 Jin-Yan Li
Jin-Yan Li Zhi-Jian Zhou
Zhi-Jian Zhou Qiong Wang
Qiong Wang Qing-Nan He
Qing-Nan He Ming-Yi Zhao
Ming-Yi Zhao Ye Qiu
Ye Qiu Xing-Yi Ge
Xing-Yi Ge- 1Hunan Provincial Key Laboratory of Medical Virology, Institute of Pathogen Biology and Immunology, College of Biology, Hunan University, Changsha, China
- 2Department of Pediatrics, The Third Xiangya Hospital, Central South University, Changsha, China
In the past two decades, coronavirus (CoV) has emerged frequently in the population. Three CoVs (SARS-CoV, MERS-CoV, SARS-CoV-2) have been identified as highly pathogenic human coronaviruses (HP-hCoVs). Particularly, the ongoing COVID-19 pandemic caused by SARS-CoV-2 warns that HP-hCoVs present a high risk to human health. Like other viruses, HP-hCoVs interact with their host cells in sophisticated manners for infection and pathogenesis. Here, we reviewed the current knowledge about the interference of HP-hCoVs in multiple cellular processes and their impacts on viral infection. HP-hCoVs employed various strategies to suppress and evade from immune response, including shielding viral RNA from recognition by pattern recognition receptors (PRRs), impairing IFN-I production, blocking the downstream pathways of IFN-I, and other evasion strategies. This summary provides a comprehensive view of the interplay between HP-hCoVs and the host cells, which is helpful to understand the mechanism of viral pathogenesis and develop antiviral therapies.
Introduction
Coronaviruses (CoVs) are enveloped positive-sense single-stranded RNA viruses infecting various mammals and birds, including humans (Zhou et al., 2021). In the last century, human coronaviruses (hCoVs) were recognized as mild respiratory pathogens, which were barely studied regarding their low pathogenicity (Zhou et al., 2021). However, highly pathogenic hCoVs (HP-hCoVs) have emerged since the year 2003 and have caused three worldwide epidemics, including severe acute respiratory syndrome (SARS) epidemic caused by SARS-CoV in 2003, Middle East Respiratory Syndrome (MERS) epidemic caused by MERS-CoV in 2012, and the recent coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2. Infections of these HP-hCoVs mainly cause acute respiratory distress syndrome (ARDS), with fatality rates of 9.5, 34.4, and 2.3% for SARS-CoV, MERS-CoV, and SARS-CoV-2, respectively (Petrosillo et al., 2020). Especially, SARS-CoV-2 shows the highest transmissibility among the three HP-hCoVs, which has led to one of the most severe global pandemics with 213 million infected cases and over 4.4 million deaths as of August 23, 2021 (WHO, n.d.). An overwhelming preponderance of cases and deaths is reported in the elderly, especially with underlying diabetes, cardiovascular, and hypertension comorbidities. By contrast, few severe cases are found in young children, whose innate immunity response is highly effective (Bajaj et al., 2020). Innate immunity is a determinant factor for disease outcome. Although 4,680 million vaccine doses have been administered, confirmed cases of COVID-19 are increasing sharply, drawing more and more attention of researchers around the world on the pathogenesis of HP-hCoV infection (WHO, n.d.). Pathogenesis is determined by the interplay between HP-hCoV and host antiviral defense. This review summarized the innate immunity evasion tactics employed by HP-hCoVs, focusing on the interactions of various viral proteins and host signaling pathways.
Highly Pathogenic Human Coronaviruses and the IFN System
All the three HP-hCoVs are single-stranded RNA (ssRNA) viruses classified into Betacoronaviruses genus of Orthocoronavirinae subfamily in Coronaviridae family (Group, 2020; Zhou et al., 2021). Specifically, SARS-CoV and SARS-CoV-2 belong to the subgenus of Sarbecovirus, while MERS-CoV is classified into the subgenus of Merbecovirus (Zhou et al., 2021). The SARS-CoV-2 genome shares around 79% identity with SARS-CoV and 30% with MERS-CoV, respectively (Zhou et al., 2020). Their typical genomic organization contains non-structural, structural, and accessory proteins flanked by a 5′-cap structure and a 3′-poly (A) tail (Yang and Leibowitz, 2015). The open reading frame (ORF) 1a and ORF1b occupy the two-third 5′ region of the viral genome and can be directly translated into two large polyproteins, pp1a and pp1b, which are further hydrolyzed into 16 non-structural proteins (NSP1 ∼ NSP16) by two viral proteases, NSP3 (papain-like protease, PL-pro) and NSP5 (3C-like protease, 3CL-pro) (Zhou et al., 2021). These NSPs take shape the replication-transcription complex (RTC), which is necessary for viral RNA transcription and replication (Li et al., 2020b). NSPs of different CoVs are evolutionarily conservative except for NSP1 and NSP2, usually virulent factors. The remaining one-third 3′ region of the viral genome encodes viral structural proteins (S, E, M, and N) and virus-specific accessory proteins, which are translated from the subgenomic RNAs (sgRNAs) synthesized in the discontinuous viral transcription process (Zhang Z. et al., 2021). Some of these ORFs are overlapping or found within a larger ORF (Zhou et al., 2021). Accessory proteins are distinct for different CoVs in their numbers, sequences, genomic locations, and functions. For instance, nine accessory proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10) of SARS-CoV-2, eight accessory proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8a, ORF8b, and ORF9b) of SARS-CoV, and five accessory proteins (ORF3a, ORF4a, ORF4b, ORF5, and ORF8b) of MERS-CoV have been identified (Figure 1). Among these proteins, many have been reported to suppress the innate immunity against HP-hCoV infection, which is considered critical for the pathogenesis of HP-hCoVs.
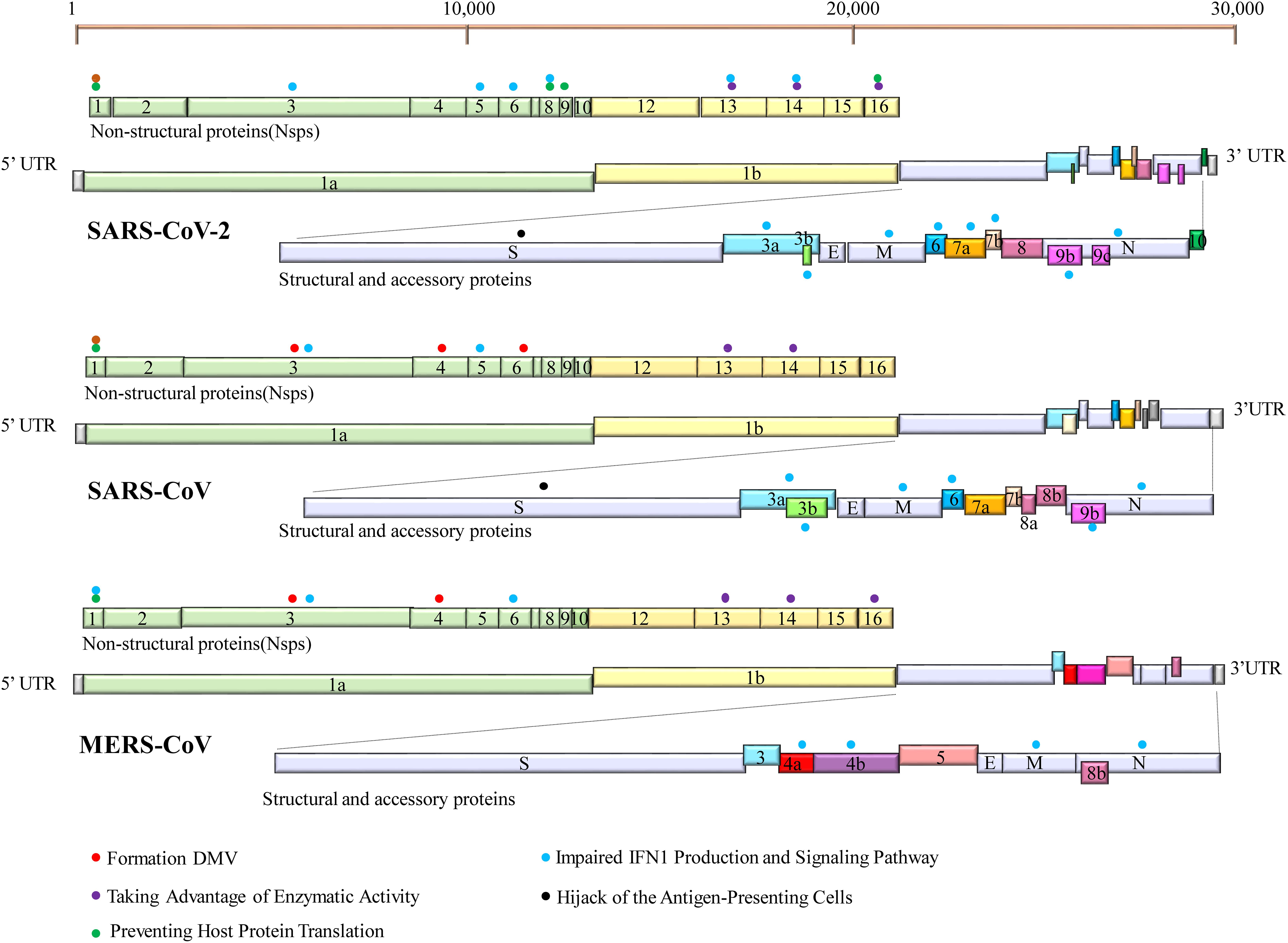
Figure 1. Genome organization of SARS-CoV-2, SARS-CoV, and MERS-CoV. The common trait is that their genomes encode two replicase polypeptides pp1a and pp1b translated from ORF1a and ORF1b. The polypeptides undergo a series of proteolytic cleavages to form 16 non-structural proteins encoded by the first two-thirds of the genome. The remaining one-third 3′ region of the viral genome encodes viral structural proteins [spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins] and virus-specific accessory proteins. Accessory proteins are interspersed within these structural proteins. Some proteins can inhibit innate immune responses by employing a variety of tactics. Their locations are indicated with specific-colored spheres. The different colored spheres represent the different immune evasive strategies. Each virus-encoded multifunctional protein, employing multiple different activities to suppress innate immunity responses.
