 Sijin Li1,2,3†
Sijin Li1,2,3† Yusi Zhou1,2,3†
Yusi Zhou1,2,3† Hairong Wang1,2,3Gexi Qu1,2,3
Hairong Wang1,2,3Gexi Qu1,2,3 Xuan Zhao1,2,3Xu Wang1,2,3Rui Hou4Zhangchun Guan1,2,3Dan Liu1,2,3*
Xuan Zhao1,2,3Xu Wang1,2,3Rui Hou4Zhangchun Guan1,2,3Dan Liu1,2,3* Junnian Zheng2,3*
Junnian Zheng2,3* Ming Shi1,2,3*
Ming Shi1,2,3*- 1Country Cancer Institute, Xuzhou Medical University, Xuzhou, China
- 2Center of Clinical Oncology, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
- 3Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Xuzhou Medical University, Xuzhou, China
- 4College of Pharmacy, Xuzhou Medical University, Xuzhou, China
Chimeric antigen receptor (CAR)-T cell therapy, which utilizes genetic engineering techniques to modify T-cells to achieve specific targeting of cancer cells, has made significant breakthroughs in cancer treatment in recent years. All marketed CAR-T products are second-generation CAR-T cells containing co-stimulatory structural domains, and co-stimulatory molecules are critical for CAR-T cell activation and function. Although CD28-based co-stimulatory molecules have demonstrated potent cytotoxicity in the clinical application of CAR-T cells, they still suffer from high post-treatment relapse rates, poor efficacy durability, and accompanying severe adverse reactions. In recent years, researchers have achieved specific results in enhancing the anti-tumor function of CD28 by mutating its signaling motifs, combining the co-stimulatory structural domains, and modifying other CAR components besides co-stimulation. This paper reviewed the characteristics and roles of CD28 in CAR-T cell-mediated anti-tumor signaling and activation. We explored potential strategies to enhance CAR-T cell efficacy and reduce side effects by optimizing CD28 motifs and CAR structures, aiming to provide a theoretical basis for further clinical CAR-T cell therapy development.
1 Introduction
CAR-T therapy has achieved remarkable results in treating hematological tumors and has become a “star therapy” for treating a variety of hematological tumors (1). In addition to the first signal provided by the T-cell receptor (TCR), the activation of T cells requires the activation of co-stimulatory molecules to activate co-stimulatory signals (2, 3). Activation of T cells by signaling through binding CD28 to its ligand is the major co-stimulatory pathway (4–7). The co-stimulatory effect of CD28 depends on its key signaling motifs (8, 9). The activated signaling pathways include the phosphoinositide 3-kinase (PI3K)- protein kinase B (AKT) pathway and the growth factor receptor-bound protein 2 (GRB2)- rat sarcoma virus oncogene (Ras) pathway, which regulate the activation and function of T cells (10, 11). The use of CD28 in CAR structures to provide co-stimulatory signals addressed the problem of the lack of activation of first-generation CAR-T cells. It became one of the most popular and widely used co-stimulatory molecules.
Although CD28-based CAR-T has significantly improved the clinical efficacy of hematological tumors, there are still challenges, such as poor in vivo durability and high recurrence rates in patients with hematological tumors and limitations in treatment and safety in solid tumors (12). Resolving these issues is critical to further enhancing CD28-based CAR-T therapies’ efficacy. Currently, optimization strategies for modulating CD28- based CAR-T include altering CD28 intrinsic signaling by mutating CD28 amino acid motifs, exploring combinatorial modes with other co-stimulatory molecules to achieve complementary co-stimulatory signaling, and optimizing CAR structural elements other than the co-stimulatory structural domains, in addition to modulation of the metabolic pathway and the use of artificial intelligence, which have also provided new ideas for optimization. This review summarizes the limitations associated with the application of CD28-based CAR-T cells, evaluates the efficacy of current optimization strategies for CD28-based CAR design, and discusses future perspectives on its clinical and therapeutic potential.
2 CD28 signaling function and problems in CAR-T application
2.1 CD28 signaling motif and function
Earlier studies have shown that CD28 is expressed in approximately 80% of CD4+ T cells and 50% of CD8+ T cells (13, 14). It consists of a transmembrane structural domain, an intracellular signaling structural domain, and a variable structural domain-like V-set structural domain connecting the transmembrane structural domain to the signaling structural domain (15). The CD28 signaling structural domain is located in the cytoplasmic structural domain and consists of 41 amino acids, including membrane-proximal YMNM (tyrosine-rich) and proline-rich PRRP and PYAP motifs. These signaling motifs activate downstream signaling pathways by either undergoing phosphorylation or binding to kinases, connexins GRB2 and GADS, and regulate T cell activation, differentiation, cell proliferation, and interleukin-2 (IL-2) secretion (16). For example, upon TCR activation or interaction with the ligand B7 protein, the tyrosine in the YMNM motif undergoes phosphorylation and binds to the p85 subunit of PI3K, which activates PI3K. PI3K can activate the nuclear factor of activated T-cells (NF-AT) transcription factor by activating AKT or regulating protein kinase C (PKC) activity through 3-phosphoinositide-dependent protein kinase-1(PDK-1) (17, 18). PRRP and PYAP bind lymphocyte-specific protein tyrosine kinas (LCK), further activating PKCθ, which shares a downstream signaling pathway with YMNM (19). In addition, PRRP binds to the interleukin-2-inducible T-cell kinase (ITK), which then activates the phospholipase C gamma, (PLCγ), Ca2+, and extracellular signal-regulated kinase (ERK)-mediated signaling pathways. For GRB2, GRB2-related adaptor protein downstream of Shc (GADS), YMNM can bind to the SH2 structural domain of GRB2 and activate Ras by bringing son of sevenless (SOS) to the plasma membrane, further activating the downstream mitogen-activated protein kinase (MAPK) pathway (20–25). GADS interacts with SH2 domain-containing leukocyte protein of 76 kDa (SLP-76), an articulatory protein for TCR signaling, through SH3, regulating PLCγ, ERK activation, and Ca2+ pathways, synergistically enhancing NF-AT activity (26–28). However, PRRP and PYAP bind SH3 of GRB2 and GADS and activate downstream signaling pathways. In addition, PYAP recruits and binds Actin protein (Filamin A), which recruits Filamin A to the T-cell membrane, and Filamin A synergistically integrates the signaling pathway with Vav guanine nucleotide exchange factor 1 (VAV1), leading to actin polymerization, mobilization of lipid rafts, and facilitation of CD28 aggregation at the immune synapse (Figure 1) (29–32).
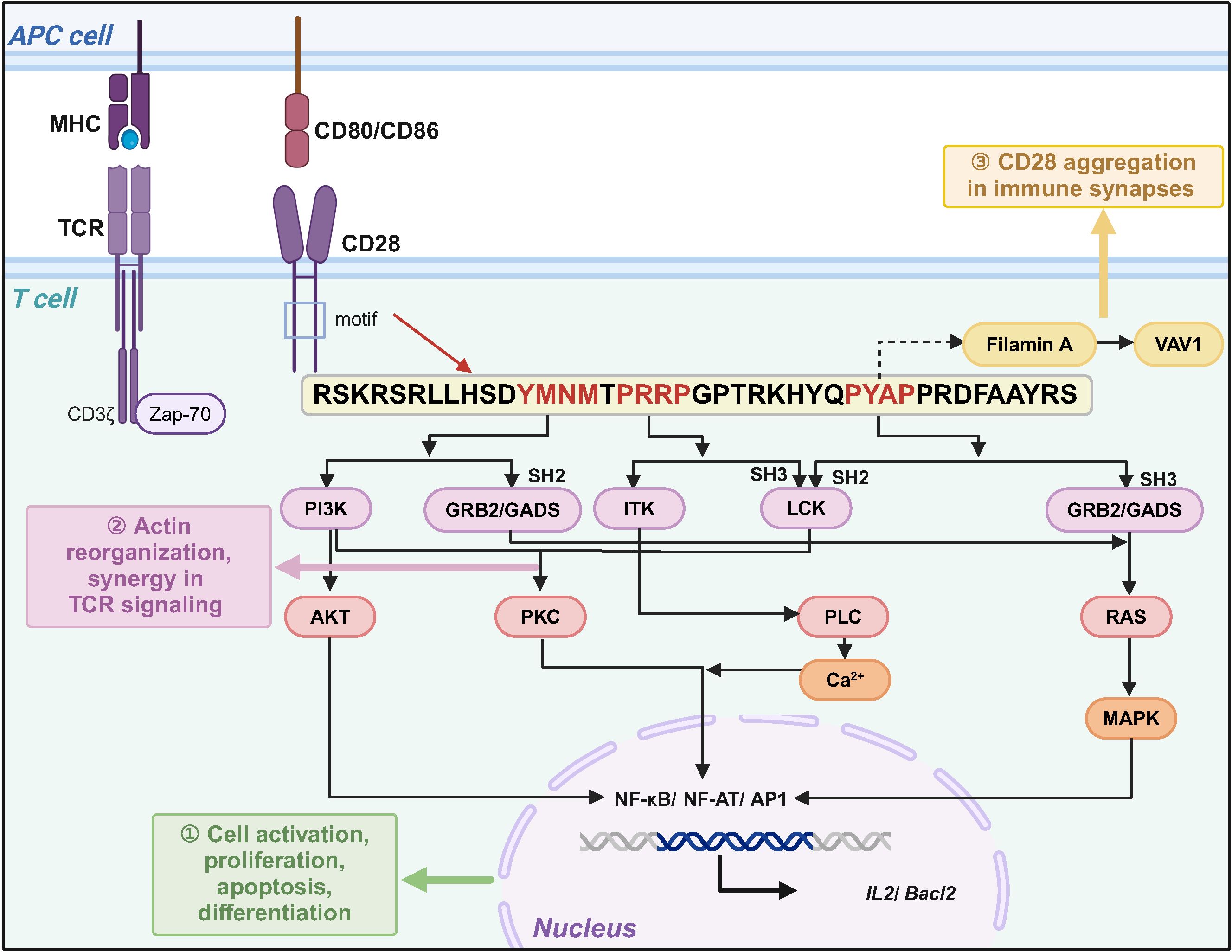
Figure 1. CD28 signaling motifs mediate co-stimulatory signaling to regulate T cell function. After CD28 binds to ligands CD80 or CD86, different signaling motifs activate downstream signaling pathways by binding to specific protein kinases or bridging proteins, which can functionally complement TCR signaling.
