


- 1Department of Neurosurgery, Mackay Memorial Hospital, Taipei, Taiwan
- 2Graduate Institute of Injury Prevention and Control, Taipei Medical University, Taipei, Taiwan
- 3Institute of Molecular Medicine, National Cheng Kung University, Tainan, Taiwan
- 4Department of Cell Biology and Anatomy, Nation Cheng Kung University, Tainan, Taiwan
- 5Department of Pathology, Nation Cheng Kung University, Tainan, Taiwan
- 6Advanced Optoelectronic Technology Center, National Cheng Kung University, Tainan, Taiwan
- 7Department of Neuroscience and Physiology, SUNY Upstate Medical University, Syracuse, NY, USA
- 8Department of Neurochemistry, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
Tumor suppressor p53 are frequently mutated in glioblastomas (GBMs) and appears to contribute, in part, to resistance to temozolomide (TMZ) and therapeutic drugs. WW domain-containing oxidoreductase WWOX (FOR or WOX1) is a proapoptotic protein and is considered as a tumor suppressor. Loss of WWOX gene expression is frequently seen in malignant cancer cells due to promoter hypermethylation, genetic alterations, and translational blockade. Intriguingly, ectopic expression of wild type WWOX preferentially induces apoptosis in human glioblastoma cells harboring mutant p53. WWOX is known to physically bind and stabilize wild type p53. Here, we provide an overview for the updated knowledge in p53 and WWOX, and postulate potential scenarios that wild type and mutant p53, or isoforms, modulate the apoptotic function of WWOX. We propose that triggering WWOX activation by therapeutic drugs under p53 functional deficiency is needed to overcome TMZ resistance and induce GBM cell death.
Mechanisms of Resistance to Temozolomide in Glioblastomas
Glioblastoma multiforme afflicts 12,500 new patients in the U.S. annually (Friedman et al., 2000; Stupp et al., 2009; Silber et al., 2012). Glioblastoma (GBM) is highly lethal, and the average survival expectancy is 14.6 months, and the overall 5-year survival rate for GBM is only 9.8% (Friedman et al., 2000; Stupp et al., 2009). High levels of resistance to current therapeutic modalities and cancer relapse are frequently seen in patients (Haar et al., 2012; Happold et al., 2012). The current standard therapy for GBM mainly includes maximum debulking surgery, radiation, and treatment with the monofunctional alkylating agent temozolomide (TMZ) (Friedman et al., 2000; Nishikawa, 2010). Multiple mechanisms are involved in the TMZ resistance, which may include cancer stem cells, microRNAs, drug efflux, DNA damage repair, tumor cells under hypoxia, histone deacetylation, epithelial-mesenchymal transition, STAT3 kinase, and many others (Haar et al., 2012; Happold et al., 2012; Johannessen and Bjerkvig, 2012; Kitange et al., 2012; Kohsaka et al., 2012; Zhang et al., 2012b).
Temozolomide induces generation of DNA lesions, including O6-methylguanine, N3-methyladenine, and N7-methylguanine (Goellner et al., 2011; Zhang et al., 2012b). The O6-methylguanine lesion is known to trigger autophagy, rather than apoptosis, to cause cell death (Kanzawa et al., 2003). Also, inhibition of antiapoptotic Bcl-2 by a pan-Bcl-2 inhibitor (−)-gossypol leads to autophagic death in gliomas and enhances the action of TMZ (Voss et al., 2010). However, a recent study demonstrated that TMZ-induced autophagy is pro-survival, and may block the eventual apoptosis in GBM cells (Knizhnik et al., 2013). Also, MAPO2 (C1orf201) gene participates in the O6-methylguanine lesion-induced apoptosis (Fujikane et al., 2012). MAPO2 gene encodes a novel 37-kDa protein. It is not determined whether this gene is involved in autophagy.
The O6-methylguanine lesion is a substrate for direct repair by O6-methylguanine-DNA methyltransferase (MGMT) (Pollack et al., 2006; Hegi et al., 2008; Fukushima et al., 2009; Zhang et al., 2012b). Without MGMT repair, O6-methylguanine initiates activation of mismatch repair-deficient (MMR) proteins or Rad3-related protein kinase that ultimately leads to apoptotic cell death (Caporali et al., 2004; Wang and Edelmann, 2006; Roos et al., 2007). High expression of MGMT or loss of MMR contributes significantly to TMZ resistance in many clinical cases (Pollack et al., 2006; Hegi et al., 2008; Sarkaria et al., 2008). The initiation of apoptotic signaling fails in the absence of the MMR system.
Sensitivity to TMZ is significantly associated with the methylation status of MGMT gene promoter in cells committed to differentiation (Villalva et al., 2012). An increase in MGMT gene promoter methylation, which blocks MGMT protein expression, prolongs cancer patient survival. Intriguingly, overexpressed microRNA-21 reduces Bax/Bcl-2 ratio and caspase-3 activity, thereby blocking TMZ-induced apoptosis (Shi et al., 2010). MicroRNA-21 is considered as a pro-survival factor for cancer cells (Li et al., 2012). Integrins play a role in the resistance of advanced cancers to radiotherapy and chemotherapy. α5β1 integrin negatively regulates p53 signaling, and the event induces glioma cell resistance to TMZ (Janouskova et al., 2012). α5β1 integrin is considered as a therapeutic target for high-grade brain tumors. The base excision repair enzyme alkylpurine-DNA-N-glycosylase (APNG), which repairs the cytotoxic lesions N3-methyladenine and N7-methylguanine, also participates in the TMZ resistance (Agnihotri et al., 2012). Upregulation of mitochondrial respiratory chain coupling to suppress the production of reactive oxygen species (ROS) regulated by cytochrome c oxidase contributes in part to TMZ resistance in gliomas (Oliva et al., 2011).
