 Chiang Wang Sun1†
Chiang Wang Sun1† Candice Willmon2†‡Li-Chen Wu1Peter Knopick3Jutta Thoerner4Richard Vile2Tim M. Townes1
Candice Willmon2†‡Li-Chen Wu1Peter Knopick3Jutta Thoerner4Richard Vile2Tim M. Townes1 David S. Terman1*
David S. Terman1*
- 1Department of Biochemistry and Molecular Genetics, University of Alabama Medical School at Birmingham, Birmingham, AL, USA
- 2Department of Molecular Medicine, Mayo Clinic Foundation, Rochester, MN, USA
- 3Department of Immunology, University of North Dakota Medical School, Grand Forks, ND, USA
- 4Hisotpathology Section, Hospital of the Monterey Peninsula, Monterey, CA, USA
Insights from the study of cancer resistance in animals have led to the discovery of novel anticancer pathways and opened new venues for cancer prevention and treatment. Sickle cells (SSRBCs) from subjects with homozygous sickle cell anemia (SCA) have been shown to target hypoxic tumor niches, induce diffuse vaso-occlusion, and potentiate a tumoricidal response in a heme- and oxidant-dependent manner. These findings spawned the hypothesis that SSRBCs and the vasculopathic microenvironment of subjects with SCA might be inimical to tumor outgrowth and thereby constitute a natural antitumor defense. We therefore implanted the B16F10 melanoma into humanized hemoglobin SS knockin mice which exhibit the hematologic and vasculopathic sequelae of human SCA. Over the 31-day observation period, hemoglobin SS mice showed no significant melanoma outgrowth. By contrast, 68–100% of melanomas implanted in background and hemoglobin AA knockin control mice reached the tumor growth end point (p < 0.0001). SS knockin mice also exhibited established markers of underlying vasculopathy, e.g., chronic hemolysis (anemia, reticulocytosis) and vascular inflammation (leukocytosis) that differed significantly from all control groups. Genetic differences or normal AA gene knockin do not explain the impaired tumor outgrowth in SS knockin mice. These data point instead to the chronic pro-oxidative vasculopathic network in these mice as the predominant cause. In related studies, we demonstrate the ability of the sickle cell component of this system to function as a therapeutic vehicle in potentiating the oncolytic/vasculopathic effect of RNA reovirus. Sickle cells were shown to efficiently adsorb and transfer the virus to melanoma cells where it induced apoptosis even in the presence of anti-reovirus neutralizing antibodies. In vivo, SSRBCs along with their viral cargo rapidly targeted the tumor and initiated a tumoricidal response exceeding that of free virus and similarly loaded normal RBCs without toxicity. Collectively, these data unveil two hitherto unrecognized findings: hemoglobin SS knockin mice appear to present a natural barrier to melanoma tumorigenesis while SSRBCs demonstrate therapeutic function as a vehicle for enhancing the oncolytic effect of free reovirus against established melanoma.
Introduction
Insights from the study of cancer resistance in animals and humans have led to the discovery of novel anticancer pathways and opened new venues for cancer prevention and treatment (1, 2). Mammals, such as elephants, blind moles, and some feral murine strains, have evolved intricate mechanisms to eradicate cancer. These include cell-cycle accelerated DNA repair, checkpoint blockade, programed cell death, and replicative senescence controlled by a network of tumor-suppressor genes and mediators such as p53, Rb, and IFN-β (1–5). In genetically deficient humans with an increased incidence of tumors, the major defects appear in DNA repair and proapoptotic pathways (6). Recently, subjects with deficiencies in growth hormone receptor and insulin-like growth factor have shown a reduced cancer mortality highlighting a role of pro-growth pathways, oxidative stress, and age-dependent genomic instability in human oncogenesis (7) in a study of cancer incidence in more than 16,000 patients with sickle cells anemia (SCA), melanoma was not listed (8). To date, the notion that SCA or an animal model thereof might constitute a natural barrier to melanoma tumorigenesis has not been explored or demonstrated.
Sickle cell anemia is a monogenic hemoglobinopathy wherein glutamic acid, the sixth amino acid in the β-globin chain, is displaced by valine (9, 10). This results in hemoglobin polymerization and sickling morphology during hemoglobin desaturation. Clinically, the disease is characterized by chronic hemolysis, intermittent vaso-occlusive events, and organ injury (11, 12). Endothelial cells are chronically activated and injured after contact with sickle cells, sickle cell-derived heme, and inflammatory mediators (9–12). Contributing to the microvascular injury and dysfunction are abundant reactive oxygen species (ROS) generated in the course of relentless ischemia–reperfusion, chronic hemolysis, and vascular inflammation (13–21).
The Townes model of SCA in knockin mice faithfully replicates the clinical sequalae of human homozygous SCA together with its pro-oxidative and thrombo-inflammatory microvascular features (22–25). Such mice exhibit a γ–βS globin configuration wherein mouse β-globin genes are replaced with human γ- and βS-globin genes and mouse α-globin genes are replaced with human α-globin genes (26, 27). They also switch human hemoglobins (HbF to HbS) at 3 weeks of age when they start to develop chronic hemolysis, severe anemia, and vascular inflammation (28).
Sickle cells (SSRBCs) from patients with SCA possess an ability to target established tumors and together with exogenous pro-oxidants produce a tumoricidal response (29). Recent studies have drawn a striking parallel between the hypoxic, pro-oxidative, and chronically upregulated microvasculature in SCA and the activated neovasculature of solid tumors (11, 22, 29–31). In both vascular networks, intravital microscopy shows that SSRBCs adhere rapidly to upregulated endothelium and recruit leukocytes and platelets forming microcellular aggregates that lead to vaso-occlusion (10, 29, 32–39). SSRBC velocity in venules of transgenic sickle mice and solid tumors is reduced resulting in SS hemoglobin polymerization and autohemolysis with consequent release of intracellular heme. The latter is a powerful cell toxin that together with ROS appear to mediate endothelial injury (14–16, 29). The consequent microvascular ischemia in SCA produces tissue injury and painful crisis while in tumors it causes tumor cell necrosis (29).
Collectively, these data spawned the hypothesis that the pro-oxidative and thrombo-inflammatory vascular microenvironment of SCA might interdict melanoma neoangiogenesis and thereby constitute a natural antitumor defense against this tumor. We therefore elected to study the outgrowth of B16F10 melanoma in transgenic mice with SCA.
In related studies, we examine the ability of human sickle cell component of the system as a therapeutic to augment oncolysis by a vasculopathic RNA virus. Previously, SSRBCs have demonstrated increased adherence to endothelium infected by RNA viruses (40). Indeed, some RNA viruses are known to target tumor neovasculature, compromise blood flow, and cause significant vascular injury (41, 42). Reovirus, a non-enveloped double stranded RNA virus, infects endothelial cells via its junctional adhesion molecule-A (JAM-A) and provides a conduit for the virus into the bloodstream (43, 44). In tumor cells, reovirus replicates independently of the Ras-EGFR pathway and exerts selective oncolysis via defective antiviral PKR (45, 46). While this agent is effective when delivered intratumorally alone or together with anti-PDI or chemotherapy, it has shown only modest ability to kill tumor when used systemically. This is largely due to binding by circulating blood cells and elimination by seroreactive neutralizing antibodies (47–51). By contrast, when the virus is adsorbed to the surface of the dendritic cells and T cells, it can be delivered to tumor niches in lymph nodes where it exhibits tumoricidal activity (52). In search of a cell carrier for systemic delivery of reovirus, we reasoned that sickle cells might be a viable candidate to protect reovirus from host defenses, target it to tumor and synergize with virus-induced tumor vascular injury. Indeed, sickle cells have been shown to carry and deliver cytotoxics into hypoxic 4T1 carcinomas and release fourfold more drug cargo into the tumor milieu than similarly loaded normal RBCs and free drug (53). We therefore loaded sickle cells with reovirus and found that the virus spontaneously translocates to melanoma cells in vitro and induces a tumoricidal response in vitro and in vivo exceeding that of similarly loaded normal RBCs (nRBCs) and free virus.
