressed on a wide range of immune cells, including monocytes. Activation of CD69 on monocytes results in nitric oxide production, and induction of phospholipase A2, cyclooxygenase, and lipoxygenase (67). CD25, an IL-2 receptor alpha chain, is expressed on activated T cells, B cells, and monocytes (67). Elevated expressions of both CD69 and CD25 were found in AP patients compared with healthy controls (67, 139).
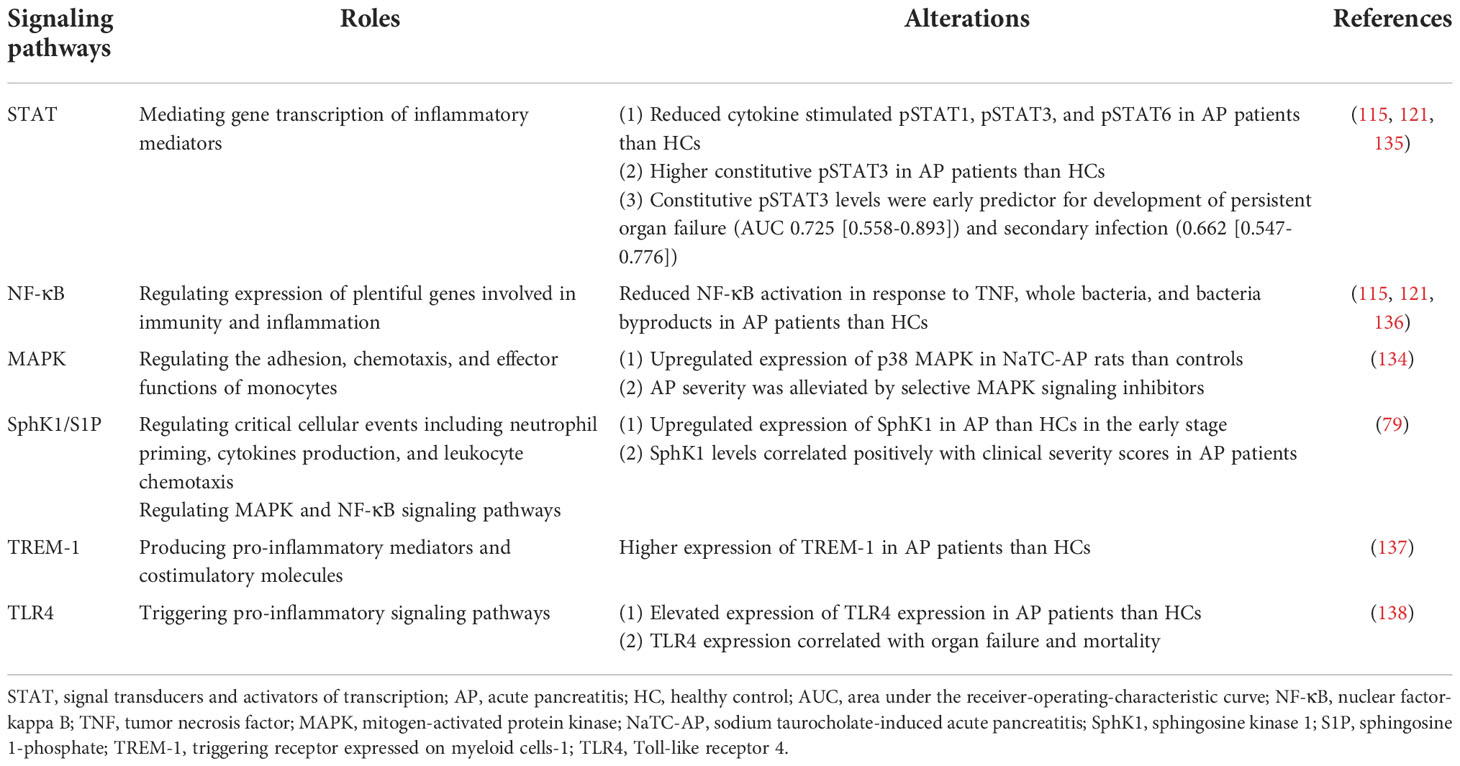
Table 2 Aberrant signaling profiles of monocytes in acute pancreatitis.
3.3.2. Adhesion
Monocyte adhesion to activated endothelial cells is an essential step for the capture, rolling and transmigration of monocytes into target tissues. This process is regulated by a number of different cell surface proteins on both monocytes and endothelial cells. For example, P-selectin glycoprotein ligand 1 (PSGL-1, CD162) modulates the migration, activation, and infiltration of immune cells through binding with E-, P-, or L-selectin (69, 140). Peripheral monocytes from AP patients exhibited increased PSGL-1 expression compared with monocytes from healthy controls (69). Genetic knockout of PSGL-1 in CER-AP mice resulted in less monocyte-endothelial interaction, fewer infiltrating pancreatic monocytes/macrophages and peripheral Ly6C+ monocytes and a milder disease course (69). Platelet endothelial cell adhesion molecule 1 (PECAM-1; CD31) and intercellular adhesion molecule 1 (ICAM-1; CD54) are adhesion molecules found on the surface of activated endothelium as well as on some immune cells including monocytes and their expression is upregulated in experimental AP (141). In patients, cellular adhesion molecules in general, but ICAM-1 in particular, have been shown to be expressed in greater amounts in patients experiencing lung injury and respiratory failure in the context of severe acute pancreatitis (67, 139, 142). CD11b, a member of the β2 integrin family of adhesion molecules, is also up-regulated on circulating monocytes in experimental AP rats at 6, 12, 24, and 48 h after disease induction (66), and in AP patients over healthy controls (139). The degree of overexpression in peripheral monocytes was again associated with AP severity and organ dysfunction (107).
3.3.3. Phagocytosis
Phagocytosis by monocytes/macrophages is critical for the clearance of microbial pathogens and apoptotic cells in order to maintain homeostasis; it is a rare event at rest, but increases during inflammation upon stimulation by bacterial products, cellular debris or cytokines (143). The phagocytic activity of monocytes is a measurable entity that may provide further insight about the innate immune response in specific disease conditions (144). Impaired phagocytosis in monocytes is associated with immunosuppression in severe trauma and sepsis (145–147). In AP patients, the phagocytotic (148, 149) and ROS generation capacity (150) of circulating monocytes is impaired and this phagocytosis potential may be a useful predictor for infectious complications. By calculating the area under the receiver-operating-characteristic curve (AUC), a statistical tool routinely used for evaluating the discriminatory ability of continuous markers, it was reported that the admission phagocytosis potential ([ROS generation index value after stimulation-basal value]/basal value) had an AUC of 0.84 with sensitivity and specificity of 76.2% and 83.6%, respectively, for predicting infectious complications in patients with MSAP/SAP (150).
3.3.4. Gaseous signaling
Activated monocytes contribute to gaseous signaling through the generation of carbon monoxide (CO) and nitric oxide (NO). Specifically, CO serves as a gaseous signaling molecule exerting anti-inflammatory effects through regulation of p38 MAPK and IL-10 pathways (151, 152), whereas NO regulates intracellular signaling molecules such as MAPK, JAK, NF-κB, and activating protein-1, mediating the production of pro-inflammatory cytokines and chemokines (153, 154).
CO is a downstream product of heme oxygenase-1 (HO-1) released together with biliverdin and ferritin as a consequence of heme metabolism, all of which contribute to an anti-inflammatory effect (155). Monocytes expressing HO-1 are anti-inflammatory and rapidly recruited to the pancreas dampening the severity of CER-AP and CDE-AP in mice (156). In MAP patients, HO-1 expression of monocytes on admission was higher than healthy controls (156). The level of HO-1 expression was highest on admission and decreased to normal levels on day 3 with clinical improvement (156). The release of CO can be achieved in a controlled way by administration of CO-releasing molecule-2 (CORM-2) without disturbing carboxyhemoglobin levels. CORM-2 suppressed TLR4/MD2 receptor complex activation and TNF-α production by macrophages in pancreas and spleen, attenuating cytokine cascade and organ injury in CER-AP and CDE-AP in mice (157). CORM-2 further inhibited pancreatic CCL2 production and blocked CCL2-triggered CCR2 endocytosis, thus impairing pathological CD11b+Ly6Chi monocyte recruitment from the blood to pancreas (94). This axis has also been effectively targeted with therapy using haemin, a HO-1 inducing agent in clinical use for the treatment of thalassemia intermedia. Haemin has been shown to upregulate HO-1 expression in peritoneal and pancreatic macrophages, which reduced severity of CER-AP and CDE-AP in mice (140, 156).
