 Bella S. Guerrouahen
Bella S. Guerrouahen Cristina Maccalli
Cristina Maccalli Chiara Cugno
Chiara Cugno Sergio Rutella2
Sergio Rutella2- 1Sidra Medicine, Member of Qatar Foundation, Research Department, Doha, Qatar
- 2John van Geest Cancer Research Centre, Nottingham Trent University, Nottingham, United Kingdom
- 3Veana Therapeutics, Inc., Portland, OR, United States
- 4Providence Cancer Center, Portland, OR, United States
Tumors employ strategies to escape immune control. The principle aim of most cancer immunotherapies is to restore effective immune surveillance. Among the different processes regulating immune escape, tumor microenvironment-associated soluble factors, and/or cell surface-bound molecules are mostly responsible for dysfunctional activity of tumor-specific CD8+T cells. These dynamic immunosuppressive networks prevent tumor rejection at several levels, limiting also the success of immunotherapies. Nevertheless, the recent clinical development of immune checkpoint inhibitors or of molecules modulating cellular targets and immunosuppressive enzymes highlights the great potential of approaches based on the selective disruption of immunosuppressive networks. Currently, the administration of different categories of immunotherapy in combination regimens is the ultimate modality for impacting the survival of cancer patients. With the advent of immune checkpoint inhibitors, designed to mount an effective antitumor immune response, profound changes occurred in cancer immunotherapy: from a global stimulation of the immune system to a specific targeting of an immune component. This review will specifically highlight the players, the mechanisms limiting an efficient antitumor response and the current immunotherapy modalities tailored to target immune suppressive pathways. We also discuss the ongoing challenges encountered by these strategies and provide suggestions for circumventing hurdles to new immunotherapeutic approaches, including the use of relevant biomarkers in the optimization of immunotherapy regimens and the identification of patients who can benefit from defined immune-based approaches.
Cancer growth and progression is controlled by the immune cells infiltrating the tumor microenvironment (TME). Several reports provide clear evidence that activation of an antitumor immune response in the host results in tumor regression and translates into better clinical outcomes in animal and human cancers (1, 2). However, more often than not, the interactions between the immunological players and the tumor cells in the TME lead to immune evasion contributing to tumor progression (3). Importantly, the immune selection inadvertently favors the emergence of tumors with reduced immunogenicity. The stromal compartment is required to create a permissive environment for the extravasation and spread of genetically and epigenetically altered tumor cells (4), and maintain the inactivation of various components of the immune system, preventing their adequate functioning (5). The major role of the immune system in oncology has been highlighted by the identification of one of the cancer hallmarks: “tumor evasion from immune surveillance” (4). Instead of using specific inhibitors to target tumor cells, immunotherapy drugs facilitate either the generation of anti-tumor immune responses or the unleashing of the patient's immune system against cancer through immune checkpoint blockade (ICB). Co-inhibitory molecules are crucial players in the regulation of T cell responses by modulating the signaling cascade initiated by T cell receptor (TCR) engagement. An antigen-independent “second signal” is required to counteract T cell activation and to induce a down-modulation of the immune system activation toward a resting state (6). ICB functions as tumor suppressing factors through the modulation of immune cell-tumor cell interaction, preventing an effective tumor attack. Immunotherapy strives to improve immune system functions, and therefore potentiate immune surveillance of cancer resulting in effective tumor control. Unfortunately, the response to treatment and the course of the disease can be influenced by several elements including the capacity of the tumor to adapt through loss of immunogenicity, the impairment of tumor antigen processing, and presentation, and the modulation of the TME toward immunosuppression. The immune contexture defined by the presence of immune-suppressive or -regulatory cell types of the adaptive and innate immune system, or the production of immunosuppressive factors may result in the impairment of local tumor effector cells that can result also in limited systemic anti-tumor immune responses and tumor progression (7). The tumor progression is primarily due to the deleterious effects of tumor- and cell- derived factors and co-inhibitory molecules present in the TME. Understanding and overcoming these tumor escape mechanisms remain a challenge for the successful treatment of cancer. In this review, factors and molecules in the TME, and the strategies that neutralize their effects will be addressed. Results from ongoing immunotherapy clinical studies and agents that have been recently identified as standard care for some type of tumors will be summarized.
Soluble Factors
Interleukin-10 (IL-10)
IL-10, identified as cytokine synthesis inhibitory factor (CSIF) is a potent anti-inflammatory cytokine which is structurally related to interferon (IFN)-γ (8). IL-10 is produced by various types of immune cells, including T regulatory cells (Tregs), T helper cells (Th)-1, Th17, B cells, activated monocytes, macrophages, mast cells, granulocytes, dendritic cells (DC) and tumor cells, thus preventing the inflammatory environment created by cancer (9, 10). Recognized as a Th2 cytokine, IL-10 secretion influences the dysfunction of innate and adaptive immunity to allow the escape of malignant cells from immune surveillance by inhibiting the Th1 immune response and the T cell cytotoxic activity (11). IL-10 signaling suppresses T cell function by stimulating the transcription of genes known to suppress toll-like receptor (TLR)-dependent and IFN-γ-dependent signaling in antigen presenting cells (APCs), and thus limiting their function. In already activated DCs, IL-10 has no effect, but in activated monocytes/macrophages IL-10 induces March-I, an ubiquitin ligase that affects antigen presentation by limiting the expression of the major histocompatibility complex (MHC)-II and CD86 (12). In a spontaneously metastatic 4T1 mammary carcinoma mouse model, the increase in myeloid-derived suppressor cells (MDSCs) production of IL-10 decreased the macrophage production of IL-12, and thereby impaired tumor immunity (13). In murine models of inflammatory bowel disease, the frequency of DNA mutations in the colon was 4- to 5-fold greater in IL-10 deficient mice than in IL-10-sufficient mice (14). Treatment of IL-10 deficient mice with a pegylated (PEG) form of recombinant human IL-10 increased the cytotoxic activity of CD8+ T cells and controlled tumor growth. Moreover, when cured by PEG IL-10, mice had a long-lasting immunity (15, 16).
A meta-analysis revealed that there is a strong correlation between high levels of circulating IL-10 and poor prognosis of various patients with most types of solid tumors and hematological malignancies (17). Using biopsies from patients with oral squamous cell carcinoma, tumor-associated macrophages (TAMs) expressing CD163+CD204+ promoted T cell apoptosis and immunosuppression via IL-10 and PD-L1 production, thus predicting an unfavorable prognosis (18). Clinical trials are ongoing for evaluating pegylated recombinant human IL-10 (AM0010, pegilodecakin) in combination with pembrolizumab in metastatic non-small-cell lung carcinoma (NSCLC) (ClinicalTrials.gov Identifier: NCT03382899). After a favorable phase Ib trial in pancreatic cancer, a phase III study is evaluating the safety and efficacy of AM0010 in combination with FOLFOX compared to FOLFOX monotherapy in metastatic pancreatic cancers as a second-line therapy (NCT02923921). Up to date, no IL-10 receptor agonist has received regulatory approval for its use.
Vascular Endothelial Growth Factor (VEGF)
The VEGF family of growth factors includes the splice variant forms: VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PLGF) (19). VEGF-A, often referred to as VEGF, is the predominant ligand for VEGF receptor 2 (VEGFR2, KDR). VEGF-A exerts a potent proangiogenic effect that stimulates endothelial cell proliferation, migration, and survival in both normal and pathological angiogenesis (19, 20). Both tumor and stromal cells in the TME can produce VEGF (19). VEGF affects lymphocyte-endothelium interactions by altering the adhesion molecule clustering process at the endothelial cells surface, thus controlling lymphocyte trafficking (21). Secondly, VEGF has a systemic immunosuppressive effect. Intratumoral VEGF production limits T cell recruitment into tumors, promote T cell exhaustion and induces accumulation of immune-regulatory cells, such as immature DCs, MDSCs, Tregs, and TAMs (22, 23). Increased VEGF serum levels or tumor expression are associated with a poor prognosis in patients with malignancies including metastatic colorectal cancer (24). Interestingly, DCs matured in the presence of VEGF express less human leukocyte antigen-DR (HLA-DR) and CD86. This expression can be restored by VEGF inhibitors, bevacizumab, and sorafenib. VEGF increases also the expression and activity of indoleamine 2,3- dioxygenase (IDO) in DCs, which has a suppressive effect on antigen (Ag)-specific and mitogen-stimulated lymphocyte proliferation (25). Tumor-produced VEGF-A attracts Nrp1-expressing Tregs and this interaction mediates Tregs infiltration into the tumor (26). In ovarian cancer patients, VEGF enhances expression of PD-1 and other inhibitory checkpoints involved in CD8+ T cell exhaustion (27). A preclinical report demonstrated in vitro inhibition of the tumor growth with a decrease in the density of vessels in tumor-bearing mice treated with monoclonal antibodies targeting and neutralizing VEGF-A (28).
Based on preclinical evidences, bevacizumab (Avastin, Genentech, Inc.) has been approved in 2004 by the U. S. Food and Drug Administration (FDA) for the first-line treatment of metastatic colorectal cancer (29). Although, several inhibitors of VEGF/VEGFR2 (i.e., bevacizumab, pazopanib, sunitinib, sorafenib) are commonly used in the clinic, they are beneficial only to a subset of patients. The limitations are due to several relapse mechanisms occurring during the anti-angiogenic therapies, including an upregulation of PD-L1 by cytotoxic T lymphocytes (CTL)-secreted IFN-γ (30), and abnormalities in the tumor endothelium (31). Multiple trials are currently investigating combinations of angiogenesis inhibitors and immunotherapies in multiple cancers [(32), NCT02443324], and in patients with advanced diseases (NCT02348008, NCT01633970). Bevacizumab treatment combined with carboplatin and paclitaxel received FDA approval in June 2018 for post-surgery treatment of patients with stage II or IV epithelial ovarian, fallopian tube, or primary peritoneal cancer, followed by single-agent bevacizumab. In metastatic melanoma patients, the combination of bevacizumab and ipilimumab induced changes in tumor vasculature, inflammation status, lymphocyte trafficking, and immune regulation. Analysis of the 46-patient cohort demonstrate a median survival >2 years with significant antitumor activity at the maximum tolerated dose (33). Maintenance nivolumab plus bevacizumab has been tested vs. nivolumab monotherapy and showed improved progression free survival (PFS) results (NCT01454102, CheckMate 012). However, in this comparison the nivolumab monotherapy arm comprise patients with squamous and non-squamous histology, while the nivolumab plus bevacizumab arm included only patients with non-squamous histology (median PFS of 16 weeks in squamous patients and 21.4 weeks in non-squamous patients in the nivolumab monotherapy arm compared to a median PFS of 37.1 weeks in the combination arm). No significant variance in the overall survival (OS) was observed in the two different treatment groups. Another phase III clinical trial, comparing the PFS and the OS of nivolumab combined with ipilimumab vs. the VEGF signaling inhibitor sunitinib in previously untreated advanced renal cell carcinoma (RCC) so far showed so far minimal toxicities and a reduction of the frequency of Tregs [NCT02231749, CheckMate 214, (34)].
