 Giovanni Barillari
Giovanni Barillari- Department of Clinical Sciences and Translational Medicine, University of Rome Tor Vergata, Rome, Italy
The growth and metastasis of malignant tumors benefit from the formation of blood vessels within the tumor area. There, new vessels originate from angiogenesis (the sprouting of pre-existing neighboring vessels) and/or vasculogenesis (the mobilization of bone marrow-derived endothelial cell precursors which incorporate in tumor vasculature and then differentiate into mature endothelial cells). These events are induced by soluble molecules (the angiogenic factors) and modulated by endothelial cell interactions with the perivascular matrix. Given angiogenesis/vasculogenesis relevance to tumor progression, anti-angiogenic drugs are often employed to buttress surgery, chemotherapy or radiation therapy in the treatment of a wide variety of cancers. Most of the anti-angiogenic drugs have been developed to functionally impair the angiogenic vascular endothelial growth factor: however, this leaves other angiogenic factors unaffected, hence leading to drug resistance and escape. Other anti-angiogenic strategies have exploited classical inhibitors of enzymes remodeling the perivascular matrix. Disappointingly, these inhibitors have been found toxic and/or ineffective in clinical trials, even though they block angiogenesis in pre-clinical models. These findings are stimulating the identification of other anti-angiogenic compounds. In this regard, it is noteworthy that drugs utilized for a long time to counteract human immune deficiency virus (HIV) can directly and effectively hamper molecular pathways leading to blood vessel formation. In this review the mechanisms leading to angiogenesis and vasculogenesis, and their susceptibility to anti-HIV drugs will be discussed.
Introduction
Human Immunodeficiency Virus (HIV) is the etiologic agent of Acquired Immune Deficiency Syndrome (AIDS), a deadly disease characterized by a profound upheaval of the immune system and the consequent increased risk of developing infectious illnesses and tumors (1).
HIV life cycle is mediated by cellular and viral proteins: among the latter, the reverse transcriptase, integrase and aspartyl protease enzymes are key for HIV replication and infectivity (2).
Briefly, HIV infection begins when the gp120 protein of the viral envelope binds to the CD4 receptor on T cell surface (2). This is followed by gp120 interaction with a co-receptor (most often a chemokine receptor), and HIV entry into the cell (2). Thereafter, the HIV reverse transcriptase copies the viral RNA genome into the viral DNA, which is integrated into host cell genome by the HIV integrase, and then transcribed into messenger RNA (2). Subsequently, HIV transcripts are translated into HIV envelope proteins and the fused precursors of HIV capsid and polymerase proteins: this gives rise to the production of immature, non-infectious viral particles that “bud” from infected cells (2). Finally, the HIV aspartyl protease cleaves the fused capsid-polymerase proteins into the functional polypeptides, and HIV matures becoming infectious (2).
Survival of HIV-positive individuals has been greatly extended by the combined Anti-Retroviral Therapy (cART), which has rendered HIV infection a chronic disease (3).
cART results from the mix of drugs inhibiting the HIV reverse transcriptase, integrase or aspartyl protease (3).
In particular, HIV-reverse transcriptase inhibitors halt the synthesis of viral DNA (3), and are available in three forms: (a) nucleoside analog reverse transcriptase inhibitors, which are modified deoxynucleotides analogs competing with natural deoxynucleotides for incorporation in the HIV DNA that is being synthesized; (b) nucleotide analog reverse transcriptase inhibitors, that are phosphonate deoxynucleotides analogs again competing the incorporation of natural deoxynucleotides in the forming HIV DNA; (c) non-nucleoside reverse transcriptase inhibitors, which bind to the allosteric sites of HIV-reverse transcriptase, thereby hampering its function (Table 1) (3). Nowadays, a HIV-reverse transcriptase inhibitors-based cART containing the nucleotide analog tenofovir, the nucleoside analog emtricitabine and the non-nucleoside efavirenz is recommended as first-line treatment of HIV infection (6).

Table 1. Anti-HIV drugs approved for clinical use, and their mechanism of action.
Among the antagonists of HIV integrase, the integrase strand transfer inhibitors impair HIV replication by preventing the insertion of HIV DNA into the host cell genome (Table 1): these drugs are in established use, especially in patients who have acquired resistance to other cART components (3).
For their part, HIV-protease inhibitors block the active site of HIV aspartyl protease, thus impeding the processing of HIV capsid-polymerase polyproteins and, consequently, the generation of mature, infectious HIV particles (4) (Table 1). To date, 10 HIV-protease inhibitors have been approved for therapeutic use in humans: saquinavir, indinavir, ritonavir, nelfinavir, lopinavir, amprenavir and its derivate fosamprenavir, atazanavir, tipranavir, and darunavir: all of them (but tipranavir) mimic HIV protease substrate (4). At the present time, darunavir or atazanavir are the most used HIV-protease inhibitors: whereas, saquinavir and indinavir are no longer employed because of their low solubility, and ritonavir is used only to increase the half-life of other HIV-protease inhibitors, due to its capability of inhibiting cytochrome P450 3A4 (4).
Consistent with chemokine receptor capability of working as HIV co-receptors, the CXC chemokine receptor 4 (CXCR4) antagonist AMD3100 efficiently reduces virus entry into target cells (Table 1), and it is then administered to HIV-positive individuals (5).
As such, these therapeutic regimens efficiently suppress HIV replication, thus improving the patients' immune functions (3).
Strikingly, results from epidemiological studies indicate that treatment of HIV-infected individuals with HIV-protease inhibitors and/or HIV-reverse transcriptase inhibitors has also reduced the incidence and/or clinical progression of AIDS-defining tumors such as Kaposi's sarcoma, non-Hodgkin lymphoma or uterine cervical carcinoma (7, 8). Moreover, chemokine receptor antagonists have been reported to be effective in the treatment of non-AIDS defining hematological malignancies affecting HIV-infected patients (9).
Definitely, the anti-tumor effects of cART are helped by the capability that this therapeutic regimen has to promote immune reconstitution by suppressing HIV replication (7, 8). Nevertheless, drugs present in cART and employed in the treatment of HIV-positive, immune-deficient patients have been found to exert anti-tumor activities also in HIV-negative, immune-competent individuals (7, 8, 10–22). Moreover, the HIV-protease inhibitors, HIV-reverse transcriptase inhibitors or chemokine receptor antagonists have been shown to directly impair the survival, growth, invasion and/or locomotion of tumor cells in HIV-free pre-clinical models (23–39). Interestingly, in some cancer cell types the cytostatic effect of HIV-protease inhibitors or HIV-reverse transcriptase inhibitors has been accompanied by the induction of cell differentiation and, eventually, by the acquisition of an immunogenic phenotype (7, 30, 31, 40). In addition, the inhibitors of HIV protease or reverse transcriptase and the antagonists of chemokine receptors have been reported to sensitize cancer cells to anti-tumor chemotherapeutics or ionizing radiations both in vitro and in vivo (32, 41–48).
In contrast, it is not currently known whether the inhibitors of HIV-integrase possess direct anti-tumor activities. Furthermore, the impact that these drugs may have on cancer incidence or progression has not been clearly established (49, 50).
The mechanisms responsible for the anti-tumor activities of HIV protease inhibitors, HIV reverse transcriptase inhibitors or chemokine receptor antagonists include the block of signaling pathways, transcription factors, enzymes, cytokines or growth factors which are deeply involved in tumor development and/or progression (23–39, 41–48).
Noteworthy, many the abovementioned molecules or mechanisms are employed by endothelial or stromal cells to generate blood vessels (51). Consistently, the inhibitors of HIV protease or reverse transcriptase and the antagonists of chemokine receptors have also been shown to counteract tumor vascularization in a variety of pre-clinical models.
In particular, results from early animal studies have indicated that the HIV protease inhibitors indinavir or saquinavir can directly block angiogenesis, that is the sprouting of new blood vessels from pre-existing ones (52). Later, also other HIV protease inhibitors including ritonavir, nelfinavir or amprenavir have been found capable of inhibiting angiogenesis in vivo (53, 54), and the anti-angiogenic effect of indinavir or saquinavir has been confirmed in mouse xenografts of highly prevalent human tumors (27). In the meantime, in vitro work has unraveled some of the molecular mechanisms responsible for the anti-angiogenic effect of HIV-protease inhibitors (55–59).
Studies evaluating the impact of HIV-reverse transcriptase inhibitors on angiogenesis are more recent than those concerning HIV-protease inhibitors. Results from those studies indicate that HIV-reverse transcriptase inhibitors including zidovudine, stavudine, efavirenz, lamivudine, emtricitabine, abacavir or tenofovir hamper endothelial cell survival, growth and locomotion in vitro and angiogenesis in vivo (60–63).
At variance with the inhibitors of the HIV protease or reverse transcriptase, the effect that HIV integrase inhibitors could have on angiogenesis has not yet been evaluated.
The discovery of the anti-angiogenic activity of chemokine receptor antagonists is quite novel. In particular, studies on this topic mostly refer to CXCR4 that, in addition to work as a co-receptor for HIV entry into target cells (5), is bound by the pro-angiogenic CXC chemokine ligand 12 (CXCL12) (64, 65). Consistent with the fact that both CXCR4 and CXCL12 are highly expressed in tumor tissues where their interaction plays a major role in the formation of new vessels, the CXCR4 antagonist AMD3100, which is employed in anti-HIV therapies, can counteract angiogenesis either in vitro or in animal models of human tumors (64–67).
Given that newly formed blood vessels nourish the growing cancer mass and furnish a portal for its metastasis, the anti-angiogenic properties of anti-HIV drugs constituting cART are likely to strongly contribute to the anti-tumor activity of this curative procedure (7, 8, 62, 68).
This considered, the present review is focused on the cellular events or molecular pathways which make HIV-protease inhibitors, HIV-reverse transcriptase inhibitors or CXCR4 antagonists capable of impairing the formation of new vessels that accompanies and favors tumor progression.
Effect of HIV-Protease Inhibitors or HIV-Reverse Transcriptase Inhibitors on Pro-Angiogenic Signaling Pathways
In the adult organism, endothelial cells lining the blood vessel lumen have a low proliferative rate: this is due to their tight intercellular junctions or anchorage to the basement membrane, and to the cytostatic stimulus they receive from vascular smooth muscle cells or pericytes surrounding the vessel externally (69, 70). Under these condition, endothelial cells can remain quiescent for years.
However, upon tissue hypoxia or damage and inflammation, endothelial cells are activated and new vessels are eventually formed. In most cases, this occurs through angiogenesis, a multi-step process affecting pre-existing neighbor vessels: there, vascular smooth muscle cells are detached and endothelial cells degrade the basement membrane, migrate in the perivascular space and proliferate, forming cellular cords that will ultimately differentiate into hollow tubes (69, 70).
Hypoxia and inflammation can occur at the same time in tumors. In particular, as the tumor grows, cells located in its central regions become distant to the neighboring vessels, being deprived of oxygen or nutrients, and undergo necrosis (69).
Necrotic tumor cells release the high-motility-group-box 1 nuclear protein, which induces the Nuclear Factor-kappa B (NF-kB) transcription factor to activate the expression of inflammatory chemokines (71) (Figure 1). The latter, in turn, recruit to the tumor site macrophages and leukocytes releasing pro-angiogenic factors (69). In this regard, it has to be highlighted that HIV-protease inhibitors including lopinavir, nelfinavir or ritonavir can impair NF-kB activation in endothelial cells (72) (Figure 1, Table 2). Moreover, saquinavir, indinavir and atazanavir are capable of inhibiting both the expression of high-mobility-group-box 1 protein and the activation of NF-kB (73, 76, 98) (Table 2). In contrast, the HIV-reverse transcriptase inhibitors tenofovir and zidovudine have no effect on NF-kB activity (101), while efavirenz even increases it (102) (Table 3).