NO is generated via the activity of nitric oxide synthase (NOS), which is inducible in inflammatory cells in response to activation (153). Increased expression of iNOS could be detected in peritoneal monocytes/macrophages in NaTC-AP but not CER-AP rats (158), and while low NO concentrations ameliorated microcirculation disturbances, large concentrations contributed to capillary leakage and micro-circulatory failure (159). Clinically, elevated iNOS expression was detected in monocytes from AP patients with systemic inflammatory response syndrome, but no significant changes were observed in those without (160).
4. Obesity and lipid modulation of monocytes in AP
Obesity, as defined by a body mass index of 30 kg/m2 or more, has reached global pandemic levels (161). Beyond merely an excess in body weight, it is a metabolic disorder associated with excessive lipid storage and chronic, low-grade inflammation (161). The relationship between obesity and AP has been gradually explored using various obesity-related AP animal models. These models include induction of NaTC-AP in high-fat diet (HFD) fed rats (162) and CER-AP (or CER/LPS-AP) in HFD fed, leptin-deficient ob/ob, or leptin receptor-defective db/db mice (163–165) as well as the administration of IL-12 plus IL-18 in ob/ob mice or HFD mice (166, 167). Most recently, our group has shown that administration of alcohol in HFD mice could induce AP with multiorgan injury mediated by adipocyte tissue lipolysis (168). Clinically, studies have linked obesity to an increased incidence and severity of AP (169). Obesity may aggravate AP severity through unsaturated fatty acids via lipase mediated lipolysis and various adipokines from adipose tissue (170–172). During AP, pancreatic lipase leaks from the injured pancreas into adipose tissue with increased amount and activity, generating excessive free fatty acids via lipolysis, which in turn worsen systemic inflammation and organ failure in AP (173, 174). Genetic knockout or pharmacological inhibition of pancreatic lipase reduced adipose tissue lipolysis, systemic inflammation, organ failure, and mortality of AP in ob/ob obese mice (170, 173, 174).
Dyslipidemias are disorders of the plasma lipid profile that are frequently related with obesity and diabetes (175, 176). With the recognition of metabolically healthy obesity and metabolically unhealthy normal weight subjects, the association of obesity and dyslipidemia becomes complex and intriguing (176). Hypertriglyceridemia (HTG) is a highly prevalent form of dyslipidemia with major health concern (175). HTG is the third most common cause of AP globally (177) and is one of the most leading etiologies in Chinese AP cohorts (178–181). Apart from elevated triglyceride levels, HTG-AP is also accompanied with maladjusted lipoprotein metabolism (177). Furthermore, the severity of AP is proportionally increased with escalating triglyceride levels (178, 182, 183).
Adipocytes release lipids through lipase-mediated lipolysis or lipid-filled exosomes, modulating the differentiation and function of monocytes/macrophages (184). Activation and polarization of monocytes/macrophages is affected by lipids via direct effects on the physical properties of plasma membranes or indirectly with lipids acting as signaling molecules (185, 186). Distinct lipid species (saturated or unsaturated fatty acids, cholesterols, and lipoproteins), lipid modifications (i.e., oxidation) and dyslipidemias (hypercholesterolemia and hypertriglyceridemia) affect monocyte responses in different ways and relevant studies are summarized in Supplementary Table 3. In extremely obese mice, adipose tissue macrophage content is over 50% of the total cell count and most are derived from circulating monocytes (187). Obesity preferentially promotes M1 polarization of monocytes/macrophages in adipose tissue of mice but the phenotypes of adipose tissue macrophages in human are not fully determined (187).
Adipose tissue, particularly areas of fat necrosis, is a vital source of inflammatory mediators promoting the pro-inflammatory M1 polarization of monocytes/macrophages and inhibition of the M2 reparative phenotype, thereby exacerbating systemic inflammation in PDL-AP or NaTC-AP rats (188, 189). It was also observed that oxidized free fatty acids in ascitic fluid interfered with the anti-inflammatory regulation of monocytes/macrophages, aggravating inflammatory responses in NaTC-AP rats (190). As aforementioned, the infiltration of macrophages in adipose tissue is increased in obese mice. After CER/LPS challenge to HFD mice, despite an overall decreased count of adipose tissue macrophages, the proportion of M1 adipose tissue macrophages increased together with enhanced expression of pro-inflammatory cytokines in adipose tissue (165). Consistent with experimental studies, CD14+CD86+ M1 monocytes increased in circulation in HTG population compared with healthy controls (68). Likewise, HTG-AP patients displayed higher M1 monocyte counts than respective HTG control patients on days 1, 3, and 7 after admission. Most importantly, the level of peripheral M1 monocytes was positively correlated with plasma triglyceride level and Ranson score (a scoring system to assess and prognosticate AP severity) in HTG-AP patients (68). However, there was no obvious difference in peripheral M2 monocytes between HTG-AP patients and HTG control patients (68). Besides, the level of M2 monocytes exhibited no association with either plasma triglyceride level or Ranson score in HTG-AP patients (68). The effects of obesity and dyslipidemias on the recruitment and polarization of monocytes/macrophages in AP are still unclear, however, the mechanism by which obesity and dyslipidemias exacerbate AP severity might, at least in part, be attributed to disturbing monocyte homeostasis (i.e., promoting M1 monocytes polarization). How far this effect is mediated by lipotoxicity of specific lipids/lipid groups warrants further investigation (Figure 3).
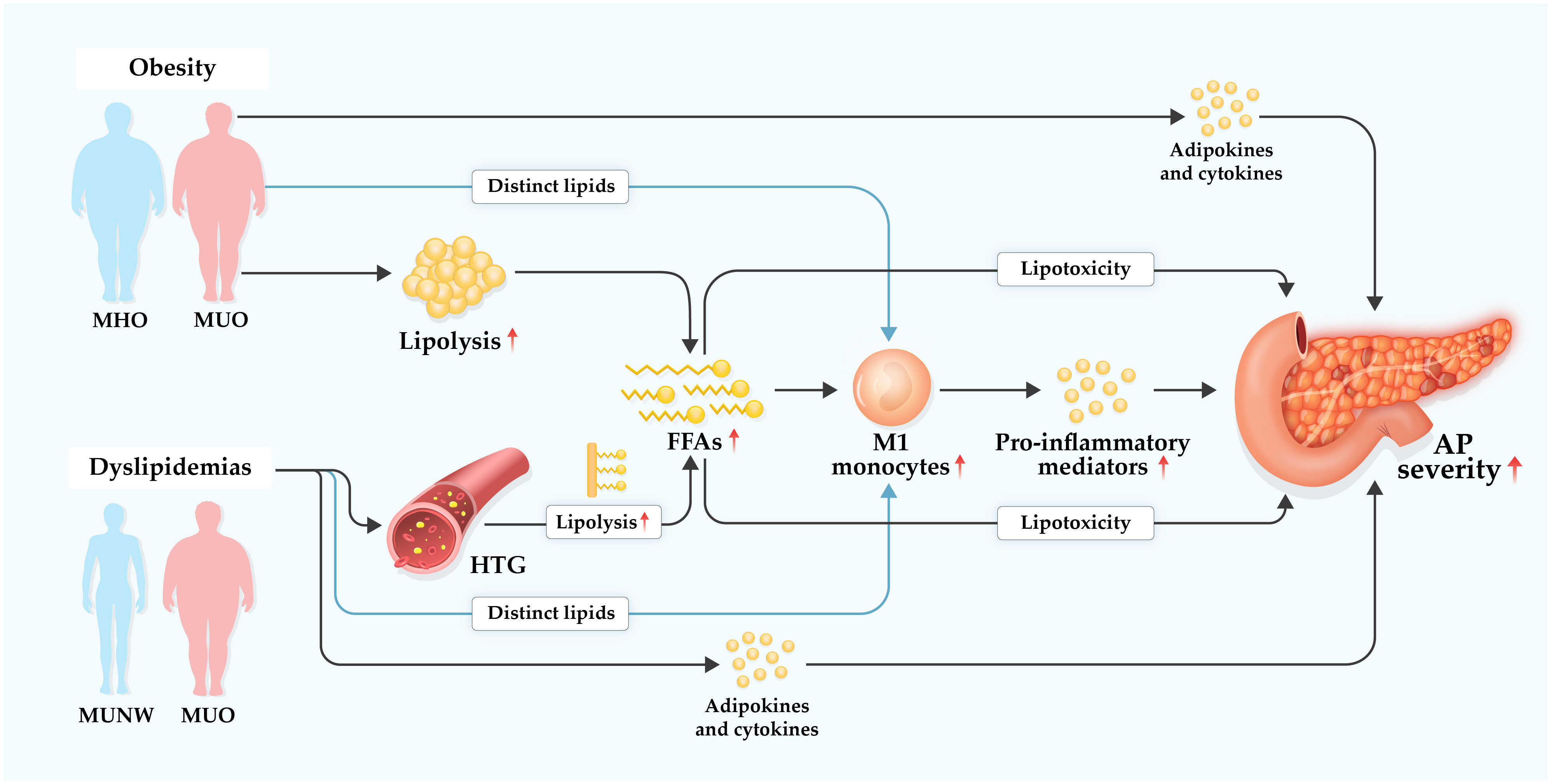
Figure 3 Hypothetical diagram of underlying mechanisms of obesity and dyslipidemias in aggravating acute pancreatitis. Dyslipidemias are prevalent in individuals with metabolically unhealthy obesity (MUO) or metabolically unhealthy normal weight (MUNM), but less prominent or even absent in individuals with metabolically healthy obesity (MHO). Hypertriglyceridemia (HTG), a common form of dyslipidemias, is one of leading etiologies of acute pancreatitis (AP) with increasing global incidence. Obesity, particularly MUO, and dyslipidemias, especially HTG, may exacerbate AP severity in several complex multifactorial ways. Excessive free fatty acids (FFAs) are accumulated via elevated lipolysis of visceral adipocyte triglycerides during AP. FFAs possess regulatory effect on monocytes and may promote their M1 polarization which induces increased production of pro-inflammatory mediators, resulting in amplified systemic inflammation. Of note, local and systemic lipotoxicity of FFAs can directly cause tissue damage and multiple organ failure in AP. Besides FFAs, other distinct lipid species may modulate generation and function of monocytes in AP. Furthermore, various adipokines and cytokines with enhanced secretion in MUO and dyslipidemia are involved in boosting systemic inflammatory response that contributes to aggravated severity of AP.
