 Michelle Ng
Michelle Ng Dirk Heckl
Dirk Heckl Jan-Henning Klusmann
Jan-Henning Klusmann- Department of Pediatrics I, Martin Luther University Halle-Wittenberg, Halle, Germany
In this post-genomic era, long noncoding RNAs (lncRNAs) are rapidly gaining recognition for their crucial roles across diverse biological processes and contexts. The human blood system is no exception, where dozens of lncRNAs have been established as regulators of normal and/or malignant hematopoiesis, and where ongoing works continue to uncover novel lncRNA functions. Our review focuses on lncRNAs that are involved in the pathogenesis of acute myeloid leukemia (AML) and the mechanisms through which they control gene expression in this disease context. We also comment on genome-wide sequencing or profiling studies that have implicated large sets of lncRNAs in AML pathophysiology.
Background: On lncRNAs and AML
Hematopoietic stem cell (HSC) homeostasis and lifelong blood formation rely on a complex interplay between many different pathways. These include growth factors, signaling cascades, and transcription factors, all of which interact to control the fine balance between self-renewal, quiescence, proliferation, and differentiation (1). Deregulation of this critical interplay can result in malignancy. One example is acute myeloid leukemia (AML), a heterogeneous hematologic disease characterized by the uncontrolled proliferation of undifferentiated myeloid precursors (blasts) (2). This phenotype can be driven by genetic abnormalities that enhance self-renewal and block differentiation, such as chromosomal translocations [e.g., t(15;17)[PML-RARA], t(9;22)[BCR-ABL], inv(16)[CBFB-MYH11], t(8;21)[RUNX1-ETO], MLL rearrangements] and gene mutations (e.g., DNMT3A, NPM1, CEBPA, IDH1/2, TET2, FLT3-ITD), the majority of which are well-defined (3–6). However, our current understanding of the underlying genetic basis of AML hinges on decades of protein-centered research. The contribution of noncoding regions to the initiation, maintenance, and evolution of AML remains to be fully revealed.
It is well-known that only about 2% of the human genome encodes protein, while 70–90% is transcribed, giving rise to an enormous number and variety of noncoding transcripts (7, 8). Among these, long noncoding RNAs (lncRNAs) comprise a substantial proportion (9) that are drawing significant attention as novel regulatory RNAs with roles in diverse physiological processes. LncRNAs are operationally defined as transcriptional products over 200 nucleotides (nt) long that ostensibly lack open reading frames (10, 11). Based on their location relative to coding loci along the genome, they can be further subdivided into four groups: (1) sense, (2) antisense, (3) intronic, or (4) intergenic (12, 13). Some mechanisms that have been described for lncRNAs, so far, include cooperating with gene regulatory circuits in the nucleus (e.g., transcription factor recruitment or chromatin looping), blocking translation, sequestering microRNAs (miRNAs), getting processed into small RNAs, and encoding micropeptides (14–18). Regulating chromatin configuration appears to represent a recurring mechanistic theme among newly characterized lncRNAs, but in general, lncRNA functions do not correspond with their current classification, and we cannot yet conclusively point to unifying pathways. Nonetheless, the picture is still incomplete, and when lncRNAs are better understood as a class, an improved, functionally defined classification system may emerge.
Given the recent rapid expansion of the lncRNA research field and its exposition of lncRNA involvement across a range of key biological processes, it is no surprise that lncRNAs also regulate the blood system. We now know a number of individual lncRNAs that influence lineage decisions along the hematopoietic hierarchy and whose deregulation can result in hematologic malignancy (19–29). These examples are discussed in comprehensive review articles that synthesize our present understanding of lncRNA in normal and malignant hematopoiesis (30–33). While these articles take a broad view on the intersection of hematology and lncRNA biology, this research area has grown to the point where targeted commentaries for different leukemia subtypes may prove beneficial—not least because of the context specificity of lncRNA expression, including between cancers (34, 35). Here, we provide a focused, updated review of lncRNAs that are specifically relevant to AML.
lncRNAs Implicated in AML Pathophysiology
In recent years, several large-scale expression studies have implicated lncRNAs in the pathogenesis of AML by demonstrating the association of distinct lncRNA profiles with genetically defined subtypes of AML (27, 36–40). In the first study of this kind, Garzon et al. profiled lncRNA expression in patients over 60 years old with untreated cytogenetically normal (CN) AML. They discovered distinctive lncRNA signatures associated with major molecular subtypes of AML including FLT3-ITD, NPM1, CEBPA, IDH2, and RUNX1 mutations. Whole-transcriptome sequencing in a cohort of younger adults (<60 years old) similarly revealed lncRNA expression profiles associated with FLT3-ITD, NPM1, and CEBPA mutations (38). Interestingly, lncRNAs located antisense to HOX genes featured among those that were upregulated in NPM1-mutated cases, particularly in the cohort of older adults. In both studies, the authors derived a prognostic score based on a small subset of survival-associated lncRNAs and demonstrated its robust performance as an independent predictor of clinical outcome. Papaioannou et al. additionally correlated their prognostic lncRNAs with messenger RNA (mRNA) and miRNA expression, thereby linking the lncRNAs to cancer-related pathways like leukocyte activation, inflammation, and apoptosis through guilt by association. Last, a smaller study of CN-AML patients uncovered an lncRNA signature that depended on the mutational status of NPM1 (37). The authors identified and validated a minimal set of 12 lncRNAs that could discriminate between NPM1-mutated and NPM1-wild type cases and initiated the preliminary characterization of one lncRNA, XLOC_109948, which they propose is involved in drug sensitivity.
Besides recurrent mutations, distinct lncRNA profiles have also been associated with certain cytogenetic subgroups of AML (27, 39). For example, using unconventional library preparation and assembly approaches, Zhang et al. uncovered a set of four noncoding transcripts that are specifically and highly expressed in patients harboring the PML-RARA translocation. These included a novel, previously unannotated lncRNA, and MEG3, an established maternally expressed lncRNA (39). Our own group has also reported subtype-specific fingerprint lncRNAs for six major subgroups of pediatric AML [inv(16), t(8;21), t(10;11), t(9;11), acute megakaryoblastic leukemia (AMKL), and Down syndrome myeloid leukemia (ML-DS)] (27). We further compared these expression profiles with those of healthy blood populations and found HSC signatures that are upregulated in AML blasts. These lists and others, spanning across the human hematopoietic lineages and pediatric AML subtypes, are publicly available as an online resource (www.lncscape.de), which aims to provide an outline of the lncRNA landscape behind normal and malignant hematopoiesis.
The molecular and cytogenetic subtype-specific expression patterns of lncRNAs in AML samples suggest their crucial contribution to the pathophysiology of this disease. In addition, this phenomenon poises lncRNAs as attractive targets for new therapeutic approaches where the specific targeting of oncogenic proteins has been unsuccessful. However, regarding the role of subgroup-associated lncRNA profiles in leukemogenesis, it remains ambiguous whether they are significant in their own right or whether they represent passenger events driven by genetic aberrations affecting coding genes. Shedding light on this question to some extent is Mer et al.'s exploration of an lncRNA-centered stratification system for AML patients (40). Based solely on lncRNA expression, the authors distinguished four molecular subtypes that differ in prognoses and active pathways and that behave independently of the European Leukemia Net (ELN) risk classification groups. However, although these lncRNA-based subgroups lacked high concordance with conventional clinical or genetic factors, there was, nonetheless, some association with traditional molecular determinants such as mutations in CEBPA, NPM1, TP53, and FLT3-ITD (40). Another recent study of CN-AML cases with leukemia stem cell (LSC)-associated core-enriched gene expression signatures (CE-GES) discovered a set of 111 lncRNAs that strongly correlate with the LSC signature (41). One of the most upregulated LSC-associated lncRNAs, DANCR, was confirmed in functionally validated LSC populations, and its knockdown was shown to reduce self-renewal capacity. The authors further demonstrated that targeting Dancr in vivo in a murine model of AML prolonged the survival of mice after secondary transplantation (41).