Reversal of TMZ resistance may be achieved by MGMT pseudosubstrates, O6-benzylguanine and lomeguatrib to sensitize tumors to TMZ (Zhang et al., 2012b). Methoxyamine-blocker of base excision repair contributes significantly to TMZ cytotoxicity particularly when O6-methylguanine adducts are repaired or tolerated (Goellner et al., 2011; Zhang et al., 2012b). Dual targeting of base excision repair and NAD(+) biosynthesis may reverse TMZ resistance in patients with resistant and recurrent GBM (Goellner et al., 2011). Interferon-β (IFN-β), levetiracetam (LEV), resveratrol, and valproic acid (VAP) increase the sensitivity of TMZ through MGMT-dependent or -independent mechanisms (Nakada et al., 2012). Resveratrol, a natural polyphenol, reverses TMZ resistance via an NF-κB-dependent mechanism (Huang et al., 2012). STAT3 inhibitor or STAT3 knockdown potentiates TMZ efficacy in resistant GBM cell lines (Kohsaka et al., 2012). Intratumoral hypoxia is common in GBMs and may be associated with the development of TMZ resistance. Induced hyperoxia can be utilized to reverse TMZ resistance in GBMs (Sun et al., 2012). Cancer stem cells are probably the key to failure in TMZ treatment. The concept of cancer stem cell survival from treatment with TMZ and other chemotherapeutic drugs has been more complicated than previously thought (Beier et al., 2011; Chen et al., 2012). CD133-positive cancer stem cells are expressed in both normal stem cells and cancer stem cells (Donovan and Pilkington, 2012). However, the role of CD133 as a marker for glioma cancer stem cells relative to its biological function has yet to be established.
WW Domain-Containing Oxidoreductase WWOX
Recently, tumor suppressors p53 and WWOX were shown to regulate the apoptosis of glioblastoma cells (Chiang et al., 2012). WW domain-containing oxidoreductase, known as WWOX, FOR, or WOX1, is encoded by human or mouse WWOX/Wwox gene. This gene is located in chromosome 16q23.3–24.1, an area known as the common fragile site FRA16D. The full-length WWOX protein is composed of two N-terminal WW domains, a C-terminal short-chain alcohol dehydrogenase/reductase (SDR) domain, and a proapoptotic C-terminal tail D3 (Chang et al., 2001, 2007, 2010; Aqeilan et al., 2004, 2007; Hong et al., 2007; Salah et al., 2012) (Figure 1). WWOX may act as an alternative receptor for sex steroid hormones, since its SDR domain possesses an NSYK motif capable of interacting with androgen and estrogen (Chang et al., 2005a; Su et al., 2012).
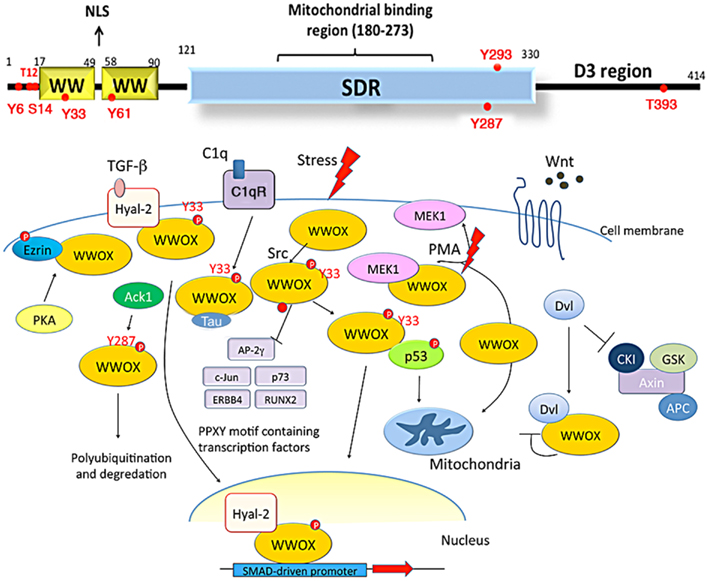
Figure 1. Schematic diagrams of WWOX structure and its role in signaling. WWOX contains two N-terminal WW domains, a C-terminal short-chain alcohol dehydrogenase/reductase (SDR) domain, and a D3 region. A nuclear localization signal (NLS) is in between the WW domains (Chang et al., 2003, 2007; Del Mare et al., 2011; Salah et al., 2012; Su et al., 2012; reviews). The SDR domain possesses a mitochondria-binding region. There are two conserved tyrosine phosphorylation sites, Tyr33 and Tyr287. Other phosphorylation sites predicted by NetPhos 2.0 Server are Tyr6, Thr12, Ser14, Tyr61, Tyr293, and Thr3B3, respectively. Upon stress stimulation, WWOX is phosphorylated in Tyr33 and binds p53. The WWOX/p53 complex then translocates to mitochondria or nucleus to induce apoptosis. WWOX also binds PPxY motif-containing transcription factors, such as RUNX2, c-Jun, and ERBB4, via its first WW domain, and the binding blocks the activity of these proteins by retaining them in the cytoplasm. WWOX binds Tau via its SDR domain. WWOX participates in the Wnt/β-catenin signal pathway by binding Dvl and inhibiting its nuclear import. Phorbol ester stimulates the dissociation of MEK/WWOX complex in Jurkat T cell, and the released WWOX translocates to the mitochondria for causing cell death. WWOX is recruited to the membrane area by association with Hyal-2 and phospho-Ezrin. Hyal-2 is an alternative receptor for TGF-β1. In response to TGF-β1, WWOX binds Hyal-2 and forms a complex, followed by relocating to the nucleus and enhancing the SMAD-driven promoter activity. Ack1, activated Cdc42-associated kinase 1; Hyal-2, hyaluronoglucosaminidase 2; CKI, casein kinase; GSK, glycogen synthase kinase; PKA, protein kinase A.