Results
B16F10 Melanoma Outgrowth Is Impaired in Sickle Cell Knockin Mice
We determined whether the B16F10 melanoma indigenous to C57Bl6 mice could grow after implantation into sickle cell knockin mice expressing human βS-globin genes and hemoglobin AA knockin mice expressing human βA-globin genes. These mice exhibit a background consisting of C57Bl/6 and 30% of 129 genes by QP analysis. For background controls, we deployed C57Bl/6 mice and also B6129SF1/J mice and B6129SF2/J, which express murine 129 genes along with B6 genes. Figure 1 shows that B16F10 melanoma grew robustly in C57Bl/6, B6129SF1/J mice, and B6129SF2/J mice with mean volumes of 1000 mm3 by days 16–18 after implantation. Tumor outgrowth to a mean of 1000 mm3 was also evident in the AA knockin mice by day 27. By contrast, melanoma outgrowth was impaired significantly in SS knockin mice relative to all controls (p < 0.0001).
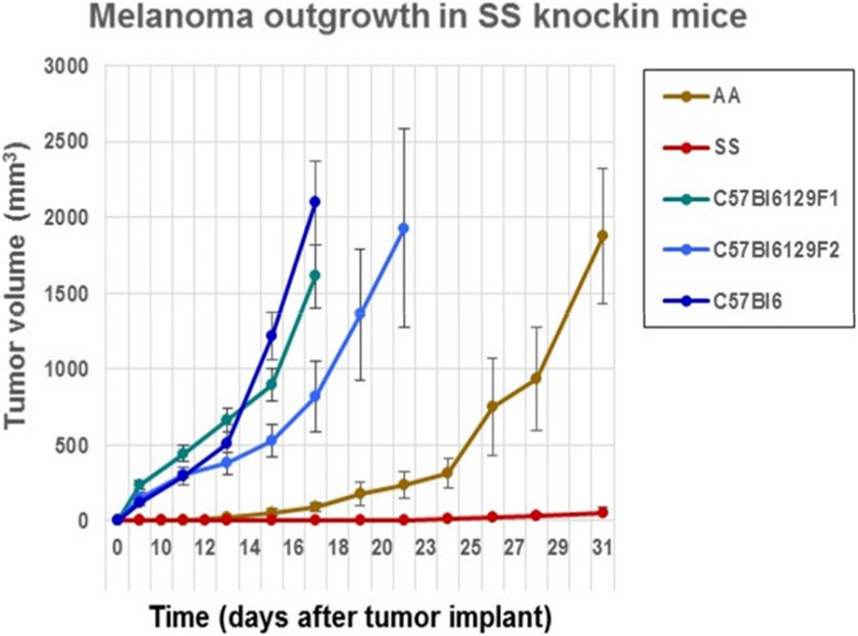
Figure 1. Comparison of the outgrowth rate of B16F10 melanoma in sickle cell knockin mice with background and AA knockin mice is shown. B16F10 melanoma outgrowth is inhibited significantly in sickle cell knockin mice compared to background and AA knockin controls (p < 0.0001).
Kaplan–Meier analysis of survival to the tumor growth end point (tumor volume of 750 mm3) similarly indicates that control groups reached median end points well before day 31 after implant, whereas in sickle knockin mice, none of the implanted tumors reached this end point during this period (p < 0.0001 compared to all controls, Figure 2).
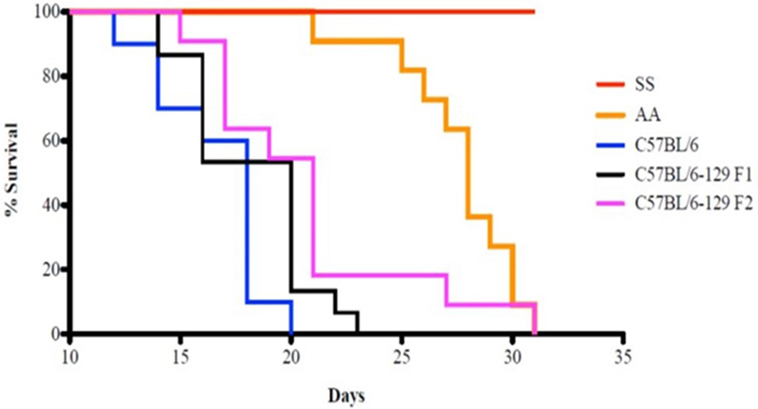
Figure 2. Kaplan–Meier analysis of sickle cell mice and controls is shown. Survival is significantly prolonged in the SS knockout mice compared to corresponding background controls and AA knockin mice. See text for details.
Figure 3 shows the percentage of tumors that exceeded 250 mm3 in each group during the 31-day observation period. In sickle cell knockin mice, only 2 of 29 (6.9%) tumors grew beyond this baseline size compared to 100% of C57Bl/6 and B6129SF1/J mice (p = < 0.0001), 81.2% of B6129SF2/J mice (p < 0.0001), and 68.7% of AA knockin mice (p < 0.0004). Notably, the two tumors in the sickle cell knockin group that grew above baseline did not reach the 750 mm3 growth end point during the 31-day observation period. Histopathologic examination of the melanomas in the latter two mice showed clusters of venules congested with SSRBCs, surrounding mononuclear cell infiltrate and adjacent tumor cell necrosis (Figure 4). These results demonstrate that the incidence of B16F10 tumorigenesis is significantly impaired in SS knockin mice relative to background and AA knockin controls.
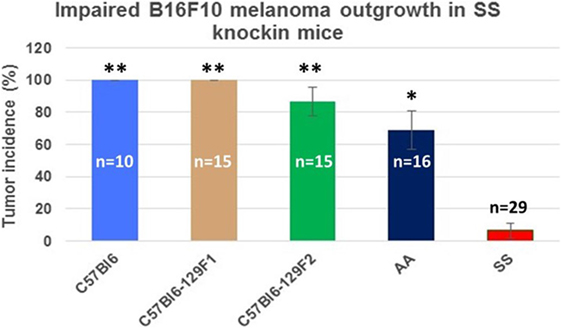
Figure 3. Comparison of the percentage of B16F10 melanomas exhibiting growth to end point tumor volume within the 31-day observation period in SS knockin mice and background/allotypic hemoglobin controls is shown. B16F10 melanoma outgrowth is significantly impaired in SS knockin mice compared with C57Bl6, C57Bl61296F1, C57Bl6129F2 (**p < 0.0001), and AA knockin (*p = 0.0004) mice.
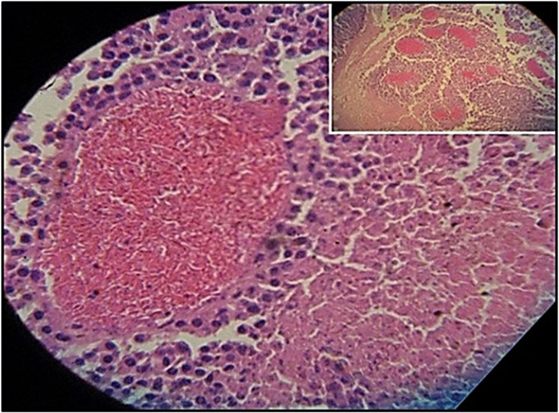
Figure 4. Histopathologic section of melanoma implanted in sickle cell knockin mice that failed to show progressive growth is shown. H&E section of melanoma implanted into SS knockin mice is shown. Insert shows cluster of eleven venules congested with sickle cells (10× magnification). Higher power shows sickle cells occluding a venule with surrounding mononuclear cell infiltrate and adjacent tumor cell necrosis (40× magnification).