All of the above studies implicate lncRNAs in the pathogenesis of AML, and De Clara et al. and Bill et al. additionally indicated their functional relevance and therapeutic potential. Other publications featuring lncRNA-based prognostic scoring systems are also beginning to emerge (42–45). Even so, extensive functional work is required to delineate the interactions between lncRNAs and known driver events and to better understand how each factor can influence the development and progression of AML. With lncRNA research continuing to grow at such a rapid rate, it will soon become crucial that we integrate this body of knowledge into our disease models and treatment practices toward ultimately improving clinical outcomes for patients.
Mechanisms of lncRNA-Mediated Gene Regulation in AML
Starting only a few years ago, the field of lncRNA research underwent—and is continuing to undergo—an exponential expansion. Dozens of articles are published per month describing lncRNA functions in a wide variety of healthy and diseased cell contexts. While this is very exciting for those of us who dream of someday being able to look up lncRNA functions as easily as we currently do for proteins, the field is still young, and in-depth mechanistic investigation is missing for many newly “characterized” lncRNAs. Further complicating the situation, some lncRNAs may produce different outcomes on gene regulation in different cell contexts or act via multiple pathways in a single context. Given the current, incomplete state of the literature—where, in many cases, one lncRNA is reported in a range of cell types along with an equal range of dissimilar mechanisms—it is difficult to distinguish between these two possibilities. By extension, it is also challenging to group lncRNAs based on their mechanisms. Nonetheless, we compiled a catalog of lncRNAs that have specifically been shown to play regulatory roles in AML cells (Table 1). While the list is not exhaustive, it summarizes the best examples of lncRNAs in AML pathogenesis and their mechanisms of action, which are discussed in detail in the rest of this review.
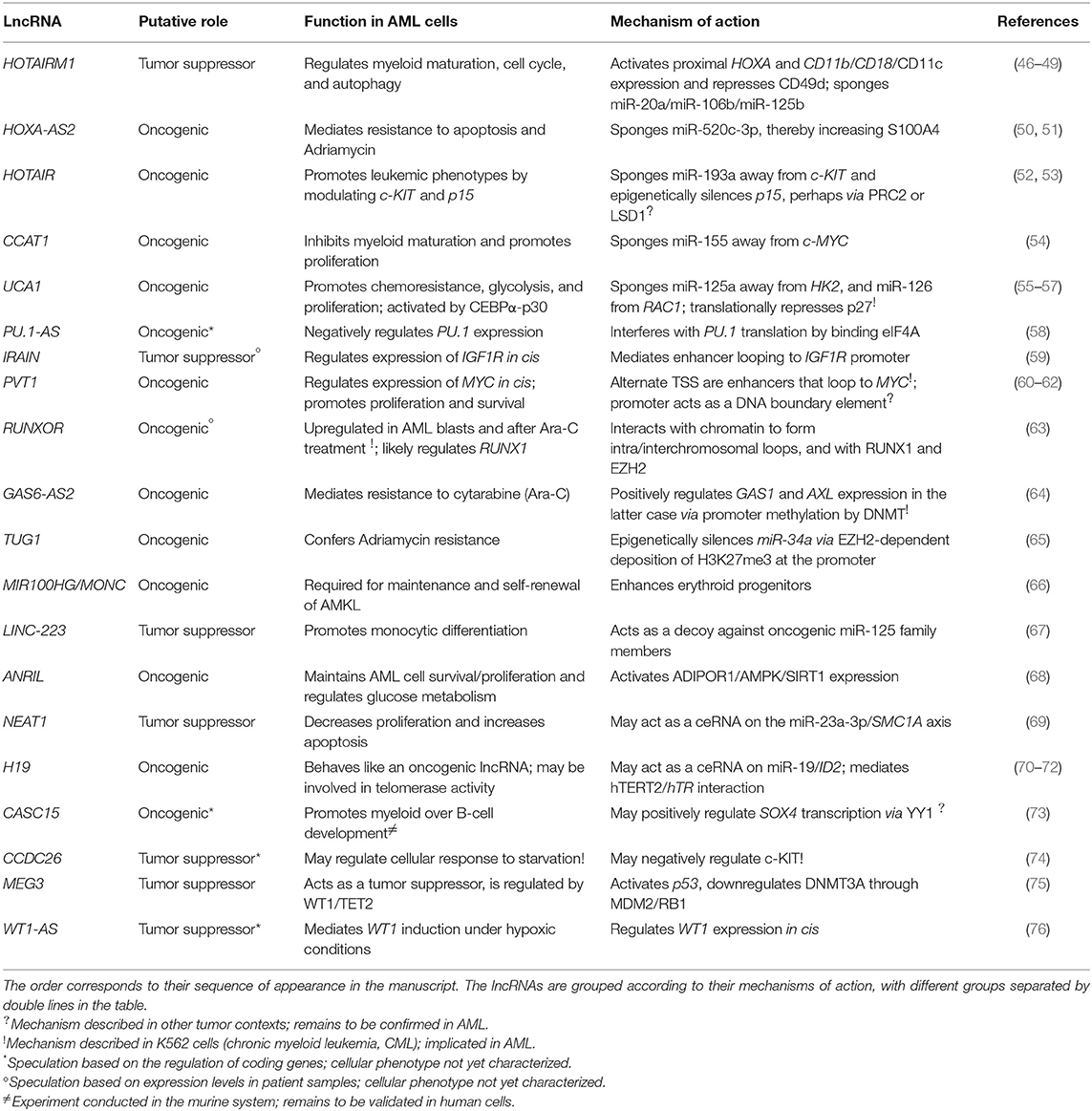
Table 1. A summary of lncRNAs that regulate gene expression in AML and their roles in leukemia cells.
Competing Endogenous RNAs
The deregulation of HOX genes has long been accepted as an important mechanism of leukemogenesis, often in connection with chromosomal translocations involving MLL (77). It is therefore unsurprising that lncRNAs transcribed from the HOX loci have also been discovered to exert regulatory roles in AML. For example, HOTAIRM1 is an intergenic lncRNA (lincRNA) located between HOXA1 and HOXA2 that is transcribed antisense to the HOXA cluster (46), and it is one of the best-studied lncRNAs in the blood system. Under ordinary circumstances, HOTAIRM1 expression is restricted to the myeloid lineage (46). In the NB-4 human acute promyelocytic leukemia (APL) cell line, it is strongly upregulated during all-trans retinoic acid (ATRA)-driven granulocytic differentiation, where it selectively modulates the induction of HOXA1/HOXA4 and of the myeloid maturation markers CD11b, CD18, and CD11c, meanwhile repressing CD49d (46, 47). HOTAIRM1 was shown to promote ATRA-driven cell cycle arrest, suggesting that HOTAIRM1-mediated gene expression changes may regulate a switch from a proliferative phase toward granulocytic maturation (47). An alternate, detailed mechanism put forward by Chen et al. contends that HOTAIRM1 regulates autophagy and thus the degradation of the PML-RARA oncoprotein, which characterizes APL and drives leukemogenesis in this subtype (48). HOTAIRM1 appears to accomplish this by acting as a competing endogenous RNA (ceRNA) and thereby sequestering miR-20a, miR-106a, and miR-125b away from their target mRNAs in the autophagy pathway—ULK1, E2F1, and DRAM2 (48) (Figure 1A).