Expression of WWOX is either altered or lost from epigenetic modification in multiple malignant cancers, such as non-small cell lung carcinoma (Donati et al., 2007), hematopoietic malignancies (Ishii and Furukawa, 2004), gastric carcinoma (Aqeilan et al., 2004), pancreatic carcinoma (Kuroki et al., 2004), breast carcinoma (Guler et al., 2004; Aqeilan et al., 2007), and glioblastoma multiforme (Kosla et al., 2011). Restoration of WWOX gene prevents the growth of lung cancer (Fabbri et al., 2005), pancreatic cancer (Nakayama et al., 2008), and prostate cancer (Hong et al., 2009).
The first WW domain of WWOX may interact with proteins possessing a PPxY motif(s) such as AP-2γ, p73, ErbB4, Ezrin, SIMPLE, c-Jun, RUNX4, and many others (Chang et al., 2007; Salah et al., 2012; Su et al., 2012; Figure 1). Transiently overexpressed WWOX binds transcription factors AP-2, p73, and c-Jun and block their nuclear relocation in vitro, which suppresses cancer cell survival (Salah et al., 2012). In stark contrast, Wwox co-migrates with proapoptotic and pro-survival transcription factors to the nuclei of neurons upon sciatic nerve axotomy in rats (Li et al., 2009). No blocking of translocation of transcription factors to the nuclei by Wwox was observed.
Under stress conditions, WWOX is activated via phosphorylation at Tyr33, and binds proteins independently of the PPxY motif (Chang et al., 2001, 2003, 2005a,b, 2007, 2010). Activated WWOX physically interacts with serine 46-phosphorylated p53 (Figure 1), which stabilizes p53 and its apoptotic function (Chang et al., 2005b). Also, WWOX binds Disheveled proteins (Dvl), which are key components in Wnt/β-catenin signaling pathway (Figure 1). No PPxY motif is in Dvl. Transiently overexpressed WWOX sequesters Dvl-2 in the cytoplasm and thereby blocks Dvl-2-mediated TCF transcriptional activity (Bouteille et al., 2009).
Overexpressed WWOX induces apoptosis and inhibits proliferation of human hepatic carcinoma cells (Hu et al., 2012) and many cancer cell types (Chang et al., 2007, 2010; Salah et al., 2012; Su et al., 2012). WWOX enhances the cytotoxic function of tumor necrosis factor by down-regulating apoptosis inhibitor Bcl-2 and Bcl-xL and up-regulating apoptotic p53 (Chang et al., 2001). Also, WWOX mediates cell death synergistically with p53. Upon exposure to chemicals or environmental stress, such as UV irradiation and chemotherapeutic drugs, WWOX undergoes phosphorylation in Tyr33 and probably others sites, followed by relocating to mitochondria or nuclei for inducing apoptosis (Chang et al., 2007, 2010).
WWOX binds MEK in Jurkat T cells (Lin et al., 2011). Phorbol myristate acetate (PMA) dissociates the MEK/WWOX complex and induces WWOX to translocate to the mitochondria to induce apoptosis, whereas MEK relocates to the lipid raft. Inhibition of MEK activity increases TMZ-induced suppression of cancer cell growth (Holt et al., 2012). Ectopic expression of WWOX in A549 cells induces procaspase-3 and procaspase-9 activation and induces cytochrome C releasing from the mitochondria (Zhang et al., 2012a). Complement C1q induces ectopic WWOX phosphorylation in Try33 and leads to cell apoptosis independently of the classical complement activation pathway (Hong et al., 2009). A portion of WWOX is anchored in the membrane/cytoskeleton area via binding with hyaluronidase Hyal-2 (Hsu et al., 2009) and Phospho-Ezrin (Jin et al., 2006). Transforming growth factor β1 binds Hyal-2 as a cognate receptor to signal the formation of the WWOX/Hyal-2/Smad4 complex to relocate to the nucleus for enhancing the SMAD-driven promoter activity (Hsu et al., 2009).
p53 and Functions
Activated p53 mediates apoptosis, cell cycle arrest, senescence, DNA repair, or metabolism (Lane and Levine, 2010). The primary structures of p53 and its isoforms are depicted (Figure 2). p53 induces cell cycle arrest by transactivating genes such as cyclin-dependent kinase inhibitor p21, or microRNA miR34. p53 induces apoptosis by transactivating proapoptotic genes such as BAX, PUMA, SCOTIN, and FAS, and inhibiting the antiapoptotic gene BCL-2 (Lane and Levine, 2010). p53 triggers pro-survival or cell death response, depending upon cell types, the intensity of the stress signal, and the extent of cellular damage (Menendez et al., 2009). Also, p53 plays a role in controlling cell motility via regulating the expression of smooth muscle α-actin (Comer et al., 1998), collagens IIα1 and VIα1 (Sun et al., 1999), and many others.