5. Conclusions and perspectives
Due to the difficulties in studying the human pancreas directly, circulating immune cells in general, and monocytes in particular, serve as excellent intermediates for assessing systemic inflammation and immune status. Monocytes play a pivotal role in the initiation and progression of AP, but important questions regarding the precise role of monocytes in the pathogenesis of AP remain. These include:
1. Dynamic changes and roles of monocyte subsets are unclear, as indeed are the degree to which cells can be encouraged into different expression patterns using environmental cues. It is still unknown how the spatially defined signals in the inflamed pancreas serve to reprogram monocyte subsets and determine their fate. Monocytes could extravasate into tissue retaining monocyte phenotypic markers without differentiating into macrophages. Increasing recognition of heterogeneity in monocyte population makes identification of tissue monocytes and deciphering the transition of monocyte subsets into particular tissue macrophages more challenging. Monocytes are remarkably multipotent cells in regulating inflammation but the pathogenetic mechanisms of monocytes in regulating AP severity are not fully investigated. More fundamentally, novel technologies have identified cells based on their distinct transcription patterns, whereby traditionally much simpler phenotypic markers were used. These can greatly compromise comparability between studies. A consensus on relevant monocyte subsets based on multiple different detection technologies, and their lineage, might be developed for better communications between laboratories and future clinical appliance.
2. The finely orchestrated balance between pro- and anti-inflammatory responses is disturbed in more severe forms of AP, which is reflected in imbalances in monocyte function. Obesity and dyslipidemias aggravate AP severity with varying degrees depending on individual factors. Accumulation of free fatty acids generated by elevated lipolysis in patients with metabolically unhealthy obesity and HTG may partially contribute to the exacerbation of AP via promoting M1 monocytes polarization, which needs further investigations. Besides free fatty acids, other lipid species are capable of regulating subsets, numbers, and functions of monocytes, but their specific involvement in dynamic changes of monocytes during AP is largely unknown. Future studies, especially those aiming to develop targeted therapy, should design methods to differentiate between causal and associative patterns in monocyte changes with disease in order to guide the development of effective therapy.
3. Immune markers on circulating monocytes provide a potential window for identifying particular inflammation types in diseases. It should be possible to refine the inflammatory signatures of immune cells and develop specific ‘immune signaling fingerprints’ to monitor the inflammatory response and disease progression during AP. These could be used to guide personalized therapeutic strategies using anti-inflammatory, immunomodulatory, immunostimulatory agents alone or in combination for the optimal treatment of individual patients using a personalized medicine approach.
Author contributions
SL, TL, WH, and QX designed this work and wrote the manuscript. PS and J-WL critically revised the manuscript. QW, RS, and LC had important intellectual input. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China (No. 81973632, No. 82270672 to WH; No. 82274321 to QX); Sichuan Science and Technology Plan Projects – Key Research and Development Program (awarded to WH); Program of Science and Technology Department of Sichuan Province (No. 2022YFS0419, to TL); National Institute for Health Research Senior Investigator Award, an EME Award (15/20/01), an MRC Award (MR/T002220/1), and the TransBioLine Consortium (all to RS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1062849/full#supplementary-material
References
1. Iannuzzi JP, King JA, Leong JH, Quan J, Windsor JW, Tanyingoh D, et al. Global incidence of acute pancreatitis is increasing over time: A systematic review and meta-analysis. Gastroenterology (2022) 162(1):122–34. doi: 10.1053/j.gastro.2021.09.043
2. Mederos MA, Reber HA, Girgis MD. Acute pancreatitis: A review. JAMA (2021) 325(4):382–90. doi: 10.1001/jama.2020.20317
3. Lee PJ, Papachristou GL. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol (2019) 16(8):479–96. doi: 10.1038/s41575-019-0158-2
4. Barreto SG, Habtezion A, Gukovskaya A, Lugea A, Jeon C, Yadav D, et al. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut (2021) 70(1):194–203. doi: 10.1136/gutjnl-2020-322163
5. Szatmary P, Grammatikopoulos T, Cai W, Huang W, Mukherjee R, Halloran C, et al. Acute Pancreatitis: Diagnosis and Treatment. Drugs. (2022) 82(12):1251–76. doi: 10.1007/s40265-022-01766-4
6. Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology (2013) 144(6):1230–40. doi: 10.1053/j.gastro.2012.12.042
7. Xue J, Sharma V, Habtezion A. Immune cells and immune-based therapy in pancreatitis. Immunol Res (2014) 58(2-3):378–86. doi: 10.1007/s12026-014-8504-5
8. Gukovskaya AS, Gukovsky I, Algül H, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology (2017) 153(5):1212–26. doi: 10.1053/j.gastro.2017.08.071
9. Peng C, Li Z, Yu X. The role of pancreatic infiltrating innate immune cells in acute pancreatitis. Int J Med Sci (2021) 18(2):534–45. doi: 10.7150/ijms.51618
10. Mayerle J. A novel role for leucocytes in determining the severity of acute pancreatitis. Gut (2009) 58(11):1440–1. doi: 10.1136/gut.2009.186692
11. Tsuji N, Watanabe N, Okamoto T, Niitsu Y. Specific interaction of pancreatic elastase and leucocytes to produce oxygen radicals and its implication in pancreatitis. Gut (1994) 35(11):1659–64. doi: 10.1136/gut.35.11.1659
12. Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology (2002) 122(4):974–84. doi: 10.1053/gast.2002.32409
13. John DS, Aschenbach J, Kruger B, Sendler M, Weiss FU, Mayerle J, et al. Deficiency of cathepsin c ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of e-cadherin. J Biol Chem (2019) 294(2):697–707. doi: 10.1074/jbc.RA118.004376
14. Mayerle J, Schnekenburger J, Krüger B, Kellermann J, Ruthenbürger M, Weiss FU, et al. Extracellular cleavage of e-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology (2005) 129(4):1251–67. doi: 10.1053/j.gastro.2005.08.002
15. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology (2015) 149(7):1920–31.e8. doi: 10.1053/j.gastro.2015.08.026
16. Abdulla A, Awla D, Thorlacius H, Regnér S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol (2011) 90(5):975–82. doi: 10.1189/jlb.0411195
17. Yang ZW, Meng XX, Xu P. Central role of neutrophil in the pathogenesis of severe acute pancreatitis. J Cell Mol Med (2015) 19(11):2513–20. doi: 10.1111/jcmm.12639
18. Wan J, Ren Y, Yang X, Li X, Xia L, Lu N. The role of neutrophils and neutrophil extracellular traps in acute pancreatitis. Front Cell Dev Biol (2020) 8:565758. doi: 10.3389/fcell.2020.565758
19. Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J, et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology (2011) 141(5):1915–26.e1-14. doi: 10.1053/j.gastro.2011.07.033
20. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med (2012) 209(9):1671–87. doi: 10.1084/jem.20111706