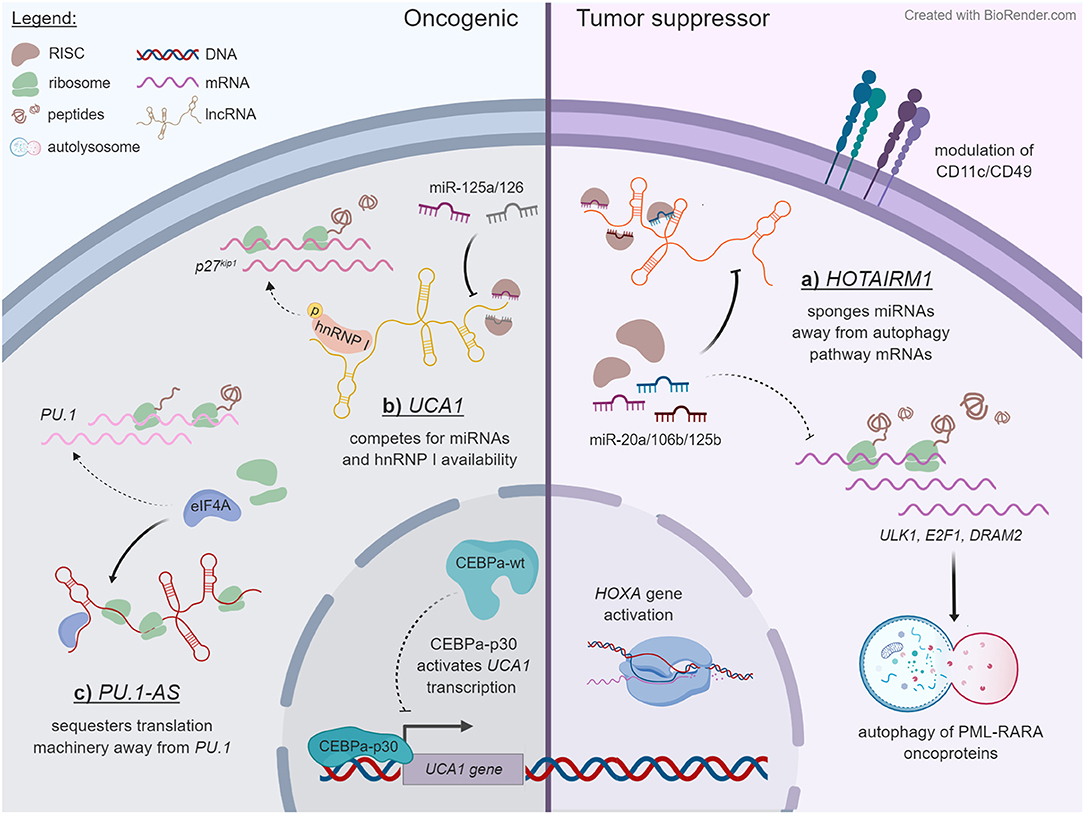
Figure 1. Some examples of lncRNA mechanisms that unfold in the cytoplasmic compartment. Dashed lines specify pathways that are reduced upon lncRNA expression, whereas solid lines indicate outcomes of lncRNA expression. The examples are ordered in a counterclockwise manner. (A) HOTAIRM1 acts as a decoy for miR-20a, miR-106b, and miR-125b in NB-4 cells, titrating them away from mRNAs that encode regulators of autophagy, and thereby enhancing the degradation of PML-RARA oncoprotein through this pathway. HOTAIRM1 also modulates the expression of HOXA genes and of cell surface lineage markers CD11c and CD49d through an undetermined mechanism. (B) In contrast to wild-type CEBPα, truncated CEBPα-p30 transactivates the UCA1 promoter in AML cells with CEBPα mutations. The UCA1 transcript functions as a sponge on miR-125a and miR-126 and additionally competes with the p27kip1 mRNA for stabilizing interactions with hnRNP I. (C) PU.1-AS lncRNAs are polysome associated and interfere with PU.1 protein expression by competitively binding the translation initiating factor eIF4A.
While these findings provide an elegant trans mechanism for HOTAIRM1, another study from around the same time frame proposed a cis-regulatory mode of action within the HOXA cluster. Wang et al. reported HOTAIRM1's involvement in controlling the three-dimensional chromatin organization behind HOXA gene activation in NT2-D1 carcinoma cells (49). Interestingly, modulating HOTAIRM1 exerted opposite effects on HOXA4/5 expression in NB-4 and NT2-D1 cells and produced chromatin conformation changes in NT2-D1 that were absent in NB-4 cells. These findings provide experimental evidence of the often-cited tissue-specific manner of lncRNA function (7–9) and offer a reminder to exercise caution when generalizing their mechanisms across cell types and contexts. Notably, both studies identified multiple HOTAIRM1 isoforms, which may explain the differences in HOTAIRM1-regulated HOXA gene expression between the two cell lines (48, 49). The primary HOTAIRM1 transcript detected in ATRA-treated NB-4 cells was ~500 nt long (48, 49), whereas ATRA-induced NT2-D1 cells produced an unspliced transcript of over 4 kb as well as an ~1.1-kb spliced form (49).
Other ceRNAs arising from the HOX loci include HOXA-AS2 (50, 51) and HOTAIR (52), which were reported to target miR-510-3p and miR-193a, respectively. In contrast to the antileukemic role of HOTAIRM1, HOXA-AS2, and HOTAIR purportedly promote AML cell survival and proliferation by freeing oncogenic pathways from the control of tumor suppressor miRNAs. HOTAIR was additionally reported to induce EZH2-dependent epigenetic silencing of the tumor suppressor gene p15 (53), implying that some lncRNAs may function through multiple pathways to coordinate multilevel gene regulation. This finding is consistent with studies in other cell contexts describing how HOTAIR recruits polycomb repressive complex 2 (PRC2) and the LSD1/coREST/REST complex to regulate chromatin dynamics and epigenetic silencing [see Ref. (78) for a review]. On the other hand, a decisive study in human embryonic kidney 293 (HEK293) cells found that artificially tethering HOTAIR to chromatin led to local conformational changes and transcriptional repression independent of PRC2 (79). The matter of whether PRC2–lncRNA interactions are promiscuous or functionally specific in nature remains highly debated, with abundant evidence for both sides of the argument meriting a separate review [or three: Refs. (80–82)].
To date, there exist two other examples of lncRNAs that appear to regulate gene expression in AML by titrating miRNAs away from their endogenous mRNA targets. First, CCAT1 was shown to inhibit myeloid maturation and promote proliferation by reducing miR-155 availability and consequently raising c-MYC levels in AML cells (54). Second, UCA1 was reported to promote chemoresistance via the miR-125b/HK2 axis (57) and sustain proliferation and survival through miR-126/RAC1 (56). Of note, UCA1 was first described in AML as a regulatory target gene of CEBPα-p30 (55)—the 30-kDa isoform that results from mutations in CEBPA. Hughes et al. showed that both wild-type CEBPα and the p30 protein bind the UCA1 promoter but produce opposite effects on UCA1 expression. Using K562 cells, they outlined UCA1's oncogenic role in suppressing translation of the cell cycle regulator p27kip1 through competing for hnRNP I—a mechanism that was originally proposed in breast cancer (83) (Figure 1B). Another notable study defines UCA1 as an RNA scaffold that is vital for normal erythrocyte development and heme biosynthesis (84).
Other Cytoplasmic Trans-Regulatory Mechanisms
As illustrated by the example of UCA1, there is evidence of lncRNAs acting at the level of translation to regulate gene expression. In AML, the remainder of this group is represented by PU.1-AS, which acts on the mRNA of PU.1, its antisense coding gene, to reduce its translation (58). The PU.1-AS transcripts originate from an intronic promoter in PU.1, which encodes a vital transcription factor for normal hematopoiesis and whose downregulation can result in leukemogenesis [for a review, see (85)]. PU.1-AS was discovered to negatively regulate PU.1 expression, through a mechanism whereby PU.1-AS antagonizes PU.1 translation by selectively binding to the initiation factor eIF4A (58) (Figure 1C). PU.1-AS also appeared to interfere with translation elongation through an unknown mechanism. The implications of these findings on the pathophysiology of AML cells remain to be elucidated as well.
Transcriptional/Epigenetic Regulation in the Nucleus
In cis
Besides acting on miRNAs or mRNAs in the cytoplasm, lncRNAs can also control gene expression in the nucleus at the level of transcription, epigenetics, or chromatin state. They can accomplish this in cis by recruiting transcription factors and epigenetic modifiers to their site of transcription or by assisting the formation of enhancer–promoter loops or other chromatin conformations. IRAIN is one such lncRNA. It is transcribed antisense to the gene encoding IGF1R—a key component of the PI3K/AKT signaling pathway that is constitutively active in AML—and expressed exclusively from the paternal allele in hematopoietic and leukemia cells (59). The transcript itself shows evidence of interacting with neighboring chromatin regions including the IGF1R promoter and an intronic enhancer (59) (Figure 2A). Indeed, Sun et al. showed that the IRAIN RNA is needed for an enhancer–promoter contact between these two elements. However, the impact of IRAIN on AML pathogenesis and whether IRAIN-dependent enhancer–promoter loop formation influences the expression of IGF1R or other genes remain to be seen. Initial hints from patient data suggest an inverse relationship between IRAIN and AML aggressiveness (59).
