 Christian Hoischen1
Christian Hoischen1 Klaus Weisshart
Klaus Weisshart Peter Hemmerich
Peter Hemmerich- 1Leibniz Institute on Aging Research, Jena, Germany
- 2Carl Zeiss Microscopy GmbH, Jena, Germany
The promyelocytic leukemia (pml) gene product PML is a tumor suppressor localized mainly in the nucleus of mammalian cells. In the cell nucleus, PML seeds the formation of macromolecular multiprotein complexes, known as PML nuclear bodies (PML NBs). While PML NBs have been implicated in many cellular functions including cell cycle regulation, survival and apoptosis their role as signaling hubs along major genome maintenance pathways emerged more clearly. However, despite extensive research over the past decades, the precise biochemical function of PML in these pathways is still elusive. It remains a big challenge to unify all the different previously suggested cellular functions of PML NBs into one mechanistic model. With the advent of genetically encoded fluorescent proteins it became possible to trace protein function in living specimens. In parallel, a variety of fluorescence fluctuation microscopy (FFM) approaches have been developed which allow precise determination of the biophysical and interaction properties of cellular factors at the single molecule level in living cells. In this report, we summarize the current knowledge on PML nuclear bodies and describe several fluorescence imaging, manipulation, FFM, and super-resolution techniques suitable to analyze PML body assembly and function. These include fluorescence redistribution after photobleaching, fluorescence resonance energy transfer, fluorescence correlation spectroscopy, raster image correlation spectroscopy, ultraviolet laser microbeam-induced DNA damage, erythrocyte-mediated force application, and super-resolution microscopy approaches. Since most if not all of the microscopic equipment to perform these techniques may be available in an institutional or nearby facility, we hope to encourage more researches to exploit sophisticated imaging tools for their research in cancer biology.
Introduction
The promyelocytic leukemia (pml) gene is a target of the t(15;17) chromosomal translocation, which fuses pml reciprocally with retinoic acid receptor α (RARα) (1). PML protein is the major building unit of the so-called PML nuclear bodies (PML NBs). PML NBs appear as nuclear dot-shaped structures that are interspersed between chromatin (2). PML NBs are heterogeneous and dynamic structures, ranging in size from 0.1 to 1.0 µm, and typically there are 5–30 bodies per nucleus, depending on the cell type, phase of cell cycle, and the cellular stress level (3–5). Overexpression of PML in cancer cell lines induces cell cycle arrest and apoptosis (6). PML knock-out mice develop a range of cancers including papillomas, carcinomas, and lymphomas after exposure to carcinogens (7). Furthermore, loss of PML is a hallmark of human cancers from diverse tissues (8). Therefore, PML is regarded as a potent tumor suppressor in in vitro (biochemistry, cell culture experiments) and in vivo (model organisms). At the physiological level, PML has been functionally linked to anti-inflammatory and antiviral response pathways, metabolism, stem cell maintenance, and aging, while more mechanistically, PML’s role in tumor suppression is linked to control of the cell cycle, apoptosis/senescence, cell migration, angiogensis, and the DNA damage response (9, 10). Since upon DNA damage PML NBs accumulate various DNA damage response factors and physically associate with damaged chromatin, they have also been suggested to play important roles in genome maintenance, probably by supporting specific aspects of DNA repair pathways (11–18).
A better understanding of the biophysical and biochemical mechanisms by which PML and/or the PML nuclear bodies participate in genome maintenance is expected to facilitate the development of therapeutic strategies for the treatment of PML-related diseases (19).
Novel microscopy methods have become key tools for studying biological systems over the past decades. Deep insight with unprecedented spatial and time resolution has been obtained for many cellular factors as a result of the rapid development of optical microscopy, fluorescent probes, and new labeling techniques (20). Since most biochemical mechanisms on the cellular level are dynamic by nature and cannot be fully understood by simply measuring fixed structures it is desirable to investigate the molecule of interest in real time, in living cells, at single-molecule, nanometer, and nanosecond resolution. Is this feasible? We think the answer is yes and the purpose of this report is explaining why.
We set out here to summarize our view of PML nuclear body function and assembly, the current status of powerful imaging methods and describe in some detail how the new imaging tools work in deciphering structural and functional aspects of PML nuclear bodies. Many of these tools may be accessible at a near-by imaging facility of most laboratories. We therefore wish to encourage those researchers in the fields of cancer biology to exploit the new methods more rigorously. Ultimately, the combination of classical biochemical approaches with dynamic methods and live cell imaging platforms may make it possible to fully elucidate the biophysical mechanisms underlying the structure, function, and networks of tumor suppressors and oncogenes, thus aiding the development of new therapeutic approaches.
PML and PML Nuclear Bodies
The pml gene product (PML) is a member of the tripartite motif (TRIM)-containing protein superfamily. In human cells, six nuclear PML isoforms (I–VI) are expressed. The various isoforms originate from alternative mRNA splicing of exons 7–9 while exons 1–6 are shared by all isoforms (Figure 1A) (21). This primary sequence complexity of PML protein expression allows for common as well as individual functional modalities among the isoforms (22). PML I is the longest isoform (882 amino acids), while PML VI (552 amino acids) is the shortest isoform in the cell nucleus. Similar to other members of the TRIM family, all nuclear PML protein isoforms contain a conserved TRIM/RBCC motif consisting of a RING domain, two B-box domains and a coiled-coil domain (RBCC) (Figure 1A) (23). A nuclear localization signal (NLS) mediates predominant nuclear localization of PML. All PML isoforms contain three well-characterized small ubiquitin-related modifier (SUMO) modification sites at position 65, 160, and 490 of the PML primary sequence (Figure 1A) (24). Generally, SUMO modification of proteins plays important roles in diverse cellular processes, including chromatin organization, transcription, DNA repair, macromolecular assembly, protein homeostasis, trafficking, and signal transduction (25). In the case of PML, SUMO-2 and SUMO-3 can form heteropolymeric poly-SUMO chains (26). PML isoforms as well as their poly-SUMOylated variants can be easily detected by Western blotting (Figure 1B) (27). Additional posttranslational modifications (PTMs) of PML include phosphorylation, acetylation, and ubiquitination, all of which may serve to fine-tune PML (nuclear body) function through multiple mechanisms (28). A common feature of TRIM/RBCC proteins is homo-multimerization which generates a variety of subcellular structures including ribbon-like structures, cytoplasmic or nucleopasmic filaments, as well as cytoplasmic or nucleoplasmic bodies (23). Indeed, all six nuclear PML isoforms, when ectopically overexpressed, individually form nuclear bodies even in the absence of endogenous PML (29), with some isoforms contributing not only to nuclear body morphology (27, 30) but also function (31–33).
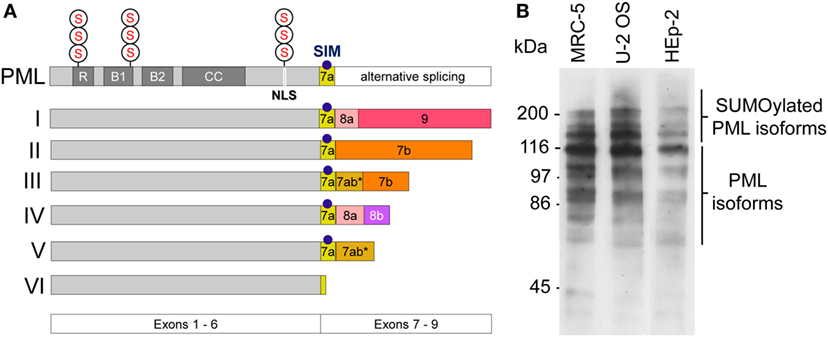
Figure 1. Promyelocytic leukemia (PML) protein isoforms. (A) Schematic depiction of the six nuclear PML isoforms (I to VI). Exons 1–6 are shared by all isoforms while their C-termini are individually different due to alternative splicing of exons 7–9. R, RING domain, B, B box; CC, coiled coil domain; NLS, nuclear localization sequence; SIM, SUMO-interacting motif; S, SUMOylation sites at arginine positions K65, K160, and K490. (B) PML protein expression in various cell lines. Western blot of whole cell lysates derived from MRC-5 (primary human lung fibroblasts), U2OS (Osteosarcoma-derived ALT cell line), and HEp-2 (human epithelial non-ALT cancer cell line) using a rabbit-anti-PML antibody (ABD-030, Jena Bioscience, Germany) at 1:500 dilution. Non-SUMOylated PML isoforms are detectable between 55 kDa and 110 kDa. Poly-SUMOylated PML isoforms are detected above 110–250 kDa.
By immunofluorescence light microscopy, normal PML bodies display as spherical structures, ranging in size from 0.2 µm up to 1 µm (Figure 2A). By electron or super-resolution light microscopy, PML protein is concentrated in a ~100 nm thick shell in the periphery of the nuclear bodies with no chromatin or RNA inside them (34–37). The shell also contains SUMO isoforms and other PML body components, such as SP100 (Figure 2B) (35). This structural arrangement provokes the question on the nature and biological function of the inner core of a PML NB. Within the nuclear body shell, PML’s branched SUMO chains stabilize protein complexes as a “molecular glue” (see next section). In functionally specialized PML bodies, such as in alternative lengthening of telomeres-associated PML nuclear bodies (APBs) and in immunodeficiency, centromeric instability, and facial dysmorphy (ICF) syndrome cells, the inner core of PML bodies contains chromatin, namely telomeric DNA in APBs or pericentric satellite heterochromatin of chromosome 1 in the giant PML bodies of ICF cells (38, 39).
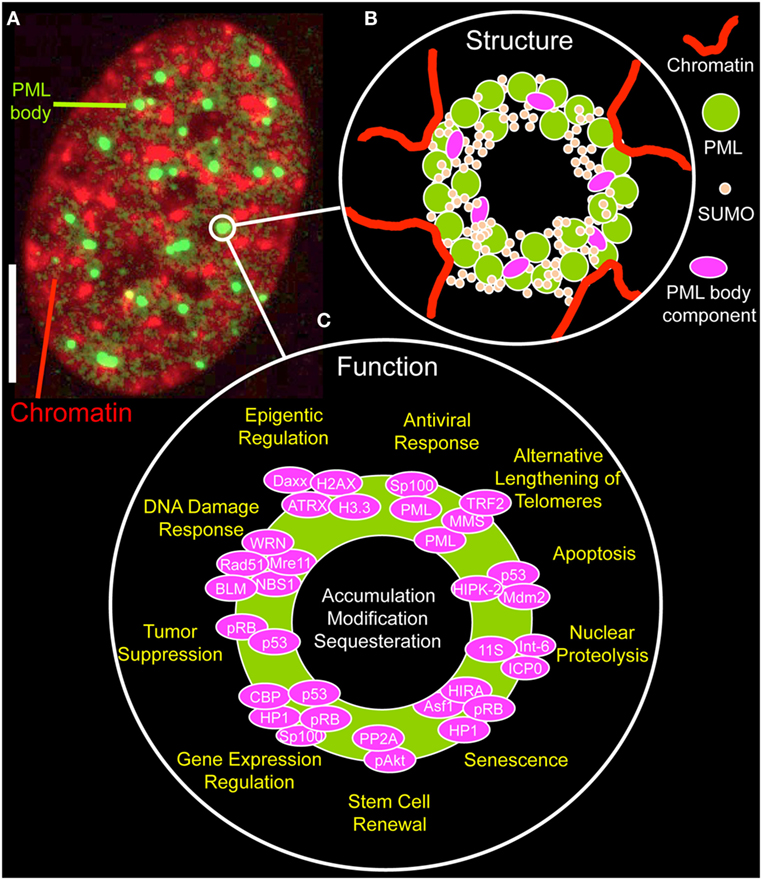
Figure 2. Structure and function of promyelocytic leukemia (PML) nuclear bodies. (A) Distribution of PML protein in a cell nucleus of a MRC-5 (primary human lung fibroblast) cell. The micrograph shows the immunofluorecence signal of an antibody directed to all PML isoforms (green, monoclonal antibody E-11, sc-377390, Santa Cruz Biotechnology, Heidelberg, Germany) along with DAPI fluorescence (red) of a mid-confocal section of the nucleus. Bar; 5 µm. (B) Structure of PML nuclear bodies. SUMOylated PML protein subunits are the building blocks of a shell-like structure in the periphery of the nuclear body. Additional PML body-interacting proteins may bind to PML, specifically to the C-termini of the various PML isoforms, to the poly-small ubiquitin-related modifier (poly-SUMO) chains or to SUMO-interaction motifs. PML nuclear bodies’ are in direct contact with chromatin fibers, which contribute to the bodies physical stability. See Figure 3 for more information on the assembly mechanism. (C) Proposed functions of PML nuclear bodies. Probably more than 100 proteins permanently or transiently bind to PML NBs. According to these protein’s function, many different physiological roles as depicted have been proposed for PML NBs.
The number of PML NBs varies between 5 and 30 depending on the cell-type, the cellular differentiation status, and the cell cycle. During interphase PML bodies are positionally stable through their physical and probably functional interplay with the surrounding chromatin (Figure 2B) (2). Yet, PML NBs are also dynamic structures that undergo significant changes in number, size, and position particularly in response to cellular stress (4). One example is fission of PML bodies into smaller bodies in early S phase (40, 41, 42). PML NBs may lose their structural integrity based on modifications or structural alterations in adjacent chromatin associated with DNA replication.
PML Nuclear Body Function
Systems biological analyses based on online repositories, most notably the Nuclear Protein Database1 (43) have predicted, that more than 150 nuclear proteins have the ability to interact with PML bodies (44, 45). The “Biological General Repository for Interaction Datasets” (BioGRID) lists 243 unique protein interactions.2 Resident factors of PML NBs include, beside all PML isoforms, SUMO paralogs, Daxx, and SP100 (46). Most other factors only transiently accumulate at PML bodies under specific stress conditions or in specialized PML bodies, such as APBs or the giant PML bodies in ICF cells (39). In addition to the telomeric chromatin and shelterin core components, APBs accumulate DNA recombination and repair factors such as the MRN complex, RAD (radiation sensitivity) family members, RPA and WRN (38, 47).
The functional diversity of transient PML NB components is likely the basis of the many different biological roles ascribed to these nuclear structures (Figure 2C) (5, 48). PML NBs have been functionally linked to apoptosis (49), nuclear proteolysis (50), senescence (51), stem cell renewal (52, 53), regulation of gene expression (54), tumor suppression (55), the DNA damage response (40, 41, 56), telomere elongation and stability (47, 57), epigenetic regulation (37, 58), and antiviral responses (59) (Figure 2C). Not surprisingly, functional annotation of PML nuclear body proteins show an enrichment of terms related to cell cycle control, cellular stress response, DNA repair, and protein modification processes (44). More globally, the various aspects of PML NB functions mainly point to their role in genome maintenance (18).