21. Dib M, Zhao X, Wang X, Andersson R. Mast cells contribute to early pancreatitis-induced systemic endothelial barrier dysfunction. Pancreatology (2002) 2(4):396–401. doi: 10.1159/000065087
22. Lopez-Font I, Gea-Sorlí S, de-Madaria E, Gutiérrez LM, Pérez-Mateo M, Closa D. Pancreatic and pulmonary mast cells activation during experimental acute pancreatitis. World J Gastroenterol (2010) 16(27):3411–7. doi: 10.3748/wjg.v16.i27.3411
23. Zhou Q, Tao X, Xia S, Guo F, Pan C, Xiang H, et al. T Lymphocytes: A promising immunotherapeutic target for pancreatitis and pancreatic cancer? Front Oncol (2020) 10:382. doi: 10.3389/fonc.2020.00382
24. Ding L, Yang Y, Li H, Wang H, Gao P. Circulating lymphocyte subsets induce secondary infection in acute pancreatitis. Front Cell Infect Microbiol (2020) 10:128. doi: 10.3389/fcimb.2020.00128
25. Shrivastava P, Bhatia M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol (2010) 16(32):3995–4002. doi: 10.3748/wjg.v16.i32.3995
26. Wu J, Zhang L, Shi J, He R, Yang W, Habtezion A, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine (2020) 58:102920. doi: 10.1016/j.ebiom.2020.102920
27. Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed Pharmacother Biomed Pharmacother (2020) 131:110693. doi: 10.1016/j.biopha.2020.110693
28. Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. Monocyte subsets: Phenotypes and function in tuberculosis infection. Front Immunol (2018) 9:1726. doi: 10.3389/fimmu.2018.01726
29. Prinyakupt J, Pluempitiwiriyawej C. Segmentation of white blood cells and comparison of cell morphology by linear and naïve bayes classifiers. BioMed Eng Online (2015) 14:63. doi: 10.1186/s12938-015-0037-1
30. Robinson A, Han CZ, Glass CK, Pollard JW. Monocyte regulation in homeostasis and malignancy. Trends Immunol (2021) 42(2):104–19. doi: 10.1016/j.it.2020.12.001
31. Patel AA, Ginhoux F, Yona S. Monocytes, macrophages, dendritic cells and neutrophils: an update on lifespan kinetics in health and disease. Immunology (2021) 163(3):250–61. doi: 10.1111/imm.13320
32. Hume DA, Irvine KM, Pridans C. The mononuclear phagocyte system: The relationship between monocytes and macrophages. Trends Immunol (2019) 40(2):98–112. doi: 10.1016/j.it.2018.11.007
33. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
34. Jung S. Macrophages and monocytes in 2017: Macrophages and monocytes: of tortoises and hares. Nat Rev Immunol (2018) 18(2):85–6. doi: 10.1038/nri.2017.158
35. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
36. Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol (2008) 8(4):290–301. doi: 10.1038/nri2279
37. Ingersoll MA, Platt AM, Potteaux S, Randolph GJ. Monocyte trafficking in acute and chronic inflammation. Trends Immunol (2011) 32(10):470–7. doi: 10.1016/j.it.2011.05.001
38. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol (2013) 14(8):821–30. doi: 10.1038/ni.2638
39. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Sci (New York NY) (2009) 325(5940):612–6. doi: 10.1126/science.1175202
40. Zhu YP, Thomas GD, Hedrick CC, Jeffrey M. 2014 Hoeg award lecture: Transcriptional control of monocyte development. Arteriosclerosis Thrombosis Vasc Biol (2016) 36(9):1722–33. doi: 10.1161/ATVBAHA.116.304054
41. Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood (1989) 74(7):2527–34. doi: 10.1182/blood.V74.7.2527.2527
42. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med (2017) 214(7):1913–23. doi: 10.1084/jem.20170355
43. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
44. Ziegler-Heitbrock L. Monocyte subsets in man and other species. Cell Immunol (2014) 289(1-2):135–9. doi: 10.1016/j.cellimm.2014.03.019
45. Wacleche VS, Tremblay CL, Routy JP, Ancuta P. The biology of monocytes and dendritic cells: Contribution to HIV pathogenesis. Viruses (2018) 10(2):65. doi: 10.3390/v10020065
46. Saluja A, Dudeja V, Dawra R, Sah RP. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology (2019) 156(7):1979–93. doi: 10.1053/j.gastro.2019.01.268
47. Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su HY, et al. Human pancreatic acinar cells: Proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am J Pathol (2017) 187(12):2726–43. doi: 10.1016/j.ajpath.2017.08.017
48. Feng YM, Chen XH, Zhang X. Roles of PECAM-1 in cell function and disease progression. Eur Rev Med Pharmacol Sci (2016) 20(19):4082–8.
49. Szatmary P, Huang W, Criddle D, Tepikin A, Sutton R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med (2018) 22(10):4617–29. doi: 10.1111/jcmm.13797
50. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
51. Cavestro GM, Zuppardo RA, Bertolini S, Sereni G, Frulloni L, Okolicsanyi S, et al. Connections between genetics and clinical data: Role of MCP-1, CFTR, and SPINK-1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am J Gastroenterol (2010) 105(1):199–206. doi: 10.1038/ajg.2009.611
52. Regnér S, Manjer J, Appelros S, Hjalmarsson C, Sadic J, Borgström A. Protease activation, pancreatic leakage, and inflammation in acute pancreatitis: differences between mild and severe cases and changes over the first three days. Pancreatology (2008) 8(6):600–7. doi: 10.1159/000161011
53. Rau B, Baumgart K, Krüger CM, Schilling M, Beger HG. CC-chemokine activation in acute pancreatitis: enhanced release of monocyte chemoattractant protein-1 in patients with local and systemic complications. Intensive Care Med (2003) 29(4):622–9. doi: 10.1007/s00134-003-1668-4
54. Brady M, Bhatia M, Christmas S, Boyd MT, Neoptolemos JP, Slavin J. Expression of the chemokines MCP-1/JE and cytokine-induced neutrophil chemoattractant in early acute pancreatitis. Pancreas (2002) 25(3):260–9. doi: 10.1097/00006676-200210000-00008
55. Ding Z, Liu J, Lin R, Hou XH. Experimental pancreatitis results in increased blood-brain barrier permeability in rats: a potential role of MCP-1. J Digestive Dis (2012) 13(3):179–85. doi: 10.1111/j.1751-2980.2011.00568.x
56. Regnér S, Appelros S, Hjalmarsson C, Manjer J, Sadic J, Borgstrom A. Monocyte chemoattractant protein 1, active carboxypeptidase b and CAPAP at hospital admission are predictive markers for severe acute pancreatitis. Pancreatology (2008) 8(1):42–9. doi: 10.1159/000114866
57. Vaquero E, Gukovsky I, Zaninovic V, Gukovskaya AS, Pandol SJ. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol (2001) 280(6):G1197–208. doi: 10.1152/ajpgi.2001.280.6.G1197
58. Yang X, Yao L, Fu X, Mukherjee R, Xia Q, Jakubowska MA, et al. Experimental acute pancreatitis models: History, current status, and role in translational research. Front Physiol (2020) 11:614591. doi: 10.3389/fphys.2020.614591
59. Schäfer C, Tietz AB, Göke B. Pathophysiology of acute experimental pancreatitis: lessons from genetically engineered animal models and new molecular approaches. Digestion (2005) 71(3):162–72. doi: 10.1159/000086138
60. Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol (2007) 69:249–69. doi: 10.1146/annurev.physiol.69.031905.161253
61. Kui B, Balla Z, Végh ET, Pallagi P, Venglovecz V, Iványi B, et al. Recent advances in the investigation of pancreatic inflammation induced by large doses of basic amino acids in rodents. Lab Invest (2014) 94(2):138–49. doi: 10.1038/labinvest.2013.143
62. Lerch MM, Aghdassi AA. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology (2010) 138(2):429–33. doi: 10.1053/j.gastro.2009.12.012