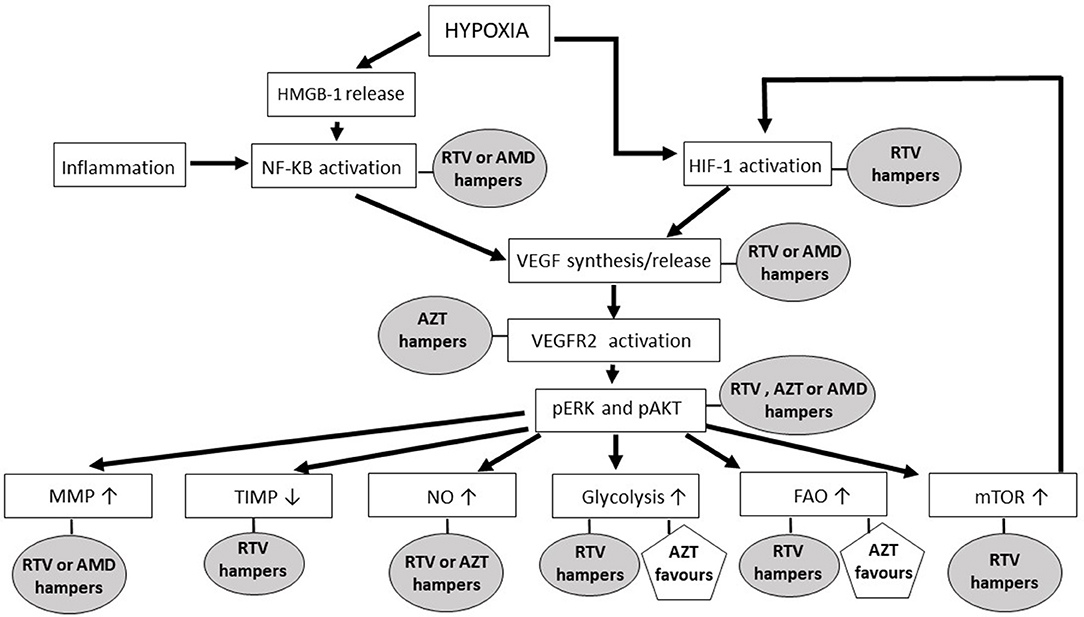
Figure 1. The impact of anti-HIV drugs on the molecular pathways of angiogenesis. In the rectangles are shown the main signaling pathways and molecules leading to new blood vessel formation; in ellipses and pentagons are reported the antagonist and agonist effects, respectively, that anti-HIV drugs exert on the angiogenic molecules or pathways. Ritonavir has been selected among the HIV-protease inhibitors, while zidovudine is the representative of the HIV-reverse transcriptase inhibitors, because both of these drugs are endowed with several anti-angiogenic activities. The chemokine receptor antagonist AMD3100 has been mentioned since it is currently employed in either anti-HIV or anti-cancer therapy. AMD, AMD3100; AZT, zidovudine; FAO, fatty acids oxidation; HIF, hypoxia-inducible factor; HMGB, high mobility group box; MMP, matrix metalloproteinase; mTOR, mammalian-target-of-rapamycin; NF-kB, nuclear factor-kappa B; NO, nitric oxide; pAKT, phosphorylated protein kinase B; pERK, phosphorylated extracellular-signal-regulated kinases; RTV, ritonavir; TIMP, tissue inhibitor of matrix metalloproteinase; VEGF, vascular endothelial growth factor; VEGFR2, type 2 vascular endothelial growth factor receptor.

Table 2. Anti-angiogenic effect of HIV-protease inhibitors: molecular targets.
Table 3. Effects of HIV-reverse transcriptase inhibitors or AMD3100 on angiogenesis regulators.
In tumor central areas also the hypoxia inducible transcription factor (HIF)-1 is activated, directly promoting the expression of angiogenic factors (69, 112) (Figure 1). While HIV-reverse transcriptase inhibitor impact on HIF-1 is currently uncharted, the HIV-protease inhibitors amprenavir, nelfinavir, saquinavir or ritonavir are known to down-regulate HIF-1 expression in both normal and tumor cells (54, 55, 59) (Figure 1, Table 2).
Vascular endothelial growth factor (VEGF)-A, basic fibroblast growth factor, platelet derived growth factor-BB and angiopoietin−1 and−2 are among the angiogenic factors produced by cancer cells, stromal cells or tumor-infiltrating immune cells upon HIF or NF-kB activation (69). By promoting vasodilatation and vascular smooth muscle cell detachment, angiopoietin-2 triggers the initiation of angiogenesis; whereas, angiopoietin-1 or platelet derived growth factor-BB mediates its termination, and VEGF-A or basic fibroblast growth factor promotes all the steps of the angiogenic process (69). Actually, the principal mediator of tumor angiogenesis is VEGF-A, which is produced either in inflammatory-driven or ischemia-induced angiogenesis, since it has response elements for both NF-kB and HIF-1 in its promoter region (69) (Figure 1). Consistent with their inhibitory effect on HIF-1 and NF-kB, the HIV-protease inhibitors ritonavir, nelfinavir or amprenavir reduces VEGF production (54, 55, 59, 77), while indinavir cannot (113) (Figure 1, Table 2).
Angiogenic factors exert their actions by binding to specific receptors whose intracellular domain contains a tyrosine kinase that auto-phosphorylates upon ligand binding (114). The most important receptor in angiogenesis is the type 2 VEGF receptor (114): noteworthy, HIV-reverse transcriptase inhibitors including zidovudine, tenofovir and lamivudine block the activation of either type 2 VEGF receptor or basic fibroblast growth factor receptor in endothelial cells (62) (Figure 1, Table 3).
The binding of an angiogenic factor to its receptor activates signaling pathways such as the mitogen-activated protein kinases (MAPK)/extracellular-signal-regulated kinases (ERK) and/or the phosphoinositide 3 kinase (PI3K)/ protein kinase B (AKT) (114) (Figure 1).
Activation of the MAPK/ERK pathway leads to endothelial cell proliferation (115). The HIV-reverse transcriptase inhibitors zidovudine, lamivudine or tenofovir weaken ERK phosphorylation promoted in endothelial cells by VEGF-A (62) (Figure 1); whereas, efavirenz activates MAPK in endothelial cells (102) (Table 3).
The HIV-protease inhibitors ritonavir, saquinavir, lopinavir or indinavir have definitely proven to counteract this signaling pathway (74, 77, 96), while nelfinavir inhibits it in some cell types but not in others (89, 90) (Figure 1, Table 2).
The AKT cascade is activated by PI3K, an enzyme having different isoforms of whom p110α is selectively required for angiogenesis (112).
PI3K generates the phosphatidylinositol (3,4,5)triphosphate which recruits AKT to the cellular membrane: there, the phosphoinositide dependent kinase-1 activates AKT by phosphorylating its kinase domain (112).
While the HIV-reverse transcriptase inhibitors zidovudine, tenofovir and lamivudine depress VEGF-promoted AKT phosphorylation in endothelial cells (62) (Figure 1, Table 3), the HIV-protease inhibitors nelfinavir, lopinavir and ritonavir decrease both total and phosphorylated AKT levels (54, 55, 81–83, 90–93, 116) (Figure 1, Table 2). Noteworthy, nelfinavir, saquinavir and amprenavir inhibit AKT phosphorylation also in leukocytes of treated individuals (78). In contrast, the HIV-protease inhibitor indinavir and the HIV-reverse transcriptase inhibitors nevirapine or efavirenz have no significant effect on AKT (83, 109, 117) (Tables 2, 3). Notwithstanding, and also in spite of its capability of promoting the activation of either NF-kB or MAPK (102), efavirenz possesses anti-angiogenic activities: in particular, this HIV-reverse transcriptase inhibitor has been found to counteract endothelial cell viability and growth via an increase in oxidative stress (61).
Results from in vitro experiments indicate that the HIV protease inhibitor nelfinavir can impair AKT phosphorylation without increasing the expression or activity of AKT negative regulators such as phosphatase-and-tensin-homolog or the protein-phosphatase-2A (94, 95). Interestingly, when combined with the anti-diabetes drug metformin, nelfinavir reduces PI3Kp110α protein levels in tumor cells (118).
AKT phosphorylation promotes endothelial cell expression of endothelial nitric oxide synthase (eNOS), this leading to the production and release of nitric oxide (112) (Figure 1). The latter is then taken up by nearby pericytes or vascular smooth muscle cells promoting vasodilatation, an early step of angiogenesis (112). Extracellular nitric oxide also enters macrophages, increasing VEGF production by these cells (69). Consistent with their inhibitory effect on AKT, the HIV-protease inhibitors nelfinavir or ritonavir (and saquinavir, lopinavir, atazanavir, or amprenavir likewise) reduces eNOS expression and nitric oxide production in both endothelial and vascular smooth muscle cells (58, 79, 84, 119) (Figure 1, Table 2). As for HIV-protease inhibitors, the HIV-reverse transcriptase inhibitors zidovudine or lamivudine hinders eNOS, while stavudine has no effect (103) (Figure 1, Table 3). In contrast, emtricitabine, tenofovir or efavirenz stimulate eNOS (6) (Table 3).
Another downstream target of AKT important for angiogenesis is the mammalian-target-of-rapamycin (mTOR), a serine/threonine protein kinase which positively regulates HIF-1α protein (112) (Figure 1). Again in accord with their inhibitory effect on AKT, either nelfinavir or ritonavir can hamper mTOR activity in normal or cancer cells (23, 74, 85, 86, 91, 120–122) (Figure 1, Table 2).
Impact of the HIV-Protease Inhibitors or HIV-Reverse Transcriptase Inhibitors on the Sequential Phases of Angiogenesis
Angiogenesis is initiated by VEGF-A and angiopoietin-2 which, by activating AKT and MAPK, induce endothelial cells to secrete proteolytic enzymes such as the matrix metalloproteinases (MMPs): the latter detach vascular smooth muscle cells or pericytes from the vessels, cleave endothelial cell-endothelial cell adhesions and degrade the vascular basement membrane, releasing angiogenic molecules which are tethered therein (114, 115) (Figure 2).
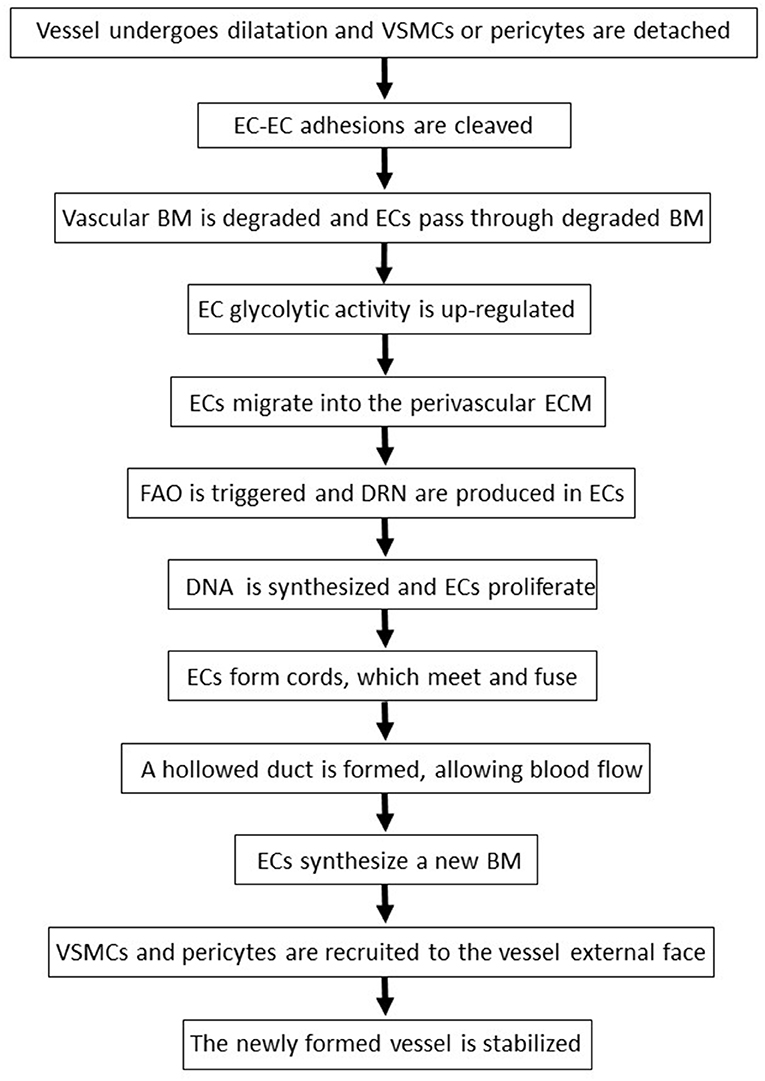
Figure 2. The sequential phases of angiogenesis. BM, basement membrane; DRN, deoxyribonucleotides; EC, endothelial cell; ECM, extracellular matrix; FAO, fatty acids oxidation; VSMC, vascular smooth muscle cells.