Hematologic Evaluation of SS Knockin Mice and Controls
We examined the notion that the pro-inflammatory and pro-oxidative vascular environment of SS knockin mice could explain the impaired melanoma outgrowth in SS knockin mice. Major causative factors of such pro-oxidative microvascular milieu in SS knockin mice are unrelenting ischemia–reperfusion, chronic hemolysis, and vascular inflammation (46, 47). In these mice, anemia and reticulocytosis are established biomarkers of underlying chronic hemolysis, while leukocytosis is an indicator of sustained vascular inflammation (48, 49). We therefore compared reticulocyte, leukocyte, and hemoglobin levels of SS knockin mice with all controls. Results shown in Table 1 indicate that the degree of reticulocytosis and leukocytosis in hemoglobin SS knockin mice was significantly different from all control mice (p < 0.00001 and p < 0.0001, respectively). Likewise, SS knockin mice were significantly more anemic than all controls (p < 0.0001). These results indicate that unlike controls, SS knockin mice exhibit severe anemia, exaggerated reticulocyte and leukocyte counts consistent with their underlying chronic hemolysis, pro-inflammatory and pro-oxidative vasculopathy.
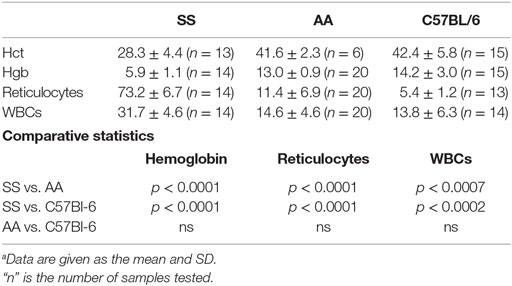
Table 1. Comparison of biomarkers of chronic hemolysis and vasculopathic inflammation in SS Knockin mice and controls.a
Sickle Cells Adsorb and Transfer Oncolytic Reovirus Vector to Melanoma Cells In Vitro and In Vivo
Reovirus is known to be oncolytic, but its delivery to tumor cells is interdicted by immune defenses and neutralizing antibodies (49–51). We therefore determined whether SSRBCs with their known tumor homing properties (29) could serve as vehicles for carriage and delivery of reovirus to tumor and protect it from neutralizing antibodies in vitro and in vivo. We first examined whether SSRBCs could bind reovirus and vesicular stomatitis virus (VSV) without replicating or inducing SSRBC cytotoxicity. Results shown in Figure 5 indicate that SSRBCs incubated with reovirus or VSV for 1 h at 37°C at various MOIs did not replicate or undergo cytotoxicity in a plaque forming assay. We then determined whether the viral-loaded SSRBCs could transfer reovirus and VSV to melanoma cells in the presence or absence of viral neutralizing antibodies. Results shown in Figure 6 demonstrate that when SSRBCs were loaded with reovirus (SSRBC-neo) and cocultured with B16tk melanoma cells, the virus translocated to melanoma cells and induced melanoma cell lysis at 24 h to a degree comparable to that of melanoma cells directly infected with virus in a standard plaque forming assay. The presence or absence of reovirus neutralizing antibody therefore did not abrogate the viral translocation from SSRBCs to melanoma cells and consequent melanoma cell lysis. VSV showed a similar ability to translocate from SSRBCs and infect melanoma cells (Figure 7). Results demonstrate that, even in the presence of neutralizing antibodies, SSRBCs efficiently adsorb and transfer reovirus and VSV to B16 melanoma cells resulting in tumor cell cytolysis.
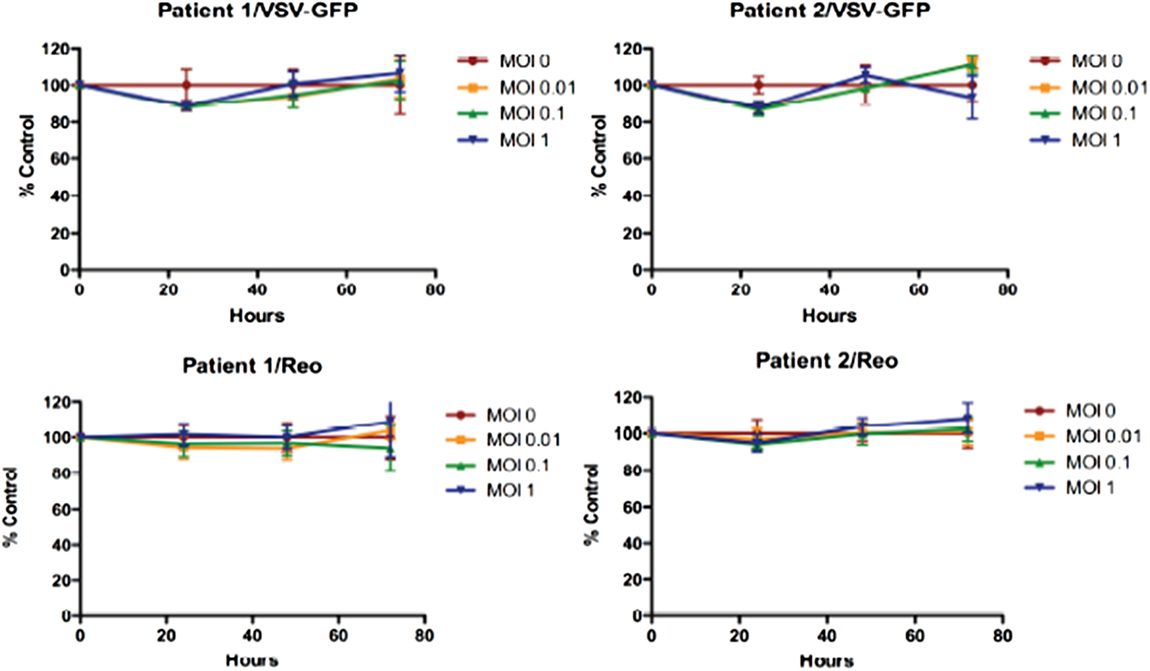
Figure 5. Reovirus and vesicular stomatitis virus bind but do not replicate in SSRBCs. Reovirus and VSV do not replicate in and are not cytotoxic to SSRBC. Human SSRBC were plated in 96-well plates and were infected with various multiplicities of infection (MOI) with reovirus and VSV. At specific time points, an MTT assay was performed according to manufacturer’s instructions. Data are plotted as a percentage of control survival with SD.
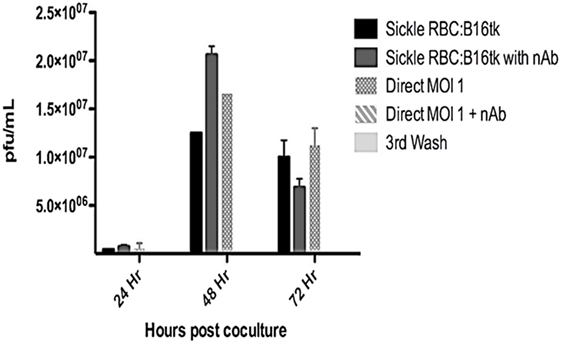
Figure 6. Reovirus is transferred to melanoma cells in vitro in the presence of neutralizing antibody. Shown above is B16tk melanoma cell lysis after co-incubation with SSRBCs that were loaded with reovirus (MOI = 1) for 2 h at 37°C, washed three times at a ratio of 1:1. In addition, at the time of coculture, reovirus neutralizing antibody was added to some of the wells. To demonstrate efficient removal of any unbound virus from the viral loaded SSRBC, an aliquot of the third wash was directly added to the B16tk cells. As a positive control, B16tk cells were directly infected with reovirus at an MOI of 1 with and without neutralizing antibodies. At specified time points, aliquots of the supernatant were collected and titered on L929 cells using a standard plaque forming unit assay. Data are plotted with SD.