2.2 Problems with CD28-based CAR-T
Despite the encouraging results of CD28-based CAR-T in treating hematologic tumors, severe toxic side effects are an essential challenge (Figure 2) (33–35). A clinical study in non-Hodgkin’s lymphoma showed that CD28-based CAR-T and 4-1BB-based CAR-T cells exhibited similar anti-tumor effects at 3 months post-treatment, but CD28-based CAR-T cells induced more severe cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (NCT03528421) (36). In addition, in patients with B-cell acute lymphoblastic leukemia (B-ALL) with a high rate of tumor load, CD28-based CAR-T induced a higher incidence of CRS and neurotoxicity, shorter long-term survival, and ineffective resistance to disease recurrence (NCT01044069) (37). Results from clinical trials for relapsed/refractory B-ALL also showed that CD19-CAR-T cells using CD28 as a co-stimulatory molecule triggered a high level of pro-inflammatory cytokine production early after infusion to the patients (NCT01593696, NCT02186860, NCT01044069) (5, 38, 39), which is also consistent with preclinical data on CD28-based CAR-T (40, 41).
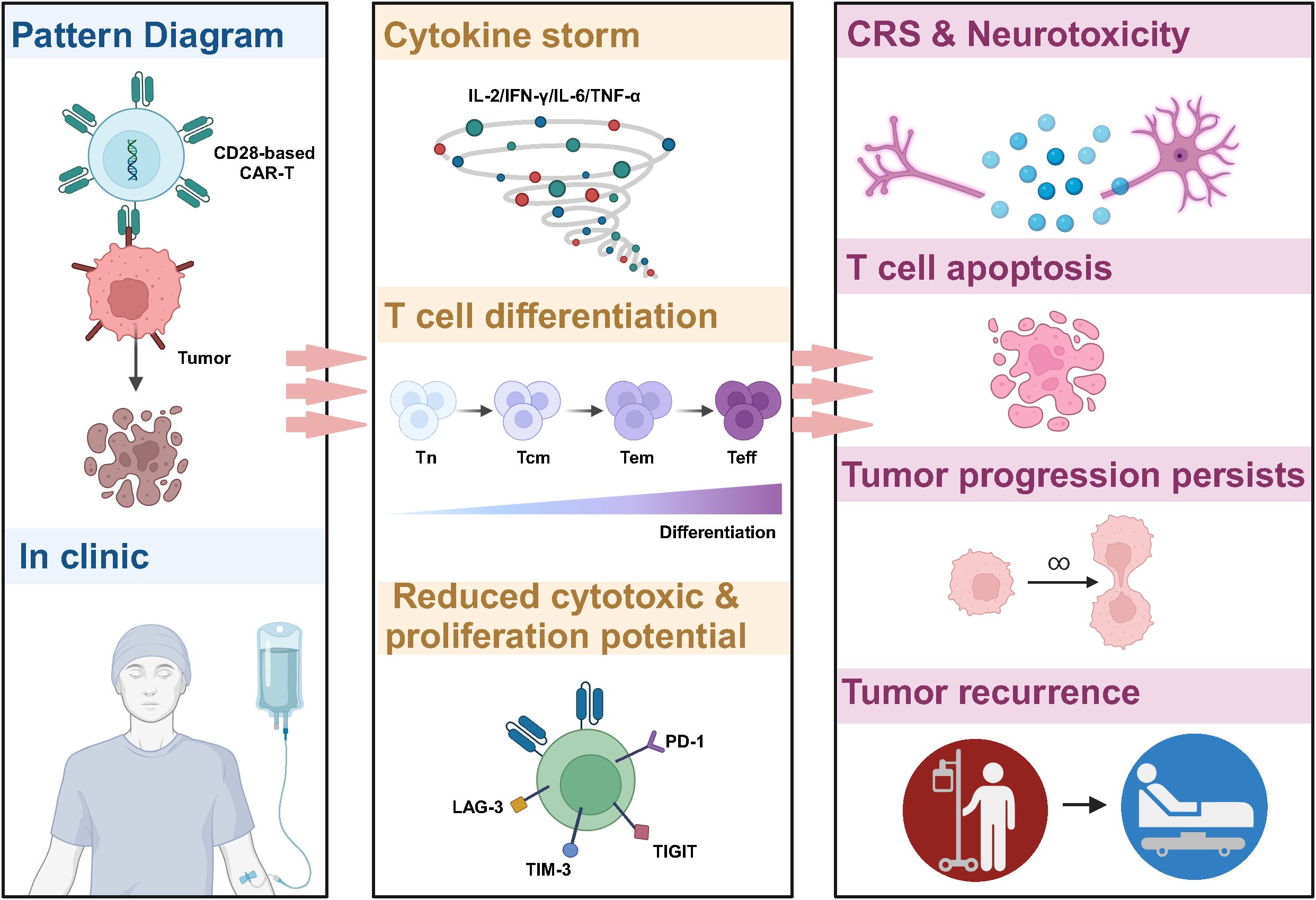
Figure 2. Limitations of CD28-based CAR-T cells in cancer therapy. CD28-based CAR-T cells tend to induce higher levels of cytokine release, triggering CRS and neurotoxicity in patients. In addition, the signaling pathway activated by CD28 leads to reprogramming CAR-T to the glycolysis pathway, which promotes the differentiation of CAR-T to effector T cells and reduces the proportion of memory T cells. This results in poor anti-tumor activity, insufficient persistence in vivo, and ineffective resistance to tumor recurrence.
The CD28 signaling pathway and its metabolic profile are important factors contributing to the poor persistence of CD28-based CAR-T cells in vivo, which often leads to cancer recurrence (37). It has been shown that activation of the PI3K-AKT pathway by CD28 induces an increase in the expression of glucose transporter 1 (Glut1) to promote glucose uptake on the one hand and, on the other hand, enhances the activity of PDK1, which inhibits the decarboxylation of pyruvate and the entry of glucose into the tricarboxylic acid cycle (TCA) cycle, and in turn increase the activity of adenosine triphosphate (ATP)-producing enzymes involved in glycolysis, resulting in cells showing glycolysis-biased metabolic reprogramming (42, 43). The glycolytic pathway promotes the transformation of CAR-T to CD45RO+ CCR7+ effector memory cells, tending to be short-term effector cells, which, in addition to increasing CAR-T expansion and interferon-gamma (IFN-γ) and IL-2 secretion, often leads to poor CAR-T persistence, which is consistent with the clinical trials showing that in vivo survival time after CD28-based CAR-T treatment in patients with hematological tumors tends to be less than three months (44–46). In vitro inhibition of PI3K in CD8+ T cells delays terminal differentiation, maintains the memory phenotype in CD8+ T cells, and may improve the in vivo anti-solid tumor therapeutic activity of adoptive CD8+ T cells (47). In the CAR-T in vitro culture phase, modulating metabolic reprogramming and increasing the percentage of memory cells by pharmacological or gene editing means has yielded promising results in the preclinical study phase, which may provide lessons for solid tumor therapy (48, 49).
3 CD28-based CAR structure optimization strategy
3.1 CD28 signaling motif optimization
Based on the role of CD28 signaling motifs in mediating CD28 signaling and initiating T-cell function, studies targeting the antitumor effects of CAR-T generated by mutations in the YMNM, PRRP, and PYAP motifs have been conducted (50). In a pancreatic tumor xenograft model, mutation of CD28 YMNM to YMFM in CD28-based CAR-T cells reduces the binding of GRB2 to CD28, decreasing VAV1 signaling, decreasing calcium in-flow, and decreasing NFAT over-activation, thereby decreasing T-cell exhaustion and dysfunction, and increasing the persistence and antitumor efficacy of CAR-T cells in a pancreatic tumor xenograft model (51). Mutating CD28-based CAR-T with PRRP based on mutated YMNM enhances the secretion of IFN-γ and tumor necrosis factor-alpha (TNF-α), reduces the expression of the exhaustion-related transcription factor Nur77, and significantly enhances the cytotoxicity of CAR-T within 48 hours of treatment, demonstrating a superior survival advantage in tumor-bearing mice (52). However, whether CAR-T with mutated YMNM has clinical efficacy needs further exploration.
Moreover, mutating the PYAP signaling motif can potentially enhance CAR-T cell functionality in both hematologic malignancies and solid tumors (53). For example, David M Kofler et al. demonstrated that mutating the PYAPP of CD28 to AYAAA eliminated the ability of PYAP to bind to LCK to eliminate IL-2-induced signaling, reduced the promotion of IL-2 to intratumor regulatory T cell (Treg), and achieved enhanced function of CD28-based CAR-T cells by decreasing the solid Treg cell infiltration in the tumor to achieve the functional enhancement of CD28-based CAR-T, which significantly improved the anti-tumor activity of solid tumors with a large number of Treg infiltration (54). In addition, this PYAP-mutated CD28 construct described above still significantly enhanced T cell proliferation, metabolism, activation, and target cell killing in FAP-targeted CAR-T cells and showed promising efficacy and durability with few side effects in conjunction with a programmed cell death protein 1 (PD-1) blocker in humanized mice suffering from tumors and the first malignant pleural mesothelioma (MPM) patients (55). These studies suggest that CD28-based CAR-T cells with PYAP mutations lacking LCK binding may optimize CAR-T functionality by reducing IL-2-mediated support for Treg cells within the tumor microenvironment, thereby enhancing therapeutic efficacy against solid tumors.