Among MMPs, MMP-2 and MMP-9 levels are positively correlated to tumor vessel density (123). Either enzymes are synthesized as zymogens: pro-MMP-2 is activated on endothelial cell surface by membrane type 1-MMP, while pro-MMP-9 is turned-on by plasmin, outside the producing cells (123). Noteworthy, the HIV-protease inhibitor indinavir blocks MMP-2 activation in endothelial cells via a reduction of membrane type 1-MMP expression: this is due to indinavir capability of inhibiting the binding of the Specificity Protein-1 transcription factor to the promoter region of membrane type 1-mmp gene (57). About MMP-9, HIV-protease inhibitors including darunavir, lopinavir, saquinavir, and ritonavir directly down-regulate its expression in normal or tumor cells and in treated patients (28, 29, 80, 97, 100, 124) (Figure 1, Table 2). In particular, by inhibiting AKT phosphorylation, saquinavir or ritonavir impairs the activation of Fra-1, a transcription factor inducing MMP-9 expression (29).
In addition to be controlled at the transcriptional level and by zymogen activation, the function of MMPs is modulated by endogenous inhibitors, the so called Tissue Inhibitors of MMPs (TIMPs): in particular, the ratio between MMPs and TIMPs rules the intensity of vascular basement membrane or perivascular matrix degradation, thereby deeply influencing the angiogenic process (125). Noteworthy, the HIV-protease inhibitors nelfinavir, indinavir and ritonavir augment the levels of anti-angiogenic TIMPs either in vitro or in treated individuals (75, 124) (Figure 1, Table 2). This important finding is in agreement with the results from pre-clinical studies conducted with other antagonists of the MAPK/ERK or PI3K/AKT pathway (126, 127).
Definitely, the inhibitory effect on MMP activity provides a molecular explanation for indinavir, saquinavir, or amprenavir capability of impairing angiogenesis and tumor growth in HIV-free animal models (27, 52, 57, 99).
Concerning HIV-reverse transcriptase inhibitors, zidovudine down-regulates MMPs in astrocytes or microglia cells (104), but not in endothelial cells (105) (Figure 1); whereas, lamivudine increases MMP and decreases TIMP serum levels in treated individuals (108) (Table 3).
Following the degradation of the blood vessel basement membrane, endothelial cells migrate into the perivascular extracellular matrix in response to VEGF-A and other angiogenic factors (69, 123) (Figure 2). The locomotion of endothelial cells depends on their adhesion to extracellular matrix molecules such as collagen-I and fibronectin, which trigger endothelial cell survival and proliferation by binding to integrin receptors expressed on the surface of these cells (69). Interestingly, indinavir impairs cellular adhesion onto fibronectin (128), while nelfinavir or atazanavir reduce collagen deposition in the extracellular matrix (98, 129).
When exposed to high VEGF-A levels, endothelial cells remodel their actin cytoskeleton acquiring a migratory phenotype characterized by filopodia or lamellipodia (70). This differentiation process requires high levels of ATP which are produced via the up-regulation of endothelial cell glycolytic activity (70) (Figure 2). Specifically, by activating AKT, VEGF increases the expression of either glucose transporter-1, which promotes glucose entry into endothelial cells, or glycolytic enzymes, including lactate dehydrogenase A (70). Most of the glucose entering endothelial cells is then converted into lactate which, in turn, stabilizes HIF-1α, this increasing VEGF synthesis by endothelial cells and promoting their migration (70). Of interest, the HIV-protease inhibitor ritonavir potently reduces glucose transporter-1 activity and glycolysis (87) (Figure 1, Table 2). In contrast, the HIV-reverse transcriptase inhibitors efavirenz or zidovudine stimulates glycolysis and increases lactate levels (106, 110) (Figure 1, Table 3).
Upon VEGF binding to type 2 VEGF receptor, migratory endothelial cells secrete the Delta-like ligand 4 which binds to the Notch receptor on the surface of neighboring endothelial cells: the latter then proliferate, maintaining connectivity with migratory endothelial cells (69). In proliferating endothelial cells, the production of deoxyribonucleotides required for DNA synthesis is sustained by the oxidation of fatty acids (70) (Figure 2). Noteworthy, fatty acids oxidation is blocked by the HIV-protease inhibitors ritonavir, atazanavir, lopinavir or darunavir, and augmented by the HIV-reverse transcriptase inhibitors zidovudine and stavudine (88, 107) (Figure 1, Tables 2, 3).
Angiogenesis is terminated when newly formed vessels meet and fuse, giving rise to a hollowed duct which allows blood flow (69) (Figure 2). Thereafter, endothelial cells stabilize the vessels by synthesizing a new basement membrane and releasing platelet-derived growth factor-BB which, in turn, recruits mural vascular smooth muscle cells and pericytes (69) (Figure 2).
In this regard, it has to be highlighted that tumor vessels display a poor vascular smooth muscle cell coverage, and that tumor endothelial cells are more responsive to growth/chemotactic factors, and more resistant to apoptosis than normal endothelial cells (70, 114, 130). Tumors are consequently provided with overgrowing, highly permeable, dilated and tortuous vessels (70, 130). As this can compromise either the arrival of chemotherapeutics to the tumor site or the efficacy of anti-cancer radiotherapy (130–132), novel anti-angiogenesis strategies are aimed at remodeling the tumor vasculature to a more normal (and functional) phenotype. In this regard, atazanavir capability of down-regulating the oxygen-sensor-prolyl-hydroxylase-domain-protein 2 (98) suggests that this HIV-protease inhibitor could normalize tumor vessels. In fact, a decrease in prolyl-hydroxylase-domain-protein 2 switches tumor endothelial cell phenotype from migratory/proliferative to quiescent (130). In contrast, the HIV-protease inhibitors amprenavir, nelfinavir and ritonavir inhibit glycogen-synthase-kinase 3β (119, 133), an enzyme that favors the differentiation of tumor endothelial cells into normal endothelial cells (134). Moreover, ritonavir also hinders the growth and migration of vascular smooth muscle cells promoted by platelet-derived growth factor-BB (135).
Regarding HIV-reverse transcriptase inhibitors, efavirenz increases vessel permeability by altering endothelial cell-endothelial cell junctions (102).
In conclusion, results from the reviewed literature indicate that both HIV-protease inhibitors and HIV-reverse transcriptase inhibitors mainly hamper the initial steps of angiogenesis. However, one should consider that a block of endothelial cell invasion such that caused by HIV-protease inhibitors is enough to halt the whole angiogenic process (57, 114).
Impact of HIV-Protease Inhibitors and Chemokine Receptor Antagonists on Vasculogenesis
The formation of tumor-associated vessels is facilitated and accelerated by vasculogenesis, a process which generally takes place during embryo life, involving the endothelial cell precursors: the latter are bone-marrow resident immature cells which, as for mature endothelial cells express both the type 2 VEGF receptor and the chemokine receptor CXCR4 (68, 136).
In tumor tissues, the activation of HIF-1 induces cancer cells, endothelial cells and fibroblasts to produce and release the angiogenic VEGF and CXCL12 (68, 69, 136) (Figure 3). In this context, nitric oxide bolsters VEGF production and release by endothelial cells (69) (Figure 3). Then, VEGF and CXCL12 reach the bone marrow and bind to type 2 VEGF receptor and CXCR4, respectively, on the surface of endothelial cell precursors (68, 69, 136) (Figure 3). Either the VEGF/type 2 VEGF receptor or the CXCL12/ CXCR4 axis triggers MAPK or AKT, sustaining the survival of endothelial cell precursors and activating MMP-9 production by those immature cells (68, 69, 136, 137) (Figure 3). MMP-9, in turn, mobilizes endothelial cell precursors from the bone marrow into the circulation, allowing them to reach the tumor (136) (Figure 3). There, because of the prolonged AKT activation triggered by VEGF, basic fibroblast growth factor and integrin-mediated adhesion onto tumor extracellular matrix, endothelial cell precursors differentiate into mature endothelial cells, then incorporating into tumor vessels (136) (Figure 3).
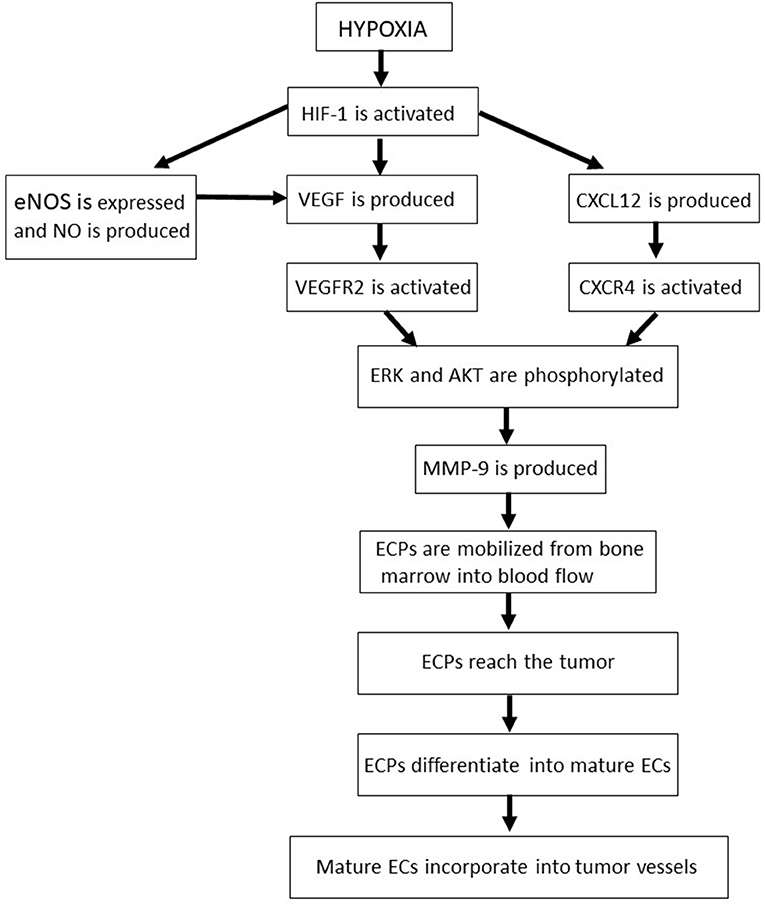
Figure 3. The molecular pathways and sequential phases of vasculogenesis. AKT, protein kinase B; CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor 4; EC, endothelial cell; ECP, endothelial cell precursor; eNOS, endothelial nitric oxide synthase; ERK, extracellular-signal-regulated kinases; HIF, hypoxia-inducible factor; MMP, matrix metalloproteinase; NO, nitric oxide; VEGF, vascular endothelial growth factor; VEGFR2, type 2 vascular endothelial growth factor receptor.
Consistent with CXCR4 role in the mobilization and survival of endothelial cell precursors, AMD3100, an antagonist of this receptor administered to HIV-infected individuals, effectively impairs tumor vasculogenesis in pre-clinical models (68, 111). Moreover, as activation of the CXCL12/CXCR4 axis stimulates VEGF and MMP production in mature endothelial cells (138), CXCR4 antagonists also inhibit tumor angiogenesis in animal models of human tumors (68).
Certainly, HIV-protease inhibitors possess all the features useful to counter tumor vasculogenesis, since they down-regulate HIF-1, AKT, MAPK, eNOS, VEGF and MMP (Figure 1, Table 2). Moreover, because of their inhibitory effect on glycogen-synthase-kinase 3β (119, 133), the HIV-protease inhibitors amprenavir, nelfinavir or ritonavir could hinder the differentiation of endothelial cell precursors into mature endothelial cells (139).
Concerning HIV-reverse transcriptase inhibitors, those stimulating eNOS activity, glycolysis and/or fatty acids oxidation (6, 106, 107, 110) (Figure 1, Table 3) could favor tumor-associated vasculogenesis. Future work will verify this possibility.
Clinical Assessment of the Anti-Tumor Activity of Anti-HIV Drugs
Based on the results of pre-clinical and epidemiological studies, clinical trials have evaluated the effects of HIV-protease inhibitors, HIV-reverse transcriptase inhibitors or chemokine receptor antagonists (alone or combined with standard anti-tumor chemotherapy and/or radiotherapy) against a variety of tumors.
Results from medical tests performed in either HIV-positive or HIV-negative patients, indicate that HIV-protease inhibitor mono-therapy is effective against early-stage tumors including the angiogenic phase of Kaposi's sarcoma (10, 140, 141) and the pre-invasive stage of uterine cervical carcinoma (11) (Table 4). In contrast, when HIV-protease inhibitors are administered alone, they are not active against highly progressed tumors such as invasive uterine cervical carcinoma (8) and recurrent gliomas or adenoid cystic carcinomas (157, 158).