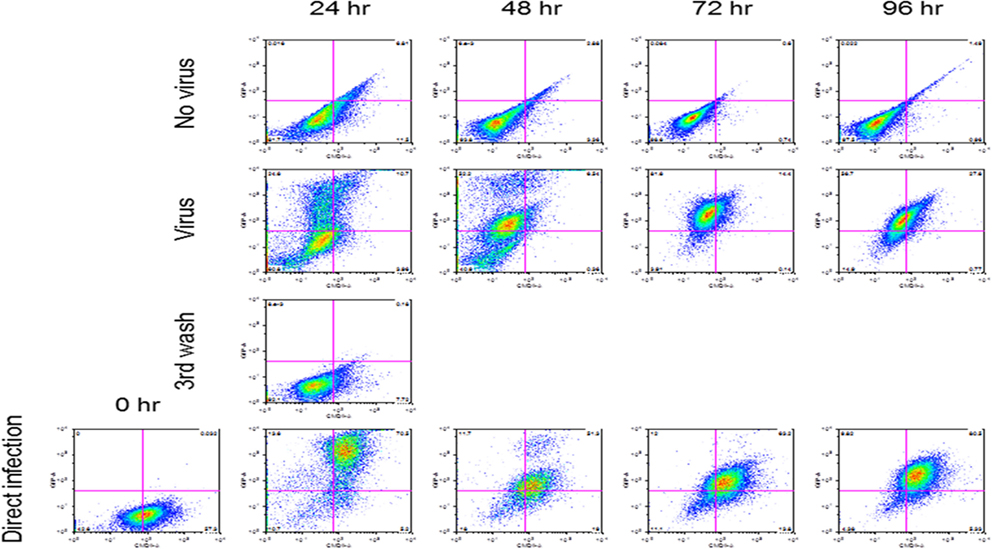
Figure 7. SSRBCs transfer vesicular stomatitis virus to melanoma cells. Human sickle RBC were incubated with or without VSV-GFP (MOI = 1) for 2 h at 37°C, washed three times, and then cocultured with B16 cells stained with Vybrant CmDiI at a ratio of 1:1. At indicated time points the B16 cells were washed and analyzed by FACS. As a positive control, B16 cells were directly infected with VSV-GFP at an MOI of 1.
SSRBCs Localize in Melanoma and Loaded with Oncolytic Reovirus Induce a Tumoricidal Response
We compared the ability of intravenously administered SSRBCs and nRBCs to localize in melanomas. Results shown in Figure 8 indicate that 30 min after injection SSRBCs accumulated in melanoma to a degree significantly greater than nRBCs. We then compared the ability of SSRBCs and nRBCs carrying oncolytic reovirus to induce a tumoricidal response in a B16 melanoma model. For this purpose, SSRBCs or nRBCs were loaded with reovirus as described in “Materials and Methods” and injected into mice bearing established B16 melanoma. As controls, mice were also injected with naked reovirus (5 × 108 TCID50) or PBS. Survival of mice receiving SSRBCs-reo was significantly prolonged compared to groups receiving nRBC-reo (p = 0.02), naked virus (p = 0.002), or PBS (p = 0.0003) (Figure 9). Mice showed no significant acute toxicity or weight loss during the study. These data demonstrate that reovirus-loaded SSRBCs can be delivered to melanoma in vivo and induce a tumoricidal response exceeding that of the nRBC-reo, PBS control and naked reovirus without significant acute toxicity.
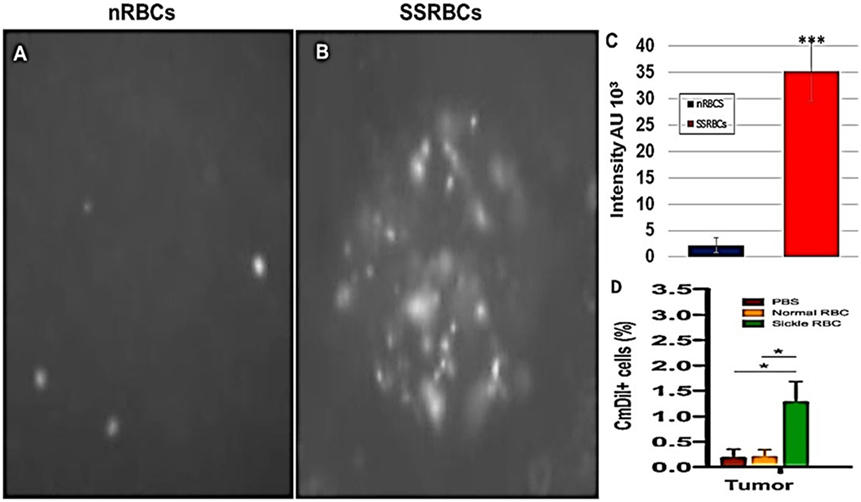
Figure 8. Sickle cells accumulation in tumor compared to normal RBCs. Athymic nude mice were injected subcutaneously with 2 × 105 B16tk melanoma cells. When tumors were palpable, nRBCs or SSRBCs stained with Vybrant CmDiI (70% hematocrit) were injected intravenously. Thirty minutes after injection, tumors were harvested and converted to single cell suspensions. Still image of CmDIl uptake in representative tumor suspensions of nRBCs (A) and SSRBCs (B) from a tumor bearing mouse is shown. Image analysis of (A,B) shown in (C) indicates a greater number of CmDil staining SSRBCs than nRBCs (p < 0.001). FACS analysis of suspensions (D) confirmed the increased uptake of CmDil-staining SSRBCs relative to nRBCs in the melanomas (n = 3) (p < 0.05).
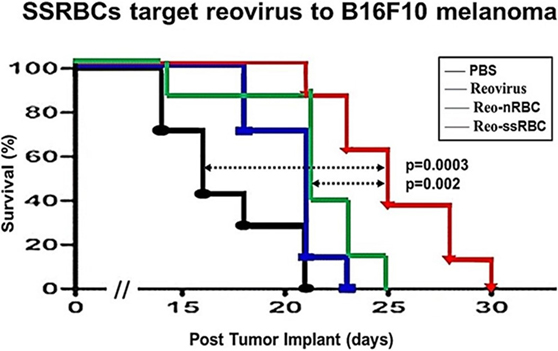
Figure 9. SSRBCs loaded with reovirus significantly improve the survival of mice bearing established B16 melanoma. Athymic nude mice were subcutaneously injected in the flank with 2 × 105 B16ova cells. Once tumors were palpable, nRBC or SSRBC were loaded with reovirus for 2 h at 37°C, washed three times and intravenously injected at a hematocrit of 70%. As controls, mice were also intravenously injected with PBS or reovirus (5 × 108 TCID50). Naked reovirus and reo-nRBC prolonged the survival of B16ova-bearing mice compared to the PBS control (p = 0.0434 for reovirus alone; p = 0.0213 for reo-nRBC). However, reo-SSRBC achieved a highly significant survival enhancement compared to the other three treatment groups (PBS, p = 0.0003; naked reovirus, p = 0.0020; reo-nRBC, p = 0.020).
