 Jun Wei1 John L. Stebbins1 Shinichi Kitada1
Jun Wei1 John L. Stebbins1 Shinichi Kitada1 Rupesh Dash2 Dayong Zhai1
Rupesh Dash2 Dayong Zhai1 William J. Placzek1 Bainan Wu1
William J. Placzek1 Bainan Wu1 Michele F. Rega1
Michele F. Rega1 Ziming Zhang1 Elisa Barile1 Li Yang1
Ziming Zhang1 Elisa Barile1 Li Yang1 Russell Dahl1
Russell Dahl1 Paul B. Fisher2 John C. Reed1
Paul B. Fisher2 John C. Reed1 Maurizio Pellecchia1*
Maurizio Pellecchia1*- 1 Infectious and Inflammatory Disease Center, Cancer Center, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA
- 2 School of Medicine, Virginia Commonwealth University, Richmond, VA, USA
Our focus in the past several years has been on the identification of novel and effective pan-Bcl-2 antagonists. We have recently reported a series of Apogossypolone (ApoG2) derivatives, resulting in the chiral compound (±) BI97D6. We report here the synthesis and evaluation on its optically pure (−) and (+) atropisomers. Compound (−) BI97D6 potently inhibits the binding of BH3 peptides to Bcl-XL, Bcl-2, Mcl-1, and Bfl-1 with IC50 values of 76 ± 5, 31 ± 2, 25 ± 8, and 122 ± 28 nM, respectively. In a cellular assay, compound (−) BI97D6 effectively inhibits cell growth in the PC-3 human prostate cancer and H23 human lung cancer cell lines with EC50 values of 0.22 ± 0.08 and 0.14 ± 0.02 μM, respectively. Similarly, compound (−) BI97D6 effectively induces apoptosis in the BP3 human lymphoma cell line in a dose-dependent manner. The compound also shows little cytotoxicity against bax−/−/bak−/− cells, suggesting that it kills cancers cells predominantly via a Bcl-2 pathway. Moreover, compound (−) BI97D6 displays in vivo efficacy in both a Bcl-2-transgenic mouse model and in a prostate cancer xenograft model in mice. Therefore, compound (−) BI97D6 represents a promising drug lead for the development of novel apoptosis-based therapies for cancer.
Introduction
Programmed cell death (apoptosis; Reed, 1999; Vaux and Korsmeyer, 1999) plays critical roles in both the onset and progress of cancer and contributes significantly to chemoresistance (Johnstone et al., 2002; Reed, 2002). B-cell lymphoma/leukemia-2 (Bcl-2) family proteins are central regulators of the apoptotic machinery (Adams and Cory, 1998; Reed, 1998; Gross et al., 1999). The Bcl-2 family is composed of anti-apoptotic and pro-apoptotic members. To date, six anti-apoptotic members of the Bcl-2 family have been identified and characterized, including Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W, and Bcl-B. Since the overexpression of anti-apoptotic Bcl-2 family proteins is associated with tumor progression, poor prognosis, and drug resistance, these proteins are representing attractive targets for anticancer drug (Reed, 1997; Wang et al., 2000; Degterev et al., 2001). X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy structural studies have elucidated a hydrophobic crevice on the surface of anti-apoptotic Bcl-2 family proteins that binds the BH3 dimerization domain of pro-apoptotic family members (Muchmore et al., 1996; Sattler et al., 1997; Reed, 1998). Thus, molecules that mimic the BH3 domain of pro-apoptotic proteins may be effective in either inducing apoptosis and/or in abrogating the ability of anti-apoptotic Bcl-2 proteins to inhibit cancer cell death (Reed, 1997, 1998; Wang et al., 2000; Degterev et al., 2001).
Our laboratories (Kitada et al., 2003) and others (Wang and Yang, 2004; Mohammad et al., 2005; Wang et al., 2006a) have reported that the natural product (±) Gossypol (Figure 1A) is a moderate inhibitor of Bcl-2, Bcl-XL, and Mcl-1, functioning as a BH3 mimic (Kitada et al., 2003; Wang and Yang, 2004; Mohammad et al., 2005; Wang et al., 2006a). (−) Gossypol (AT101, Ascenta Pharmaceuticals) is currently in phase II clinical trials as single-agent (Mohammad et al., 2005; Wang et al., 2006a; Meng et al., 2008). However, (±) Gossypol may target other proteins due to its two reactive aldehyde groups. Hence, we designed (±)Apogossypol (Figure 1A), a compound lacking these undesired functional groups, but retains activity against anti-apoptotic Bcl-2 family proteins in vitro and in cell (Becattini et al., 2004). Accordingly, we found that (±) Apogossypol had superior efficacy and markedly reduced toxicity compared to (±) Gossypol (Kitada et al., 2008). Furthermore, single-dose pharmacokinetic characteristics of (±) Apogossypol in mice displayed superior blood concentrations over time compared to compound (±) Gossypol, due to slower clearance (Coward et al., 2006). Recently, we reported on the separation and characterization of the atropisomers of (±) Apogossypol (Wei et al., 2009c). We further reported the synthesis and evaluation of (±) 5, 5′ alkyl, ketone, and amide substituted (±) Apogossypol derivatives, with the best compounds (±) BI97D10 (Wei et al., 2009a) and (RS, ±, RS) 8r (Wei et al., 2009b; Figure 1A) displaying improved in vitro and in vivo efficacy compared to (±) Apogossypol. Moreover, we reported the optically pure compound (R, −, R) 8r (BI97C1, Sabutoclax; Figure 1A; Wei et al., 2010b; Dash et al., 2011) showed marked enhanced efficacy in vivo compared to diastereomer mixtures (RS, ±, RS) 8r (Wei et al., 2010b).
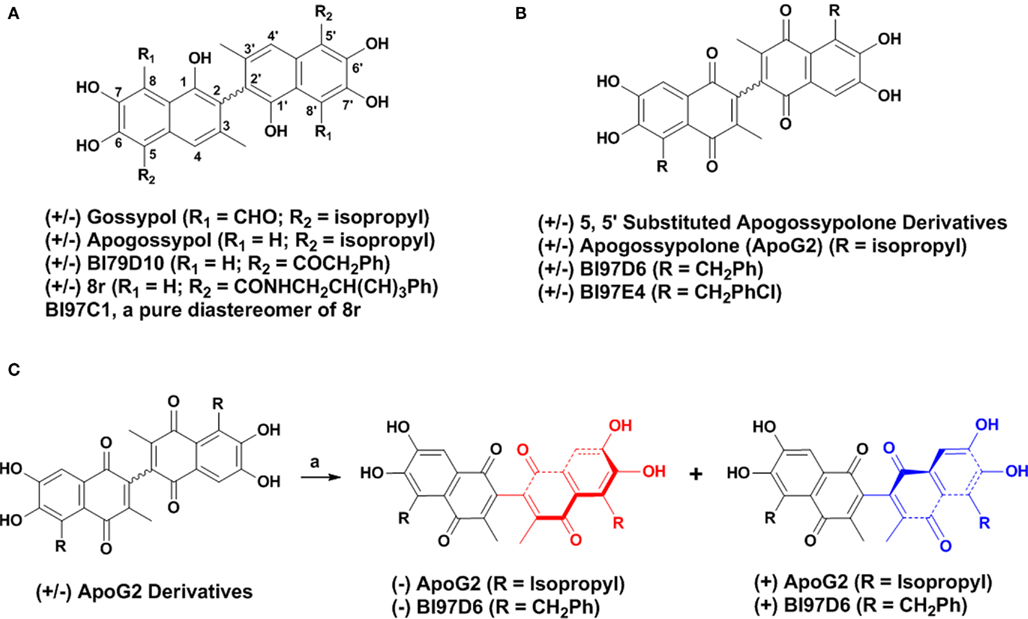
Figure 1. (A) Structure of (±) Gossypol, Apogossypol, BI79D10, 8r, and BI97C1. (B) Structure of 5, 5′ substituted Apogossypolone derivatives, Apogossypolone (ApoG2), BI97D6, and BI97E4. (C) Preparation of pure (−) and (+) BI97D6 and ApoG2 atropisomers using a normal phase liquid chiral column chromatography.
(±) Apogossypolone (ApoG2, Figure 1B) is a Gossypol derivative designed by Ascenta Pharmaceuticals to reduce the toxicity of Gossypol. (±) ApoG2 has been reported as a potent inhibitor of Mcl-1, Bcl-2, and Bcl-XL. (±) ApoG2 blocks binding of Bim and Bcl-2 and induces apoptosis in a number of human cancer cell lines (Wang et al., 2006b; Arnold et al., 2008; Hu et al., 2008; Mi et al., 2008; Lin et al., 2009; Sun et al., 2009). In addition, (±) ApoG2 induces regression in several tumor xenograft models, and its maximum tolerated dose (MTD) appeared to be less toxic than compound (−) Gossypol, with oral MTD values of >240 mg/kg and ∼50 mg/kg, respectively (Wang et al., 2006b; Mi et al., 2008). We have recently reported on the synthesis and biological evaluation of novel (±) ApoG2 derivatives which replaced the isopropyl groups of (±) ApoG2 with various alkyl groups at 5, 5′ positions (Figure 1B; Wei et al., 2010a). Some 5, 5′ substituted ApoG2 derivatives 6i (BI97D6) and 6l (BI97E4; Figure 1B) displayed improved in intro and in vivo efficacy compared to (±) ApoG2. For example, compounds (±) BI97D6 and BI97E4 induced 43 ± 4 and 38 ± 7% reduction of spleen size at dose of 30 μmol/kg in B6Bcl-2-transgenic mice model, whereas compound (±) ApoG2 induced only 15 ± 2% reduction at even a higher dose (60 μmol/kg) in the same assay (Wei et al., 2010a). However, compounds (±) BI97D6 and ApoG2 are mixtures of (+) and (−) atropisomers (Figure 1B). In principle, (+) and (−) atropisomers should be treated as different compounds because they have different physical, chemical, and pharmacology properties. Indeed, the (−) Gossypol displayed a marked differential activity compared to its natural racemic mixture (Wang and Yang, 2004). Therefore, in this current work, we focus our attention on preparing pure atropisomers of (±) ApoG2 and its most potent derivative (BI97D6) followed by further investigation of their in vitro and in vivo activities (Figure 1C).