Prostaglandin E2 (PGE2)
The bioactive lipid PGE2, product of the conversion of arachidonic acid by cyclooxygenase 2 (COX-2) is synthesized by various cell types, including cancer, stromal, and infiltrating myeloid cells. In the TME, PGE2 mediates its effects by acting on a group of G protein-coupled receptors (EP1-EP4) (35). The involvement of each receptor in regard to immunosuppression has been studied and revealed that EP1 and EP2 are low-affinity receptors and require significantly higher concentrations of PGE2 for effective signaling. EP3 and EP4 are high affinity receptors (35). Most of the immunomodulatory effects of PGE2 on immune cells are mediated through EP2 and EP4 receptors. EP2 and EP4 are Gαs coupled protein and stimulate adenylyl cyclase to raise the intracellular level of cAMP, and thus protein kinase A (PKA) which activate various types of signaling molecules. However, only EP4, mainly expressed on myeloid cells, T lymphocytes, and tumor cells is known to induce T cell factor–mediated transcriptional activity through phosphatidylinositol 3-kinase (PI3K) as well as PKA (36). EP4, additionally contributes to PGE2-mediated enhancement of tumor survival pathways and suppression of antitumor immune responses. PGE2 induces immunosuppression by inhibiting effector functions of macrophages, neutrophils, CTL, Th1 and natural killer (NK) cell-mediated immunity and by directly downregulating the production of Th1 cytokines. PGE2 also stimulates the development of suppressive types of Tregs, Th17, MDSCs and upregulates Th2-associated cytokines (36). PGE2 has the ability to suppress the production of IL-12 in monocytes and DCs, which is essential to Th1 responses (37). In RCC patients, PGE2 induces arginase I production by MDSCs. Subsequently arginine is depleted and T cell signal transduction and function impaired (38). A positive feedback loop between PGE2 and COX2 leads to redirect the differentiation of DCs toward monocytic MDSCs and locally promote the development of suppressive MDSCs (39). Moreover, in the ascites, MDSC precursors locally accumulate in a CXCR4-dependent manner that requires COX2 activity and autocrine PGE2 production (40). Mice deficient in EP2 significantly develop less tumors compared with tumors in wild-type mice (41). In animal models, E7046, a selective small-molecule inhibitor of EP4 showed an immune dependent growth inhibitory activity, and moreover showed synergistic antitumor activity when combined with anti-CTLA-4 antibodies (42).
The recently completed phase I study NCT02540291 assessed the potential of E7046 in patients with cancers that harbor high levels of myeloid infiltrate cells in the TME. The results showed that although the maximum tolerated dose was not reached, E7046 demonstrated an antitumor activity, and immune modulation in tumors and peripheral blood. Another EP4 antagonist, AAT 008, more potent in pharmacokinetic and pharmacodynamic modeling than Grapiprant (AT-001, AAT-007, CJ-023,423) has not yet been tested in clinic (43).
Colony-Stimulating Factor (CSF-1)
The proinflammatory cytokine CSF-1, also known as macrophage-CSF (M-CSF) binds to the CSF-1 receptor (CSF1R), which belongs to the Type III protein tyrosine kinase receptors. The axis CSF-1/CSF1R is crucial for the differentiation and survival of the mononuclear phagocyte system, and in particular macrophages. Its expression correlates with diminished survival in some cancers, including breast cancer (44, 45). Moreover, elevated CSF-1 levels in the serum of early breast cancer patients predicts poor survival (46). Macrophages are recruited into tumors following activation of CSF1R by either CSF1 or IL-34, two high-affinity ligands. Interestingly, in a cohort of lung cancer patients, the co-expression of CSF-1 with IL-34 in primary tissues correlates with advanced disease stages and poor survival (47). In addition to TAMs, CSF1R is expressed on DCs, neutrophils, and MDSCs. In murine models of cervical and breast carcinomas, inhibition of CSF1R by a highly selective small molecule inhibitor attenuates the turnover rate of TAMs while increasing the number of CD8+T cells that infiltrate the tumors (48). Other preclinical studies suggest that targeting of CSF1R pathway in combination with standard of care treatment may strategically be more effective for activating antitumor responses (49).
Inhibition of CSF-1/CSF-1R pathways suppresses the recruitment of circulating monocytes at the tumor site, and the blocking of TAMs activation, as the major survival factors for these macrophages. Small molecules or clinical antibodies against CSF-1, and CSF-1R belong to a new class of immune-modulatory drugs. Several clinical trials evaluating those inhibitors (PLX3397, RG7155, Durvalumab) alone or in combination with immunotherapy agents (anti-PD-1, -PD-L1) or chemotherapies (paclitaxel, fluorouracil, gemcitabine, oxaliplatin, irinotecan) have been completed (NCT02452424, NCT01494688, NCT01346358, NCT02718911). Recently, a study was launched to measure PFS for patients with advanced pancreatic cancer with poor prognosis treated with the IgG4 monoclonal antibody (mAb) blocking CSF-1R (cabiralizumab) combined with nivolumab, with or without chemotherapy (NCT03336216).
Transforming Growth Factor (TGF)-β
TGF-β is the prototype of the superfamily containing over forty members, including TGF-βs, Nodal, Activin, and bone morphogenetic proteins (BMPs) (50). TGF-βs are composed of a group of three members (TGF-β1, -β2, and -β3) normally involved in a variety of biological processes (50). Frequently, TGF-β refers to the isoform TGF-β1, which represents the most widely studied. Produced in large amounts within the TME by every leukocyte lineage, including lymphocytes, macrophages, and DCs, TGF-β1 is crucial for protecting the body from the development of excessive immune responses, therefore maintaining immune homeostasis (51). Active TGF-β binds to dimeric TGF-β type 2 receptor (TβRII), which recruits and activates a second dimeric type 1 receptor (TβRI) through its serine/threonine kinase to form a complex. The activated receptor initiates the signaling pathways and phosphorylates the transcription factors SMAD2 and SMAD3 that subsequently form a complex with SMAD4 and cofactors to translocate into the nucleus and modulate the expression of target genes. In addition, depending on the pleiotropic nature of TGF-β actions, activated TGF-β receptor complexes can also trigger a SMAD-independent pathway, including MAPK, PI3K/AKT, and Rho-like GTPase signaling pathways. (52). In the context of cancer, TGF-β play a pivotal role depending on the stage of the tumor (50). In the phase of pre-malignant cells, the tumor suppressive role of TGF-β interferes with tumor proliferation and progression. TGF-β promotes the immunological tolerance by directly suppressing the cytolytic activity of NK and CTLs, repressing the transcription of genes encoding key proteins (such as perforin, granzymes, and cytotoxins), and controlling the inflammatory responses through the regulation of chemotaxis, activation, and survival of lymphocytes, macrophages, granulocytes, NK cells, DCs and mast cells (53, 54). TGF-β is also instrumental for converting conventional CD4+ T cells to “induced” FoxP3+ Tregs (55), recruiting and stimulating the expansion of the mediators of in vivo suppression, MDSCs and Tregs. Recently, it has also been suggested that tumor immune evasion can occur by TGF-β-driven conversion of NK cells into type 1 innate lymphoid cells by an unknown mechanism (56). However, in late stage of cancer development, elevated levels of TGF-β favors tumor progression via effects on the stroma, induction of angiogenesis and/or promotion of the epithelial-to-mesenchymal transition (57–59). The explanation for these contradictory roles is that some tumors develop TGF-β-inactivating mutations and progress in a TGF-β-independent manner, while other tumors accumulate mutations in tumor suppressor genes downstream of TGF-β signaling. Cancer cells that acquire these mutations gain a great advantage over their non-mutated counterparts, as they can exploit the wide range of pro-tumorigenic effectors downstream of TGF-β stimulation (60). Increased systemic ligand levels of TGF-β and aberrant TGFβ signaling are often correlated with aggressive disease and poor prognosis (61). In preclinical studies, targeting the TGF-β pathway may be achieved through the use of several agents, including antisense oligonucleotides, TGF-β-neutralizing antibodies and TGF-β receptor kinase inhibitors (52). In some models of tumor-bearing mice, blockade of the immunomodulator TGF-β with antibodies or genetic manipulation decrease the number of induced Tregs (62). In mice expressing the four main mutations associated with colorectal cancer, the blockade of TGF signaling sensitized the tumor to the action of anti–PD-L1 antibodies (63). In a model replicating the immune-excluded phenotype, co-administration of anti-PDL1 and anti-TGF-β therapies reduced TGFβ signaling in stromal cells, and thus increased the ability of T cells to infiltrate tumors (64). Although, the microenvironment-targeted strategy targeting TGF-β pathway is being well tolerated in preclinical studies, translation into patients remains challenging and raises concerns, which include the development of autoimmune toxicities, the targeting of other homeostatic functions such as angiogenesis, and the risk of developing new malignancies.
Early phases clinical trials using TGF-β monotherapy have yielded conflicting results. While, one study using a monoclonal blocking antibody specific for TGF-β1 reported no clear evidences of antitumor effects in patients despite no major adverse side effects (65), other evaluations showed encouraging results. An mAb neutralizing all isoforms of TGF-β1, GC1008 (Fresolimumab) demonstrated preliminary evidence of anti-tumor effects in a subgroup of patients with advanced malignant melanoma and RCC (66). Interestingly, metastatic breast cancer patients treated with high dose fresolimumab during radiotherapy had a favorable systemic immune response, and thus, a better prognosis (67). Preliminary data from a phase I dose-escalation study suggested that M7824 (MSB0011359C), a bifunctional fusion protein composed of a chimeric version of an anti-PD-L1 mAb with a fragment of TGF-βR4 to entrap active TGF-β, has demonstrated a favorable safety profile in patients with pre-treated advanced solid tumors [NCT02517398, (68)]. Galunisertib (LY2157299), a small molecule antagonist of TGFβ-R1 is being studied in combination with an anti-PD-L1 in NSCLC, hepatocellular carcinoma, pancreatic cancer (NCT02423343, NCT02734160) and with paclitaxel in triple negative breast cancer (NCT02672475).
Enzymes
Indolamin 2, 3-Dioxygenase
IDO is an IFN-γ-inducible metabolic enzyme localized in the cell cytoplasm. IDO catalyzes the breakdown of tryptophan (Trp), an essential amino acid for lymphocyte proliferation, to kynurenine (Kyn), highly toxic for effector cells. This enzyme exists in two isoforms (IDO-1, IDO-2). IDO-1 is mainly responsible for tryptophan degradation and highly expressed in multiple types of human cancer, including acute myeloid leukemia (69, 70). Although IDO-2 is expressed in some human tumors, its function still remains to be clarified (71). IDO-1 can be induced in immune cells recruited by the tumor, especially APCs through canonical and non-canonical pathways including NF-κB, Jak/STAT, PKC and TGF-β signaling pathways (72). The rise in the Kyn/Trp ratio in cancer patients suggested an increase in IDO activity and low concentrations of tryptophan in serum/plasma is a reflection of the chronic activation of IDO-1 in the TME, which correlates with tumor progression and poor patient outcomes (73). The Trp shortage, which results in mTORC1 inhibition and general control non-derepressible 2 (GCN2) activation, leads to an anergic status of effector T cells (72). IDO-1 also reduces cytokine release, favors the expansion of Tregs and MDSCs, (74), regulates the differentiation of tolerogenic DCs (75), and along with PGE2, mediates the inhibitory effect of major NK receptors (NCRs and NKG2D), creating a consequent impairment of NK cell-mediated cytolytic activity (76). Using a mouse pregnancy model, IDO-1 highly expressed in the placenta, or products of tryptophan catabolism play a role in maternal T-cell activity suppression, hence protecting the mouse fetus from the maternal immune rejection (77). This effect of rejection was observed in the context of cancer. The expression of IDO by immunogenic murine tumor cells prevents their rejection by preimmunized mice due to a lack of specific T cell accumulation at the tumor site. This can be reverted by systemic treatment of mice with an inhibitor of IDO (78). These preclinical data led to a rapid clinical development of the first generation IDO-1 inhibitors (indoximod, 1-MT, NLG8189), which has been demonstrated to relieve IDO-mediated immunosuppression in vitro and in vivo by the creation of a critical Trp-sufficiency signal that bypasses activation of GCN2 and inhibition of mTOR in conditions of Trp deprivation (79).