Discussion
Here, we show that melanoma outgrowth is impaired significantly in transgenic SS knockin mice relative to C57BL/6, B6129SF1/J, and B6129SF2/J background controls and transgenic hemoglobin AA mice. The B16F10 melanoma is syngeneic in C57BL/6 mice but grows comparably in B6129SF1/J and B6129SF2/J mice, which express 129 genes together with B6 genes. This suggests that the 129 genetic component did not interfere significantly with melanoma outgrowth. Likewise, SS and AA mice not only express 129 and B6 genes but also synthesize human hemoglobin allele(s). Since tumor outgrowth occurred in a significantly higher percentage of hemoglobin AA mice relative to SS mice, the presence of the human hemoglobin AA alleles did not appear to interfere significantly with tumor outgrowth or tumor incidence. The restricted tumor outgrowth in SS mice can therefore be explained by the presence of hemoglobin SS alleles in SS knockin rather than by genetic differences between the strains.
As shown herein, impaired melanoma outgrowth in sickle cell knockin mice was accompanied by significant anemia, reticulocytosis, and leukocytosis relative to controls. In sickle cell knockin mice, reticulocytosis is an established marker of underlying chronic hemolysis, while leukocytosis is indicative of a chronic thrombo-inflammatory microvasculopathy induced by recruitment of neutrophils and monocytes to sites of vaso-occlusion (14, 48, 49). In these mice, hemolysis is a consequence of repeated episodes of oxidatively stressful sickling/unsickling, spontaneous SS hemoglobin auto-oxidation, and recurrent episodes of ischemia–reperfusion injury (19, 52, 53). The latter activates vascular pro-oxidant mitochondrial networks (19), while hemolytic by-products arginase and heme reduce bioavailability of antioxidant nitrous oxide (13). SS heme, also activates nuclear factor-kB (Nf-kB) leading to expression of endothelial adhesins VCAM-1, ICAM-1 that bind cognate ligands on SSRBCs leading to formation of cellular microaggregates, blood stasis, vaso-occlusion, and organ injury (54, 55). Figure 4 showing clusters of tumor venules occluded by SSRBCs with surrounding tumor cell necrosis depicts the late phase of tumor microvascular injury. Hemolysis and leukocytosis in SS knockin mice, denoted herein by reticulocytosis and leukocytosis, therefore, play a central role in the biogenesis of the thrombo-inflammatory microvasculopathy. Such an abnormal vascular microenvironment is the likely basis of melanoma growth inhibition in SS knockin mice.
In both SCA and sickle knockin mice, ROS generation results from unrelenting ischemia–reperfusion and nitrous oxide insufficiency. This leads to elaboration of microvascular pro-oxidants NADH, endothelial xanthine oxidase, and peroxynitrites (16, 54). Excessive mitochondrial ROS generated during reperfusion depolarizes mitochondrial membranes (56–59) while microvascular ROS signaling leads to endothelial activation. Intravital microscopy of activated vascular networks in sickle cell mice and solid tumors perfused with SSRBCs shows that microvascular blood flow stagnates leading to vaso-occlusion and autohemolysis with consequent release of excessive ROS (10–12, 29). This self-perpetuating pro-oxidant network is a major determinant of the microvascular injury in SCA and the tumor endothelium (12, 19, 20, 29).
The notion that this highly pro-oxidative microvasculature in SS knockin mice is antithetical to neoangiogensis after implantation of the B16F10 melanoma is supported by recent studies showing that powerful pro-oxidants doxorubicin and zinc protoporphyrin potentiate the tumoricidal effect of SSRBC infusions against the drug resistant, triple negative 4T1 breast carcinoma (29). In addition, SSRBC pro-oxidant surrogates hemin and H2O2 were shown to induce tumor cell cytotoxicity in vitro suggesting that heme, ROS, and heme-nitrosyl complexes generated by interaction of trapped SSRBCs with tumor endothelium are capable of lysing adjacent tumor cells (29). Okuno et al. demonstrated that silencing of the endothelial cell antioxidant mATM gene produced a pro-oxidative microvasculature, which suppressed B16F10 angiogenesis and significantly delayed tumor outgrowth (60). Furthermore, inhibition of GSH and thioredoxin antioxidant pathways or selective activation of the Akt pro-oxidant pathways led to synergistic tumor regression confirming the remarkable potency of excessive ROS when harnessed for tumor eradication (61, 62). In this context, melanoma cells, in particular, have shown an increased sensitivity to exogenous H2O2 relative to other solid tumor cells (63).
In light of these data, it is likely that melanoma angiogenesis is interrupted in the pro-oxidative vascular microenvironment of sickle knockin mice. During angiogenesis, embryonic tumor blood vessels rely on host endothelial cells to sprout and grow, promoted by VEGF and stabilized by interactions with the extracellular matrix (64, 65). VEGF receptors are activated early in tumor neoangiogenesis and their expression and activation may be impaired in the pro-oxidative vascular milieu of the SS knockin mice (64, 65). Indeed, Subtinib a biologic that competitively blocks VEGF receptors inhibited B16F10 melanoma outgrowth (66). Circulating SSRBCs are likely trapped in the network of sprouting tumor microvessels. Heme and ROS generated from such trapped/hemolyzed SSRBCs may promote free radical generation resulting in tumor endothelial injury, vaso-occlusion, and regression of neoangiogenesis. It is therefore plausible that melanoma neoangiogenesis requiring a coordinated interaction between endothelial cells of SS knockin mice and B16F10 melanoma cells is compromised in the pro-oxidative, thrombo-inflammatory microvascular environment of SS knockin mice.
In related studies, we assessed the ability of SSRBCs to act as a therapeutic to carry and deliver oncolytic reovirus to tumors and induce a tumoricidal effect. Previously, we showed that SSRBCs could be loaded with and release fourfold more drug into hypoxic tumors than free drug or similarly loaded nRBCs (53). Some viral carriers have shown an ability to localize in tumors and induce a tumoricidal effect, while others are hampered by their tendency to sequester in liver, lungs, and reticuloendothelial system (42, 67–69). Although a proportion of virus-loaded SSRBCs are also removed by non-tumor organs, we show here that a larger fraction homes to tumors and extends survival relative to nRBCs and free virus. SSRBCs therefore appear to direct the virus to the tumor where it induces a tumoricidal response.
We further show that reovirus loaded onto SSRBCs can spontaneously translocate to melanoma cells. This transfer may be explained in part by the presence on melanoma cells of high-affinity sialic acid residues along with JAM-A receptors, which promote dissociation of reovirus from its low affinity receptor on SSRBCs (70–72). Notably, adherence of the reovirus to SSRBCs does not kill these cells whereas viral binding to melanoma cells after viral transfer results in melanoma cytolysis. Primary viral adhesion to melanoma cells is followed by a secondary higher affinity interaction between the sigma head of the virus and JAM-A receptors, which promotes virus internalization and melanoma cell apoptosis (73). That neutralizing antibodies did not displace the bound virus from the SSRBCs or the melanoma cells may likewise be ascribed to a higher affinity of the virus for SSRBCs or melanoma cells. Alternatively, the dominant reovirus sigma head epitope recognized by neutralizing antibodies may be sterically screened from recognition by the topology of the virus when bound to SSRBC receptors.
We previously reported that infusion of human SSRBCs along with pro-oxidants into mice bearing established 4T1 breast carcinomas resulted in a tumoricidal response without significant toxicity or histopathologic tissue injury (29). In this context sections of all organs including brains of the treated mice were devoid of inflammation, thrombosis, or necrosis. The lack of toxicity of SSRBC infusions in non-SS tumor bearing mice may be explained by the absence of a procoagulant and inflammatory vascular phenotype in non-tumor organs and repeated cycles of ischemia–reperfusion that predispose SS knockin mice and SCA patients to vaso-occlusion.