Materials and Methods
Molecular Modeling
Molecular modeling studies were conducted on a Linux workstation and a 64 3.2-GHz CPUs Linux cluster. Docking studies were performed using the crystal structure of Bcl-XL in complex with a BH3 mimetic ligand (Protein Data Bank code 2YXJ; Oltersdorf et al., 2005; Bruncko et al., 2007; Lee et al., 2007). The ligand was extracted from the protein structure and was used to define the binding site for small molecules. Compounds (−) and (+) BI97D6 were docked into the Bcl-XL protein by the GOLD (Jones et al., 1997) docking program using ChemScore (Eldridge et al., 1997) as the scoring function. The active site radius was set at 10 Å and 10 GA solutions were generated for each molecule. The GA docking procedure in GOLD (Jones et al., 1997) allowed the small molecules to flexibly explore the best binding conformations whereas the protein structure was static. The protein surface was prepared with the program MOLCAD (Teschner et al., 1994) as implemented in Sybyl (Tripos, St. Louis) and was used to analyze the binding poses for studied small molecules.
Chemicals
The synthesis of pure (−) and (+) atropisomers of ApoG2 and BI97D6 is outlined in Figure 1C. The preparation of racemic (±) ApoG2 and (±) BI97D6 has been previously reported by us (Wei et al., 2009b). These racemic compounds were readily resolved using a liquid normal phase chiral column chromatography to afford (−) and (+) optically pure atropisomers of ApoG2 and BI97D6 (Figure 1C). The optical configuration and enantiomeric purity of each atropisomer were determined using a combination of polarimeter and liquid normal phase chiral column chromatography (Table A1; Figure A1A in Appendix). The optical rotation ([α]) generated by the atropisomery in ApoG2 and BI97D6 was approximately from ±40°, depending on different substitutions on 5, 5′ positions of ApoG2 (Table A1 in Appendix). The separation and characterization of the compound (−) BI97D6 are given as an example. The detailed experiment and characterization of all other compounds are in Appendix. The (±) BI97D6 was resolved to afford pure (−) and (+) BI97D6 (Figure 1C) using a RegisCell 5 μM 250 mm × 10 mm liquid normal phase semi-preparative chiral column from Regis Technologies Inc. Mobile phase A was 0.1% TFA in 2-propanol and mobile phase B was 0.1% TFA in hexane. Flow rate was 4 mL/min. The run duration was 25 min. The separation was done using constant 30% A and 70% B in 15 min followed by 10 min at 100% A. The optical rotation ([α]) of (−) BI97D6 is −38.75°. 1H NMR (600 MHz, CD3OD) δ 7.448 (s, 2H), 7.224 (d, J = 7.8 Hz, 4H), 7.178 (t, J1 = 7.8 Hz, J2 = 7.2 Hz, 4H), 7.084 (t, J1 = J2 = 7.2 Hz, 2H), 4.643 (dd, J1 = 14.4 Hz, J2 = 13.8 Hz, 4H), 1.867 (s, 6H); 13C NMR (600 MHz, (CD3)2SO) δ 185.44, 182.61, 150.04, 149.83, 146.59, 140.95, 138.56, 130.07, 128.66, 128.39, 126.52, 125.82, 123.76, 111.74, 31.74, 14.84; HRMS calculated for [C36H26O8 + H] 587.1700, Found 587.1723. High-performance liquid chromatography is 98% pure.
NMR Experiments
Nuclear magnetic resonance-based binding assays have been conducted by acquiring one-dimensional 1H experiments with 500 μL solution of Bcl-XL at 20 μM concentration, in absence and presence of added compounds, each at 100 μM concentration. By observing the aliphatic region of the spectra, binding could be readily detected due to chemical shift changes in active site methyl groups of Ile, Leu, Thr, Val, or Ala (region between −0.8 and 0.3 ppm). The Mcl-1 binding mode was characterized by recording [15N, 1H]-HSQC experiments with 500 μL solution of uniformly 15N-labeled Mcl-1 (25 μM concentration) in absence and presence of added compounds, each at 25 and 125 μM concentration, respectively. Bcl-XL and Mcl-1 samples were prepared and purified as described previously (Sattler et al., 1997; Day et al., 2005). All experiments were performed with a 600-MHz spectrometer Bruker Advance 600 equipped with four rf channels and z-axis pulse-field gradients.
Fluorescence Polarization Assays
A Bak BH3 peptide (F-BakBH3) (GQVGRQLAIIGDDINR) was labeled at the NH2 terminus with fluorescein isothiocyanate (FITC; Molecular Probes) and purified by HPLC. For competitive binding assays, 20 nM GST–Bcl-XL ΔTM protein was preincubated with the tested compound at varying concentrations in 47.5 μL phosphate-buffered saline (PBS; pH = 7.4) in 96-well black plates at room temperature for 5 min, then 2.5 μL of 15 nM FITC-labeled Bim BH3 (FITC-Ahx-DMRPEIWIAQELRRIGDEFNAYYAR) peptide was added to produce a final volume of 50 μL. The wild-type and mutant Bim BH3 peptides were included in each assay plate as positive and negative controls, respectively. After 10 min incubation at room temperature, the polarization values in millipolarization units (Sattler et al., 1997) were measured at excitation/emission wavelengths of 480/535 nm with a multilabel plate reader (PerkinElmer). IC50 was determined by fitting the experimental data to a sigmoidal dose–response non-linear regression model (SigmaPlot 10.0.1; Systat Software, Inc., San Jose, CA, USA). Data reported are mean of three independent experiments ± SE. Performance of Bcl-2, Mcl-1, and Bfl-1 FPA are similar. Briefly, 20 nM of GST–Bcl-2 or –Mcl-1, or –Bfl-1 were incubated with various concentrations of compounds (−), (+), (±) ApoG2 and BI97D6 for 2 min, then 15 nM FITC-conjugated-Bim BH3 peptide (Ramjaun et al., 2007) was added in PBS buffer. Fluorescence polarization (FP) was measured after 10 min.
Cell Viability Assays
The activity of the compounds against human cancer cell lines (PC-3, H460, and H23) were assessed by using the ATP-LITE assay (PerkinElmer). All cells were seeded in either 12F2 or RPMI1640 medium with 5 mM L-glutamine supplemented with 5% fetal bovine serum (Mediatech Inc.), penicillin, and streptomycin (Omega). For maintenance, cells were cultured in 5% FBS. Cells plated into 96-well plates at varying initial densities depending on doubling time. H460 and H23 plated at 2000 cells/well and PC-3 at 3000 cells/well. Compounds were diluted to final concentrations with 0.1% DMSO. Prior to dispensing compounds onto cells, fresh 5% media was placed into wells. Administration of compounds occurred 24 h after seeding into the fresh media. Cell viability was evaluated using ATP-LITE reagent (PerkinElmer) after 72 h of treatment. Data were normalized to the DMSO control-treated cells using Prism version 5.01 (GraphPad Software). Data were reported as mean of three independent experiments ± SE. The activity of compounds (−) and (+) BI97D6 against mouse embryonic fibroblast wild-type cells (MEF/WT) and mouse embryonic fibroblast BAX/Bak double knockout cells (DKO/MEF) was assessed by 1 day ATP-LITE assay. Wild-type MEF and DKO/MEF were seeded in 96-well plate at a seeding density of 10,000 cells per well. The next day, compounds (−) and (+) BI97D6 were added to wild-type and DKO cells. Cell viability was evaluated using ATP-LITE reagent (PerkinElmer) after 24 h of treatment. Data were normalized to the DMSO control-treated cells using Prism version 5.01 (GraphPad Software). Data were reported as mean of three independent experiments ± SE.
The apoptotic activity of the compounds against BP3 cells was assessed by staining with Annexin V-FITC and propidium iodide (PI). BP3 cells were cultured in RPMI 1640 medium (Mediatech Inc., Herndon, VA 20171, USA) containing 10% fetal bovine serum (Mediatech Inc., Herndon, VA 20171, USA) and Penicillin/Streptomycin (Mediatech Inc., Herndon, VA 20171, USA). Cells were cultured with various concentrations of (±), (−), and (+) ApoG2 and its 5, 5′ substituted derivative (BI97D6) for 1 day. The percentage of viable cells was determined by FITC-Annexin V- and propidium iodide (PI)-labeling, using an Apoptosis Detection kit (BioVision Inc.), and analyzing stained cells by flow-cytometry (FACSort; Bectin-Dickinson, Inc., Mountain View, CA, USA). Cells that were annexin-V-negative and PI-negative were considered viable. Data reported are mean of two independent experiments ± SE.
In vitro ADME studies
Liver microsomal stability
Pooled rat liver microsomes (BD Biosciences, # 452701) were preincubated with test compounds at 37.5°C for 5 min in the absence of NADPH. The reaction was initiated by addition of NADPH and then incubated under the same conditions. The final incubation concentrations were 4 μM test compound, 2 mM NADPH, and 1 mg/mL (total protein) liver microsomes in PBS at pH 7.4. One aliquot (100 μL) of the incubation mixture was withdrawn at 0, 15, 30, and 60 min and combined immediately with 200 μL of ACN/MeOH containing an internal standard. After mixing, the sample was centrifuged at approximately 13,000 rpm for 12 min. The supernatant was transferred into an auto sampler vial and the amount of test compound was quantified using the Shimadzu LCMS-2010EV mass spectrometer. The change of the AUC (area under the curve) of the parent compound as function of time was used as a measure of microsomal stability. Data reported are mean of two independent experiments ± SE.
Plasma stability
A 20-μL aliquot of a 10-mM solution in DMSO of the test compound was added to 2.0 mL of heparinized rat plasma (Lampire, P1-150N) to obtain a 100-μM final solution. The mixture was incubated for 1 h at 37.5°C. Aliquots of 100 μL were taken (0, 30 min, 1 h) and diluted with 200 μL of MeOH containing internal standard. After mixing, the sample was centrifuged at approximately 13,000 rpm for 12 min. The supernatant was transferred into an autosampler vial and the amount of test compound was quantified using the Shimadzu LCMS-2010EV system. The change of the AUC (area under the curve) of the parent compound as function of time was used as a measure of microsomal stability. Data reported are mean of two independent experiments ± SE.