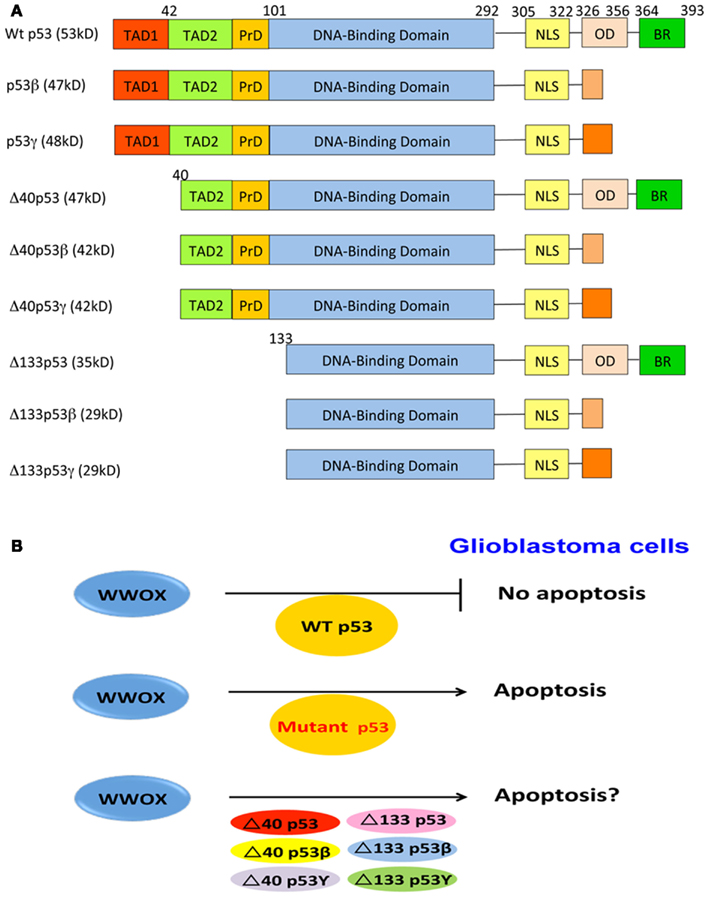
Figure 2. p53 and WWOX in GBM cell apoptosis. (A) Full-length p53 possesses two N-terminal transactivation acidic domains, a proline-rich domain, a central DNA-binding region, and a C-terminal domain, containing a nuclear localization signal, an oligomerization domain, and a basic region. N-terminal deletion mutants of p53 are also shown. (B) Three likely scenarios are proposed for WWOX and p53 to regulate GBM cell death. First, ectopic WWOX fails to induce apoptosis of GBM cells expressing endogenous wild type p53. It appears that ectopic WWOX binds and functionally antagonizes p53, and both proteins nullify each other’s function in inducing apoptosis. Second, no physical interactions between mutant p53 proteins and WWOX are expected in GBM cells. Thus, ectopic WWOX induce apoptosis in mutant p53-expressing cells. Finally, whether WWOX binds p53 isoforms is unknown. Also, whether ectopic WWOX induction of apoptosis in GBM cells expressing p53 isoforms remains to be established. TAD, transactivation domain; PrD, proline domain; NLS, nuclear localization signal; OD, oligomerization domain; BR, basic region.
p53 Isoforms
At least nine isoforms of p53 have been identified due to alternative mRNA splicing, multiple gene promoters, and alternative initiation sites of translation (Ghosh et al., 2004; Ray et al., 2006; Figure 2). In cancers, aberrant expression of p53 isoforms occurs frequently (Bourdon et al., 2005; Bourdon, 2007). Full-length p53, Δ133p53, and p53β are localized mainly in the nucleus, and only few of them are in cytoplasm. p53γ and Δ133p53β are localized in the nucleus and cytoplasm. Δ133p53γ is localized in the cytoplasm (Bourdon et al., 2005). Co-transfection of p53 and p53β increases p53-mediated apoptosis, whereas co-expression of p53 with Δ133p53 suppresses p53-mediated apoptosis (Bourdon et al., 2005). Δ133p53 differentially regulates gene expression in p53-dependent and -independent manners (Aoubala et al., 2011). Co-expression of Δ133p53β or Δ133p53γ with p53 does not affect p53 transcriptional activity on p21 and Bax promoters, as well as apoptosis.
p53 and WWOX Alterations in GBMs
p53 mutants are considered as unfavorable factors for the effectiveness in radiotherapy and TMZ treatment in glioma cells (Gjerset et al., 1995; Hirose et al., 2001; Squatrito et al., 2010; Blough et al., 2011). Loss of functional p53 confers sensitivity to TMZ in glioma cells, whereas wild type p53 increases the TMZ resistance (Blough et al., 2011). p53 mutants reduce TMZ sensitivity in gliomas (Blough et al., 2011).
Altered WWOX expression is shown in GBMs, in which downregulation of WWOX is associated with loss of heterozygosity and promoter methylation (Kosla et al., 2011). Recently, we demonstrated that overexpressed WWOX induces apoptosis of glioblastoma U373-MG cells harboring mutant p53 by causing hypoploidy and DNA fragmentation (Chiang et al., 2012). However, ectopic WWOX has no effect with U87-MG expressing wild type p53. Unlike TMZ, WWOX induces apoptosis of U373-MG cells via a mitochondria-independent and caspase-3-independent pathway (Chiang et al., 2012). While the underlying mechanisms are unknown, it is reasonable to assume that the survival of human glioblastoma cells depends upon interactions between the gain-of-function of p53 mutants and WWOX. Activated WWOX binds wild type p53 with Ser46 phosphorylation (Chang et al., 2005b). UV irradiation enhances the binding interactions. Despite the presence of Ser46 in Δ40p53, binding of this protein with WWOX remains to be determined. We postulate that in GBM cells, both wild type p53 and ectopic WWOX proteins appear to have a functional antagonism, thereby nullifying each other’s function in inducing apoptosis (Figure 2). Mutant p53 proteins cannot bind ectopic WWOX in GBM cells, and WWOX is able to induce apoptosis. Whether WWOX causes apoptosis in GBM expressing p53 isoforms is unknown and remains to be established.
Perspectives
Whether WWOX affects TMZ sensitivity has never been determined. Binding proteins for WWOX and/or p53 are likely to affect apoptosis and TMZ sensitivity in GBM cells. WWOX binds MEK, and that PMA dissociates this complex for causing apoptosis of T leukemia cells (Lin et al., 2011). Thus, appropriate chemicals, which break apart the WWOX/MEK complex, are expected to cause GBM cell death. Preliminary studies from our screening of chemicals have selected certain small molecules that induce apoptosis of many types of cancer cells (Lu et al., unpublished). Indeed, specific inhibition of MEK by selumetinib enhances TMZ-induced cancer cell death in vivo (Holt et al., 2012). Serum factors can be utilized for blocking cancer growth. For example, serum complement C1q induces apoptosis of prostate cancer and neuroblastomas cells without participation of downstream proteins in the classical activation pathway (Hong et al., 2009). In this event, ectopic WWOX is activated for inducing apoptosis.