The innate immunity acts as a frontline of defense characterized mainly by type I interferon (IFN-I) response, including recognizing pathogen-associated molecular patterns (PAMPs), IFN induction, and IFN signal transduction (Figure 2). Upon CoV infection, viral RNA could be recognized by the RIG-I-like receptors (RLRs), including retinoic acid-inducible gene I (RIG-I), melanoma differentiation gene 5 (MDA5), and toll-like receptors (TLRs) (Ivashkiv and Donlin, 2014). RIG-I and MDA5 trigger the downstream adaptor mitochondrial antiviral signaling protein (MAVS) on mitochondria. MAVS subsequently recruits the two IKKε and TANK-binding kinase 1 (TBK1), leading to phosphorylation and nuclear translocation of IFN-regulatory factor 3 (IRF3), which induces the expression and secretion of IFNs (Kawai et al., 2005). In addition, MAVS recruits IKK-related kinases (IKKα, IKKβ, and IKKγ) and activates the NF-κB pathway by promoting phosphorylation and nuclear translocation of p65, leading to cytokine production (Kawai et al., 2005). The secreted IFN-I can turn on the antiviral status in infected or neighboring cells through autocrine or paracrine. In brief, once the secreted IFN-I bind to their receptors, IFNAR (interferon alpha/beta receptor), on the cell surface, the downstream Janus kinase (Jak)/signal transducer and activator of transcription (STAT) signal pathway will be initiated by the activation of receptor-associated Jak1/TYK2 (tyrosine kinase 2). Then, phosphorylated STAT1 and STAT2 form heterodimers, interacting with IFN regulatory factor 9 (IRF9) to form an IFN-stimulated gene factor 3 (ISGF3) transcription complex, which translocates to the nucleus and binds to IFN-stimulated response elements (ISREs) in gene promoters, thereby activating the expression of interferon-stimulated genes (ISGs) to establish the host antiviral status (Ivashkiv and Donlin, 2014). Therefore, IFN-I is a strong immune modulator. It has a wide range of antiviral functions: (1) it can induce the expression of various antiviral proteins and restrict the synthesis of viral proteins, thus impairing virus replication; (2) it can induce apoptosis of infected cells, eliminating the “virus production factory”; (3) it can promote the maturation and activation of dendritic cells, promoting the activation of the adaptive immune response.
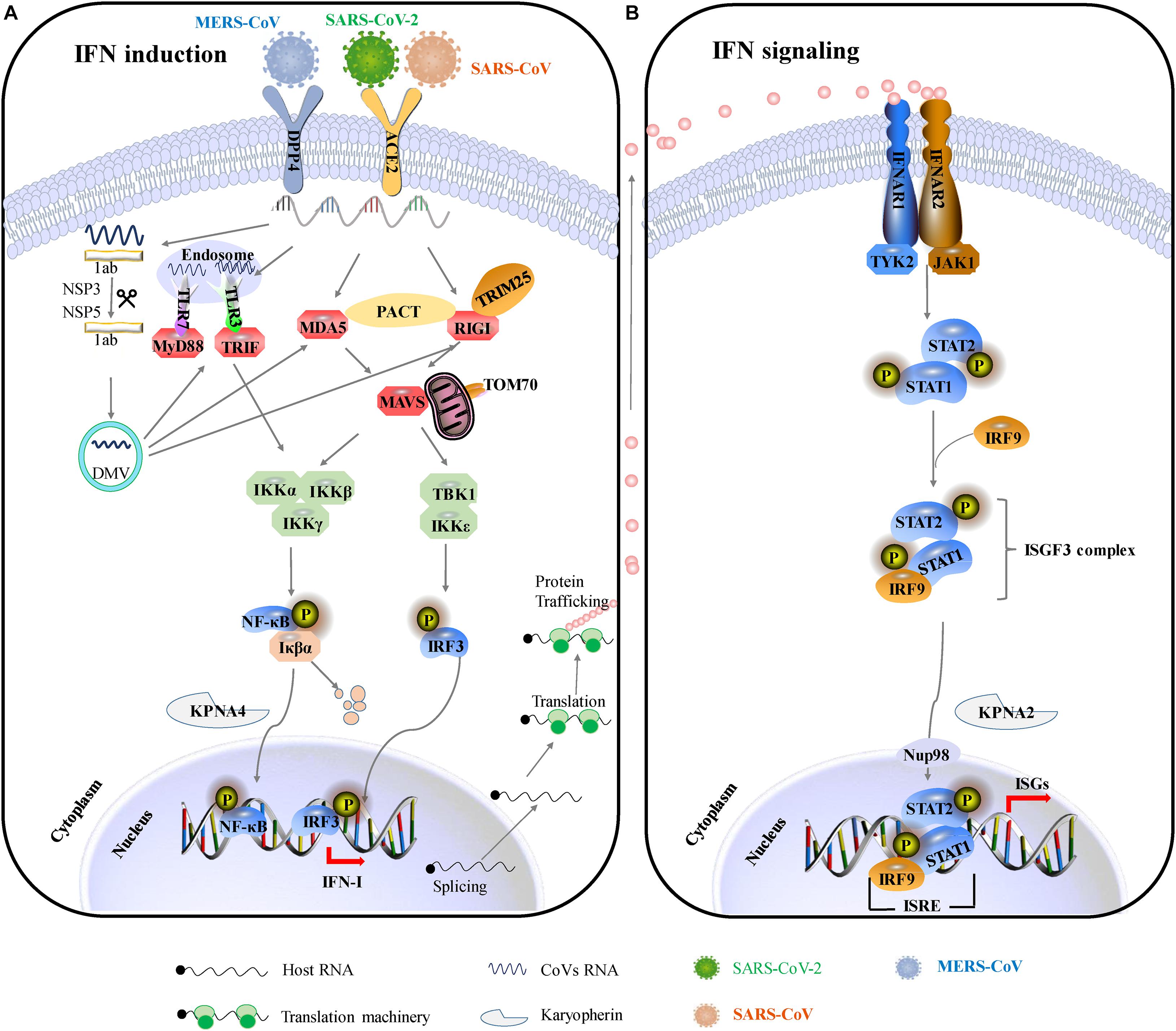
Figure 2. IFN-mediated antiviral responses. (A) SARS-CoV and SARS-CoV-2 use the same receptor ACE2 for host cellular entry, while MERS-CoV utilizes DPP4 for host cellular entry. They are most likely recognized by PRRs, including cytoplasmic RIG-I and MDA5 and by endosomal TLR3 and TLR7. RIG-I/MDA5 conveys signal through a mitochondrial adaptor MAVS, while TLR signals through TRIF/MyD88. Subsequently, activating the shared downstream kinases and transcription factors. Both pathways can activate the key transcription factors (IRF3 and NF-κB) phosphorylation and subsequent dimerization, which are translocated into the nucleus to promote IFN-I expression. (B) IFNs bind to the heterodimeric receptor complexes IFNAR1/IFNAR2, initiating JAK/STAT signaling. Receptor-associated tyrosine kinases Jak1 and Tyk2 are triggered for self-phosphorylation and activation, leading to the phosphorylation of STATs. Phosphorylated STAT1/2 with IRF9 forms a complex ternary ISGF3 (STAT1/STAT2/IRF9), which translocates into the nucleus and binds to IFN-stimulated response elements (ISREs), promoting the transcription of hundreds of IFN-stimulated genes (ISGs). ACE2, angiotensin-converting enzyme 2; DPP4, dipeptidyl peptidase 4; DMVs, double-membraned vesicles; RIG-1, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated protein 5; MyD88, myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing IFN-β; MAVS, mitochondria antiviral signaling protein; PACT, protein activator of protein kinase R; Iκβα, inhibitor of NF-κB; IRF3, interferon regulatory factor 3; TBK1, TANK-binding kinase 1; IKKε, I-kappa-B kinase ε; Nup93, nuclear pore complex protein 93; KPNA4, karyopherin-α4; KPNA2, karyopherin-α2; ISGF3, IFN-stimulated gene factor 3; IFNAR1, interferon-alpha/beta receptor alpha chain KPNA2, karyopherin-α2; ISG, interferon gene expression; JAK/STAT, Janus kinases/signal transducer and activator transcription proteins.
Based on previous therapeutic interventions of IFNs during SARS-CoV or MERS-CoV infection, IFN-β was the most potent IFN-I subtype (Antonelli et al., 2003; Hensley et al., 2004; Omrani et al., 2014). Compared to SARS-CoV and MERS-CoV, SARS-CoV-2 is substantially more sensitive to IFN-I. IFNs do alleviate the pathogenesis of those HP-hCoV-infected patients. Two therapeutic regimens of IFN-α + lopinavir/ritonavir and IFN-α + lopinavir/ritonavir + ribavirin are beneficial for COVID-19 patients (Yuan et al., 2020). Inhaled nebulized interferon beta-1a (SNG001) has greater odds of improvement, and treated patients recovered more rapidly from SARS-CoV-2 infection (Monk et al., 2021). Numerous independent clinical trials confirm that IFN therapy could attenuate the clinical consequences of COVID-19 in the early stages of infection (Sallard et al., 2020). These facts strongly indicate that impaired IFN-I expression may at least partially contribute to the severity of the disease. Indeed, CoVs do counter the IFN-I response by employing multipronged strategies to survive in the host. Given the importance of IFN-I responses in the pathogenesis of HP-hCoVs, the mechanisms of these viruses to antagonize IFN-I responses are discussed in more detail below and graphical summary is shown in Figure 3.