63. Wan MH, Huang W, Latawiec D, Jiang K, Booth DM, Elliott V, et al. Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB Off J Int Hepato Pancreato Biliary Assoc (2012) 14(2):73–81. doi: 10.1111/j.1477-2574.2011.00408.x
64. Manohar M, Jones EK, Rubin SJS, Subrahmanyam PB, Swaminathan G, Mikhail D, et al. Novel circulating and tissue monocytes as well as macrophages in pancreatitis and recovery. Gastroenterology (2021) 161(6):2014–29.e14. doi: 10.1053/j.gastro.2021.08.033
65. Meriläinen S, Mäkelä J, Anttila V, Koivukangas V, Kaakinen H, Niemelä E, et al. Acute edematous and necrotic pancreatitis in a porcine model. Scand J Gastroenterol (2008) 43(10):1259–68. doi: 10.1080/00365520802158580
66. de Dios I, Perez M, de la Mano A, Sevillano S, Orfao A, Ramudo L, et al. Contribution of circulating leukocytes to cytokine production in pancreatic duct obstruction-induced acute pancreatitis in rats. Cytokine (2002) 20(6):295–303. doi: 10.1006/cyto.2002.2011
67. Dabrowski A, Osada J, Dabrowska MI, Wereszczynska-Siemiatkowska U. Monocyte subsets and natural killer cells in acute pancreatitis. Pancreatology (2008) 8(2):126–34. doi: 10.1159/000123605
68. Zheng J, Fan J, Huang C, Lu Y, Huang Z, Wang X, et al. Dynamic detection of monocyte subsets in peripheral blood of patients with acute hypertriglyceridemic pancreatitis. Gastroenterol Res Practice (2019) 2019:5705782. doi: 10.1155/2019/5705782
69. Zhang X, Zhu M, Jiang XL, Liu X, Liu X, Liu P, et al. P-selectin glycoprotein ligand 1 deficiency prevents development of acute pancreatitis by attenuating leukocyte infiltration. World J Gastroenterol (2020) 26(41):6361–77. doi: 10.3748/wjg.v26.i41.6361
70. Li Y, Zhao Y, Feng L, Guo R. Comparison of the prognostic values of inflammation markers in patients with acute pancreatitis: a retrospective cohort study. BMJ Open (2017) 7(3):e013206. doi: 10.1136/bmjopen-2016-013206
71. Liu G, Tao J, Zhu Z, Wang W. The early prognostic value of inflammatory markers in patients with acute pancreatitis. Clinics Res Hepatol Gastroenterol (2019) 43(3):330–7. doi: 10.1016/j.clinre.2018.11.002
72. Mubder M, Dhindsa B, Nguyen D, Saghir S, Cross C, Makar R, et al. Utility of inflammatory markers to predict adverse outcome in acute pancreatitis: A retrospective study in a single academic center. Saudi J Gastroenterol Off J Saudi Gastroenterol Assoc (2020) 26(4):216–21. doi: 10.4103/sjg.SJG_49_20
73. Junare PR, Debnath P, Nair S, Chandnani S, Udgirkar S, Thange R, et al. Complete hemogram: simple and cost-effective in staging and predicting outcome in acute pancreatitis. Wien Klin Wochenschr (2021) 133(13-14):661–8. doi: 10.1007/s00508-021-01821-2
74. Khan NA, Haider Kazmi SJ, Asghar MS, Singh M, Iqbal S, Jawed R, et al. Hematological indices predicting the severity of acute pancreatitis presenting to the emergency department: A retrospective analysis. Cureus (2021) 13(7):e16752. doi: 10.7759/cureus.16752
75. Djordjevic D, Rondovic G, Surbatovic M, Stanojevic I, Udovicic I, Andjelic T, et al. Neutrophil-to-Lymphocyte ratio, monocyte-to-Lymphocyte ratio, platelet-to-Lymphocyte ratio, and mean platelet volume-to-Platelet count ratio as biomarkers in critically ill and injured patients: Which ratio to choose to predict outcome and nature of bacteremia? Mediators Inflammation (2018) 2018:3758068. doi: 10.1155/2018/3758068
76. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, et al. Classification of acute pancreatitis–2012: revision of the Atlanta classification and definitions by international consensus. Gut (2013) 62(1):102–11. doi: 10.1136/gutjnl-2012-302779
77. Kolber W, Kuśnierz-Cabala B, Maraj M, Kielar M, Mazur P, Maziarz B, et al. Neutrophil to lymphocyte ratio at the early phase of acute pancreatitis correlates with serum urokinase-type plasminogen activator receptor and interleukin 6 and predicts organ failure. Folia Med Cracoviensia (2018) 58(4):57–74. doi: 10.24425/fmc.2018.125704
78. Pian G, Li H, Piao Y. Clinical significance of inflammation markers in predicting the severity of acute pancreatitis. Pancreas (2021) 50(2):201–5. doi: 10.1097/MPA.0000000000001749
79. Li Q, Wang C, Zhang Q, Tang C, Li N, Li J. The role of sphingosine kinase 1 in patients with severe acute pancreatitis. Ann Surg (2012) 255(5):954–62. doi: 10.1097/SLA.0b013e31824d2ca4
80. Sendler M, Dummer A, Weiss FU, Krüger B, Wartmann T, Scharffetter-Kochanek K, et al. Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut (2013) 62(3):430–9. doi: 10.1136/gutjnl-2011-300771
81. Mikami Y, Takeda K, Shibuya K, Qiu-Feng H, Shimamura H, Yamauchi J, et al. Do peritoneal macrophages play an essential role in the progression of acute pancreatitis in rats? Pancreas (2003) 27(3):253–60. doi: 10.1097/00006676-200310000-00011
82. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arteriosclerosis Thrombosis Vasc Biol (2017) 37(1):35–42. doi: 10.1161/ATVBAHA.116.308198
83. Perides G, Weiss ER, Michael ES, Laukkarinen JM, Duffield JS, Steer ML. TNF-alpha-dependent regulation of acute pancreatitis severity by ly-6C(hi) monocytes in mice. J Biol Chem (2011) 286(15):13327–35. doi: 10.1074/jbc.M111.218388
84. Zhou GX, Zhu XJ, Ding XL, Zhang H, Chen JP, Qiang H, et al. Protective effects of MCP-1 inhibitor on a rat model of severe acute pancreatitis. Hepatobiliary Pancreat Dis Int (2010) 9(2):201–7. doi: 10.1016/j.intimp.2020.106900
85. Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol (2005) 288(6):G1259–65. doi: 10.1152/ajpgi.00435.2004
86. Saeki K, Kanai T, Nakano M, Nakamura Y, Miyata N, Sujino T, et al. CCL2-induced migration and SOCS3-mediated activation of macrophages are involved in cerulein-induced pancreatitis in mice. Gastroenterology (2012) 142(4):1010–20.e9. doi: 10.1053/j.gastro.2011.12.054
87. Meuret G, Bammert J, Hoffmann G. Kinetics of human monocytopoiesis. Blood (1974) 44(6):801–16. doi: 10.1182/blood.V44.6.801.801
88. Potteaux S, Ait-Oufella H, Mallat Z. Role of splenic monocytes in atherosclerosis. Curr Opin Lipidol (2015) 26(5):457–63. doi: 10.1097/MOL.0000000000000223
89. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest (2007) 117(4):902–9. doi: 10.1172/JCI29919
90. Kamei M, Carman CV. New observations on the trafficking and diapedesis of monocytes. Curr Opin Hematol (2010) 17(1):43–52. doi: 10.1097/MOH.0b013e3283333949
91. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A (2012) 109(7):2491–6. doi: 10.1073/pnas.1113744109
92. Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arteriosclerosis Thrombosis Vasc Biol (2015) 35(5):1066–70. doi: 10.1161/ATVBAHA.114.304652
93. Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, et al. Extramedullary hematopoiesis generates ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation (2012) 125(2):364–74. doi: 10.1161/CIRCULATIONAHA.111.061986
94. Wu J, Zhang R, Hu G, Zhu HH, Gao WQ, Xue J. Carbon monoxide impairs CD11b(+)Ly-6C(hi) monocyte migration from the blood to inflamed pancreas via inhibition of the CCL2/CCR2 axis. J Immunol (2018) 200(6):2104–14. doi: 10.4049/jimmunol.1701169
95. Yasuda T, Takeyama Y, Ueda T, Takase K, Nishikawa J, Kuroda Y. Splenic atrophy in experimental severe acute pancreatitis. Pancreas (2002) 24(4):365–72. doi: 10.1097/00006676-200205000-00007
96. Zhou R, Zhang J, Bu W, Zhang W, Duan B, Wang X, et al. A new role for the spleen: Aggravation of the systemic inflammatory response in rats with severe acute pancreatitis. Am J Pathol (2019) 189(11):2233–45. doi: 10.1016/j.ajpath.2019.07.008