The US FDA has approved the clinical registration applications of IDO inhibitors, in 2016, PF-06840003 in the completed trial NCT02764151; in 2017, NLG802 in NCT03164603 and HTI-1090, respectively, in NCT03208959. Furthermore, combinational regimens with other treatment modalities are under evaluation (80), and some of them such as the phase I/II trial of indoximod combined with temozolomide, have shown promising results in patients with primary malignant brain tumors (NCT02052648). The completion of the safety and efficacy study evaluating indoximod in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (NCT02077881) revealed a promising activity as shown by the increased in intratumoral CD8+ T-cell density (poster presentation at the annual meeting of the American Society of Clinical Oncology 2018). In a phase II trial, indoximod plus ICB in patients with advanced melanoma (NCT02073123) showed a favorable overall response rate compared to pembrolizumab alone. The combination of the potent and selective oral inhibitor of IDO-1, epacadostat (INCB024360) with nivolumab demonstrated safety, tolerability, and efficacy for treatment of patients with naïve advanced melanoma and head and neck squamous cell carcinoma (HNSCC) in a phase I/II trial (ECHO-204, NCT02327078). Another phase I/II trial evaluating epacadostat plus pembrolizumab (ECHO-202, NCT02178722) demonstrated activity in patients with advanced NSCLC. Though in a phase III clinical trial, epacadostat plus pembrolizumab did not meet the primary endpoint of improving PFS in patients with unresectable or metastatic melanoma when compared to pembrolizumab monotherapy (ECHO-301/KEYNOTE-252, NCT02752074). Despite some promising results, concerns have been raised regarding the use of IDO inhibitors which can result in severe autoimmune reactions. Indeed, as IDO can be activated by many stimuli such as IFN-γ and tumor necrosis factor (TNF)-α, IDO inhibitors may not work in a patient with a cold tumor, not infiltrated by T cells.
Arginase and Inducible Nitric Oxide Synthase (iNOS)
The semi-essential amino acid arginine, also known as L-arginine is a precursor for several metabolites and a critical regulator of lymphocyte proliferation and function. The arginases (Arg1 and 2) and nitric oxide synthetases (NOS1-3) are the major enzymes responsible for arginine metabolism in inflammatory immune responses (81, 82). Arg1 and 2 induce the same reaction but differ in tissue distribution and intracellular localization (83). Tumor arginase deprives immune cells within the local TME of arginine by catalyzing the hydrolytic conversion of arginine into L-ornithine and urea, resulting in dysfunctional immune cells (81, 84). This degradation of extracellular arginine affects CD3 ζ chain, the main signaling chain of the TCR, resulting in T cell anergy (84). The depletion of L-arginine also induces a blockade in infiltrating T-cells and cell cycle progression (81). The immunosuppressive enzyme from the NOS family, NOS2 (inducible NOS or iNOS) is inducible by inflammatory cytokines and metabolizes L-arginine to produce reactive free radical NO and L-citrulline (81). The cellular signaling molecule NO was found to be actively associated with tumors as well as the tumor environment and in addition to its role in cancer initiation and progression, NO contributes to the anti-tumor immune response and limits T cell proliferation and activity by promoting apoptosis and by inhibiting cytokine and chemokine production (85, 86). NOS2 is expressed by various cell types involved in inflammation, including neutrophils, M2 macrophages, MDSCs, DCs, NK cells, endothelial and tumor cells (87). Upregulation of arginase prevents NOS2 activity, whereas arginase inhibition causes enhanced NOS2 expression and leads to increased NO production. TAMs and MDSCs overexpress and secrete Arg1 and NOS2 to deplete intracellular and extracellular arginine facilitating the T cell impairment. Moreover, in MDSCs, the expression of ARG1 and iNOS is regulated by cyclic GMP levels, which is in turn controlled by the activity of phosphodiesterase type 5 (PDE5). Therefore, the agents that can elevate intracellular cGMP levels, such as PDE5 inhibitors, reduce MDSC-mediated immune suppression (88).
Decreased circulating arginine levels in cancer patients are considered indicative of elevated plasma levels and high Arg1 expression in tumors (89). Up to date, only a few studies have been conducted with arginase inhibitors in different human cancers, mostly to avoid complications related to the conversion of highly toxic ammonia to urea which will be excreted. However, in late 2017, the first cohort of patients with advanced/metastatic solid tumors was treated with INCB01158 (an Arg1 inhibitor formerly known as CB-1158) monotherapy or combined with pembrolizumab (NCT02903914). PDE-5, initially used for the treatment of erectile dysfunction, brings clinical benefits in a range of cancers by acting on the NO/cyclic guanosine monophosphate (cGMP) signaling pathway (90). Tadalafil, a PDE-5 inhibitor decreases circulating MDSCs, lowers iNOS and arginase expression in these cells, and enriches tumor-specific T cells in HNSCC patients (91) and it is being clinically tested in patients with multiple myeloma in combination with lenalidomide (NCT01858558) and in patients with HNSCC in combination with mucin 1 vaccine (NCT02544880).
Ectonucleotidases CD39 and CD73
The accumulation of adenosine, an extracellular immunosuppressive metabolite, is a strategy used by tumors to evade immunosurveillance. Extracellular adenosine triphosphate (ATP) and adenosine are abundant metabolites and have an important autocrine/paracrine role. ATP is catabolized to adenosine in the TME by two ectonucleotidases: CD39 and CD73, anchored in cancer cells, regulatory immune and endothelial cells from the vasculature (88). CD39 and CD73 can be recognized as “immune checkpoint mediators” since they interfere with anti-tumor immune responses (92). CD39 reversibly produces AMP from ATP or ADP, which is subsequently converted into extracellular adenosine by CD73 (93). The hypoxic TME maintains elevated levels of adenosine due to the high-level expression of CD73 by tumor cells, which results in a chronic suppression of immune cells (88). In addition to tumor cells, adenosine can also be the product of immune cells. CD56bright NK cells release adenosine in the presence of autologous CD4+ T cells (94). Tregs overexpress CD39 and CD73 responsible of the sequential conversion of pro-inflammatory extracellular ATP into AMP and adenosine (95). Among the four adenosine-binding G protein-coupled receptors, A2A receptor is the most expressed subtype on immune cells: T, NK, NKT, macrophages, and DCs (96). Upregulation and activation of A2A receptor switches macrophages from an M1 to an M2 phenotype and induces the production of VEGF and IL-10 (97). When binding to A2B receptor, adenosine promotes the expansion and functions of MDSCs (98), and induces high expression levels of angiogenic, proinflammatory, immune suppressor, and tolerogenic factors (99). Using an A2aR-null mouse model, several melanoma and T cell lymphoma lines were rejected in a CD8+T cell dependent manner, and addition of a pharmacologic blockade of A2aR could enhance T cell mediated tumor regression in a sarcoma and LL-LCMV tumor model (100). Other preclinical studies have focused on blocking the adenosine pathway by targeting CD73 and/or CD39. CD73- null mice significantly induce tumor rejection in a variety of syngeneic tumor models (101) and, CD39-null mice were resistant to tumor metastases in B16/F10 mouse melanoma and MCA-38 colorectal models (102). In addition, weakening of upstream tumor hypoxia by supplemental oxygenation decreases the intensity of downstream A2AR-mediated immunosuppression in mice (103). Co-inhibition of CD73 and A2ARs in mice with spontaneous or transplantable tumors improve the inhibition of tumor initiation, growth, and metastasis (104).
It has been reported that CD39-expressing-melanoma cells inhibit both T cell proliferation and generation of cytotoxic effectors in an adenosine-dependent manner (105). Recently, in a phase I/Ib clinical trial CPI-444, an oral small molecule targeting the adenosine-A2A receptor combined with the intravenous PD-L1 inhibitor atezolizumab demonstrated an OS of 88% at more than 20 months follow-up in treatment-refractory RCC patients (NCT02655822). Another agent targeting A2A receptor, NIR178 (PBF-509) in combination with an anti-PD1 is currently under investigation for the treatment of advanced NSCLC. Clinical benefits were reported in immunotherapy-exposed and -naïve patients irrespective of PD-L1 status (NCT02403193).
Co-inhibitory Molecules
Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4)
Member of the immunoglobulin superfamily, CTLA-4 (CD152) was the first discovered co-inhibitory receptor (106). CTLA-4 shares the same ligands with CD28, namely CD80 (B7.1) and CD86 (B7.2) but with less affinity, thus counteracting the stimulatory effects of CD28 ligation (107). Mice deficient in CTLA-4 develop generalized lymphoproliferative syndrome with a lymphocytic infiltration of all organs (108). Using mice bearing partially immunogenic tumors, the Allison group showed that CTLA4 blockade could enhance the endogenous anti-tumor response after tumor implantation (109). CTLA-4 was then clinically targeted, and the following relevant investigations of its role in modulation of the amplitude of the early stages of T cell activation represent the breakthrough of cancer immunology (110). The proposed mechanism is the direct inhibition at the TCR immune synapse, inhibition of CD28 or its signaling pathway, or increase in mobility of T cells that are less prone to interact with APCs (111). CTLA-4 is expressed by effector T cells and constitutively expressed by Tregs (112). To prevent T cell activation, Tregs primarily target APC via the engagement of CTL-4 with CD80 and/or expression on APCs and transmitting inhibitory signals (113). The mobilization of CTLA-4 from the intracellular protein stores to the cell surface happen as early as an hour after antigen engagement, thus allowing the occurrence of a feedback inhibition. The balance of CD28 and CTLA-4-derived signals is critical for maintaining the equilibrium between T cell activation or tolerance. When administered to patients, mAbs that block the binding of CTLA-4 to its ligands were able to unleash antitumor responses (114).
A pivotal phase III clinical trial launched in 2010 showed that ipilimumab, a fully humanized anti-CTLA-4 mAb, alone or in combination with gp100 peptide vaccine, improved survival in metastatic melanoma patients (115). Ipilimumab was the first FDA-approved checkpoint immunotherapy in patients with advanced melanoma. Afterwards, this agent was approved by the European Medicines Agency (EMA) and is currently indicated for the first line treatment of melanoma and in adjuvant settings.
Programmed Death 1 (PD-1)/PD-L
PD-1 (CD279) negatively regulates the activity of effector T cell within tissues and tumors, where the immune response is already ongoing, while CTLA-4 is expressed only expressed in T and pro-B cells, the member of the immunoglobulin superfamily is more broadly expressed. (116). PD-1 can bind two ligands from the B7 protein family: PD-L1 (B7-H1, CD274) expressed by macrophages, DCs, activated T and B cells, tumor cells, and tissues, such as heart, lung, spleen and PD-L2 (B7-DC, CD273), which is mainly expressed on DCs and tumor tissues. Unlike CTLA-4, PD-1 acts in the secondary immune response and its expression on the surface of activated T cells is delayed (6–12 h) due to the need of transcriptional activation. PD-1 binds to its ligands directly overexpressed on cancer cells, clusters with TCR and recruits the inhibitory phosphatase SHP2 (Src homology 2 domain-containing tyrosine phosphatase 2) via its immunoreceptor tyrosine inhibitory motif, which induces dephosphorylation of the proximal TCR signaling molecules, thus suppressing T cell activation (117). High levels of PD-1 expression on T cells induces a state of anergy and exhaustion, as shown by in vitro and in vivo models (118). PD-L1 is expressed by different tumor types (breast, ovary and colon carcinomas), and its expression is up-regulated in the presence of IFN-γ that is released in the TME. The binding of PD-1 to PD-L1 generates an immunosuppressive effect, impairs T cell activation (119) and in addition, increases the proliferation of the tumor infiltrating Tregs (120). A preclinical study in glioblastoma showed that targeting factors responsible for the myeloid PD-L1 upregulation, such as IL-6 enhance the anti-tumor activity exerted by PD-1 therapy. Thus, interfering with IL-6 signaling diminished myeloid immunosuppression, tumor growth, and increase mice survival (121).