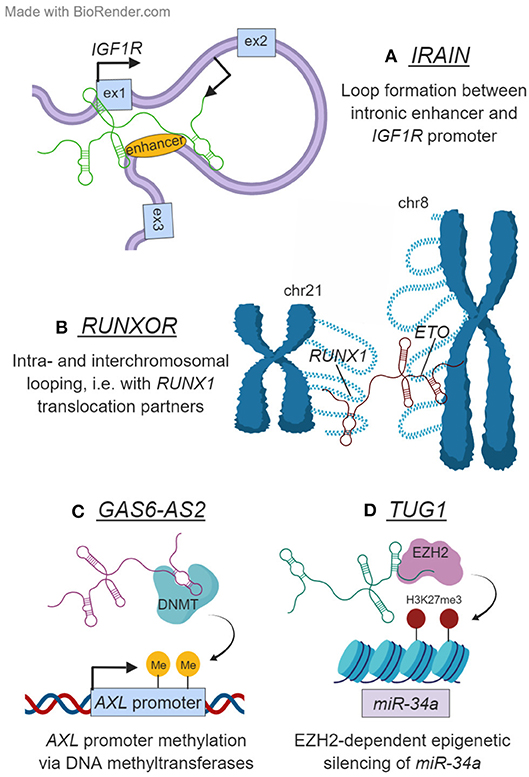
Figure 2. Examples of lncRNAs that function in the nucleus. (A) IRAIN is transcribed antisense to IGF1R and is necessary for the formation of an enhancer–promoter loop in this locus. (B) RUNXOR originates from the RUNX1 locus on chromosome 21. Besides mediating intrachromosomal looping in this region, RUNXOR also contacts distant loci on other chromosomes—including the frequent RUNX1 translocation partner ETO on chromosome 8. (C) GAS6-AS2 modulates the expression of its head-to-head coding gene GAS6, as well as that of GAS6's target receptor AXL. It accomplishes the latter by depositing repressive marks on the AXL promoter DNA, likely with the help of a DNA methyltransferase (DNMT). (D) TUG1 epigenetically regulates miR-34a transcription in an EZH2-dependent manner.
Another example of a cis-regulatory lncRNA is PVT1. The PVT1 gene lies within the human chromosome 8q24 region, which is frequently disrupted in human cancers (i.e., by rearrangements or amplifications) and which harbors numerous MYC enhancer elements—including several within the PVT1 gene locus (86). A landmark paper by Tseng et al. demonstrated PVT1 dependence in breast cancer cells with MYC copy number increase (87). Consistent with the oncogenic role of PVT1 in other cancers, its knockdown reduced proliferation and induced apoptosis in AML cell lines (60, 62, 88) and was accompanied by MYC downregulation in some (60, 88). A CRISPRi tiling screen covering 3 Mb of the locus identified alternative transcription start sites of two PVT1 isoforms as enhancers that loop to the MYC promoter in K562 cells (61). Interestingly, in breast cancer cells, a tumor suppressor role was recently ascribed to the PVT1 promoter, which is proposed to act as a DNA boundary element that insulates MYC from its downstream enhancers (89).
The RUNX1 genetic locus on human chromosome 21 also plays a critical role in normal and malignant hematopoiesis [for a review, see Ref. (90)]. It is frequently disrupted by chromosomal aberrations in AML, such as the t(8;21) translocation that occurs in 30–40% of AML cases. In addition, the locus produces a rather unconventional lncRNA named RUNXOR, which is transcribed as a 216-kb unspliced sense RNA from a promoter several kilobases upstream of RUNX1 and which, thus, overlaps the RUNX1 introns and exons (63). Wang et al. found RUNXOR to be upregulated in AML patient samples compared to healthy controls, and in K562 cells following cytarabine (Ara-C) treatment. In KG-1 cells, the RUNXOR transcript showed evidence of interacting via its 3' end with the RUNX1 promoters and enhancers (63). It also appeared to form an intrachromosomal contact with the most common RUNX1 intronic translocation breakpoint, as well as interchromosomal contacts with distant loci including EVI1 and ETO—recurrent RUNX1 translocation partners in hematologic malignancies (Figure 2B). These latter findings suggest a revolutionary model where RUNXOR may physically mediate the chromosomal translocation process. Lastly, Wang et al. showed EZH2 and RUNX1 binding to the RUNXOR RNA, thereby implying its involvement in recruiting and directing these transcription factors (63).
Bidirectional Promoters
The last few years have seen a steep rise in the application of large-scale CRISPR/Cas9 screens toward functionalizing lncRNAs (64, 91–98). These have been carried out in various cell systems with unified outcomes in several respects: (1) lncRNAs are indeed important in a range of cellular contexts; (2) whether they are essential or not appears to be cell type specific, sometimes even between cell lines derived from the same cancer type; and (3) the screens tend to have low hit identification rates, which may be due in part to the context-restricted manner of lncRNA expression and function.
A recent study by Bester et al. utilized a CRISPR activation (CRISPRa)-based approach to identify lncRNAs that influence cytarabine (Ara-C) resistance in AML cell lines (64). Their screen identified GAS6/GAS6-AS2 as an enriched coding–noncoding gene pair in Ara-C-resistant cells. GAS6/GAS6-AS2 are transcribed in opposite directions from a bidirectional promoter—a “head-to-head” configuration that qualifies GAS6-AS2 as a divergent lncRNA. Divergent lncRNAs have been linked with the genetic loci of essential developmental transcription factors and show evidence of regulating their expression in cis (99–101). Consistent with this model, GAS6-AS2 and GAS6 expression were strongly correlated across 760 cancer cell lines, and antisense oligonucleotide-based GAS6-AS2 knockdown also caused a significant downregulation of GAS6 (64). In addition, AXL, the receptor of the GAS6 ligand in the TYRO3–AXL–MERTK pro-survival signaling cascade, was reduced at the mRNA level, suggesting a coexisting trans mechanism for GAS6-AS2. Based on changes in AXL promoter methylation upon GAS6-AS2 knockdown and on RNA immunoprecipitation assays in K562 cells, the authors speculate that GAS6-AS2 coordinates the activity of DNA methyltransferase proteins at the AXL promoter, on top of regulating GAS6 expression (64) (Figure 2C).
In Trans
As exemplified by GAS6-AS2, lncRNA transcripts can diffuse away from their genomic origin to regulate distant loci by interfacing with nuclear regulatory complexes or by acting as a scaffold for the assembly of transcriptional machinery. TUG1, unlike GAS6-AS2, seems to function exclusively in trans, at least so far [refer to reviews (102, 103)]. Its expression is elevated in AML patients and cell lines (65, 104) and may be linked to aurora kinase (AURKA) protein levels, leading to protection against apoptosis (104). Recent work by Li et al. moreover provides compelling evidence of TUG1's role in promoting chemoresistance (65). Not only was TUG1 upregulated in AML patients compared to healthy controls, but it was also particularly high in Adriamycin (ADR)-resistant leukemia cells relative to ADR-sensitive samples (65). In a series of complementary overexpression and knockdown experiments in HL-60 (sensitive) and HL-60/ADR (resistant) cells, Li et al. robustly demonstrated that TUG1 confers ADR resistance through EZH2-mediated epigenetic silencing of miR-34a (65) (Figure 2D).
Small RNA Host Genes
A number of miRNA host genes have been described to carry out independent regulatory functions as lncRNAs, separate from their miRNA products. Our group demonstrated this for MONC and MIR100HG, each of which harbors a three-miRNA cluster that is transcribed as a single polycistron (tricistron): miR-99a~125b-2 on chromosome 21 and miR-100~125b-1 on chromosome 11, respectively (66). The tricistrons are homologs of each other and were shown to protect AML blasts from transforming growth factor-β1 (TGF-β1)-mediated cell cycle arrest and apoptosis by shifting the overall balance of cellular signaling toward the Wnt pathway (105). This finding has since been corroborated by Lu et al., who showed that MIR100HG-derived miR-100 and miR-125b promote cetuximab resistance through the coordinated repression of five negative regulators of Wnt/β-catenin (106). They additionally discovered a double-negative feedback loop between MIR100HG and GATA6, in which GATA6-mediated MIR100HG repression is relieved through miR-125b targeting of the GATA6 mRNA. Meanwhile, miR-125b appears to act as an oncomiR in megakaryoblastic leukemias (107). Returning to the lncRNA host genes, MIR100HG and MONC expressions are elevated in AML cell lines of the megakaryoblastic subtype (AMKL) (66) and are required for the self-renewal and maintenance of AMKL cells. When overexpressed in CD34+ cord blood cells, we saw that ectopic MONC expression interfered with hematopoietic lineage decisions and enhanced the growth of immature erythroid progenitors.