97. Rahman SH, Salter G, Holmfield JH, Larvin M, McMahon MJ. Soluble CD14 receptor expression and monocyte heterogeneity but not the c-260T CD14 genotype are associated with severe acute pancreatitis. Crit Care Med (2004) 32(12):2457–63. doi: 10.1097/01.CCM.0000148008.99716.9C
98. Zhang R, Shi J, Zhang R, Ni J, Habtezion A, Wang X, et al. Expanded CD14(hi)CD16(-) immunosuppressive monocytes predict disease severity in patients with acute pancreatitis. J Immunol (2019) 202(9):2578–84. doi: 10.4049/jimmunol.1801194
99. Nalisa M, Nweke EE, Smith MD, Omoshoro-Jones J, Devar JW, Metzger R, et al. Chemokine receptor 8 expression may be linked to disease severity and elevated interleukin 6 secretion in acute pancreatitis. World J Gastrointestinal Pathophysiol (2021) 12(6):115–33. doi: 10.4291/wjgp.v12.i6.115
100. Crane MJ, Daley JM, van Houtte O, Brancato SK, Henry WL Jr., Albina JE. The monocyte to macrophage transition in the murine sterile wound. PloS One (2014) 9(1):e86660. doi: 10.1371/journal.pone.0086660
101. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res (2014) 114(10):1611–22. doi: 10.1161/CIRCRESAHA.114.303204
102. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med (2015) 212(4):447–56. doi: 10.1084/jem.20141539
103. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
104. Volk HD, Reinke P, Krausch D, Zuckermann H, Asadullah K, Müller JM, et al. Monocyte deactivation–rationale for a new therapeutic strategy in sepsis. Intensive Care Med (1996) 22 Suppl 4:S474–81. doi: 10.1007/BF01743727
105. Richter A, Nebe T, Wendl K, Schuster K, Klaebisch G, Quintel M, et al. HLA-DR expression in acute pancreatitis. Eur J Surg (1999) 165(10):947–51. doi: 10.1080/110241599750008053
106. Gotzinger P, Sautner T, Spittler A, Barlan M, Wamser P, Roth E, et al. Severe acute pancreatitis causes alterations in HLA-DR and CD14 expression on peripheral blood monocytes independently of surgical treatment. Eur J Surg (2000) 166(8):628–32. doi: 10.1080/110241500750008286
107. Kylanpaa-Back ML, Takala A, Kemppainen E, Puolakkainen P, Kautiainen H, Jansson SE, et al. Cellular markers of systemic inflammation and immune suppression in patients with organ failure due to severe acute pancreatitis. Scand J Gastroenterol (2001) 36(10):1100–7. doi: 10.1080/003655201750422738
108. Satoh A, Miura T, Satoh K, Masamune A, Yamagiwa T, Sakai Y, et al. Human leukocyte antigen-DR expression on peripheral monocytes as a predictive marker of sepsis during acute pancreatitis. Pancreas (2002) 25(3):245–50. doi: 10.1097/00006676-200210000-00006
109. Mentula P, Kylanpaa-Back ML, Kemppainen E, Takala A, Jansson SE, Kautiainen H, et al. (human leucocyte antigen)-DR expression on peripheral blood monocytes predicts the development of organ failure in patients with acute pancreatitis. Clin Sci (Lond) (2003) 105(4):409–17. doi: 10.1042/CS20030058
110. Mentula P, Kylanpaa ML, Kemppainen E, Jansson SE, Sarna S, Puolakkainen P, et al. Plasma anti-inflammatory cytokines and monocyte human leucocyte antigen-DR expression in patients with acute pancreatitis. Scand J Gastroenterol (2004) 39(2):178–87. doi: 10.1080/00365520310008278
111. Yu WK, Li WQ, Li N, Li JS. Mononuclear histocompatibility leukocyte antigen-DR expression in the early phase of acute pancreatitis. Pancreatology (2004) 4(3-4):233–43. doi: 10.1159/000078748
112. Kylanpaa ML, Mentula P, Kemppainen E, Puolakkainen P, Aittomaki S, Silvennoinen O, et al. Monocyte anergy is present in patients with severe acute pancreatitis and is significantly alleviated by granulocyte-macrophage colony-stimulating factor and interferon-gamma in vitro. Pancreas (2005) 31(1):23–7. doi: 10.1097/01.mpa.0000164449.23524.94
113. Ho YP, Sheen IS, Chiu CT, Wu CS, Lin CY. A strong association between down-regulation of HLA-DR expression and the late mortality in patients with severe acute pancreatitis. Am J Gastroenterol (2006) 101(5):1117–24. doi: 10.1111/j.1572-0241.2006.00495.x
114. Mentula P, Kylanpaa ML, Kemppainen E, Repo H, Puolakkainen P. Early inflammatory response in acute pancreatitis is little affected by body mass index. Scand J Gastroenterol (2007) 42(11):1362–8. doi: 10.1080/00365520701427086
115. Oiva J, Mustonen H, Kylänpää ML, Kyhälä L, Alanärä T, Aittomäki S, et al. Patients with acute pancreatitis complicated by organ failure show highly aberrant monocyte signaling profiles assessed by phospho-specific flow cytometry. Crit Care Med (2010) 38(8):1702–8. doi: 10.1097/CCM.0b013e3181e7161c
116. Ho YP, Chiu CT, Sheen IS, Tseng SC, Lai PC, Ho SY, et al. Tumor necrosis factor-alpha and interleukin-10 contribute to immunoparalysis in patients with acute pancreatitis. Hum Immunol (2011) 72(1):18–23. doi: 10.1016/j.humimm.2010.10.002
117. Oiva J, Mustonen H, Kylänpää ML, Kuuliala K, Siitonen S, Kemppainen E, et al. Patients with acute pancreatitis complicated by organ dysfunction show abnormal peripheral blood polymorphonuclear leukocyte signaling. Pancreatology (2013) 13(2):118–24. doi: 10.1016/j.pan.2013.01.010
118. Li JP, Yang J, Huang JR, Jiang DL, Zhang F, Liu MF, et al. Immunosuppression and the infection caused by gut mucosal barrier dysfunction in patients with early severe acute pancreatitis. Front Biosci-Landmrk (2013) 18(3):892–900. doi: 10.2741/4150
119. Lin ZQ, Guo J, Xia Q, Yang XN, Huang W, Huang ZW, et al. Human leukocyte antigen-DR expression on peripheral monocytes may be an early marker for secondary infection in severe acute pancreatitis. Hepatogastroenterology (2013) 60(128):1896–902. doi: 10.5754/hge.13313
120. Qiu L, Zhou Y, Yu Q, Yu J, Li Q, Sun R. Decreased levels of regulatory b cells in patients with acute pancreatitis: association with the severity of the disease. Oncotarget (2018) 9(90):36067–82. doi: 10.18632/oncotarget.23911
121. Turunen A, Kuuliala A, Penttilä A, Kaukonen KM, Mustonen H, Pettilä V, et al. Time course of signaling profiles of blood leukocytes in acute pancreatitis and sepsis. Scandinavian J Clin Lab Invest (2020) 80(2):114–23. doi: 10.1080/00365513.2019.1700548
122. Minkov G, Dimitrov E, Yovtchev Y, Enchev E, Lokova R, Halacheva K. Prognostic value of peripheral blood CD14+HLA-DR+ monocytes in patients with acute pancreatitis. J Immunoassay Immunochem (2021) 42(5):478–92. doi: 10.1080/15321819.2021.1903491
123. Yu ZX, Chen XC, Zhang BY, Liu N, Gu Q. Association between HLA-DR expression and multidrug-resistant infection in patients with severe acute pancreatitis. Curr Med Sci (2018) 38(3):449–54. doi: 10.1007/s11596-018-1899-9
124. Sharma D, Jakkampudi A, Reddy R, Reddy PB, Patil A, Murthy HVV, et al. Association of systemic inflammatory and anti-inflammatory responses with adverse outcomes in acute pancreatitis: Preliminary results of an ongoing study. Dig Dis Sci (2017) 62(12):3468–78. doi: 10.1007/s10620-017-4813-6
125. Chen Y, Li M, Liu J, Pan T, Zhou T, Liu Z, et al. sPD-L1 expression is associated with immunosuppression and infectious complications in patients with acute pancreatitis. Scand J Immunol (2017) 86(2):100–6. doi: 10.1111/sji.12564
126. Zawada AM, Fell LH, Untersteller K, Seiler S, Rogacev KS, Fliser D, et al. Comparison of two different strategies for human monocyte subsets gating within the large-scale prospective CARE FOR HOMe study. Cytometry A (2015) 87(8):750–8. doi: 10.1002/cyto.a.22703