In 2014, nivolumab (OPDIVO, Bristol-Myers Squibb Company), an anti-PD-1 mAb was approved by the FDA for melanoma patients and marked the start of several other approvals in other cancer types. In 2015, the FDA approved nivolumab for the management of advanced metastatic RCC after progression on first-line therapy or following prior anti-angiogenic therapy (approval based on an extension in OS in the CheckMate-025 trial, NCT668784), squamous NSCLC as a second-line treatment across all histologies (approval based on data from the phase III CheckMate-057 trial, NCT01673867), advanced or metastatic urothelial carcinoma (phase II CheckMate-275 trial, NCT02387996) and hepatocellular carcinoma after sorafenib treatment (CheckMate 040 trial, NCT01658878). Nivolumab became the first and only immuno-oncology treatment option for patients with metastatic small cell lung cancer progressed after platinum-based chemotherapy and at least one other line of therapy. Interestingly, in the adjuvant setting, results from the phase III, NCT02388906 (CheckMate 238) comparing the efficacy of nivolumab vs. ipilimumab in patients with resected stage III/IV melanoma at high risk of recurrence showed that nivolumab provides superior safety and survival compared to ipilimumab, regardless of tumor PD-L1 status. Several trials combined nivolumab with other cancer therapies. Combination with gemcitabine, which induces the killing of MDSCs, increases the efficacy of nivolumab in metastatic NSCLC (NCT03302247). The trial NCT02922764 is evaluating the combination of RGX-104, agonist of the nuclear receptor liver X receptor (LXR) with nivolumab in advanced solid malignancies and lymphoma. This interaction between LXR and RGX-104 induces depletion of both MDSCs and endothelial cells. The clinical study NCT01454102 (CheckMate 012) testing the combination of nivolumab plus ipilimumab as first line treatment for advanced NSCLC patients showed durable efficacy and resulted in a significantly longer OS than ipilimumab alone in a phase III trial involving patients with advanced melanoma [CheckMate 067, NCT01844505, (122)]. The trial NCT01472081 (CheckMate 016) evaluated the safety and efficacy of nivolumab combined with ipilimumab in patients with metastatic RCC (123). And in April 2018, nivolumab has been approved in combination with ipilimumab as first-line treatment for patients with advanced RCC. Another anti-PD-1 agent, pembrolizumab (KEYTRUDA, Merck & Co., Inc.) was approved for advanced melanoma in 2014, followed by several other approvals in other cancer types, such as advanced NSCLC in 2015, Hodgkin lymphoma and urothelial carcinoma in 2017. Interestingly, in May 2017, for the first time, the FDA approved a cancer drug based on tumor genetics rather than tissue type or tumor site. Indeed, pembrolizumab was granted approval for the treatment of any unresectable or metastatic solid tumor with specific genetic features (mismatch repair deficiency or microsatellite instability). In 2018, the FDA granted the approval of pembrolizumab for treating recurrent or metastatic cervical cancer whose tumor expresses PD-L1 and refractory or relapsed mediastinal large B-cell lymphoma. The clinical trial identified as NCT03241927 is currently investigating the effect of pembrolizumab on NK cell exhaustion in melanoma based on the idea that releasing the PD-1 immune checkpoint in NK cells may help them to infiltrate the tumor and exert their effector functions against the tumor. Results from the phase III study (NCT02252042) assessing the antitumor activity and toxicity of pembrolizumab in patients with recurrent or metastatic HNSCC showed a lower risk of death in patients with high tumor PD-L1 expression, and this despite missing predetermined endpoints. The combination of the histone deacetylase inhibitor entinostat with pembrolizumab is currently under investigation in patients with advanced solid tumors (NCT02909452), and so far, resulted in a reduction in monocytic MDSCs across all the treatment arms. Hypothetically, treating patients resistant to anti-PD-1 with a TLR agonist injected into the tumor may allow the TME to be more immunogenic and thus, more sensitive to PD-1 inhibition. In this context, pembrolizumab combined with CPM-001, an agent that activates TLR-9 is being tested in patients with advanced melanoma resistant to PD-1 inhibition (NCT02680184). Reports are showing objective and durable tumor responses. A phase I/II study combining pembrolizumab with targeted molecule inhibitors (BRAF plus MEK) is ongoing in melanoma and other solid tumor patients (NCT02130466). Other clinical studies are evaluating the efficacy of pembrolizumab plus chemotherapy (cisplatin, capecitabine or 5-fluorouracil) (NCT02494583). Recently, the FDA granted approval for pembrolizumab in combination with chemotherapy (pemetrexed and platinum) for first line treatment of metastatic non-squamous NSCLC, with no EGFR or ALK genomic tumor aberrations. Several anti-PD-L1 mAbs are under investigation in various cancers including melanoma, multiple myeloma, leukemia, lymphoma, glioblastoma as well as gastric, renal cell, bladder, colorectal, hepatocellular, cutaneous, breast and NSCLC cancers. Atezolizumab (Tecentriq, Genentech) is currently being tested in combination with bevacizumab and/or with chemotherapy in patients with locally advanced or metastatic solid tumors (NCT01633970). In 2016, the FDA approved atezolizumab as first line treatment for cisplatin resistant metastatic urothelial carcinoma and metastatic NSCLCs. In April 2017, atezolizumab became the first cancer immunotherapy approved by the FDA for patients with advanced bladder cancer and thus is employed as a standard of care. Avelumab (Bavencio, EMD Serono, Inc.) is a fully human anti-PD-L1 mAb which received FDA approvals in March and May 2017 for the treatment of patients with metastatic Merkel-cell carcinoma and advanced or metastatic urothelial carcinoma during or after treatment with platinum-containing chemotherapy administered in neoadjuvant or adjuvant setting. Recently discovered through a novel genome-scale T cell activity array, the immune suppressor Siglec-15 which is highly expressed on tumor cells and tumor-infiltrating myeloid cells can serve as a biomarker for predicting the outcomes of anti-PD-1/PD-L1 therapy. Siglec-15 has been shown to continuously inhibit T cell activity. Thus, using anti-Siglec-15 therapy may offer an alternative strategy to PD-1/PD-L1 pathway and lead to tumor immune normalization (124).
Lymphocyte-Activated Gene-3 (LAG-3, CD223)
LAG-3 is a surface molecule structurally related to CD4 (125). LAG-3 consists of four extracellular immunoglobulin superfamily-like domains (D1-D4) and binds to MHC class II molecules with greater affinity than CD4, utilizing an additional 30 amino acid loop (126). Like the CTLA-4/CD28 subfamily, the LAG-3/CD4 subfamily represents an inhibitory/stimulatory receptor subfamily modulating TCR signaling. LAG-3 is expressed in activated CD4+ and CD8+ T cells 3–4 days post activation (127), on a subset of NK cells (128) and on activated Tregs (129). LAG-3 does not feature an immunoreceptor tyrosine-based inhibition motif (ITIM) but possesses in its cytoplasmic tail two distinct motifs mediating the intrinsic negative inhibitory signal: a repetitive “EP” motif consisting of a series of glutamic acid-proline dipeptide repeats, and a single lysine residue (K468) within the conserved KIEELE motif in the cytoplasmic domain (130). The interaction between highly constitutive expression of MHC class II molecules at the surface of melanoma cells and LAG-3 greatly expressed on melanoma-specific CD4+ T cells elicits a local TNF-rich inflammatory environment, reducing the cytotoxic CD8+ T cell responses (131). LSECtin, expressed on melanoma is a type II transmembrane protein which belongs to the C-type lectin receptor superfamily and has been identified as a ligand of LAG-3. When LSECtin interact with the co-regulatory molecule LAG-3, and limits CD8+ T cell- specific responses in a LAG-3-dependent fashion (132). Galectin-3, an S-type lectins is a carbohydrate-binding protein that plays a key role in tumor escape from immunosurveillance (133, 134). Galectin-3-mediated suppression of CD8+T cells occurs upon binding to LAG-3 (135). Interestingly, co-expression of LAG3 with CD49b defines a subset of peripherally induced CD4+ Th1 regulatory cells secreting high amounts of IL-10 (136), and it is also involved in the maturation and activation of DCs (137) and plasmacytoid DCs (138). Recently, fibrinogen-like protein 1 (FGL1) has been shown to be a key ligand of LAG-3. Abundantly produced by cancer cells, FGL1 plasma level is increased in cancer patients and correlates with poor prognosis (139). Preclinical studies exploring the immune regulatory role of LAG-3 on various types of lymphocytes have showed its cooperation with other inhibitory receptors, such as PD-1/PD-L1. The association of LAG-3 and PD1 contributes to their rapid trafficking to the immunological synapse, leading to the synergistic inhibitory effect on T cell signaling in several tumor murine models (140, 141). In addition, LAG-3 and PD-1 are co-expressed on tumor infiltrating lymphocytes (TIL)-specific CD8+ T cells in the peripheral blood and tumors of ovarian cancer patients resulting in T cell dysfunction (142).
LAG-3-targeted immunotherapy started in 2006 with a toxicology study aiming to determine the dosage and frequency of the preclinical grade human soluble LAG-3 protein named IMP321 (143). In 2013, a phase I clinical trial with the anti-LAG3 mAb (BMS-986016) was initiated. There are now several LAG3 modulators at various stages of preclinical and clinical development for the treatment of relapsed or refractory hematologic malignancies (NCT02061761), and advanced cancers, alone or in combination with anti-PD1 in patients with (NCT02966548, NCT03005782). A phase I/IIa study (NCT01968109) evaluating efficacy and safety of the combination therapy with the anti-LAG-3 antibody BMS-986016 (relatlimab) and nivolumab in patients with melanoma that progressed during or after anti-PD-1/PD-L1 immunotherapy has reported a safety profile similar to that of nivolumab alone, but with superior efficacy.
T Cell Immunoglobulin and Mucin 3 (TIM-3)
TIM-3 is a surface negative regulator of CD4+ Th1, CD8+ T cytotoxic 1 cells, and innate immune cells and contain an immunoglobulin and a mucin-like domain (144, 145). TIM-3 regulates Th1 and Th17 responses when interacting with its ligands, thus inhibiting the expression of proinflammatory cytokines such as INF-γ and TNF-α (145, 146). Galectin-9 (Gal-9), expressed in various tumors has a key role in tumor immunity (147). Gal-9 was the first TIM-3 identified ligand and binds the Tim-3 immunoglobulin variable domain to regulate Th1 immunity in TIM-3-deficient mice (148). Other identified ligands of TIM-3 include: carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1), high-mobility group protein B1 (HMGB1), and phosphatidylserine (PS) (149). TIM-3 contain two of its more membrane-proximal cytoplasmic tail tyrosines which can directly bind to the Src family tyrosine kinase Fyn and the PI3K adaptor but no any known inhibitory signaling motifs. Expressed on tumor-infiltrating DCs, TIM-3 plays a critical role in suppressing innate antitumor immune responses through the recognition of tumor-derived nucleic acids (150). Interestingly, the inhibition of the antitumor responses via TIM-3 mediates T cell exhaustion, a phenomenon that was first identified in vitro in patients with HIV-1 infection (151), and later in cancer patients (152).