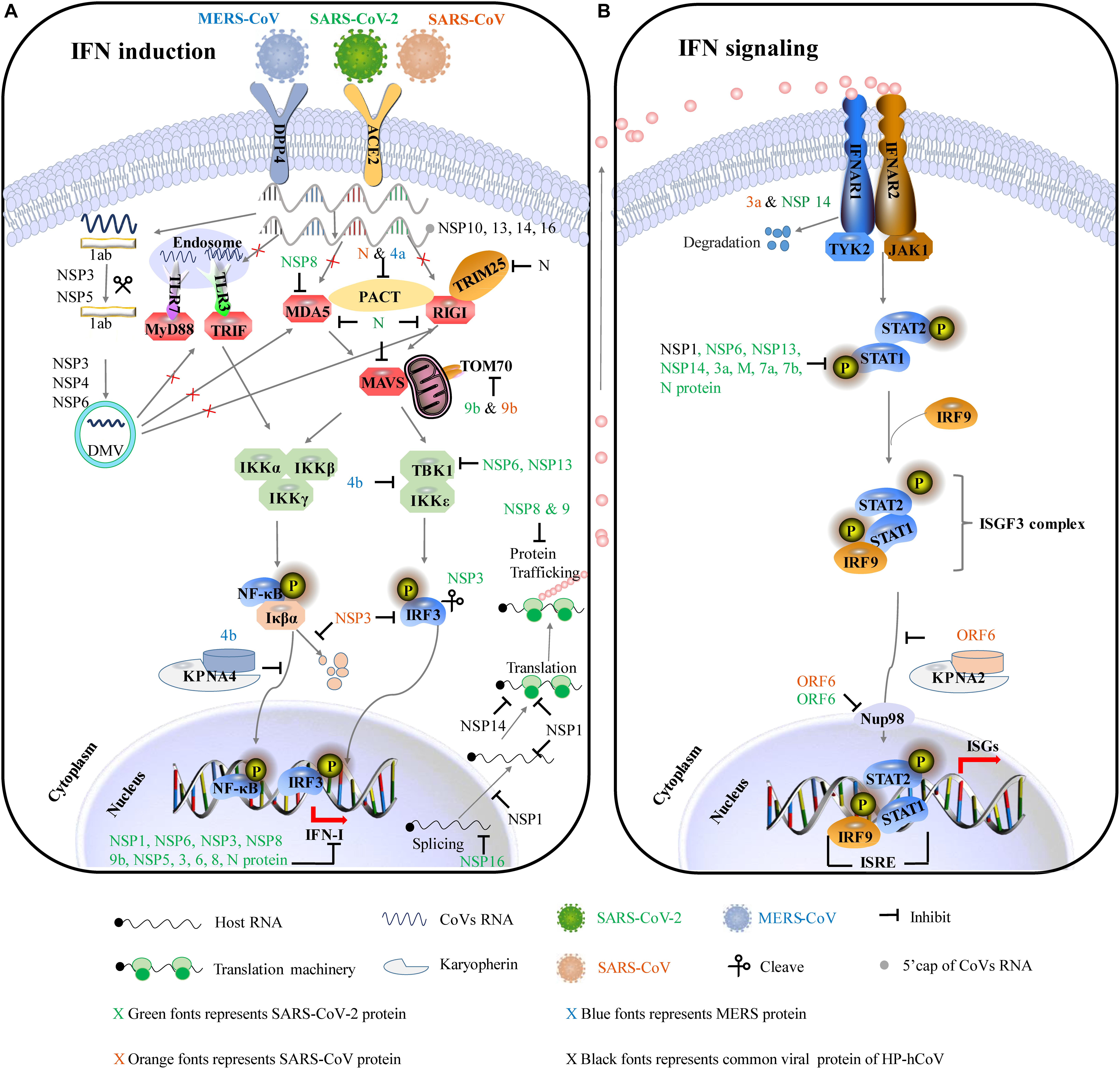
Figure 3. The major immune evasive strategies by HP-hCoVs as discussed in this review. The CoV-encoded proteins inhibit multiple aspects of the host innate immune signaling from sustaining viral replication and propagation. Different colors represent the proteins encoded by different viruses. Green represents SARS-CoV-2 encoded proteins, orange represents SARS-CoV encoded proteins, blue represents MERS-CoV encoded proteins, and black represents their common proteins. Virus antagonistic tactics are shown with black lines and arrows. (A) HP-hCoVs uses multiple gene products to impair IFN induction. To shield viral RNA (ssRNA and dsRNA) from recognition by PRRs, CoV replication takes place and DMVs are formed by NSP3, NSP4, and NSP6. In addition, NSP10, NSP13, NSP14, and NSP16 can modify the 5′-cap structure of viral RNA to mask viral PAMPs. HP-hCoVs interfere with the transmission of signals at almost every step. HP-hCoV N proteins interact with TRIM25, interfering with RIG-I signaling. MERS-CoV 4a and SARS-CoV N associate with PACT, sequestering the association of PACT and RIG-I/MDA5. SARS-CoV-2 NSP8 interacts with MDA5 to interfere with IFN induction. SARS-CoV-2 N is reported to associate with RIG-I, MDA5, and MAVS. SARS-CoV-2 and SARS-CoV M proteins are reported to associate with multiple adapters (not shown). SARS-CoV-2 and SARS-CoV 9b proteins interact with human TOM70 to block signaling downstream of MAVS. MERS-CoV ORF4b could specifically interact with TBK1 and IKKε, thereby blocking IRF3 phosphorylation. In addition, MERS-CoV ORF4b can associate karyopherin-α4 (KPNA4), out-competing NF-κB for KPNA4 binding and suppressing NF-κB nuclear transport. SARS-CoV NSP3 binds to IRF3 and inhibits the degradation of IκBα. SARS-CoV-2 NSP3 can cleave IRF3 directly. HP-hCoV NSP1 efficiently interferes with the cellular translation machinery. SARS-CoV-2 NSP16 disrupts mRNA splicing. SARS-CoV-2 NSP8 and NSP9 interfere with host protein trafficking. (B) HP-hCoVs use multiple gene products to impair IFN signaling. SARS-CoV 3a and SARS-CoV-2 NSP14 can degrade IFNAR1. SARS-CoV and SARS-CoV-2 ORF6 bind directly to Nup98 and Rae1 to prevent bidirectional nucleocytoplasmic transport. SARS-CoV ORF6 associates karyopherin-α2 (KPNA2), retaining KPNA2 in the cytoplasm and suppressing STAT1 nuclear import. SARS-CoV-2 NSP6, NSP13, NSP14, 3a, M, 7a, 7b, and N protein inhibit the phosphorylation of STAT1. HP-hCoV NSP1 inhibits the phosphorylation of STAT.
Shielding Viral RNA From Recognition by Pattern Recognition Receptors
Protecting the ‘self’ from the ‘non-self’ is an important part of innate immunity. Upon CoV infection, distinguishing cellular RNA from external RNA is essential for living organisms to maintain life. Although organisms employ a well-developed mechanism for recognizing harmful external factors, HP-hCoVs have evolved three means to evade detection by the innate immunity RNA sensors: (1) double-membrane vesicles (DMV) hiding nascent viral RNAs from PRRs; (2) modification of the 5′-cap structure to mask viral PAMPs; (3) manipulation of stress granule formation. HP-hCoVs utilize the above diverse strategies to avoid their RNA recognition by PRRs, concomitantly guaranteeing their mRNAs to be recognized felicitously by the translation machinery of the host (Table 1).

Table 1. Strategies of highly pathogenic human coronaviruses (HP-hCoVs) to shield viral RNA from recognition by pattern recognition receptors (PRRs).
Double-Membrane Vesicles Hiding Nascent Viral RNAs From Pattern Recognition Receptors
The replication of CoV RNA is processed at cytoplasmic membranous replication organelles (ROs). DMVs are a prominent type of virus-related ROs. CoV infection induces host endoplasmic reticulum (ER) alterations and rearranges, leading to the formation of DMVs, which shield the nascent viral RNAs from recognition by cytosolic PRRs such as RIG-I/MDA5 and endosomal PRRs such as TLR3/TLR7 (Taefehshokr et al., 2020). SARS-CoV-2 infection induces an intense membrane remodeling and forms the double-lipid bilayer in electron micrographs (Klein et al., 2020). Surprisingly, a triple transfection of NSP3, NSP4, and NSP6 of SARS yielded DMV, similar to those induced in SARS coronavirus infected cells (Angelini et al., 2013). Similarly, co-expression of MERS-CoV NSP3 and NSP4 either as individual proteins or as a self-cleaving NSP3-4 precursor induced the formation of DMVs, whereas MERS-CoV NSP6 did not noticeably affect DMV formation (Oudshoorn et al., 2017).
Modification of the 5′-Cap Structure to Mask Viral Pathogen-Associated Molecular Patterns
The genomes of HP-hCoVs contain the standard eukaryotic 5′-terminal 7-methylguanosine cap structure and a 3′ poly (A) tail, mimicking cellular mRNA to shield the pathogen-associated molecular patterns (PAMP) on the viral genome from the recognition of PRRs. The 5′ capping of eukaryotic mRNA is an important strategy to distinguish cellular RNA from external RNA. Uncapped viral RNAs could be recognized by a series of PRRs, initiating host immune response. To mimic eukaryotic RNA structures and evade the recognition by PRRs, CoVs process post-translational capping of viral mRNA catalyzed by the capping enzymes in their polymerase complexes, including NSP13 functions as RNA helicase and 5′-triphosphatase, NSP14 as an exoribonuclease and N7-MTase, and NSP16 as a 2′-O methyltransferase (2′-O-MTase) (Chang and Chen, 2021). The 2′-O-methyltransferase function is associated with K-D-K-E (lysine-aspartate-lysine-glutamate) motif in NSP13 conserved among all HP-hCoVs (von Grotthuss et al., 2003). In addition, NSP14 of HP-hCoVs possesses guanine N7-MTase activity coupled with exonuclease activity, involving RNA cap formation and cleaving RNA-PAMPs, also contributing to immune evasion (Chen et al., 2009; Ramasamy and Subbian, 2021). NSP16 of all HP-hCoVs, with its activating cofactor nsp10, can form a 2′-O-methylated cap for immune evasion in which the conserved D130 is critical (Gorkhali et al., 2021). Taken together, the 5′ cap modification of viral RNA impairs the recognition by cytosolic PRRs, thereby resisting the IFN-mediated antiviral response. The functions of polymerase complexes are common to CoVs.
Manipulation of Stress Granule Formation
Stress granules (SGs) are membraneless cytoplasmic RNA granules responding to various stresses, including virus infection. Upon virus infection, the host can form SGs by wrapping viral RNA, transcription and translation-related proteins. The accumulation of viral RNA provides a pool of substrates for PRRs such as RIG-I and MDA5, triggering RIG-I like receptor signaling pathway (Onomoto et al., 2012; Kim et al., 2019; Kikkert, 2020). Indeed, SGs form a platform for innate immunity and play a significant role in antiviral response. However, HP-hCoVs can inhibit the formation of SGs to antagonize innate immunity responses for benefiting virus replication. MERS-CoV encoded 4a protein could prevent stress granule formation by binding to viral dsRNA and impairing dsRNA-mediated PKR (protein kinase R) activation, promoting viral translation and virus replication (Nakagawa et al., 2018). In addition, NSP15 from SARS-CoV and SARS-CoV-2, harboring the conserved function, can antagonize the formation of chemical-induced SGs (Gao et al., 2021). SARS-CoV-2 and SARS-CoV N proteins could associate with the protein kinase PKR and stress granule protein G3BP1 (the Ras-GTPase-activating protein SH3 domain-binding protein 1), impairing dsRNA-triggered SG formation (Gordon et al., 2020; Zheng et al., 2021). In contrast, MERS-CoV N can associate with PKR but not G3BP1 to block SG formation (Zheng et al., 2021).