Failure of ectopic WWOX in inducing apoptosis of glioma cells possessing wild type p53 is unusual (Chiang et al., 2012). In most cases, we have shown that p53 functionally interacts with WWOX, and both proteins induce apoptosis in a synergistic manner (Chang et al., 2001, 2003, 2005a,b, 2007, 2010, 2012; Su et al., 2012). A likely scenario is that p53-binding proteins, which are present in GBMs, may interfere with the apoptotic function of WWOX and p53. Functional antagonism among tumor suppressors has never been documented in the literature. However, it is not surprising to find that many tumor suppressor proteins, e.g., p53, WWOX, Smad4, PTEN, PP2A, and etc., are significantly upregulated during the early stage of cancer progression (Lai et al., 2005; Chang et al., unpublished). Do these proteins act synergistically in blocking cancer progression but lose control eventually? Or, do they counteract each other’s function, thus allowing cancer growth? Whether endogenous WWOX counteracts with the function of endogenous p53 is unknown and remains to be established.
One of the unique characteristics for malignant gliomas is their diffuse infiltration into distant brain tissue. Signal pathways, involving PI3K, Akt, mTOR, NF-κB, and autophagy, are believed to confer these migrating cells resistant to apoptotic death (Lefranc et al., 2005). Glioma cells possess CD44 as a receptor for interacting with brain matrix hyaluronan (Murai et al., 2004; Yoshida et al., 2012), and secrete hyaluronidases and metalloproteinases to facilitate their migration (Delpech et al., 2002; Junker et al., 2003; Hagemann et al., 2012). Also, lack of WWOX expression is expected to enhance cell migration. For example, loss of WWOX facilitates migration of ovarian cancer and osteosarcoma cells (Gourley et al., 2009; Del Mare et al., 2011). Ectopic expression of TIAF1 (TGF-β-induced antiapoptotic factor), p53, and WWOX suppresses anchorage-independent growth and cell migration and causes apoptosis in cancer cells (Chang et al., 2012). We believe that glioma cells, with stem cell-like properties, migrate individually rather than collectively. This assumption is based upon our observations that mouse Wwox gene knockout cells (e.g., embryonic fibroblasts) migrate individually and aggressively. In contrast, wild type cells migrate collectively (Chou et al., unpublished). While a portion of WWOX is anchored on the cell membrane/cytoskeleton, WWOX-negative cells lose recognition by parental WWOX-positive cells, and this increases the mobility of WWOX-negative cells to move away from the WWOX-positive cells. Conceivably, WWOX-negative glioma cells migrate individually and turn away from WWOX-expressing brain cells to low WWOX expression areas. Apparently, the migratory behavior of WWOX-negative glioma cells may account for the diffuse invasion into distant brain tissue (Chou et al., unpublished).
Metabolic alterations have been shown in Wwox knockout mice, including postnatal lethality, bone metabolism defects, ataxia, steroidogenesis, and generation of osteosarcomas (Del Mare et al., 2011; Salah et al., 2012). In a Drosophila model, Wwox is shown to participate in pathways involving aerobic metabolism and oxidative stress for generation of ROS (O’Keefe et al., 2011). Under UV stress, functional Wwox gene expression induces ROS production in Drosophila. Cancer cells are known to overly utilize glycolysis for growth advantage – the so-called Warburg (Moncada et al., 2012). Conceivably, WWOX is likely to override glucose consumption in cancer cells and exerts generation of ROS to curb the cancer cell growth and invasion.
In summary, in this perspective article we have discussed the potential role of tumor suppressors p53 and WWOX in regulating TMZ sensitivity in GBM cells. We have shown the role of WWOX in controlling cell migration and metabolic alterations, and discussed the effects of WWOX deficiency and TMZ resistance in cancer cells.
Authors Contribution
Ming-Fu Chiang and Chun-I Sze wrote the sections dealing with TMZ; Pei-Yi Chou wrote the review sections for p53 and WWOX and designed graphs; Wan-Jen Wang managed citations; Nan-Shan Chang conceived ideas, wrote and thoroughly revised the manuscript, and discussed with coauthors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported, in part, by the Department of Defense USA (W81XWH-08-1-0682), the National Science Council, Taiwan, ROC (NSC96-2320-B-006-014, 98-2628-B-006-045-MY3, 98-2628-B-006-041-MY3, and 99-2320-B-006-012-MY3), the National Health Research Institute, Taiwan, ROC (NHRI-EX99-9704BI), and the Department of Health, Taiwan, ROC (DOH101-TD-PB-111-TM010; to Nan-Shan Chang).
References
Agnihotri, S., Gajadhar, A. S., Ternamian, C., Gorlia, T., Diefes, K. L., Mischel, P. S., et al. (2012). Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J. Clin. Invest. 122, 253–266.
Aoubala, M., Murray-Zmijewski, F., Khoury, M. P., Fernandes, K., Perrier, S., Bernard, H., et al. (2011). p53 directly transactivates Delta133p53alpha, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 18, 248–258.
Aqeilan, R. I., Donati, V., Gaudio, E., Nicoloso, M. S., Sundvall, M., Korhonen, A., et al. (2007). Association of Wwox with ErbB4 in breast cancer. Cancer Res. 67, 9330–9336.
Aqeilan, R. I., Kuroki, T., Pekarsky, Y., Albagha, O., Trapasso, F., Baffa, R., et al. (2004). Loss of WWOX expression in gastric carcinoma. Clin. Cancer Res. 10, 3053–3058.
Beier, D., Schulz, J. B., and Beier, C. P. (2011). Chemoresistance of glioblastoma cancer stem cells–much more complex than expected. Mol. Cancer 10, 128.
Blough, M. D., Beauchamp, D. C., Westgate, M. R., Kelly, J. J., and Cairncross, J. G. (2011). Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neurooncol. 102, 1–7.