127. Waller K, James C, de Jong A, Blackmore L, Ma Y, Stagg A, et al. ADAM17-mediated reduction in CD14(++)CD16(+) monocytes ex vivo and reduction in intermediate monocytes with immune paresis in acute pancreatitis and acute alcoholic hepatitis. Front Immunol (2019) 10:1902. doi: 10.3389/fimmu.2019.01902
128. Babu S, Kumaraswami V, Nutman TB. Alternatively activated and immunoregulatory monocytes in human filarial infections. J Infect Dis (2009) 199(12):1827–37. doi: 10.1086/599090
129. Medeiros LT, Peraçoli JC, Bannwart-Castro CF, Romão M, Weel IC, Golim MA, et al. Monocytes from pregnant women with pre-eclampsia are polarized to a M1 phenotype. Am J Reprod Immunol (2014) 72(1):5–13. doi: 10.1111/aji.12222
130. Satoh N, Shimatsu A, Himeno A, Sasaki Y, Yamakage H, Yamada K, et al. Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients: effect of pioglitazone. Diabetes Care (2010) 33(1):e7. doi: 10.2337/dc09-1315
131. Zhang ML, Jiang YF, Wang XR, Ding LL, Wang HJ, Meng QQ, et al. Different phenotypes of monocytes in patients with new-onset mild acute pancreatitis. World J Gastroenterol (2017) 23(8):1477–88. doi: 10.3748/wjg.v23.i8.1477
132. Zhang M, Ding L, Wang X, Hou J, Li M, Jiang Y, et al. Circulating CD14(+)CD163(+)CD115(+) M2 monocytes are associated with the severity of new onset severe acute pancreatitis in Chinese patients. Int Immunopharmacol (2018) 57:181–9. doi: 10.1016/j.intimp.2018.02.018
133. Zawada AM, Rogacev KS, Schirmer SH, Sester M, Böhm M, Fliser D, et al. Monocyte heterogeneity in human cardiovascular disease. Immunobiology (2012) 217(12):1273–84. doi: 10.1016/j.imbio.2012.07.001
134. Liu HS, Pan CE, Liu QG, Yang W, Liu XM. Effect of NF-kappaB and p38 MAPK in activated monocytes/macrophages on pro-inflammatory cytokines of rats with acute pancreatitis. World J Gastroenterol (2003) 9(11):2513–8. doi: 10.3748/wjg.v9.i11.2513
135. Turunen A, Kuuliala A, Mustonen H, Puolakkainen P, Kylänpää L, Kuuliala K. Blood leukocyte signaling pathways as predictors of severity of acute pancreatitis. Pancreas (2021) 50(5):710–8. doi: 10.1097/MPA.0000000000001832
136. Shi C, Zhao X, Lagergren A, Sigvardsson M, Wang X, Andersson R. Immune status and inflammatory response differ locally and systemically in severe acute pancreatitis. Scand J Gastroenterol (2006) 41(4):472–80. doi: 10.1080/00365520500318965
137. Ferat-Osorio E, Wong-Baeza I, Esquivel-Callejas N, Figueroa-Figueroa S, Duarte-Rojo A, Guzman-Valdivia-Gomez G, et al. Triggering receptor expressed on myeloid cells-1 expression on monocytes is associated with inflammation but not with infection in acute pancreatitis. Crit Care (2009) 13(3):R69. doi: 10.1186/cc7876
138. Li HG, Zhou ZG, Li Y, Zheng XL, Lei S, Zhu L, et al. Alterations of toll-like receptor 4 expression on peripheral blood monocytes during the early stage of human acute pancreatitis. Dig Dis Sci (2007) 52(8):1973–8. doi: 10.1007/s10620-006-9211-4
139. Bhatnagar A, Wig JD, Majumdar S. Expression of activation, adhesion molecules and intracellular cytokines in acute pancreatitis. Immunol Lett (2001) 77(3):133–41. doi: 10.1016/S0165-2478(01)00210-3
140. Tinoco R, Otero DC, Takahashi AA, Bradley LM. PSGL-1: A new player in the immune checkpoint landscape. Trends Immunol (2017) 38(5):323–35. doi: 10.1016/j.it.2017.02.002
141. Zhao X, Dib M, Wang X, Widegren B, Andersson R. Influence of mast cells on the expression of adhesion molecules on circulating and migrating leukocytes in acute pancreatitis-associated lung injury. Lung (2005) 183(4):253–64. doi: 10.1007/s00408-004-2538-8
142. Chooklin S. Pathogenic aspects of pulmonary complications in acute pancreatitis patients. Hepatobiliary Pancreat Dis Int (2009) 8(2):186–92. doi: 10.1111/j.1523-5378.2009.00663.x
143. Uribe-Querol E, Rosales C. Phagocytosis: Our current understanding of a universal biological process. Front Immunol (2020) 11:1066. doi: 10.3389/fimmu.2020.01066
144. Döring M, Cabanillas Stanchi KM, Erbacher A, Haufe S, Schwarze CP, Handgretinger R, et al. Phagocytic activity of monocytes, their subpopulations and granulocytes during post-transplant adverse events after hematopoietic stem cell transplantation. Immunobiology (2015) 220(5):605–13. doi: 10.1016/j.imbio.2014.12.002
145. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13(3):260–8. doi: 10.1016/S1473-3099(13)70001-X
146. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis (2019) 10(10):782. doi: 10.1038/s41419-019-2015-1
147. Janicova A, Becker N, Xu B, Simic M, Noack L, Wagner N, et al. Severe traumatic injury induces phenotypic and functional changes of neutrophils and monocytes. J Clin Med (2021) 10(18):4139. doi: 10.3390/jcm10184139
148. Larvin M, Alexander DJ, Switala SF, McMahon MJ. Impaired mononuclear phagocyte function in patients with severe acute pancreatitis: evidence from studies of plasma clearance of trypsin and monocyte phagocytosis. Dig Dis Sci (1993) 38(1):18–27. doi: 10.1007/BF01296768
149. Liras G, Carballo F. An impaired phagocytic function is associated with leucocyte activation in the early stages of severe acute pancreatitis. Gut (1996) 39(1):39–42. doi: 10.1136/gut.39.1.39
150. Susak YM, Dirda OO, Fedorchuk OG, Tkachenko OA, Skivka LM. Infectious complications of acute pancreatitis is associated with peripheral blood phagocyte functional exhaustion. Dig Dis Sci (2021) 66(1):121–30. doi: 10.1007/s10620-020-06172-y
151. Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med (2002) 8(3):240–6. doi: 10.1038/nm0302-240
152. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med (2000) 6(4):422–8. doi: 10.1038/74680
153. Kobayashi Y. The regulatory role of nitric oxide in proinflammatory cytokine expression during the induction and resolution of inflammation. J Leukoc Biol (2010) 88(6):1157–62. doi: 10.1189/jlb.0310149
154. Taylor EL, Megson IL, Haslett C, Rossi AG. Nitric oxide: a key regulator of myeloid inflammatory cell apoptosis. Cell Death Diff (2003) 10(4):418–30. doi: 10.1038/sj.cdd.4401152
155. Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol (2003) 24(8):449–55. doi: 10.1016/S1471-4906(03)00181-9
156. Habtezion A, Kwan R, Yang AL, Morgan ME, Akhtar E, Wanaski SP, et al. Heme oxygenase-1 is induced in peripheral blood mononuclear cells of patients with acute pancreatitis: a potential therapeutic target. Am J Physiol Gastrointest Liver Physiol (2011) 300(1):G12–20. doi: 10.1152/ajpgi.00231.2010
157. Xue J, Habtezion A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J Clin Invest (2014) 124(1):437–47. doi: 10.1172/JCI71362
158. Satoh A, Shimosegawa T, Kimura K, Moriizumi S, Masamune A, Koizumi M, et al. Nitric oxide is overproduced by peritoneal macrophages in rat taurocholate pancreatitis: the mechanism of inducible nitric oxide synthase expression. Pancreas (1998) 17(4):402–11. doi: 10.1097/00006676-199811000-00012
159. Zhang XP, Lin Q, Zhou YF. Progress of study on the relationship between mediators of inflammation and apoptosis in acute pancreatitis. Dig Dis Sci (2007) 52(5):1199–205. doi: 10.1007/s10620-006-9388-6
160. Tanjoh K, Tomita R, Izumi T, Kinoshita K, Kawahara Y, Moriya T, et al. The expression of the inducible nitric oxide synthase messenger RNA on monocytes in severe acute pancreatitis. Hepatogastroenterology (2007) 54(75):927–31.
161. Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol (2019) 15(5):288–98. doi: 10.1038/s41574-019-0176-8
162. Segersvard R, Sylvan M, Herrington M, Larsson J, Permert J. Obesity increases the severity of acute experimental pancreatitis in the rat. Scandinavian J Gastroenterol (2001) 36(6):658–63. doi: 10.1080/003655201750163213
163. Araki H, Nishihara T, Matsuda M, Fukuhara A, Kihara S, Funahashi T, et al. Adiponectin plays a protective role in caerulein-induced acute pancreatitis in mice fed a high-fat diet. Gut (2008) 57(10):1431–40. doi: 10.1136/gut.2007.135665
164. Zyromski NJ, Mathur A, Pitt HA, Lu D, Gripe JT, Walker JJ, et al. A murine model of obesity implicates the adipokine milieu in the pathogenesis of severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol (2008) 295(3):G552–8. doi: 10.1152/ajpgi.90278.2008
165. Yu Q, Xu T, Ding F, Ding Z, Lin R. Decreased infiltration of adipose tissue macrophages and amplified inflammation of adipose tissue in obese mice with severe acute pancreatitis. Pancreatology (2021) S1424-3903(21):00156–3. doi: 10.1016/j.pan.2021.05.005
166. Sennello JA, Fayad R, Pini M, Gove ME, Ponemone V, Cabay RJ, et al. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci U S A (2008) 105(23):8085–90. doi: 10.1073/pnas.0804091105
167. Pini M, Sennello JA, Cabay RJ, Fantuzzi G. Effect of diet-induced obesity on acute pancreatitis induced by administration of interleukin-12 plus interleukin-18 in mice. Obes (Silver Spring) (2010) 18(3):476–81. doi: 10.1038/oby.2009.263
168. Yang X, Yao L, Dai L, Yuan M, He W, Liu T, et al. Alcohol predisposes obese mice to acute pancreatitis via adipose triglyceride lipase-dependent visceral adipocyte lipolysis. Gut (2022). doi: 10.1136/gutjnl-2022-326958
169. Aune D, Mahamat-Saleh Y, Norat T, Riboli E. High body mass index and central adiposity is associated with increased risk of acute pancreatitis: A meta-analysis. Dig Dis Sci (2021) 66(4):1249–67. doi: 10.1007/s10620-020-06275-6
170. Navina S, Acharya C, DeLany JP, Orlichenko LS, Baty CJ, Shiva SS, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med (2011) 3(107):107ra10. doi: 10.1126/scitranslmed.3002573
171. Sempere L, Martinez J, de Madaria E, Lozano B, Sanchez-Paya J, Jover R, et al. Obesity and fat distribution imply a greater systemic inflammatory response and a worse prognosis in acute pancreatitis. Pancreatology (2008) 8(3):257–64. doi: 10.1159/000134273
172. Khatua B, El-Kurdi B, Singh VP. Obesity and pancreatitis. Curr Opin Gastroenterol (2017) 33(5):374–82. doi: 10.1097/MOG.0000000000000386
173. de Oliveira C, Khatua B, Noel P, Kostenko S, Bag A, Balakrishnan B, et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J Clin Invest (2020) 130(4):1931–47. doi: 10.1172/JCI132767
174. Patel K, Trivedi RN, Durgampudi C, Noel P, Cline RA, DeLany JP, et al. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am J Pathol (2015) 185(3):808–19. doi: 10.1016/j.ajpath.2014.11.019
175. Pirillo A, Casula M, Olmastroni E, Norata GD, Catapano AL. Global epidemiology of dyslipidaemias. Nat Rev Cardiol (2021) 18(10):689–700. doi: 10.1038/s41569-021-00541-4
176. Vekic J, Zeljkovic A, Stefanovic A, Jelic-Ivanovic Z, Spasojevic-Kalimanovska V. Obesity and dyslipidemia. Metabol: Clin Experimental (2019) 92:71–81. doi: 10.1016/j.metabol.2018.11.005
177. Yang AL, McNabb-Baltar J. Hypertriglyceridemia and acute pancreatitis. Pancreatology (2020) 20(5):795–800. doi: 10.1016/j.pan.2020.06.005
178. Zhang R, Deng L, Jin T, Zhu P, Shi N, Jiang K, et al. Hypertriglyceridaemia-associated acute pancreatitis: diagnosis and impact on severity. HPB Off J Int Hepato Pancreato Biliary Assoc (2019) 21(9):1240–9. doi: 10.1016/j.hpb.2019.01.015
179. Shi N, Liu T, de la Iglesia-Garcia D, Deng L, Jin T, Lan L, et al. Duration of organ failure impacts mortality in acute pancreatitis. Gut (2020) 69(3):604–5. doi: 10.1136/gutjnl-2019-318241
180. Ke L, Zhou J, Mao W, Chen T, Zhu Y, Pan X, et al. Immune enhancement in patients with predicted severe acute necrotising pancreatitis: a multicentre double-blind randomised controlled trial. Intensive Care Med (2022) 48(7):899–909. doi: 10.1007/s00134-022-06745-7
181. Su N, Zhu Y, Peng Y, Xia W, Chen L, Yu H, et al. Triglycerides to high-density lipoprotein cholesterol (TG/HDL-c) ratio is an independent predictor of the severity of hyperlipidaemic acute pancreatitis. J Hepatobiliary Pancreat Sci (2022). doi: 10.1002/jhbp.1281
182. Nawaz H, Koutroumpakis E, Easler J, Slivka A, Whitcomb DC, Singh VP, et al. Elevated serum triglycerides are independently associated with persistent organ failure in acute pancreatitis. Am J Gastroenterol (2015) 110(10):1497–503. doi: 10.1038/ajg.2015.261
183. Mosztbacher D, Hanák L, Farkas N, Szentesi A, Mikó A, Bajor J, et al. Hypertriglyceridemia-induced acute pancreatitis: A prospective, multicenter, international cohort analysis of 716 acute pancreatitis cases. Pancreatology (2020) 20(4):608–16. doi: 10.1016/j.pan.2020.03.018
184. Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science (2019) 363(6430):989–93. doi: 10.1126/science.aaw2586
187. Dalmas E, Clément K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol (2011) 32(7):307–14. doi: 10.1016/j.it.2011.04.008
188. Franco-Pons N, Gea-Sorlí S, Closa D. Release of inflammatory mediators by adipose tissue during acute pancreatitis. J Pathol (2010) 221(2):175–82. doi: 10.1002/path.2691
189. Bonjoch L, Gea-Sorlí S, Closa D. Lipids generated during acute pancreatitis increase inflammatory status of macrophages by interfering with their M2 polarization. Pancreatology (2015) 15(4):352–9. doi: 10.1016/j.pan.2015.04.007
Keywords: acute pancreatitis, monocytes, immunity, inflammation, animal models, lipotoxicity, obesity, hypertriglyceridemia
Citation: Liu S, Szatmary P, Lin J-w, Wang Q, Sutton R, Chen L, Liu T, Huang W and Xia Q (2022) Circulating monocytes in acute pancreatitis. Front. Immunol. 13:1062849. doi: 10.3389/fimmu.2022.1062849
Received: 06 October 2022; Accepted: 28 November 2022;
Published: 12 December 2022.
Edited by:
Giamila Fantuzzi, University of Illinois at Chicago, United StatesReviewed by:
Rand T. Akasheh, American University of Madaba, JordanYasushi Sawanobori, Dokkyo Medical University, Japan
Matthias Sendler, Universitätsmedizin Greifswald, Germany
Copyright © 2022 Liu, Szatmary, Lin, Wang, Sutton, Chen, Liu, Huang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingting Liu, bGl1dGluZ3RpbmdAd2Noc2N1LmNu; Wei Huang, ZHJfd2VpX2h1YW5nQHNjdS5lZHUuY24=; Qing Xia, eGlhcWluZ0BtZWRtYWlsLmNvbS5jbg==