In RCC, TIM-3 expression has been shown to be elevated on tumor and myeloid cells of patients (153), and its upregulation on CD8+T cells lead to immune evasion at relatively early stage (154). Furthermore, prostate-specific antigen cells expressing high levels of TIM-3 are exhausted, influencing a patient's response to therapy (155). In patients with advanced melanoma, a majority of CD8+ TILs co-express PD-1 and TIM-3 (156). The co-expression of both checkpoint molecules reflected a more exhausted phenotype characterized by reduced T cell proliferation and IFN-γ, IL-2, and TNF-α secretion (116). In addition, overexpression of TIM-3 can function as an NK-cell exhaustion marker in advanced melanoma and is associated with a poor prognosis (157). A phase I study is evaluating TSR-022, an anti-human TIM-3 blocking antibody as monotherapy and in combination with an anti-PD-1 antibody for patients with advanced solid tumors who had limited available treatment options (NCT02817633). The safety and effectiveness of the anti-TIM-3 mAb (MBG453) is being evaluated in patients with solid tumors, either alone or combined with another immunotherapy (NCT02608268) and the combination MBG453-decitabine in patients with hematologic cancers (NCT03066648). The clinical results are not yet reported.
V-Domain Ig-Containing Suppressor of T Cell Activation (VISTA)
VISTA, also known as PD1 homolog (PD-1H), belongs to the B7 family members. VISTA contains a single IgV domain with three additional cysteine residues, which differs structurally from the other B7 family members (158). VISTA is known to play a central role in the regulation of T cell responses and its extracellular domain has some similarities with PD-L1 (159). While VISTA does not contain ITIM/ immunoreceptor tyrosine-based activation motif (ITAM), it has a conserved Src homology 2 (SH2) and its cytoplasmic tail domain contains two potential protein kinase C binding sites and proline residues that could function as docking sites for adaptor proteins. VISTA transcription is partially controlled by p53 (160). VISTA has been shown to have a dual functionality as a co-inhibitory receptor on T cells (161) and as a co-inhibitory ligand for T cells (158, 159). Recently reported, blocking the interaction of VISTA with its ligand VSIG-3 inhibits human T cell functions in vitro, as well as cytokine and chemokine production in colorectal, gastric cancers, and hepatocellular carcinomas (162). In humans and mice, VISTA is expressed mainly on hematopoietic cells with high levels on myeloid APCs, a weak density on T cells and NK cells, and no expression on B cells (158, 159, 163). In addition to immune cells, it has been demonstrated that high levels of VISTA are also expressed on human and murine tumor cells (158, 164). Anti-VISTA monotherapy with an mAb significantly reduces the growth of the tumor in multiple transplantable or inducible tumor models of melanoma and bladder carcinoma (163). Furthermore, combining blockades of VISTA together with CTLA-4 is more efficient than the PD-1 and VISTA combination in the HNSCC model (165). While in primary cutaneous melanoma patients, VISTA expression is considered as an independent negative prognostic factor (166), its overexpression on immune cells, especially macrophages, that infiltrated pancreatic tumors has been highlighted as a potential immunotherapeutic target (167). VISTA upregulation in prostate cancer may represent a compensatory inhibitory pathway after ipilimumab administration (168).
Blocking the interactions between PD-1/PD-L1 and VISTA using CA-170, a small molecule that antagonizes the PD-L1/PD-L2 and VISTA pathways improves the anti-tumor responses in certain tumor models and highlights their distinct and non-redundant functions in regulating the immune response to tumors (169). Preclinical data demonstrated VISTA's ability to independently suppress T cell responses, thus supporting the starting of a phase I dose escalation trial in patients with advanced tumors and lymphomas in 2016 (NCT02812875). So far, in toxicology studies, CA-170 showed a favorable safety profile when multiple dose levels were administered orally once daily, and preliminary evidence of antitumor activity. Interestingly, a recent immunotherapy clinical trial using CA-170 has been launched in mesothelioma patients. Currently, CA-170 is the only human mAb against VISTA being studied in a clinical trial in advanced cancers, since the recruitment for the trial NCT02671955 evaluating JNJ-61610588 (Onvatilimab) has been stopped due to a business decision done by Janssen.
T Cell Immunoglobulin and Immunoreceptor Tyrosine-Based Inhibitory Domain (TIGIT)
TIGIT (also known as WUCAM, Vstm3, VSIG9) is a type 1 transmembrane protein containing an IgV extracellular domain and two ITIMs in its cytoplasmic tail. These motifs mediate the recruitment of the phosphatase SHIP-1, thus providing a mechanism by which TIGIT can dampen activating signals (170). Engagement of CD155 (poliovirus receptor, PVR) ligand with the co-inhibitory receptor TIGIT leads to T cell inhibition responses, while the interaction of CD155 with the co-stimulatory receptor CD226 (DNAM-1) leads to T cell activation (171). TIGIT is associated with human cancers and expressed on NK and lymphoid cell populations (activated and memory T cells, and a subset of Treg). This provide an opportunity to target both the adaptive and innate arms of the immune system (172, 173). In murine cancer models, TIGIT+ Tregs may drive a dysfunctional phenotype in CD8+T cells via their high production of IL-10 (174). Interestingly, due to cellular stress within the TME, the expression of CD155 increase at the surface of APCs during the malignant transformation (175). Moreover, the interaction between TIGIT and CD155 on mature DCs induces a switch into a tolerogenic phenotype in DCs (171). TIGIT deficiency in NK cells alone has been reported to be sufficient to delay tumor growth in multiple tumor-bearing mouse models, and anti-TIGIT mAbs reverse NK cell exhaustion (176). Other preclinical data demonstrated that anti-TIGIT treatment reduced the abundance of Tregs within tumors in animal models (Abstract 5627, AACR annual Meeting 2018).
Preclinical studies demonstrated its efficacy in in vivo tumor models and higher effectiveness when combined with other checkpoint inhibitors. Initial evaluation of the anti-TIGIT mAb, OMP-31M32 (Etigilimab) in a phase Ia/b showed preclinical in vivo anti-tumor effects as a single agent and in combination with anti-PD-1 (NCT03119428) results presented at the Society for Immunotherapy of Cancer 2018). In an ongoing phase I/II trial, the experimental medication BMS-986207 is evaluated for its safety and effectiveness as monotherapy or in combination with nivolumab for the treatment of advanced or metastatic solid cancers (NCT02913313). Soon, EOS884448, an anti-TIGIT antibody disrupting the immunosuppressive binding of CD155 to TIGIT in the TME and mediating the restoration of T cell effector functions and the preferentially depleting Tregs is expected to enter the clinic.
The main immunological abnormalities conferring tumor evasion are illustrated in Figure 1. Some of the ongoing clinical trials aiming to assess the role of soluble factor inhibitors, enzymes and metabolic inhibitors and immune checkpoint inhibitors alone or in various combination with other cancer therapeutics are summarized in the Tables 1–3.
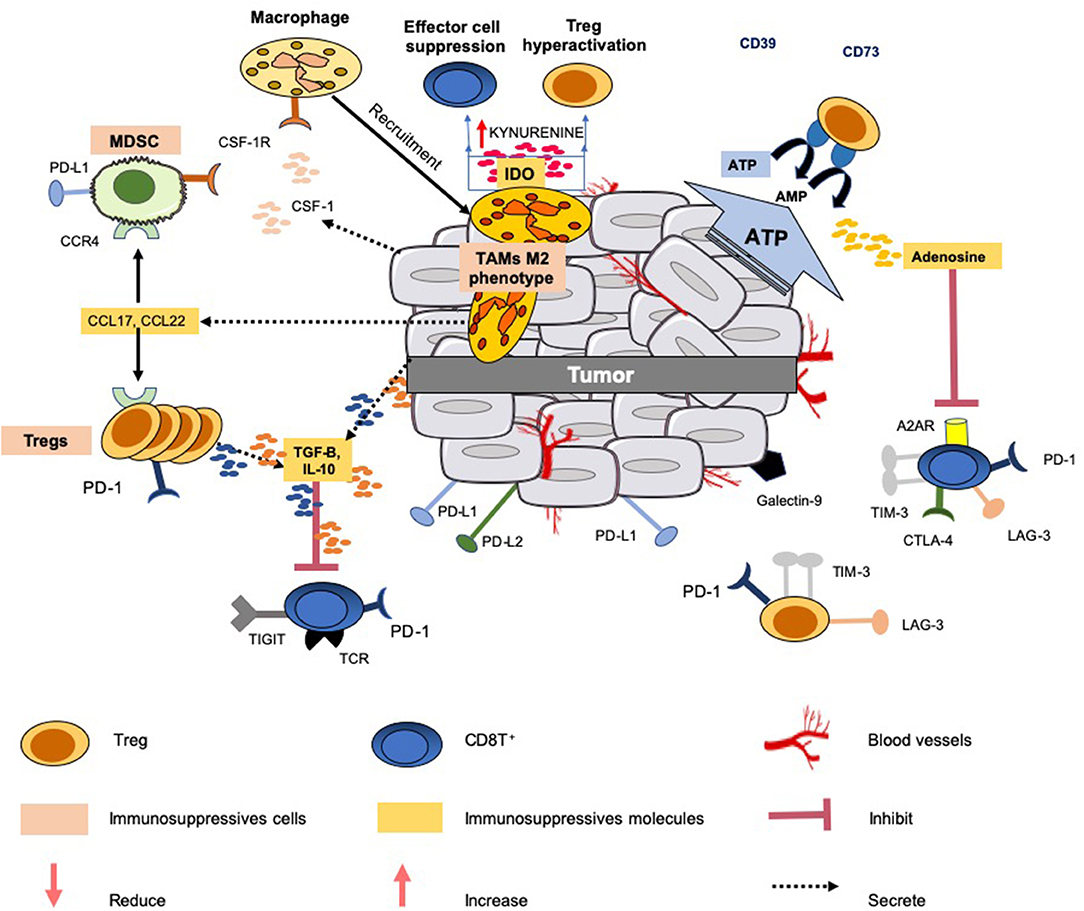
Figure 1. Immunosuppressive mechanisms common in the tumor microenvironment. Several mechanisms are developed by the tumor to limit an efficient tumor immunosurveillance, and therefore an unfavorable environment. This explains that a portion of epithelial cancers displays modest responses to immune checkpoint blockade therapy and other modulators of immunity. All the mechanisms known to interfere with these immunotherapies do not operate simultaneously in these cancers. This figure illustrates only a few common mechanisms of immune resistance to different tumors: (1) production and secretion of immunosuppressive factors into the microenvironment (such as TGF-β, IL-10, adenosine); (2) co-expression and/or upregulation of inhibitory receptors (LAG3, TIGIT, TIM-3,TIGIT) by immunosuppressive cells (Tregs, MSDCs and TAMs) and effector cells (CD8T+ and NK); (3) release of the chemokines CCL17 and CCL22 by the tumor which triggers the accumulation of Tregs and MDSCs to tumor sites; (4) release of IL-10 and TGF-β by Tregs which inhibit the functions of CD8+ T cells; (5) IDO expression by TAMs metabolizes tryptophan to kynurenine and limits T-cell function. Additionally, the tumor can also gain additional immunosuppressive properties, such as the expression of PD-L1, PD-L2, and secretion of suppressive cytokines (e.g., IL-10, TGF-β).