Impaired IFN-I Production and Signaling Pathway
Dysregulated IFN-I responses contribute to the robust early HP-hCoV replication and trigger a cytokine storm. The HP-hCoVs employ extensive measures to interfere with the key host signaling factors to counteract the IFN system by fully using certain proteins, including accessory proteins, NSPs, and structural proteins. Recent studies report that SARS-CoV-2 NSP1, NSP3, NSP6, NSP12, NSP13, NSP14, NSP15, M, ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF9b, and N can interfere with the key host signaling factors of IFN signaling pathway. It has been documented that more than half of SARS-CoV-2 proteins have antagonistic effects on IFN responses by either targeting viral sensors or blocking downstream antiviral signaling molecules. The detailed antagonistic effects on IFN responses by NSPs, accessory proteins, and structural proteins are summarized one by one below (Table 2).
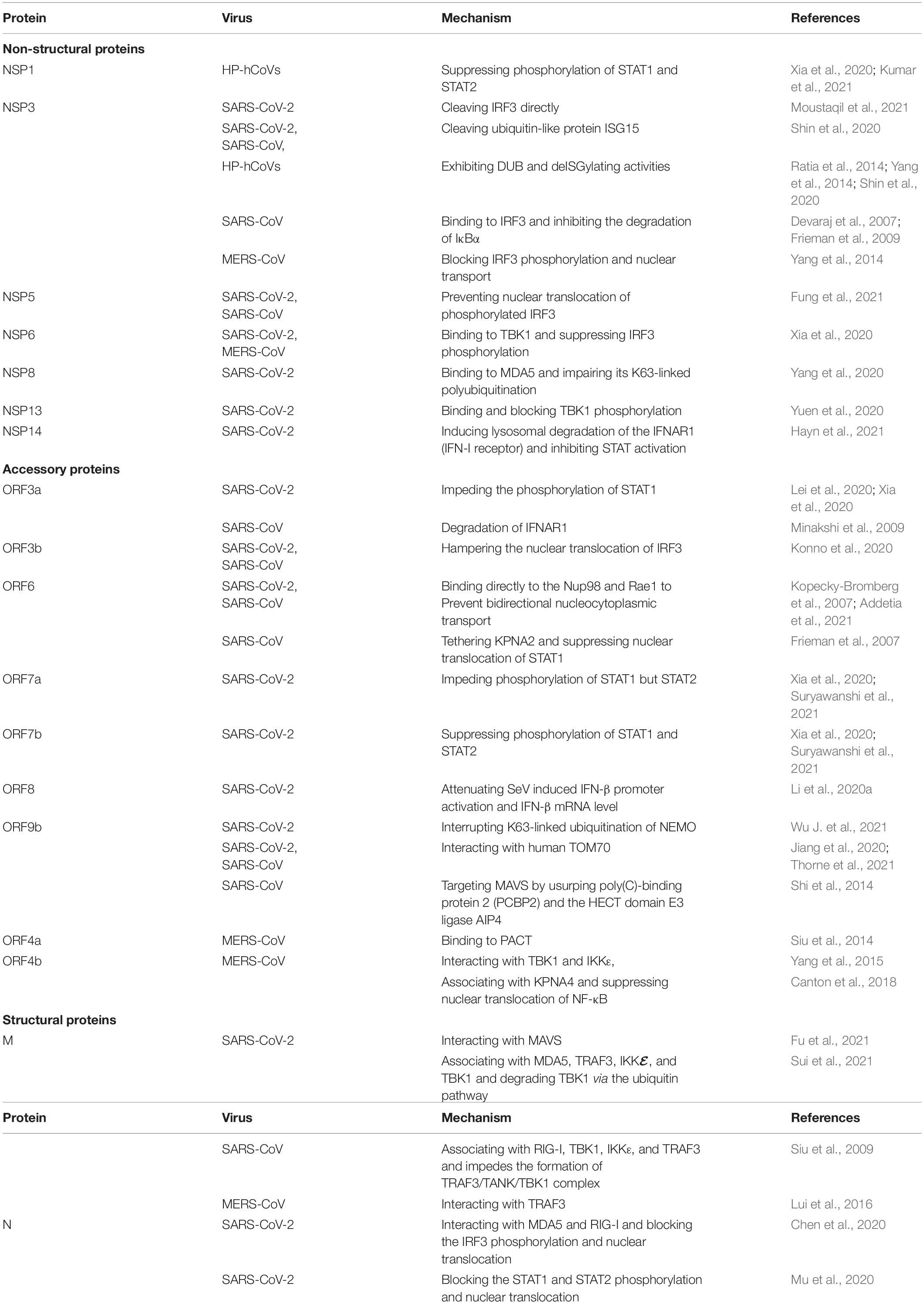
Table 2. Evasion IFN-I system by HP-hCoV-encoded proteins.
Non-structural Proteins
The CoVs NSPs have multiple functions, including viral transcription and replication and antagonizing IFN responses. SARS-CoV-2 NSP1 has been confirmed to block IRF3 phosphorylation and nuclear transport. Moreover, SARS-CoV-2 NSP1 causes the depletion of antiviral factors Tyk2 and STAT2, showing a decrease in STAT1 and STAT2 phosphorylation (Kumar et al., 2021). SARS-CoV-2 NSP1 owns 84% amino acid sequence identity with SARS-CoV NSP1, showing a similar mechanism to antagonize IFN-I response. Furthermore, comparing with SARS-CoV and MERS-CoV NSP1, SARS-CoV-2 NSP1 suppresses phosphorylation of STAT1 and STAT2 more efficiently (Xia et al., 2020). The CoV proteases NSP3 and NSP5 are multifunctional proteins, playing an essential role in the proteolytic processing of the viral polyproteins, maturation, and assembly of the RTC and cleaving proteinaceous post-translational modifications on host proteins. The cleavage of innate immunity factors, antagonizing IFN and its downstream JAK-STAT signal transduction, is a particularly effective strategy to evade the host innate immunity.
SARS-CoV-2 NSP3 directly cleaves IRF3, resulting in reduced IFN production. SARS-CoV and SARS-CoV-2 NSP3 cleave ubiquitin-like protein ISG15, a typical regulator of host innate immune signaling pathways, decreasing IFN production and enhancing virus replication and spread (Moustaqil et al., 2021). Compared with SARS-CoV NSP3, SARS-CoV-2 NSP3 preferentially reduces ISG15-conjugated (ISGylated) protein substrates (Shin et al., 2020). Besides proteolytic activity, CoVs NSP3 acts as a deubiquitinating enzyme (DUB) processing deISGylating activity, efficiently degrading mono-polyubiquitin, di-polyubiquitin, and branched-polyubiquitin chains. The deubiquitinating modifications from host cell proteins disturb the host innate immune responses to viral infection (Lindner et al., 2007; Ratia et al., 2014; Baez-Santos et al., 2015; Clasman et al., 2020). SARS-CoV-2 NSP3 has 83% sequence identity with SARS-CoV NSP3 but exhibits different host substrate preferences. SARS-CoV-2 NSP3 preferentially cleaves the ubiquitin-like interferon-stimulated 15 protein (ISG15), whereas SARS-CoV NSP3 predominantly targets ubiquitin chains (Shin et al., 2020). SARS NSP3 removes Lys63-linked ubiquitin chains of TRAF3 and TRAF6, but not Lys48-linked ubiquitin chains, suppressing the Toll-like receptor 7 (TLR7) signaling pathway (Li et al., 2016). Similarly, MERS-CoV NSP3 exhibits DUB activity and acts on both K48- and K63-linked ubiquitination and ISG15-linked ISGylation, and the catalytic sites C1592 and H1759 are required for deISGylation (Yang et al., 2014). Expect for cleavage and deISGylating activities, HP-hCoV NSP3 could interfere with the signaling factors of the interferon signaling pathway. SARS-CoV NSP3 interacts with STING (stimulator of interferon genes, also known as MITA/ERIS/MYPS), inhibiting the phosphorylation and dimerization of IRF3 (Sun et al., 2012; Chen et al., 2014; Matthews et al., 2014).
In addition, SARS-CoV NSP3 can associate with IRF3 and block the phosphorylation and nuclear translocation of IRF3 (Devaraj et al., 2007). Moreover, SARS-CoV Nsp3 can inhibit the degradation of IκBα, an inhibitor of NF-κB, suppressing the NF-κB signaling pathway (Frieman et al., 2009). MERS-CoV NSP3 acts as an IFN antagonist by blocking the phosphorylation and nuclear translocation of IFN regulatory factor 3 (IRF3) (Yang et al., 2014). Confirmed by the above, CoV NSP3 is a multifunctional protein, so is CoVs NSP5. NSP5 proteins of SARS-CoV and SARS-CoV-2, sharing 96% amino acid sequence identity, suppress IFN-I production by perturbing nuclear translocation of phosphorylated IRF3 without affecting the phosphorylation of IRF3 (Fung et al., 2021).
SARS-CoV-2 NSP6 associates with TANK binding kinase 1 (TBK1) without affecting TBK1 phosphorylation, and the NSP6/TBK1 interaction suppresses IRF3 phosphorylation (Xia et al., 2020). SARS-CoV-2 NSP6 inhibits IFN-I production more efficiently than MERS-CoV NSP6. However, SARS-CoV NSP6 does not inhibit the IFN-I response (Xia et al., 2020). SARS-CoV-2 NSP8 inhibits the expression of IFN-I, IFN-stimulated genes, and proinflammatory cytokines by associating with MDA5 and impairing its K63-linked polyubiquitination (Yang et al., 2020). SARS-CoV-2 NSP12, the viral RNA-dependent RNA polymerase (RdRp), attenuates SeV or Poly(I:C) induced IFN-β promoter activation by suppressing the nuclear translocation of IRF3 but does not impair IRF3 phosphorylation (Wang W. et al., 2021). Paradoxically, another article reported that SARS-CoV-2 NSP12 protein is not an IFN-β antagonist, owing to NSP12 inhibits neither IFN-β production nor downstream IFN-β signaling pathway (Li et al., 2021). SARS-CoV-2 NSP13 binds and blocks TBK1 phosphorylation, decreasing IRF3 activation (Yuen et al., 2020). Moreover, SARS-CoV-2 NSP13 can suppress IFN-I signaling by inhibiting STAT1 and STAT2 activation, leading to the retention of STAT1 in the cytoplasm and compromising stimulation of the ISRE promoter (Lei et al., 2020). SARS-CoV-2 NSP14 induces lysosomal degradation of the IFNAR1, thereby inhibiting STAT activation (Hayn et al., 2021).