LINC-223, the precursor of miR-223, is a second example of a miRNA host transcript with independent lncRNA functions (67). Its expression is induced in HL-60 cells upon vitamin D3-driven monocytic differentiation, without a concomitant increase in miR-223, and similar effects were observed during monocytic differentiation of CD34+ cord blood progenitors (67). In order to interrogate LINC-223 specifically, without contribution from miR-223, the authors generated a LINC-223 derivative lacking the requisite region for DROSHA cleavage and miRNA processing. In HL-60 cells, ectopic expression of this construct impaired proliferation and cell cycle progression, meanwhile promoting monocytic differentiation (67). Mechanistically, the authors robustly demonstrated LINC-223's role as a ceRNA decoy of the oncogenic miR-125 family members (miR-125a, miR-125b-1, and miR-125b-2) and implicated this lncRNA in the regulation of the transcription factor IFN4, which is typically targeted by miR-125.
Other Regulatory lncRNAs
In this section we describe lncRNAs that show experimental evidence of regulating gene expression, but that remain ambiguous as to their precise mechanisms of action. These lncRNAs lack defined interaction partners and/or functional rescue data, making it difficult to robustly discriminate cis from trans modes of action or determine direct influence on chromatin architecture, RNA stability, translation, or regulation on any other level. Given the depth of data needed to conclusively resolve lncRNA mechanisms, many newly characterized lncRNAs (or well-known lncRNAs that are investigated in a new cell context) predictably fall in this category.
ANRIL, an lncRNA transcribed from the INK4A–ARF–INK4B cluster on chromosome 9, is known for acting through PRC1/2 to regulate the expression of p15INK4B and p16INK4A in cis and for repressing distant genes in trans [for a review, refer to Ref. (108)]. Polymorphisms affecting ANRIL have been associated with risk for various diseases including cancer, albeit with a wide range of reported impacts on the transcript itself, and ANRIL gain of function also gives rise to diverse cell type-dependent results despite universally increasing oncogenic phenotypes like proliferation and survival (108). This observation again highlights how lncRNAs may have context-dependent functions and discourages their generalization (108). With respect to AML, ANRIL was described to be essential for AML maintenance and to positively regulate glucose metabolism through ADIPOR1 and its targets, AMPK and SIRT1 (68).
The NEAT1 lncRNA is an essential structural component of paraspeckles and plays an indispensable role in their spatial organization, possibly through long-range interactions between NEAT1 transcripts (109). Dysregulation of NEAT1 has been reported in human cancers: interestingly, it is upregulated in various solid tumors, whereas it is downregulated in hematologic malignancies [for recent reviews, refer to Refs. (110, 111)]. Consistent with this, NEAT1 overexpression led to decreased proliferation and increased apoptosis in primary AML blasts and THP-1 cells (69). Zhao et al. suggested the miR-23a-3p/SMC1A axis as a downstream effector of NEAT1 and showed that its perturbation recapitulates the NEAT1 phenotype. Another earlier study in the APL cell line NB-4 found a subtle inverse correlation between NEAT1 and PML-RARA expression and showed the upregulation of NEAT1 upon ATRA-induced differentiation (112).
H19 was among the first-discovered lncRNAs and has since been shown to play essential roles during development and tumorigenesis [see reviews (113, 114)]. H19 is also quite established in the blood system, where it is one of the few lncRNAs that have been characterized in both normal and malignant hematopoiesis. The most recent data come from a single-cell resolution lncRNA landscape of murine HSC development, which identified H19 as a critical regulator of endothelial-to-HSC transition, independent of its derivative mir-675 (115). This study further demonstrated how H19 acts in trans to modulate promoter methylation of key hematopoietic transcription factors like Runx1 and Spi1. In human AML cell lines, H19 knockdown caused a decrease in proliferation and increase in apoptosis (70, 72), while elevated expression correlated with poor treatment response and overall survival in patients (72). Since its expression was observed to reciprocate that of miR-19, H19 was proposed to act as a ceRNA upon the miR-19/ID2 axis (70). It has also been linked to telomerase activity in the NB-4 cell line, where elevated H19 disrupted the interaction between TERT and TR (71). Additional studies are required to determine whether H19 regulates both circuits at once and to resolve the mechanistic details surrounding its mode of action.
Moving this discussion to less renowned lncRNAs, CASC15 was originally reported as a tumor suppressor in neuroblastoma (116), though it has since been found to promote oncogenic processes in melanoma (117) and other solid tumors. A study from Fernando et al. found elevated CASC15 levels in AML and B-cell acute lymphoid leukemia (B-ALL) patients with RUNX1 rearrangements (respectively, t(8;21)[RUNX1-ETO] and t(12;21)[RUNX1-ETV6]) compared to other cytogenetic profiles (73). Enforced expression of Casc15 in normal murine bone marrow cells impaired their engraftment and led to a myeloid differentiation bias concurrent with defective B-cell development. The CASC15 gene lies next to that of the B-cell transcription factor SOX4, and the pair show coordinated expression across AML and B-ALL cell lines—once again, with elevated expression in instances with RUNX1 translocations (REH, Kasumi-1, and SKNO-1) (73). Using several strategies to deplete CASC15 in B-ALL cell lines, Fernando et al. demonstrated its regulatory action on SOX4 and mechanistic synergy with the YY1 transcription factor. It remains unclear whether this occurs in cis or in trans, and the mechanism has yet to be validated in AML cells.
Along with MYC and PVT1, the CCDC26 lncRNA gene lies in the human chromosome 8q21 region that is frequently amplified in cancer (86). Despite being expressed at roughly one copy per cell in myeloid leukemia cell lines, it is upregulated in these cells compared to other cancer types—including lymphoid malignancies—in which CCDC26 expression is not detectable (74). Knockdown clones derived from K562 cells showed increased proliferation rates in serum-depleted media, as well as reduced cell death compared to control cells. This oncogenic phenotype was attributed to the induction of KIT, which was among the most upregulated genes in CCDC26 knockdown cells (74).
MEG3 is a well-known tumor suppressor lncRNA, and its expression is lost—mostly through promoter methylation—in a growing list of human cancers (118). MEG3 was first shown to activate p53 transcription in carcinoma cell lines (119), but a more recent study also discovered a p53-independent pathway where MEG3 downregulates DNMT3A via MDM2/RB signaling to suppress leukemogenesis (75). The latter study further connected the transcriptional regulation of MEG3 to WT1/TET2 (75), two frequently inactivated genes in AML. Notably, the WT1 gene locus produces its own antisense lncRNA, WT1-AS, which is frequently disrupted by aberrant splicing in AML patients (120). WT1-AS and WT1 expression have been linked to hypoxia-induced, TET2-dependent demethylation of the CpG island in intron 1 of WT1 (76). WT1-AS additionally appears to be necessary for WT1 expression but failed to induce WT1 in trans (76). Altogether, these data afford a glimpse into how complex gene regulatory networks can be steered by lncRNAs.
Concluding Remarks
There is obviously substantial work that remains before we can claim an in-depth understanding of how lncRNAs contribute to AML biology or, indeed, to biology in general. Nevertheless, individual examples provide important insights into the regulatory roles of a few lncRNAs in the context of AML. From these, it is apparent that lncRNAs are crucial players in coordinating both novel as well as established oncogenic gene expression pathways across this heterogeneous genetic disease, whether they act upon spatial chromatin organization, epigenetics, competitive miRNA binding, or other processes. We look forward to witnessing how this new layer of regulatory complexity mediated by lncRNAs will integrate into our current protein-centric view of human health and disease and to unraveling the exquisite gene networks that will surely emerge.
Author Contributions
MN drafted the manuscript. DH and J-HK revised the content and approved the manuscript for publication.