One hypothesis for the integration of all of these functions in a unifying concept is based on the idea that PML NBs provide a stable protein scaffold onto which binding partners associate for their efficient PTM or sequestration (Figure 2C) (28, 60, 61). Controlled accumulation at or release of specific nuclear factors from the nuclear bodies may enhance their functional interaction based on mass-law action, thereby fine-tuning signaling cascades through the nucleoplasm. This mechanism may enable chemical reactions or complex formation between low-abundance nuclear factors, as was also suggested for other subnuclear structures such as Cajal bodies or nucleoli (62). More specifically, PML NBs may be SUMOylation hot spots. This hypothesis is driven by the observation that most components of the SUMOylation machinery concentrate in PML NBs (45).
PML Nuclear Body Assembly
The formation and structural integrity of PML NBs relies on at least five basic mechanistic principles: (i) oxidation-driven intermolecular disulfide cross-linking of PML, (ii) the self-oligomerizing properties of PML’s RBCC motif, (iii) the poly-SUMO chains on the three major target lysines, (iv) the non-covalent interaction of SUMO with SUMO interacting motifs (SIM) in nuclear body-associated factors, and (v) specific sequences in various PML protein isoforms (Figure 3). In the initial step of nuclear body assembly, oxidized PML monomers allow the formation of disulfide-crosslinked covalent multimers that self-organize into the NB outer shell (9, 63). Non-covalent homodimerization mediated by the RBCC domain may be similarly important for the early PML NB assembly step, since the isolated RING domain of PML very efficiently forms multimeres in vitro (64). Subsequently, UBC9-mediated poly-SUMOylation, SUMO/SIM interactions (9, 65) and addition of SUMO and/or SIM-containing binding partners create a mature PML body with a peripheral scaffold consisting of the six different PML isoforms, their SIM motifs and the poly-SUMO chains (Figure 3). Recently, it was demonstrated that certain regions in the C-terminal domains of specific PML isoforms are also important for NB assembly and function (32, 33). These findings add an additional layer of complexity in the structural and functional maintenance of PML NB integrity. The PML nuclear body scaffold offers a multitude of potential sites to which an assortment of PML-interacting, SIM-containing, and/or SUMOylated partner proteins may bind transiently to a more or less extent. The varying residence times (Rts) of binding partners at PML NBs would be expected to depend on the number and strength of their individual interaction modules (66). This is in line with the presence of several SUMOylation sites and SIMs in major PML-NB components including PML, SP100, DAXX, HIPK2, UBC9, PIASy, and RNF4 (9).
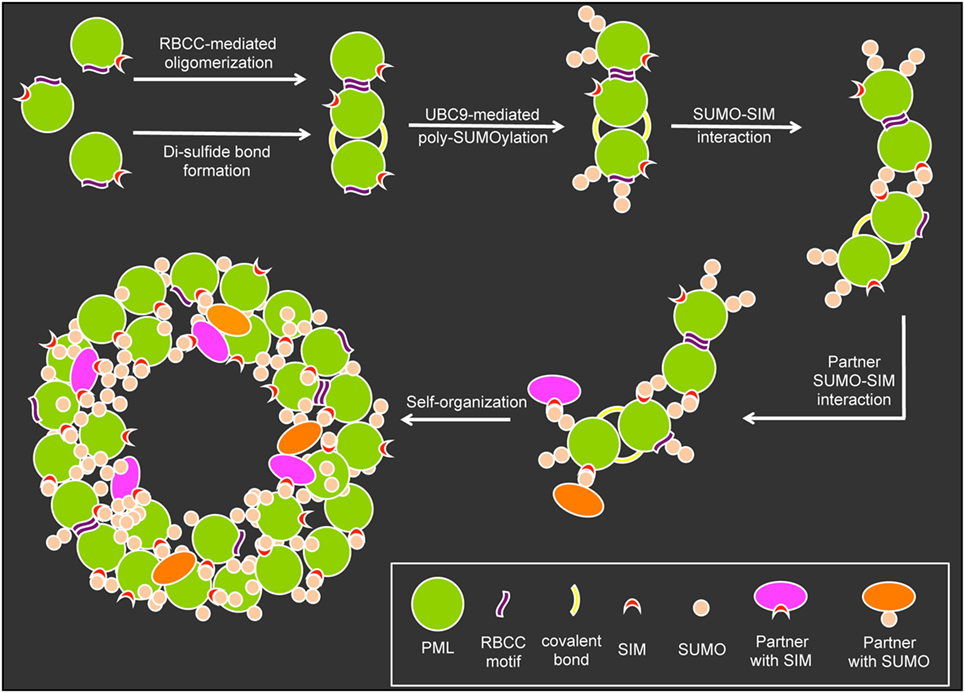
Figure 3. Assembly of promyelocytic leukemia (PML) nuclear bodies. The assembly of PML nuclear bodies is initiated by oligomerization of non-SUMOylated PML monomers. Oligomerization occurs via weak non-covalent interactions through the RBCC motif and covalent di-sulfide bonds between cystein residues. The E2-small ubiquitin-related modifier (SUMO) ligase UBC9 then promotes (poly-)SUMOylation of the PML moieties which allows for multiple SUMO–SUMO interacting motifs (SIM) interaction possibilities to form larger aggregates. Binding partners carrying SIMs and or SUMO residues can bind to the preassembled aggregates to form a normal PML body based on self-organization.
The iterative nature of the multiple binding sites creates a multivalency, which has now been suggested to be responsible for the compartmentalization activity of PML NBs through the biophysical mechanism of phase-separation (67). Although only inferred as probable from GFP-SUMO/RFP-SIM phase-separation data obtained in vitro, the Banani et al. report suggests that that the polySUMO/polySIM interfaces in PML NBs may form phase-separated liquid droplet structures in living cells (68). Thus PML NBs belong to the family of viscous liquid, membrane less nuclear compartments, which may function as phase separating condensates equivalent to lipid droplets (69). The biochemical environment within a phase-separating PML body is different from that in the surrounding nucleoplasm, and this difference could enable unique strategies for regulating nuclear response pathways, including (a) regulation of enzyme reaction kinetics (i.e., posttranslational modifications), (b) regulation of the specificity of biochemical reactions, (c) sequestration of molecules, and (d) buffering cellular concentration of molecules (67).
Cell cycle-dependent disassembly of PML NBs begins upon de-SUMOylation of PML at the onset of mitosis. The spherical shell structure of PML NBs breaks down and other NB components such as SUMO, SP100, and DAXX detach or are removed. During mitosis PML aggregates into so-called mitotic accumulations of PML protein (MAPPs) (40, 41). Interestingly, PML bodies form stable interactions with early endosomes throughout mitosis and the two compartments dissociate in the cytoplasm of newly divided daughter cells (70). When followed through the telophase/G1 transition, Chen et al. observed that GFP-tagged MAPPs become trapped in the newly formed nuclei but also that many PML NBs are formed de novo at different sites in daughter nuclei. This suggests that PML NBs can assemble from both, MAPPs as well as soluble PML monomers in G1 (71). At the M/G1 border of the cell cycle, MAPPs also complex with FG repeat-containing peripheral components of the nuclear pore complex to become CyPNs (cytoplasmic assemblies of PML and nucleoporins) (72). Within CyPNs, PML appears to be instrumental in a novel, nuclear pore-independent, mechanism of nucleoporin and nuclear cargo protein targeting to the reforming G1 cell nucleus (73). The recruitment of SP100 and DAXX into newly formed PML NBs occurs considerably (ca. 30 min) later than PML NB genesis itself, suggesting that tightly controlled PTMs are required for full maturation of functional PML bodies in early G1 (71).
PML in Tumorigenesis
So far, we have summarized some aspects of PML NB biology derived from microscopic, cell, and molecular biology approaches. Another branch of PML research has tackled questions on PML protein function by means of genetics. These approaches uncovered PML’s role in cancer biology. PML was originally identified as a potential gene of interest in tumorigenesis due to its association with acute promyelocytic leukemia (APL). APL is a rare but aggressive subtype of white blood cell cancer, characterized by an accumulation of promyelocytes in the bone marrow and peripheral blood (74). The majority of APL patients are characterized by the t(15;17) chromosomal translocation that reciprocally joins the PML and retinoic acid receptor α (RARα) genes, resulting in balanced expression of PML-RARα and RARα-PML fusion proteins (1). While PML-RARα blocks differentiation of promyelocytes by suppressing the transcriptional function of RARα, PML-RARα disrupts the structure of PML nuclear bodies through formation of PML-RARα/PML heterodimers. This phenotype was observed in 99% of APL patients (75). Treatment of APL for many years was retinoic acid, arsenic trioxide or a combination of the two, which, fortunately, seemed to cure most APL patients. It is now known that the mechanism of this therapy involves targeting of the PML/RARα fusion protein to proteasomal degradation (76). Strikingly, drug treatment reverses the pathological microspeckled PML distribution in the nucleus of APL cells toward the regular morphology of PML nuclear bodies (77).
Another link between cancer and PML became evident by comparing PML protein expression in normal and neoplastic human tissues. Such studies documented loss of PML expression in breast carcinoma (78), gastric cancer (79), small cell lung carcinoma (80), and in invasive epithelial tumors (81). Furthermore, microarray analyses of PML mRNA expression showed complete loss of or strongly reduced PML transcript expression in many different human neoplasms, including colon, prostate, and breast adenocarcinomas, as well as in lung, CNS, germ cell, and non-Hodgkin’s tumors/lymphomas (8). The same study reported that PML protein is also frequently overexpressed in carcinomas of larynx and thyroid, epithelial thymomas, Kaposi’s sarcoma, and in Hodgkin cells, a tumor of cytokine-producing cells. The latter phenomenon may be attributable to strong upregulation of the PML gene after Interferon induction (82). Taken together, loss of expression in many (but not all) cancer types have suggested that PML works as a tumor suppressor (83).
Tumor Suppressor and Oncogenic Functions of PML
Beside the correlative connection between carcinogenesis and PML expression, there is plenty of experimental evidence for a direct tumor-suppressive role of PML. Several independent studies have demonstrated that overexpression of PML can slow down or block cell cycle progression in a variety of cancer cell lines (6, 81, 84). Likewise, in primary human or mouse fibroblasts overexpression of PML isoform IV induces a stable senescence-associated cell cycle arrest (85, 86). Further analyses of typical stress-response pathways revealed the involvement of the tumor suppressors pRb and p53 in PML overexpression-induced cellular senescence (86, 87). However, the molecular details of PML action along the pRB and/or p53 tumor suppressive pathways remain elusive. Besides in cellular senescence, PML has an essential functional role in apoptosis (49). This is based on initial observations on the first reported PML knock-out mouse model, where splenic lymphocytes and thymocytes from Pml−/− mice show barely half the capacity of wild-type cells to initiate apoptosis after ionizing radiation or after induction of the cytokine death-receptor pathway (88). As already pointed out, PML loss correlates with the progression of many cancers and in most cases low PML expression is associated with poor prognosis. The tumor suppressor function of PML NBs may be linked to their ability to accumulate many proteins involved in DNA damage response and repair pathways, which is believed to stabilize DNA repair complexes and enhance their activities (4, 13, 60). In support of this hypothesis, it was shown recently in a knock-in mouse model, that intact PML bodies are critical for DNA damage responses. Functional assays in mice expressing PML but lacking PML NBs showed impaired homologous recombination (HR) and non-homologous end-joining repair pathways, with defective localization of Brca1 and Rad51 to sites of DNA damage (89). Thus, although the physiological function of PML and the nuclear bodies have not been thoroughly elucidated, their tumor-suppressive role by supporting DNA damage response pathways may be common to all of these potential functions (19, 89).
The lack of PML is not necessarily a tumor-promoting event. Functional analysis of the hematopoietic stem cell compartment in mice have uncovered that PML is required for leukemia initiating cell maintenance (90). The authors suggest a new therapeutic approach for eradication of cancer-initiating cells in leukemia through pharamacological inhibition of PML. This and other reports have lead to the suggestion that PML may act as both a tumor suppressor and an oncogene, depending on the cellular context (91). Along these lines it was also demonstrated that PML targeting impacts on breast cancer (BCa)-initiating cell function, and hence on cancer initiation and dissemination in BCa (92). Furthermore, in triple-negative breast cancer cell and mouse models PML promotes cell migration, invasion, and metastasis through binding to regulatory regions of HIF1A target genes (93). These initially unexpected findings clearly suggest a previously underestimated importance of PML in the maintenance of some tumors.
Laser-Based Fluorescence Imaging and Manipulation Approaches to Analyze Nuclear Protein Function
Inter- and intracellular mechanisms of molecular communication may be better understood through direct visualization. In the past decades, advancements in imaging technologies have expanded our ability to access and analyze in living specimen the morphology of tissues and cellular components. These enabled analyses of fine-structural features at the nanoscale level, precise localization, and the dynamic interplay of single and macromolecular assemblies that drive cell growth, the cell cycle, differentiation, and cell death (20–37). Super-resolution light microscopy delivered images with unprecedented sensitivity and clarity allowing the exploration of interactions between individual molecules with a distance resolution as low as 20 nm (94). New fluorescence fluctuation microscopy (FFM) approaches provided the basis for determining the biophysical and interaction properties of single molecules in living cells (95, 96). Laser-based FFM analysis tools are outlined below but many more exist, all of which unfortunately cannot be covered by this overview, including single particle tracking (SPT), light sheet microscopy, total internal reflection microscopy just to name a few. To acquire the full picture of live cell laser-based imaging technologies, we refer to recent excellent reviews on these topics (20, 97–99). Altogether, a plethora of new live cell imaging techniques have been developed which even large research groups are unable to establish to a broad extent in their departments. To address this, dedicated advanced light microscopy imaging facilities are extremely helpful as their members are usually microscope experts (100). However, we believe that research laboratories are still reluctant in exploiting the full potential of microscopy facilities. We therefore provide an introductory overview on some imaging instrumentation which are covered by such facilities and provide specific examples in PML biology to encourage cancer cell biologists and biochemists to extend their experimental approaches toward the exciting new imaging technologies.
Fluorescence Recovery after Photobleaching (FRAP)
Arguably, one of the most commonly used approaches to study dynamic cellular processes in living cells is FRAP (101). FRAP is able to access average dynamics of diffusing molecules within the observation volume. The original description of FRAP was coined continuous fluorescence microphotolysis, which itself has been established for more than three decades (102). When subjected to repeated cycles of excitation and emission, fluorescent molecules eventually lose their ability to emit fluorescence, enabling the creation of photobleached spots of fluorescent molecules in solution or in living cells by the application of a laser beam. By monitoring the redistribution of the fluorescent molecules from the unbleached volume, their diffusion or transport properties can be assessed (101). FRAP and related techniques such as point continuous photobleaching, fluorescence loss in photobleaching, inverse-FRAP, and photoactivation/conversion have been developed in the past, each suitable to quantitatively assess specific biophysical properties of the molecule under investigation (97). However, the limitations and pitfalls of FRAP experiments, in particular when they are employed to extract biophysical parameters also, have to be considered. Things to consider include the complete set-up of the FRAP experiment (103), knowledge on the bleach volume profile (104), as well as the potential phototoxic effects elicited by the bleaching laser beam (105).