Accessory Proteins
Accessory proteins of CoVs are not required for replication, but they play critical roles during infection and pathogenesis, owing to antagonizing the host response. Different studies have reported that SARS-CoV-2 ORF3a inhibits IFN signaling by impeding STAT1 phosphorylation (Lei et al., 2020; Xia et al., 2020). At the same time, SARS-CoV ORF3a induces the degradation of IFNAR1 to increase IFNAR1 ubiquitination (Minakshi et al., 2009). SARS-CoV-2 ORF3b has only 22 amino acids (69 bp, including the stop codon), which could inhibit the induction of IFN more efficiently than its SARS-CoV ortholog (153 amino acids on average), and its anti-IFN activity is increased by a naturally occurring elongation variant (Konno et al., 2020). SARS-CoV and SARS-CoV-2 ORF6 bind directly to Nup98 and Rae1 to prevent bidirectional nucleocytoplasmic transport, namely, nuclear export of host mRNA and nuclear import of various host factors, blocking IRF3 and STAT nuclear import (Kopecky-Bromberg et al., 2007; Addetia et al., 2021). Moreover, compared to SARS-CoV ORF6, SARS-CoV-2 ORF6 may more dramatically suppress protein expression through a stronger interaction with the Rae1and Nup98 (Addetia et al., 2021). Consistently, another study confirmed that SARS-CoV-2 ORF6 causes the accumulation of heterogeneous ribonucleoprotein A (hnRNPA1) in the nucleus (Kato et al., 2021).
In line with the above result, SARS-CoV-2 ORF6 interferes less efficiently with human interferon induction and interferon signaling than SARS-CoV ORF6 using reverse genetics (Schroeder et al., 2021). SARS-CoV ORF6 can associate karyopherin-α2 (KPNA2), retaining KPNA2 in the cytoplasm, and suppressing STAT1 nuclear import. Furthermore, recombinant SARS-CoV lacking ORF6 did not tether KPNA2 to the ER/Golgi membrane and allowed the import of the STAT1 complex into the nucleus (Frieman et al., 2007). SARS-CoV-2 ORF7a could impede the phosphorylation of STAT1 but STAT2, while SARS-CoV-2 ORF7b could suppress STAT1 and STAT2 phosphorylation (Xia et al., 2020; Suryawanshi et al., 2021). SARS-CoV-2 ORF8 can attenuate SeV induced IFN-β promoter activation and IFN-β mRNA level (Li et al., 2020a).
SARS-CoV-2 ORF9b has been reported to impede the host innate immune system by targeting multiple molecules. SARS-CoV-2 ORF9b interrupts K63-linked ubiquitination of NEMO upon VSV stimulation, thereby inhibiting the canonical IκB kinase alpha (IKKα)/β/γ-NF-κB signaling and subsequent IFN production (Wu J. et al., 2021). SARS-CoV-2 and SARS-CoV ORF9b block signaling downstream of MAVS by interacting with human TOM70, and that this process is regulated by phosphorylation (Jiang et al., 2020; Thorne et al., 2021). SARS-CoV ORF9b targets MAVS by usurping poly(C)-binding protein 2 (PCBP2) and the HECT domain E3 ligase AIP4, leading to the degradation of MAVS, TRAF3, and TRAF6 (Shi et al., 2014). MERS-CoV ORF4a protein binds with protein kinase R (PACT) protein activator, thereby inhibiting PACT-induced activation of RIG-I and MDA5 (Siu et al., 2014). MERS-CoV ORF4b could specifically interact with TBK1 and IKKε, thereby blocking IRF3 phosphorylation (Yang et al., 2015). In addition, MERS-CoV ORF4b can bind to karyopherin-α4 (KPNA4), out-competing NF-κB for KPNA4 binding and suppressing NF-κB nuclear transport (Canton et al., 2018).
Structural Proteins
Coronaviruses structural proteins are mainly responsible for viral assembly, coating, entry into host cells, and packaging of the RNA genome (Endriyas Kelta et al., 2020). In addition, M and N proteins can block the IFN-I response. CoV M protein is a glycosylated structural protein with three membrane-spanning domains. M protein predominantly localizes to the Golgi complex and is necessary for the assembly of viral particles. SARS-CoV-2 M, blocking RIG-I and MAVS, but not TBK1, IKKε, and IRF3-5D triggered IFN-β promoter activation, inhibits the innate antiviral response by interacting with MAVS, and its TM1/2 domains are essential for this inhibitory function (Fu et al., 2021). Paradoxically, another study reported that SARS-CoV-2 M associates with MDA5, TRAF3, IKKℰ, and TBK1 and degrades TBK1 via ubiquitin pathway (Sui et al., 2021). However, the SARS-CoV M protein physically associates with RIG-I, TBK1, IKKε, and TRAF3 and impedes the formation of TRAF3/TANK/TBK1 complex (Siu et al., 2009), whereas the MERS CoV M protein interacts with TRAF3 and disrupts TRAF3-TBK1 association, leading to reduced phosphorylation of IRF3 (Lui et al., 2016).
HP-hCoVs M proteins collectively possess the common and conserved mechanism of IFN-I expression attenuation, but they target different signal adapters. SARS-CoV-2 N protein is confirmed as the interferon antagonist by different research teams, targeting multiple factors. It interacts with MDA5 and RIG-I through the DExD/H domain of RIG-I, leading to the IRF3 phosphorylation and nuclear translocation and STAT1 and STAT2 (Chen et al., 2020; Mu et al., 2020). Besides, SARS-CoV-2 N protein, undergoing liquid–liquid phase separation with RNA, inhibits Lys63-linked poly-ubiquitination and aggregation of MAVS and thereby suppresses the innate antiviral immune response (Wang S. et al., 2021). SARS-CoV-2 N protein targets the initial step and interferes with TRIM25-mediated RIG-I ubiquitination (Gori Savellini et al., 2021). Like SARS-CoV-2 N protein, SARS-CoV and MERS-CoV N proteins interfere with RIG-I signaling by interacting with TRIM25 (Hu et al., 2017; Chang et al., 2020). Another literature reported that SARS-CoV N protein could associate with protein activator of protein kinase R (PACT), activating RIG-I and MDA5. The N-PACT association sequestered the binding between PACT and RIG-I/MDA5, which in turn inhibited IFN-β production (Ding et al., 2017).
Furthermore, the different studies reported that SARS-CoV-2 N protein could induce inflammatory responses. SARS-CoV-2 N protein promotes the activation of NF-κB signaling by recruiting TAK1 and IKK complex, the key kinases of NF-κB signaling (Wu Y. et al., 2021). Another article reported that SARS-CoV-2 N protein associates directly with NLRP3 protein, enhancing the interaction between NLRP3 and ASC and promoting NLRP3 inflammasome activation to induce hyper inflammation (Pan et al., 2021). Taken together, SARS-CoV-2 N protein could inhibit IFN production and then prevent host innate immunity system from recognizing and combating infection in the first stages. When the host innate immune system sensed a virus, SARS-CoV-2 N protein could promote NLRP3 inflammasome activation, exuberant chemokines, and inflammatory cytokine production and flood the bloodstream faster than normal, resulting in a cytokine storm and disseminated damage to the host. It is consistent with the previous clinical results that low levels of types I and III interferons in the serum of patients with COVID-19 juxtaposed to elevated amounts of chemokines and proinflammatory cytokines (Arunachalam et al., 2020; Blanco-Melo et al., 2020; Huang et al., 2020).
Other Evasion Strategies
Preventing Host Protein Translation
Coronaviruses, as obligate intracellular parasites, hijack host cell components for viral translation and assembly by disrupting host mRNA splicing, translation, and protein trafficking to allow the translation of viral mRNA and concomitantly suppress host antiviral immune responses (Finkel et al., 2021). CoV NSP1 is a major viral virulence factor, which represses multiple steps of host protein expression. NSP1 of α- and β-CoVs, regardless of their low sequence identity, show similar biological function in inducing endonucleolytic cleavage of host mRNAs but viral mRNAs and inhibition of host translation (Nakagawa and Makino, 2021). SARS-CoV-2 NSP1 associates with the host mRNA export receptor heterodimer NXF1-NXT1, preventing proper interaction of NXF1 with mRNA export adaptors. As a result, many cellular mRNAs are withheld in the nucleus and cannot evade from the nucleus, and the infected cells do not release lots of IFN, alerting the immune system (Zhang K. et al., 2021). Besides, SARS-CoV-2 NSP1 binds to 18S ribosomal RNA in the mRNA entry channel of the 40S ribosomal subunit, and structural analysis by cryo-electron microscopy of in vitro-reconstituted nsp1-40S and various native nsp1-40S and nsp1-80S complexes revealed that the nsp1 C terminus binds to and obstructs the mRNA entry tunnel (Schubert et al., 2020; Thoms et al., 2020).
Furthermore, the C-terminal domain of NSP1 is located at the entrance of ribosomal mRNA, which prevents the entry of some but not all of the mRNAs. Its N-terminal can stabilize the binding between its C-terminal and ribosome acting as a non-specific barrier to block the mRNA channel, thus exerting a more thorough blocking effect and abrogating host mRNA translation (Zhao et al., 2021). Similarly, SARS-CoV NSP1 localizes exclusively in the cytoplasm and associates with the 40S ribosome to block the assembly of the translationally competent ribosome (Banerjee et al., 2020). Another study reported that SARS-CoV NSP1 prevents mRNA nuclear export by disrupting localization of nuclear pore complex protein 93 (Nup93) and altering the composition of the nuclear pore complex (NPC), thus subsequently suppressing protein synthesis (Gomez et al., 2019). MERS-CoV NSP1, distributed in the nucleus and the cytoplasm, selectively targets nuclear-transcribed host mRNAs for suppression, but mRNAs of cytoplasmic origin (Lokugamage et al., 2015).
Furthermore, SARS-CoV and MERS-CoV evade the NSP1-mediated translational inhibition by inducing a specific association between SL1 in the 5′ UTR of viral RNA and NSP1 (Terada et al., 2017). In brief, human HP-hCoV NSP1 efficiently disturb the cellular translation machinery. In addition, SARS-CoV-2 NSP16 binds mRNA recognition domains of the spliceosome’s U1 and U2 RNA components and disrupts mRNA splicing. SARS-CoV-2 NSP14 can shut down host protein translation, and the formation of the NSP14-NSP10 complex enhances translation inhibition executed by NSP14, suppressing host protein synthesis. This role of NSP14-mediated translation inhibition is conservative among three highly pathogenic Betacoronaviruses (Hsu et al., 2021). SARS-CoV-2 NSP8 and NSP9 interfere with host protein trafficking to the cell membrane by binding to discrete regions on the 7SL RNA component of the signal recognition particle (SRP) (Banerjee et al., 2020; Table 3).