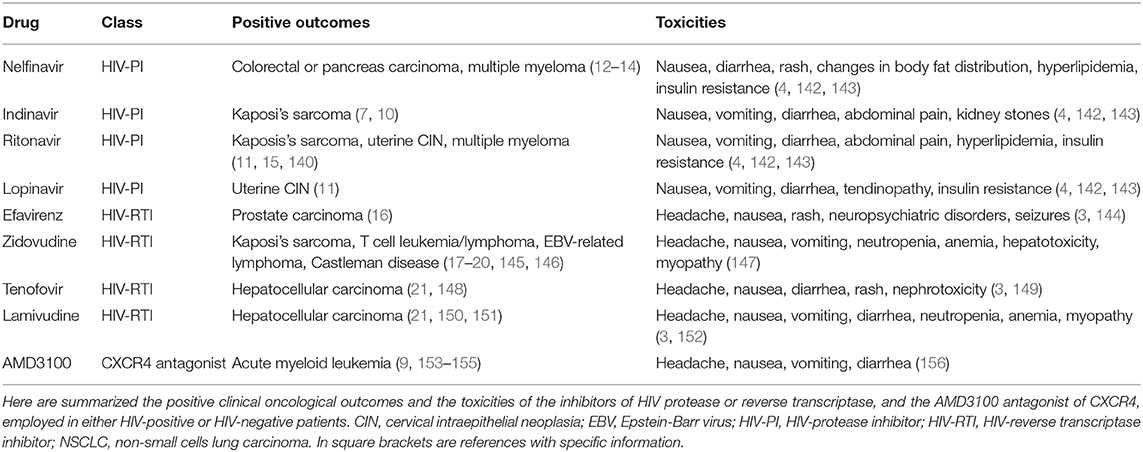
Table 4. Clinical outcomes and toxicities of anti-HIV drugs endowed with anti-tumor activities.
Concerning the combination of HIV-protease inhibitors and classical anti-cancer radiotherapy and/or chemotherapy, the great majority of clinical trials has been performed for nelfinavir. Results from these trials indicate that, in addition to be safe and well-tolerated, combination therapy with nelfinavir is associated with the regression of rectal or pancreatic carcinoma (Table 4), this being accompanied by the “normalization” of the aberrant tumor vasculature (12, 13). In addition, nelfinavir has also been reported to overcome the resistance of multiple myeloma to proteasome inhibitors (14) (Table 4).
Regarding ritonavir-based or saquinavir-based combinatory therapies, the inhibitory effect that these HIV-protease inhibitors exert on cytochrome P450 has been shown to impair the clearance of anti-cancer chemotherapeutics: this has led to severe toxicities in some clinical trials (159–161), but not in others (15).
Concerning the toxicity that HIV-protease inhibitors have in themselves, results from large observational studies conducted on HIV-infected subjects, indicate that HIV-protease inhibitor-based cART elevates cholesterol, triglycerides and glucose plasma levels (142, 143). These side effects have occurred mostly with first generation HIV-protease inhibitors such as ritonavir, nelfinavir or lopinavir (Table 4), which increase lipemia by inhibiting adiponectin expression, and/or cause insulin resistance via an impairment of glucose transporter-4 activity (4). In contrast, second generation HIV-protease inhibitors including darunavir and atazanavir display only minor toxicities and have no effect on lipemia or insulin sensitivity (4).
Regarding the HIV-reverse transcriptase inhibitors, most of the clinical trials have been performed for zidovudine. Results indicate that when combined with antivirals (e.g., interferon α or valgaciclovir) or cytotoxic drugs (e.g., bleomycin or methotrexate), zidovudine is effective against HIV-associated Kaposi's sarcoma (145), T cell leukemia (17, 18, 20), Epstein-Barr virus related-lymphomas (19) or Castleman lymphoproliferative disease (146) (Table 4). Other clinical studies have shown that hepatitis B virus-infected cancer patients have a reduced incidence of hepatocellular carcinoma when they undergo prophylactic treatment with HIV reverse transcriptase inhibitors including lamivudine, entecavir or tenofovir (21, 148, 150, 151) (Table 4). Finally, high plasma concentrations of efavirenz have been reported to slow the progression of prostate carcinoma (16) (Table 4).
However, in the majority of the abovementioned diseases, the anti-tumor activity of HIV-reverse transcriptase inhibitors is not likely to depend on the impairment of angiogenesis, but rather on the capability that these anti-HIV drugs have to hamper the replication of tumorigenic viruses (17–21, 145, 146, 148, 150, 151).
Observational studies have shown that the most common adverse effects which can occur in HIV-infected patients taking HIV-reverse transcriptase inhibitors include headache, nausea, vomiting, diarrhea and/or rash (147, 149, 152) (Table 4). In addition, long-term use or high-dose treatment with HIV-reverse transcriptase inhibitors is associated with severe, therapy-limiting side effects such as anemia, neutropenia, myopathy, impaired liver function or nephrotoxicity (147, 149, 152) (Table 4). Furthermore, some of the efavirenz-treated patients have experienced neuropsychiatric disorders and/or seizures (144) (Table 4).
About the use of chemokine receptor antagonists in cancer patients, the CXCR4 antagonist AMD3100 is the only one approved for clinical use: at the present time, the drug is being employed mostly in order to mobilize hematopoietic stem cells from the bone marrow, hence allowing their autologous transplantation in patients affected by non-Hodgkin lymphoma or multiple myeloma (153, 154). In addition, AMD3100 has recently been used to mobilize bone marrow-resident leukemic cells into the blood flow, this leading to the sensitization of leukemic cells to cytotoxic drugs (155) (Table 4). Interestingly, results from pre-clinical studies support AMD3100 combination with radio/chemotherapy to counteract lymph node or distant metastases of solid tumors (38, 48, 162, 163). This strategy is also sustained by observational studies (164, 165).
Headache, nausea, vomiting, diarrhea or muscle/bone pains are relatively frequent in patients treated with AMD3100 (Table 4): however, in most cases these adverse effects are mild and transient (156).
Classical Anti-Angiogenic Drugs: The VEGF Antagonists
Combining angiogenesis antagonists with standard anti-tumor therapies has ameliorated the prognosis of a variety of human tumors (70).
In particular, inhibitors of the VEGF pathway have provided survival benefits to cancer patients (114).
At the present time, the VEGF antagonists which have been approved for clinical use include: (a) BEVACIZUMAB, a recombinant humanized monoclonal antibody which binds to VEGF-A and prevents its interaction with the type 2 VEGF receptor (166); (b) RAMUCIRUMAB, a blocking antibody directed against type-2 VEGF receptor that impedes its binding by VEGF (167); (c) AFLIBERCEPT, a human recombinant fusion protein that acts as a soluble decoy receptor sequestering various members of the VEGF family (168); (d) the inhibitors of receptor kinase activity, small molecules which pass through the cell membrane and prevent ATP binding to type-2 VEGF receptor (169) (Table 5).
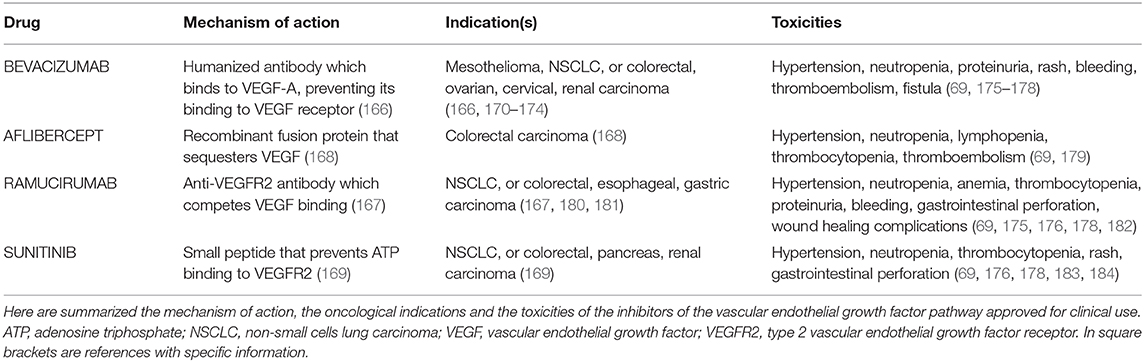
Table 5. Indications and toxicities of anti-VEGF compounds approved for clinical use.
Results from clinical trials combining VEGF pathway inhibitors and standard anti-tumor cytotoxic drugs indicate that BEVACIZUMAB augments overall survival and/or progression-free survival in patients affected by mesothelioma, non-small cell lung cancer or colon, renal, ovarian or uterine cervical carcinoma (166, 170–174), while RAMUCIRUMAB provides survival benefits in individuals with gastric cancer, non-small cell lung cancer or colon carcinoma (167, 180, 181) (Table 5). For their part, either AFLIBERCEPT or the receptor kinase inhibitors SUNITINIB and PAZOPANIB are effective against colorectal carcinoma (168, 169) (Table 5).
However, long-term use of VEGF antagonists can lead to side effects including bone marrow toxicity (this resulting in neutropenia and, eventually, lymphopenia, thrombocythemia and/or anemia), hypertension, proteinuria, liver malfunction, diarrhea, gastrointestinal perforations, reduced wound healing, disturbances in blood clotting, skin rash or discoloration, hyponatremia and/or hyperglycemia (69, 175–179, 182–184) (Table 5). Moreover, VEGF inhibitors trigger hypoxia, which exacerbates tumor aggressiveness (70). In addition, treated patients can develop drug resistance over time: this occurs via the up-regulation of the angiogenic basic fibroblast growth factor, angiopoietin-2 or CXCL12 (68).
Altogether, these clinical findings are stimulating the continual search for other antagonists of angiogenesis.
Conclusions and Perspectives
Cancer growth and dissemination are favored and sustained by the formation of new blood vessels within the tumor area (69, 70, 114). Consistently, compounds counteracting the development of tumor vasculature can limit or slow cancer clinical progression (69, 70, 114).
In this regard, antagonists of the highly angiogenic VEGF have been found effective against different types of human tumors (166–174, 180, 181) (Table 5).
However, the finding that patients treated with these drugs can undergo severe adverse effects (69, 70, 175–179, 182–184) (Table 5), and/or develop drug resistance (68), is prompting the identification of anti-angiogenesis drugs alternative to VEGF antagonists.
In this context, it has to reminded that clinical trials assessing the anti-tumor efficacy of synthetic MMP inhibitors impairing tumor angiogenesis in pre-clinical models have failed: this has been due to the poor solubility, lack of specificity and/or inefficacy of the drugs (123).
In view of chemokine role in inflammation-driven, pathological angiogenesis, antagonists of the chemokine receptors are currently being evaluated for their efficacy in countering tumor growth and metastases (69). Indeed, drugs targeting single chemokine receptors have been found effective against hematological malignancies (9, 153, 154). Moreover, CXCR4 antagonists have been shown to reduce tumor angiogenesis in animal models of human tumors (68). However, one should consider that a given chemokine can bind to different receptors, which are all capable of triggering the AKT-MMP pathway starting angiogenesis (136). In this regard, HIV-protease inhibitors effectively inhibit the AKT-MMP as well as other pathways which lead to both new blood vessel formation (Figure 1, Table 2) and cancer cell survival, growth or locomotion (7, 8).
In contrast, based on the reviewed literature, the anti-angiogenic actions of HIV-reverse transcriptase inhibitors appear limited: in fact, these drugs hamper some pro-angiogenic pathways, while favoring others (Figure 1, Table 3).
Among HIV-protease inhibitors, ritonavir and nelfinavir have proven to be particularly effective in inhibiting tumor-associated new blood vessel formation (Figure 1, Table 2). Repositioning of these anti-HIV drugs in cancer therapy has been feasible, as they are employed since many years, and their pharmacokinetic and tissue distribution are well known (4). Actually, clinical trials combining ritonavir or nelfinavir with standard anti-cancer therapeutics have given good results (13–15). However, as for other first-generation HIV-protease inhibitors, either nelfinavir or ritonavir increases lipid and glucose plasma levels in treated patients (4, 142, 143). Though novel HIV-protease inhibitors such as darunavir and atazanavir do not affect lipemia or glycemia (4), information on their anti-angiogenic activities is narrow (Table 2).