Funding
This work was supported by the European Research Council (ERC) funds granted to J-HK through the Horizon 2020 research and innovation program (#714226), and by the St. Baldrick's Foundation Robert J. Arceci Innovation Award, of which J-HK is a recipient.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SG declared a past co-authorship with the authors to the handling editor.
Acknowledgments
We thank all the members of the J-HK/DH group for their support and discussions.
Abbreviations
ADR, Adriamycin; AML, acute myeloid leukemia; AMKL, acute megakaryoblastic leukemia; APL, acute promyelocytic leukemia; Ara-C, Cytarabine; ATRA, all-trans retinoic acid; ceRNA, competing endogenous RNA; CML, chronic myeloid leukemia; CN, cytogenetically normal; CRISPR, clustered regularly interspaced short palindromic repeats; DNMT, DNA methyltransferase; FLT3-ITD, FLT3 internal tandem duplications; HSC, hematopoietic stem cell; lncRNA, long noncoding RNA; LSC, leukemic stem cell; miRNA, microRNA; ML-DS, myeloid leukemia of Down syndrome; PRC, polycomb repressive complex.
References
2. Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW. SnapShot: acute myeloid leukemia. Cancer Cell. (2012) 22:698–98. doi: 10.1016/j.ccr.2012.10.017e691.
3. Cancer Genome Atlas Research, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
4. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
5. Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:103–12. doi: 10.1038/nm.4439
6. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. (2018) 562:526–31. doi: 10.1038/s41586-018-0623-z
7. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. (2005) 309:1559–63. doi: 10.1126/science.1112014
8. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. (2012) 489:101–8. doi: 10.1038/nature11233
9. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. (2011) 25:1915–27. doi: 10.1101/gad.17446611
10. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. (2009) 458:223–7. doi: 10.1038/nature07672
11. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. (2009) 10:155–9. doi: 10.1038/nrg2521
12. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. (2012) 489:57–74. doi: 10.1038/nature11247
13. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
14. Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. (2013) 20:300–7. doi: 10.1038/nsmb.2480
15. Flynn RA, Chang HY. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell. (2014) 14:752–61. doi: 10.1016/j.stem.2014.05.014
16. Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. (2014) 15:423–37. doi: 10.1038/nrg3722
17. Huarte M. The emerging role of lncRNAs in cancer. Nat Med. (2015) 21:1253–61. doi: 10.1038/nm.3981
18. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. (2016) 29:452–63. doi: 10.1016/j.ccell.2016.03.010
19. Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. (2013) 152:727–42. doi: 10.1016/j.cell.2013.01.034
20. Sehgal L, Mathur R, Braun FK, Wise JF, Berkova Z, Neelapu S, et al. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. (2014) 28:2376–87. doi: 10.1038/leu.2014.126
21. Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. (2014) 158:593–606. doi: 10.1016/j.cell.2014.05.049
22. Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. (2014) 344:310–3. doi: 10.1126/science.1251456
23. Luo M, Jeong M, Sun D, Park HJ, Rodriguez BA, Xia Z, et al. Long non-coding RNAs control hematopoietic stem cell function. Cell Stem Cell. (2015) 16:426–38. doi: 10.1016/j.stem.2015.02.002
24. Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. (2015) 16:318–25. doi: 10.1038/ni.3093
25. Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell. (2016) 165:1672–85. doi: 10.1016/j.cell.2016.05.075
26. Alvarez-Dominguez JR, Knoll M, Gromatzky AA, Lodish HF. The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell Rep. (2017) 19:2503–14. doi: 10.1016/j.celrep.2017.05.082
27. Schwarzer A, Emmrich S, Schmidt F, Beck D, Ng M, Reimer C, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun. (2017) 8:218. doi: 10.1038/s41467-017-00212-4
28. Delas MJ, Jackson BT, Kovacevic T, Vangelisti S, Munera Maravilla E, Wild SA, et al. lncRNA Spehd regulates hematopoietic stem and progenitor cells and is required for multilineage differentiation. Cell Rep. (2019) 27:719–29. doi: 10.1016/j.celrep.2019.03.080e716.
29. Wu Z, Gao S, Zhao X, Chen J, Keyvanfar K, Feng X, et al. Long noncoding RNAs of single hematopoietic stem and progenitor cells in healthy and dysplastic human bone marrow. Haematologica. (2019) 104:894–906. doi: 10.3324/haematol.2018.208926
30. Alvarez-Dominguez JR, Hu W, Gromatzky AA, Lodish HF. Long noncoding RNAs during normal and malignant hematopoiesis. Int J Hematol. (2014) 99:531–41. doi: 10.1007/s12185-014-1552-8
31. Morlando M, Ballarino M, Fatica A. Long non-coding RNAs: new players in hematopoiesis and leukemia. Front Med. (2015) 2:23. doi: 10.3389/fmed.2015.00023
32. Jeong M, Goodell MA. Noncoding regulatory RNAs in hematopoiesis. Curr Top Dev Biol. (2016) 118:245–70. doi: 10.1016/bs.ctdb.2016.01.006
33. Alvarez-Dominguez JR, Lodish HF. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood. (2017) 130:1965–75. doi: 10.1182/blood-2017-06-788695
34. Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. (2015) 28:529–40. doi: 10.1016/j.ccell.2015.09.006
35. Chiu HS, Somvanshi S, Patel E, Chen TW, Singh VP, Zorman B, et al. Pan-cancer analysis of lncRNA regulation supports their targeting of cancer genes in each tumor context. Cell Rep. (2018) 23:297–312 e212. doi: 10.1016/j.celrep.2018.03.064
36. Garzon R, Volinia S, Papaioannou D, Nicolet D, Kohlschmidt J, Yan PS, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci USA. (2014) 111:18679–84. doi: 10.1073/pnas.1422050112
37. De Clara E, Gourvest M, Ma H, Vergez F, Tosolini M, Dejean S, et al. Long non-coding RNA expression profile in cytogenetically normal acute myeloid leukemia identifies a distinct signature and a new biomarker in NPM1-mutated patients. Haematologica. (2017) 102:1718–26. doi: 10.3324/haematol.2017.171645
38. Papaioannou D, Nicolet D, Volinia S, Mrozek K, Yan P, Bundschuh R, et al. Prognostic and biologic significance of long non-coding RNA profiling in younger adults with cytogenetically normal acute myeloid leukemia. Haematologica. (2017) 102:1391–400. doi: 10.3324/haematol.2017.166215
39. Zhang J, Griffith M, Miller CA, Griffith OL, Spencer DH, Walker JR, et al. Comprehensive discovery of noncoding RNAs in acute myeloid leukemia cell transcriptomes. Exp Hematol. (2017) 55:19–33. doi: 10.1016/j.exphem.2017.07.008
40. Mer AS, Lindberg J, Nilsson C, Klevebring D, Wang M, Gronberg H, et al. Expression levels of long non-coding RNAs are prognostic for AML outcome. J Hematol Oncol. (2018) 11:52. doi: 10.1186/s13045-018-0596-2
41. Bill M, Papaioannou D, Karunasiri M, Kohlschmidt J, Pepe F, Walker CJ, et al. Expression and functional relevance of long non-coding RNAs in acute myeloid leukemia stem cells. Leukemia. (2019). doi: 10.1038/s41375-019-0429-5. [Epub ahead of print].