Table 3. Other innate immune evasion strategies employed by HP-hCoVs.
Hijack of the Antigen-Presenting Cells
Dendritic cells (DCs), the most potent antigen-presenting cells (APCs), are capable of entering peripheral tissues, taking up antigens, migrating to lymphoid tissues, and activating helper T cells (Anselmi et al., 2020). DCs link the innate and the adaptive immunity and can serve as an important target of viral replication and a vehicle for dissemination. However, the virus could exploit the function of DCs to evade from immune surveillance and facilitate cell-to-cell dissemination. Recent research reported that DC-SIGN serves as receptor, mediating SARS-CoV-2 entry into human target cells and leading to hypercoagulability, inflammatory response, and multiorgan dysfunction (Amraie et al., 2020; Campana et al., 2020). SARS-CoV spike protein could bind DC-SIGN receptor on DCs. Although SARS-CoV could not infect DCs, DCs can take up SARS-CoV and transfer it to susceptible target cells, which is useful for the viruses to break through the epithelial barrier and evade antiviral immune responses (Yang et al., 2004). MERS-CoV could productively infect DCs, leading to produce a large number of cytokines and chemokines and accelerating viral replication and dissemination (Chu et al., 2014). SARS-CoV-2 ORF8 directly interacts with major histocompatibility complex I (MHC-I) and disrupts antigen presentation for immune activation by downregulating MHC-I expression on the surface of cells, and therefore may help in immune evasion (Zhang Y. et al., 2021). However, SARS-CoV ORF8 does not affect MHC-I downregulation (Zhang Y. et al., 2021). Taken together, the SARS-CoV-2 adopts various strategies to evade the host innate immunity and the acquired immune response (humoral immunity and cellular immunity). Recent research reported that immune-compromised individuals with prolonged viral replication occurred a broader range of viral evolution (Kemp et al., 2020; Stanevich et al., 2021; Williamson et al., 2021; Table 3).
Cytokine Storm Syndrome Induced by Highly Pathogenic Human Coronaviruses
In the initial stage of HP-hCoV infection, the virus evades the host’s innate immune response by using diverse strategies to obtain effective replication and establish a window of opportunity for infection. When the immune system detects the virus, the virus titer has reached a high level, which induces highly concentrated and prolonged proinflammatory cytokines and chemokines (IL-2, IL-10, GSCF, IP10, MCP-1, TNF-α, etc.) known as “cytokine storm” (Huang et al., 2005; Zhou et al., 2014; Fan et al., 2020). Cytokine storm is the uncontrolled release, fast-developing, and life-threatening, which can accelerate the depletion and exhaustion of T cells, leading to complicated medical symptoms such as fever, septic shock, capillary leak syndrome, acute respiratory distress syndrome (ARDS), disseminated intravascular coagulation, and multiple organ failure, and ultimately death in the most severe cases (Song et al., 2020). Early administration of IFN could help to reduce viral load and rarely induce cytokine storm, thus ameliorating the patient clinical feature. By comparison, delayed administration of IFN did not provide any advantage compared to placebo controls (Channappanavar and Perlman, 2017). Cytokine storm is responsible for the deterioration of HP-hCoV patients. Therefore, besides controlling viral load with early administration of IFN, the timing of attenuating cytokine storm could be life-saving. The current interventions for cytokine storm include traditional anti-inflammatory drugs (corticosteroids, chloroquine, and colchicines) and intravenous immunoglobulin, traditional Chinese medicine, and corticosteroids (Yang et al., 2021). However, the clinical treatment of cytokine storms has been proved challenging. It urgently needs a safe and effective drug to tailor treatment of cytokine storm at specific disease stages, ultimately ameliorating disease severity and return to homeostasis. In addition, not all HP-hCoV patients develop the same symptoms, but the immunological determinants of a poor prognosis are unknown, which remains to be explored further.
Conclusion
In recent years, great progress has been achieved in studying the mechanism of the immune evasion of viruses. However, there are still many basic problems remaining to be solved. Most of the studies were conducted with overexpression experiments in cellular models. The experimental results drawn with different research methods are often inconsistent. This phenomenon is probably due to the difference in expression levels and phases of viral proteins expressed by plasmids and real viruses. Moreover, experimental results may be affected by different experimental systems, including protein tags and plasmid backbones. Validations should be conducted in multiple systems, especially under viral replication and even by animal physiological experiments.
When the worldwide COVID-19 pandemic emerged, vaccines and drugs were expected to end the pandemic. However, breakthrough infections occur even after vaccination because SARS-CoV-2 constantly mutates under various selective pressures, either naturally acquired post-exposure or vaccine acquired immunity, mainly due to the RNA polymerase lacking proofreading capability of the virus. The key mutant viral strains, including Alpha (first appeared in the United Kingdom), Beta (first appeared in South Africa), Gamma, and Delta (first appeared in Brazil), have been sweeping the world. Before the Delta strain was lifted, the Lambda strain has emerged. At present, asymptomatic infection allows the virus to continue spreading and mutating, making it more difficult to prevent and control the epidemic. The longer SARS-CoV-2 circulates, the more mutant strains emerge, which would challenge the current drug or vaccine. The changes of mutant strains and mutant genes of SARS-CoV-2 related to escaping host immunity should be continuously tracked and analyzed.
Author Contributions
All authors contributed to the work. J-YL designed and wrote the manuscript. Z-JZ and QW helped with the figures. X-YG, YQ, Q-NH, and M-YZ revised the whole manuscript. All authors approved the final manuscript.
Funding
This work was jointly funded by the National Natural Science Foundation of China (32041001 and 81902070), the Provincial Natural Science Foundation of Hunan Province (2019JJ20004 and 2019JJ50035), the Hunan Innovative Province Construction Project (2019SK2211), and the Fundamental Research Funds for the Central Universities (531118010008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Addetia, A., Lieberman, N. A. P., Phung, Q., Hsiang, T. Y., Xie, H., Roychoudhury, P., et al. (2021). SARS-CoV-2 ORF6 disrupts bidirectional nucleocytoplasmic transport through interactions with Rae1 and Nup98. mBio 12:e00065-21. doi: 10.1128/mBio.00065-21
Amraie, R., Napoleon, M. A., Yin, W., Berrigan, J., Suder, E., Zhao, G., et al. (2020). CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv [Preprint]. doi: 10.1101/2020.06.22.165803
Angelini, M. M., Akhlaghpour, M., Neuman, B. W., and Buchmeier, M. J. (2013). Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4:e00524-13. doi: 10.1128/mBio.00524-13
Anselmi, G., Helft, J., and Guermonprez, P. (2020). Development and function of human dendritic cells in humanized mice models. Mol. Immunol. 125, 151–161. doi: 10.1016/j.molimm.2020.07.005
Antonelli, G., Scagnolari, C., Vicenzi, E., and Clementi, M. (2003). Treatment of SARS with human interferons. Lancet 362, 293–294. doi: 10.1016/s0140-6736(03)14482-0
Arunachalam, P. S., Wimmers, F., Mok, C. K. P., Perera, R., Scott, M., Hagan, T., et al. (2020). Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science (New York, N.Y.) 369, 1210–1220. doi: 10.1126/science.abc6261
Baez-Santos, Y. M., St John, S. E., and Mesecar, A. D. (2015). The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 115, 21–38. doi: 10.1016/j.antiviral.2014.12.015
Bajaj, V., Gadi, N., Spihlman, A. P., Wu, S. C., Choi, C. H., and Moulton, V. R. (2020). Aging, immunity, and COVID-19: how age influences the host immune response to coronavirus infections? Front. Physiol. 11:571416. doi: 10.3389/fphys.2020.571416
Banerjee, A. K., Blanco, M. R., Bruce, E. A., Honson, D. D., Chen, L. M., Chow, A., et al. (2020). SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21. doi: 10.1016/j.cell.2020.10.004
Blanco-Melo, D., Nilsson-Payant, B. E., Liu, W. C., Uhl, S., Hoagland, D., Moller, R., et al. (2020). Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9. doi: 10.1016/j.cell.2020.04.026
Campana, P., Parisi, V., Leosco, D., Bencivenga, D., Della Ragione, F., and Borriello, A. (2020). Dendritic Cells and SARS-CoV-2 infection: still an unclarified connection. Cells. 9:2046. doi: 10.3390/cells9092046
Canton, J., Fehr, A. R., Fernandez-Delgado, R., Gutierrez-Alvarez, F. J., Sanchez-Aparicio, M. T., Garcia-Sastre, A., et al. (2018). MERS-CoV 4b protein interferes with the NF-kappaB-dependent innate immune response during infection. PLoS Pathog. 14:e1006838. doi: 10.1371/journal.ppat.1006838
Chang, C. Y., Liu, H. M., Chang, M. F., and Chang, S. C. (2020). Middle east respiratory syndrome coronavirus nucleocapsid protein suppresses Type I and Type III interferon induction by targeting RIG-I signaling. J. Virol. 94:e00099-20. doi: 10.1128/jvi.00099-20
Chang, L. J., and Chen, T. H. (2021). NSP16 2′-O-MTase in coronavirus pathogenesis: possible prevention and treatments strategies. Viruses. 13:538. doi: 10.3390/v13040538
Channappanavar, R., and Perlman, S. (2017). Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 39, 529–539. doi: 10.1007/s00281-017-0629-x