Parallel artificial membrane permeation assay
A 96-well microtiter plate (Millipore, # MSSACCEPTOR) was completely filled with aqueous buffer solution (pH 7.2) and covered with a microtiter filterplate (Millipore, # MAPBMN310). The hydrophobic filter material was impregnated with a 10% solution of hexadecane in hexane and the organic solvent was allowed to completely evaporate. Permeation studies were started by the transfer of 200 μL of a 100-μM test compound solution on top of the filterplate. In general phosphate buffer at pH 7.2 buffer was used. The maximum DMSO content of the stock solutions was <5%. In parallel, an equilibrium solution lacking a membrane was prepared using the exact concentrations and specifications but lacking the membrane. The concentrations of the acceptor and equilibrium solutions were determined using the Shimadzu LCMS-2010EV and AUC methods. The permeation of a compound through the membrane layer is described by the percentage permeation (% flux). The flux values were calculated considering the concentration of the acceptor compartment after 8 h and that of a reference well with the same concentration containing no membrane barrier.
Bcl-2-Transgenic Mice Studies
Transgenic mice expressing Bcl-2 have been described as the B6 line (Katsumata et al., 1992). The BCL-2 transgene represents a minigene version of a t(14;18) translocation in which the human BCL-2 gene is fused with the immunoglobulin heavy-chain (IgH) locus and associated IgH enhancer. The transgene was propagated on the Balb/c background. These mice develop polyclonal B-cell hyperplasia with asynchronous transformation to monoclonal aggressive lymphomas beginning at approximately 6 months of age, with approximately 90% of mice undergoing transformation by the age of 12–24 months. All animals used here had not yet developed aggressive lymphoma. Compounds dissolved in 500 μL of solution (Ethanol:Cremophor EL:Saline = 10:10:80) were injected intraperitoneally to age- and sex-matched B6Bcl-2 mouse, while control-mice were injected intraperitoneally with 500 μL of the same formulation without compound. After 24 h, B6Bcl-2 mice were sacrificed by intraperitoneal injection of lethal dose of Avertin. Spleen was removed and weighed. The spleen weight of mice is used as an end-point for assessing activity as we determined that spleen weight is highly consistent in age- and sex-matched Bcl-2-transgenic mice in preliminary studies (Kitada et al., 2008). Variability of spleen weight was within ±2% among control-treated age-matched, sex-matched B6Bcl-2 mice. Data reported in Figure 3C are mean of five independent experiments ± SE.
Human Prostate Cancer Xenografts in Athymic Nude Mice
PC-3-Luc cells (1 × 106) were injected s.c. in 100 μL of PBS in the left flank of male athymic nude mice (NCRnu/nu, 4 weeks old, 20 g body weight). After establishing visible tumors of ∼75-mm3, requiring ∼5–6 days, compound dissolved in 500 μL of solvent (ethanol/Cremophor EL/saline = 10:10:80) were injected intraperitoneally (i.p.). The injections were given every 2 days for a total of nine injections. Seven treatment groups were established for the experiment, i.e., DMSO only, 3 mg/kg of (±) BI97D6, (−) BI97D6, (+) BI97D6, 5 mg/kg of (± ) BI97D6, (−) BI97D6, (+) BI97D6, respectively. A minimum of five animals was used per experimental condition. Data reported are mean of five independent experiments ± SE. For in vivo imaging of tumors, the mice were anesthetized and injected i.p. with 150 mg/kg luciferin and light emitted from each tumor was determined using a Xenogen system with CCD camera with an integration time of 1 min. Luminescence measurements were made using Living Image software (version 2.50.1; Xenogen).
Results and Discussion
We had recently reported that (±) BI97D6 (Figure 1B) was a promising inhibitor of Bcl-XL, Bcl-2, and Mcl-1 with improved in vitro and in vivo efficacy compared to (±) ApoG2 (Wei et al., 2010a). However, by using chiral chromatography we found that, similar to what was known about (±) Gossypol and Apogossypol (Wang and Yang, 2004; Wei et al., 2009c), compound ApoG2 and its derivative (BI97D6) also displayed axial chirality due to restricted rotation around the binaphtyl bond (Figure 1B). Therefore, it was attractive to explore whether optically pure (−) and (+) atropisomers of ApoG2 and BI97D6 presented different in vitro and in vivo activities. In order to predict binding poses of (−) and (+) BI97D6 into the BH3 binding groove of Bcl-XL, molecular docking studies were performed (Figures 2A,B). Analysis of the predicted binding models indicated that both atropisomers (−) BI97D6 and (+) BI97D6 could fit well into the BH3 binding groove of Bcl-XL (Figures 2A,B), with the left half components of (−) and (+) atropisomers bound to Bcl-XL in a similar orientation. However, their right substituted naphthalene rings were predicted to present rather different binding modes (Figures 2A,B). The (−) BI97D6 was predicted to form hydrogen bonding with residue Asn 136 in Bcl-XL through its 1′ oxygen on the right naphthalene ring (Figure 2A), whereas the (+) BI97D6 could not form the hydrogen bonding (Figure 2B). The chemscore of (−) BI97D6 was 28.54, which was greater than 20.25 for (+) BI97D6. Therefore, we anticipated that the atropisomer (−) BI97D6 might have higher binding affinity for Bcl-XL compared to its (+) atropisomer.
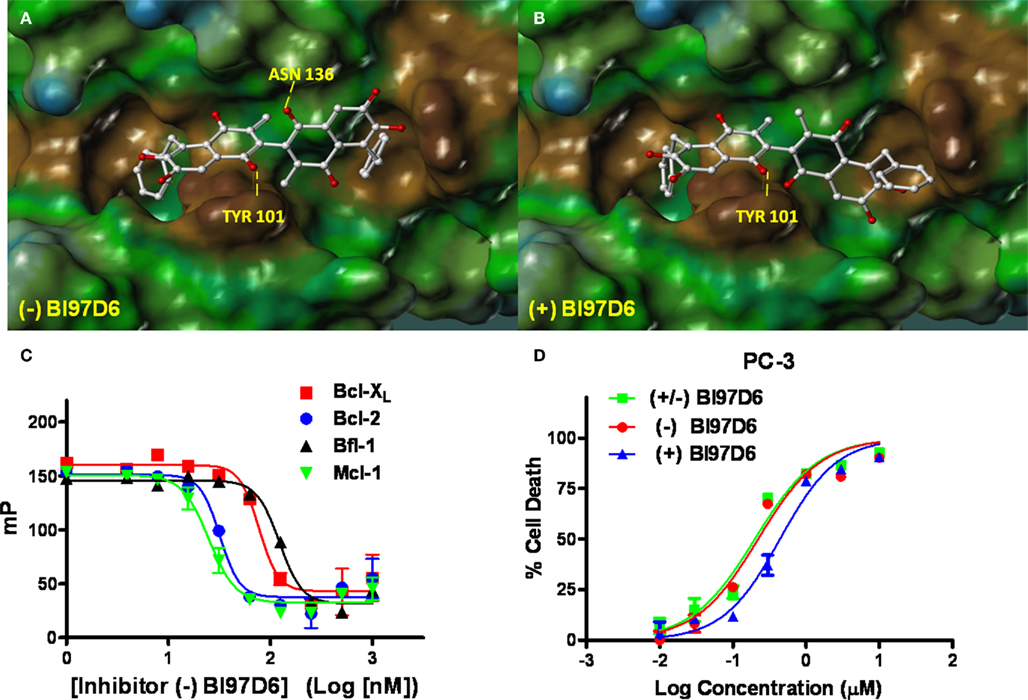
Figure 2. Molecular docking studies. Stereo views of docked structures of (−) BI97D6 (A) and (+) BI97D6 (B) into the BH3 peptide binding groove in Bcl-XL (PDB ID: 2YXJ). (C) Fluorescence polarization-based competitive dose–response curves of (−) BI97D6 to Bcl-XL (red square), Bcl-2 (blue dot), Bfl-1 (dark up triangle), and Mcl-1 (green down triangle). (D) Inhibition of cell growth by compounds (± ) BI97D6 (Green Square), (−) BI97D6 (red dot), and (+) BI97D6 (blue up triangle) in the PC-3 human prostate cancer cell line. Cells were treated for 3 days and cell viability was evaluated using ATP-LITE assay.
The pure atropisomers, namely compounds (−) BI97D6, (+) BI97D6, (−) ApoG2, and (+) ApoG2 were then evaluated by NMR spectroscopy binding assays (Figure A1B in Appendix), competitive FPAs (Table 1) and cell viability assays (Table 2). Compounds (−) BI97D6 and (−) ApoG2 induced larger chemical shift perturbations in the aliphatic region (region between −0.38 and 0.42 ppm) compared to (+) BI97D6 and (+) ApoG2 in the one-dimensional 1H NMR spectroscopy (1D-1H NMR) binding assays against Bcl-XL (Figure A1B in Appendix). To confirm the result of the NMR binding data, we next evaluated the binding property of these compounds against Bcl-XL using FP assays (Table 1 and Figure 2C). Consistent with the NMR data, compound (−) BI97D6 bound tightly to Bcl-XL with an IC50 value of 76 ± 5 nM and and is more potent compared to (+) BI97D6 (125 ± 7 nM), (−) ApoG2 (88 ± 3 nM), and (+) ApoG2 (92 ± 3 nM) in FP assay (Table 1). In agreement, compound (−) BI97D6 was more effective in inhibiting growth of PC-3 cells, which expressed high levels of Bcl-XL. The EC50 value of (−) BI97D6 in killing PC-3 cells was 0.22 ± 0.08 μM, hence more potent than (+) BI97D6 (0.44 ± 0.10 μM), (+) ApoG2 (5.41 ± 0.19 μM), and (−) ApoG2 (4.39 ± 0.28 μm; Figure 2D and Table 2). This is in agreement with previous observations that lead to the selection of (−) Gossypol (AT101) for clinical trials (Wang and Yang, 2004). In fact, (−) Gossypol and (+) Gossypol bound to Bcl-XL with IC50 values of 0.48 and 0.54 μM, respectively, in FPA assays while their EC50 values in killing PC-3 cells were 3.3 and 17.8 μM, respectively.