Therefore, added work should dissect darunavir or atazanavir impact on angiogenesis, and then design and test atazanavir, darunavir, ritonavir or nelfinavir analogs endowed with selective anti-angiogenic effects. To this end, further molecular modeling approaches and protein-ligand studies are needed in order to identify more precisely the targets of HIV-protease inhibitors.
Finally, given that inflammation plays a major role in new blood vessel formation (69, 70), additional clinical investigations should evaluate whether the anti-angiogenic effect of anti-HIV drugs could be potentiated by anti-inflammatory drugs, including cyclooxygenase 2 inhibitors which have been shown to promote tumor vessel normalization in pre-clinical models (185).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This study was supported by the Italian Ministry of Health (grant no. OR/0906).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank B. Ensoli, C. Sgadari, E. Toschi, P. Monini, A. Iovane, and I. Bacigalupo (National Institute of Health, Rome, Italy) for the work done together in order to assess HIV-protease inhibitor impact on angiogenesis.
Abbreviations
AIDS, Acquired Immune Deficiency Syndrome; cART, combined Anti-Retroviral Therapy; CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor 4; eNOS, endothelial nitric oxide synthase; HIF, hypoxia inducible transcription factor; HIV, human immunodeficiency virus; MAPK/ERK, mitogen-activated protein kinases/extracellular-signal-regulated kinases; MMP, matrix metalloproteinase; NF-kB, Nuclear Factor-kappa B; PI3K/AKT, phosphoinositide3-kinase/protein kinases B; TIMP, tissue inhibitor of matrix metalloproteinase; VEGF, vascular endothelial growth factor.
References
1. Casadellà M, Cozzi-Lepri A, Phillips A, Noguera-Julian M, Bickel M, Sedlacek D, et al. Plasma HIV-1 tropism and the risk of short-term clinical progression to AIDS or death. PLoS ONE. (2017) 12:e0166613. doi: 10.1371/journal.pone.0166613
2. Li G, De Clercq E. HIV Genome-Wide Protein Associations: a Review of 30 Years of Research. Microbiol Mol Biol Rev. (2016) 80:679–731. doi: 10.1128/MMBR.00065-15
3. Johnson SC. Antiretroviral therapy for HIV Infection: when to initiate therapy, which regimen to use, and how to monitor patients on therapy. Top Antivir Med. (2017) 23:161–7.
4. Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS. (2015) 7:95–104. doi: 10.2147/HIV.S79956
5. De Clercq E. Mozobil (Plerixafor, AMD3100), 10 years after its approval by the US food and drug administration. Antivir Chem Chemother. (2019) 27:2040206619829382. doi: 10.1177/2040206619829382
6. Everson F, Genis A, Ogundipe T, De Boever P, Goswami N, Lochner A, et al. Treatment with a fixed dose combination antiretroviral therapy drug containing tenofovir, emtricitabine and efavirenz is associated with cardioprotection in high calorie diet-induced obese rats. PLoS ONE. (2018) 13:e0208537. doi: 10.1371/journal.pone.0208537
7. Monini P, Sgadari C, Toschi E, Barillari G Ensoli B. Antitumour effects of antiretroviral therapy. Nature Rev Cancer. (2004) 4:861–75. doi: 10.1038/nrc1479
8. Barillari G, Monini P, Sgadari, Ensoli B. The impact of human papilloma viruses, matrix metalloproteinases and HIV protease inhibitors on the onset and progression of uterine cervix epithelial tumors: a review of preclinical and clinical studies. Int J Mol Sci. (2018) 19:1418–42. doi: 10.3390/ijms19051418
9. Bandera A, Verga L, Perseghin P, Incontri A, Pioltelli PE, Gori A. Use of CXCR4-antagonist for haematopoietic stem cell mobilization in HIV-infected patients with haematological malignancies. AIDS. (2013) 27:1037–9. doi: 10.1097/QAD.0b013e32835ecbcd
10. Monini P, Sgadari C, Grosso MG, Bellino S, Di Biagio A, Toschi E, et al. Clinical course of classic Kaposi's sarcoma in HIV-negative patients treated with the HIV protease inhibitor indinavir. AIDS. (2009) 23:534–8. doi: 10.1097/QAD.0b013e3283262a8d
11. Hampson L, Maranga IO, Masinde MS, Oliver AW, Batman G, He X, et al. A single-arm, proof-of-concept trial of lopimune (lopinavir/ritonavir) as a treatment for HPV-related pre-invasive cervical disease. PLoS ONE. (2016) 11:e0147917, doi: 10.1371/journal.pone.0147917
12. Hill EJ, Roberts C, Franklin JM, Enescu M, West N, MacGregor TP, et al. Clinical trial of oral nelfinavir before and during radiation therapy for advanced rectal cancer. Clin Cancer Res. (2016) 22:1922–31. doi: 10.1158/1078-0432.CCR-15-1489
13. Wilson JM, Fokas E, Dutton SJ, Patel N, Hawkins MA, Eccles C, et al. ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother Oncol. (2016) 119:306–11. doi: 10.1016/j.radonc.2016.03.021
14. Driessen C, Müller R, Novak U, Cantoni N, Betticher D, Mach N, et al. Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma. Blood. (2018) 132:2097–100. doi: 10.1182/blood-2018-05-851170
15. de Weger VA, Stuurman FE, Hendrikx JJMA, Moes JJ, Sawicki E, Huitema ADR, et al. A dose-escalation study of bi-daily once weekly oral docetaxel either as ModraDoc001 or ModraDoc006 combined with ritonavir. Eur J Cancer. (2017) 86:217–25. doi: 10.1016/j.ejca.2017.09.010
16. Houédé N, Pulido M, Mourey L, Joly F, Ferrero JM, Bellera C, et al. A phase II trial evaluating the efficacy and safety of efavirenz in metastatic castration-resistant prostate cancer. Oncologist. (2014) 19:1227–8. doi: 10.1634/theoncologist.2014-0345
17. Katsuya H, Ishitsuka K. Treatment advances and prognosis for patients with adult T-cell leukemia-lymphoma. J Clin Exp Hematop. (2017) 57:87–97. doi: 10.3960/jslrt.17008
18. Nasr R, Marçais A, Hermine O, Bazarbachi A. Overview of targeted therapies for adult T-cell leukemia/lymphoma. Methods Mol Biol. (2017) 1582:197–216. doi: 10.1007/978-1-4939-6872-5_15
19. Bayraktar UD, Diaz LA, Ashlock B, Toomey N, Cabral L, Bayraktar S, et al. Zidovudine-based lytic-inducing chemotherapy for Epstein-Barr virus-related lymphomas. Leuk Lymphoma. (2014) 55:786–94. doi: 10.3109/10428194.2013.818142
20. Kchour G, Tarhini M, Kooshyar MM, El Hajj H, Wattel E, Mahmoudi M, et al. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood. (2009) 113:6528–32. doi: 10.1182/blood-2009-03-211821
21. Yoo J, Hann HW, Coben R, Conn M, DiMarino AJ. Update Treatment for HBV Infection and Persistent Risk for Hepatocellular Carcinoma: Prospect for an HBV Cure. Diseases. (2018) 6:E27. doi: 10.3390/diseases6020027
22. Kim WR, Loomba R, Berg T, Aguilar Schall RE, Yee LJ, Dinh PV, et al. Impact of long-term tenofovir disoproxil fumarate on incidence of hepatocellular carcinoma in patients with chronic hepatitis B. Cancer. (2015) 121:3631–8. doi: 10.1002/cncr.29537
23. Okubo K, Isono M, Asano T, Sato A. Lopinavir-ritonavir combination induces endoplasmic reticulum stress and kills urological cancer cells. Anticancer Res. (2019) 39:5891–901. doi: 10.21873/anticanres.13793
24. Jiang W, Li X, Li T, Wang H, Shi W, Qi P, et al. Repositioning of amprenavir as a novel extracellular signal-regulated kinase-2 inhibitor and apoptosis inducer in MCF-7 human breast cancer. Int J Oncol. (2017) 50:823–34. doi: 10.3892/ijo.2017.3860
25. Kariya R, Taura M, Suzu S, Kai H, Katano H, Okada S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-κB pathway. Cancer Lett. (2014) 342:52–9. doi: 10.1016/j.canlet.2013.08.045
26. McLean K, VanDeVen NA, Sorenson DR, Daudi S, Liu JR. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol Oncol. (2009) 112:623–30. doi: 10.1016/j.ygyno.2008.11.028
27. Toschi E, Sgadari C, Malavasi L, Bacigalupo I, Chiozzini C, Carlei D, et al. Human immunodeficiency virus protease inhibitors reduce the growth of human tumors via proteasome-independent block of angiogenesis and matrix metalloproteinases. Int J Cancer. (2011) 128:82–93. doi: 10.1002/ijc.25550
28. Barillari G, Iovane A, Bacigalupo I, Palladino C, Bellino S, Leone P, et al. Ritonavir or saquinavir impairs the invasion of cervical intraepithelial neoplasia cells via a reduction of MMP expression and activity. AIDS. (2012) 26:909–19. doi: 10.1097/QAD.0b013e328351f7a
29. Bacigalupo I, Palladino C, Leone P, Toschi E, Sgadari C, Ensoli B, et al. Inhibition of MMP-9 expression by ritonavir or saquinavir is associated with inactivation of the AKT/Fra-1 pathway in cervical intraepithelial neoplasia cells. Oncol Lett. (2017) 13:2903–8. doi: 10.3892/ol.2017.583
30. Landriscina M, Fabiano A, Altamura S, Bagalà C, Piscazzi A, Cassano A, et al. Reverse transcriptase inhibitors down-regulate cell proliferation in vitro and in vivo and restore thyrotropin signaling and iodine uptake in human thyroid anaplastic carcinoma. J Clin Endocrinol Metab. (2005) 90:5663–71. doi: 10.1210/jc.2005-0367
31. Landriscina M, Altamura SA, Roca L, Gigante M, Piscazzi A, Cavalcanti E, et al. Reverse transcriptase inhibitors induce cell differentiation and enhance the immunogenic phenotype in human renal clear-cell carcinoma. Int J Cancer. (2008) 122:2842–50. doi: 10.1002/ijc.23197
32. Brüning A, Burger P, Gingelmaier A, Mylonas I. The HIV reverse transcriptase inhibitor tenofovir induces cell cycle arrest in human cancer cells. Invest New Drugs. (2012) 30:1389–95. doi: 10.1007/s10637-011-9704-7
33. Brüning A, Jückstock J, Kost B, Tsikouras P, Weissenbacher T, Mahner S, et al. Induction of DNA damage and apoptosis in human leukemia cells by efavirenz. Oncol Rep. (2017) 37:617–21. doi: 10.3892/or.2016.5243