3.2 Separating CD28 and CD3ζ signaling
Few tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) are available for solid tumors, and targeting a single TAA may lead to off-target toxicity of CAR-T to target lowly expressed antigens in healthy tissues (56). Based on this, researchers proposed dual-targeting CAR-T that separates the CD3ζ and the co-stimulatory molecules, which means the CD3ζ-CAR provides suboptimal activation signals upon binding one antigen, and the chimeric co-stimulatory structural domain containing the co-stimulatory receptor provides T-cell co-stimulatory signals upon recognition of the second antigen (57). This design enhances the responsiveness of CAR-T cells to double-positive tumor cells and reduces the risk of off-targeting and tumor antigen escape. For example, Evripidis Lanitis developed the mesothelin (Meso) single chain variable fragment (scFv)-CD3ζ and folate receptor alpha (FRα) scFv-CD28-based CAR, which had no potent activity against normal tissues expressing only mesothelin; meanwhile, they showed potent anti-solid tumor activity and persistence in vivo (58). Another study of the glypican 3 (GPC3)-CD3ζ and asialoglycoprotein receptor 1 (ASGR1)-CD28-41BB- against hepatocellular carcinoma (HCC) CAR-T cells study also demonstrated that CAR-T cells separating CD28 and CD3ζ signals had potent antitumor activity and safety against tumor cells carrying both antigens. However, the design depends on recognizing the two antigens, and its complex engineering may increase the production cost. Its effectiveness in solid tumor microenvironment (TME) still needs to be further explored, and the discovery of novel tumor antigens may promote the application of this strategy in solid tumors.
3.3 Combination of CD28 and other co-stimulatory molecules
Second-generation CARs, with the addition of co-stimulatory molecules, have significantly enhanced in mediating T cell proliferation, cytokine release, and in vivo antitumor function compared with first-generation CARs, illustrating the importance of co-stimulatory molecules. However, different co-stimulatory molecules have advantages and disadvantages in cell activation, cell differentiation, proliferation, metabolism, and in vivo safety (59). Based on this, researchers have proposed combining different costimulatory molecules to achieve complementary benefits (Figure 3 and Table 1).
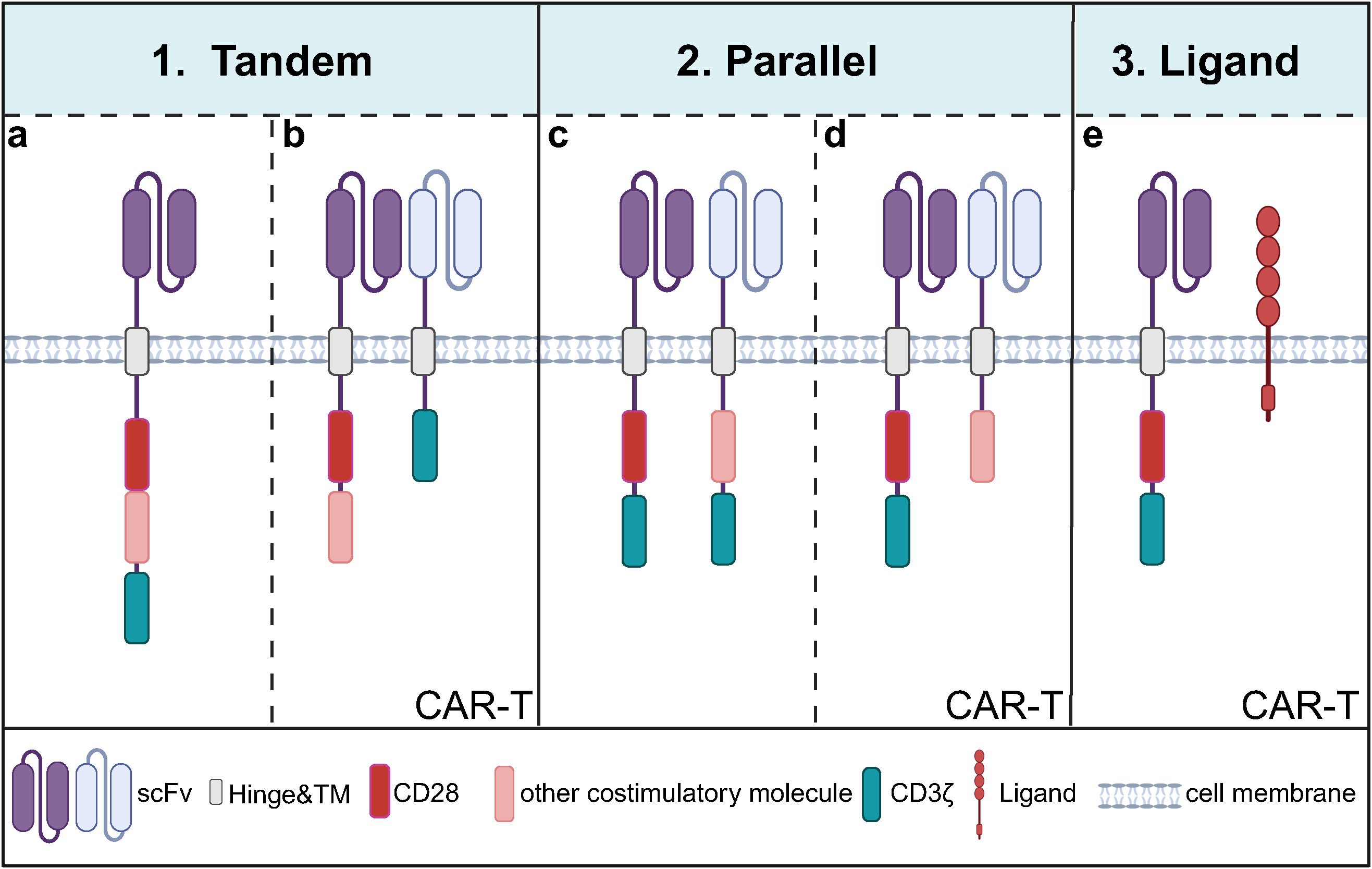
Figure 3. Current modes of combining co-stimulatory molecules of CARs containing CD28. (a) A common tandem mode of co-stimulatory molecule involves linearly fusing CD28 with other costimulatory molecules, such as 4-1BB or OX40, within a CAR that targets a single antigen; (b) Another tandem mode entails T cells carrying two CARs: one CAR contains two linearly fused co-stimulatory molecules but lacks the CD3ζ domain, while the other is a first-generation CAR targeting a different antigen utilizing the CD3ζ domain for intracellular signaling but without costimulatory molecules; (c) One of the co-stimulatory molecules in parallel mode involves T cells carrying two second-generation CAR that recognizes the two antigens; (d) Another parallel mode consists of T cells expressing a second-generation CAR with CD28 while simultaneously expressing another CAR that lacks the CD3ζ domain but recognizes a different antigen; (e) T cells express a second- or third-generation CAR containing CD28 and simultaneously express a ligand for other co-stimulatory molecules.
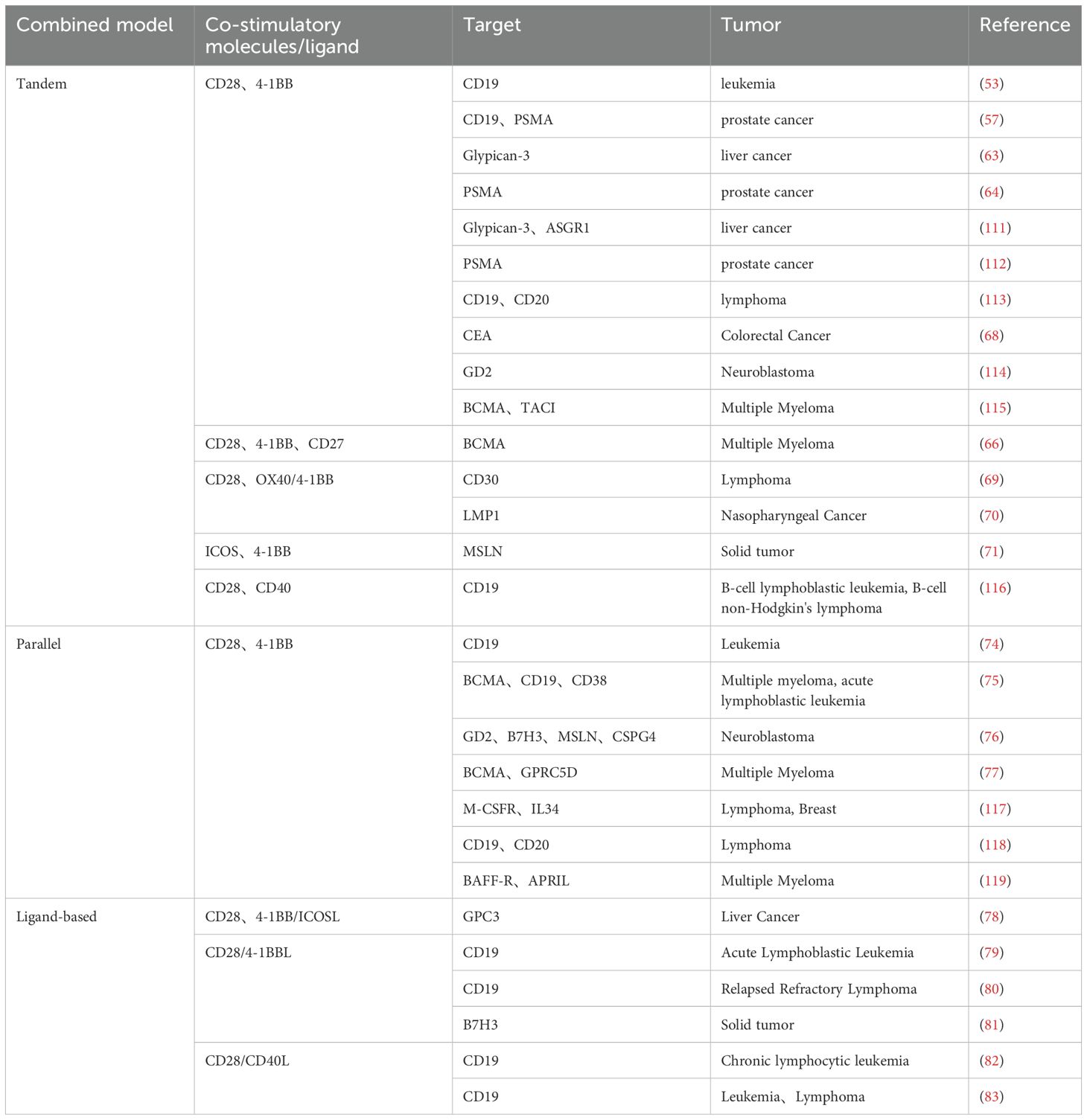
Table 1. Optimization strategies for combinatorial modes targeting CD28 with other co-stimulatory molecules.