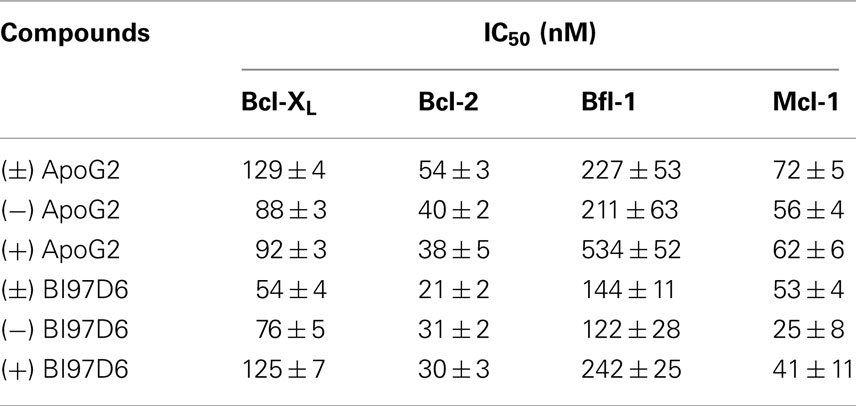
Table 1. Cross-activity of (±), (+), and (−) ApoG2 and BI97D6 against Bcl-XL, Bcl-2, Bfl-1, and Mcl-1.
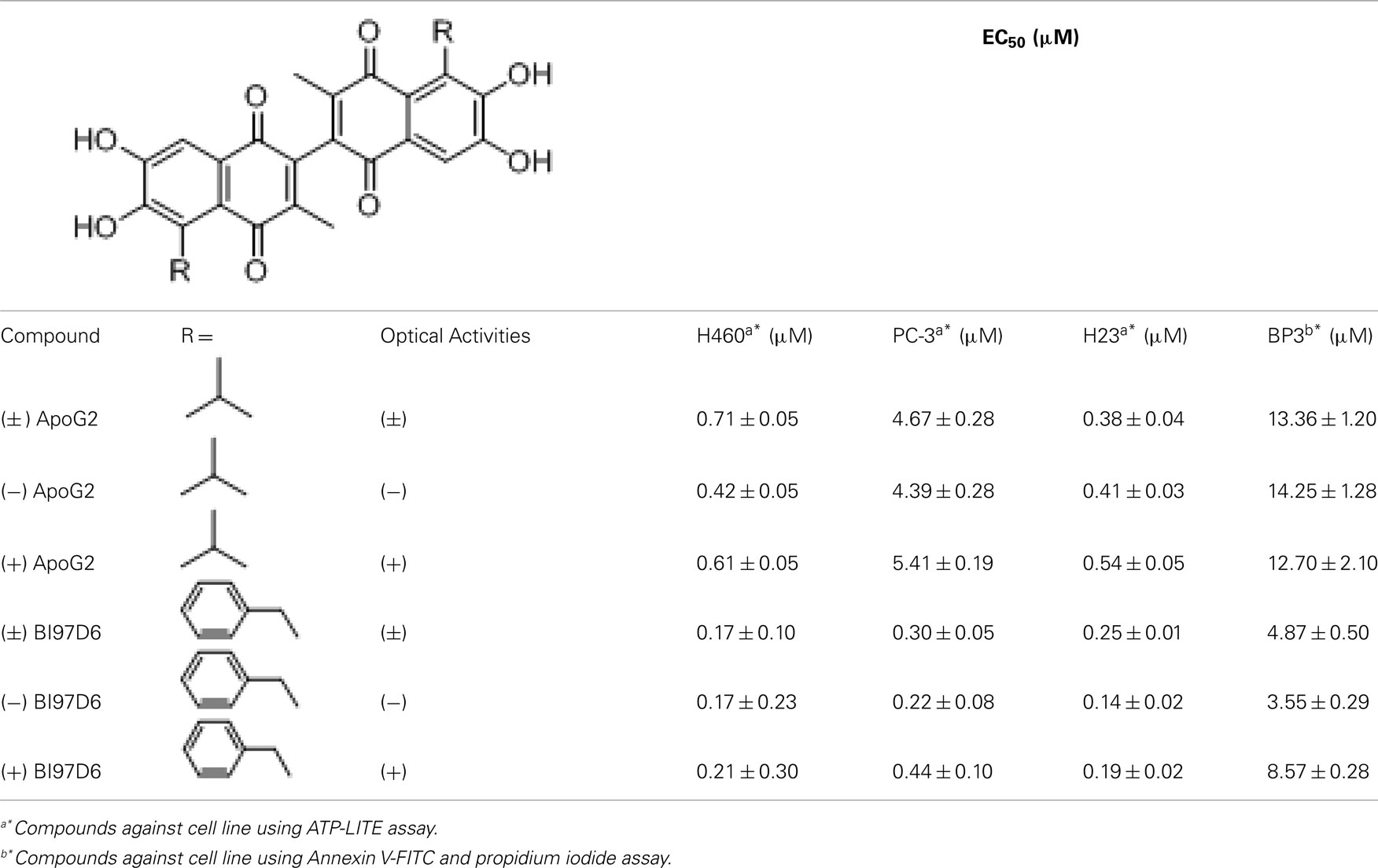
Table 2. Evaluation of (±), (+), and (−) ApoG2 and BI97D6 using cell viability assays.
In addition to Bcl-XL, other members of the Bcl-2 family were known to play critical roles in tumor survival (Brien et al., 2007; Wesarg et al., 2007; Placzek et al., 2010). Therefore, we further evaluated the binding properties and specificity of (−) and (+) atropisomers (BI97D6, ApoG2) against Mcl-1, Bcl-2, and Bfl-1 using a similar FP assay (Table 2). Compound (−) BI97D6 displayed highly potent binding affinity against Mcl-1 (IC50 = 25 ± 8 nM), Bcl-2 (IC50 = 31 ± 2 nM), and Bfl-1 (IC50 = 122 ± 28 nM) in these assays (Table 2). To confirm these results, we also produced uniformly 15N-labeled Mcl-1 protein and measured 2D [15N,1H]-TROSY correlation spectra in absence and presence of compound (−) BI97D6 (Figure A1C in Appendix). Compound (−) BI97D6 displayed a significant binding to Mcl-1, as qualitatively evaluated by the nature of significant changes in chemical shifts at the ligand/protein ratio of 5:1. Although (+) atropisomer BI97D6 was a potent inhibitor of Mcl-1 (IC50 = 41 ± 11 nM) and Bfl-1 (IC50 = 242 ± 25 nM), it is one- to two-fold less potent than its (−) atropisomer BI97D6 in same FP assays (Table 1). However, compound (+) BI97D6 has similar inhibitory activity against Bcl-2 (IC50 = 30 ± 3 nM) compared to its (−) atropisomer (IC50 = 31 ± 2 nM). Compound (−) BI97D6 was further evaluated against H460 and H23 cancer cell lines, which express high levels of Bcl-2 and Mcl-1, respectively (Table 2; Brien et al., 2007; Voortman et al., 2007; Li et al., 2008; Placzek et al., 2010). Consistent with the FP data, compound (−) BI97D6 also displayed potent efficacy in inhibiting cell growth in human cancer H460 and H23 cell lines (Ferreira et al., 2000; Voortman et al., 2007; Li et al., 2008) in a 3-day ATP-Lite assay, with EC50 values of 170 ± 23 and 140 ± 2 nM, respectively (Table 2 and Figure A2A in Appendix). (−) and (+) atropisomers of ApoG2 also displayed strong inhibitory properties against Bcl-XL, Bcl-2, Mcl-1, and Bfl-1 (Tables 1 and 2). Although (−) BI97D6 and (−) ApoG2 have comparable inhibitory properties against Bcl-2, Mcl-1, and Bfl-1 in FP assays (Table 1), (−) BI97D6 are approximately two to four times more potent than (−) ApoG2 in inhibiting cell growth in human cancer H460 and H23 cell lines (Table 2).
We further evaluated the ability of (−) and (+) atropisomers (BI97D6 and ApoG2) to induce apoptosis of the human BP3 cell line (which express high levels of Bfl-1 and Mcl-1), which originates from a human diffuse large B-cell lymphoma (DLBCL; Brien et al., 2007; Wei et al., 2009b). For these assays, we used a 1-day Annexin V-FITC and propidium iodide (PI) double staining, followed by flow-cytometry analysis (Table 2). All compounds effectively induced apoptosis of the BP3 cell line in a dose-dependent manner (Table 2). In particular, compound (−) BI97D6 was effective with an IC50 value of 3.55 ± 0.29 μM, which was approximately 2.5 times more potent than its (+) atropisomer BI97D6 (EC50 = 8.57 ± 0.28 μM; Table 2). The mRNA ratio of Bfl-1, Bcl-XL, and Mcl-1 is approximately 10:3:1 in BP3 cell lines (Brien et al., 2007). However, we determined that BP3 cells express high levels of both Bfl-1 and Mcl-1 by Western blot analysis (Wei et al., 2009a). In agreement with these observations, the potent dual Bcl-XL and Bcl-2 antagonist ABT-737 (Oltersdorf et al., 2005) displayed no cytotoxic activity against BP3 cell lines, presumably because ABT-737 is not effective against Mcl-1 and Bfl-1 (Cory and Adams, 2005; Oltersdorf et al., 2005; Wesarg et al., 2007). Consistent with previous results obtained with human PC-3, H460, and H23 cancer cell lines, (−), (+) atropisomers of ApoG2 also induced apoptosis of the BP3 cell lines in a dose-dependent manner (Table 2). Again, compounds (−) BI97D6 are approximately four times more potent than (−) ApoG2 (EC50 = 14.25 ± 1.28 μM) in inducing apoptosis of the BP3 cell line (Table 2).