34. Hecht M, Erber S, Harrer T, Klinker H, Roth T, Parsch H, et al. Efavirenz has the highest anti-proliferative effect of non-nucleoside reverse transcriptase inhibitors against pancreatic cancer cells. PLoS ONE. (2015) 10:e0130277. doi: 10.1371/journal.pone.0130277
35. Shang H, Zhao J, Yao J, Wang H, Wang S, Dong J, et al. Nevirapine inhibits migration and invasion in dedifferentiated thyroid cancer cells. Thorac Cancer. (2019) 10:2243–52. doi: 10.1111/1759-7714.13211
36. Benedicto A, Romayor I, Arteta B. CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol Rep. (2018) 39:2022–30. doi: 10.3892/or.2018.6254
37. Jung YH, Lee DY, Cha W, Kim BH, Sung MW, Kim KH, et al. Antitumor effect of CXCR4 antagonist AMD3100 on the tumorigenic cell line of BHP10-3 papillary thyroid cancer cells. Head Neck. (2016) 38:1479–86. doi: 10.1002/hed.24461
38. Werner TA, Forster CM, Dizdar L, Verde PE, Raba K, Schott M, et al. CXCR4/CXCR7/CXCL12-axis in follicular thyroid carcinoma. J Cancer. (2018) 9:929–40. doi: 10.7150/jca.23042
39. Xi Y, Qi Z, Ma J, Chen Y. PTEN loss activates a functional AKT/CXCR4 signaling axis to potentiate tumor growth and lung metastasis in human osteosarcoma cells. Clin Exp Metastasis. (2019) 37:173–85. doi: 10.1007/s10585-019-09998-7
40. Perna A, Lucariello A, Sellitto C, Agliata I, Carleo MA, Sangiovanni V, et al. Different cell cycle modulation in SKOV-3 ovarian cancer cell line by anti-HIV drugs. Oncol Res. (2017) 25:1617–24. doi: 10.3727/096504017X14905635363102
41. Liu W, Meng Q, Sun Y, Wang C, Huo X, Liu Z, et al. Targeting P-glycoprotein: nelfinavir reverses adriamycin resistance in K562/ADR Cells. Cell Physiol Biochem. (2018) 51:1616–31. doi: 10.1159/000495650
42. Kim JY, Park YJ, Lee BM, Yoon S. Co-treatment with HIV protease inhibitor nelfinavir greatly increases late-phase apoptosis of drug-resistant KBV20C cancer cells independently of P-glycoprotein inhibition. Anticancer Res. (2019) 39:3757–65. doi: 10.21873/anticanres.13524
43. Chen X, Wang C, Guan S, Liu Y, Han L, Cheng Y. Zidovudine, abacavir and lamivudine increase the radiosensitivity of human esophageal squamous cancer cell lines. Oncol Rep. (2016) 36:239–46. doi: 10.3892/or.2016.4819
44. Mayr C, Neureiter D, Pichler M, Berr F, Wagner A, Kiesslich T, et al. Cytotoxic effects of chemokine receptor 4 inhibition by AMD3100 in biliary tract cancer cells: Potential drug synergism with gemcitabine. Mol Med Rep. (2015) 12:2247–52. doi: 10.3892/mmr.2015.3589
45. Hecht M, Harrer T, Körber V, Sarpong EO, Moser F, Fiebig N, et al. Cytotoxic effect of Efavirenz in BxPC-3 pancreatic cancer cells is based on oxidative stress and is synergistic with ionizing radiation. Oncol Lett. (2018) 15:1728–36. doi: 10.3892/ol.2017.7523
46. Zhou KX, Xie LH, Peng X, Guo QM, Wu QY, Wang WH, et al. CXCR4 antagonist AMD3100 enhances the response of MDA-MB-231 triple-negative breast cancer cells to ionizing radiation. Cancer Lett. (2018) 418:196–203. doi: 10.1016/j.canlet.2018.01.009
47. Reeves PM, Abbaslou MA, Kools FRW, Poznansky MC. CXCR4 blockade with AMD3100 enhances Taxol chemotherapy to limit ovarian cancer cell growth. Anticancer Drugs. (2017) 28:935–42. doi: 10.1097/CAD.0000000000000518
48. Lecavalier-Barsoum M, Chaudary N, Han K, Koritzinsky M, Hill R, Milosevic M. Targeting the CXCL12/CXCR4 pathway and myeloid cells to improve radiation treatment of locally advanced cervical cancer. Int J Cancer. (2018) 143:1017–28. doi: 10.1002/ijc.31297
49. Moraes Filho AV, Carvalho CJ, Carneiro CC, Vale CR, Lima DC, Carvalho WF, et al. Genotoxic and cytotoxic effects of antiretroviral combinations in mice bone marrow. PLoS ONE. (2016) 11:e0165706. doi: 10.1371/journal.pone.0165706
50. Cozzi-Lepri A, Zangerle R, Machala L, Zilmer K, Ristola M, Pradier C, et al. Incidence of cancer and overall risk of mortality in individuals treated with raltegravir-based and non-raltegravir-based combination antiretroviral therapy regimens. HIV Med. (2018) 19:102–17. doi: 10.1111/hiv.12557
51. Cruz da Silva E, Dontenwill M, Choulier L, Lehmann M. Role of integrins in resistance to therapies targeting growth factor receptors in cancer. Cancers. (2019) 11:E692. doi: 10.3390/cancers11050692
52. Sgadari C, Barillari G, Toschi E, Carlei D, Bacigalupo I, Baccarini S, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi's sarcoma. Nature Med. (2002) 8:225–32. doi: 10.1038/nm0302-225
53. Maggiorella L, Wen B, Frascogna V, Opolon P, Bourhis J, Deutsch E. Combined radiation sensitizing and anti-angiogenic effects of ionizing radiation and the protease inhibitor ritonavir in a head and neck carcinoma model. Anticancer Res. (2005) 25:4357–62.
54. Pore N, Gupta AK, Cerniglia GJ, Maity A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia. (2006) 8:889–95. doi: 10.1593/neo.06535
55. Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, et al. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. (2006) 66:9252–9. doi: 10.1158/0008-5472.CAN-06-1239
56. Cuneo KC, Tu T, Geng L, Fu A, Hallahan DE, Willey CD. HIV protease inhibitors enhance the efficacy of irradiation. Cancer Res. (2007) 67:4886–93. doi: 10.1158/0008-5472.CAN-06-3684
57. Barillari G, Iovane A, Bacigalupo I, Labbaye C, Chiozzini C, Sernicola L, et al. The HIV protease inhibitor indinavir down-regulates the expression of the pro-angiogenic MT1-MMP by human endothelial cells. Angiogenesis. (2014) 7:831–8. doi: 10.1007/s10456-014-9430-9
58. Auclair M, Afonso P, Capel E, Caron-Debarle M, Capeau J. Impact of darunavir, atazanavir and lopinavir boosted with ritonavir on cultured human endothelial cells: beneficial effect of pravastatin. Antivir Ther. (2014) 19:773–82. doi: 10.3851/IMP2752
59. Vadlapatla RK, Vadlapudi AD, Pal D, Mukherji M, Mitra AK. Ritonavir inhibits HIF-1α-mediated VEGF expression in retinal pigment epithelial cells in vitro. Eye. (2014) 28:93–101. doi: 10.1038/eye.2013.240
60. Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y, et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science. (2014) 346:1000–13. doi: 10.1126/science.1261754
61. Weiß M, Kost B, Renner-Müller I, Wolf E, Mylonas I, Brüning A. Efavirenz Causes Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy in Endothelial Cells. Cardiovasc Toxicol. (2016) 16:90–9. doi: 10.1007/s12012-015-9314-2
62. Song L, Ding S, Ge Z, Zhu X, Qiu C, Wang Y, et al. Nucleoside/nucleotide reverse transcriptase inhibitors attenuate angiogenesis and lymphangiogenesis by impairing receptor tyrosine kinases signalling in endothelial cells. Br J Pharmacol. (2018) 175:1241–59. doi: 10.1111/bph.14036
63. Chen YF, Stampley JE, Irving BA, Dugas TR. Chronic nucleoside reverse transcriptase inhibitors disrupt mitochondrial homeostasis and promote premature endothelial senescence. Toxicol Sci. (2019) 172:445–56. doi: 10.1093/toxsci/kfz203
64. Yoshida S, Kawai H, Eguchi T, Sukegawa S, Oo MW, Anqi C, et al. Tumor angiogenic inhibition triggered necrosis (taitn) in oral cancer. Cells. (2019) 8:E761. doi: 10.3390/cells8070761
65. He W, Yang T, Gong XH, Qin RZ, Zhang XD, Liu WD. Targeting CXC motif chemokine receptor 4 inhibits the proliferation, migration and angiogenesis of lung cancer cells. Oncol Lett. (2018) 16:3976–82. doi: 10.3892/ol.2018.9076
66. Sun X, Charbonneau C, Wei L, Yang W, Chen Q, Terek RM. CXCR4-targeted therapy inhibits VEGF expression and chondrosarcoma angiogenesis and metastasis. Mol Cancer Ther. (2013) 12:1163–70. doi: 10.1158/1535-7163.MCT-12-1092
67. Newey SE, Tsaknakis G, Khoo CP, Athanassopoulos T, Camicia R, Zhang Y, et al. The hematopoietic chemokine CXCL12 promotes integration of human endothelial colony forming cell-derived cells into immature vessel networks. Stem Cells Dev. (2014) 23:2730–43. doi: 10.1089/scd.2014.0005
68. Arbab AS. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol Histopathol. (2012) 27:549–57. doi: 10.14670/hh-27.549
69. Ridiandries A, Tan JT, Bursill CA. The role of CC-chemokines in the regulation of angiogenesis. Int J Mol Sci. (2016) 17:E1856. doi: 10.3390/ijms17111856
70. Petrillo S, Tolosano E, Munaron L, Genova T. Targeting metabolism to counteract tumor angiogenesis: a review of patent literature. Rec Pat Anti-cancer Drug Disc. (2018) 13:422–7. doi: 10.2174/1574892813666180528105023
71. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. (2019) 10:85. doi: 10.3389/fimmu.2019.00085
72. Equils O, Shapiro A, Madak Z, Liu C, Lu D. Human immunodeficiency virus type 1 protease inhibitors block toll-like receptor 2 (TLR2)- and TLR4-Induced NF-kappa B activation. Antimicrob Agents Chemother. (2004) 48:3905–11. doi: 10.1128/AAC.48.10.3905-3911.2004
73. Ji Y, Zhang G, Zhu H, Li D, Jiang W. Indinavir plus methylprednisolone ameliorates experimental acute lung injury in vitro and in vivo. Shock. (2018) 49:196–204. doi: 10.1097/SHK.0000000000000911
74. Hong-Brown LQ, Brown CR, Lang CH. Indinavir impairs protein synthesis and phosphorylations of MAPKs in mouse C2C12 myocytes. Am J Physiol Cell Physiol. (2004) 287:1482–92. doi: 10.1152/ajpcell.00038.2004
75. Malizia AP, Cotter E, Chew N, Powderly WG, Doran PP. HIV protease inhibitors selectively induce gene expression alterations associated with reduced calcium deposition in primary human osteoblasts. AIDS Res Hum Retroviruses. (2007) 23:243–50. doi: 10.1089/aid.2006.0084
76. Zhang G, Zhang X, Huang H, Ji Y, Li D, Jiang W. Saquinavir plus methylprednisolone ameliorates experimental acute lung injury. Braz J Biol Med Res. (2018) 51:e7579. doi: 10.1590/1414-431X20187579
77. Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol Cancer Ther. (2004) 3:473–9.