3.3.1 Tandem mode
The tandem mode of co-stimulatory molecules is characterized by constructing two or more co-stimulatory molecules in a single CAR by tandem mode. The most studied mode is the tandem combination of CD28 and 4-1BB (60). A CAR containing CD28 triggers rapid and high-intensity lysis of target cells, while a CAR containing 4-1BB triggers a low-intensity and long-lasting response (61, 62). However, preclinical studies of CAR-T cells tandeming CD28 and 4-1BB have shown that the effect of tandem combination is not a simple signaling superposition. The effect of tandeming is related to both tumor type and programmed death-ligand 1 (PD-L1) expression level (63–65). For example, tandem mode in combination with blockade of PD-L1/PD-1 or secretion of anti-PD-L1 scFv enhances the killing activity of tumor cells targeting high levels of PD-L1. It reduces the exhaustion of CAR-T cells (66). In addition, other co-stimulatory molecules tandemly associated with CD28 are OX40 and inducible T-cell costimulator (ICOS) (67). CAR-T cells containing tandem CD28-OX40 showed significantly higher T-cell expansion and IL-2 secretion levels than CD28-based CAR-T cells. The mechanism is that OX40 signaling inhibits the secretion of IL-10 by CAR-T cells and reduces the inhibition of other T-cell functions in the TME (68). However, comparing the antitumor activity of the tandem mode of CD28-4-1BB versus CD28-OX40 has shown inconsistent results in different studies (69, 70). ICOS is a co-stimulatory molecule belonging to the CD28 family, and the CAR-T antitumor activity of ICOS in tandem with other co-stimulatory molecules may be related to other structures in the CAR. For example, studies have shown that CD28 in tandem with ICOS significantly enhances the persistence of CD8+ CAR-T cells, and ICOS and 4-1BB tandem have an antitumor advantage in solid tumors (71, 72). ICOS-OX40 tandem maintains high target cytolysis toxicity despite multiple rounds of tumor stimulation in vitro (73). Overall, the combination of co-stimulatory molecules targeting CD28 is influenced by tumor type, tumor microenvironment, and CAR structure, and the optimal mode of co-stimulatory molecule tandem will need to be determined in the future based on specific tumors.
3.3.2 Parallel mode
Parallel mode refers to the mode of CAR carrying two different co-stimulatory molecules in a single T cell (74, 75). Studies have shown that”two co-stimulatory molecules sharing one CD3ζ” has better antitumor efficacy than the parallel mode of “two co-stimulatory molecules sharing two CD3ζs”. Separate activation of CD3ζ by dual-targeted CAR resulted in the overactivation of CAR-T cells and induced cellular exhaustion, possibly a reason for its poor effectiveness (76, 77). In addition, for the parallel mode of CD28 and 4-1BB, it was demonstrated that dual-targeted CAR-T carrying CD28 and 4-1BB sharing one CD3ζ had higher tumor-killing activity, dividing and proliferating ability and durability than the parallel mode of dual co-stimulatory molecules sharing two CD3ζ (76). The problem with this model is that if two individual CARs are transduced into T cells, it cannot be guaranteed that both CARs are expressed at the same level in all the transduced cells, and if it is a single double cis-trans vector containing two individual CARs, the transduction efficiency of the double CAR may be affected due to the large size of the vector. Moreover, the current co-stimulatory molecules in parallel combination are only CD28 and 4-1BB, and the antitumor potential of other co-stimulatory molecules in parallel combination remains to be explored.
3.3.3 Combined patterns of co-stimulatory molecules and ligands
In addition, simultaneously expressing a co-stimulatory molecule with a ligand for another co-stimulatory molecule in T cells can generate synergistic co-stimulatory signaling (78). For example, the combination of CD28-4-1BBL can continuously complement 4-1BB signaling after CD28 signaling activation. The concurrently activated interferon regulatory factor 7 (IRF7)/IFNβ signaling pathway initiates DCs in the TME and inhibits Treg activation. CD28-4-1BBL-based CAR-T has shown safe and effective results in targeting CD19 hematologic tumors in both preclinical and clinical trials (NCT03085173) (79, 80), and even effective anti-tumor effects in solid tumors targeting B7-H3 (81). These results illustrate that the combination of CD28 and 4-1BBL can spatiotemporally and spatially differentially activate both CD28 and 4-1BB co-stimulatory signaling and that this synergistic co-stimulatory signaling may surpass CAR target antigens and activate the host immune system, which provides a new strategy for modulating TME to enhance CAR-T function. Based on the role of the CD40 pathway in improving anti-tumor response, some researchers combined CD28 with CD40L in CD19 CAR-T, which up-regulate the proliferation of T cells, the expression of CD80 and CD40 of dendritic cells after co-culture with CD40-positive tumor cells, increase the recruitment of endogenous effector T cells by DC, and enhance the immunogenicity of CD40-expressing tumor cells. However, clinical safety needs to be investigated as the CD40L/CD40 pathway may induce a systemic inflammatory response (82, 83).
3.4 Optimization of CD28 upstream and downstream structural domains
Single-chain variable fragments are the antigen-binding domains of CAR structures, and it has been shown that lowering the affinity of scFV for antigen binding can reduce reactivity to antigens expressed at physiological levels while maintaining potent antitumor activity, thereby attenuating off-target toxicity (84). Common CAR hinge domains/spacer regions consist of the IgG1 or IgG4 hinge regions and the CH2-CH3 structural domains of immunoglobulin G fragment crystallizable (IgG Fc). The length and composition of the hinge domain is an important factor in designing CARs with optimal antitumor activity or low toxicity (85–87). Although no reports show the relationship between CD28 and the function of the scFV and hinge region, CAR is a combinatorial entity, and exploring CD28 optimization strategies for specific scFV and hinge region patterns is expected to improve the application of CAR-T. The transmembrane structural domain anchors CAR to the T cell membrane and transduces ligand recognition signals to the cytoplasmic signals of the TCR, which plays a critical role in CAR expression or structural stability (72). Studies have shown that both extracellular and transmembrane structural domains of CD28 can partially induce T cell activation (88), providing more choices of CAR transmembrane domains. CD3ζ instead of FcRγ has been widely used as a CAR signaling structural domain (89). The cytoplasmic tail of CD3ζ contains three immunoreceptor tyrosine-based activation motifs (ITAMs), and CD28-based CAR studies have shown that the number and position of ITAMs are related to CAR-T cell function (90, 91). For example, retaining only the second position of ITAMs in CD3ζ reduces apoptosis of CD28-based CAR-T cells that target ErbB2 (92). For CD19-CAR, placing ITAM1 or ITAM3 of CD3ζ in CD28-based CAR close to the proximal membrane position of CARs targeting CD19 or inserting an IL-2Rβ structural domain between CD28 and CD3ζ, and inserting a YXXQ motif in the distal region of the CD3ζ structural domain can improve the persistence and therapeutic efficacy of CAR-T cells (2, 93). In summary, the scFV, hinge region, transmembrane region, and signal transduction region can be comprehensively optimized in combination with tumor specificity and individual patient differences to develop a CD28-based CAR with the best overall performance.
4 Conclusion and prospective
CD28 has been widely used in preclinical and clinical studies (Figure 4). CAR-T cells with CD28 as a co-stimulatory structure suffer from rapid exhaustion and poor durability in therapy, accompanied by more significant adverse effects and higher relapse rates. Studies have shown that optimizing the CD28-based CAR structure—through approaches such as mutating the CD28 signaling motif, combining it with other co-stimulatory molecules, and modifying its upstream and downstream structures—is expected to improve the durability and anti-tumor efficacy of CAR-T cells by enhancing their ability to regulate T cell proliferation and survival, with a relatively optimistic safety profile (Figure 5, Table 2). Currently, the existing CD28 modifications have been successful, but CD28-based CAR-T cells face much greater complexity in real human tumor environments compared to in vitro or mouse tumor models. Moving forward, there is a need to develop more accurate CAR-T evaluation models to assess the anti-tumor efficacy of CD28-modified CAR-T cells, including metrics such as tumor-killing ability, differentiation, persistence, and exhaustion. In the future, leveraging the latest advancements in synthetic biology and gene editing technologies will help optimize logic-gate CARs and develop more rational combinations of CD28 mutations or co-stimulatory molecules.