We next explored whether (−), (+), and (±) BI97D6 had cytotoxic properties against wild-type mouse embryonic fibroblast cells (MEF/WT) and transformed Bax/Bak double knockout MEF cells (MEF/DKO) in which anti-apoptotic Bcl-2 family proteins lack a cytoprotective phenotype (Wei et al., 2001). Compound (−) BI97D6 displayed slight toxicity in MEF/DKO cells at 30 μM (Figure 3A) while it killed almost 76 ± 3% MEF/WT cells at the same concentration (Figure 3A), implying that the compound (−) BI97D6 only displayed minor off-target effects. Although Compound (+) BI97D6 has similar cytotoxicity in MEF/DKO cells at 3−30 μM compared to (−) BI97D6, it is less potent than its (−) isomer and killed 66 ± 5% MEF/WT cells at 30 μM (Figure A2B in Appendix). (±) BI97D6 displayed similar cytotoxic properties against MEF/WT and MEF/DKO compared to (+) BI97D6 (Figure A2C in Appendix).
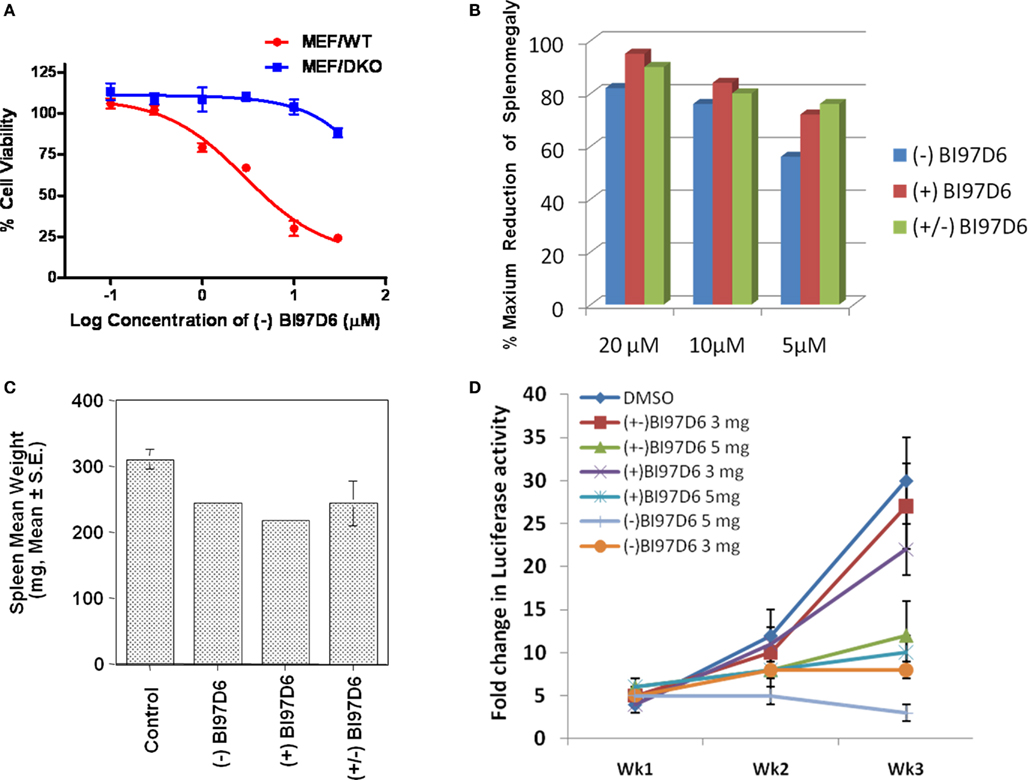
Figure 3. (A) Mouse embryonic fibroblast cells with wild-type (MEF/WT; red dot) or bax−/−bak−/− double knockout (MEF/DKO, blue square) genotypes were treated with (−) BI97D6 at various concentrations using ATP-LITE assay. (B–D) Characterization of (−), (+), and (±) BI97D6 in vivo. (B) Dose-dependent effects of (−) BI97D6 (blue), (+) BI97D6 (red), and (±) BI97D6 (green) on shrinkage of Bcl-2-transgenic mice spleen at a single intraperitoneal injection (i.p.) dose of 20, 10, and 5 μmol/kg, respectively. All shrinkage data are reported as percentage of maximum reduction of mice spleen size. (C) Effects of compounds (−), (+), and (±) BI97D6, respectively, on shrinkage of six B6Bcl-2-transgenic mice spleen at a single i.p. injection dose of 5 μmol/kg. (D) Human prostate cancer xenograft in athymic nude mice. PC-3-Luc cells (1 × 106) were injected s.c. in 100 μL of PBS in the left flank of male athymic nude mice (NCRnu/nu, 4 weeks old, 20 g body weight). After establishing visible tumors of ∼75-mm3, compounds were injected intraperitoneally (i.p.) every 2 days for a total of nine injections. Seven treatment groups were established for the experiment, i.e., DMSO only (blue dot), 3 mg/kg of (±) BI97D6 (red square), 5 mg/kg of (±) BI97D6 (green up triangle), 3 mg/kg of (+) BI97D6 (purple × ), 5 mg/kg of (+) BI97D6 (deep blue star), 3 mg/kg of (−) BI97D6 (orange dot), and 5 mg/kg of (−) BI97D6 (light blue cross). A minimum of five animals was used per experimental condition. For in vivo imaging of tumors, the mice were anesthetized and injected i.p. with 150 mg/kg luciferin and light emitted from each tumor was determined using a Xenogen system with CCD camera with an integration time of 1 min. Luminescence measurements were made using Living Image software (version 2.50.1; Xenogen).
In order to test the pharmacological properties of compounds (−), (+), and (±) BI97D6, we determined their in vitro plasma stability, microsomal stability, cell membrane permeability, and chemical stability (Table 3). From these studies, we could conclude that compounds (−), (+), and (±) BI97D6 displayed good rat plasma stability and only degraded 2.4 ± 0.8, 5.5 ± 0.3, and 1.0 ± 0.5%, respectively, after 1 h incubation in rat plasma. By comparison, (±) ApoG2 degraded 23% after 40 min incubation in rat plasma (Wei et al., 2010a). In addition, compounds (−), (+), and (±) BI97D6 demonstrated relatively good microsomal stability and degraded 20.3 ± 2.0, 23.7 ± 1.5, and 19.3 ± 5.4%, respectively, after 1 h incubation in rat microsomal preparations, whereas (±) ApoG2 degraded 53% after 40 min incubation in rat microsomal preparations (Wei et al., 2010a). Racemic BI97D6 and its (+), (−) isomers displayed similar cell membrane permeability (Table 3). The chemical stability of solid compounds (−), (+), and (±) BI97D6 was also evaluated at room temperature. Although those compounds have a potentially unstable polyphenol scaffold with four hydroxyl groups, these compounds showed excellent chemical stability (Table 3). In particular, compounds (−) BI97D6 and (+) BI97D6 were only 5 ± 0.5% degraded after 100 days at room temperature (Table 3), presumably because electron withdrawing ketone groups stabilized the polyphenol scaffold (Figure 1B).
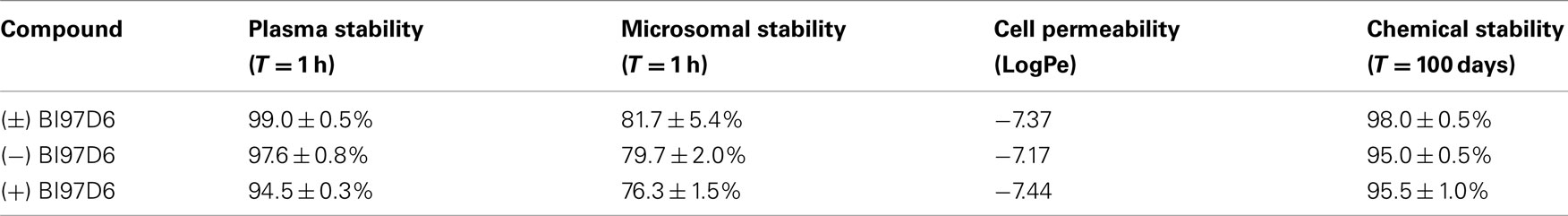
Table 3. Plasma stability, microsomal stability, cell permeability, and chemical stability of (±), (−), and (+) BI97D6 atropisomers.
Taken together, these observations suggested that unlike currently available antagonists (van de Donk et al., 2003; Oltersdorf et al., 2005), our compounds (−) BI97D6, (+) BI97D6, and (±) BI97D6 were effective in inhibiting several anti-apoptotic Bcl-2 proteins, and therefore were expected to display in vivo efficacy against a variety of in vivo models that relied on different Bcl-2 proteins for growth and progression (Kitada et al., 2008; Dash et al., 2010; Placzek et al., 2010). To test this hypothesis, we selected two different in vivo models: a Bcl-2-transgenic mouse model and a prostate cancer xenograft model (Kitada et al., 2008; Dash et al., 2010). B-cells of B6 transgenic mice overexpressed human Bcl-2 proteins and accumulated in the spleen resulting of a measurable weight enlargement. Because we had determined that the spleen weight was highly consistent in age- and sex-matched Bcl-2-transgenic mice, varying by only ±2% among control Bcl-2 mice (Kitada et al., 2008), the spleen weight was used as an end-point for assessing in vivo activity of anti-Bcl-2 compounds (Kitada et al., 2008). B6Bcl-2-transgenic mice were treated with compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 in doses of 5, 10, and 20 μmol/kg (i.p.), respectively (Figure 3B). In agreement with our in vitro and cell data, compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 induced significant spleen weight reduction in mice in a dose-dependent manner. In particular, compound (−) BI97D6 induced 27, 38, and 41% spleen weight reduction in a single-dose of 5, 10, and 20 μmol/kg, respectively (Figure 3B). Since the maximum spleen shrinkage would be no more than 50% in this experimental model (Kitada et al., 2008), compound (−) BI97D6 induced near 54, 76, and 82% maximal biological activity. Given that compounds (−), (+), and (±) BI97D6 has comparable binding affinity against Bcl-2 in FP assays (Table 1), compounds (+) and (±) BI97D6 has similar activities as (−) BI97D6 and induced 81, 77% maximal biological activity in a single-dose of 10 μmol/kg, respectively (Figure 3B). However, compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 also exhibited toxicity profiles that were more pronounced at higher doses. For instance, mice treated with (−) BI97D6 in a dose of 10 and 20 μmol/kg displayed mild to moderate hepato-toxicity, respectively, whereas mice treated with (−) BI97D6 in a dose of 5 μmol/kg displayed no evident sign of toxicity. To confirm the results of the single transgenic mouse experiment at a dose of 5 μmol/kg, we next evaluated the in vivo activity of compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 in groups of six B6Bcl-2-transgenic mice each at a dose of 5 μmol/kg. Consistent with the single mouse experiment, compounds (−), (±), and (+) BI97D6 treatment resulted in 47 ± 2, 45 ± 7, and 52 ± 2%, respectively, maximal reduction of spleen weight compared to the control group of six mice at a dose of 5 μmol/kg (Figure 3C). All mice tolerated the treatment well, with no evident signs of toxicity.