78. Plastaras JP, Vapiwala N, Ahmed MS, Gudonis D, Cerniglia GJ, Feldman MD, et al. Validation and toxicity of PI3K/AKT pathway inhibition by HIV protease inhibitors in humans. Cancer Biol Ther. (2008) 7:628–35. doi: 10.4161/cbt.7.5.5728
79. Chai H, Yang H, Yan S, Li M, Lin PH, Lumsden AB, et al. Effects of 5 HIV protease inhibitors on vasomotor function and superoxide anion production in porcine coronary arteries. J Acquir Immune Defic Syndr. (2005) 40:12–19. doi: 10.1097/01.qai.0000172368.05327.7b
80. De Barros S, Zakaroff-Girard A, Lafontan M, Galitzky J, Bourlier V. Inhibition of human preadipocyte proteasomal activity by HIV protease inhibitors or specific inhibitor lactacystin leads to a defect in adipogenesis, which involves matrix metalloproteinase-9. J Pharmacol Exp Ther. (2007) 320:291–9. doi: 10.1124/jpet.106.111849
81. Srirangam A, Mitra R, Wang M, Gorski JC, Badve S, Baldridge L, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. (2006) 12:1883–96. doi: 10.1158/1078-0432.CCR-05-1167
82. Batchu R, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, et al. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals. (2014) 7:46–57. doi: 10.3390/ph7010046
83. Pasquereau S, Kumar A, Abbas W, Herbein G. Counteracting AKT activation by HIV protease inhibitors in monocytes/macrophages. Viruses. (2018) 10:E190. doi: 10.3390/v10040190
84. Lü JM, Jiang J, Jamaluddin MS, Liang Z, Yao Q, Chen C. Ginsenoside Rb1 blocks ritonavir-induced oxidative Stress and eNOS down-regulation through activation of estrogen receptor-beta and up-regulation of SOD in human endothelial cells. Int J Mol Sci. (2019) 20:E294. doi: 10.3390/ijms20020294
85. Isono M, Sato A, Asano T, Okubo K, Asano T. Delanzomib Interacts with Ritonavir Synergistically to Cause Endoplasmic Reticulum Stress in Renal Cancer Cells. Anticancer Res. (2018) 38:3493–500. doi: 10.21873/anticanres.12620
86. Sato A, Asano T, Okubo K, Isono M, Asano T. Nelfinavir and Ritonavir Kill Bladder Cancer Cells Synergistically by Inducing Endoplasmic Reticulum Stress. Oncol Res. (2018) 26:323–32. doi: 10.3727/096504017X14957929842972
87. Hresko RC, Hruz PW. HIV Protease Inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS ONE. (2011) 6:e25237. doi: 10.1371/journal.pone.0025237
88. Richmond SR, Carper MJ, Lei X, Zhang S, Yarasheski KE, Ramanadham S. HIV-protease inhibitors suppress skeletal muscle fatty acid oxidation by reducing CD36 and CPT1 fatty acid transporters. Biochim Biophys Acta. (2010) 1801:559–66. doi: 10.1016/j.bbalip.2010.01.007
89. Wallet MA, Reist CM, Williams JC, Appelberg S, Guiulfo GL, Gardner B, et al. The HIV-1 protease inhibitor nelfinavir activates PP2 and inhibits MAPK signaling in macrophages: a pathway to reduce inflammation. J Leukoc Biol. (2012) 92:795–805. doi: 10.1189/jlb.0911447
90. Jensen K, Bikas A, Patel A, Kushchayeva Y, Costello J, McDaniel D, et al. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr Relat Cancer. (2017) 24:147–56. doi: 10.1530/ERC-16-0568
91. Zeng J, See AP, Aziz K, Thiyagarajan S, Salih T, Gajula RP, et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol Ther. (2011) 12:657–63. doi: 10.4161/cbt.12.7.17172
92. Gupta AK, Lee JH, Wilke WW, Quon H, Smith G, Maity A, et al. Radiation response in two HPV-infected head-and-neck cancer cell lines in comparison to a non-HPV-infected cell line and relationship to signaling through AKT. Int J Radiat Oncol Biol Phys. (2009) 74:928–33. doi: 10.1016/j.ijrobp.2009.03.004
93. Jiang Z, Pore N, Cerniglia GJ, Mick R, Georgescu MM, Bernhard EJ, et al. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. (2007) 67:4467–73. doi: 10.1158/0008-5472.CAN-06-3398
94. Ben-Romano R, Rudich A, Tirosh A, Potashnik R, Sasaoka T, Riesenberg K, et al. Nelfinavir-induced insulin resistance is associated with impaired plasma membrane recruitment of the PI3-kinase effectors AKT/PKB and PKC-zeta. Diabetologia. (2004) 47:1107–17. doi: 10.1007/s00125-004-1408-5
95. Kachko I, Maissel A, Mazor L, Ben-Romano R, Watson RT, Hou JC, et al. Post-receptoral adipocyte insulin resistance induced by nelfinavir is caused by insensitivity of PKB/AKT to phosphatidylinositol-3,4,5-trisphosphate. Endocrinology. (2009) 150:2618–26. doi: 10.1210/en.2008-1205
96. Ravaud C, Paré M, Yao X, Azoulay S, Mazure NM, Dani C, et al. Resveratrol and HIV-protease inhibitors control UCP1 expression through opposite effects on p38 MAPK phosphorylation in human adipocytes. J Cell Physiol. (2019) 235:1184–96. doi: 10.1002/jcp.29032
97. Kennedy A, Hennessy M, Bergin C, Mulcahy F, Hopkins S, Spiers JP. Ribavirin and interferon alter MMP-9 abundance in vitro and in HIV-HCV-coinfected patients. Antivir Ther. (2011) 16:1237–47 doi: 10.3851/IMP1867
98. Song S, Ji Y, Zhang G, Zhang X, Li B, Li D, et al. Protective effect of atazanavir sulphate against pulmonary fibrosis in vivo and in vitro. Basic Clin Pharmacol Toxicol. (2018) 122:199–207. doi: 10.1111/bcpt.12871
99. Esposito V, Verdina A, Manente L, Spugnini EP, Viglietti R, Parrella R, et al. Amprenavir inhibits the migration in human hepatocarcinoma cell and the growth of xenografts. J Cell Physiol. (2013) 228:640–5. doi: 10.1002/jcp.24173
100. Latronico T, Pati I, Ciavarella R, Fasano A, Mengoni F, Lichtner M, et al. In vitro effect of antiretroviral drugs on cultured primary astrocytes: analysis of neurotoxicity and matrix metalloproteinase inhibition. J Neurochem. (2018) 144:271–84. doi: 10.1111/jnc.14269
101. Melchjorsen J, Risør MW, Søgaard OS, O'Loughlin KL, Chow S, Paludan SR, et al. Tenofovir selectively regulates production of inflammatory cytokines and shifts the IL-12/IL-10 balance in human primary cells. J Acquir Immune Defic Syndr. (2011) 57:265–75. doi: 10.1097/QAI.0b013e3182185276
102. Jamaluddin MS, Lin PH, Yao Q, Chen C. Non-nucleoside reverse transcriptase inhibitor efavirenz increases monolayer permeability of human coronary artery endothelial cells. Atherosclerosis. (2010) 208:104–11. doi: 10.1016/j.atherosclerosis.2009.07.029
103. Wang X, Chai H, Lin PH, Yao Q, Chen C. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti-human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am J Pathol. (2009) 174:771–81. doi: 10.2353/ajpath.2009.080157
104. Liuzzi GM, Mastroianni CM, Latronico T, Mengoni F, Fasano A, Lichtner M, et al. Anti-HIV drugs decrease the expression of matrix metalloproteinases in astrocytes and microglia. Brain. (2004) 127:398–407. doi: 10.1093/brain/awh049
105. Jörg A, Grubert T, Grimm T, Guenzi E, Naschberger E, Samson E, et al. Maternal HIV type 1 infection suppresses MMP-1 expression in endothelial cells of uninfected newborns: non-viral vertical transmission of HIV type 1-related effects. AIDS Res Hum Retroviruses. (2005) 21:940–4. doi: 10.1089/aid.2005.21.940
106. Pan-Zhou XR, Cui L, Zhou XJ, Sommadossi JP, Darley-Usmar VM. Differential effects of antiretroviral nucleoside analogs on mitochondrial function in HepG2 cells. Antimicrob Agents Chemother. (2000) 44:496–503. doi: 10.1128/AAC.44.3.496-503.2000
107. Maisonneuve C, Igoudjil A, Begriche K, Lettéron P, Guimont MC, Bastin J, et al. Effects of zidovudine, stavudine and beta-aminoisobutyric acid on lipid homeostasis in mice: possible role in human fat wasting. Antivir Ther. (2004) 9:801–10.
108. Lebensztejn DM, Skiba E, Sobaniec-Łotowska ME, Kaczmarski M. Matrix metalloproteinases and their tissue inhibitors in children with chronic hepatitis B treated with lamivudine. Adv Med Sci. (2007) 52:114–9.
109. Hecht M, Harrer T, Büttner M, Schwegler M, Erber S, Fietkau R, et al. Cytotoxic effect of efavirenz is selective against cancer cells and associated with the cannabinoid system. AIDS. (2013) 27:2031–40. doi: 10.1097/QAD.0b013e3283625444
110. Arend C, Rother A, Stolte S, Dringen R. Consequences of a chronic exposure of cultured brain astrocytes to the anti-retroviral drug efavirenz and its primary metabolite 8-hydroxy efavirenz. Neurochem Res. (2016) 41:3278–88. doi: 10.1007/s11064-016-2059-x
111. Petit I, Jin D, Rafii S. The SDF-1–CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis Trends Immunol. (2007) 28:299–307. doi: 10.1016/j.it.2007.05.007
112. Zhang Z, Yao L, Yang J, Zhenkang W, Du G. PI3K/AKT and HIF-1 signaling pathway in hypoxia-ischemia. Mol Med Rep. (2018) 18:3547–54. doi: 10.3892/mmr.2018.9375
113. Esposito V, Palescandolo E, Spugnini EP, Montesarchio V, De Luca A, Cardillo I, et al. Evaluation of antitumoral properties of the protease inhibitor indinavir in a murine model of hepatocarcinoma. Clin Cancer Res. (2006) 12:2634–9. doi: 10.1158/1078-0432.CCR-05-2188
114. Loizzi V, Del Vecchio V, Gargano G, De Liso M, Kardashi A, Naglieri E, et al. Biological pathways involved in tumor angiogenesis and bevacizumab based anti-angiogenic therapy with special references to ovarian cancer. Int J Mol Sci. (2017) 18:E1967. doi: 10.3390/ijms18091967
115. Liang Z, Chi YJ, Lin GQ, Luo SH, Jiang QY, Chen YK. MiRNA-26a promotes angiogenesis in a rat model of cerebral infarction via PI3K/AKT and MAPK/ERK pathway. Eur Rev Med Pharmacol Sci. (2018) 22:3485–92. doi: 10.26355/eurrev_201806_15175
116. Kumar S, Bryant CS, Chamala S, Qazi A, Seward S, Pal J, et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. (2009) 8:26. doi: 10.1186/1476-4598-8-26
117. Nolte LA, Yarasheski KE, Kawanaka K, Fisher J, Le N, Holloszy JO. The HIV protease inhibitor indinavir decreases insulin- and contraction-stimulated glucose transport in skeletal muscle. Diabetes. (2001) 50:1397–401. doi: 10.2337/diabetes.50.6.1397
118. Xia C, Chen R, Chen J, Qi Q, Pan Y, Du L, et al. Combining metformin and nelfinavir exhibits synergistic effects against the growth of human cervical cancer cells and xenograft in nude mice. Sci Rep. (2017) 7:43373. doi: 10.1038/srep43373
119. Gary-Bobo G, Houssaini A, Amsellem V, Rideau D, Pacaud P, Perrin A, et al. Effects of HIV protease inhibitors on progression of monocrotaline- and hypoxia-induced pulmonary hypertension in rats. Circulation. (2010) 122:1937–47. doi: 10.1161/CIRCULATIONAHA.110.973750
120. Brüning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol. (2013) 7:1012–8. doi: 10.1016/j.molonc.2013.07.010
121. Johnson MD, O'Connell M, Pilcher W. Lopinavir inhibits meningioma cell proliferation by Akt independent mechanism. J Neuroncol. (2011) 101:441–8. doi: 10.1007/s11060-010-0281-y
122. Tian J, Liu R, Qu Q. Role of endoplasmic reticulum stress on cisplatin resistance in ovarian carcinoma. Oncol Lett. (2017) 13:1437–443. doi: 10.3892/ol.2017.5580
123. Liu J, Khalil RA. Matrix metalloproteinase inhibitors as investigational and therapeutic tools in unrestrained tissue remodeling and pathological disorders. Prog Mol Biol Transl Sci. (2017) 148:355–420. doi: 10.1016/bs.pmbts.2017.04.003
124. Latronico T, Liuzzi GM, Riccio P, Lichtner M, Mengoni F, D'Agostino C, et al. Antiretroviral therapy inhibits matrix metalloproteinase-9 from blood mononuclear cells of HIV-infected patients. AIDS. (2007) 21:677–84. doi: 10.1097/QAD.0b013e328018751d
125. Wang X, Khalil RA. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol. (2018) 81:241–330. doi: 10.1016/bs.apha.2017.08.002
126. Zhang L, Chen C, Duanmu J, Wu Y, Tao J, Yang A, et al. Cryptotanshinone inhibits the growth and invasion of colon cancer by suppressing inflammation and tumor angiogenesis through modulating MMP/TIMP system, PI3K/Akt/mTOR signaling and HIF-1α nuclear translocation. Int Immunopharmacol. (2018) 65:429–37. doi: 10.1016/j.intimp.2018.10.035
127. Kwak HJ, Park MJ, Cho H, Park CM, Moon SI, Lee HC, et al. Transforming growth factor-beta1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol Cancer Res. (2006) 4:209–20. doi: 10.1158/1541-7786.MCR-05-0140