Figure 4A shows a typical FRAP experiment for GFP-tagged PML (isofom V) at nuclear bodies. Measuring the redistribution of fluorescence into the bleached region then yields the FRAP recovery curve (Figure 4B). During the observation time of the FRAP experiment, the fluorescence in the bleached region may return to the prebleach value (Figure 4B, curve A) or not (Figure 4B, curve B). Incomplete recovery even after long observation (>1 h) has been observed for some chromatin-binding proteins, which suggests the presence of immobile or very slow exchanging populations of molecules (106, 107). FRAP data can be analyzed using mathematical models to yield kinetic parameters (108). Today, many FRAP models of processes in the cell nucleus assume that the proteins undergo diffusion as well as binding/unbinding events at chromatin or other more static subnuclear structures such as nuclear bodies. Importantly, both diffusion and binding/unbinding events contribute to the spatial dynamics of nuclear proteins (109, 110).
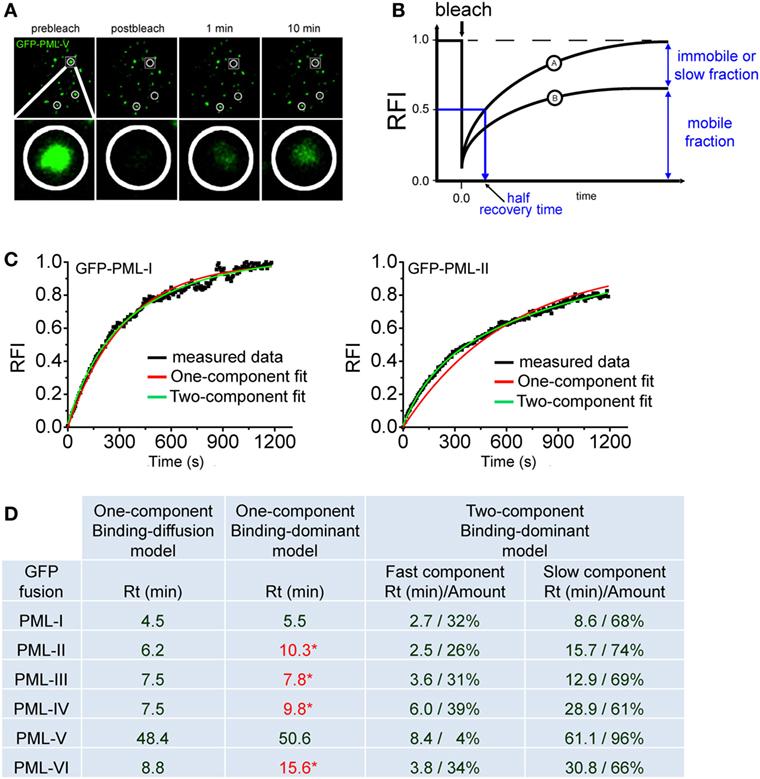
Figure 4. Fluorescence recovery after photobleaching (FRAP) to assess component exchange at promyelocytic leukemia (PML) nuclear bodies. (A) A typical FRAP experiment is shown. Two circular regions in the nucleus of a GFP-PML-V expressing U2OS cell were exposed to a short 488 nm laser bleach pulse and fluorescence redistribution was monitored over time. A third circled unbleached region at the bottom of this nucleus is shown as a positive control. One particular bleach spot is shown in a magnified view in the bottom panels. (B) Quantitation of FRAP experiments. After background subtraction, compensation for imaging-induced photobleaching and normalization, typical FRAP curves are obtained. FRAP curve A shows full recovery to prebleach fluorescence values indicating complete exchange of the GFP-tagged protein in the bleached spot within the observation time. In FRAP curve B, fluorescence recovery is not complete within observation time indicating an immobile fraction of molecules or a fraction with a very slow exchange rate. (C) FRAP curve fitting using exponential functions. FRAP curves for GFP-tagged PML-I (left) and PML-II (right) were fitted with one-component (red) or two-component (green) exponential functions. (D) Table showing the residence time (Rt) in minutes of GFP-tagged PML isoforms I to VI derived from fitting FRAP curves employing different mathematical models (See text for details). * numbers in red letters represent Rt values derived from one component modeling which failed to precisely fit to the measured FRAP curve as shown for GFP-PML-II in (C).
With respect to PML protein exchange at nuclear bodies it is safe to assume a binding-dominant behavior because of the very slow exchange rates as observed by FRAP (Figure 4A). Previously, the residence time (Rt) at nuclear bodies of all PML isoforms had been determined by FRAP using a binding-diffusion model based on differential equations (29, 66). It was therefore interesting to compare different modeling approaches. Figure 4C shows two examples of fitting FRAP data obtained for GFP-tagged PML isoforms I and II. Interestingly both, one- and two-exponential functions delivered good fits to the FRAP curve for GFP-PML-I but not for GFP-PML-II, where only a two-component exponential function gave good fit results (Figure 4C).
To obtain a complete picture we collected the Rts of all PML isoforms after fitting to one- and two-component exponential functions (Figure 4D). This approach delivers the Rt of the protein under investigation (110). One-component exponential fits were successful for GFP-tagged PML-I and PML-V, while FRAP curves for the other isoforms could only be fitted with two-component exponential fits (Figure 4C and data not shown). For comparison, the table includes the data we previously obtained by application of a binding-diffusion model based on more sophisticated differential equation modeling to analyze the FRAP curves (66). The table shows that the Rts of PML isoforms at nuclear bodies as deduced from one-component exponential fits, is convincingly close to those obtained from assuming a binding-diffusion model (Figure 4D) although the fits are not satisfactory for PML-II, -III, -IV, and -VI (values in red letters). In particular, the very long Rt of PML-V (~50 min) is confirmed. This observation is fully consistent with the presence of a strong homo-dimerization domain we found in the unique C-terminus of PML isofom V (32). Obviously, this domain in PML-V confers additional binding strength toward PML bodies. Fitting with two-component exponential functions assumes the presence of two populations of molecules exchanging at PML bodies with different on/off rates. These functions provided perfect fits for all PML isoforms (Figure 4C, green curves, and data not shown), and the Rts are shown in Figure 4D. Interestingly, the two-component fits deliver considerably large populations of PML isoforms IV (61%) and VI (66%) with a Rt of ~half an hour (Figure 4D). This suggests that a subfraction of these isoforms may contribute to the structural integrity of nuclear bodies through stable incorporation.
In conclusion, Figure 4 suggests that different FRAP modeling approaches, despite subtle differences, arrive at overall similar Rts for PML isoforms at nuclear bodies. It should be noted however that these long Rts do not necessarily reflect the time in which one PML molecule stays bound to one and the same specific binding site. Long Rts may also originate from PML molecules undergoing rapid binding and unbinding events at multiple adjacent binding sites (in our case at the nuclear body) without leaving the observation volume (110). If binding/unbinding events do not occur on well-separated time scales, the interaction parameters may not be readily extractable from the FRAP curves (111). A combination of different FFM approaches may be required for accurate determination of binding parameters (112). To assess binding/unbinding events at PML NBs at higher resolution, the tool kit should be extended to single particle tracking (SPT) since this approach is able to quantitatively describe several populations of molecules with distinct binding properties (113).
Fluorescence (Cross) Correlation Spectroscopy
Fluorescence correlation spectroscopy (FCS) is an in vivo method that analyses diffusing particles in a diffraction-limited illumination ellipsoid (114, 115). The FCS detection volume is created by a laser beam in a pinhole-adjustable confocal optical system focused through a high numerical aperture objective (Figure 5A). The FCS detection volume is defined by the point spread function of the objective and the confocal pinhole. The excitation laser beam determines how much of the detection volume is excited and the final observation volume is determined by the overlap of excitation and detection volumes. For objectives with a high numerical aperture (i.e., NA = 1.4) the effective measuring volume is ~1 fl (116). Photons emitted from diffusing fluorescent particles are counted continuously over time through the same optics using sensitive avalanche photodiodes (APDs) or galliumarsenidephosphide (GaAsP) hybrid detectors at single molecule resolution (Figure 5A) (117). The fluorescence intensity fluctuations are recorded over time (Figure 5B). Particle concentration is reflected by the fluctuation amplitude, whereas the frequency gives information on the diffusion times of the fluorescent particles. For quantitative evaluation, the photon trace is correlated with a time-shifted replica of itself (autocorrelation) at different time values (Figure 5C). The amplitude of the autocorrelation curve is inversely proportional to the average number of fluorescent molecules in the confocal volume allowing determination of particle concentration (Figure 5C). A more detailed overview on the theory, history, and application of FCS can be found here: (118, 119).
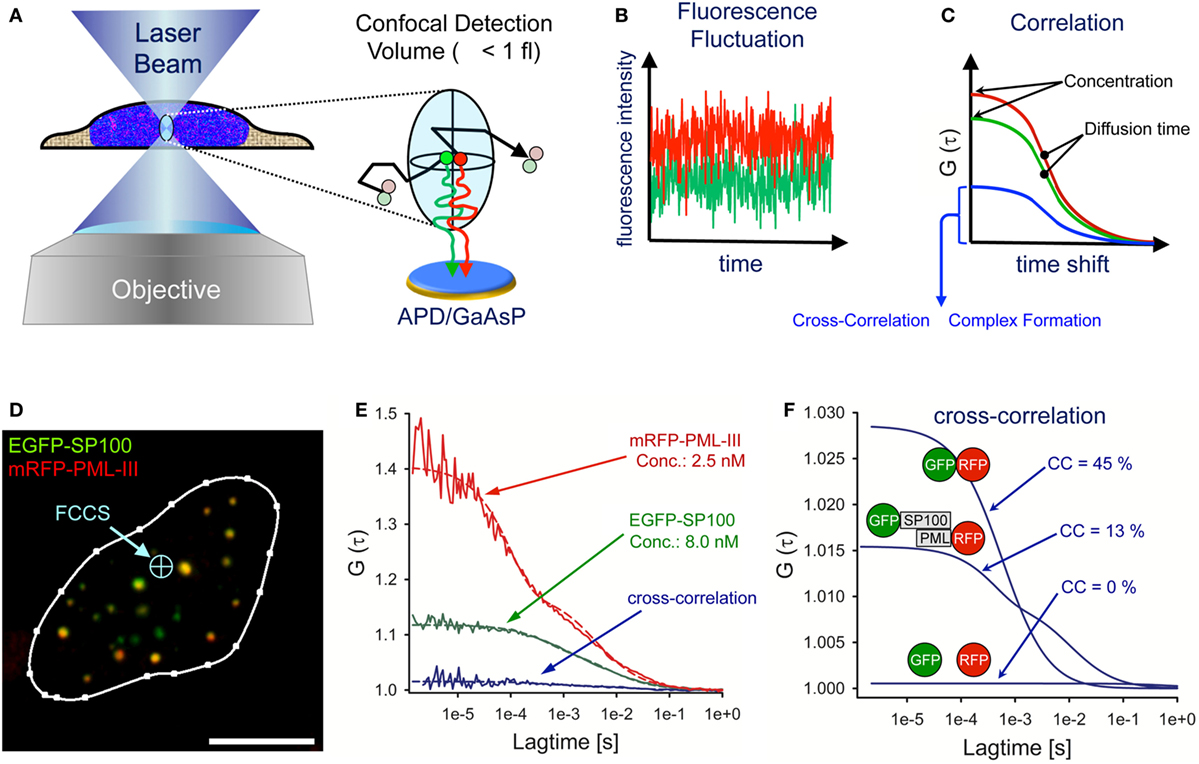
Figure 5. Fluorescence cross-correlation spectroscopy (FCCS) analysis of promyelocytic leukemia (PML) nuclear body components. (A) Schematic side view of a living cell with the FCS laser beam focused to a position within the nucleus. The objective creates a laser light-illuminated subfemtoliter measuring volume in which single fluorescent molecules are excited to emit photons. The photons are counted on an avalanche photodiode (APD) or a galliumarsenidphosphid (GaAsP) detector as a time series of fluorescence intensity (B). These statistical fluctuations are mathematically processed using an autocorrelation algorithm, from which biophysical parameters such as the particle concentration, the diffusion coefficient and complex formation properties can readily be assessed (C). (D) Confocal live cell image of a U2OS nucleus coexpressing EGFP-SP100 and mRFP-PML-III (bar: 5 µm). The FCCS laser beam (light-blue) can be positioned anywhere in the cell. (E) By fitting the measured FCS data points (solid lines) to appropriate diffusion models (dashed lines), one can extract from the reciprocal of the amplitude and the decay half-time value the number of particles in the detection volume (concentration) and the diffusion time, respectively. The cross-correlation (CC) result of EGFP-SP100 and mRFP-PML-III are also shown. (F) CC results in the nucleus of living cells for a GFP–RFP fusion protein (positive control, high CC), GFP and RFP as individual proteins (negative control, no CC) and the measurement performed in (D,E).
In fluorescence cross-correlation spectroscopy (FCCS), two spectrally distinct fluorophores are measured in the same detection volume at the same time (Figures 5A,B, red and green lines) and correlated by cross-correlation (CC) (Figure 5C, blue curve). The amplitude of the CC curve is directly proportional to the degree of complex formation and/or direct interaction between the two fluorescent particles (120). A practical guide to set up FCS and FCCS experiments in living cells can be found here (121–123).
An example of FCCS measurements of PML body components is shown in Figure 5D. The image shows a live-cell confocal snapshot of a U2OS cell nucleus transiently expressing EGFP-SP100 and mRFP-PML III. The FCS laser spot was parked at a position in the nucleoplasm where the fluorescence signals of the fusion proteins are extremely low (Figure 5D, blue arrow). EGFP and mRFP fluorescence fluctuation was then recorded over time (10 × 10 s measurements) and the fluctuation data correlated for each fluorophore (Figure 5E). By fitting theoretical model functions to the measured autocorrelation curves, the diffusion coefficient and the concentration of the diffusing species can be extracted. In this particular cell nucleus (Figure 5D), the concentration of EGFP-SP100 and mRFP-PML-III was 8.0 nM and 2.5 nM, respectively, demonstrating the power of FCS to work at extremely low expression levels (Figure 5E). The diffusion coefficient in the nucleoplasm outside nuclear bodies for these PML body components had been determined previously (DSP100 = 1.23 µm2 s−1, DPML-III = 1.63 µm2 s−1) (66).