Since SSRBCs largely target hypoxic vasculature with upregulated adhesion receptors, cancer patients with vascular diseases bearing an underlying chronic oxidative stress signature such as hypertension and atherosclerotic cardio- and cerebrovascular disease (74) might be at risk higher risk of SSRBC treatment. However, the risk in these settings may be no greater than that of many widely used cancer cytotoxics (e.g., doxorubicin), antiangiogenic agents (e.g., bevacizumab), and biologics (e.g., IL-2), which are known to exacerbate the same vascular conditions. The risk assessment protocol for prospective patients receiving SSRBC treatment would be similar to that used for patients at risk of chemotherapy-associated venous thromboembolism. Particular attention would be directed to parameters predictive of an inflammatory and procoagulant phenotype such as chronic leukocytosis with increased monocytes and PMNs along with high levels of acute phase proteins and cytokines to include TNFα, IL1β, IL6, IL8, IFNγ, CRP, CCL3, CCL2, lactoferrin/elastase, thrombin-anti-thrombin complexes (TAT), and D-dimer (75–80). With appropriate precautions, SSRBC infusions may therefore be useful against several human tumor types without undue toxicity.
In conclusion, these data provide two hitherto unrecognized properties of SSRBCs. First, SS knockin mice restrict melanoma outgrowth relative to background and beta globin controls suggesting that their pro-oxidative microenvironment may constitute a natural host defense system against this tumor. The dissection of this network is likely to lead to pharmacologic mimicry and therapeutic integration of the relevant pro-oxidant systems for prevention and treatment of melanoma. Second, we show that the SSRBC component of this system can function as a therapeutic in its ability to carry and transfer oncolytic reovirus to melanoma cells and induce a tumoricidal response against established melanoma in vivo. SSRBCs and the natural microvasculopathic environment of SCA therefore provide novel insight into a potent natural anticancer network and offer promising new tools that can be harnessed for prevention and treatment malignant melanoma.
Materials and Methods
Mice
All animal procedures were approved by the UAB and Mayo Foundation Institutional Animal Care and Use Committees or the Animal Use Committees in compliance with the Guide for the Care and Use of Laboratory Animals. Male and female mice 8–12 weeks of age weighing 19–26 g were used. Hemoglobin SS knockin mice (B6; 129-Hbatm1(HBA/Tow) Hbbtm2(HBG1,HBB)/Tow/Hbbtm3(HBG1,HBB)Tow/J) and homozygous hemoglobin AA knockin mice were obtained from a breeding colony maintained at UAB animal research facility. QTL evaluation of SS and AA knockin mice indicated that they are predominantly B6 and exhibit up to 30% of 129 genes. C57BL/6, B6129SF1/J, and B6129SF2/J mice were purchased from Jackson (Bar Harbor, Me). Female athymic homozygous nude mice (nu−/nu–), between 8–12 weeks of age weighing 19–26 g, were obtained from Charles River Laboratories (Wilmington, MA, USA) or Harlan Laboratories (Indianapolis, IN, USA). The mice were housed five animals per cage in a 12-h light–dark cycle with water, food ad libitum. All infusions were performed using the retro-orbital route or tail vein.
Tumors, Cells, and Viruses
The B16/F10 melanoma (CRL-6475) was obtained from ATCC, Manassas, VA, USA. B16ova murine melanoma cell line was grown in Dubelcco’s modified Eagle medium supplemented with 10% FBS and 5 mg/mL of G418. B16ova cells were derived from the parental cells by transduction of a cDNA encoding chicken ovalbumin gene. B16tk cells were derived from B16 cells by transducing them with a cDNA encoding the herpes simplex virus thymidine kinase (tk) gene (81) and were grown in DMEM supplemented with 10% FBS and 1.25 μg/mL puromycin. All cell lines were monitored routinely and found to be free of mycoplasma infection. Reovirus Dearing Type 3 was provided by Oncolytics Biotech Inc. (Calgary, AB, Canada). Vesicular stomatitis virus (VSV) was generated as previously described (82). Virus titers were measured by a standard plaque assay on L929 cells for reovirus and BHK-21 cells for VSV.
Tumor Outgrowth and Established Tumor Studies
Mice were injected in the right flank with 1 × 105 B16F10 melanoma cells in volume of 50 μL containing 25 μL of Matrigel (Fisher Scientific). For the studies in mice bearing B16F10 melanoma, tumor volumes were measured with standard calipers and volumes were calculated as length × width2/2 where length is the long axis and width the short axis. Mice were monitored for toxicity. During the 32 day observation period, a tumor was considered to show progressive growth only if its volume exceeded 250 mm3. The end point was a tumor volume of 750–1500 mm3.
With the approval by the Institutional Review Board of Mayo Foundation normal and sickle red blood cells were obtained from patients with homozygous SCA or normal healthy volunteers. Informed consent was obtained from each donor. For B16ova melanoma studies, subcutaneous (s.c.) tumors were established by injecting 2 × 105 or 5 × 105 B16ova melanoma cells into the flank of C57Bl/6 mice. SSRBC or nRBC infusion studies were started when s.c. tumors reached approximately 200 mm3. SSRBCs or nRBCs were loaded with reovirus for 2 h at 37°C, washed three times, and injected intravenously at a hematocrit of 70% in 100 μL. Control mice were injected with PBS or free reovirus (5 × 108 TCID50). Prior to injection, cell smears were stained with DiffQuick, examined under a light microscope, and photographed to ensure sickle cell or normal erythrocyte morphology. Tumors were measured twice a week with standard calipers, and mice were monitored for toxicity. Mice were euthanized if toxicity was evident or tumor burden exceeded 1500 mm3.
Viral Loading of SSRBCs and NRBCs
For the survival studies, human (nude mice) or murine (C57Bl/6 mice) nRBC or SSRBC were loaded with reovirus for 2 h at 37°C, washed three times, and injected intravenously at a hematocrit of 70%. As controls, mice were also injected intravenously with PBS or reovirus (5 × 108 TCID50). Prior to injection, cell smears were stained with Diff-Quick and examined under a light microscope to ensure that sickle cell or normal RBC morphology was maintained.
Viral Transfer Studies from SSRBCs to Melanoma Cells In Vitro
Sickle cells (SSRBC) were incubated with reovirus (MOI = 1) for 2 h at 37°C, washed three times, and then cocultured with B16tk at a ratio of 1:1. In addition, during coculture of reovirus with SSRBC, neutralizing antibody was added to selected wells. In order to demonstrate efficient removal of any unbound virus from the loaded SSRBC, an aliquot of the third wash was directly added to the B16tk cells. Additionally, B16tk cells were directly infected with reovirus at an MOI of 1 for comparison of B16tk cells incubated with and without reovirus neutralizing antibody. At specified time points, aliquots of the supernatant were collected and assayed on L929 cells in a standard plaque forming unit assay.
MTT Assay
MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assays were performed as follows: Target cells were plated in quadruplicate on 96-well plates and were infected with reovirus at various MOIs (ranging from 0.1 to 100). Cell viability was assessed at the indicated time points according to manufacturer’s protocol (Promega, Madison, WI, USA). Optical density was read at 570 nm using an enzyme-linked immunosorbent assay plate reader corrected for background.
Tumor Uptake Studies
Athymic nude mice were injected subcutaneously with 2 × 105 B16tk melanoma cells. When tumors were palpable, nRBCs or SSRBCs stained with Vybrant CmDiI (70% hematocrit) were injected intravenously. Thirty minutes after injection tumors were harvested and tumor cells were dissociated into single cell suspensions and photographed. Fluorescence intensity of still images was quantified using Adobe Photoshop CS2 software (Adobe Systems Inc., San Jose, CA, USA). Ten determinations of pixel intensity from four quadrants of the image were obtained and averaged to yield mean fluorescence intensity. The difference in uptake of the dissociated cells was confirmed by FACS analysis.