42. Marcucci G, Yan P, Maharry K, Frankhouser D, Nicolet D, Metzeler KH, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene score. J Clin Oncol. (2014) 32:548–56. doi: 10.1200/JCO.2013.50.6337
43. Yao CY, Chen CH, Huang HH, Hou HA, Lin CC, Tseng MH, et al. A 4-lncRNA scoring system for prognostication of adult myelodysplastic syndromes. Blood Adv. (2017) 1:1505–16. doi: 10.1182/bloodadvances.2017008284
44. Beck D, Thoms JAI, Palu C, Herold T, Shah A, Olivier J, et al. A four-gene LincRNA expression signature predicts risk in multiple cohorts of acute myeloid leukemia patients. Leukemia. (2018) 32:263–72. doi: 10.1038/leu.2017.210
45. Tsai CH, Yao CY, Tien FM, Tang JL, Kuo YY, Chiu YC, et al. Incorporation of long non-coding RNA expression profile in the 2017 ELN risk classification can improve prognostic prediction of acute myeloid leukemia patients. EBioMedicine. (2019) 40:240–50. doi: 10.1016/j.ebiom.2019.01.022
46. Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, et al. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood. (2009) 113:2526–34. doi: 10.1182/blood-2008-06-162164
47. Zhang X, Weissman SM, Newburger PE. Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle progression during myeloid maturation in NB4 human promyelocytic leukemia cells. RNA Biol. (2014) 11:777–87. doi: 10.4161/rna.28828
48. Chen ZH, Wang WT, Huang W, Fang K, Sun YM, Liu SR, et al. The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ. (2017) 24:212–24. doi: 10.1038/cdd.2016.111
49. Wang XQ, Dostie J. Reciprocal regulation of chromatin state and architecture by HOTAIRM1 contributes to temporal collinear HOXA gene activation. Nucleic Acids Res. (2017) 45:1091–104. doi: 10.1093/nar/gkw966
50. Zhao H, Zhang X, Frazao JB, Condino-Neto A, Newburger PE. HOX antisense lincRNA HOXA-AS2 is an apoptosis repressor in all trans retinoic acid treated NB4 promyelocytic leukemia cells. J Cell Biochem. (2013) 114:2375–83. doi: 10.1002/jcb.24586
51. Dong X, Fang Z, Yu M, Zhang L, Xiao R, Li X, et al. Knockdown of long noncoding RNA HOXA-AS2 suppresses chemoresistance of acute myeloid leukemia via the miR-520c-3p/S100A4 axis. Cell Physiol Biochem. (2018) 51:886–96. doi: 10.1159/000495387
52. Xing CY, Hu XQ, Xie FY, Yu ZJ, Li HY, Bin Z, et al. Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett. (2015) 589:1981–87. doi: 10.1016/j.febslet.2015.04.061
53. Gao S, Zhou B, Li H, Huang X, Wu Y, Xing C, et al. Long noncoding RNA HOTAIR promotes the self-renewal of leukemia stem cells through epigenetic silencing of p15. Exp Hematol. (2018) 67:32–40. doi: 10.1016/j.exphem.2018.08.005e33.
54. Chen L, Wang W, Cao L, Li Z, Wang X. Long non-coding RNA CCAT1 acts as a competing endogenous RNA to regulate cell growth and differentiation in acute myeloid leukemia. Mol Cells. (2016) 39:330–6. doi: 10.14348/molcells.2016.2308
55. Hughes JM, Legnini I, Salvatori B, Masciarelli S, Marchioni M, Fazi F, et al. C/EBPalpha-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget. (2015) 6:18534–44. doi: 10.18632/oncotarget.4069
56. Sun MD, Zheng YQ, Wang LP, Zhao HT, Yang S. Long noncoding RNA UCA1 promotes cell proliferation, migration and invasion of human leukemia cells via sponging miR-126. Eur Rev Med Pharmacol Sci. (2018) 22:2233–45. doi: 10.26355/eurrev_201804_14809
57. Zhang Y, Liu Y, Xu X. Knockdown of LncRNA-UCA1 suppresses chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA-125a/hexokinase 2 pathway. J Cell Biochem. (2018) 119:6296–308. doi: 10.1002/jcb.26899
58. Ebralidze AK, Guibal FC, Steidl U, Zhang P, Lee S, Bartholdy B, et al. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev. (2008) 22:2085–92. doi: 10.1101/gad.1654808
59. Sun J, Li W, Sun Y, Yu D, Wen X, Wang H, et al. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. (2014) 42:9588–601. doi: 10.1093/nar/gku549
60. Zeng C, Yu X, Lai J, Yang L, Chen S, Li Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J Hematol Oncol. (2015) 8:126. doi: 10.1186/s13045-015-0223-4
61. Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. (2016) 354:769–73. doi: 10.1126/science.aag2445
62. Salehi M, Sharifi M. Induction of apoptosis and necrosis in human acute erythroleukemia cells by inhibition of long non-coding RNA PVT1. Mol Biol Res Commun. (2018) 7:89–96. doi: 10.22099/mbrc.2018.29081.1316
63. Wang H, Li W, Guo R, Sun J, Cui J, Wang G, et al. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer. (2014) 135:2783–94. doi: 10.1002/ijc.28922
64. Bester AC, Lee JD, Chavez A, Lee YR, Nachmani D, Vora S, et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell. (2018) 173:649–64. doi: 10.1016/j.cell.2018.03.052e620.
65. Li Q, Song W, Wang J. TUG1 confers adriamycin resistance in acute myeloid leukemia by epigenetically suppressing miR-34a expression via EZH2. Biomed Pharmacother. (2019) 109:1793–801. doi: 10.1016/j.biopha.2018.11.003
66. Emmrich S, Streltsov A, Schmidt F, Thangapandi VR, Reinhardt D, Klusmann JH. LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol Cancer. (2014) 13:171. doi: 10.1186/1476-4598-13-171
67. Mangiavacchi A, Sorci M, Masciarelli S, Larivera S, Legnini I, Iosue I, et al. The miR-223 host non-coding transcript linc-223 induces IRF4 expression in acute myeloid leukemia by acting as a competing endogenous RNA. Oncotarget. (2016) 7:60155–68. doi: 10.18632/oncotarget.11165
68. Sun LY, Li XJ, Sun YM, Huang W, Fang K, Han C, et al. LncRNA ANRIL regulates AML development through modulating the glucose metabolism pathway of AdipoR1/AMPK/SIRT1. Mol Cancer. (2018) 17:127. doi: 10.1186/s12943-018-0879-9
69. Zhao C, Wang S, Zhao Y, Du F, Wang W, Lv P, et al. Long noncoding RNA NEAT1 modulates cell proliferation and apoptosis by regulating miR-23a-3p/SMC1A in acute myeloid leukemia. J Cell Physiol. (2019) 234:6161–72. doi: 10.1002/jcp.27393
70. Zhao TF, Jia HZ, Zhang ZZ, Zhao XS, Zou YF, Zhang W, et al. LncRNA H19 regulates ID2 expression through competitive binding to hsa-miR-19a/b in acute myelocytic leukemia. Mol Med Rep. (2017) 16:3687–93. doi: 10.3892/mmr.2017.7029
71. El Hajj J, Nguyen E, Liu Q, Bouyer C, Adriaenssens E, Hilal G, et al. Telomerase regulation by the long non-coding RNA H19 in human acute promyelocytic leukemia cells. Mol Cancer. (2018) 17:85. doi: 10.1186/s12943-018-0835-8
72. Zhang TJ, Zhou JD, Zhang W, Lin J, Ma JC, Wen XM, et al. H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin Epigenetics. (2018) 10:47. doi: 10.1186/s13148-018-0486-z
73. Fernando TR, Contreras JR, Zampini M, Rodriguez-Malave NI, Alberti MO, Anguiano J, et al. The lncRNA CASC15 regulates SOX4 expression in RUNX1-rearranged acute leukemia. Mol Cancer. (2017) 16:126. doi: 10.1186/s12943-017-0692-x
74. Hirano T, Yoshikawa R, Harada H, Harada Y, Ishida A, Yamazaki T. Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression. Mol Cancer. (2015) 14:90. doi: 10.1186/s12943-015-0364-7
75. Lyu Y, Lou J, Yang Y, Feng J, Hao Y, Huang S, et al. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia. (2017) 31:2543–51. doi: 10.1038/leu.2017.116
76. McCarty G, Loeb DM. Hypoxia-sensitive epigenetic regulation of an antisense-oriented lncRNA controls WT1 expression in myeloid leukemia cells. PLoS ONE. (2015) 10:e0119837. doi: 10.1371/journal.pone.0119837
77. Rice KL, Licht JD. HOX deregulation in acute myeloid leukemia. J Clin Invest. (2007) 117:865–8. doi: 10.1172/JCI31861
78. Bhan A, Mandal SS. LncRNA HOTAIR: a master regulator of chromatin dynamics and cancer. Biochim Biophys Acta. (2015) 1856:151–64. doi: 10.1016/j.bbcan.2015.07.001