Cross-correlation analysis between EGFP-SP100 and mRFP-PML-III revealed a small but significant amplitude above the value of 1.0 (Figure 5E, blue curve indicated with GFP-SP100 and RFP-PML), indicating the formation of complexes between these fusion proteins. To evaluate this observation, the CC was compared with values obtained for individually expressed GFP and RFP molecules (negative control) as well as a GFP-RFP fusion protein (positive control) (Figures 5E,F). Experiments with these fluorochromes determine the dynamic range of the FCCS set-up. The mathematical delineation of the CC values is described elsewhere (124). Analyzing EGFP and mRFP as single molecules in our system resulted in CC = 1.001, indicating 0% complex formation while for the mRFP-EGFP fusion protein we observed a CC amplitude of 1.029, corresponding to 45% complex formation. The CC value for EGFP-SP100 and mRFP-PML-III was CC = 1.010, indicating that in this cell nucleus ca. 13% of SP100 molecules reside in a complex with PML (Figures 5E,F). These analyses demonstrate that FCS and FCCS, although technically somewhat more demanding than for example FRAP experiments, provide an extremely powerful tool to precisely extract biophysical and interaction data on diffusing molecules of interest in living cells.
Raster Image Correlation Spectroscopy (RICS)
Ideally, in the assessment of biophysical parameters of mobile molecules in living specimens, one wants to know the space-resolved behavior of single molecules in terms of their kinetics and interactions and without the disturbance of the equilibrium state, as occurs in FRAP. All of these parameters are provided by RICS (125). Data acquisition in RICS is quite simple as only a 2D confocal image or time series analysis is required. The scanning encodes dynamic information within a single image, which can then be extracted using RICS. The processing of the resulting images, however, is not trivial: RICS data are computed from the power spectrum of the spatial autocorrelation function that is obtained from the intensity images by 2D fast Fourier transformation algorithms (125).
Raster image correlation spectroscopy thereby expands the accessible timescales of FCS as it can resolve dynamics in the range of microseconds to seconds with still a sufficient spatial resolution (125). Data in cells are most conveniently acquired as a time series stack by raster scanning of images of selected cell areas. Due to its broad dynamic access by analyzing the fluctuations between neighboring pixels in the x- and y-direction, nearly all diffusion processes that take place in cellular subregions can be studied (125). A major advantage of the RICS technology is that it can be used in principle on any commercial confocal microscope with analog detection (126). The software and application tutorials developed by the Enrico Gratton lab can be found as downloads here.3
To measure RICS we have used a Zeiss LSM710 which conveniently provides a built-in RICS module in the ZEN microscope software. Some examples of RICS measurements are shown in Figure 6. As a positive control, a confocal time series was acquired in a subregion of a U2OS cell expressing EGFP (Figure 6A). RICS analysis then delivers a spatial correlation of this region (Figure 6B). By fitting of the correlation data with a 3D-free diffusion model, a spatially resolved diffusion coefficient map is generated (Figure 6C). This map shows that EGFP diffuses throughout the cellular volume with variable diffusion coefficients ranging between 5 µm2 s−1 and 50 µm2 s−1. The mean value for EGFP in the nucleus by RICS was 25 (± 5) μm2 s−1 (n = 12) consistent with FCS data (127). The RICS approach also delivers a map of the number of detected mobile molecules in the diffusion analysis (Figure 6D). RICS was then applied to a U2OS cell nucleus expressing EGFP-PML (isoform IV) (Figures 6E–M). Two subregions of the 2D confocal time stack were selected for RICS analysis. RICS analysis in the nucleoplasm (Figures 6F–I) showed that the diffusion coefficient of GFP-tagged PML-IV is about one order of magnitude smaller than that of GFP alone (Figure 6H), consistent with FCS measurements (66). This suggests incorporation of PML into larger complexes and/or interaction with an immobile structure (i.e., chromatin), or both. RICS can also be performed on large (but not small) PML nuclear bodies (Figures 6J–M). The resulting diffusion map reveals very slow diffusion of GFP-PML-IV at or within PML NBs (Figure 6L), suggesting that binding events predominante PML molecule mobility at or in the nuclear body.
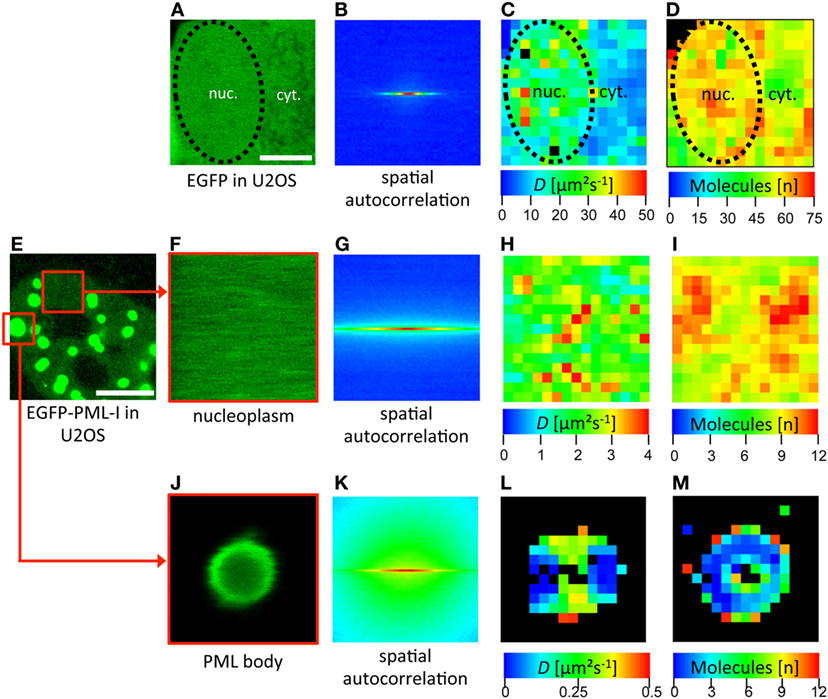
Figure 6. Spatial mapping of promyelocytic leukemia (PML) protein mobility in the nucleus by raster image correlation spectroscopy (RICS). (A–D) As an introductory example, RICS was performed in a U2OS cell expressing EGFP alone. (A) Shows a subregion of the cell containing nuclear (nuc.) and cytoplasmic (cyt.) parts. The nuclear outshape is indicated by a black dashed line. For RICS analysis, a time series of GFP fluorescence images was acquired by confocal microscopy. Thus, the fluorescence intensity of each pixel is collected and the spatial autocorrelation is obtained per image (B). The image stack serves to increase the SNR and to be able to remove immobile and slow molecules. This analysis generates spatial maps of the diffusion coefficient (C) and the number of free molecules which contributed to the assessment (D). Since the shape of the spatial autocorrelation image indicates mostly freely diffusing species, the RICS data were fitted with a one-component 3D-free diffusion model yielding diffusion coefficients for EGFP in the nucleus between 10 µm2 s−1 and 50 µm2 s−1 (C). RICS was then performed similarly in a EGFP-PML-I-expressing U2OS cell (E–M). In the nucleoplasm, the diffusion coefficient of EGFP-tagged PML-I ranged between 1 µm2 s−1 and 4 µm2 s−1 (H). RICS in a region containing a large PML NB (J) still delivered acceptable spatial autocorrelation quality (K). This approach revealed a diffusion coefficient of EGFP-PML-I in or at PML bodies which was one order of magnitude lower than in the nucleoplasm (L). Bars; 5 µm.
The examples shown illustrate the power of RICS to determine spatial maps of concentrations, aggregation, diffusion and binding of mobile molecules in living cells using readily accessible instrumentation (125).
Förster Resonance Energy Transfer (FRET)
The FRET process is a dipole–dipole interaction in which an excited donor fluorophore transfers energy to an acceptor molecule in nanometer vicinity without absorption and emission of a photon (128). FRET is therefore commonly employed to measure the spatial distance between two fluorophores in fixed as well as in living cells (129). The FRET efficiency depends on the distance between two adjacent fluorescent molecules. At the Förster radius distance between a FRET pair (typically around 5 nm), the FRET efficiency is 50% (130). This size regime is comparable to the size of many proteins, the distance within which proteins interact, and the distance between sites on multisubunit proteins. Therefore, FRET can deliver parameters on the distance between two distinct sites on a macromolecule, the distance between two fluorophore-tagged proteins, and hence if and how these two proteins interact (131). Basically five different FRET detection methods have been developed for light microscopy, including acceptor photobleaching, donor photobleaching, ratio imaging, sensitized emission, and fluorescence lifetime measurements (132).
In the past, we have mainly used acceptor photobleaching FRET (abFRET) to analyze spatial proximities within chromatin-interacting complexes (133). In abFRET, the acceptor chromophore is photobleached, thereby preventing FRET from the donor to the acceptor (Figure 7A). If the donor and acceptor were in sufficient proximity for energy transfer, photobleaching the acceptor results in an observable increase in donor fluorescence (Figure 7B). The measurement of abFRET only generates positive values when the distance between the donor and acceptor (in our case EGFP and mRFP, respectively) is between 3 and 8 nm. An abFRET example is shown in Figure 7C, where EGFP-Sumo-1 (green) and mRFP-PML I (red) are coexpressed in living cells. Two PML bodies were selected for analysis and acceptor photobleaching performed in region 1 but not in region 2 which served as internal control for non-FRET effects (Figure 7C). For quantitation, FRET efficiencies in bleached and unbleached regions are then plotted in a bar diagram (Figure 7D). The plot indicates that FRET between EGFP-SUMO-1 and mRFP-PML-I occurred in most of the cases (mean of FRET = 5.5%; black bars in Figure 7D), while the unbleached control spots show a FRET mean value of −1.4% (gray bars in Figure 7C). The FRET efficiency distribution is significantly different from that in control regions (p < 0.001, n = 88) (Figure 7D). Thus, Sumo-1 is in close proximity to PML I at PML NBs which was expected because of the covalent conjugation of SUMO-1 to PML in nuclear bodies (134). A similar abFRET approach has previously been used to document the functional interaction between the CHFR mitotic checkpoint protein and PML within PML NBs (135).
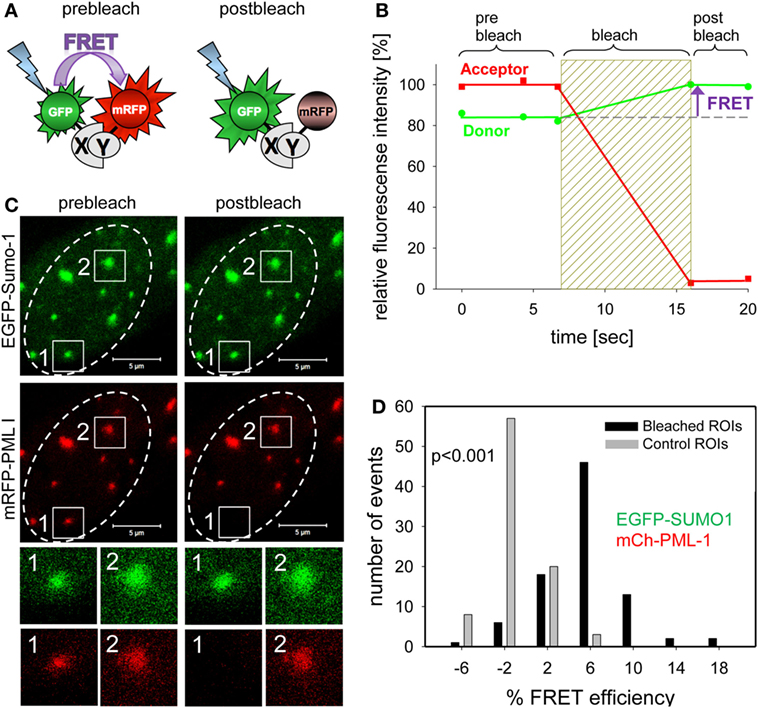
Figure 7. Complex formation assessment of promyelocytic leukemia (PML) body components by acceptor-photobleaching Förster resonance energy transfer (FRET). (A) Schematic explanation of acceptor-photobleaching Förster resonance energy transfer (abFRET). The energy donor EGFP and the energy acceptor mRFP are fused to proteins X and Y, which are sufficiently close (<10 nm) to each other to allow for FRET. Left side: the acceptor mRFP absorbs radiation-free energy from the exited donor EGFP resulting in decreased donor fluorescence intensity. Right side: acceptor mRFP is bleached and energy is no longer transferred from donor EGFP to acceptor mRFP resulting in an increase of donor fluorescence intensity. (B) Time courses of the fluorescence intensity of donor and acceptor during abFRET. Bleaching of the acceptor results in a fluorescence intensity increases of the donor indicating FRET (arrow). (C) Cell nucleus showing the location of EGFP-Sumo-1 and mRFP-PML I in PML-bodies. Two of them, spot1 and spot2, were selected for fluorescence intensity analysis before and after acceptor-photobleaching (see enlargements below). At spot 1, the acceptor fluorophore mRFP was bleached (compare prebleach and postbleach), whereas spot 2 was not bleached and served as control. (D) The donor fluorescence intensity variation observed during acceptor-photobleaching was determined for 89 not bleached control PML-bodies (see spot 2) yielding Evar (gray bars) and for 89 acceptor-photobleaching PML bodies (see spot 1) yielding EFRET (black bars). The numbers of observed single cases (grouped into Evar or EFRET value ranges of 4%) are displayed versus the values of Evar and EFRET.
By expanding this abFRET approach, the individual spatial relationships between many PML NB components and probably even the degree of PML SUMOylation could now be determined to obtain a full picture of the molecular interaction landscape within PML NBs. This kind of approach proved successful in detecting the spatial inter-relationships within the large human kinetochore complex (136) as well as in smaller complexes such as the nucleosome (137). Thus, adding FRET techniques to the experimental tool kits in many laboratories would significantly increase the understanding of protein-protein interaction networks in cancer biology.
Laser Microirradiation
Experimental induction of DNA damage foci in living cells became an ideal method to analyze in time and space the recruitment and binding properties of repair factors at DNA lesions (138). Live cell imaging of DNA damage foci has been used to reveal the mobility of repair proteins, their assembly timing into repair sites, and the movement of damaged chromatin (139–141). DNA damage can be induced globally by ionizing radiation or radiomimetic drugs allowing for bulk analysis of the DNA damage response at multiple sites of DNA damage in the nucleus (142). Generation of single focal spots of DNA damage is instrumental to analyze single repair sites and became possible by targeted expression of endonucleases or microirradiation (143). Coupling of UV-A light-emitting lasers into confocal microscopes resulted in the development of laser-microirradiation technologies (Figure 8A) (144, 145). Laser lines in the visible range spectrum (405–514 nm) as well as multiphoton excitation (>750 nm) have been implemented in such devices (146). The advantages and disadvantages of the different laser systems to study cellular responses to DNA damage has been assessed (147). By fine-tuning the microirradiation system it is possible to discriminate between induction of base lesions, single–strand and double-strand DNA breaks. Special UV-suitable lenses (i.e., quartz glass) must be used to avoid energy loss and destroying the lenses. Objectives with a high numerical aperture should be employed to achieve diffraction-limited focusing and fine micromanipulation. A starting point to plan the application of microirradiation techniques can be found here: (148, 149).