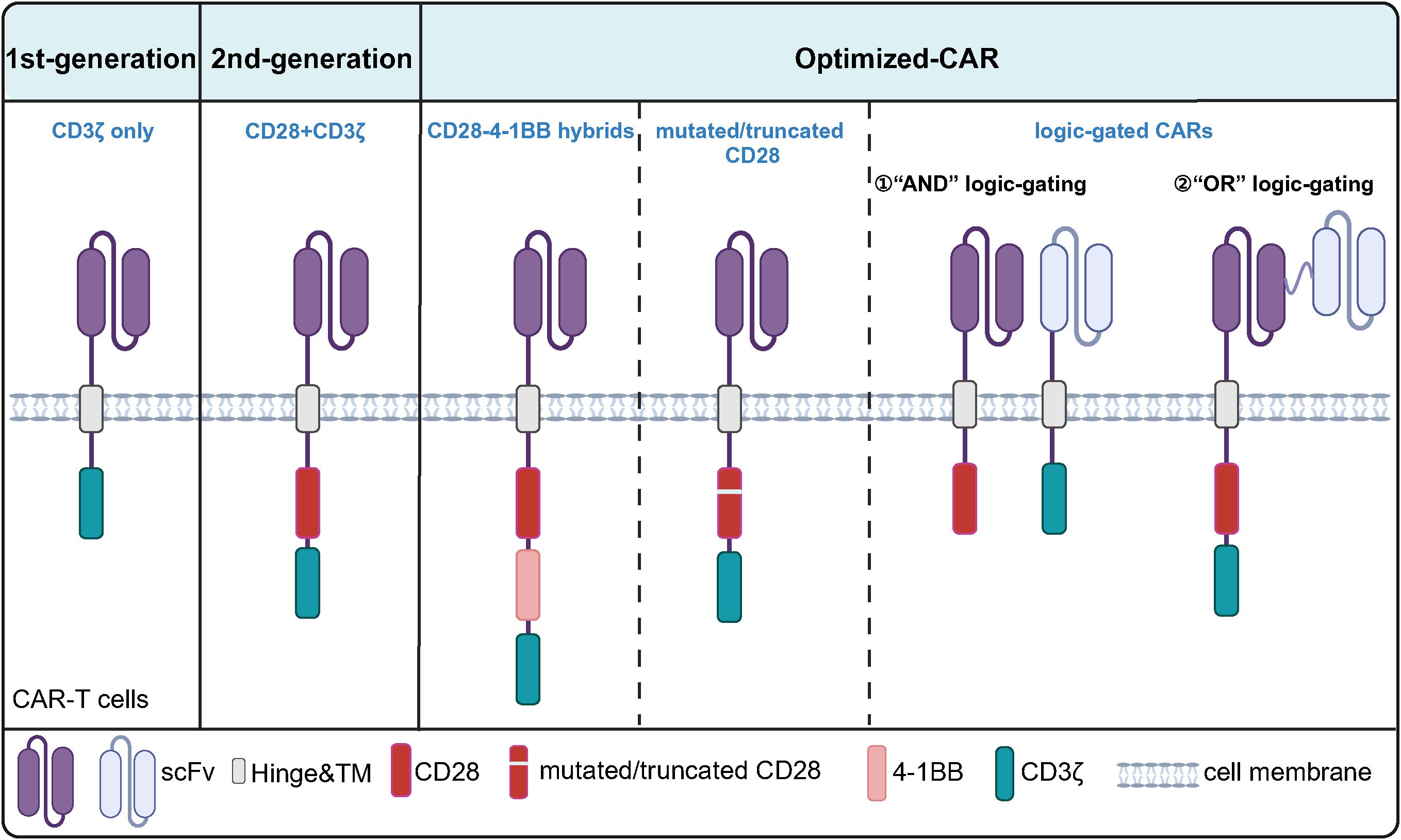
Figure 4. Structural Evolution of CD28-Based CARs. The first-generation CARs contain only the CD3ζ chain as the signaling domain. The second-generation CARs, built upon the first-generation design, incorporate CD28 as a co-stimulatory domain. Further optimization of CD28-based CARs includes combinations of CD28 with other co-stimulatory molecules such as 4-1BB, as well as mutated/truncated CD28 and "AND" or "OR" logic-gated CARs.
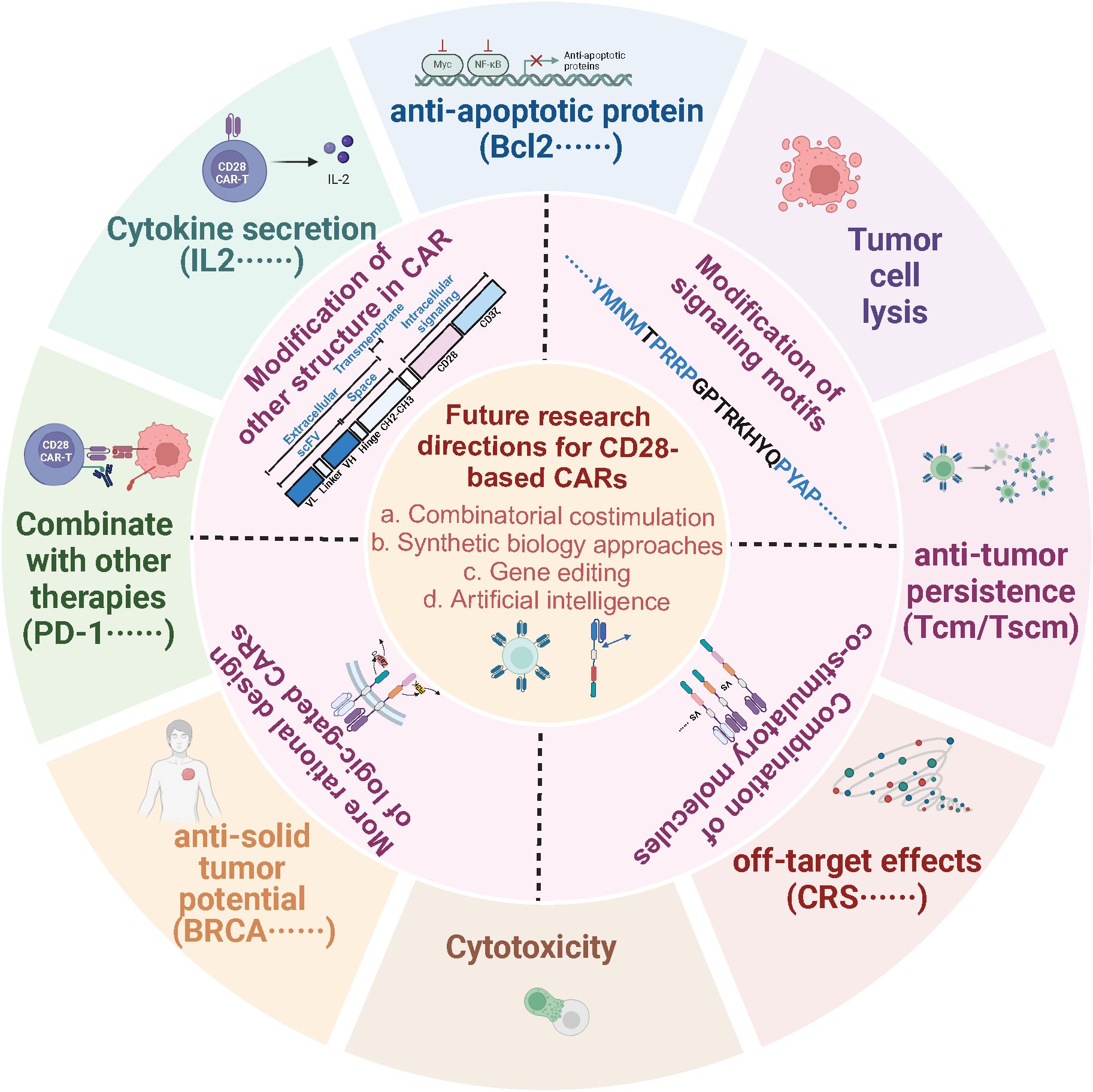
Figure 5. Optimization schemes for CD28-based CAR structures. Strategies to optimize the CD28-based CAR structure include mutating the signaling motif of CD28, combining it with other costimulatory molecules, separating the CD3ζ and CD28 signaling motifs or optimizing other modules in the CAR structure to regulate the activation, differentiation, survival, proliferation, and antitumor activity of CAR-T cells to enhance the durability and antitumor efficacy of CAR-T cell therapy, which offers potential to expand the application of CAR -T therapy in hematological or solid tumor applications.
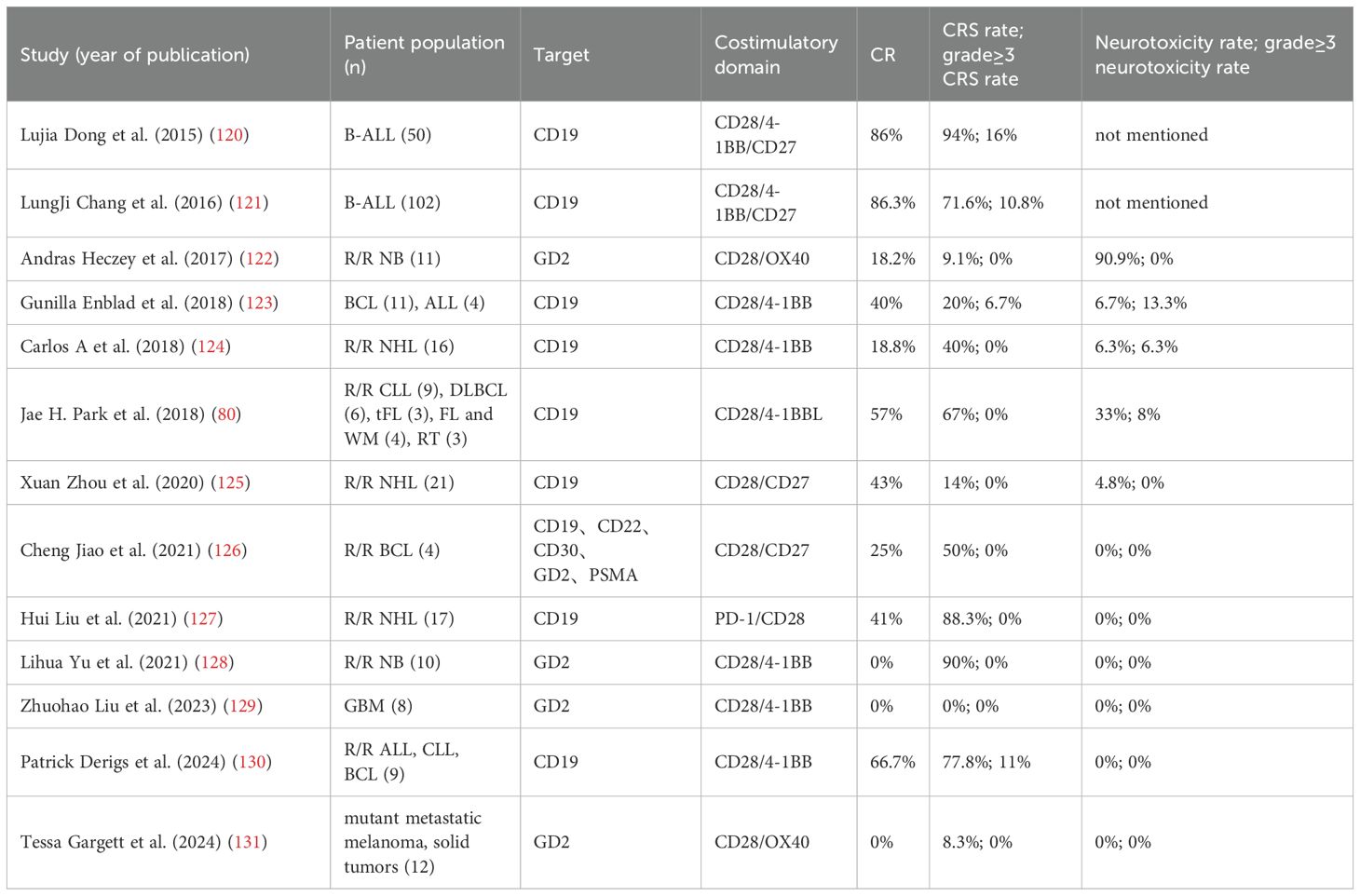
Table 2. The CRS and toxicity levels of CD28-based CARs following structure optimization in clinical trials.