Chen, K., Xiao, F., Hu, D., Ge, W., Tian, M., Wang, W., et al. (2020). SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-Mediated IFN-beta production. Viruses. 13:47. doi: 10.3390/v13010047
Chen, X., Yang, X., Zheng, Y., Yang, Y., Xing, Y., and Chen, Z. (2014). SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5, 369–381. doi: 10.1007/s13238-014-0026-3
Chen, Y., Cai, H., Pan, J., Xiang, N., Tien, P., Ahola, T., et al. (2009). Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 106, 3484–3489. doi: 10.1073/pnas.0808790106
Chu, H., Zhou, J., Wong, B. H., Li, C., Cheng, Z. S., Lin, X., et al. (2014). Productive replication of middle east respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology. 454-455, 197–205. doi: 10.1016/j.virol.2014.02.018
Clasman, J. R., Everett, R. K., Srinivasan, K., and Mesecar, A. D. (2020). Decoupling deISGylating and deubiquitinating activities of the MERS virus papain-like protease. Antiviral Res. 174:104661. doi: 10.1016/j.antiviral.2019.104661
Devaraj, S. G., Wang, N., Chen, Z., Chen, Z., Tseng, M., Barretto, N., et al. (2007). Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282, 32208–32221. doi: 10.1074/jbc.M704870200
Ding, Z., Fang, L., Yuan, S., Zhao, L., Wang, X., Long, S., et al. (2017). The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-β antagonizing mechanism: attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 8, 49655–49670. doi: 10.18632/oncotarget.17912
Endriyas Kelta, W., Abebe Dukessa, D., Tariku Sime, G., and Urge Gerema, K. (2020). Role of structural and functional proteins of SARS -COV-2. GSC Biol. Pharm. Sci. 12, 117–129. doi: 10.30574/gscbps.2020.12.3.0275
Fan, E., Beitler, J. R., Brochard, L., Calfee, C. S., Ferguson, N. D., Slutsky, A. S., et al. (2020). COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir. Med. 8, 816–821. doi: 10.1016/S2213-2600(20)30304-0
Finkel, Y., Gluck, A., Nachshon, A., Winkler, R., Fisher, T., Rozman, B., et al. (2021). SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature 594, 240–245. doi: 10.1038/s41586-021-03610-3
Frieman, M., Ratia, K., Johnston, R. E., Mesecar, A. D., and Baric, R. S. (2009). Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83, 6689–6705. doi: 10.1128/JVI.02220-08
Frieman, M., Yount, B., Heise, M., Kopecky-Bromberg, S. A., Palese, P., and Baric, R. S. (2007). Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824. doi: 10.1128/JVI.01012-07
Fu, Y. Z., Wang, S. Y., Zheng, Z. Q., Yi, H., Li, W. W., Xu, Z. S., et al. (2021). SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell. Mol. Immunol. 18, 613–620. doi: 10.1038/s41423-020-00571-x
Fung, S. Y., Siu, K. L., Lin, H., Yeung, M. L., and Jin, D. Y. (2021). SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 17, 1547–1554. doi: 10.7150/ijbs.59943
Gao, B., Gong, X., Fang, S., Weng, W., Wang, H., Chu, H., et al. (2021). Inhibition of antiviral stress granule formation by coronavirus endoribonuclease nsp15 ensures efficient virus replication. PLoS Pathog. 17:e1008690. doi: 10.1371/journal.ppat.1008690
Gomez, G. N., Abrar, F., Dodhia, M. P., Gonzalez, F. G., and Nag, A. (2019). SARS coronavirus protein nsp1 disrupts localization of Nup93 from the nuclear pore complex. Biochem. Cell Biol. 97, 758–766. doi: 10.1139/bcb-2018-0394
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. doi: 10.1038/s41586-020-2286-9
Gori Savellini, G., Anichini, G., Gandolfo, C., and Cusi, M. G. (2021). SARS-CoV-2 N Protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses 13:1439. doi: 10.3390/v13081439
Gorkhali, R., Koirala, P., Rijal, S., Mainali, A., Baral, A., and Bhattarai, H. K. (2021). Structure and function of major SARS-CoV-2 and SARS-CoV proteins. Bioinform. Biol. Insights 15:11779322211025876. doi: 10.1177/11779322211025876
Group, I. C. S. (2020). The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Hayn, M., Hirschenberger, M., Koepke, L., Nchioua, R., Straub, J. H., Klute, S., et al. (2021). Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 35:109126. doi: 10.1016/j.celrep.2021.109126
Hensley, L. E., Fritz, L. E., Jahrling, P. B., Karp, C. L., Huggins, J. W., and Geisbert, T. W. (2004). Interferon-beta 1a and SARS coronavirus replication. Emerg. Infect. Dis. 10, 317–319. doi: 10.3201/eid1002.030482
Hsu, J. C., Laurent-Rolle, M., Pawlak, J. B., Wilen, C. B., and Cresswell, P. (2021). Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. U.S.A. 118:e2101161118. doi: 10.1073/pnas.2101161118
Hu, Y., Li, W., Gao, T., Cui, Y., Jin, Y., Li, P., et al. (2017). The severe acute respiratory syndrome coronavirus nucleocapsid inhibits Type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 91:e02143-16. doi: 10.1128/JVI.02143-16
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395, 497–506. doi: 10.1016/s0140-6736(20)30183-5
Huang, K. J., Su, I. J., Theron, M., Wu, Y. C., Lai, S. K., Liu, C. C., et al. (2005). An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 75, 185–194. doi: 10.1002/jmv.20255
Ivashkiv, L. B., and Donlin, L. T. (2014). Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. doi: 10.1038/nri3581
Jiang, H. W., Zhang, H. N., Meng, Q. F., Xie, J., Li, Y., Chen, H., et al. (2020). SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 17, 998–1000. doi: 10.1038/s41423-020-0514-8
Kato, K., Ikliptikawati, D. K., Kobayashi, A., Kondo, H., Lim, K., Hazawa, M., et al. (2021). Overexpression of SARS-CoV-2 protein ORF6 dislocates RAE1 and NUP98 from the nuclear pore complex. Biochem. Biophys. Res. Commun. 536, 59–66. doi: 10.1016/j.bbrc.2020.11.115
Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., et al. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988. doi: 10.1038/ni1243
Kemp, S. A., Collier, D. A., Datir, R., Ferreira, I., Gayed, S., Jahun, A., et al. (2020). Neutralising antibodies in Spike mediated SARS-CoV-2 adaptation. medRxiv [Preprint]. doi: 10.1101/2020.12.05.20241927
Kikkert, M. (2020). Innate immune evasion by human respiratory RNA viruses. J. Innate Immun. 12, 4–20. doi: 10.1159/000503030
Kim, S. S., Sze, L., Liu, C., and Lam, K. P. (2019). The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J. Biol. Chem. 294, 6430–6438. doi: 10.1074/jbc.RA118.005868
Klein, S., Cortese, M., Winter, S. L., Wachsmuth-Melm, M., Neufeldt, C. J., Cerikan, B., et al. (2020). SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 11:5885. doi: 10.1038/s41467-020-19619-7
Konno, Y., Kimura, I., Uriu, K., Fukushi, M., Irie, T., Koyanagi, Y., et al. (2020). SARS-CoV-2 ORF3b Is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 32:108185. doi: 10.1016/j.celrep.2020.108185
Kopecky-Bromberg, S. A., Martinez-Sobrido, L., Frieman, M., Baric, R. A., and Palese, P. (2007). Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81, 548–557. doi: 10.1128/JVI.01782-06
Kumar, A., Ishida, R., Strilets, T., Cole, J., Lopez-Orozco, J., Fayad, N., et al. (2021). SARS-CoV-2 Nonstructural protein 1 inhibits the interferon response by causing depletion of key host signaling factors. J. Virol. 95:e0026621. doi: 10.1128/JVI.00266-21
Lei, X., Dong, X., Ma, R., Wang, W., Xiao, X., Tian, Z., et al. (2020). Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11:3810. doi: 10.1038/s41467-020-17665-9
Li, A., Zhao, K., Zhang, B., Hua, R., Fang, Y., Jiang, W., et al. (2021). SARS-CoV-2 NSP12 protein is not an interferon-β antagonist. J. Virol. 95:e0074721. doi: 10.1128/JVI.00747-21
Li, J. Y., You, Z., Wang, Q., Zhou, Z. J., Qiu, Y., Luo, R., et al. (2020b). The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 22, 80–85. doi: 10.1016/j.micinf.2020.02.002
Li, J. Y., Liao, C. H., Wang, Q., Tan, Y. J., Luo, R., Qiu, Y., et al. (2020a). The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 286:198074. doi: 10.1016/j.virusres.2020.198074
Li, S. W., Wang, C. Y., Jou, Y. J., Huang, S. H., Hsiao, L. H., Wan, L., et al. (2016). SARS coronavirus papain-like protease inhibits the TLR7 signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6. Int. J. Mol. Sci. 17:678. doi: 10.3390/ijms17050678
Lindner, H. A., Lytvyn, V., Qi, H., Lachance, P., Ziomek, E., and Menard, R. (2007). Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 466, 8–14. doi: 10.1016/j.abb.2007.07.006
Lokugamage, K. G., Narayanan, K., Nakagawa, K., Terasaki, K., Ramirez, S. I., Tseng, C. T., et al. (2015). Middle East respiratory syndrome coronavirus nsp1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J. Virol. 89, 10970–10981. doi: 10.1128/JVI.01352-15
Lui, P. Y., Wong, L. Y., Fung, C. L., Siu, K. L., Yeung, M. L., Yuen, K. S., et al. (2016). Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg. Microbes Infect. 5:e39. doi: 10.1038/emi.2016.33
Matthews, K., Schäfer, A., Pham, A., and Frieman, M. (2014). The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol. J. 11:209. doi: 10.1186/s12985-014-0209-9
Minakshi, R., Padhan, K., Rani, M., Khan, N., Ahmad, F., and Jameel, S. (2009). The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 4:e8342. doi: 10.1371/journal.pone.0008342
Monk, P. D., Marsden, R. J., Tear, V. J., Brookes, J., Batten, T. N., Mankowski, M., et al. (2021). Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 9, 196–206. doi: 10.1016/s2213-2600(20)30511-7
Moustaqil, M., Ollivier, E., Chiu, H. P., Van Tol, S., Rudolffi-Soto, P., Stevens, C., et al. (2021). SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg. Microbes Infect. 10, 178–195. doi: 10.1080/22221751.2020.1870414
Mu, J., Fang, Y., Yang, Q., Shu, T., Wang, A., Huang, M., et al. (2020). SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 6:65. doi: 10.1038/s41421-020-00208-3
Nakagawa, K., and Makino, S. (2021). Mechanisms of coronavirus Nsp1-mediated control of host and viral gene expression. Cells. 10:300. doi: 10.3390/cells10020300