As anticipated earlier, we and others had recently realized the importance of Mcl-1 inhibition in designing Bcl-2 antagonist (van de Donk et al., 2003; Oltersdorf et al., 2005; Placzek et al., 2010; Wei et al., 2010a,b; Dash et al., 2011). For example, the potent Bcl-XL/Bcl-2 antagonist ABT-737 and the Bcl-2 antisense Genasense (Genta) were not effective against cancer cells that overexpressed Mcl-1 (van de Donk et al., 2003; Oltersdorf et al., 2005; Placzek et al., 2010). Therefore, to further examine the therapeutic potential of our molecules as single agents against tumors, compounds (−) BI97D6 and (+) BI97D6 were evaluated side by side with compound (±) BI97D6 in a prostate cancer xenograft using PC-3 cell line in which Mcl-1 and Bcl-XL were overexpressed (Placzek et al., 2010). A quantity of 1 × 106 PC-3 cells were injected subcutaneously in the left flank of male athymic nude mice, and the tumors were allowed to grow to an average size of ≈75 mm3 prior to initiation of therapy. Compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 were administrated (i.p.) every 2 days (total of nine injections) at two doses of 3 and 5 mg/kg (5 and 8 μmol/kg), respectively. All tested compounds, (±) BI97D6, (−) BI97D6, and (+) BI97D6, displayed a marked inhibitory effect of tumor size compared with the control group, particularly at the dose of 5 mg/kg (Figure 3D; Figure A2D in Appendix). In fact, the most potent compound (−) BI97D6 induced near complete inhibition of tumor growth at the dose of 5 mg/kg compared with the control group (Figure 3D). Since (−) BI97D6 displayed better activities against Bcl-XL and Mcl-1 in FP and PC-3 cell assays compared to its (+) isomer, the (−) atropisomer BI97D6 displayed better PC-3 tumor growth inhibitory effect compared to its (+) atropisomer at both doses of 3 and 5 mg/kg (Figure 3D). All mice tolerated the treatment well with no apparent signs of toxicity in this in vivo assay. The most potent (−) BI97D6 is currently undergoing pharmacokinetic and toxicity studies to decide whether it has scientific merit for further development as a new apoptosis-based cancer drug.
Conclusions
In summary, (−) and (+) atropisomers of compounds BI97D6 and ApoG2 were synthesized and evaluated in a variety of in vitro and in vivo assays. The optically pure compound (−) BI97D6 was found to bind to Bcl-XL, Bcl-2, Mcl-1, and Bfl-1 with IC50 values of 76 ± 5, 31 ± 2, 25 ± 8, and 122 ± 28 nM, respectively in FP assays. The compound also potently inhibited growth in culture of the PC-3 human prostate cancer and H23 human lung cancer cell lines with EC50 values of 0.22 ± 0.08 and 0.14 ± 0.02 μM, respectively. Compound (−) BI97D6 effectively induced apoptosis of the BP3 human lymphoma cell line in a dose-dependent manner and potently killed MEF/WT cell while it showed little cytotoxicity against MEF bax−/−/bak−/− cells, suggesting that it killed cancers cells predominantly via the intended mechanism. Finally, (−) BI97D6 demonstrated favorable in vitro ADME properties and superior in vivo efficacy in transgenic mice, in which Bcl-2 is overexpressed in splenic B-cells and further demonstrated greater single-agent antitumor efficacy in a prostate cancer mouse xenograft model at the dose of 5 mg/kg. Given the critical roles of anti-apoptotic Bcl-2 family proteins in tumorigenesis, chemoresistance, and the potent inhibitory effect of (−) BI97D6 against anti-apoptotic Bcl-2 family proteins, we conclude that the reported (−) BI97D6 represent a viable drug candidate for the development of novel apoptosis-based cancer therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank NIH (Grant CA 149668 to Maurizio Pellecchia) for financial support.
References
Adams, J. M., and Cory, S. (1998). The Bcl-2 protein family: arbiters of cell survival. Science 281, 1322–1326.
Arnold, A. A., Aboukameel, A., Chen, J., Yang, D., Wang, S., Al-Katib, A., and Mohammad, R. M. (2008). Preclinical studies of apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in follicular small cleaved cell lymphoma model. Mol. Cancer 7, 20.
Becattini, B., Kitada, S., Leone, M., Monosov, E., Chandler, S., Zhai, D., Kipps, T. J., Reed, J. C., and Pellecchia, M. (2004). Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem. Biol. 11, 389–395.
Brien, G., Trescol-Biemont, M. C., and Bonnefoy-Berard, N. (2007). Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene 26, 5828–5832.
Bruncko, M., Oost, T. K., Belli, B. A., Ding, H., Joseph, M. K., Kunzer, A., Martineau, D., Mcclellan, W. J., Mitten, M., Ng, S. C., Nimmer, P. M., Oltersdorf, T., Park, C. M., Petros, A. M., Shoemaker, A. R., Song, X., Wang, X., Wendt, M. D., Zhang, H., Fesik, S. W., Rosenberg, S. H., and Elmore, S. W. (2007). Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 50, 641–662.
Cory, S., and Adams, J. M. (2005). Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell 8, 5–6.
Coward, L., Gorman, G., Noker, P., Kerstner-Wood, C., Pellecchia, M., Reed, J. C., and Jia, L. (2006). Quantitative determination of apogossypol, a pro-apoptotic analog of gossypol, in mouse plasma using LC/MS/MS. J. Pharm. Biomed. Anal. 42, 581–586.
Dash, R., Azab, B., Quinn, B. A., Shen, X., Wang, X. Y., Das, S. K., Rahmani, M., Wei, J., Hedvat, M., Dent, P., Dmitriev, I. P., Curiel, D. T., Grant, S., Wu, B., Stebbins, J. L., Pellecchia, M., Reed, J. C., Sarkar, D., and Fisher, P. B. (2011). Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc. Natl. Acad. Sci. U.S.A. 108, 8785–8790.
Dash, R., Richards, J. E., Su, Z. Z., Bhutia, S. K., Azab, B., Rahmani, M., Dasmahapatra, G., Yacoub, A., Dent, P., Dmitriev, I. P., Curiel, D. T., Grant, S., Pellecchia, M., Reed, J. C., Sarkar, D., and Fisher, P. B. (2010). Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10-related cytokine. Cancer Res. 70, 5034–5045.
Day, C. L., Chen, L., Richardson, S. J., Harrison, P. J., Huang, D. C., and Hinds, M. G. (2005). Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem. 280, 4738–4744.
Degterev, A., Lugovskoy, A., Cardone, M., Mulley, B., Wagner, G., Mitchison, T., and Yuan, J. (2001). Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat. Cell Biol. 3, 173–182.
Eldridge, M. D., Murray, C. W., Auton, T. R., Paolini, G. V., and Mee, R. P. (1997). Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 11, 425–445.
Ferreira, C. G., Span, S. W., Peters, G. J., Kruyt, F. A., and Giaccone, G. (2000). Chemotherapy triggers apoptosis in a caspase-8-dependent and mitochondria-controlled manner in the non-small cell lung cancer cell line NCI-H460. Cancer Res. 60, 7133–7141.
Gross, A., Mcdonnell, J. M., and Korsmeyer, S. J. (1999). BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911.
Hu, Z. Y., Zhu, X. F., Zhong, Z. D., Sun, J., Wang, J., Yang, D., and Zeng, Y. X. (2008). ApoG2, a novel inhibitor of antiapoptotic Bcl-2 family proteins, induces apoptosis and suppresses tumor growth in nasopharyngeal carcinoma xenografts. Int. J. Cancer 123, 2418–2429.
Johnstone, R. W., Ruefli, A. A., and Lowe, S. W. (2002). Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153–164.
Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997). Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748.
Katsumata, M., Siegel, R. M., Louie, D. C., Miyashita, T., Tsujimoto, Y., Nowell, P. C., Greene, M. I., and Reed, J. C. (1992). Differential effects of Bcl-2 on T and B cells in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 89, 11376–11380.
Kitada, S., Kress, C. L., Krajewska, M., Jia, L., Pellecchia, M., and Reed, J. C. (2008). Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2-transgenic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048). Blood 111, 3211–3219.
Kitada, S., Leone, M., Sareth, S., Zhai, D., Reed, J. C., and Pellecchia, M. (2003). Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J. Med. Chem. 46, 4259–4264.
Lee, E. F., Czabotar, P. E., Smith, B. J., Deshayes, K., Zobel, K., Colman, P. M., and Fairlie, W. D. (2007). Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 14, 1711–1713.
Li, J., Viallet, J., and Haura, E. B. (2008). A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 61, 525–534.
Lin, J., Wu, Y.-J., Yang, D.-J., and Zhao, Y.-Q. (2009). Effect of apogossypolone on induction apoptosis in multiple myeloma cells and its mechanisms. Zhongguo Shi Yan Xue Ye Xue Za Zhi 17, 92–98.