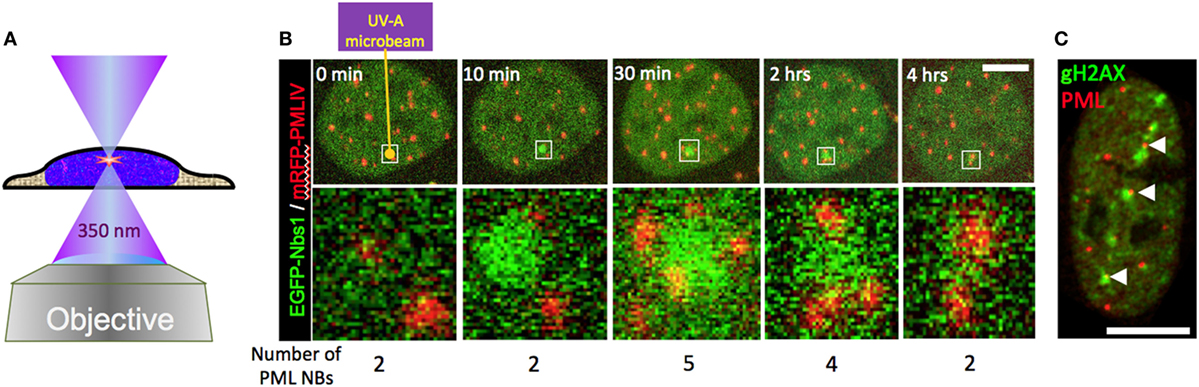
Figure 8. Behavior of promyelocytic leukemia (PML) bodies at UV-A microbeam induced DNA damage foci. (A) For laser damage induction, a pulsed 350 nm Nd:YLF (neodymium-doped yttrium lithium fluoride) UV-A laser (Spectra Physics) was coupled into a confocal laser scanning microscope (LSM 510) via the epifluorescence illumination path. The laser-microbeam is focused into the middle of the field of view by a 100×, NA 1.3 Plan Neofluar oil immersion objective (Zeiss). The Nd:YLF laser can be frequency-tripled delivering 20 ns duration pulses at 350 nm with user-defined energies from 1 µJ to 200 µJ at user defined repetition rates 1–1000 Hz. (B) A living U2OS cell expressing EGFP-tagged NBS1 and mRFP-tagged PML-IV was irradiated at a single defined spot with approx. 5 µJ of energy using the set-up described above (yellow arrow). Confocal stacks were acquired across the nucleus before and at indicated time points after the damage pulse. In (C), U2OS cells were microirradiated at multiple positions with high laser power (40 µJ per site). Twenty-four hours after DNA damage induction cells were fixed and stained to detect the DNA damage marker gH2AX (green) and endogenous PML (red). Scale bars, 5 µm.
The involvement of PML NBs in cellular DNA damage response/repair pathways became evident upon the demonstration of their colocalization with experimentally induced DNA damage foci (12, 13, 56, 150). UV laser microirradiation was also used in the initial studies to analyze in more detail the behavior of PML NBs in the vicinity of DNA damage in live cells (40, 41). These studies revealed intriguing morphological changes of PML NBs near the damaged chromatin, including moving toward the breaks, coalescing, and loss of positional stability. A recapitulation of these observations is shown in Figure 8B. In this experiment, a short pulse of 350 nm UV light was focused into the nucleus of a U2OS cell expressing GFP-tagged NBS1 (A DNA damage sensor protein) (green) and mRFP-tagged PML (red). As expected, NBS1 accumulates focally at the microirradiated chromatin spot within minutes. Shortly after, new PML bodies appear at the periphery of the damage focus (30 min). After 4 h, NBS1 has detached from the irradiated area and the number of PML NBs returned to preirradiation levels (Figure 8B, 4 h). The repair process has most likely been successfully completed by that time although direct evidence for successful repair is lacking. All these observations are consistent with previously reported data (40, 41). When DNA damage becomes irreparable, repair foci become permanent, as demonstrated for damaged telomeres (151). PML bodies stay stably associated with such irreparable DNA breaks (152). This phenomenon is illustrated in Figure 8C, where a U2OS cell was microirradiated at multiple locations in the nucleus with a high UV-A laser dose (40 μJ pulse−1) and stained for PML and gH2AX 24 h after damage induction. Interestingly, UV-induced DNA damage foci that colocalize with PML NBs are positionally more stable than non-colocalizing (14), suggesting that PML NBs may function to support topographic stability of DNA repair foci within chromatin.
Of course, these fascinating microscopic observations remain descriptive without supporting functional studies. Previously it was shown that depletion of PML indeed decreases the ability to perform homologeous recombination (HR) repair (15, 16), and it should also be mentioned that permanent lack of PML induces genomic instability and increased susceptibility to cancer (11). Thus it would be now interesting to fine-dissect the molecular mechanisms by which the presence of a PML NB is supportive to DNA repair events at particular DNA damage foci. A straightforward model would be a scaffold or platform function of the bodies for efficient biochemical repair activities nearby damaged chromatin. A combination of super-resolution techniques with live cell imaging after microirradiation is an attractive approach to further study this phenomenon.
Optical Tweezer (OT): Erythrocyte-Mediated Force Application (EMFA)
Since their introduction in 1986 by Ashkin et al. (153), OTs have developed rapidly over the past decades (154). OTs are today widely used tools in physics, chemistry, biological, and medical research (155). OTs are applicable to objects at nanometer up to several micrometer size ranges. The simplest form to use OTs is by focusing a laser beam using an objective lens of high numerical aperture (Figure 9A). As the rear pupil of the objective must be entirely illuminated, the diameter of the laser beam is expanded by telescope optics before directed to the microscope. Dielectric particles such as small biological objects near the focus will mainly experience two forces: radiation pressure in the direction of light propagation and gradient forces in the direction of the spatial light gradient. The balancing of both forces is required. The equilibrium position of particles in the focus is given if gradient force dominates over the scattering force.
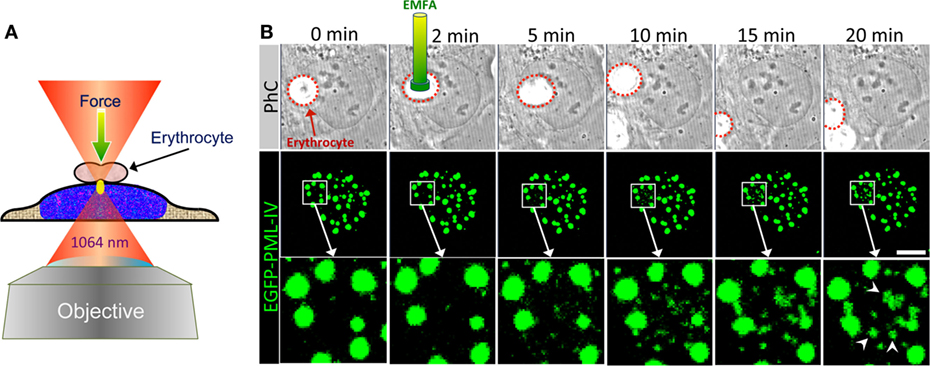
Figure 9. Optical tweezer (OT) as a tool to analyze PML nuclear body assembly. (A) Schematic depiction of erythrocyte-mediated force application (EMFA) based on OTs. Polyethylenimine coated erythrocytes attach unspecifically to the surfaces of the adherent target cells. Erythrocytes serve as very efficient “force transmitting devices” for axial force application on cells. The cell layer is moved into the region of the desired position in such a way that the laser focus (yellow ellipse) locates slightly below the erythrocyte. Immediately after switching on the laser the erythrocyte is pulled toward the focus due to the gradient force of the laser light, which causes a brief physical force onto the cell. The experimental setup used here consists of a continuous wave (cw) diode pumped Nd-YAG-laser (Spectra Physics) emitting at 1064 nm. The laser beam is coupled into an inverted confocal laser scanning microscope (LSM 510, Carl Zeiss Jena) and was focused via a high numerical aperture objective (100×, 1.30 NA) into the object plane. (B) U2OS cells expressing EGFP-tagged PML-IV were subjected to EMFA as shown in (A). In phase contrast (PhC) imaging, the position of the nucleus relative to erythrocytes can be monitored during the course of the experiment (upper panels). The behavior of PML nuclear bodies was monitored by confocal sectioning (middle panels, images show maximum intensity projections). The nuclear region of force application is shown as a magnified view in the bottom panels. Arrowheads indicate de novo formation of PML NBs. Scale bar, 5 µm.
There are also several setup variants: conventional OT with standard Gaussian laser beam, non-Gaussian laser beams based on a Bessel beam or a Laguerre–Gaussian mode, dual beams, and multiple traps or as optical strecher (156–159). OTs are excellent nanotools with which manipulation in a living cell or a living organism is possible without perforating the cell membrane. Further information on OT technologies can be found in Ref. (160–162). Generally OTs are used either to trap biological objects directly with light or as indirect force transducers to exert linear forces via trapped microbeads. In EMFA, polyethylenimine-coated erythrocytes are used instead of beads as the “force transmitting device” for axial force application on cells (Figure 9A) (161).
Here, we used EMFA to recapitulate some of the published data on PML NB behavior after global nuclear stress. Previously it had been observed that PML NBs disintegrate into many small PML-containing structures during heat shock or exposure to Cadmium2+ ions, implying that these structures undergo a stress response to altered chromatin organization or topology (163, 164). When EMFA is applied on living cells expressing GFP-tagged PML, force is applied on chromatin located just below the erythrocyte (Figure 9A). This physical pressure induces the appearance of PML-containing microstructures within the region of force application (Figure 9B). Eventually, such microstructures fuse with each other to form larger structures, while the native PML NBs remain positionally stable (Figure 9B). This behavior of PML microstructures occurs on a minute scale and was interpreted as evidence for a supramolecular assembly/disassembly model in which PML NBs are not a uniform, homogeneous polymer, but rather are composed of units or modules that are linked together as supramolecular assemblies (4, 41). This view is supported by super-resolution analyses of the PML NB architecture which revealed distinct occupation rather than uniform distribution of various PML body components in a shell-like structure (35) (Figure 10). Rapid disassembly/reassembly cycles of PML nuclear bodies upon cellular stress may be instrumental in their function as damage sensors and in genome maintenance.
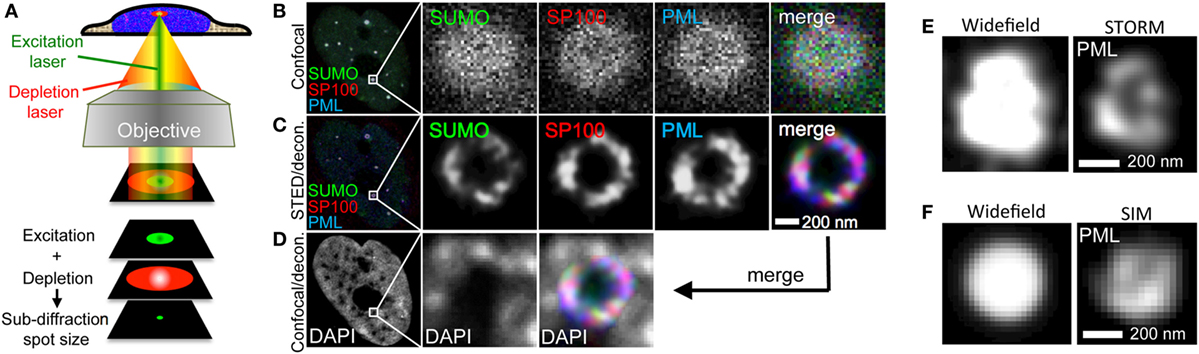
Figure 10. Super-resolution imaging of promyelocytic leukemia (PML) nuclear bodies. (A) Principle of stimulated emission depletion (STED) microscopy. In STED, two lasers are focused through a high numerical aperture objective lens. The excitation laser (green) serves to excite the fluorophore of interest similar to confocal imaging. Excitation light pulses are immediately followed by a high energy red-shifted STED beam with circularly polarized light (red). The STED light de-excites the excited fluorescence except for a small central spot due to the donut-like shape of the STED beam. This results in a subdiffraction size illumination excitation beam which can be scanned across the sample with a confocal scanner to produce super-resolved images. (B–D) Example of 3-color STED imaging of PML NBs using a Leica STED microscope. Fixed U2OS cells were immunofluorescently labeled to detect SUMO, SP100, and PML with secondary antibodies coupled to STAR-635P (green), STAR-580 (red), and Atto-490LS (blue), respectively. All dyes were depleted using the 770 nm STED laser. (B) shows one confocal section in the center of the nucleus recorded in confocal mode. (C) shows the same focal section as in (B) but recorded with the depletion laser switched on followed by deconvolution of the fluorescence signals using Huygens software (STED/decon.). (D) The DAPI signal was also acquired in the same focal section employing the HyVolution II mode of the Leica LSM (= confocal mode with the pinhole closed to 0.5 Airy units followed by deconvolution) (Confocal/decon.). (E) 3D-STORM imaging of a PML nuclear body in U2OS cells immunofluorescently labeled with an anti-PML antibody (Secondary antibody: Alexa-647N). (F) Super-resolution SIM imaging of a PML nuclear body in U2OS cells immunofluorescently labeled with an anti-PML antibody (Secondary antibody: Cy3). STORM and SIM imaging was performed using a Zeiss Elyra system.
Super-Resolution Microscopy (SRM)
The resolution of a light microscope is limited to about 200 nm by diffraction (165). The microscopic images of small cellular organelles or nuclear bodies in this size range therefore appear blurred and their morphological details go undetected. Fortunately, several SRM approaches have been established over past decade which improve resolution by a factor of 2–10, depending on the technique. Meanwhile, three main super-resolution technologies are commercially available, namely structured illumination microscopy (SIM), single molecule localization (SML), and stimulated emission depletion (STED) (94).
Structured illumination microscopy is a versatile and the most gentle super-resolution approach which increases the resolution by up to twofold in lateral and axial direction (166, 167). This is achieved by illuminating the sample with a grid pattern. The pattern can for example be generated by laser light passing through a movable optical grating which is projected via the objective onto the sample (168). The interference of the pattern with sample structures allows access to high frequency or in other words high-resolution information that would be otherwise obscured in a normal wide field image. SIM requires at least 9 images (2D-SIM) or 15 images (3D-SIM) to be taken for each optical section, whereby the illumination pattern is phase shifted and rotated in order to access the high-resolution information by sophisticated algorithms (168). The advantage of SIM is that it is compatible with all fluorescent dyes, making even super-resolved multicolor live-cell imaging feasible (169).
In SML switching of molecules between two distinct fluorescent states, normally an “on” and an “off” state is used to determine the exact position of a fluorescent molecule by determining the center of mass within the blurry fluorescent spot. The blinking is thereby adjusted to have at average only one molecule in its fluorescent state within the diffraction limited spot. The concept of blinking was realized using photoactivatable dyes, such as paGFP in photoactivated localization microscopy (PALM) and fluorescence PALM, or by using photoswitchable dye pairs (such as Cy3–Cy5 or EosFPs) as in stochastic optical reconstruction microscopy (STORM). Although PALM was established using fluorescence proteins, it was soon realized that any organic dye under appropriate reducing conditions can be brought to on and off switch cycles, a technology termed dSTORM (170).