Hematology
Reticulocyte, leukocyte, and hemoglobin determinations were carried out on tail vein or retro-orbital samples of mouse blood using a Heska HemaTrue analyzer (Loveland, CO, USA).
Histology
Sections of tumors were stained with hematoxylin and eosin.
Statistical Analyses
For tumor outgrowth studies, tumor growth curves were analyzed and compared by the method of Kruskal Wallis. Survival curves were plotted according to the Kaplan–Meier method, and statistical significance between the different treatment groups was determined using the log-rank (Mantel–Cox) and Gehan–Breslow–Wilcoxon tests. For comparison of individual data points, two-sided Student’s t-test was applied to determine statistical significance.
Author Contributions
Study concept and design: DT and RV. Acquisition of data: CS, CW, L-CW, JT, and PK. Analysis of data: DT, RV, TT, CS, and CW. Drafting of manuscript: DT. Study supervision: DT and RV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH grants R01HL057619, CA175386-01A1 and CA108961-10P3, Azore (NP) Foundation, the Mayo Foundation, Oncolytics Biotech (Calgary). Dr. DST is recipient of the Doctors Cancer Research Foundation Award for 2016. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA (2015) 314:1850–60. doi:10.1001/jama.2015.13134
2. Gorbunova V, Hine C, Tian X, Ablaeva J, Gudkov AV, Nevo E, et al. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc Natl Acad Sci U S A (2012) 109:19392–6. doi:10.1073/pnas.1217211109
3. Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature (2013) 499:346–9. doi:10.1038/nature12234
4. Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, et al. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell (2008) 7:813–23. doi:10.1111/j.1474-9726.2008.00431.x
5. Balmain A, Nagase H. Cancer resistance genes in mice: models for the study of tumor modifiers. Trends Genet (1998) 14:139–44. doi:10.1016/S0168-9525(98)01422-X
6. Klein G. Toward a genetics of cancer resistance. Proc Natl Acad Sci U S A (2009) 106:859–63. doi:10.1073/pnas.0811616106
7. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng C-W, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans science. Sci Transl Med (2011) 3:1–9. doi:10.1126/scitranslmed.3001845
8. Schultz WH, Ware RE. Malignancy in patients with sickle cell disease. Am J Hematol (2003) 74:249–53. doi:10.1002/ajh.10427
9. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet (2010) 376:2018–31. doi:10.1016/S0140-6736(10)61029-X
10. Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation (2009) 16:97–111. doi:10.1080/10739680802279394
11. Belcher JD, Mahaseth H, Welch TE, Vilback AE, Sonbol KM, Kalambur VS, et al. Critical role of endothelial cell activation in hypoxia-induced vasoocclusion in transgenic sickle mice. Am J Physiol Heart Circ Physiol (2005) 288:H2715–25. doi:10.1152/ajpheart.00986.2004
12. Belcher JD, Chunsheng Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood (2014) 123:377–90. doi:10.1182/blood-2013-04-495887
13. Hidalgo A, Chang J, Jang J, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med (2009) 15:384–91. doi:10.1038/nm.1939
14. Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin N Am (2014) 28:181–98. doi:10.1016/j.hoc.2013.11.005
15. Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A (2001) 98:15215–20. doi:10.1073/pnas.221292098
16. Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy: a state of nitric oxide resistance. Free Radic Biol Med (2008) 44:1506–28. doi:10.1016/j.freeradbiomed.2008.01.008
17. Aslan M, Thornley-Brown D, Freeman BA. Reactive species in sickle cell disease. Ann N Y Acad Sci (2000) 899:375–91. doi:10.1111/j.1749-6632.2000.tb06201.x
18. Klings ES, Farber HW. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir Res (2001) 2:280–5. doi:10.1186/rr70
19. Hebbel RP, Morgan WT, Eaton JW, Hedlund BE. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci U S A (1988) 85:237241. doi:10.1073/pnas.85.1.237
20. Nagababu E, Fabry ME, Nagel RL, Rifkind JM. Heme degradation and oxidative stress in murine models for hemoglobinopathies: thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol Dis (2008) 41:6166. doi:10.1016/j.bcmd.2007.12.003
21. Akohoue SA, Shankar S, Milne GL, Morrow J, Chen KY, Ajayi WU, et al. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res (2007) 61:233–8. doi:10.1203/pdr.0b013e31802d7754
22. Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, et al. Transgenic sickle mice have vascular inflammation. Blood (2003) 101:3953–9. doi:10.1182/blood-2002-10-3313
23. Nath KA, Grande JP, Croatt AJ, Frank E, Caplice NM, Hebbel RP, et al. Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol (2005) 166:963–72. doi:10.1016/S0002-9440(10)62318-8
24. Kaul DK, Liu X, Zhang X, Ma L, Hsia C, Nagel RL. Inhibition of sickle red cell adhesion and vasoocclusion in the microcirculation by antioxidants. Am J Physiol Heart Circ Physiol (2006) 291:H167–75. doi:10.1152/ajpheart.01096.2005
25. Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest (2000) 106:411–20. doi:10.1172/JCI9225E1
26. Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science (1997) 278:873–8. doi:10.1126/science.278.5339.873
27. Ryan TM, Townes TM, Reilly MP, Asakura T, Palmiter RD, Brinster RL, et al. Human sickle hemoglobin in transgenic mice. Science (1990) 247:566–8. doi:10.1126/science.2154033
28. Behringer RR, Ryan TM, Palmiter RD, Brinster RL, Townes TM. Human Aγ- to β-globin switching in transgenic mice. Gene Dev (1990) 4:380–9. doi:10.1101/gad.4.3.380
29. Terman DS, Viglianti BL, Zennadi R, Fels D, Boruta RJ, Yuan H, et al. Sickle erythrocytes target cytotoxics to hypoxic tumor microvessels and potentiate a tumoricidal response. PLoS One (2013) 8(1):e52543. doi:10.1371/journal.pone.0052543
30. Dewhirst MW. Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat Res (2009) 172:653–65. doi:10.1667/RR1926.1
31. Gorini C, Harris IS, Mak TW. Modulation of oxidative stress as an antitumor strategy. Nat Rev Drug Discov (2013) 12:931–47. doi:10.1038/nrd4002
32. Faller D. Vascular modulation. In: Embury S, Hebbel R, Mohandas N, Steinberg M, editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York, NY: Raven (1994). p. 235–46.
33. Brown MD, Wick TM, Eckman JR. Activation of vascular endothelial cell adhesion molecule expression by sickle blood cells. Pediatr Pathol Mol Med (2001) 20:47–72. doi:10.1080/15513810109168817
34. Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood (1998) 92:3924–35.