79. Portoso M, Ragazzini R, Brencic Z, Moiani A, Michaud A, Vassilev I, et al. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. (2017) 36:981–94. doi: 10.15252/embj.201695335
80. Brockdorff N. Noncoding RNA and polycomb recruitment. RNA. (2013) 19:429–42. doi: 10.1261/rna.037598.112
81. Davidovich C, Cech TR. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA. (2015) 21:2007–22. doi: 10.1261/rna.053918.115
82. Yan J, Dutta B, Hee YT, Chng WJ. Towards understanding of PRC2 binding to RNA. RNA Biol. (2019) 16:176–84. doi: 10.1080/15476286.2019.1565283
83. Huang J, Zhou N, Watabe K, Lu Z, Wu F, Xu M, et al. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. (2014) 5:e1008. doi: 10.1038/cddis.2013.541
84. Liu J, Li Y, Tong J, Gao J, Guo Q, Zhang L, et al. Long non-coding RNA-dependent mechanism to regulate heme biosynthesis and erythrocyte development. Nat Commun. (2018) 9:4386. doi: 10.1038/s41467-018-06883-x
85. van Riel B, Rosenbauer F. Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol Chem. (2014) 395:1265–74. doi: 10.1515/hsz-2014-0195
86. Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: an oasis of non-coding transcriptional activity. Front Genet. (2012) 3:69. doi: 10.3389/fgene.2012.00069
87. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. (2014) 512:82–6. doi: 10.1038/nature13311
88. Houshmand M, Yazdi N, Kazemi A, Atashi A, Hamidieh AA, Anjam Najemdini A, et al. Long non-coding RNA PVT1 as a novel candidate for targeted therapy in hematologic malignancies. Int J Biochem Cell Biol. (2018) 98:54–64. doi: 10.1016/j.biocel.2018.03.001
89. Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, et al. Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell. (2018) 173:1398–412 e1322. doi: 10.1016/j.cell.2018.03.068
90. Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci. (2012) 17:1120–39. doi: 10.2741/3977
91. McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest. (2016) 126:639–52. doi: 10.1172/JCI83265
92. Sanjana NE, Wright J, Zheng K, Shalem O, Fontanillas P, Joung J, et al. High-resolution interrogation of functional elements in the noncoding genome. Science. (2016) 353:1545–9. doi: 10.1126/science.aaf7613
93. Zhu S, Li W, Liu J, Chen CH, Liao Q, Xu P, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat Biotechnol. (2016) 34:1279–86. doi: 10.1038/nbt.3715
94. Covarrubias S, Robinson EK, Shapleigh B, Vollmers A, Katzman S, Hanley N, et al. CRISPR/Cas-based screening of long non-coding RNAs (lncRNAs) in macrophages with an NF-kappaB reporter. J Biol Chem. (2017) 292:20911–20. doi: 10.1074/jbc.M117.799155
95. Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. (2017) 548:343–6. doi: 10.1038/nature23451
96. Koirala P, Huang J, Ho TT, Wu F, Ding X, Mo YY. LncRNA AK023948 is a positive regulator of AKT. Nat Commun. (2017) 8:14422. doi: 10.1038/ncomms14422
97. Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. (2017) 355:eaah7111. doi: 10.1126/science.aah7111
98. Liu Y, Cao Z, Wang Y, Guo Y, Xu P, Yuan P, et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat Biotechnol. (2018). doi: 10.1038/nbt.4283
99. Lepoivre C, Belhocine M, Bergon A, Griffon A, Yammine M, Vanhille L, et al. Divergent transcription is associated with promoters of transcriptional regulators. BMC Genomics. (2013) 14:914. doi: 10.1186/1471-2164-14-914
100. Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci USA. (2013) 110:2876–81. doi: 10.1073/pnas.1221904110
101. Luo S, Lu JY, Liu L, Yin Y, Chen C, Han X, et al. Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell. (2016) 18:637–52. doi: 10.1016/j.stem.2016.01.024
102. Li Z, Shen J, Chan MT, Wu WK. TUG1: a pivotal oncogenic long non-coding RNA of human cancers. Cell Prolif. (2016) 49:471–5. doi: 10.1111/cpr.12269
103. Wang WY, Wang YF, Ma P, Xu TP, Shu YQ. Taurine upregulated gene 1: a vital long noncoding RNA associated with cancer in humans (Review). Mol Med Rep. (2017) 16:6467–71. doi: 10.3892/mmr.2017.7472
104. Wang X, Zhang L, Zhao F, Xu R, Jiang J, Zhang C, et al. Long non-coding RNA taurine-upregulated gene 1 correlates with poor prognosis, induces cell proliferation, and represses cell apoptosis via targeting aurora kinase A in adult acute myeloid leukemia. Ann Hematol. (2018) 97:1375–89. doi: 10.1007/s00277-018-3315-8
105. Emmrich S, Rasche M, Schoning J, Reimer C, Keihani S, Maroz A, et al. miR-99a/100~125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFbeta and Wnt signaling. Genes Dev. (2014) 28:858–74. doi: 10.1101/gad.233791.113
106. Lu Y, Zhao X, Liu Q, Li C, Graves-Deal R, Cao Z, et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat Med. (2017) 23:1331–41. doi: 10.1038/nm.4424
107. Klusmann JH, Li Z, Bohmer K, Maroz A, Koch ML, Emmrich S, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. (2010) 24:478–90. doi: 10.1101/gad.1856210
108. Kong Y, Hsieh CH, Alonso LC. ANRIL: a lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front Endocrinol. (2018) 9:405. doi: 10.3389/fendo.2018.00405
109. Lin Y, Schmidt BF, Bruchez MP, McManus CJ. Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res. (2018) 46:3742–52. doi: 10.1093/nar/gky046
110. Yu X, Li Z, Zheng H, Chan MT, Wu WK. NEAT1: a novel cancer-related long non-coding RNA. Cell Prolif. (2017) 50:e12329. doi: 10.1111/cpr.12329
111. Ghaforui-Fard S, Taheri M. Nuclear enriched abundant transcript 1 (NEAT1): a long non-coding RNA with diverse functions in tumorigenesis. Biomed Pharmacother. (2018) 111:51–9. doi: 10.1016/j.biopha.2018.12.070
112. Zeng C, Xu Y, Xu L, Yu X, Cheng J, Yang L, et al. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer. (2014) 14:693. doi: 10.1186/1471-2407-14-693
113. Gabory A, Jammes H, Dandolo L. The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays. (2010) 32:473–80. doi: 10.1002/bies.200900170
114. Raveh E, Matouk IJ, Gilon M, Hochberg A. The H19 long non-coding RNA in cancer initiation, progression and metastasis—a proposed unifying theory. Mol Cancer. (2015) 14:184. doi: 10.1186/s12943-015-0458-2
115. Zhou J, Xu J, Zhang L, Liu S, Ma Y, Wen X, et al. Combined single-cell profiling of lncRNAs and functional screening reveals that H19 is pivotal for embryonic hematopoietic stem cell development. Cell Stem Cell. (2019) 24:285–298 e285. doi: 10.1016/j.stem.2018.11.023
116. Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M, et al. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. (2015) 75:3155–66. doi: 10.1158/0008-5472.CAN-14-3613
117. Lessard L, Liu M, Marzese DM, Wang H, Chong K, Kawas N, et al. The CASC15 long intergenic noncoding RNA locus is involved in melanoma progression and phenotype switching. J Invest Dermatol. (2015) 135:2464–74. doi: 10.1038/jid.2015.200
118. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. (2012) 48:R45–53. doi: 10.1530/JME-12-0008
119. Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. (2007) 282:24731–42. doi: 10.1074/jbc.M702029200
Keywords: acute myeloid leukemia, long noncoding RNA, regulatory RNA, antisense transcripts, competing endogenous RNA, chromatin looping, lncRNA profiles
Citation: Ng M, Heckl D and Klusmann J-H (2019) The Regulatory Roles of Long Noncoding RNAs in Acute Myeloid Leukemia. Front. Oncol. 9:570. doi: 10.3389/fonc.2019.00570
Received: 02 April 2019; Accepted: 12 June 2019;
Published: 09 July 2019.
Edited by:
Sheng F. Cai, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Francesco Fazi, Sapienza University of Rome, ItalySarah Grasedieck, Ulm University Medical Center, Germany
Copyright © 2019 Ng, Heckl and Klusmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan-Henning Klusmann, SmFuLUhlbm5pbmcuS2x1c21hbm5AdWstaGFsbGUuZGU=