In PALM/STORM, a series of several thousand images from the blinking specimen are recorded and mathematically processed into high-resolution images reaching resolutions below 30 nm in the lateral direction (171, 172). SML approaches have the inherent disadvantage that typically (ten)thousands of frames need to be acquired to reconstruct a single super-resolved image. The entailed low temporal resolution, extended exposure with high excitation power and associated phototoxicity render these methods less suitable for live cell imaging.
More recently developed fluctuation microscopy (SOFI, SIRF) approaches in part overcome these limitations at the expense of much lower resolution increase (173, 174). Nevertheless, live cell imaging using SML has been reported (175). Optical resolution in STED usually is well below 50 nm in fixed samples and ca. 70 nm in live-cell experiments (94). A more in depth explanation on the theory and on practical applications of SIM, SML, and STED can be found here: (176, 177). Possible practical limitations and compromises that must be considered when designing super-resolution experiments have been pointed out by Lambert and Waters (178).
Stimulated emission depletion is based on the application of two laser beams in a confocal (point-scanning) set-up. The STED depletion laser is delivered into the optical path through a phase filter, which creates a donut-shaped beam on the confocal fluorescence spot by controlled de-excitation of the previously excited fluorophore (Figure 10A). The high intensity STED beam extinguishes the peripheral fluorescence signal, leaving a subdiffraction-sized fluorescence spot in the center of the donut (179, 180) (Figure 10A). Interestingly, the first report on super-resolution light microscopy of PML nuclear bodies was not based on the three SRM methods described above but was realized with the so-called 4Pi microscope developed by Hell et al. (179). Four-Pi fluorescence laser-scanning microscopy studies revealed that during interphase PML NBs adopt a spherical organization characterized by the assembly of different PML body components into distinct, partially overlapping patches within a 50–100-nm thick shell (35). The spherical organization of PML NBs had been observed already before by electron microscopy (36, 48), but 4Pi allowed for simultaneous pair-wise detection of two PML body components.
One example of STED nanoscopy on PML NBs is shown in Figures 10B–D. The three major PML NB constituents SUMO, SP100 and PML were immunolabeled in U2OS cells with different fluorophores and imaged in confocal as well as in STED mode to visualize the improvement in optical resolution through STED (Figures 10B,C, respectively). As expected, STED reveals that these proteins decorate distinct, yet partially overlapping patches in the peripheral shell of the PML NB (Figure 10C). Super-imposition of the STED image with the DAPI pattern of the same confocal section confirms the absence of chromatin in the core of normal PML NBs (Figure 10D) (36). We also applied STORM and SIM imaging of PML in U2OS cells. STORM is similarly well suited to reveal the shell morphology of PML protein distribution (Figure 10E) while the resolution in SIM is, as expected, considerably lower than in STED or STORM (Figure 10F). However, since the laser load is much less, SIM would be better suited for live cell super-resolved imaging of PML nuclear body morphology, i.e., in the analyses of fission and fusion events of PML microstructures in stress conditions (Figure 9) or DNA at damage foci (Figure 8). In conclusion, this section shows that with commercially available SRM microscopes the analysis of biomolecules can be lifted to a new optical dimension.
Outlook
We believe that many biochemical or molecular biology oriented research labs are still not aware of the multitude of new and exciting microscopic methods and their capabilities. The aim of this contribution was to present recent advances in bioimaging in combination with selected application examples in PML nuclear body biology. Here we have illustrated the power of imaging methods and provide a guide to these techniques to make them more accessible to a larger number of labs involved in oncogene or tumor suppressor research. We have presented several experimental examples feasible in our imaging facility, yet the number of additional techniques is much higher. Bioimaging facility networks have been established in several countries worldwide and these can be approached with specific imaging requests. A source for comprehensive bioimaging methodology is available Europe-wide4 and a global bioimaging network project may be realized in the near future.5
Local, regional, national, and supranational imaging networks will continue to develop with the aim to provide access, service and training to state-of-the-art imaging technologies. Only such dedicated facility infrastructures and/or very specialized imaging research labs will be able to cope with the fast development of novel microscopy techniques. Although probably a demanding task, both, the facility members as well as basic research scientists are now in charge to synergistically work together to fully exploit the powerful imaging tools in the study of molecular and cellular mechanisms.
We have also summarized the current knowledge on the potential functions and assembly of PML nuclear bodies. PML has been analyzed using wet-lab and genetic techniques on one hand and imaging methods on the other. With the new microscopy methods now at hand it will be exciting to see the two different approaches merging. For example, a combination of STED and FCS (STED-FCS) (181), should make it possible to assess biophysical and binding properties of PML-interacting partners within PML NBs. This would help (i) to understand the molecular/biochemical events occurring molecularly at PML NBs at sites of DNA damage and (ii) to better visualize/understand the proposed phase separation function of PML NBs (68). For example, single-molecule tracking at nanoscale resolution has recently been employed to demonstrate the liquid droplet nature of stress granules in the cell nucleus (182). As nanoscopy will become less phototoxic in the future (183), super-resolution imaging of APBs in living cells will shed more light on the mechanisms of DNA recombination events which occur in PML NBs during telomere elongation in ALT cancer cells (184).
More physiologically, it would be seminal to investigate PML NBs in their most physiological setting, the living model organism. A combination of confocal microscopy and/or nanoscopy with adaptive optics for better tissue penetration (185, 186) would enable monitoring of fluorescent PML NBs in living tissue such as skin or brain of GFP-PML knock-in mice under normal vs. stress conditions (irradiation, chemicals). Since PML has an established role in certain stem cell niches, these nuclear bodies could be imaged and functionally analyzed in various living spheroid or organoid stem cell systems using a combination of multicolor lightsheet and super-resolution approaches (187, 188). In the same experimental setting, laser-assisted ablation of single PML NB-expressing cells could help to identify PML-mediated mechanisms of stem cell plasticity (189). Seeing is believing and therefore we look forward to monitor PML nuclear body biochemistry through new imaging set-ups in real time in living cells in the future.
Author Note
This contribution is dedicated to Jörg Langowski.
Author Contributions
CH, SM, KW, and PH prepared the figures and wrote the text.
Conflict of Interest Statement
KW was employed by Carl Zeiss Microscopy GmbH (ZEISS Group, Carl-Zeiss-Promenade 1007745 Jena, Germany). All other authors declare no competing interests.
Acknowledgments
We apologize to all authors who’s excellent articles in the field of PML body biology and microscopy techniques could not be cited in this contribution due to space limitations. We would like to thank the following colleagues for their time and effort in helping to establish the microscopy techniques described in our facility: Stephanie Weidtkamp-Peters, Lars Schmiedeberg, Almut Horch, Sandra Münch, Sandra Orthaus, Karolin Klement, Paulius Grigaravicius, Daniela Hellwig, Tobias Ulbricht, Volker Döring, Otto Greulich, Friedrich Haubensak, Eberhard Schmitt, Frank Große, and Stephan Diekmann. We would like to thank Debra Weih for proof-reading of the manuscript.
Funding
This work was supported by grant HE 2484/3-1 to PH from the Deutsche Forschungsgemeinschaft.
Footnotes
- ^http://npd.hgu.mrc.ac.uk/ (Accessed: November 17, 2017).
- ^https://thebiogrid.org/111384/summary/homo-sapiens/pml.html (Accessed: November 17, 2017).
- ^http://www.lfd.uci.edu/globals/ (Accessed: November 17, 2017).
- ^http://www.eurobioimaging.eu/ (Accessed: January 12, 2018).
- ^http://www.eurobioimaging.eu/global-bioimaging (Accessed: January 12, 2018).
References
1. Melnick A, Licht JD. Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood (1999) 93:3167–215.
2. Eskiw CH, Dellaire G, Bazett-Jones DP. Chromatin contributes to structural integrity of promyelocytic leukemia bodies through a SUMO-1-independent mechanism. J Biol Chem (2004) 279:9577–85. doi:10.1074/jbc.M312580200
3. Hodges M, Tissot C, Howe K, Grimwade D, Freemont PS. Structure, organization, and dynamics of promyelocytic leukemia protein nuclear bodies. Am J Hum Genet (1998) 63:297–304. doi:10.1086/301991
4. Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays (2004) 26:963–77. doi:10.1002/bies.20089
5. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol (2007) 8:1006–16. doi:10.1038/nrm2277
6. Mu ZM, Le XF, Vallian S, Glassman AB, Chang KS. Stable overexpression of PML alters regulation of cell cycle progression in HeLa cells. Carcinogenesis (1997) 18:2063–9. doi:10.1093/carcin/18.11.2063
7. Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, et al. Role of PML in cell growth and the retinoic acid pathway. Science (1998) 279:1547–51. doi:10.1126/science.279.5356.1547
8. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst (2004) 96:269–79. doi:10.1093/jnci/djh043
9. Sahin U, Ferhi O, Jeanne M, Benhenda S, Berthier C, Jollivet F, et al. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J Cell Biol (2014) 204(6):931–45. doi:10.1083/jcb.201305148
10. Guan D, Kao HY. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci (2015) 5:60. doi:10.1186/s13578-015-0051-9
11. Zhong S, Hu P, Ye TZ, Stan R, Ellis NA, Pandolfi PP. A role for PML and the nuclear body in genomic stability. Oncogene (1999) 18:7941–7. doi:10.1038/sj.onc.1203367
12. Carbone R, Pearson M, Minucci S, Pelicci PG. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene (2002) 21(11):1633–40. doi:10.1038/sj.onc.1205227
13. Bøe SO, Haave M, Jul-Larsen A, Grudic A, Bjerkvig R, Lønning PE. Promyelocytic leukemia nuclear bodies are predetermined processing sites for damaged DNA. J Cell Sci (2006) 119(Pt 16):3284–95. doi:10.1242/jcs.03068
14. Foltánková V, Matula P, Sorokin D, Kozubek S, Bártová E. Hybrid detectors improved time-lapse confocal microscopy of PML and 53BP1 nuclear body colocalization in DNA lesions. Microsc Microanal (2013) 19:360–9. doi:10.1017/S1431927612014353
15. Boichuk S, Hu L, Makielski K, Pandolfi PP, Gjoerup OV. Functional connection between Rad51 and PML in homology-directed repair. PLoS One (2011) 6(10):e25814. doi:10.1371/journal.pone.0025814
16. Yeung PL, Denissova NG, Nasello C, Hakhverdyan Z, Chen JD, Brenneman MA. Promyelocytic leukemia nuclear bodies support a late step in DNA double-strand break repair by homologous recombination. J Cell Biochem (2012) 113:1787–99. doi:10.1002/jcb.24050
17. Legartová S, Sehnalová P, Malyšková B, Küntziger T, Collas P, Cmarko D, et al. Localized movement and levels of 53BP1 protein are changed by γ-irradiation in PML deficient cells. J Cell Biochem (2016) 117:2583–96. doi:10.1002/jcb.25551
18. Chang HR, Munkhjargal A, Kim MJ, Park SY, Jung E, Ryu JH, et al. The functional roles of PML nuclear bodies in genome maintenance. Mutat Res (2017). doi:10.1016/j.mrfmmm.2017.05.002
19. Gamell C, Jan Paul P, Haupt Y, Haupt S. PML tumour suppression and beyond: therapeutic implications. FEBS Lett (2014) 588:2653–62. doi:10.1016/j.febslet.2014.02.007
20. Liu Z, Lavis LD, Betzig E. Imaging live-cell dynamics and structure at the single-molecule level. Mol Cell (2015) 58:644–59. doi:10.1016/j.molcel.2015.02.033
21. Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene (2001) 20:7223–33. doi:10.1038/sj.onc.1204765
22. Nisole S, Maroui MA, Mascle XH, Aubry M, Chelbi-Alix MK. Differential roles of PML isoforms. Front Oncol (2013) 3:1–17. doi:10.3389/fonc.2013.00125
23. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J (2001) 20:2140–51. doi:10.1093/emboj/20.9.2140
24. Kamitani T, Kito K, Nguyen HP, Wada H, Fukuda-Kamitani T, Yeh ET. Identification of three major sentrinization sites in PML. J Biol Chem (1998) 273:26675–82.
25. Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem (2013) 82:357–85. doi:10.1146/annurev-biochem-061909-093311
26. Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, et al. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem (2001) 276:35368–74. doi:10.1074/jbc.M104214200
27. Condemine W, Takahashi Y, Zhu J, Puvion-Dutilleul F, Guegan S, Janin A, et al. Characterization of endogenous humanpromyelocyticl eukemia isoforms. Cancer Res (2006) 66:6192–8. doi:10.1158/0008-5472.CAN-05-3792
28. Schmitz ML, Grishina I. Regulation of the tumor suppressor PML by sequential post-translational modifications. Front Oncol (2012) 2:204. doi:10.3389/fonc.2012.00204
29. Brand P, Lenser T, Hemmerich P. Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys (2010) 3:3. doi:10.1186/1757-5036-3-3
30. Beech SJ, Lethbridge KJ, Killick N, McGlincy N, Leppard KN. Isoforms of the promyelocytic leukemia protein differ in their effects on ND10 organization. Exp Cell Res (2005) 307:109–17. doi:10.1016/j.yexcr.2005.03.012
31. Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, et al. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J Cell Sci (2011) 124:280–91. doi:10.1242/jcs.075390
32. Geng Y, Monajembashi S, Shao A, Cui D, He W, Chen Z, et al. Contribution of the C-terminal regions of promyelocytic leukemia protein (PML) isoforms II and V to PML nuclear body formation. J Biol Chem (2012) 287(36):30729–42. doi:10.1074/jbc.M112.374769
33. Li C, Peng Q, Wan X, Sun H, Tang J. C-terminal motifs in promyelocytic leukemia protein isoforms critically regulate PML nuclear body formation. J Cell Sci (2017) 130:3496–506. doi:10.1242/jcs.202879
34. de Thè G, Riviére M, Bernhard W. Examen au microscope e’lectronique de la tumeur VX2 du lapin domestique de´rive´e du papillome de Shope. Bull Cancer (1960) 47:570–84.
35. Lang M, Jegou T, Chung I, Richter K, Münch S, Udvarhelyi A, et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J Cell Sci (2010) 123:392–400. doi:10.1242/jcs.053496
36. Boisvert FM, Hendzel MJ, Bazett-Jones DP. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol (2000) 148:283–92. doi:10.1083/jcb.148.2.283
37. Torok D, Ching RW, Bazett-Jones DP. PML nuclear bodies as sites of epigenetic regulation. Front Biosci (2009) 14:1325–36. doi:10.2741/3311
38. Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res (1999) 59:4175–9.