CAR optimization strategies based on mutated CD28 signaling motifs indicate that CD28 participates in multiple signaling pathways. Selective manipulation of these pathways can achieve long-term persistence and anti-tumor activity of CAR-T cells, which is attributed to the application of gene editing technology in CAR structure design, which is expected to provide further space for mutation or modification of CD28 structural domains. Various combination modes of CD28 with other co-stimulatory molecules have been developed, demonstrating promising effects by synergizing multiple co-stimulatory signals. Multiple co-stimulatory signals have mobilized the host’s immune response by complementing each other’s strengths and even modulating dendritic cells (DCs), Treg cells, and immunosuppressive PD-L1/PD-1 in TMEs, increasing the potential of CAR-T therapies to be applied in solid tumors.
An important reason for the poor treatment of CAR-T solid tumors is the presence of multiple metabolic inhibitors (lactate, reactive oxygen species, prostaglandin E2) in the TME (94–97), which severely impairs CAR-T cells’ antitumor activity. Strategies to overcome this obstacle are mainly to express relevant molecules or enzymes resistant to metabolic inhibitors in TME (catalase) on CAR-T cells through gene editing (98, 99) or to express genes that enhance mitochondrial biogenesis and function (PGC1α) (100). Current modification strategies engineering CD28-based CAR-T resistance to TME are primarily limited to incorporating TME-resistant response elements, attenuation of PD-L1/PD-1 inhibitory signaling, and combination of metabolic regulatory drugs to orchestrate CD28 signaling to enhance CAR-T cell activity, and no modification of CD28 signaling or sequence has been reported. In the future, exploring the antitumor efficacy of resistance to TME triggered by CD28 mutation strategies in the construction of ex vivo TME models that mimic the hypoxic, high-lactate, glucose-competitive, and immunosuppressive features of TME (101, 102) could further develop TME-induced enhanced CD28, or design response elements targeting the inhibitory signaling on the basis thereof, offering the possibility of CAR-T therapy for broader application to cancer and improved efficacy.
Combining metabolic regulation with strategies to increase resistance to the tumor microenvironment can effectively enhance CAR-T cells’ survival and anti-tumor ability in the tumor microenvironment. A deeper understanding of the mechanisms of metabolic regulation can help to tailor tumor immunotherapy regimens to individual patients. For instance, considering the differences in the tumor microenvironment and T-cell status of different patients and selecting the most suitable CD28-based CAR design and metabolic regulation strategies to improve therapeutic efficacy and reduce adverse effects, may offer hope for CAR-T precision therapy. However, we must also be concerned about the possible toxicity risks of adding multiple co-stimulatory molecules. For example, a clinical trial treating 11 patients with non-Hodgkin’s lymphoma showed that tandem of a Toll-like receptor 2 (TLR2) TIR structural domain in a CD28-based CAR structure targeting CD19 resulted in severe CRS in 2 patients (18%) and severe ICANS in 1 patient (9%) (NCT04049513) (103). With the help of new technological tools, dynamic regulation of CD28 and other signaling intensities might provide safety for the combination of co-stimulatory molecules.
Currently, the application of artificial intelligence (AI) in CAR-T is at the frontier of exploration. The application of AI in CAR-T cancer therapy mainly focuses on AI-assisted prognosis of clinical efficacy of patients (104), establishment of toxicity prediction models, and assessment of side effects after CAR-T treatment (105, 106). Studies directly applying AI technology to optimize the structure of CARs have only focused on designing antibodies against cancer targets by using the protein design tool RFdiffusion to create antibodies against cancer targets (107) or using AI to design protein conjugates with high affinity for cancer antigens to replace scFV (108). AI has accelerated the development of CAR-T products by designing candidate proteins for binding to the target antigens of CAR-T. However, whether the antibodies or protein conjugates can be safely applied to humans without inducing immune responses requires further validation through experiments. In addition, for optimizing CAR signaling, including prediction and optimization of CAR-T tonic signaling using CAR-Toner’s AI tools (109), prediction of unnatural combinations of specific signaling motifs combinations and configurations affecting T-cell phenotypes (110). However, AI-based prediction of optimized CD28 signaling has not yet been reported. In the future, perhaps it may be possible to use the AI algorithms to construct virtual models for predicting CAR-T functional effects due to mutated CD28 signaling sites, accelerating the understanding of CD28 signaling mechanisms, based on which, combined with existing databases, may be able to provide personalized CD28-based CAR optimization solutions for cancer patients. Through AI’s learning of large amounts of data and the development of additional AI algorithms, it may be possible to predict the effects of different molecular combinations or modifications on CD28 co-stimulatory activity and further expand to predict the efficacy of changes including modification of scFV, hinge region, transmembrane region, and CD3ζ, which is expected to provide a basis for optimizing CAR-T cell therapy.
Author contributions
SL: Writing – original draft, Writing – review & editing. YZ: Writing – original draft, Writing – review & editing. HW: Writing – review & editing. GQ: Writing – review & editing. XZ: Validation, Visualization, Writing – review & editing. XW: Validation, Visualization, Writing – review & editing. RH: Validation, Visualization, Writing – review & editing. ZG: Validation, Visualization, Writing – review & editing. DL: Supervision, Writing – review & editing. JZ: Supervision, Writing – review & editing. MS: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the National Natural Science Foundation of China (No.: 82403846, 82273207, 82471378, 82204541 and 82304535), Natural Science Foundation of Jiangsu Province (No.: BK20210910, BK20243034 and BK20220671), Young academic leaders of Jiangsu Qinglan Project, National science research in Universities of Jiangsu Province (No.:22KJB320009).
Acknowledgments
Figures were created with biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. (2022) 33:259–75. doi: 10.1016/j.annonc.2021.12.003
2. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
3. Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science. (2022) 378:eaba1624. doi: 10.1126/science.aba1624
4. Wu L, Brzostek J, Sakthi Vale PD, Wei Q, Koh CKT, Ong JXH, et al. CD28-CAR-T cell activation through FYN kinase signaling rather than LCK enhances therapeutic performance. Cell Rep Med. (2023) 4:100917. doi: 10.1016/j.xcrm.2023.100917
5. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
6. Li C, Zhou F, Wang J, Chang Q, Du M, Luo W, et al. Novel CD19-specific γ/δ TCR-T cells in relapsed or refractory diffuse large B-cell lymphoma. J Hematol Oncol. (2023) 16:5. doi: 10.1186/s13045-023-01402-y
7. Zhao Y, Caron C, Chan YY, Lee CK, Xu X, Zhang J, et al. cis-B7:CD28 interactions at invaginated synaptic membranes provide CD28 co-stimulation and promote CD8(+) T cell function and anti-tumor immunity. Immunity. (2023) 56:1187–203.e12. doi: 10.1016/j.immuni.2023.04.005
8. Ford ML. T cell cosignaling molecules in transplantation. Immunity. (2016) 44:1020–33. doi: 10.1016/j.immuni.2016.04.012
9. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. . doi: 10.1158/2159-8290.Cd-19-0945
10. Holling GA, Chavel CA, Sharda AP, Lieberman MM, James CM, Lightman SM, et al. CD8+ T cell metabolic flexibility elicited by CD28-ARS2 axis-driven alternative splicing of PKM supports antitumor immunity. Cell Mol Immunol. (2024) 21:260–74. doi: 10.1038/s41423-024-01124-2
11. Humblin E, Korpas I, Lu J, Filipescu D, van der Heide V, Goldstein S, et al. Sustained CD28 costimulation is required for self-renewal and differentiation of TCF-1(+) PD-1(+) CD8 T cells. Sci Immunol. (2023) 8:eadg0878. doi: 10.1126/sciimmunol.adg0878
12. Rotolo A, Atherton MJ. Applications and opportunities for immune cell CAR engineering in comparative oncology. Clin Cancer Res. (2024) 30:2359–69. doi: 10.1158/1078-0432.CCR-23-3690
13. Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. (1993) 11:191–212. doi: 10.1146/annurev.iy.11.040193.001203
14. June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today. (1990) 11:211–6. doi: 10.1016/0167-5699(90)90085-n
15. Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. (2002) 20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806
16. Gomez-Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec family kinases Itk and Rlk/Txk in T lymphocytes: cross-regulation of cytokine production and T-cell fates. FEBS J. (2011) 278:1980–9. doi: 10.1111/j.1742-4658.2011.08072.x
17. Andres PG, Howland KC, Nirula A, Kane LP, Barron L, Dresnek D, et al. Distinct regions in the CD28 cytoplasmic domain are required for T helper type 2 differentiation. Nat Immunol. (2004) 5:435–42. doi: 10.1038/ni1044
18. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. (2001) 1:220–8. doi: 10.1038/35105024
19. Holdorf AD, Green JM, Levin SD, Denny MF, Straus DB, Link V, et al. Proline residues in CD28 and the Src homology (SH)3 domain of Lck are required for T cell costimulation. J Exp Med. (1999) 190:375–84. doi: 10.1084/jem.190.3.375
20. Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: implications for T-cell costimulation. Proc Natl Acad Sci U.S.A. (1995) 92:8891–5. doi: 10.1073/pnas.92.19.8891
21. Kim HH, Tharayil M, Rudd CE. Growth factor receptor-bound protein 2 SH2/SH3 domain binding to CD28 and its role in co-signaling. J Biol Chem. (1998) 273:296–301. doi: 10.1074/jbc.273.1.296
22. zur Hausen JD, Burn P, Amrein KE. Co-localization of Fyn with CD3 complex, CD45 or CD28 depends on different mechanisms. Eur J Immunol. (1997) 27:2643–9. doi: 10.1002/eji.1830271025
23. Marengere LE, Okkenhaug K, Clavreul A, Couez D, Gibson S, Mills GB, et al. The SH3 domain of Itk/Emt binds to proline-rich sequences in the cytoplasmic domain of the T cell costimulatory receptor CD28. J Immunol. (1997) 159:3220–9. doi: 10.4049/jimmunol.159.7.3220
24. Okkenhaug K, Rottapel R. Grb2 forms an inducible protein complex with CD28 through a Src homology 3 domain-proline interaction. J Biol Chem. (1998) 273:21194–202. doi: 10.1074/jbc.273.33.21194
25. Carey KD, Dillon TJ, Schmitt JM, Baird AM, Holdorf AD, Straus DB, et al. CD28 and the tyrosine kinase lck stimulate mitogen-activated protein kinase activity in T cells via inhibition of the small G protein Rap1. Mol Cell Biol. (2000) 20:8409–19. doi: 10.1128/MCB.20.22.8409-8419.2000
26. Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. (1991) 251:313–6. doi: 10.1126/science.1846244
27. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol. (1987) 7:4472–81. doi: 10.1128/mcb.7.12.4472-4481.1987
28. Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, et al. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci U.S.A. (1989) 86:1333–7. doi: 10.1073/pnas.86.4.1333
29. Tavano R, Contento RL, Baranda SJ, Soligo M, Tuosto L, Manes S, et al. CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse. Nat Cell Biol. (2006) 8:1270–6. doi: 10.1038/ncb1492
30. Muscolini M, Sajeva A, Caristi S, Tuosto L. A novel association between filamin A and NF-kappaB inducing kinase couples CD28 to inhibitor of NF-kappaB kinase alpha and NF-kappaB activation. Immunol Lett. (2011) 136:203–12. doi: 10.1016/j.imlet.2011.01.011
31. Sanchez-Lockhart M, Graf B, Miller J. Signals and sequences that control CD28 localization to the central region of the immunological synapse. J Immunol. (2008) 181:7639–48. doi: 10.4049/jimmunol.181.11.7639
32. Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, et al. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity. (2008) 29:589–601. doi: 10.1016/j.immuni.2008.08.011
33. Neelapu SS, Jacobson CA, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. (2023) 141:2307–15. doi: 10.1182/blood.2022018893
34. Westin JR, Oluwole OO, Kersten MJ, Miklos DB, Perales MA, Ghobadi A, et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. N Engl J Med. (2023) 389:148–57. doi: 10.1056/NEJMoa2301665
35. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. Three-year follow-up of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma, including high-risk subgroups, in the ZUMA-2 study. J Clin Oncol. (2023) 41:555–67. doi: 10.1200/JCO.21.02370
36. Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell non-hodgkin’s lymphoma. Mol Ther Oncolytics. (2019) 15:60–8. doi: 10.1016/j.omto.2019.08.002
37. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
38. Tang XY, Sun Y, Zhang A, Hu GL, Cao W, Wang DH, et al. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ Open. (2016) 6:e013904. doi: 10.1136/bmjopen-2016-013904
39. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
40. Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight. (2018) 3(18):e121322. doi: 10.1172/jci.insight.121322
41. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
42. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
43. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/s1074-7613(02)00323-0
44. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
45. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U.S.A. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
46. Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell Malignancies in phase I clinical trials: a meta-analysis. Oncotarget. (2015) 6:33961–71. doi: 10.18632/oncotarget.5582
47. Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced therapeutic efficacy and memory of tumor-specific CD8 T cells by ex vivo PI3K-delta inhibition. Cancer Res. (2017) 77:4135–45. doi: 10.1158/0008-5472.CAN-16-1925
48. Chan JD, Scheffler CM, Munoz I, Sek K, Lee JN, Huang YK, et al. FOXO1 enhances CAR T cell stemness, metabolic fitness and efficacy. Nature. (2024) 629:201–10. doi: 10.1038/s41586-024-07242-1
49. Zhu M, Han Y, Gu T, Wang R, Si X, Kong D, et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. (2024) 43:114065. doi: 10.1016/j.celrep.2024.114065
50. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. (2018) 11(544):eaat6753. doi: 10.1126/scisignal.aat6753
51. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. (2020) 130:3087–97. doi: 10.1172/JCI133215
52. Boucher JC, Li G, Kotani H, Cabral ML, Morrissey D, Lee SB, et al. CD28 costimulatory domain-targeted mutations enhance chimeric antigen receptor T-cell function. Cancer Immunol Res. (2021) 9:62–74. doi: 10.1158/2326-6066.CIR-20-0253
53. Fu Z, Huang Z, Xu H, Liu Q, Li J, Song K, et al. IL-2-inducible T cell kinase deficiency sustains chimeric antigen receptor T cell therapy against tumor cells. J Clin Invest. (2024) 135(4):e178558. doi: 10.1172/JCI178558
54. Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. (2011) 19:760–7. doi: 10.1038/mt.2011.9
55. Gulati P, Ruhl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, et al. Aberrant lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. (2018) 24:3981–93. doi: 10.1158/1078-0432.CCR-17-1788
56. Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. (2023) 12:14. doi: 10.1186/s40164-023-00373-7
57. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
58. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. (2013) 1:43–53. doi: 10.1158/2326-6066.CIR-13-0008
59. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
60. Guedan S, Calderon H, Posey AD Jr., Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
61. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
62. Feucht J, Sadelain M. Function and evolution of the prototypic CD28zeta and 4-1BBzeta chimeric antigen receptors. Immunooncol Technol. (2020) 8:2–11. doi: 10.1016/j.iotech.2020.09.001
63. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. (2014) 20:6418–28. doi: 10.1158/1078-0432.CCR-14-1170
64. Zuccolotto G, Penna A, Fracasso G, Carpanese D, Montagner IM, Dalla Santa S, et al. PSMA-specific CAR-engineered T cells for prostate cancer: CD28 outperforms combined CD28-4-1BB “Super-stimulation. Front Oncol. (2021) 11:708073. doi: 10.3389/fonc.2021.708073
65. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. (2016) 7:690. doi: 10.3389/fimmu.2016.00690
66. Yuti P, Sawasdee N, Natungnuy K, Rujirachaivej P, Luangwattananun P, Sujjitjoon J, et al. Enhanced antitumor efficacy, proliferative capacity, and alleviation of T cell exhaustion by fifth-generation chimeric antigen receptor T cells targeting B cell maturation antigen in multiple myeloma. BioMed Pharmacother. (2023) 168:115691. doi: 10.1016/j.biopha.2023.115691
67. Honikel MM, Olejniczak SH. Co-stimulatory receptor signaling in CAR-T cells. Biomolecules. (2022) 12(9):1303. doi: 10.3390/biom12091303
68. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. (2012) 1:458–66. doi: 10.4161/onci.19855
69. Guercio M, Orlando D, Di Cecca S, Sinibaldi M, Boffa I, Caruso S, et al. CD28.OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica. (2021) 106:987–99. doi: 10.3324/haematol.2019.231183
70. Tang X, Tang Q, Mao Y, Huang X, Jia L, Zhu J, et al. CD137 co-stimulation improves the antitumor effect of LMP1-specific chimeric antigen receptor T cells in vitro and in vivo. Onco Targets Ther. (2019) 12:9341–50. doi: 10.2147/OTT.S221040
71. Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
72. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:712. doi: 10.1016/j.immuni.2016.02.023
73. Moreno-Cortes E, Franco-Fuquen P, Garcia-Robledo JE, Forero J, Booth N, Castro JE. ICOS and OX40 tandem co-stimulation enhances CAR T-cell cytotoxicity and promotes T-cell persistence phenotype. Front Oncol. (2023) 13:1200914. doi: 10.3389/fonc.2023.1200914
74. Halim L, Das KK, Larcombe-Young D, Ajina A, Candelli A, Benjamin R, et al. Engineering of an avidity-optimized CD19-specific parallel chimeric antigen receptor that delivers dual CD28 and 4-1BB co-stimulation. Front Immunol. (2022) 13:836549. doi: 10.3389/fimmu.2022.836549
75. Katsarou A, Sjostrand M, Naik J, Mansilla-Soto J, Kefala D, Kladis G, et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Transl Med. (2021) 13:eabh1962. doi: 10.1126/scitranslmed.abh1962
76. Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fuca G, et al. Dual targeting CAR-T cells with optimal costimulation and metabolic fitness enhance antitumor activity and prevent escape in solid tumors. Nat Cancer. (2021) 2:904–18. doi: 10.1038/s43018-021-00244-2
77. Fernandez de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discovery. (2020) 1:146–54. doi: 10.1158/2643-3230.BCD-20-0020
78. Hu W, Huang X, Huang X, Chen W, Hao L, Chen Z. Chimeric antigen receptor modified T cell (CAR-T) co-expressed with ICOSL-41BB promote CAR-T proliferation and tumor rejection. BioMed Pharmacother. (2019) 118:109333. doi: 10.1016/j.biopha.2019.109333
79. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