Bourdon, J. C., Fernandes, K., Murray-Zmijewski, F., Liu, G., Diot, A., Xirodimas, D. P., et al. (2005). p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137.
Bouteille, N., Driouch, K., Hage, P. E., Sin, S., Formstecher, E., Camonis, J., et al. (2009). Inhibition of the Wnt/beta-catenin pathway by the WWOX tumor suppressor protein. Oncogene 28, 2569–2580.
Caporali, S., Falcinelli, S., Starace, G., Russo, M. T., Bonmassar, E., Jiricny, J., et al. (2004). DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol. Pharmacol. 66, 478–491.
Chang, J. Y., Chiang, M. F., Lin, S. R., Lee, M. H., He, H., Chou, P. Y., et al. (2012). TIAF1 self-aggregation in peritumor capsule formation, spontaneous activation of SMAD-responsive promoter in p53-deficient environment, and cell death. Cell Death Dis. 3, e302.
Chang, J. Y., He, R. Y., Lin, H. P., Hsu, L. J., Lai, F. J., Hong, Q., et al. (2010). Signaling from membrane receptors to tumor suppressor WW domain-containing oxidoreductase. Exp. Biol. Med. (Maywood) 235, 796–804.
Chang, N. S., Doherty, J., Ensign, A., Lewis, J., Heath, J., Schultz, L., et al. (2003). Molecular mechanisms underlying WOX1 activation during apoptotic and stress responses. Biochem. Pharmacol. 66, 1347–1354.
Chang, N. S., Hsu, L. J., Lin, Y. S., Lai, F. J., and Sheu, H. M. (2007). WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol. Med. 13, 12–22.
Chang, N. S., Pratt, N., Heath, J., Schultz, L., Sleve, D., Carey, G. B., et al. (2001). Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J. Biol. Chem. 276, 3361–3370.
Chang, N. S., Schultz, L., Hsu, L. J., Lewis, J., Su, M., and Sze, C. I. (2005a). 17beta-Estradiol upregulates and activates WOX1/WWOXv1 and WOX2/WWOXv2 in vitro: potential role in cancerous progression of breast and prostate to a premetastatic state in vivo. Oncogene 24, 714–723.
Chang, N. S., Doherty, J., Ensign, A., Schultz, L., Hsu, L. J., and Hong, Q. (2005b). WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 280, 43100–43108.
Chen, J., Li, Y., Yu, T. S., McKay, R. M., Burns, D. K., Kernie, S. G., et al. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526.
Chiang, M. F., Yeh, S. T., Liao, H. F., Chang, N. S., and Chen, Y. J. (2012). Overexpression of WW domain-containing oxidoreductase WOX1 preferentially induces apoptosis in human glioblastoma cells harboring mutant p53. Biomed. Pharmacother. 66, 433–438.
Comer, K. A., Dennis, P. A., Armstrong, L., Catino, J. J., Kastan, M. B., and Kumar, C. C. (1998). Human smooth muscle alpha-actin gene is a transcriptional target of the p53 tumor suppressor protein. Oncogene 16, 1299–1308.
Del Mare, S., Kurek, K. C., Stein, G. S., Lian, J. B., and Aqeilan, R. I. (2011). Role of the WWOX tumor suppressor gene in bone homeostasis and the pathogenesis of osteosarcoma. Am. J. Cancer Res. 1, 585–594.
Delpech, B., Laquerriere, A., Maingonnat, C., Bertrand, P., and Freger, P. (2002). Hyaluronidase is more elevated in human brain metastases than in primary brain tumours. Anticancer Res. 22, 2423–2427.
Donati, V., Fontanini, G., Dell’Omodarme, M., Prati, M. C., Nuti, S., Lucchi, M., et al. (2007). WWOX expression in different histologic types and subtypes of non-small cell lung cancer. Clin. Cancer Res. 13, 884–891.
Donovan, L. K., and Pilkington, G. J. (2012). CD133: holy of grail of neuro-oncology or promiscuous red-herring? Cell Prolif. 45, 527–537.
Fabbri, M., Iliopoulos, D., Trapasso, F., Aqeilan, R. I., Cimmino, A., Zanesi, N., et al. (2005). WWOX gene restoration prevents lung cancer growth in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 15611–15616.
Friedman, H. S., Kerby, T., and Calvert, H. (2000). Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 6, 2585–2597.
Fujikane, R., Sanada, M., Sekiguchi, M., and Hidaka, M. (2012). The identification of a novel gene, MAPO2, that is involved in the induction of apoptosis triggered by O6-methylguanine. PLoS ONE 7:e44817. doi:10.1371/journal.pone.0044817
Fukushima, T., Takeshima, H., and Kataoka, H. (2009). Anti-glioma therapy with temozolomide and status of the DNA-repair gene MGMT. Anticancer Res. 29, 4845–4854.
Ghosh, A., Stewart, D., and Matlashewski, G. (2004). Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell. Biol. 24, 7987–7997.
Gjerset, R. A., Turla, S. T., Sobol, R. E., Scalise, J. J., Mercola, D., Collins, H., et al. (1995). Use of wild-type p53 to achieve complete treatment sensitization of tumor cells expressing endogenous mutant p53. Mol. Carcinog. 14, 275–285.
Goellner, E. M., Grimme, B., Brown, A. R., Lin, Y. C., Wang, X. H., Sugrue, K. F., et al. (2011). Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 71, 2308–2317.
Gourley, C., Paige, A. J., Taylor, K. J., Ward, C., Kuske, B., Zhang, J., et al. (2009). WWOX gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin alpha3. Cancer Res. 69, 4835–4842.
Guler, G., Uner, A., Guler, N., Han, S. Y., Iliopoulos, D., Hauck, W. W., et al. (2004). The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer 100, 1605–1614.