39. Luciani JJ, Depetris D, Usson Y, Metzler-Guillemain C, Mignon-Ravix C, Mitchell MJ, et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J Cell Sci (2006) 119:2518–31. doi:10.1242/jcs.02965
40. Dellaire G, Eskiw CH, Dehghani H, Ching RW, Bazett-Jones DP. Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J Cell Sci (2006) 119(Pt 6):1034–42. doi:10.1242/jcs.02817
41. Dellaire G, Ching RW, Ahmed K, Jalali F, Tse KC, Bristow RG, et al. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol (2006) 175:55–66. doi:10.1083/jcb.200604009
42. Ching G, Dehghani RW, Ren HY, Bazett-Jones DP. The number of PML nuclear bodies increases in early S phase by a fission mechanism. J Cell Sci (2006) 119:1026–33. doi:10.1242/jcs.02816
43. Dellaire G, Farrall R, Bickmore WA. The Nuclear Protein Database (NPD): sub-nuclear localisation and functional annotation of the nuclear proteome. Nucleic Acids Res (2003) 31:328–30. doi:10.1093/nar/gkg018
44. Mohamad N, Bodén M. The proteins of intra-nuclear bodies: a data-driven analysis of sequence, interaction and expression. BMC Syst Biol (2010) 4:44. doi:10.1186/1752-0509-4-44
45. Van Damme E, Laukens K, Dang TH, Van Ostade X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci (2010) 6:51–67. doi:10.7150/ijbs.6.51
46. Negorev D, Maul GG. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene (2001) 20:7234–42. doi:10.1038/sj.onc.1204764
47. Chung I, Osterwald S, Deeg KI, Rippe K. PML body meets telomere: the beginning of an ALTernate ending? Nucleus (2012) 3:263–75. doi:10.4161/nucl.20326
48. Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol (2010) 2(5):a000661. doi:10.1101/cshperspect.a000661
49. Bernardi R, Papa A, Pandolfi PP. Regulation of apoptosis by PML and the PMLNBs. Oncogene (2008) 27:6299–631. doi:10.1038/onc.2008.305
50. Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honorè N, Doubeikovsky A, et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med (2001) 193(12):1361–71. doi:10.1084/jem.193.12.1361
51. Ivanschitz L, De Thè H, Le Bras M. PML, SUMOylation, and senescence. Front Oncol (2013) 3:171. doi:10.3389/fonc.2013.00171
52. Salomoni P. Stemming out of a new PML era? Cell Death Differ (2009) 16(8):1083–92. doi:10.1038/cdd.2009.63
53. Zhou W, Bao S. PML-mediated signaling and its role in cancer stem cells. Oncogene (2014) 33(12):1475–84. doi:10.1038/onc.2013.111
54. Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol (2000) 2(5):E85–90. doi:10.1038/35010583
55. Hofmann TG, Will H. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ (2003) 10(12):1290–9. doi:10.1038/sj.cdd.4401313
56. Varadaraj A, Dovey CL, Laredj L, Ferguson B, Alexander CE, Lubben N, et al. Evidence for the receipt of DNA damage stimuli by PML nuclear domains. J Pathol (2007) 211(4):471–80. doi:10.1002/path.2126
57. Marchesini M, Matocci R, Tasselli L, Cambiaghi V, Orleth A, Furia L, et al. PML is required for telomere stability in non-neoplastic human cells. Oncogene (2016) 35:1811–21. doi:10.1038/onc.2015.246
58. Salomoni P. The PML-interacting protein DAXX: histone loading gets into the picture. Front Oncol (2013) 3:152. doi:10.3389/fonc.2013.00152
59. Tavalai N, Stamminger T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta (2008) 1783(11):2207–21. doi:10.1016/j.bbamcr.2008.08.004
60. Dellaire G, Bazett-Jones DP. Beyond repair foci: subnuclear domains and the cellular response to DNA damage. Cell Cycle (2007) 15:1864–72. doi:10.4161/cc.6.15.4560
61. Borden KL, Culjkovic B. Perspectives in PML: a unifying framework for PML function. Front Biosci (Landmark Ed) (2009) 14:497–509. doi:10.2741/3258
62. Rajendra TK, Praveen K, Matera AG. Genetic analysis of nuclear bodies: from nondeterministic chaos to deterministic order. Cold Spring Harb Symp Quant Biol (2010) 75:365–74. doi:10.1101/sqb.2010.75.043
63. Sahin U, Lallemand-Breitenbach V, de The H. PML nuclear bodies: regulation, function and therapeutic perspectives. J Pathol (2014) 234:289–91. doi:10.1002/path.4426
64. Kentsis A, Gordon RE, Borden KL. Control of biochemical reactions through supramolecular RING domain self-assembly. Proc Natl Acad Sci U S A (2002) 99(24):15404–9. doi:10.1073/pnas.202608799
65. Matunis MJ, Zhang XD, Ellis NA. SUMO: the glue that binds. Dev Cell (2006) 11(5):596–7. doi:10.1016/j.devcel.2006.11.011
66. Weidtkamp-Peters S, Lenser T, Negorev D, Gerstner N, Hofmann TG, Schwanitz G, et al. Dynamics of component exchange at PMLnuclear bodies. J Cell Sci (2008) 121:2731–43. doi:10.1242/jcs.031922
67. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol (2017) 18(5):285–98. doi:10.1038/nrm.2017.7
68. Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, et al. Compositional control of phase-separated cellular bodies. Cell (2016) 166(3):651–63. doi:10.1016/j.cell.2016.06.010
69. Uversky VN. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol (2017) 44:18–30. doi:10.1016/j.sbi.2016.10.015
70. Palibrk V, Lång E, Lång A, Schink KO, Rowe AD, Bøe SO. Promyelocytic leukemia bodies tether to early endosomes during mitosis. Cell Cycle (2014) 13(11):1749–55. doi:10.4161/cc.28653
71. Chen YC, Kappel C, Beaudouin J, Eils R, Spector DL. Live cell dynamics of promyelocytic leukemia nuclear bodies upon entry into and exit from mitosis. Mol Biol Cell (2008) 19(7):3147–62. doi:10.1091/mbc.E08-01-0035
72. Jul-Larsen A, Grudic A, Bjerkvig R, Bøe SO. Cell cycle regulation and dynamics of cytoplasmic compartments containing the promyelocytic leukemia protein and nucleoporins. J Cell Sci (2009) 122:1201–10. doi:10.1242/jcs.040840
73. Lång A, Eriksson J, Schink KO, Lång E, Blicher P, Połeć A, et al. Visualization of PML nuclear import complexes reveals FG-repeat nucleoporins at cargo retrieval sites. Nucleus (2017) 8:404–20. doi:10.1080/19491034.2017.1306161
74. Hillestad LK. Acute promyelocytic leukemia. Acta Med Scand (1957) 159:189–94. doi:10.1111/j.0954-6820.1957.tb00124.x
75. Goddard AD, Borrow J, Freemont PS, Solomon E. Characterization of a zinc finger gene disrupted by the t(15,17) in acute promyelocytic leukemia. Science (1991) 254:1371–4. doi:10.1126/science.1720570
76. de Thè H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer (2010) 10:775–83. doi:10.1038/nrc2943
77. Zhu J, Koken MHM, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, et al. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc Natl Acad Sci U S A (1997) 94:3978–83. doi:10.1073/pnas.94.8.3978
78. Gambacorta M, Flenghi L, Fagioli M, Pileri S, Leoncini L, Bigerna B, et al. Heterogeneous nuclear expression of the promyelocytic leukemia (PML) protein in normal and neoplastic human tissues. Am J Pathol (1996) 149:2023–35.
79. Lee HE, Jee CD, Kim MA, Lee HS, Lee YM, Lee BL, et al. Loss of promyelocytic leukemia protein in human gastric cancers. Cancer Lett (2007) 247:103–9. doi:10.1016/j.canlet.2006.03.034
80. Zhang P, Chin W, Chow LT, Chan AS, Yim AP, Leung SF, et al. Lack of expression for the suppressor PML in human small cell lung carcinoma. Int J Cancer (2000) 85:599–605. doi:10.1002/(SICI)1097-0215(20000301)85:5<599::AID-IJC1>3.0.CO;2-#
81. Koken MH, Linares-Cruz G, Quignon F, Viron A, Chelbi-Alix MK, Sobczak-Thépot J, et al. The PML growthsuppressor has an altered expression in human oncogenesis. Oncogene (1995) 10:1315–24.
82. Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, et al. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene (1995) 11(5):871–6.
83. Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell (2002) 108:165–70. doi:10.1016/S0092-8674(02)00626-8
84. Le XF, Yang P, Chang KS. Analysis of the growth and transformation suppressor domains of promyelocytic leukemia gene, PML. J Biol Chem (1996) 271:130–5. doi:10.1074/jbc.271.1.130
85. Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PMLinduced premature senescence. EMBO J (2002) 21:3358–69. doi:10.1093/emboj/cdf341
86. Mallette FA, Goumard S, Gaumont-Leclerc MF, Moiseeva O, Ferbeyre G. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene (2004) 23:91–9. doi:10.1038/sj.onc.1206886
87. de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell (2004) 13:523–35. doi:10.1016/S1097-2765(04)00062-0
88. Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, et al. PML is essential for multiple apoptotic pathways. Nat Genet (1998) 20:266–72. doi:10.1038/3073
89. Voisset E, Moravcsik E, Stratford EW, Jaye A, Palgrave CJ, Hills RK, et al. Pml nuclear body disruption cooperates in APL pathogenesis and impairs DNA damage repair pathways in mice. Blood (2018) 131:636–48. doi:10.1182/blood-2017-07-794784
90. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature (2008) 453:1072–8. doi:10.1038/nature07016
91. Mazza M, Pelicci PG. Is PML a tumor suppressor? Front Oncol (2013) 3:174. doi:10.3389/fonc.2013.00174
92. Martín-Martín N, Piva M, Urosevic J, Aldaz P, Sutherland JD, Fernández-Ruiz S, et al. Stratification and therapeutic potential of PML in metastatic breast cancer. Nat Commun (2016) 7:12595. doi:10.1038/ncomms12595
93. Ponente M, Campanini L, Cuttano R, Piunti A, Delledonne GA, Coltella N, et al. PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight (2017) 2:e87380. doi:10.1172/jci.insight.87380
94. Sahl SJ, Hell SW, Jakobs S. Fluorescence nanoscopy in cell biology. Nat Rev Mol Cell Biol (2017) 18:685–701. doi:10.1038/nrm.2017.71
95. Weidemann T, Mücksch J, Schwille P. Fluorescence fluctuation microscopy: a diversified arsenal of methods to investigate molecular dynamics inside cells. Curr Opin Struct Biol (2014) 28:69–76. doi:10.1016/j.sbi.2014.07.008
96. Bag N, Wohland T. Imaging fluorescence fluctuation spectroscopy: new tools for quantitative bioimaging. Annu Rev Phys Chem (2014) 65:225–48. doi:10.1146/annurev-physchem-040513-103641
97. Wachsmuth M, Conrad C, Bulkescher J, Koch B, Mahen R, Isokane M, et al. High-throughput fluorescence correlation spectroscopy enables analysis of proteome dynamics in living cells. Nat Biotechnol (2015) 33(4):384–9. doi:10.1038/nbt.3146
98. Thorn K. A quick guide to light microscopy in cell biology. Mol Biol Cell (2016) 27(2):219–22. doi:10.1091/mbc.E15-02-0088
99. Follain G, Mercier L, Osmani N, Harlepp S, Goetz JG. Seeing is believing – multi-scale spatio-temporal imaging towards in vivo cell biology. J Cell Sci (2017) 130(1):23–38. doi:10.1242/jcs.189001
100. Ferrando-May E, Hartmann H, Reymann J, Ansari N, Utz N, Fried HU, et al. Advanced light microscopy core facilities: balancing service, science and career. German BioImaging network. Microsc Res Tech (2016) 79(6):463–79. doi:10.1002/jemt.22648
101. Carrero G, McDonald D, Crawford E, de Vries G, Hendzel MJ. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods (2003) 29:14–28.
102. Peters R, Brünger A, Schulten K. Continuous fluorescence microphotolysis: a sensitive method for study of diffusion processes in single cells. Proc Natl Acad Sci U S A (1981) 78(2):962–6. doi:10.1073/pnas.78.2.962
103. Mueller F, Mazza D, Stasevich TJ, McNally JG. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know? Curr Opin Cell Biol (2010) 22(3):403–11. doi:10.1016/j.ceb.2010.03.002
104. Blumenthal D, Goldstien L, Edidin M, Gheber LA. Universal approach to FRAP analysis of arbitrary bleaching patterns. Sci Rep (2015) 5:11655. doi:10.1038/srep11655
105. Dobrucki JW, Feret D, Noatynska A. Scattering of exciting light by live cells in fluorescence confocal imaging: phototoxic effects and relevance for FRAP studies. Biophys J (2007) 93:1778–86. doi:10.1529/biophysj.106.096636
106. Hemmerich P, Schmiedeberg L, Diekmann S. Dynamic as well as stable protein interactions contribute to genome function and maintenance. Chromosome Res (2011) 19(1):131–51. doi:10.1007/s10577-010-9161-8
107. van Royen ME, Zotter A, Ibrahim SM, Geverts B, Houtsmuller AB. Nuclear proteins: finding and binding target sites in chromatin. Chromosome Res (2011) 19(1):83–98. doi:10.1007/s10577-010-9172-5
108. McNally JG. Quantitative FRAP in analysis of molecular binding dynamics in vivo. Methods Cell Biol (2008) 85:329–51. doi:10.1016/S0091-679X(08)85014-5
109. Beaudouin J, Mora-Bermúdez F, Klee T, Daigle N, Ellenberg J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys J (2006) 90:1878–94. doi:10.1529/biophysj.105.071241
110. Sprague BL, Pego RL, Stavreva DA, McNally JG. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys J (2004) 86:3473–95. doi:10.1529/biophysj.103.026765
111. Erdel F, Müller-Ott K, Baum M, Wachsmuth M, Rippe K. Dissecting chromatin interactions in living cells from protein mobility maps. Chromosome Res (2011) 19(1):99–115. doi:10.1007/s10577-010-9155-6
112. Baum M, Erdel F, Wachsmuth M, Rippe K. Retrieving the intracellular topology from multi-scale protein mobility mapping in living cells. Nat Commun (2014) 24(5):4494. doi:10.1038/ncomms5494
113. Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, et al. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods (2013) 10(5):421–6. doi:10.1038/nmeth.2411
114. Magde D, Elson E, Webb WW. Thermodynamic fluctuations in a reacting system – measurement by fluorescence correlation spectroscopy. Phys Rev Lett (1972) 29:705–8. doi:10.1103/PhysRevLett.29.705
115. Rigler R, Widengren J. Ultrasensitive detection of single molecules by fluorescence correlation spectroscopy. Bioscience (1990) 3:180–3.