80. Park JH, Palomba ML, Batlevi CL, Riviere I, Wang X, Senechal B, et al. A phase I first-in-human clinical trial of CD19-targeted 19-28z/4-1BBL “Armored” CAR T cells in patients with relapsed or refractory NHL and CLL including richter’s transformation. Blood. (2018) 132:224. doi: 10.1182/blood-2018-99-117737
81. Nguyen P, Okeke E, Clay M, Haydar D, Justice J, O’Reilly C, et al. Route of 41BB/41BBL costimulation determines effector function of B7-H3-CAR.CD28zeta T cells. Mol Ther Oncolytics. (2020) 18:202–14. doi: 10.1016/j.omto.2020.06.018
82. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. (2015) 23:769–78. doi: 10.1038/mt.2015.4
83. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell. (2019) 35:473–88.e6. doi: 10.1016/j.ccell.2019.02.006
84. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned erbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
85. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
86. Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. (2016) 5:e1253656. doi: 10.1080/2162402X.2016.1253656
87. Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. (2015) 22:391–403. doi: 10.1038/gt.2015.4
88. Morin SO, Giroux V, Favre C, Bechah Y, Auphan-Anezin N, Roncagalli R, et al. In the absence of its cytosolic domain, the CD28 molecule still contributes to T cell activation. Cell Mol Life Sci. (2015) 72:2739–48. doi: 10.1007/s00018-015-1873-7
89. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. (2003) 3:939–51. doi: 10.1038/nri1248
90. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, et al. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol. (2001) 166:182–7. doi: 10.4049/jimmunol.166.1.182
91. James JR. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci Signal. (2018) 11(531):eaan1088. doi: 10.1126/scisignal.aan1088
92. Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. (2009) 183:5563–74. doi: 10.4049/jimmunol.0900447
93. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
94. Cheng H, Qiu Y, Xu Y, Chen L, Ma K, Tao M, et al. Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat Metab. (2023) 5:314–30. doi: 10.1038/s42255-022-00730-6
95. Kennel KB, Greten FR. Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol. (2021) 42:101891. doi: 10.1016/j.redox.2021.101891
96. Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. (2015) 21:327–34. doi: 10.1038/nm.3831
97. Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. (2015) 4:e1016700. doi: 10.1080/2162402X.2015.1016700
98. Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. (2018) 78:4692–703. doi: 10.1158/0008-5472.CAN-18-0030
99. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. (2016) 196:759–66. doi: 10.4049/jimmunol.1401710
100. Peng JJ, Wang L, Li Z, Ku CL, Ho PC. Metabolic challenges and interventions in CAR T cell therapy. Sci Immunol. (2023) 8:eabq3016. doi: 10.1126/sciimmunol.abq3016
101. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
102. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–18.e9. doi: 10.1016/j.ccell.2022.01.001
103. George P, Dasyam N, Giunti G, Mester B, Bauer E, Andrews B, et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: a phase I clinical trial protocol (ENABLE). BMJ Open. (2020) 10:e034629. doi: 10.1136/bmjopen-2019-034629
104. Naghizadeh A, Tsao WC, Hyun Cho J, Xu H, Mohamed M, Li D, et al. In vitro machine learning-based CAR T immunological synapse quality measurements correlate with patient clinical outcomes. PloS Comput Biol. (2022) 18:e1009883. doi: 10.1371/journal.pcbi.1009883
105. Bogatu A, Wysocka M, Wysocki O, Butterworth H, Pillai M, Allison J, et al. Meta-analysis informed machine learning: Supporting cytokine storm detection during CAR-T cell Therapy. J BioMed Inform. (2023) 142:104367. doi: 10.1016/j.jbi.2023.104367
106. Wei Z, Zhao C, Zhang M, Xu J, Xu N, Wu S, et al. Meta-DHGNN: method for CRS-related cytokines analysis in CAR-T therapy based on meta-learning directed heterogeneous graph neural network. Brief Bioinform. (2024) 25(3):bbae104. doi: 10.1093/bib/bbae104
107. Callaway E. ‘A landmark moment’: scientists use AI to design antibodies from scratch. Nature. (2024). doi: 10.1038/d41586-024-00846-7
108. Xia Z, Jin Q, Long Z, He Y, Liu F, Sun C, et al. Targeting overexpressed antigens in glioblastoma via CAR T cells with computationally designed high-affinity protein binders. Nat BioMed Eng. (2024) 8(12):1634–50. doi: 10.1038/s41551-024-01258-8
109. Qiu S, Chen J, Wu T, Li L, Wang G, Wu H, et al. CAR-Toner: an AI-driven approach for CAR tonic signaling prediction and optimization. Cell Res. (2024) 34:386–8. doi: 10.1038/s41422-024-00936-1
110. Daniels KG, Wang S, Simic MS, Bhargava HK, Capponi S, Tonai Y, et al. Decoding CAR T cell phenotype using combinatorial signaling motif libraries and machine learning. Science. (2022) 378:1194–200. doi: 10.1126/science.abq0225
111. Chen C, Li K, Jiang H, Song F, Gao H, Pan X, et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol Immunother. (2017) 66:475–89. doi: 10.1007/s00262-016-1949-8
112. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
113. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. (2021) 9(10):e003354. doi: 10.1136/jitc-2021-003354
114. Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. (2005) 12:933–41. doi: 10.1016/j.ymthe.2005.04.016
115. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. (2018) 131:746–58. doi: 10.1182/blood-2017-05-781351
116. Khopanlert W, Choochuen P, Maneechai K, Jangphattananont N, Ung S, Okuno S, et al. Co-stimulation of CD28/CD40 signaling molecule potentiates CAR-T cell efficacy and stemness. Mol Ther Oncol. (2024) 32:200837. doi: 10.1016/j.omton.2024.200837
117. Muliaditan T, Halim L, Whilding LM, Draper B, Achkova DY, Kausar F, et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep Med. (2021) 2:100457. doi: 10.1016/j.xcrm.2021.100457
118. Khaniya A, Rad S, Halpin J, Tawinwung S, McLellan A, Suppipat K, et al. Development of a compact bidirectional promoter-driven dual chimeric antigen receptor (CAR) construct targeting CD19 and CD20 in the Sleeping Beauty (SB) transposon system. J Immunother Cancer. (2024) 12(4):e008555. doi: 10.1136/jitc-2023-008555
119. Ng BD, Rajagopalan A, Kousa AI, Fischman JS, Chen S, Massa A, et al. IL-18-secreting multiantigen targeting CAR T cells eliminate antigen-low myeloma in an immunocompetent mouse model. Blood. (2024) 144:171–86. doi: 10.1182/blood.2023022293
120. Dong L, Chang L-J, Gao Z, Lu D-P, Zhang J-P, Wang J-B, et al. Chimeric antigen receptor 4SCAR19-modified T cells in acute lymphoid leukemia: a phase II multi-center clinical trial in China. Blood. (2015) 126:3774. doi: 10.1182/blood.V126.23.3774.3774
121. Chang L-J, Dong L, Liu Y-C, Tsao S-T, Li Y-C, Liu L, et al. Safety and efficacy evaluation of 4SCAR19 chimeric antigen receptor-modified T cells targeting B cell acute lymphoblastic leukemia - three-year follow-up of a multicenter phase I/II study. Blood. (2016) 128:587. doi: 10.1182/blood.V128.22.587.587
122. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
123. Enblad G, Karlsson H, Gammelgard G, Wenthe J, Lovgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. (2018) 24:6185–94. doi: 10.1158/1078-0432.CCR-18-0426
124. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin’s lymphomas. Mol Ther. (2018) 26:2727–37. doi: 10.1016/j.ymthe.2018.09.009
125. Zhou X, Tu S, Wang C, Huang R, Deng L, Song C, et al. Phase I trial of fourth-generation anti-CD19 chimeric antigen receptor T cells against relapsed or refractory B cell non-hodgkin lymphomas. Front Immunol. (2020) 11:564099. doi: 10.3389/fimmu.2020.564099
126. Jiao C, Zvonkov E, Lai X, Zhang R, Liu Y, Qin Y, et al. 4SCAR2.0: a multi-CAR-T therapy regimen for the treatment of relapsed/refractory B cell lymphomas. Blood Cancer J. (2021) 11:59. doi: 10.1038/s41408-021-00455-x
127. Liu H, Lei W, Zhang C, Yang C, Wei J, Guo Q, et al. CD19-specific CAR T cells that express a PD-1/CD28 chimeric switch-receptor are effective in patients with PD-L1-positive B-cell lymphoma. Clin Cancer Res. (2021) 27:473–84. doi: 10.1158/1078-0432.CCR-20-1457
128. Yu L, Huang L, Lin D, Lai X, Wu L, Liao X, et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol. (2022) 148:2643–52. doi: 10.1007/s00432-021-03839-5
129. Liu Z, Zhou J, Yang X, Liu Y, Zou C, Lv W, et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. (2023) 22:3. doi: 10.1186/s12943-022-01711-9
130. Derigs P, Schubert ML, Dreger P, Schmitt A, Yousefian S, Haas S, et al. Third-generation anti-CD19 CAR T cells for relapsed/refractory chronic lymphocytic leukemia: a phase 1/2 study. Leukemia. (2024) 38:2419–28. doi: 10.1038/s41375-024-02392-7
131. Gargett T, Truong NTH, Gardam B, Yu W, Ebert LM, Johnson A, et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J Immunother Cancer. (2024) 12(5):e008659. doi: 10.1136/jitc-2023-008659
Keywords: CD28, CAR, co-stimulatory molecules, adoptive immune cell therapy, cancer therapy
Citation: Li S, Zhou Y, Wang H, Qu G, Zhao X, Wang X, Hou R, Guan Z, Liu D, Zheng J and Shi M (2025) Advances in CAR optimization strategies based on CD28. Front. Immunol. 16:1548772. doi: 10.3389/fimmu.2025.1548772
Received: 20 December 2024; Accepted: 26 February 2025;
Published: 13 March 2025.
Edited by:
Fabio Grizzi, Humanitas Research Hospital, ItalyReviewed by:
Yuedi Wang, Fudan University, ChinaShivani Srivastava, Yale University, United States
Liqiong Liu, The 6th Affiliated Hospital of Shenzhen University Health Science Center, Shenzhen, China
Copyright © 2025 Li, Zhou, Wang, Qu, Zhao, Wang, Hou, Guan, Liu, Zheng and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Liu, bGl1ZGFuX2JkQHNpbmEuY29t; Junnian Zheng, am56aGVuZ0B4emhtdS5lZHUuY24=; Ming Shi, bWluZ3NoaUB4emhtdS5lZHUuY24=
†These authors have contributed equally to this work