35. Terada LS. Oxidative stress and endothelial activation. Crit Care Med (2002) 30:S186–91. doi:10.1097/00003246-200205001-00003
36. Kalambur VS, Mahaseth H, Bischof JC, Kielbik MC, Welch TE, Vilbäck A, et al. Microvascular blood flow and stasis in transgnic sickle mice: utility of a dorsal skin fold chamber for intravital microscopy. Am J Hematol (2004) 77:117–25. doi:10.1002/ajh.20143
37. Telen MJ. Cellular adhesion and the endothelium: E-selectin, L-selectin, and pan-selectin inhibitors. Hematol Oncol Clin North Am (2014) 28:341–54. doi:10.1016/j.hoc.2013.11.010
38. Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med (1993) 177:1277–86. doi:10.1084/jem.177.5.1277
39. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell (2004) 5:429–41. doi:10.1016/S1535-6108(04)00115-1
40. Hebbel RP, Visser MR, Goodman JL, Jacob HS, Vercellotti GM. Potentiated adherence of sickle erythrocytes to endothelium infected by virus. J Clin Invest (1987) 80:1503–6. doi:10.1172/JCI113233
41. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther (2011) 19:886–94. doi:10.1038/mt.2011.26
42. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature (2011) 477:99–102. doi:10.1038/nature10358
43. Lai CM, Boehme KW, Pruijssers AJ, Parekh VV, Van Kaer L, Parkos CA, et al. Endothelial JAM-A promotes reovirus viremia and bloodstream dissemination. J Infect Dis (2015) 211:383–93. doi:10.1093/infdis/jiu476
44. Annukka ARA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, et al. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe (2009) 5:59–71. doi:10.1016/j.chom.2008.12.001
45. Thirukkumaran CM, Nodwell MJ, Hirasawa K, Shi ZQ, Diaz R, Luider J, et al. Oncolytic viral therapy for prostate cancer: efficacy of reovirus as a biological therapeutic. Cancer Res (2010) 70:2435–44. doi:10.1158/0008-5472.CAN-09-2408
46. Twigger K, Roulstone V, Kyula J, Karapanagiotou EM, Syrigos KN, Morgan R, et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer (2012) 12:368. doi:10.1186/1471-2407-12-368
47. Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther (2015) 24(1):166–74. doi:10.1038/mt.2015.156
48. Pandha HS, Heinemann L, Simpson GI, Melcher A, Prestwich R, Errington F, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res (2009) 15:6158–66. doi:10.1158/1078-0432.CCR-09-0796
49. Errington F, White CL, Twigger KR, Rose A, Scott K, Steele L, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther (2008) 15:1257–70. doi:10.1038/gt.2008.58
50. White CL, Twigger KR, Vidal L, De Bono JS, Coffey M, Heinemann L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther (2008) 15:911–20. doi:10.1038/gt.2008.21
51. Vidal L, Pandha HS, Yap TA, White CL, Twigger K, Vile RG, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res (2008) 14:7127–37. doi:10.1158/1078-0432.CCR-08-0524
52. Ilett EJ, Prestwich RJ, Kottke T, Errington F, Thompson JM, Harrington KJ, et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther (2009) 16:689–99. doi:10.1038/gt.2009.29
53. Choe S, Terman DS, Rivers AE, Rivera J, Lottenberg R, Sorg BS. Drug-loaded sickle cells programmed ex vivo for delayed hemolysis target hypoxic tumor microvessels and augmenttumor drug delivery. J Control Release (2013) 171:184–92. doi:10.1016/j.jconrel.2013.07.008
54. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation (2004) 11:129–51. doi:10.1080/mic.11.2.129.151
55. Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets (2009) 9:271–92. doi:10.2174/1871529X10909040271
56. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
57. Sabharwal S, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer (2014) 14:709–19. doi:10.1038/nrc3803
58. Trachootham D, Jerome Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Dis (2009) 8:579–88. doi:10.1038/nrd2803
59. Nur E, Biemond BJ, Otten HM, Brandjes DP, Schnog JJ; CURAMA Study Group. Oxidative stress in sickle cell disease: pathophysiology and potential implications for disease management. Am J Hematol (2011) 86:484–9. doi:10.1002/ajh.22012
60. Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat Med (2012) 18:1208–16. doi:10.1038/nm.2846
61. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell (2015) 27:211–22. doi:10.1016/j.ccell.2014.11.019
62. Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell (2008) 14:458–70. doi:10.1016/j.ccr.2008.11.003
63. Meyskins FR, Chau HV, Tohidan N, Buckmeier J. Luminol-enhanced chemiluminescent response of human melanocytes and melanoma cells to hydrogen peroxide stress. Pigment Cell Res (1997) 10:184–9. doi:10.1111/j.1600-0749.1997.tb00482.x
64. Carmeliet P, Jain R. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473(7347):298–307. doi:10.1038/nature10144
65. Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene (1999) 18:5356–62.
66. Farbod S, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res (2010) 70:10090–100. doi:10.1158/0008-5472.CAN-10-0489
67. Roy DG, Bell JC. Cell carriers for oncolytic viruses: current challenges and future directions. Oncolytic Virother (2013) 2:47–56.
68. Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, Fuller M, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med (2012) 4:138ra77. doi:10.1126/scitranslmed.3003578
69. Peng KW, Dogan A, Vrana J, Liu C, Ong HT, Kumar S, et al. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol (2009) 84:401–7. doi:10.1002/ajh.21444
70. Danthi P, Holm GH, Stehle T, Dermody TS. Reovirus reovirus receptors, cell entry and proapoptotic signaling. In: Pöhlmann S, Simmons G, editors. Viral Entry into Host Cells. Austin, TX: Landes Bioscience and Springer Science Business Media (2013). p. 42–67.
71. Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F. Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro pigment cell. Melanoma Res (2011) 24:504–11. doi:10.1111/j.1755-148X.2011.00856.x
72. Reisfeld RA, Cherish DA. Human tumor antigens. Adv Immunol (1987) 40:323–37. doi:10.1016/S0065-2776(08)60242-4
73. Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem (2001) 276(3):2200–11. doi:10.1074/jbc.M004680200
74. Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol (2005) 25:29–38. doi:10.1161/01.ATV.0000150649.39934.13
75. Khorana AA, Streiff MB, Farge D, Mandala M, Debourdeau P, Cajfinger F, et al. Venous thromboembolism prophylaxis and treatment in cancer: a consensus statement of major guidelines panels and call to action. J Clin Oncol (2009) 27:4919–26. doi:10.1200/JCO.2009.22.3214
76. Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood (1997) 89:1787–92.
77. Pathare A, Al Kindi S, Alnaqdy AA, Daar S, Knox-Macaulay H, Dennison D. Cytokine profile of sickle cell disease. Am J Hematol (2004) 77:323–8. doi:10.1002/ajh.20196
78. Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clin Appl Thromb Hemost (2012) 18:195–200. doi:10.1177/1076029611420992
79. Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol (1999) 66:411–5.
80. Ataga KI, Brittain JE, Desai P, May R, Jones S, Delaney J, et al. Association of coagulation activation with clinical complications in sickle cell disease. PLoS One (2012) 7:e29786. doi:10.1371/journal.pone.0029786
81. Vile RG, Hart IR. Use of tissue-specific expression of the herpes simplex virus thymidine kinase gene to inhibit growth of established murine melanomas following direct intratumoral injection of DNA. Cancer Res (1993) 53:3860–4.
Keywords: sickle cells, melanoma, neoangiogenesis, oncolytic viruses, reactive oxygen species
Citation: Sun CW, Willmon C, Wu L-C, Knopick P, Thoerner J, Vile R, Townes TM and Terman DS (2016) Sickle Cells Abolish Melanoma Tumorigenesis in Hemoglobin SS Knockin Mice and Augment the Tumoricidal Effect of Oncolytic Virus In Vivo. Front. Oncol. 6:166. doi: 10.3389/fonc.2016.00166
Received: 19 March 2016; Accepted: 20 June 2016;
Published: 08 July 2016
Edited by:
Giuseppe Giaccone, Georgetown University, USAReviewed by:
Soldano Ferrone, University of Pittsburgh Cancer Institute, USAMin Hee Kang, School of Medicine, Texas Tech University Health Sciences Center, USA
Copyright: © 2016 Sun, Willmon, Wu, Knopick, Thoerner, Vile, Townes and Terman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Terman, ZHN0QHNiY2dsb2JhbC5uZXQ=
†Chiang Wang Sun and Candice Willmon contributed equally to this work.
‡Present address: Candice Willmon, Seattle Genetics, Inc., Bothell, WA, USA