Haar, C. P., Hebbar, P., Wallace, G. C. IV, Das, A., Vandergrift, W. A. III, Smith, J. A., et al. (2012). Drug resistance in glioblastoma: a mini review. Neurochem. Res. 37, 1192–1200.
Hagemann, C., Anacker, J., Ernestus, R. I., and Vince, G. H. (2012). A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 3, 67–79.
Happold, C., Roth, P., Wick, W., Schmidt, N., Florea, A. M., Silginer, M., et al. (2012). Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 122, 444–455.
Hegi, M. E., Liu, L., Herman, J. G., Stupp, R., Wick, W., Weller, M., et al. (2008). Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 26, 4189–4199.
Hirose, Y., Berger, M. S., and Pieper, R. O. (2001). p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 61, 1957–1963.
Holt, S. V., Logié, A., Odedra, R., Heier, A., Heaton, S. P., Alferez, D., et al. (2012). The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br. J. Cancer 106, 858–866.
Hong, Q., Hsu, L. J., Schultz, L., Pratt, N., Mattison, J., and Chang, N. S. (2007). Zfra affects TNF-mediated cell death by interacting with death domain protein TRADD and negatively regulates the activation of NF-kappaB, JNK1, p53 and WOX1 during stress response. BMC Mol. Biol. 8:50. doi:10.1186/1471-2199-8-50
Hong, Q., Sze, C. I., Lin, S. R., Lee, M. H., He, R. Y., Schultz, L., et al. (2009). Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS ONE 4:e5755. doi:10.1371/journal.pone.0005755
Hsu, L. J., Schultz, L., Hong, Q., Van Moer, K., Heath, J., Li, M. Y., et al. (2009). Transforming growth factor beta1 signaling via interaction with cell surface Hyal-2 and recruitment of WWOX/WOX1. J. Biol. Chem. 284, 16049–16059.
Hu, B. S., Tan, J. W., Zhu, G. H., Wang, D. F., Zhou, X., and Sun, Z. Q. (2012). WWOX induces apoptosis and inhibits proliferation of human hepatoma cell line SMMC-7721. World J. Gastroenterol. 18, 3020–3026.
Huang, H., Lin, H., Zhang, X., and Li, J. (2012). Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-kappaB-dependent pathway. Oncol. Rep. 27, 2050–2056.
Ishii, H., and Furukawa, Y. (2004). Alterations of common chromosome fragile sites in hematopoietic malignancies. Int. J. Hematol. 79, 238–242.
Janouskova, H., Maglott, A., Leger, D. Y., Bossert, C., Noulet, F., Guerin, E., et al. (2012). Integrin alpha5beta1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 72, 3463–3470.
Jin, C., Ge, L., Ding, X., Chen, Y., Zhu, H., Ward, T., et al. (2006). PKA-mediated protein phosphorylation regulates ezrin-WWOX interaction. Biochem. Biophys. Res. Commun. 341, 784–791.
Johannessen, T. C., and Bjerkvig, R. (2012). Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 12, 635–642.
Junker, N., Latini, S., Petersen, L. N., and Kristjansen, P. E. (2003). Expression and regulation patterns of hyaluronidases in small cell lung cancer and glioma lines. Oncol. Rep. 10, 609–616.
Kanzawa, T., Bedwell, J., Kondo, Y., Kondo, S., and Germano, I. M. (2003). Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J. Neurosurg. 99, 1047–1052.
Kitange, G. J., Mladek, A. C., Carlson, B. L., Schroeder, M. A., Pokorny, J. L., Cen, L., et al. (2012). Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 18, 4070–4079.
Knizhnik, A. V., Roos, W. P., Nikolova, T., Quiros, S., Tomaszowski, K. H., Christmann, M., et al. (2013). Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 8:e55665. doi:10.1371/journal.pone.0055665
Kohsaka, S., Wang, L., Yachi, K., Mahabir, R., Narita, T., Itoh, T., et al. (2012). STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 11, 1289–1299.
Kosla, K., Pluciennik, E., Kurzyk, A., Jesionek-Kupnicka, D., Kordek, R., Potemski, P., et al. (2011). Molecular analysis of WWOX expression correlation with proliferation and apoptosis in glioblastoma multiforme. J. Neurooncol. 101, 207–213.
Kuroki, T., Yendamuri, S., Trapasso, F., Matsuyama, A., Aqeilan, R. I., Alder, H., et al. (2004). The tumor suppressor gene WWOX at FRA16D is involved in pancreatic carcinogenesis. Clin. Cancer Res. 10, 2459–2465.
Lai, F. J., Cheng, C. L., Chen, S. T., Wu, C. H., Hsu, L. J., Lee, J. Y., et al. (2005). WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin. Cancer Res. 11, 5769–5777.
Lane, D., and Levine, A. (2010). p53 Research: the past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2, a000893.
Lefranc, F., Brotchi, J., and Kiss, R. (2005). Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 23, 2411–2422.
Li, M. Y., Lai, F. J., Hsu, L. J., Lo, C. P., Cheng, C. L., Lin, S. R., et al. (2009). Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE 4:e7820. doi:10.1371/journal.pone.0007820
Li, S., Liang, Z., Xu, L., and Zou, F. (2012). MicroRNA-21: a ubiquitously expressed pro-survival factor in cancer and other diseases. Mol. Cell. Biochem. 360, 147–158.
Lin, H. P., Chang, J. Y., Lin, S. R., Lee, M. H., Huang, S. S., Hsu, L. J., et al. (2011). Identification of an in vivo MEK/WOX1 complex as a master switch for apoptosis in T cell leukemia. Genes Cancer 2, 550–562.
Menendez, D., Inga, A., and Resnick, M. A. (2009). The expanding universe of p53 targets. Nat. Rev. Cancer 9, 724–737.
Moncada, S., Higgs, E. A., and Colombo, S. L. (2012). Fulfilling the metabolic requirements for cell proliferation. Biochem. J. 446, 1–7.