Nakagawa, K., Narayanan, K., Wada, M., and Makino, S. (2018). Inhibition of stress granule formation by middle east respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J. Virol. 92:e00902-18. doi: 10.1128/JVI.00902-18
Omrani, A. S., Saad, M. M., Baig, K., Bahloul, A., Abdul-Matin, M., Alaidaroos, A. Y., et al. (2014). Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect. Dis. 14, 1090–1095. doi: 10.1016/s1473-3099(14)70920-x
Onomoto, K., Jogi, M., Yoo, J. S., Narita, R., Morimoto, S., Takemura, A., et al. (2012). Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031
Oudshoorn, D., Rijs, K., Limpens, R., Groen, K., Koster, A. J., Snijder, E. J., et al. (2017). Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio 8:e01658-17. doi: 10.1128/mBio.01658-17
Pan, P., Shen, M., Yu, Z., Ge, W., Chen, K., Tian, M., et al. (2021). SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 12:4664. doi: 10.1038/s41467-021-25015-6
Petrosillo, N., Viceconte, G., Ergonul, O., Ippolito, G., and Petersen, E. (2020). COVID-19, SARS and MERS: are they closely related? Clin. Microbiol. Infect. 26, 729–734. doi: 10.1016/j.cmi.2020.03.026
Ramasamy, S., and Subbian, S. (2021). Critical determinants of cytokine storm and Type I interferon response in COVID-19 pathogenesis. Clin. Microbiol. Rev. 34:e00299-20. doi: 10.1128/CMR.00299-20
Ratia, K., Kilianski, A., Baez-Santos, Y. M., Baker, S. C., and Mesecar, A. (2014). Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 10:e1004113. doi: 10.1371/journal.ppat.1004113
Sallard, E., Lescure, F. X., Yazdanpanah, Y., Mentre, F., and Peiffer-Smadja, N. (2020). Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 178:104791. doi: 10.1016/j.antiviral.2020.104791
Schroeder, S., Pott, F., Niemeyer, D., Veith, T., Richter, A., Muth, D., et al. (2021). Interferon antagonism by SARS-CoV-2: a functional study using reverse genetics. Lancet Microbe 2, e210–e218. doi: 10.1016/s2666-5247(21)00027-6
Schubert, K., Karousis, E. D., Jomaa, A., Scaiola, A., Echeverria, B., Gurzeler, L. A., et al. (2020). SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 27, 959–966. doi: 10.1038/s41594-020-0511-8
Shi, C. S., Qi, H. Y., Boularan, C., Huang, N. N., Abu-Asab, M., Shelhamer, J. H., et al. (2014). SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089. doi: 10.4049/jimmunol.1303196
Shin, D., Mukherjee, R., Grewe, D., Bojkova, D., Baek, K., Bhattacharya, A., et al. (2020). Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662. doi: 10.1038/s41586-020-2601-5
Siu, K. L., Kok, K. H., Ng, M. J., Poon, V. K. M., Yuen, K. Y., Zheng, B. J., et al. (2009). Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 284, 16202–16209. doi: 10.1074/jbc.M109.008227
Siu, K. L., Yeung, M. L., Kok, K. H., Yuen, K. S., Kew, C., Lui, P. Y., et al. (2014). Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88, 4866–4876. doi: 10.1128/JVI.03649-13
Song, P., Li, W., Xie, J., Hou, Y., and You, C. (2020). Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 509, 280–287. doi: 10.1016/j.cca.2020.06.017
Stanevich, O., Alekseeva, E., Sergeeva, M., Fadeev, A., Komissarova, K., Ivanova, A., et al. (2021). SARS-CoV-2 escape from cytotoxic T cells during long-term COVID-19. Research Square [Preprint]. doi: 10.21203/rs.3.rs-750741/v1
Sui, L., Zhao, Y., Wang, W., Wu, P., Wang, Z., Yu, Y., et al. (2021). SARS-CoV-2 membrane protein inhibits Type I interferon production through ubiquitin-mediated degradation of TBK1. Front. Immunol. 12:662989. doi: 10.3389/fimmu.2021.662989
Sun, L., Xing, Y., Chen, X., Zheng, Y., Yang, Y., Nichols, D. B., et al. (2012). Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7:e30802. doi: 10.1371/journal.pone.0030802
Suryawanshi, R. K., Koganti, R., Agelidis, A., Patil, C. D., and Shukla, D. (2021). Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 29, 224–237. doi: 10.1016/j.tim.2020.12.007
Taefehshokr, N., Taefehshokr, S., Hemmat, N., and Heit, B. (2020). Covid-19: perspectives on innate immune evasion. Front Immunol. 11:580641. doi: 10.3389/fimmu.2020.580641
Terada, Y., Kawachi, K., Matsuura, Y., and Kamitani, W. (2017). MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 511, 95–105. doi: 10.1016/j.virol.2017.08.026
Thoms, M., Buschauer, R., Ameismeier, M., Koepke, L., Denk, T., Hirschenberger, M., et al. (2020). Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science (New York, N.Y.). 369, 1249–1255. doi: 10.1126/science.abc8665
Thorne, L. G., Bouhaddou, M., Reuschl, A. K., Zuliani-Alvarez, L., Polacco, B., Pelin, A., et al. (2021). Evolution of enhanced innate immune evasion by the SARS-CoV-2 B.1.1.7 UK variant. bioRxiv [Preprint]. doi: 10.1101/2021.06.06.446826
von Grotthuss, M., Wyrwicz, L. S., and Rychlewski, L. (2003). mRNA cap-1 methyltransferase in the SARS genome. Cell 113, 701–702. doi: 10.1016/s0092-8674(03)00424-0
Wang, S., Dai, T., Qin, Z., Pan, T., Chu, F., Lou, L., et al. (2021). Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 23, 718–732. doi: 10.1038/s41556-021-00710-0
Wang, W., Zhou, Z., Xiao, X., Tian, Z., Dong, X., Wang, C., et al. (2021). SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Immunol. 18, 945–953. doi: 10.1038/s41423-020-00619-y
WHO (n.d.). Available online at: https://covid19.who.int/
Williamson, M. K., Hamilton, F., Hutchings, S., Pymont, H. M., Hackett, M., Arnold, D., et al. (2021). Chronic SARS-CoV-2 infection and viral evolution in a hypogammaglobulinaemic individual. medRxiv [Preprint]. doi: 10.1101/2021.05.31.21257591
Wu, J., Shi, Y., Pan, X., Wu, S., Hou, R., Zhang, Y., et al. (2021). SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 34:108761. doi: 10.1016/j.celrep.2021.108761
Wu, Y., Ma, L., Cai, S., Zhuang, Z., Zhao, Z., Jin, S., et al. (2021). RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-kappaB hyper-activation and inflammation. Signal. Transduct. Target Ther. 6:167. doi: 10.1038/s41392-021-00575-7
Xia, H., Cao, Z., Xie, X., Zhang, X., Chen, J. Y., Wang, H., et al. (2020). Evasion of Type I interferon by SARS-CoV-2. Cell Rep. 33:108234. doi: 10.1016/j.celrep.2020.108234
Yang, D., and Leibowitz, J. L. (2015). The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 206, 120–133. doi: 10.1016/j.virusres.2015.02.025
Yang, L., Xie, X., Tu, Z., Fu, J., Xu, D., and Zhou, Y. (2021). The signal pathways and treatment of cytokine storm in COVID-19. Signal. Transduct. Target Ther. 6:255. doi: 10.1038/s41392-021-00679-0
Yang, X., Chen, X., Bian, G., Tu, J., Xing, Y., Wang, Y., et al. (2014). Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 95(Pt 3), 614–626. doi: 10.1099/vir.0.059014-0
Yang, Y., Ye, F., Zhu, N., Wang, W., Deng, Y., Zhao, Z., et al. (2015). Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci. Rep. 5:17554. doi: 10.1038/srep17554
Yang, Z. Y., Huang, Y., Ganesh, L., Leung, K., Kong, W. P., Schwartz, O., et al. (2004). pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78, 5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004
Yang, Z., Zhang, X., Wang, F., Wang, P., Kuang, E., and Li, X. (2020). Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. bioRxiv [Preprint]. doi: 10.1101/2020.08.12.247767
Yuan, J., Zou, R., Zeng, L., Kou, S., Lan, J., Li, X., et al. (2020). The correlation between viral clearance and biochemical outcomes of 94 COVID-19 infected discharged patients. Inflamm. Res. 69, 599–606. doi: 10.1007/s00011-020-01342-0
Yuen, C. K., Lam, J. Y., Wong, W. M., Mak, L. F., Wang, X., Chu, H., et al. (2020). SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 9, 1418–1428. doi: 10.1080/22221751.2020.1780953
Zhang, K., Miorin, L., Makio, T., Dehghan, I., Gao, S., Xie, Y., et al. (2021). Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 7:eabe7386. doi: 10.1126/sciadv.abe7386
Zhang, Y., Chen, Y., Li, Y., Huang, F., Luo, B., Yuan, Y., et al. (2021). The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc. Natl. Acad. Sci. U.S.A. 118:e2024202118. doi: 10.1073/pnas.2024202118
Zhang, Z., Liu, Q., Sun, Y., Li, J., Liu, J., Pan, R., et al. (2021). Live attenuated coronavirus vaccines deficient in N7-Methyltransferase activity induce both humoral and cellular immune responses in mice. Emerg. Microbes Infect. 10, 1626–1637. doi: 10.1080/22221751.2021.1964385
Zhao, K., Ke, Z., Hu, H., Liu, Y., Li, A., Hua, R., et al. (2021). Structural basis and function of the N terminus of SARS-CoV-2 nonstructural protein 1. Microbiol. Spect. 9:e0016921. doi: 10.1128/Spectrum.00169-21
Zheng, Z. Q., Wang, S. Y., Xu, Z. S., Fu, Y. Z., and Wang, Y. Y. (2021). SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. 7:38. doi: 10.1038/s41421-021-00275-0
Zhou, J., Chu, H., Li, C., Wong, B. H., Cheng, Z. S., Poon, V. K., et al. (2014). Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 209, 1331–1342. doi: 10.1093/infdis/jit504
Zhou, P., Yang, Y. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. doi: 10.1038/s41586-020-2012-7
Keywords: SARS-CoV-2, highly pathogenic coronaviruses, IFN signaling pathway, host–virus interaction, innate immunity
Citation: Li J-Y, Zhou Z-J, Wang Q, He Q-N, Zhao M-Y, Qiu Y and Ge X-Y (2021) Innate Immunity Evasion Strategies of Highly Pathogenic Coronaviruses: SARS-CoV, MERS-CoV, and SARS-CoV-2. Front. Microbiol. 12:770656. doi: 10.3389/fmicb.2021.770656
Received: 04 September 2021; Accepted: 16 September 2021;
Published: 29 October 2021.
Edited by:
Chunfu Zheng, University of Calgary, CanadaReviewed by:
Jungang Chen, University of Arkansas for Medical Sciences, United StatesYi-Quan Wu, National Cancer Institute (NCI), United States
Copyright © 2021 Li, Zhou, Wang, He, Zhao, Qiu and Ge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ye Qiu, cWl1eWVAaG51LmVkdS5jbg==; Xing-Yi Ge, eHlnZUBobnUuZWR1LmNu