Meng, Y., Tang, W., Dai, Y., Wu, X., Liu, M., Ji, Q., Ji, M., Pienta, K., Lawrence, T., and Xu, L. (2008). Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. Cancer Ther. 7, 2192–2202.
Mi, J. X., Wang, G. F., Wang, H. B., Sun, X. Q., Ni, X. Y., Zhang, X. W., Tang, J. M., and Yang, D. J. (2008). Synergistic antitumoral activity and induction of apoptosis by novel pan Bcl-2 proteins inhibitor apogossypolone with adriamycin in human hepatocellular carcinoma. Acta Pharmacol. Sin. 29, 1467–1477.
Mohammad, R. M., Wang, S., Aboukameel, A., Chen, B., Wu, X., Chen, J., and Al-Katib, A. (2005). Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(-)-gossypol] against diffuse large cell lymphoma. Mol. Cancer Ther. 4, 13–21.
Muchmore, S. W., Sattler, M., Liang, H., Meadows, R. P., Harlan, J. E., Yoon, H. S., Nettesheim, D., Chang, B. S., Thompson, C. B., Wong, S. L., Ng, S. L., and Fesik, S. W. (1996). X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381, 335–341.
Oltersdorf, T., Elmore, S. W., Shoemaker, A. R., Armstrong, R. C., Augeri, D. J., Belli, B. A., Bruncko, M., Deckwerth, T. L., Dinges, J., Hajduk, P. J., Joseph, M. K., Kitada, S., Korsmeyer, S. J., Kunzer, A. R., Letai, A., Li, C., Mitten, M. J., Nettesheim, D. G., Ng, S., Nimmer, P. M., O’connor, J. M., Oleksijew, A., Petros, A. M., Reed, J. C., Shen, W., Tahir, S. K., Thompson, C. B., Tomaselli, K. J., Wang, B., Wendt, M. D., Zhang, H., Fesik, S. W., and Rosenberg, S. H. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681.
Placzek, W. J., Wei, J., Kitada, S., Zhai, D., Reed, J. C., and Pellecchia, M. (2010). A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 1, 1–9.
Ramjaun, A. R., Tomlinson, S., Eddaoudi, A., and Downward, J. (2007). Upregulation of two BH3-only proteins, Bmf and Bim, during TGF beta-induced apoptosis. Oncogene 26, 970–981.
Reed, J. C. (1997). Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv. Pharmacol. (San Diego, Calif.) 41, 501–532.
Sattler, M., Liang, H., Nettesheim, D., Meadows, R. P., Harlan, J. E., Eberstadt, M., Yoon, H. S., Shuker, S. B., Chang, B. S., Minn, A. J., Thompson, C. B., and Fesik, S. W. (1997). Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986.
Sun, J., Li, Z.-M., Hu, Z.-Y., Zeng, Z.-L., Yang, D.-J., and Jiang, W.-Q. (2009). Apogossypolone inhibits cell growth by inducing cell cycle arrest in U937 cells. Oncol. Rep. 22, 193–198.
Teschner, M., Henn, C., Vollhardt, H., Reiling, S., and Brickmann, J. (1994). Texture mapping: a new tool for molecular graphics. J. Mol. Graph. 12, 98–105.
van de Donk, N. W., Kamphuis, M. M., Van Dijk, M., Borst, H. P., Bloem, A. C., and Lokhorst, H. M. (2003). Chemosensitization of myeloma plasma cells by an antisense-mediated downregulation of Bcl-2 protein. Leukemia 17, 211–219.
Voortman, J., Checinska, A., Giaccone, G., Rodriguez, J. A., and Kruyt, F. A. (2007). Bortezomib, but not cisplatin, induces mitochondria-dependent apoptosis accompanied by up-regulation of noxa in the non-small cell lung cancer cell line NCI-H460. Mol. Cancer Ther. 6, 1046–1053.
Wang, G., Nikolovska-Coleska, Z., Yang, C.-Y., Wang, R., Tang, G., Guo, J., Shangary, S., Qiu, S., Gao, W., Yang, D., Meagher, J., Stuckey, J., Krajewski, K., Jiang, S., Roller, P. P., Abaan, H. O., Tomita, Y., and Wang, S. (2006a). Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J. Med. Chem. 49, 6139–6142.
Wang, S., Nikolovska-Coleska, Z., Yang, C.-Y., and Chen, J. (2006b). Apogossypolone and the Uses Thereof. PCT Int. Appl, WO 2006050447 A2 20060511.
Wang, J. L., Liu, D., Zhang, Z. J., Shan, S., Han, X., Srinivasula, S. M., Croce, C. M., Alnemri, E. S., and Huang, Z. (2000). Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. U.S.A. 97, 7124–7129.
Wang, S., and Yang, D. (2004). Small Molecular Antagonists of Bcl-2 Family Proteins. US patent applications series no. 2004/0214902 A1. 10/729,156.
Wei, J., Kitada, S., Rega, M. F., Emdadi, A., Yuan, H., Cellitti, J., Stebbins, J. L., Zhai, D., Sun, J., Yang, L., Dahl, R., Zhang, Z., Wu, B., Wang, S., Reed, T. A., Lawrence, N., Sebti, S., Reed, J. C., and Pellecchia, M. (2009a). Apogossypol derivatives as antagonists of antiapoptotic Bcl-2 family proteins. Mol. Cancer Ther. 8, 904–913.
Wei, J., Kitada, S., Rega, M. F., Stebbins, J. L., Zhai, D., Cellitti, J., Yuan, H., Emdadi, A., Dahl, R., Zhang, Z., Yang, L., Reed, J. C., and Pellecchia, M. (2009b). Apogossypol derivatives as pan-active inhibitors of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem. 52, 4511–4523.
Wei, J., Rega, M. F., Kitada, S., Yuan, H., Zhai, D., Risbood, P., Seltzman, H. H., Twine, C. E., Reed, J. C., and Pellecchia, M. (2009c). Synthesis and evaluation of apogossypol atropisomers as potential Bcl-xL antagonists. Cancer Lett. 273, 107–113.
Wei, J., Kitada, S., Stebbins, J. L., Placzek, W., Zhai, D., Wu, B., Rega, M. F., Zhang, Z., Cellitti, J., Yang, L., Dahl, R., Reed, J. C., and Pellecchia, M. (2010a). Synthesis and biological evaluation of apogossypolone derivatives as pan-active inhibitors of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem. 53, 8000–8011.
Wei, J., Stebbins, J. L., Kitada, S., Dash, R., Placzek, W., Rega, M. F., Wu, B., Cellitti, J., Zhai, D., Yang, L., Dahl, R., Fisher, P. B., Reed, J. C., and Pellecchia, M. (2010b). BI-97C1, an optically pure apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem. 53, 4166–4176.
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., Macgregor, G. R., Thompson, C. B., and Korsmeyer, S. J. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730.
Wesarg, E., Hoffarth, S., Wiewrodt, R., Kroll, M., Biesterfeld, S., Huber, C., and Schuler, M. (2007). Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int. J. Cancer 121, 2387–2394.
Appendix
General Synthetic Procedures
Unless otherwise indicated, all reagents and anhydrous solvents (CH2Cl2, THF, diethyl ether, etc.,) were obtained from commercial sources and used without purification. All reactions were performed in oven-dried glassware. All reactions involving air or moisture sensitive reagents were performed under a nitrogen atmosphere. Silica gel or reverse phase chromatography was performed using prepacked silica gel or C-18 cartridges (RediSep), respectively. All final compounds were purified to >95% purity, as determined by a HPLC Breeze from Waters Co. using an Atlantis T3 3 μM 4.6 mm × 150 mm reverse phase column. Method A: The eluant was a linear gradient with a flow rate of 1 mL/min from 50% A and 50% B to 5% A and 95% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: ACN with 0.1% TFA). Compounds were detected at λ = 254 nm. 1H NMR spectra were recorded on Varian 300 or Bruker 600 MHz instruments. Chemical shifts are reported in ppm (δ) relative to 1H (Me4Si at 0.00 ppm). Coupling constant (J) are reported in Hz throughout. Mass spectral data were acquired on Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for high resolution.
The syntheses of compounds (±), (−), and (+) BI97D6 and ApoG2 have been previously described by us (Wei et al., 2009). Racemic (±) BI97D6 and ApoG2 were resolved to afford pure (−) and (+) BI97D6 and ApoG2 atropisomers, respectively, using a RegisCell 5 μM 250 mm × 10 mm normal phase semi-preparative chiral column from Regis Technologies Inc. Results were analyzed using 5 μM 250 mm × 4.6 mm normal phase analytical chiral column. Mobile phase A was 0.1% TFA in 2-propanol and mobile phase B was 0.1% TFA in hexane. Flow rate was 4 mL/min for preparative and 1 mL/min for analytical analysis, respectively. The run duration was 25 min. The separation of (±) ApoG2 and BI97D6 were accomplished using different ratio of phase A and B (Methods A, B, and C), respectively. Method A: constant 8% A and 92% B in 15 min followed by 10 min at 100% A. Method B: constant 30% A and 70% B in 15 min followed by 10 min at 100% A. Method C: constant 25% A and 75% B in 15 min followed by 10 min at 100% A.
(−) ApoG2: (−)-6,6′,7,7′-tetrahydroxy-5,5′-diisopropyl-3,3′-dimethylo-2,2′-binaphthyl-1,1′,4,4′-tetraone 1H NMR (600 MHz, (CD3)2SO) δ 10.942 (s, 2H), 9.426 (s, 2H), 7.322 (s, 2H), 4.312 (m, 2H), 1.837 (s, 6H), 1.359 (d, J = 6.6 Hz, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 187.10, 182.50, 150.91, 149.53, 147.60, 137.78, 137.11, 126.24, 125.01, 111.01, 27.07, 20.50, 20.35, 15.00. HPLC purity 95.0%, tR = 10.43 min. HRMS calcd for C28H26O8 491.1700 (M + H), found 491.1720.