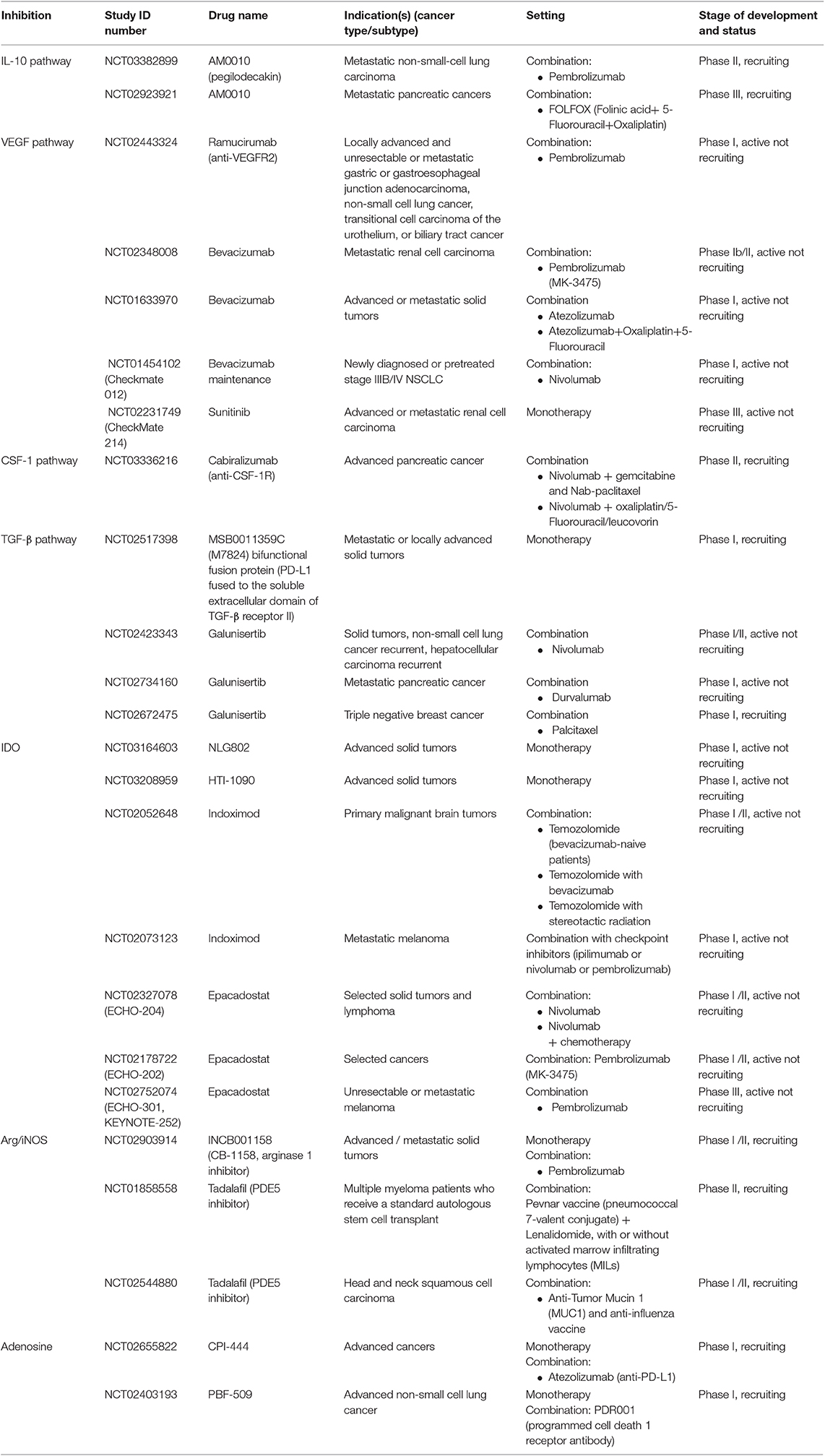
Table 1. Clinical studies testing agents targeting soluble factors, enzymes and metabolic inhibitors.
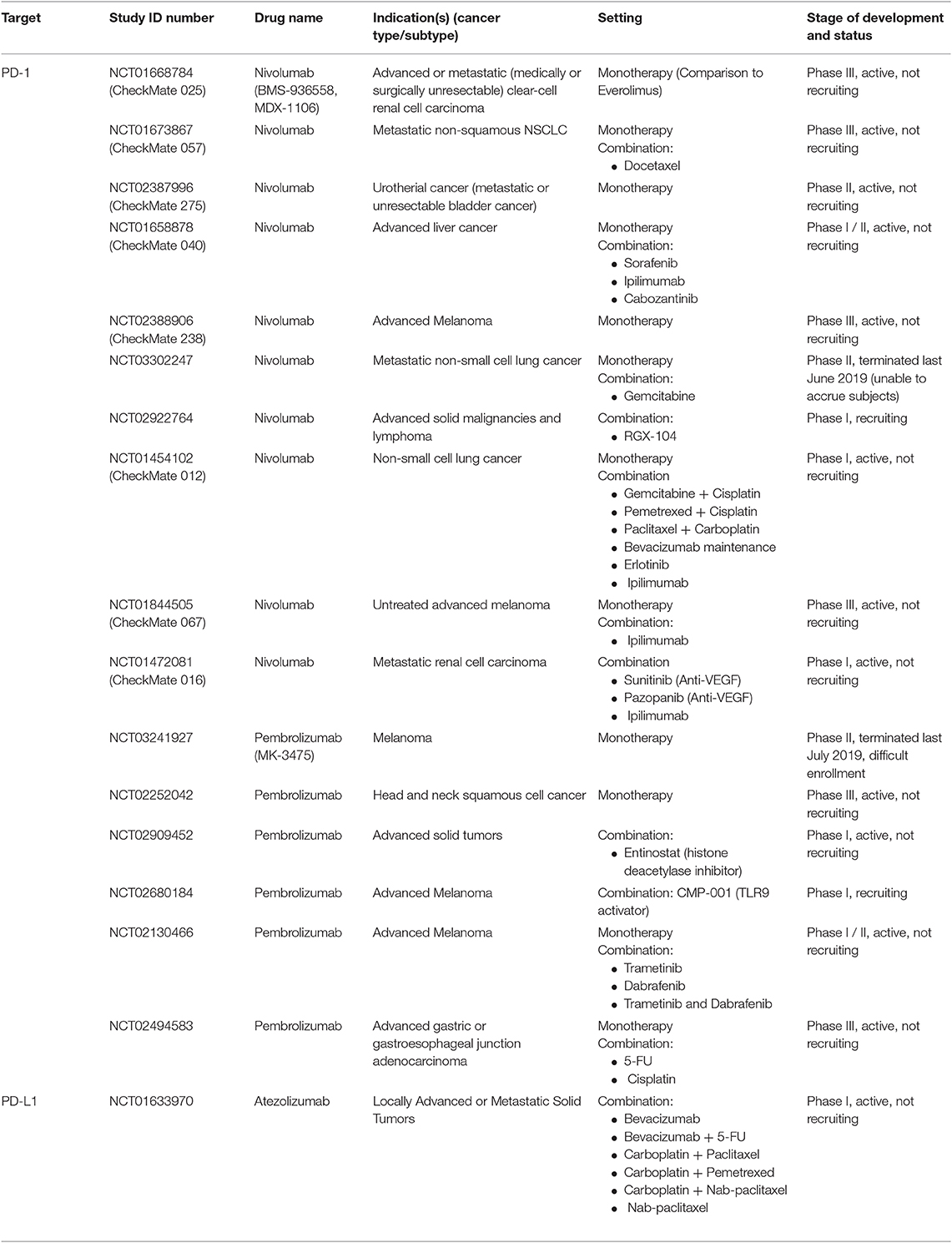
Table 2. Anti-PD-1 and anti-PD-L1 agents in clinical trials as mono- and/or combination therapies.
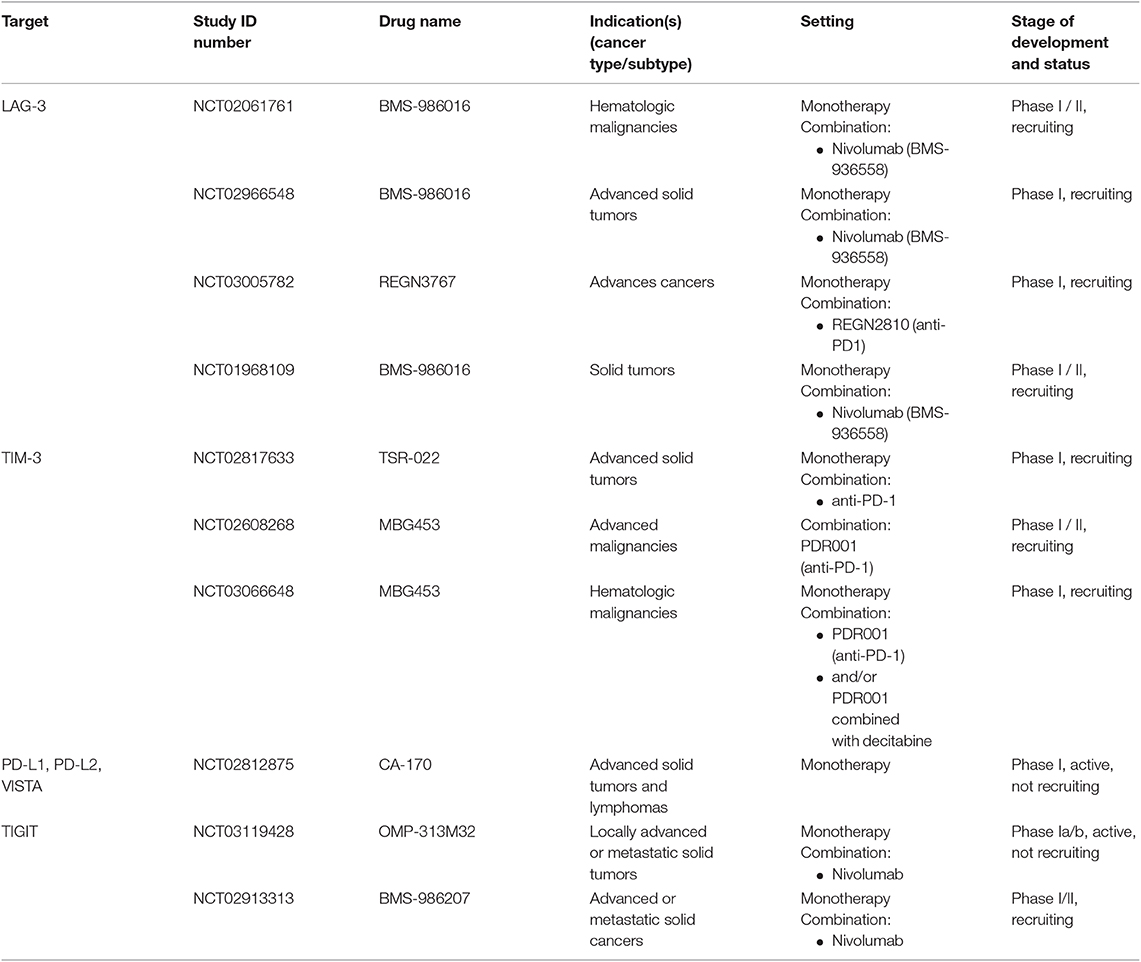
Table 3. Anti-LAG-3, anti-TIM-3, anti-PD-L1, -L2, -VISTA and anti-TIGIT immune checkpoint blockade tested in clinical trials as mono- and/or combination therapies.