Murai, T., Miyazaki, Y., Nishinakamura, H., Sugahara, K. N., Miyauchi, T., Sako, Y., et al. (2004). Engagement of CD44 promotes Rac activation and CD44 cleavage during tumor cell migration. J. Biol. Chem. 279, 4541–4550.
Nakada, M., Furuta, T., Hayashi, Y., Minamoto, T., and Hamada, J. (2012). The strategy for enhancing temozolomide against malignant glioma. Front. Oncol. 2:98. doi:10.3389/fonc.2012.00098
Nakayama, S., Semba, S., Maeda, N., Aqeilan, R. I., Huebner, K., and Yokozaki, H. (2008). Role of the WWOX gene, encompassing fragile region FRA16D, in suppression of pancreatic carcinoma cells. Cancer Sci. 99, 1370–1376.
Nishikawa, R. (2010). Standard therapy for glioblastoma–a review of where we are. Neurol. Med. Chir. (Tokyo) 50, 713–719.
O’Keefe, L. V., Colella, A., Dayan, S., Chen, Q., Choo, A., Jacob, R., et al. (2011). Drosophila orthologue of WWOX, the chromosomal fragile site FRA16D tumour suppressor gene, functions in aerobic metabolism and regulates reactive oxygen species. Hum. Mol. Genet. 20, 497–509.
Oliva, C. R., Moellering, D. R., Gillespie, G. Y., and Griguer, C. E. (2011). Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 6:e24665. doi:10.1371/journal.pone.0024665
Pollack, I. F., Hamilton, R. L., Sobol, R. W., Burnham, J., Yates, A. J., Holmes, E. J., et al. (2006). O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 Cohort. J. Clin. Oncol. 24, 3431–3437.
Ray, P. S., Grover, R., and Das, S. (2006). Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 7, 404–410.
Roos, W. P., Batista, L. F., Naumann, S. C., Wick, W., Weller, M., Menck, C. F., et al. (2007). Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 26, 186–197.
Salah, Z., Alian, A., and Aqeilan, R. I. (2012). WW domain-containing proteins: retrospectives and the future. Front. Biosci. 17, 331–348.
Sarkaria, J. N., Kitange, G. J., James, C. D., Plummer, R., Calvert, H., Weller, M., et al. (2008). Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 14, 2900–2908.
Shi, L., Chen, J., Yang, J., Pan, T., Zhang, S., and Wang, Z. (2010). MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 1352, 255–264.
Silber, J. R., Bobola, M. S., Blank, A., and Chamberlain, M. C. (2012). O(6)-Methylguanine-DNA methyltransferase in glioma therapy: promise and problems. Biochim. Biophys. Acta 1826, 71–82.
Squatrito, M., Brennan, C. W., Helmy, K., Huse, J. T., Petrini, J. H., and Holland, E. C. (2010). Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 18, 619–629.
Stupp, R., Hegi, M. E., Mason, W. P., Van Den Bent, M. J., Taphoorn, M. J., Janzer, R. C., et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466.
Su, W. P., Chen, S. H., Chen, S. J., Chou, P. Y., Huang, C. C., and Chang, N. S. (2012). “WW Domain-containing oxidoreductase is a potential receptor for sex steroid hormones,” in Sex Hormones, ed. R. K. Dubey (Rijeka: InTech), 333–351.
Sun, S., Lee, D., Lee, N. P., Pu, J. K., Wong, S. T., Lui, W. M., et al. (2012). Hyperoxia resensitizes chemoresistant human glioblastoma cells to temozolomide. J. Neurooncol. 109, 467–475.
Sun, Y., Wenger, L., Rutter, J. L., Brinckerhoff, C. E., and Cheung, H. S. (1999). p53 down-regulates human matrix metalloproteinase-1 (Collagenase-1) gene expression. J. Biol. Chem. 274, 11535–11540.
Villalva, C., Cortes, U., Wager, M., Tourani, J. M., Rivet, P., Marquant, C., et al. (2012). O6-methylguanine-methyltransferase (MGMT) promoter methylation status in glioma stem-like cells is correlated to temozolomide sensitivity under differentiation-promoting conditions. Int. J. Mol. Sci. 13, 6983–6994.
Voss, V., Senft, C., Lang, V., Ronellenfitsch, M. W., Steinbach, J. P., Seifert, V., et al. (2010). The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 8, 1002–1016.
Wang, J. Y., and Edelmann, W. (2006). Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell 9, 417–418.
Yoshida, T., Matsuda, Y., Naito, Z., and Ishiwata, T. (2012). CD44 in human glioma correlates with histopathological grade and cell migration. Pathol. Int. 62, 463–470.
Zhang, P., Jia, R., Ying, L., Liu, B., Qian, G., Fan, X., et al. (2012a). WWOX-mediated apoptosis in A549 cells mainly involves the mitochondrial pathway. Mol. Med. Report 6, 121–124.
Keywords: tumor suppressor, p53, WWOX, glioblastoma multiforme, temozolomide resistance
Citation: Chiang M-F, Chou P-Y, Wang W-J, Sze C-I and Chang N-S (2013) Tumor suppressor WWOX and p53 alterations and drug resistance in glioblastomas. Front. Oncol. 3:43. doi: 10.3389/fonc.2013.00043
Received: 30 November 2012; Accepted: 17 February 2013
Published online: 04 March 2013.
Edited by:
Gaspar Kitange, Mayo Clinic, USAReviewed by:
Sydney M. Evans, University of Pennsylvania School of Medicine, USAArnab Chakravarti, Ohio State University Medical Center/James Cancer Hospital, USA
Copyright: © 2013 Chiang, Chou, Wang, Sze and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Nan-Shan Chang, Institute of Molecular Medicine, National Cheng Kung University, 1 University Road, Tainan 70101, Taiwan. e-mail:Y2hhbmduc0BtYWlsLm5ja3UuZWR1LnR3
†Ming-Fu Chiang and Pei-Yi Chou have contributed equally to this work.