116. Rüttinger S, Buschmann V, Krämer B, Erdmann R, Macdonald R, Koberling F. Comparison and accuracy of methods to determine the confocal volume for quantitative fluorescence correlation spectroscopy. J Microsc (2008) 232:343–52. doi:10.1111/j.1365-2818.2008.02105.x
117. Becker W, Su B, Holub O, Weisshart K. FLIM and FCS detection in laser-scanning microscopes: increased efficiency by GaAsP hybrid detectors. Microsc Res Tech (2011) 74(9):804–11. doi:10.1002/jemt.20959
118. Elson EL. Fluorescence correlation spectroscopy: past, present, future. Biophys J (2011) 101(12):2855–70. doi:10.1016/j.bpj.2011.11.012
119. Ries J, Schwille P. Fluorescence correlation spectroscopy. Bioessays (2012) 34(5):361–8. doi:10.1002/bies.201100111
120. Schwille P, Meyer-Almes FJ, Rigler R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys J (1997) 72(4):1878–86. doi:10.1016/S0006-3495(97)78833-7
121. Thews E, Gerken M, Eckert R, Zäpfel J, Tietz C, Wrachtrup J. Cross talk free fluorescence cross correlation spectroscopy in live cells. Biophys J (2005) 89:2069–76. doi:10.1529/biophysj.104.057919
122. Bacia K, Kim SA, Schwille P. Fluorescence cross-correlation spectroscopy in living cells. Nat Methods (2006) 3(2):83–9. doi:10.1038/nmeth822
123. Weidtkamp-Peters S, Weisshart K, Schmiedeberg L, Hemmerich P. Fluorescence correlation spectroscopy to assess the mobility of nuclear proteins. Methods Mol Biol (2009) 464:321–41. doi:10.1007/978-1-60327-461-6_18
124. Bacia K, Schwille P. A dynamic view of cellular processes by in vivo fluorescence auto- and cross-correlation spectroscopy. Methods (2003) 29(1):74–85. doi:10.1016/S1046-2023(02)00291-8
125. Digman MA, Brown CM, Sengupta P, Wiseman PW, Horwitz AR, Gratton E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys J (2005) 89(2):1317–27. doi:10.1529/biophysj.105.062836
126. Brown CM, Dalal RB, Hebert B, Digman MA, Horwitz AR, Gratton E. Raster image correlation spectroscopy (RICS) for measuring fast protein dynamics and concentrations with a commercial laser scanning confocal microscope. J Microsc (2008) 229:78–91. doi:10.1111/j.1365-2818.2007.01871.x
127. Chen Y, Müller JD, Ruan Q, Gratton E. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys J (2002) 82(1 Pt 1):133–44. doi:10.1016/S0006-3495(02)75380-0
128. Förster T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys (1948) 437:55–75. doi:10.1002/andp.19484370105
129. Sun Y, Rombola C, Jyothikumar V, Periasamy A. Förster resonance energy transfer microscopy and spectroscopy for localizing protein-protein interactions in living cells. Cytometry A (2013) 83(9):780–93. doi:10.1002/cyto.a.22321
130. Clegg RM. FRET tells us about proximities, distances, orientations and dynamic properties. J Biotechnol (2002) 82(3):177–9. doi:10.1016/S1389-0352(01)00044-7
131. Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Curr Opin Chem Biol (2006) 10(5):409–16. doi:10.1016/j.cbpa.2006.08.021
132. Padilla-Parra S, Tramier M. FRET microscopy in the living cell: different approaches, strengths and weaknesses. Bioessays (2012) 34(5):369–76. doi:10.1002/bies.201100086
133. Diekmann S, Hoischen C. Biomolecular dynamics and binding studies in the living cell. Phys Life Rev (2014) 11(1):1–30. doi:10.1016/j.plrev.2013.11.011
134. Müller S, Matunis MJ, Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J (1998) 17(1):61–70. doi:10.1093/emboj/17.1.61
135. Daniels MJ, Marson A, Venkitaraman AR. PML bodies control the nuclear dynamics and function of the CHFR mitotic checkpoint protein. Nat Struct Mol Biol (2004) 11(11):1114–21. doi:10.1038/nsmb837
136. Hellwig D, Hoischen C, Ulbricht T, Diekmann S. Acceptor-photobleaching FRET analysis of core kinetochore and NAC proteins in living human cells. Eur Biophys J (2009) 38(6):781–91. doi:10.1007/s00249-009-0498-x
137. Bui M, Dimitriadis EK, Hoischen C, An E, Quénet D, Giebe S, et al. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell (2012) 150(2):317–26. doi:10.1016/j.cell.2012.05.035
138. Karanam K, Loewer A, Lahav G. Dynamics of the DNA damage response: insights from live-cell imaging. Brief Funct Genomics (2013) 12(2):109–17. doi:10.1093/bfgp/els059
139. Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, et al. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J (2002) 21(8):2030–7. doi:10.1093/emboj/21.8.2030
140. Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) (2010) 9(12):1219–28. doi:10.1016/j.dnarep.2010.09.010
141. Lemaître C, Soutoglou E. DSB (Im)mobility and DNA repair compartmentalization in mammalian cells. J Mol Biol (2015) 427(3):652–8. doi:10.1016/j.jmb.2014.11.014
142. Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA damage foci: meaning and significance. Environ Mol Mutagen (2015) 56(6):491–504. doi:10.1002/em.21944
143. Polo SEP, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev (2011) 25(5):409–33. doi:10.1101/gad.2021311
144. Kim JS, Heale JT, Zeng W, Kong X, Krasieva TB, Ball AR Jr, et al. In situ analysis of DNA damage response and repair using laser microirradiation. Methods Cell Biol (2007) 82:377–407. doi:10.1016/S0091-679X(06)82013-3
145. Ferrando-May E, Tomas M, Blumhardt P, Stöckl M, Fuchs M, Leitenstorfer A. Highlighting the DNA damage response with ultrashort laser pulses in the near infrared and kinetic modeling. Front Genet (2013) 16(4):135. doi:10.3389/fgene.2013.00135
146. Gassman NR, Wilson SH. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amst) (2015) 31:52–63. doi:10.1016/j.dnarep.2015.05.001
147. Kong X, Mohanty SK, Stephens J, Heale JT, Gomez-Godinez V, Shi LZ, et al. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res (2009) 37(9):68. doi:10.1093/nar/gkp221
148. Holton NW, Andrews JF, Gassman NR. Application of laser micro-irradiation for examination of single and double strand break repair in mammalian cells. J Vis Exp (2017) (127). doi:10.3791/56265
149. Lukas C, Bartek J, Lukas J. Imaging of protein movement induced by chromosomal breakage: tiny ‘local’ lesions pose great ‘global’ challenges. Chromosoma (2005) 114(3):146–54. doi:10.1007/s00412-005-0011-y
150. Bischof O, Kim SH, Irving J, Beresten S, Ellis NA, Campisi J. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J Cell Biol (2001) 153(2):367–80. doi:10.1083/jcb.153.2.367
151. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol (2012) 14(4):355–65. doi:10.1038/ncb2466
152. Münch S, Weidtkamp-Peters S, Klement K, Grigaravicius P, Monajembashi S, Salomoni P, et al. The tumor suppressor PML specifically accumulates at RPA/Rad51-containing DNA damage repair foci but is nonessential for DNA damage-induced fibroblast senescence. Mol Cell Biol (2014) 34:1733–46. doi:10.1128/MCB.01345-13
153. Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S. Observation of a single-beam gradient force optical trap for dielectric paricles. Opt Lett (1986) 11:288–90. doi:10.1364/OL.11.000288
154. Greulich KO. Selected applications of laser scissors and tweezers and new applications in heart research. Methods Cell Biol (2008) 82:59–80. doi:10.1016/S0091-679X(06)82002-9
155. Greulich KO, Pilarczyk G, Hoffmann A, Meyer Zu Hörste G, Schäfer B, Uhl V, et al. Micromanipulation by laser microbeam and optical tweezers: from plant cells to single molecules. J Microsc (2000) 198:182–7. doi:10.1046/j.1365-2818.2000.00698.x
156. MCGloin D, Dholakia K. Bessel beams: diffraction in a new light. Contemp Phys (2005) 46:15–28. doi:10.1080/0010751042000275259
157. Nie Z, Shi G, Li D, Zhang X, Wang Y, Song Y. Tight focusing of a radially polarized Laguerre–Bessel–Gaussian beam and its application to manipulation of two types of particles. Phys Lett A (2015) 379:857–63. doi:10.1016/j.physleta.2014.11.029
158. Diekmann R, Wolfson DL, Spahn C, Heilemann M, Schuttpelz M, Huser T. Nanoscopy of bacterial cells immobilized by holographic optical tweezers. Nat Commun (2016) 7:13711–4. doi:10.1038/ncomms13711
159. Guck J, Ananthakrishnan R, Mahmood H, Moon TJ, Cunningham CC, Käs J. The optical stretcher: a novel laser tool to micromanipulate cells. Biophys J (2001) 81:767–84. doi:10.1016/S0006-3495(01)75740-2
160. Greulich KO. Manipulation of cells with laser microbeam scissors and optical tweezers: a review. Rep Prog Phys (2017) 80(2):026601. doi:10.1088/1361-6633/80/2/026601
161. Grigaravicius P, Greulich KO, Monajembashi S. Laser microbeams and optical tweezers in ageing research. Chemphyschem (2009) 10:79–85. doi:10.1002/cphc.200800725
162. Zhang H, Liu K-K. Optical tweezers for single cells. J R Soc Interface (2008) 5:671–90. doi:10.1098/rsif.2008.0052
163. Maul GG, Yu E, Ishov AM, Epstein AL. Nuclear domain 10 (ND10) associated proteins are also present in nuclear bodies and redistribute to hundreds of nuclear sites after stress. J Cell Biochem (1995) 59:498–513. doi:10.1002/jcb.240590410
164. Eskiw CH, Dellaire G, Mymryk JS, Bazett-Jones DP. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J Cell Sci (2003) 116:4455–66. doi:10.1242/jcs.00758
165. Abbe E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch für Mikroskopische Anat (1873) 9:413–8. doi:10.1007/BF02956173
166. Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc (2000) 198:82–7. doi:10.1046/j.1365-2818.2000.00710.x
167. Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, et al. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J (2008) 94:4957–70. doi:10.1529/biophysj.107.120345
168. Heintzmann R, Huser T. Super-resolution structured illumination microscopy. Chem Rev (2017) 117(23):13890–908. doi:10.1021/acs.chemrev.7b00218
169. Shao L, Kner P, Rego EH, Gustafsson MG. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods (2011) 8:1044–6. doi:10.1038/nmeth.1734
170. van de Linde S, Löschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc (2011) 6:991–1009. doi:10.1038/nprot.2011.336
171. Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science (2006) 313:1642–5. doi:10.1126/science.1127344
172. Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J (2006) 91:4258–72. doi:10.1529/biophysj.106.091116
173. Dertinger T, Colyer R, Vogel R, Heilemann M, Sauer M, Enderlein J, et al. Superresolution optical fluctuation imaging (SOFI). Adv Exp Med Biol (2012) 733:17–21. doi:10.1007/978-94-007-2555-3_2
174. Gustafsson N, Culley S, Ashdown G, Owen DM, Pereira PM, Henriques R. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radialfluctuations. Nat Commun (2016) 7:12471. doi:10.1038/ncomms12471
175. Hess ST, Gould TJ, Gudheti MV, Maas SA, Mills KD, Zimmerberg J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc Natl Acad Sci U S A (2007) 104:17370–5. doi:10.1073/pnas.0708066104
176. Blom H, Widengren J. Stimulated emission depletion microscopy. Chem Rev (2017) 117:7377–427. doi:10.1021/acs.chemrev.6b00653
177. Fornasiero EF, Opazo F. Super-resolution imaging for cell biologists: concepts, applications, current challenges and developments. Bioessays (2015) 37:436–51. doi:10.1002/bies.201400170
178. Lambert TJ, Waters JC. Navigating challenges in the application of superresolution microscopy. J Cell Biol (2017) 216:53–63. doi:10.1083/jcb.201610011
179. Hell SW, Stelzer EH, Lindek S, Cremer C. Confocal microscopy with an increased detection aperture: type-B 4Pi confocal microscopy. Opt Lett (1994) 19:222–32. doi:10.1364/OL.19.000222
180. Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett (1994) 19:780–2. doi:10.1364/OL.19.000780
181. Mueller V, Honigmann A, Ringemann C, Medda R, Schwarzmann G, Eggeling C. FCS in STED microscopy: studying the nanoscale of lipid membrane dynamics. Methods Enzymol (2013) 519:1–38. doi:10.1016/B978-0-12-405539-1.00001-4
182. Niewidok B, Igaev M, Pereira da Graca A, Strassner A, Lenzen C, Richter CP, et al. Single-molecule imaging reveals dynamic biphasic partition of RNA-binding proteins in stress granules. J Cell Biol (2018) 217(4):1303–18. doi:10.1083/jcb.201709007
183. Balzarotti F, Eilers Y, Gwosch KC, Gynnå AH, Westphal V, Stefani FD, et al. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science (2017) 355:606–12. doi:10.1126/science.aak9913
184. Osterwald S, Deeg KI, Chung I, Parisotto D, Wörz S, Rohr K, et al. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J Cell Sci (2015) 128:1887–900. doi:10.1242/jcs.148296
185. Booth M, Andrade D, Burke D, Patton B, Zurauskas M. Aberrations and adaptive optics in super-resolution microscopy. Microscopy (Oxf) (2015) 64:251–61. doi:10.1093/jmicro/dfv033
186. Heine J, Reuss M, Harke B, D’Este E, Sahl SJ, Hell SW. Adaptive-illumination STED nanoscopy. Proc Natl Acad Sci U S A (2017) 114:9797–802. doi:10.1073/pnas.1708304114
187. Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, et al. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature (2017) 546:162–7. doi:10.1038/nature22369
188. Gustavsson AK, Petrov PN, Lee MY, Shechtman Y, Moerner WE. 3D single-molecule super-resolution microscopy with a tilted light sheet. Nat Commun (2018) 9:123. doi:10.1038/s41467-017-02563-4
Keywords: live cell imaging, fluorescence fluctuation microscopy, super-resolution, promyelocytic leukemia, tumor suppressor, oncogene
Citation: Hoischen C, Monajembashi S, Weisshart K and Hemmerich P (2018) Multimodal Light Microscopy Approaches to Reveal Structural and Functional Properties of Promyelocytic Leukemia Nuclear Bodies. Front. Oncol. 8:125. doi: 10.3389/fonc.2018.00125
Received: 17 November 2017; Accepted: 05 April 2018;
Published: 25 May 2018
Edited by:
Elisa Ferrando-May, Universität Konstanz, GermanyReviewed by:
Lothar Schermelleh, University of Oxford, United KingdomGraham Dellaire, Dalhousie University, Canada
Don C. Lamb, Ludwig-Maximilians-Universität München, Germany
Copyright: © 2018 Hoischen, Monajembashi, Weisshart and Hemmerich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Hemmerich, cGV0ZXIuaGVtbWVyaWNoQGxlaWJuaXotZmxpLmRl