Conclusion
In contrast to chemotherapy or oncogene-targeted therapies, cancer immunotherapy relies on promoting an anti-cancer immune response, a dynamic process involving several mechanisms and cross-talks between different cellular compartments. When neutralizing immune factors, one of the major issues is the off-target effects, which lead to unforeseen complications for patients. Many soluble factors are involved in several signaling pathways, thus the targeting of one specific soluble factor may disturb signaling pathways that were not initially meant to be targeted. Moreover, they are not unique to the tumor and TME, but mandatory for maintaining homeostasis. Following a series of breakthroughs, we are witnessing an acceleration of the research in the field of cancer immunotherapy, which can simultaneously target several TME abnormalities in the clinical setting. For instance, optimal cancer therapy with inhibition of the PD-1/PD-L1 axis should include: (1) modulation at the tumor site due to the localized expression pattern of PD-L1 in the TME, (2) targeting of elevated immune inhibitory cytokines (IL10, TGF-β), tumor metabolites and regulatory cells, and (3) rescue of the tolerated tumor immunity (177). Since 2011, the FDA has approved several agents as a standard of care therapy in oncology and hematology (Supplementary Table 1). Active research focuses on the integration of combinations that may boost the response rate to immunotherapy, which still remains low. An unmet need is the identification of predictive biomarkers for immunotherapy that could optimize patients' stratification and administration of combination therapies. This can be done through the discovery of “biomarkers of response” in patients that showed clinical benefit. Predictive factors for immunotherapy are being actively investigated for establishing an “immune signature” of tumors that defines genetic, molecular and functional profiles of immune cells present in the TME (178). The patient-specific landscape of the TME can be appreciated by using “immunograms” as integrated biomarkers, obtained by capturing the immune profile with next-generation sequencing data. The development of personalized biomarker profiles, and thus, the characterization of microenvironmental features and their changes during treatment, represents a comprehensive knowledge which will become a valuable resource for optimal personalized immunotherapy and patient monitoring (179). In the era of precision medicine, tailoring cancer immune-interventions combined with other comprehensive approaches may pave the way toward an appropriate modulation of anti-cancer immune responses, on the basis of the genetic and immunological analyses in each patient. Combination approaches altering the TME and/or decreasing immunosuppression along with strategies that are countervailing insufficient tumor immunogenicity and antigenicity may be required to achieve effective tumor control.
Author Contributions
BG and EA designed the manuscript. BG wrote the manuscript with inputs from CM and EA. CC, CM, SR, EA, and BG reviewed and approved the manuscript.
Funding
This manuscript was funded by a National Priorities Research Program (NPRP #8-2297-3-494) grant from the Qatar National Research Fund (QNRF).
Conflict of Interest
EA is co-founder and CEO of Veana Therapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Heba Sidahmed for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01554/full#supplementary-material
Abbreviations
ICB, Immune checkpoint blockade; TME, Tumor microenvironment; Tregs, regulatory T cells; CTL, cytotoxic T lymphocytes IFN-γ: interferon-γ; IDO, indoleamine 2,3- dioxygenase; CCL, C-C Motif Chemokine Ligands; TGF- β, Transforming growth factor- β; NK, natural killer; DC, dendritic cells; NSCLC, non-small-cell lung carcinoma; MDSCs, myeloid-derived suppressor cells; mAb, monoclonal antibodies; OS, overall survival; IL, Interleukin; APC, antigen presenting cells; TLR, Toll-like receptor; MHC-II, major histocompatibility complex II; TAMs, tumor-associated macrophages; VEGF, Vascular endothelial growth factor; FDA, Food and Drug Administration; PFS, progression free survival; NSCLC, Non-small-cell lung carcinoma; M-CSF, macrophage colony-stimulating factor; CAF, cancer-associated fibroblasts; ICIs, immune checkpoint inhibitors; TNF-α, tumor necrosis factor; iNOS, inducible nitric oxide synthase; PDE-5, Phosphodiesterase type 5 inhibitors; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; HNSCC, Head and neck squamous cell carcinomaSHP2: Src homology 2 domain-containing tyrosine phosphatase 2; PD-1, Programmed cell death protein 1; PD-L1, programmed death-ligand 1; RCC, renal cell carcinoma; LXR, liver X receptor; TIM-3, T cell immunoglobulin and mucin 3; LAG-3, Lymphocyte-activated gene-3; ITIM, immunoreceptor tyrosine-based inhibition motif; VISTA, V-domain Ig-containing suppressor of T cell activation or PD-1 homologue; TIGIT, T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory domain.
References
1. Budhu S, Wolchok J, Merghoub T. The importance of animal models in tumor immunity and immunotherapy. Curr Opin Genet Dev. (2014) 24:46–51. doi: 10.1016/j.gde.2013.11.008
2. Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. (2016) 4:261. doi: 10.21037/atm.2016.04.01
3. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) 35(Suppl.):S185–98. doi: 10.1016/j.semcancer.2015.03.004
4. Hanahan D, Weinberg RA. Hallmarks of cancer. the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting. the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. (2006) 90:1–50. doi: 10.1016/S0065-2776(06)90001-7
6. Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, Saldmann A, Gey A, Oudard S, Tartour E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. (2017) 2:e000213. doi: 10.1136/esmoopen-2017-000213
7. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. (2014) 192:5451–58. doi: 10.4049/jimmunol.1490019
8. Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. (2008) 226:205–18. doi: 10.1111/j.1600-065X.2008.00706.x
9. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
10. Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. (2004) 14:433–9. doi: 10.1016/j.semcancer.2004.06.006
11. Mocellin S, Marincola FM, Young HA. Interleukin-10 and the immune response against cancer: a counterpoint. J Leukoc Biol. (2005) 78:1043–51. doi: 10.1189/jlb.0705358
12. Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol. (2015) 34:22–7. doi: 10.1016/j.coi.2014.12.009
13. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
14. Sato Y, Takahashi S, Kinouchi Y, Shiraki M, Endo K, Matsumura Y, et al. IL-10 deficiency leads to somatic mutations in a model of IBD. Carcinogenesis. (2006) 27:1068–73. doi: 10.1093/carcin/bgi327
15. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
16. Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. (2012) 72:3570–81. doi: 10.1158/0008-5472.CAN-12-0721
17. Zhao S, Wu D, Wu P, Wang Z, Huang J. Serum IL-10 Predicts worse outcome in cancer patients: a meta-analysis. PLoS ONE. (2015) 10:e0139598. doi: 10.1371/journal.pone.0139598
18. Kubota K, Moriyama M, Furukawa S, Rafiul H, Maruse Y, Jinno T T, et al. CD163(+)CD204(+) tumor-associated macrophages contribute to T cell regulation via interleukin-10 and PD-L1 production in oral squamous cell carcinoma. Sci Rep. (2017) 7:1755. doi: 10.1038/s41598-017-01661-z
19. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. (2012) 2:a006502. doi: 10.1101/cshperspect.a006502
20. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. (2013) 13:871–82. doi: 10.1038/nrc3627
21. Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol. (2007) 178:1505–11. doi: 10.4049/jimmunol.178.3.1505
22. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. (2018) 9:978. doi: 10.3389/fimmu.2018.00978
23. Lapeyre-Prost A, Terme M, Pernot S, Pointet AL, Voron T, Tartour E, et al. Immunomodulatory activity of VEGF in Cancer. Int Rev Cell Mol Biol. (2017) 330:295–342. doi: 10.1016/bs.ircmb.2016.09.007
24. Zhao L, Zhang D, Ma H, Jin M, Huang F, Zhang T. High VEGF-A level at baseline predicts poor treatment effect of bevacizumab-based chemotherapy in metastatic colorectal cancer: a meta-analysis. Panminerva Med. (2016) 58:48–58.
25. Marti LC, Pavon L, Severino P, Sibov T, Guilhen D, Moreira-Filho CA. Vascular endothelial growth factor-A enhances indoleamine 2,3-dioxygenase expression by dendritic cells and subsequently impacts lymphocyte proliferation. Mem Inst Oswaldo Cruz. (2014) 109:70–9. doi: 10.1590/0074-0276130252
26. Hansen W. Neuropilin 1 guides regulatory T cells into VEGF-producing melanoma. Oncoimmunology. (2013) 2:e23039. doi: 10.4161/onci.23039
27. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. (2015) 212:139–48. doi: 10.1084/jem.20140559
28. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. (1993) 362:841–4. doi: 10.1038/362841a0
29. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. (2004) 350:2335–42. doi: 10.1056/NEJMoa032691
30. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. (2017) 9:eaak9679. doi: 10.1126/scitranslmed.aak9679
31. Guerrouahen BS, Pasquier J, Kaoud NA, Maleki M, Beauchamp MC, Yasmeen A, et al. Akt-activated endothelium constitutes the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol Cancer Therape. (2014) 13:3123–36. doi: 10.1158/1535-7163.MCT-13-1053
32. Manegold C, Dingemans AC, Gray JE, Nakagawa K, Nicolson M, Peters S, et al. The potential of combined immunotherapy and antiangiogenesis for the synergistic treatment of advanced NSCLC. J Thorac Oncol. (2017) 12:194–207. doi: 10.1016/j.jtho.2016.10.003
33. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. (2014) 2:632–42. doi: 10.1158/2326-6066.CIR-14-0053
34. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
35. Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today. (2017) 22:57–71. doi: 10.1016/j.drudis.2016.08.003
36. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. (2012) 188:21–8. doi: 10.4049/jimmunol.1101029
37. van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J, Aarden LA. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. (1995) 181:775–9. doi: 10.1084/jem.181.2.775
38. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clinical Cancer Res. (2007) 13(2 Pt 2):721s−6s. doi: 10.1158/1078-0432.CCR-06-2197
39. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
40. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
41. O'Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br J Pharmacol. (2015) 172:5239–50. doi: 10.1111/bph.13331
42. Albu DI, Wang Z, Huang KC, Wu J, Twine N, Leacu S, et al. EP4 Antagonism by E7046 diminishes myeloid immunosuppression and synergizes with Treg-reducing IL-2-Diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology. (2017) 6:e1338239. doi: 10.1080/2162402X.2017.1338239
43. Okumura Y, Yamagishi T, Nukui S, Nakao K. Discovery of AAT-008, a novel, potent, and selective prostaglandin EP4 receptor antagonist. Bioorg Med Chem Lett. (2017) 27:1186–92. doi: 10.1016/j.bmcl.2017.01.067
44. Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. (2016) 44:333–41. doi: 10.1042/BST20150245
45. Richardsen E, Uglehus RD, Johnsen SH, Busund LT. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. (2015) 35:865–74.
46. Aharinejad S, Salama M, Paulus P, Zins K, Berger A, Singer CF. Elevated CSF1 serum concentration predicts poor overall survival in women with early breast cancer. Endocr Relat Cancer. (2013) 20:777–83. doi: 10.1530/ERC-13-0198
47. Baghdadi M, Endo H, Takano A, Ishikawa K, Kameda Y, Wada H, et al. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci Rep. (2018) 8:418. doi: 10.1038/s41598-017-18796-8
48. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. (2013) 2:e26968. doi: 10.4161/onci.26968
49. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
50. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. (2012) 13:616–30. doi: 10.1038/nrm3434
51. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
52. Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-beta pathway for cancer therapy. Clinical Cancer Res. (2012) 18:4514–21. doi: 10.1158/1078-0432.CCR-11-3224
53. Hahn T, Akporiaye ET. Targeting transforming growth factor beta to enhance cancer immunotherapy. Curr Oncol. (2006) 13:141–3.
54. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. (2010) 10:554–67. doi: 10.1038/nri2808
55. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. (2007) 178:2018–27. doi: 10.4049/jimmunol.178.4.2018
56. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
57. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. (2006) 6:506–20. doi: 10.1038/nrc1926
58. Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. (2009) 19:89–102. doi: 10.1038/cr.2008.316
60. Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. (2000) 1:169–78. doi: 10.1038/35043051
61. Ivanovic V, Demajo M, Krtolica K, Krajnovic M, Konstantinovic M, Baltic V, Prtenjak G, et al. Elevated plasma TGF-beta1 levels correlate with decreased survival of metastatic breast cancer patients. Clin Chim Acta. (2006) 371:191–3. doi: 10.1016/j.cca.2006.02.027
62. Petrausch U, Jensen SM, Twitty C, Poehlein CH, Haley DP, Walker EB, Fox BA. Disruption of TGF-beta signaling prevents the generation of tumor-sensitized regulatory T cells and facilitates therapeutic antitumor immunity. J Immunol. (2009) 183:3682–9. doi: 10.4049/jimmunol.0900560
63. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
64. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
65. Cohn A, Lahn MM, Williams KE, Cleverly AL, Pitou C, Kadam SK, et al. A phase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int J Oncol. (2014) 45:2221–31. doi: 10.3892/ijo.2014.2679
66. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. (2014) 9:e90353. doi: 10.1371/journal.pone.0090353
67. Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal Irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clinical Cancer Res. (2018) 24:2493–504. doi: 10.1158/1078-0432.CCR-17-3322
68. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, in Advanced Solid Tumors. Clinical Cancer Res. (2018) 24:1287–95. doi: 10.1158/1078-0432.CCR-17-2653
69. Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, Terness P. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. (2009) 58:153–7. doi: 10.1007/s00262-008-0513-6
70. Mangaonkar A, Mondal AK, Fulzule S, Pundkar C, Park EJ, Jillella A, et al. A novel immunohistochemical score to predict early mortality in acute myeloid leukemia patients based on indoleamine 2,3 dioxygenase expression. Sci Rep. (2017) 7:12892. doi: 10.1038/s41598-017-12940-0
71. Witkiewicz AK, Costantino CL, Metz R, Muller AJ, Prendergast GC, Yeo CJ, Brody JR. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J Am Coll Surg. (2009) 208:781–7. doi: 10.1016/j.jamcollsurg.2008.12.018
72. Li F, Zhang R, Li S, Liu J. IDO1. An important immunotherapy target in cancer treatment. Int Immunopharmacol. (2017) 47:70–7. doi: 10.1016/j.intimp.2017.03.024
73. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clinical Cancer Res. (2011) 17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331
74. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
75. Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic Phenotype of IFN-gamma-Induced IDO+ dendritic cells is maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J Immunol. (2016) 197:962–70. doi: 10.4049/jimmunol.1502615
76. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. (2012) 72:1407–15. doi: 10.1158/0008-5472.CAN-11-2544
77. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
78. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
79. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. (2012) 1:1460–8. doi: 10.4161/onci.21716
80. Molinier-Frenkel V, Castellano F. Immunosuppressive enzymes in the tumor microenvironment. FEBS Lett. (2017) 591:3135–57. doi: 10.1002/1873-3468.12784
81. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive Immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
82. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
83. Durante W. Role of arginase in vessel wall remodeling. Front Immunol. (2013) 4:111. doi: 10.3389/fimmu.2013.00111
84. Munder M. Arginase. an emerging key player in the mammalian immune system. Br J Pharmacol. (2009) 158:638–51. doi: 10.1111/j.1476-5381.2009.00291.x
85. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
86. Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. (2015) 36:161–78. doi: 10.1016/j.it.2015.01.003
87. Vannini F, Kashfi K, Nath N. The dual role of iNOS in cancer. Redox Biol. (2015) 6:334–3. doi: 10.1016/j.redox.2015.08.009
88. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. (2015) 14:603–22. doi: 10.1038/nrd4596
89. de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. Oncoimmunology. (2012) 1:1305–12. doi: 10.4161/onci.21678
90. Peak TC, Richman A, Gur S, Yafi FA, Hellstrom WJ. The Role of PDE5 Inhibitors and the NO/cGMP Pathway in Cancer. Sex Med Rev. (2016) 4:74–84. doi: 10.1016/j.sxmr.2015.10.004
91. Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, G, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clinical Cancer Res. (2015) 21:30–8. doi: 10.1158/1078-0432.CCR-14-1716
92. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
93. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. (2014) 4:879–88. doi: 10.1158/2159-8290.CD-14-0341
94. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol. (2015) 195:965–72. doi: 10.4049/jimmunol.1500591
95. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. (2010) 285:7176–86. doi: 10.1074/jbc.M109.047423
96. Ohta A. A metabolic immune checkpoint. adenosine in tumor microenvironment. Front Immunol. (2016) 7:109. doi: 10.3389/fimmu.2016.00109
97. Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. (2009) 175:2439–53. doi: 10.2353/ajpath.2009.090290
98. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
99. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
100. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
101. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.CAN-10-4246
102. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
103. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, Sethumadhavan S, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. (2015) 7:277ra230. doi: 10.1126/scitranslmed.aaa1260
104. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
105. Bonnefoy N, Bastid J, Alberici G, Bensussan A, Eliaou JF. CD39. A complementary target to immune checkpoints to counteract tumor-mediated immunosuppression. Oncoimmunology. (2015) 4:e1003015. doi: 10.1080/2162402X.2014.1003015
106. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity. (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
107. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
108. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8. doi: 10.1126/science.270.5238.985
109. van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. (1999) 190:355–66. doi: 10.1084/jem.190.3.355
110. Littman DR. Releasing the Brakes on Cancer Immunotherapy. Cell. (2015) 162:1186–90. doi: 10.1016/j.cell.2015.08.038
111. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
112. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
113. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. (2011) 11:852–63. doi: 10.1038/nri3108
114. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA. (2003) 100:4712–7. doi: 10.1073/pnas.0830997100
115. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
116. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
117. Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. (2014) 40:235–47. doi: 10.1016/j.immuni.2013.11.017
118. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
119. Pardoll DM. Immunology beats cancer: a blueprint for successful translation. Nat Immunol. (2012) 13:1129–32. doi: 10.1038/ni.2392
120. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
121. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-Derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clinical Cancer Res. (2019) 25:3643–57. doi: 10.1158/1078-0432.CCR-18-2402
122. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
123. Amin A, Plimack ER, Ernstoff MS, Lewis LD, Bauer TM, McDermott DF, et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: the CheckMate 016 study. J Immunother Cancer. (2018) 6:109. doi: 10.1186/s40425-018-0420-0
124. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. (2019) 25:656–66. doi: 10.1038/s41591-019-0374-x
125. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
126. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. (2017) 276:80–96. doi: 10.1111/imr.12519
127. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics. (1994) 39:213–7. doi: 10.1007/BF00241263
128. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37. doi: 10.1084/jem.176.2.327
129. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
130. Workman CJ, Dugger KJ, Vignali DA. Cutting edge. molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. (2002) 169:5392–5. doi: 10.4049/jimmunol.169.10.5392
131. Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, et al. Aberrant Expression of MHC Class II in melanoma attracts inflammatory tumor-specific CD4+ T- Cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res. (2015) 75:3747–59. doi: 10.1158/0008-5472.CAN-14-2956
132. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, Du X, Tang L, He F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
133. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. (2005) 5:29–41. doi: 10.1038/nrc1527
134. Rabinovich GA, Toscano MA. Turning 'sweet' on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. (2009) 9:338–52. doi: 10.1038/nri2536
135. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 Shapes antitumor immune responses by suppressing CD8+ T Cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
136. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. (2013) 19:739–46. doi: 10.1038/nm.3179
137. Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J Immunol. (2002) 168:3874–80. doi: 10.4049/jimmunol.168.8.3874
138. Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol. (2014) 134:1893–902. doi: 10.1038/jid.2014.29
139. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. (2019) 176:334–47 e312. doi: 10.1016/j.cell.2018.11.010
140. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. (2015) 6:27359–77. doi: 10.18632/oncotarget.4751
141. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
142. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci USA. (2010) 107:7875–80. doi: 10.1073/pnas.1003345107
143. Fougeray S, Brignone C, Triebel F. A soluble LAG-3 protein as an immunopotentiator for therapeutic vaccines: Preclinical evaluation of IMP321. Vaccine. (2006) 24:5426–33. doi: 10.1016/j.vaccine.2006.03.050
144. Li Z, Ju Z, Frieri M. The T-cell immunoglobulin and mucin domain (Tim) gene family in asthma, allergy, and autoimmunity. Allergy Asthma Proc. (2013) 34:e21–6. doi: 10.2500/aap.2013.34.3646
145. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
146. Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. (2009) 39:2492–501. doi: 10.1002/eji.200939274
147. Rabinovich GA, Liu FT, Hirashima M, Anderson A. An emerging role for galectins in tuning the immune response: lessons from experimental models of inflammatory disease, autoimmunity and cancer. Scand J Immunol. (2007) 66:143–58. doi: 10.1111/j.1365-3083.2007.01986.x
148. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
149. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
150. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
151. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. (2008) 205:2763–79. doi: 10.1084/jem.20081398
152. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE. (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
153. Komohara Y, Morita T, Annan DA, Horlad H, Ohnishi K, Yamada S, et al. The coordinated actions of TIM-3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol Res. (2015) 3:999–1007. doi: 10.1158/2326-6066.CIR-14-0156
154. Cai C, Xu YF, Wu ZJ, Dong Q, Li MY, Olson JC, et al. Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J Urol. (2016) 34:561–7. doi: 10.1007/s00345-015-1656-7
155. Japp AS, Kursunel MA, Meier S, Malzer JN, Li X, Rahman NA, et al. Dysfunction of PSA-specific CD8+ T cells in prostate cancer patients correlates with CD38 and Tim-3 expression. Cancer Immunol Immunother. (2015) 64:1487–94. doi: 10.1007/s00262-015-1752-y
156. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
157. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
158. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
159. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
160. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. (2015) 349:1261669. doi: 10.1126/science.1261669
161. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, Chen L. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest. (2014) 124:1966–75. doi: 10.1172/JCI74589
162. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. (2019) 156:74–85. doi: 10.1111/imm.13001
163. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
164. Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K, et al. VISTA expressed in tumour cells regulates T cell function. Br J Cancer. (2019) 120:115–27. doi: 10.1038/s41416-018-0313-5
165. Kondo Y, Ohno T, Nishii N, Harada K, Yagita H, Azuma M. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. (2016) 57:54–60. doi: 10.1016/j.oraloncology.2016.04.005
166. Kuklinski LF, Yan S, Li Z, Fisher JL, Cheng C, Noelle RJ, et al. Ernstoff MS. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol Immunother. (2018) 67:1113–21. doi: 10.1007/s00262-018-2169-1
167. Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci USA. (2019) 116:1692–7. doi: 10.1073/pnas.1811067116
168. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. (2017) 23:551–5. doi: 10.1038/nm.4308
169. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA. (2015) 112:6682–7. doi: 10.1073/pnas.1420370112
170. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J Biol Chem. (2014) 289:17647–57. doi: 10.1074/jbc.M114.572420
171. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
172. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
173. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. (2011) 41:902–15. doi: 10.1002/eji.201041136
174. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
175. Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer. (2004) 4:73. doi: 10.1186/1471-2407-4-73
176. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
177. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. (2015) 125:3384–91. doi: 10.1172/JCI80011
178. Bogunovic D, O'Neill DW, Belitskaya-Levy I, Vacic V, Yu YL, Adams S, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA. (2009) 106(48):20429–34. doi: 10.1073/pnas.0905139106
Keywords: immunotherapy, immunosuppression, tumor escape, soluble factors, tumor microenvironment, immune checkpoint inhibitors, immunosuppressive enzymes
Citation: Guerrouahen BS, Maccalli C, Cugno C, Rutella S and Akporiaye ET (2020) Reverting Immune Suppression to Enhance Cancer Immunotherapy. Front. Oncol. 9:1554. doi: 10.3389/fonc.2019.01554
Received: 04 September 2019; Accepted: 23 December 2019;
Published: 21 January 2020.
Edited by:
Jason Roszik, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Theresa L. Whiteside, University of Pittsburgh, United StatesFernando Aranda, August Pi i Sunyer Biomedical Research Institute (IDIBAPS), Spain
Fabio Malavasi, University of Turin, Italy
Copyright © 2020 Guerrouahen, Maccalli, Cugno, Rutella and Akporiaye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bella S. Guerrouahen, YmVsbGEucy5ndWVycm91YWhlbkBnbWFpbC5jb20=; Emmanuel T. Akporiaye, ZXRha3BvcmlheWVAZ21haWwuY29t