(−) BI97D6: (−)-5,5′-dibenzyl-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone 1H NMR (600 MHz, CD3OD) δ 7.448 (s, 2H), 7.224 (d, J = 7.8 Hz, 4H), 7.178 (t, J1 = 7.8 Hz, J2 = 7.2 Hz, 4H), 7.084 (t, J1 = J2 = 7.2 Hz, 4H), 4.643 (dd, J1 = 14.4 Hz, J2 = 13.8 Hz, 4H), 1.867 (s, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.44, 182.61, 150.04, 149.83, 146.59, 140.95, 138.56, 130.07, 128.66, 128.39, 126.52, 125.82, 123.76, 111.74, 31.74, 14.84. HPLC purity 98.0%, tR = 10.18 min. HRMS calcd for C36H26O8 587.1700 (M + H), found 587.1723.
(+) ApoG2: (+)-6,6′,7,7′-tetrahydroxy-5,5′-diisopropyl-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone 1H NMR (600 MHz, (CD3)2SO) δ 10.941 (s, 2H), 9.428 (s, 2H), 7.322 (s, 2H), 4.312 (m, 2H), 1.881 (s, 6H), 1.358 (d, J = 6.6 Hz, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 187.10, 182.50, 150.91, 149.53, 147.60, 137.78, 137.11, 126.24, 125.01, 111.02, 27.07, 20.50, 20.35, 15.00. HPLC purity 94.9%, tR = 10.36 min. HRMS calcd for C28H26O8 491.1700 (M + H), found 491.1706.
(+) BI97D6: (+)-5,5′-dibenzyl-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone. 1H NMR (600 MHz, CD3OD) δ 7.448 (s, 2H), 7.225 (d, J = 7.2 Hz, 4H), 7.178 (t, J1 = 7.8 Hz, J2 = 7.2 Hz, 4H), 7.084 (t, J1 = J2 = 7.2 Hz, 4H), 4.642 (dd, J1 = 14.4 Hz, J2 = 13.8 Hz, 4H), 1.867 (s, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.44, 182.61, 150.04, 149.84, 146.59, 140.95, 138.56, 130.07, 128.66, 128.39, 126.52, 125.82, 123.75, 111.75, 31.74, 14.84. HPLC purity 98.3%, tR = 10.15 min. HRMS calcd for C36H26O8 587.1700 (M + H), found 587.1719.
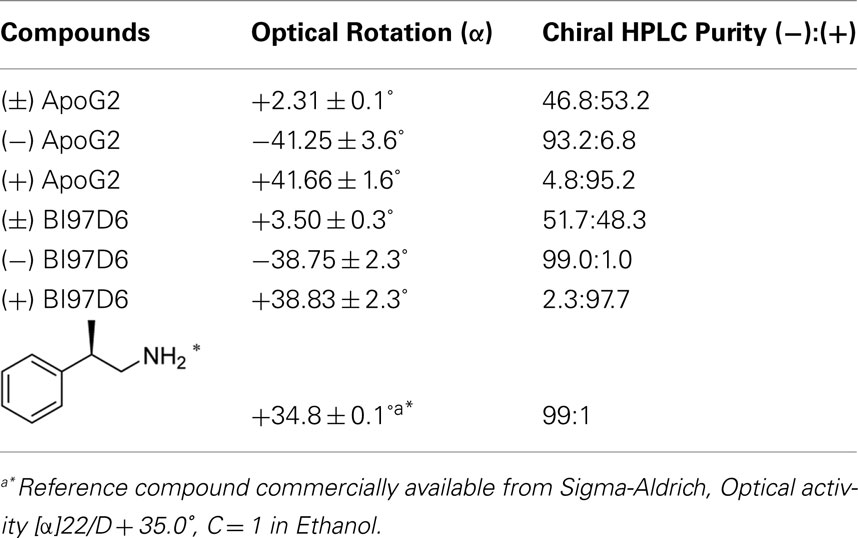
Table A1. Optical and chiral HPLC purity of (±), (−), and (+) ApoG2 and BI97D6.
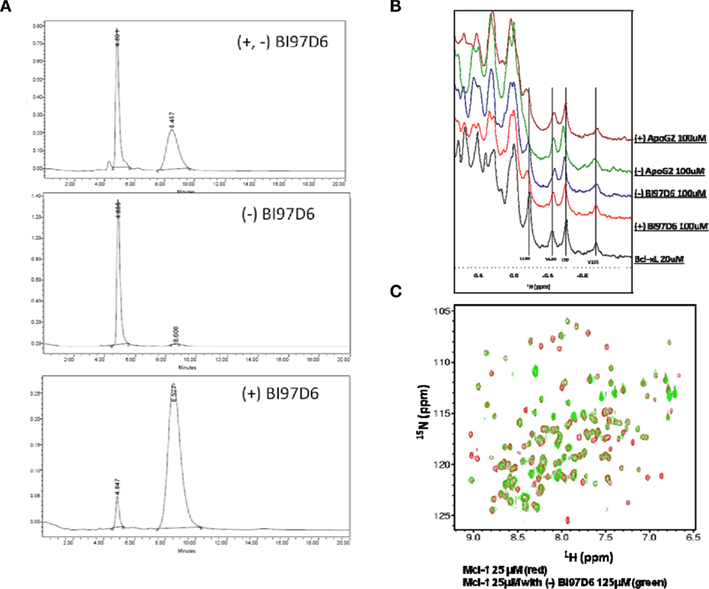
Figure A1. (A) HPLC purity of (±), (−), and (+) BI97D6 using normal phase chiral column chromatography. (B) NMR binding studies. Aliphatic region of the 1H-NMR spectrum of Bcl-XL (20 μM, black) and Bcl-XL in the presence of compound (+) ApoG2 (100 μM, pink), (−) ApoG2 (100 μM, Green), (−) BI97D6 (100 μM, blue), and (+) BI97D6 (100 μM, red). (C) Superposition of 2D [15N,1H]-TROSY spectra of hMcl-1 (25 μM; red) before and after addition of compound (−) BI97D6 (125 μM; green).
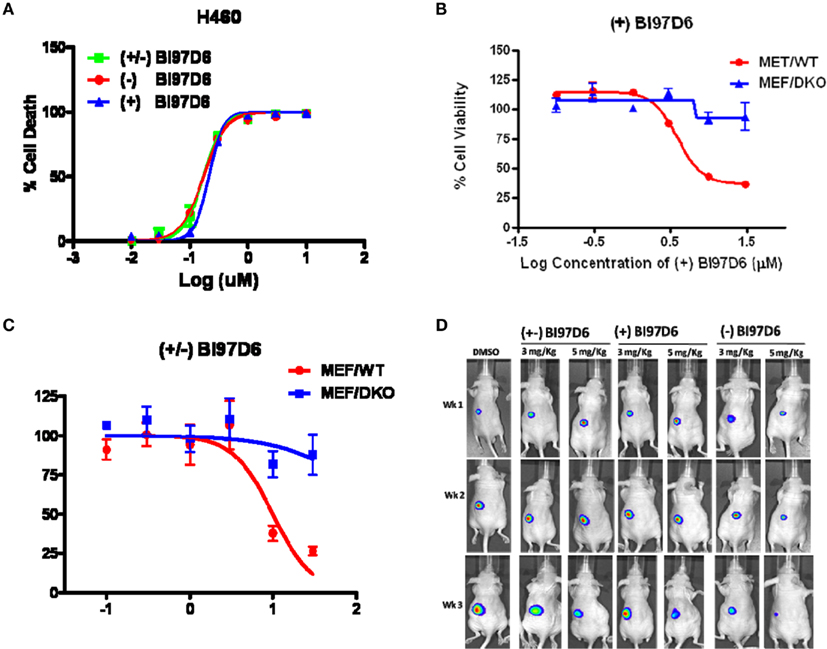
Figure A2. (A) Inhibition of cell growth by (−) BI97D6 (red dot), (±) (green square), and (+) BI97D6 (blue up triangle) in the H460 human lung cancer cell line. Cells were treated for 3 days and cell viability was evaluated using ATP-LITE assay. (B) Mouse embryonic fibroblast cells with wild-type (MEF/WT; red dot) or bax−/−bak−/− double knockout (MEF/DKO, blue up triangle) genotypes were treated with compounds (+) BI97D6 at various concentrations using ATP-LITE assay. (C) Mouse embryonic fibroblast cells with wild-type (MEF/WT; red dot) or bax−/−bak−/− double knockout (MEF/DKO, blue up triangle) genotypes were treated with compounds (±) BI97D6 at various concentrations using ATP-LITE assay. (D) Tumor xenografts from PC-3-Luc cells were established in athymic nude mice on the left flanks. After establishing visible tumors of ∼75-mm3, requiring ∼5–6 days, intraperitoneal (i.p.) injections of DMSO or compounds (±) BI97D6, (−) BI97D6, and (+) BI97D6 (3 or 5 mg/kg) were given every 2 days (total of nine injections). For in vivo imaging the mice were anesthetized and injected i.p. with 150 mg/kg luciferin and light emitted from each tumor determined in a Xenogen system with CCD camera with an integration time of 1 min.
Keywords: apoptosis, anti-apoptotic Bcl-2, cancer, apogossypolone, 5, 5′ apogossypolone derivatives
Citation: Wei J, Stebbins JL, Kitada S, Dash R, Zhai D, Placzek WJ, Wu B, Rega MF, Zhang Z, Barile E, Yang L, Dahl R, Fisher PB, Reed JC and Pellecchia M (2011) An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic Bcl-2 family proteins. Front. Oncol. 1:28. doi: 10.3389/fonc.2011.00028
Received: 13 June 2011; Accepted: 30 August 2011;
Published online: 30 September 2011.
Edited by:
William Douglas Figg, National Cancer Institute, USAReviewed by:
Douglas K. Price, National Institutes of Health, USAYongmei Song, Peking Union Medical College, China
Copyright: © 2011 Wei, Stebbins, Kitada, Dash, Zhai, Placzek, Wu, Rega, Zhang, Barile, Yang, Dahl, Fisher, Reed and Pellecchia. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Maurizio Pellecchia, Infectious and Inflammatory Disease Center, Cancer Center, Sanford-Burnham Medical Research Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA. e-mail:bXBlbGxlY2NoaWFAc2FuZm9yZGJ1cm5oYW0ub3Jn