128. Whelan KT, Lin CL, Cella M, McMichael AJ, Austyn JM, Rowland-Jones SL. The HIV protease inhibitor indinavir reduces immature dendritic cell trans-endothelial migration. Eur J Immunol. (2003) 33:2520–30. doi: 10.1002/eji.200323646
129. Sanchez CG, Molinski SV, Gongora R, Sosulski M, Fuselier T, MacKinnon SS, et al. The antiretroviral agent nelfinavir mesylate: a potential therapy for systemic sclerosis. Arthritis Rheumatol. (2018) 70:115–26. doi: 10.1002/art.40326
130. Hamming LC, Slotman BJ, Verheul HMW, Thijssen VL. The clinical application of angiostatic therapy in combination with radiotherapy: past, present, future. Angiogenesis. (2017) 20:217–32. doi: 10.1007/s10456-017-9546-9
131. Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. (2017) 20:409–26. doi: 10.1007/s10456-017-9562-9
132. Goedegebuure RSA, de Klerk LK, Bass AJ, Derks S, Thijssen VLJL. Combining radiotherapy with anti-angiogenic therapy and immunotherapy; a therapeutic triad for cancer? Front Immunol. (2019) 9:3107. doi: 10.3389/fimmu.2018.03107
133. Yang Y, Ikezoe T, Nishioka C, Bandobashi K, Takeuchi T, Adachi Y, et al. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced AKT signaling, apoptosis and docetaxel sensitization in NSCLC cell lines. Br J Cancer. (2006) 95:1653–62. doi: 10.1038/sj.bjc.6603435
134. Kim SH, Song Y, Seo HR. GSK-3β regulates the endothelial-to-mesenchymal transition via reciprocal crosstalk between NSCLC cells and HUVECs in multicellular tumor spheroid models. J Exp Clin Cancer Res. (2019) 38:46. doi: 10.1186/s13046-019-1050-1
135. Kappert K, Caglayan E, Bäumer AT, Südkamp M, Fätkenheuer G, Rosenkranz S. Ritonavir exhibits anti-atherogenic properties on vascular smooth muscle cells. AIDS. (2004) 18:403–11. doi: 10.1097/00002030-200402200-00005
136. Tamma R, Ribatti D. Bone niches, hematopoietic stem cells, and vessel formation. Int J Mol Sci. (2017) 18:E151. doi: 10.3390/ijms18010151
137. Arlier S, Murk W, Guzeloglu-Kayisli O, Semerci N, Larsen K, Tabak MS, et al. The extracellular signal-regulated kinase 1/2 triggers angiogenesis in human ectopic endometrial implants by inducing angioblast differentiation and proliferation. Am J Reprod Immunol. (2017) 78. doi: 10.1111/aji.12760
138. Song ZY, Wang F, Cui SX, Qu XJ. Knockdown of CXCR4 inhibits CXCL12-induced angiogenesis in HUVECs through down-regulation of the MAPK/ERK and PI3K/AKT and the Wnt/β-Catenin Pathways. Cancer Invest. (2018) 36:10–8. doi: 10.1080/07357907.2017.1422512
139. Aoki H, Yamashita M, Hashita T, Nakayama M, Yagi M, Iwao T, et al. Isolation of induced pluripotent stem cell-derived endothelial progenitor cells from sac-like structures. Biochem Biophys Res Commun. (2019) 515:672–8. doi: 10.1016/j.bbrc.2019.05.179
140. Conant MA, Opp KM, Poretz D, Mills RG. Reduction of Kaposi's sarcoma lesions following treatment of AIDS with ritonavir. AIDS. (1997) 11:1300–1. doi: 10.1097/00002030-199710001-00007
141. Lebbé C, Blum L, Pellet C, Blanchard G, Vérola O, Morel P, et al. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi's sarcoma. AIDS. (1998) 12:F45–9. doi: 10.1097/00002030-199807000-00002
142. Mulligan K, Grunfeld C, Tai VW, Algren H, Pang M, Chernoff DN, et al. Hyperlipidemia and insulin resistance are induced by protease inhibitors independent of changes in body composition in patients with HIV infection. J Acquir Immune Defic Syndr. (2000) 23:35–43. doi: 10.1097/00042560-200001010-00005
143. Anastos K, Lu D, Shi Q, Tien PC, Kaplan RC, Hessol NA, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr. (2007) 45:34–42. doi: 10.1097/QAI.0b013e318042d5fe
144. Ma Q, Vaida F, Wong J, Sanders CA, Kao YT, Croteau D, et al. Long-term efavirenz use is associated with worse neurocognitive functioning in HIV-infected patients. J Neurovirol. (2016) 22:170–8. doi: 10.1007/s13365-015-0382-7
145. Opravil M, Hirschel B, Bucher HC, Lüthy R. A randomized trial of interferon-alpha2a and zidovudine versus bleomycin and zidovudine for AIDS-related Kaposi's sarcoma. Swiss HIV cohort study. Int J STD AIDS. (1999) 10:369–75. doi: 10.1258/0956462991914285
146. Uldrick TS, Polizzotto MN, Aleman K, O'Mahony D, Wyvill KM, Wang V, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. (2011) 117:6977–86. doi: 10.1182/blood-2010-11-317610
147. Leroi C, Balestre E, Messou E, Minga A, Sawadogo A, Drabo J, et al. Incidence of Severe Neutropenia in HIV-Infected People Starting Antiretroviral Therapy in West Africa. PLoS ONE. (2017) 12:e0170753. doi: 10.1371/journal.pone.0170753
148. Buti M, Manzano ML, Morillas RM, García-Retortillo M, Martín L, Prieto M, et al. Randomized prospective study evaluating tenofovir disoproxil fumarate prophylaxis against hepatitis B virus reactivation in anti-HBc-positive patients with rituximab-based regimens to treat hematologic malignancies: The Preblin study. PLoS ONE. (2017) 12:e0184550. doi: 10.1371/journal.pone.0184550
149. Matłosz B, Kowalska JD, Bakowska E, Firlag-Burkacka E, Vassilenko A, Horban A. Discontinuation of tenofovir due to nephrotoxicity: insight into 12 years of clinical practice. Przegl Epidemiol. (2019) 73:249–55. doi: 10.32394/pe.73.24
150. Long M, Jia W, Li S, Jin L, Wu J, Rao N, et al. A single-center, prospective and randomized controlled study: Can the prophylactic use of lamivudine prevent hepatitis B virus reactivation in hepatitis B s-antigen seropositive breast cancer patients during chemotherapy? Breast Cancer Res Treat. (2011) 127:705–12. doi: 10.1007/s10549-011-1455-9
151. Lin GN, Peng JW, Xiao JJ, Liu DY, Xia ZJ. Hepatitis B virus reactivation in hepatitis B surface antigen seropositive patients with metastatic non-small cell lung cancer receiving cytotoxic chemotherapy: the efficacy of preemptive lamivudine and identification of risk factors. Med Oncol. (2014) 31:119. doi: 10.1007/s12032-014-0119-0
152. Quercia R, Perno CF, Koteff J, Moore K, McCoig C, St Clair M, et al. Twenty-Five Years of Lamivudine: Current and Future Use for the Treatment of HIV-1 Infection. J Acquir Immune Defic Syndr. (2018) 78:125–35. doi: 10.1097/QAI.0000000000001660
153. Bhamidipati PK, Fiala MA, Grossman BJ, DiPersio JF, Stockerl-Goldstein K, Gao F, et al. Results of a prospective randomized, open-label, noninferiority study of tbo-filgrastim (granix) versus filgrastim (neupogen) in combination with plerixafor for autologous stem cell mobilization in patients with multiple myeloma and non-hodgkin lymphoma. Biol Blood Marrow Transplant. (2017) 23:2065–9. doi: 10.1016/j.bbmt.2017.07.023
154. Matsue K, Kumagai K, Sugiura I, Ishikawa T, Igarashi T, Sato T, et al. Plerixafor for mobilization and collection of haematopoietic stem cells for autologous transplantation in Japanese patients with non-Hodgkin lymphoma: a randomized phase 2 study. Int J Hematol. (2018) 108:524–34. doi: 10.1007/s12185-018-2505-4
155. Martínez-Cuadrón D, Boluda B, Martínez P, Bergua J, Rodríguez-Veiga R, Esteve J, et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann Hematol. (2018) 97:763–72. doi: 10.1007/s00277-018-3229-5
156. Cashen AF, Rettig M, Gao F, Smith A, Abboud C, Stockerl-Goldstein K, et al. Phase I/II study of intravenous Plerixafor added to a mobilization regimen of granulocyte colony–stimulating factor in lymphoma patients undergoing autologous stem cell collection. Biol Blood Marrow Transplant. (2017) 23:1282–9. doi: 10.1016/j.bbmt.2017.04.024
157. Ahluwalia MS, Patton C, Stevens G, Tekautz T, Angelov L, Vogelbaum MA, et al. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J Neurooncol. (2011) 102:317–21. doi: 10.1007/s11060-010-0325-3
158. Hoover AC, Milhem MM, Anderson CM, Sun W, Smith BJ, Hoffman HT, et al. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: Results of a phase II clinical trial. Head Neck. (2015) 37:722–6. doi: 10.1002/hed.23664
159. Koolen SL, Oostendorp RL, Beijnen JH, Schellens JH, Huitema AD. Population pharmacokinetics of intravenously and orally administered docetaxel with or without co-administration of ritonavir in patients with advanced cancer. Br J Clin Pharmacol. (2010) 69:465–74. doi: 10.1111/j.1365-2125.2010.03621.x
160. Rudek MA, Moore PC, Mitsuyasu RT, Dezube BJ, Aboulafia D, Gerecitano J, et al. A phase 1/pharmacokinetic study of sunitinib in combination with highly active antiretroviral therapy in human immunodeficiency virus-positive patients with cancer: AIDS Malignancy Consortium trial AMC 061. Cancer. (2014) 120:1194–202. doi: 10.1002/cncr.28554
161. Sparano JA, Wiernik PH, Hu X, Sarta C, Henry DH, Ratech H. Saquinavir enhances the mucosal toxicity of infusional cyclophosphamide, doxorubicin, and etoposide in patients with HIV-associated non-Hodgkin's lymphoma. Med Oncol. (1998) 15:50–7. doi: 10.1007/BF02787345
162. Chaudary N, Pintilie M, Jelveh S, Lindsay P, Hill RP, Milosevic M. Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clin Cancer Res. (2017) 23:1242–9. doi: 10.1158/1078-0432.CCR-16-1730
163. Greco SJ, Patel SA, Bryan M, Pliner LF, Banerjee D, Rameshwar P. AMD3100-mediated production of interleukin-1 from mesenchymal stem cells is key to chemosensitivity of breast cancer cells. Am J Cancer Res. (2011) 1:701–15.
164. Rios A, Hsu SH, Blanco A, Buryanek J, Day AL, McGuire MF, et al. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience. (2016) 3:156–63. doi: 10.18632/oncoscience.311
165. Tabouret E, Tchoghandjian A, Denicolai E, Delfino C, Metellus P, Graillon T, et al. Recurrence of glioblastoma after radio-chemotherapy is associated with an angiogenic switch to the CXCL12-CXCR4 pathway. Oncotarget. (2015) 6:11664–75. doi: 10.18632/oncotarget.3256
166. Gridelli C, de Castro Carpeno J, Dingemans AC, Griesinger F, Grossi F, Langer C, et al. Safety and efficacy of bevacizumab plus standard-of-care treatment beyond disease progression in patients with advanced non-small cell lung cancer: the avaall randomized clinical trial. JAMA Oncol. (2018) 4:e183486. doi: 10.1001/jamaoncol.2018.3486
167. Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. (2015) 16:499–508. doi: 10.1016/S1470-2045(15)70127-0
168. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. (2012) 30:3499–506. doi: 10.1200/JCO.2012.42.8201
169. Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. (2013) 369:722–31. doi: 10.1056/NEJMoa1303989
170. Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the eastern cooperative oncology group study E3200. J Clin Oncol. (2007) 25:1539–44. doi: 10.1200/JCO.2006.09.6305
171. Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. (2011) 365:2484–96. doi: 10.1056/NEJMoa1103799
172. Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. (2014) 370:734–43. doi: 10.1056/NEJMoa1309748
173. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma avastin cisplatin pemetrexed study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. (2016) 387:1405–14. doi: 10.1016/S0140-6736(15)01238-6
174. Rini BI, Bellmunt J, Clancy J, Wang K, Niethammer AG, Hariharan S, et al. Randomized phase III trial of temsirolimus and bevacizumab versus interferon alfa and bevacizumab in metastatic renal cell carcinoma: INTORACT trial. J Clin Oncol. (2014) 32:752–9. doi: 10.1200/JCO.2013.50.5305
175. Alshangiti A, Chandhoke G, Ellis PM. Antiangiogenic therapies in non-small-cell lung cancer. Curr Oncol. (2018) 25:S45–58. doi: 10.3747/co.25.3747
176. Mawalla B, Yuan X, Luo X, Chalya PL. Treatment outcome of anti-angiogenesis through VEGF-pathway in the management of gastric cancer: a systematic review of phase II and III clinical trials. BMC Res Notes. (2018) 11:21. doi: 10.1186/s13104-018-3137-8
177. Ohno M, Miyakita Y, Takahashi M, Igaki H, Matsushita Y, Ichimura K, et al. Survival benefits of hypofractionated radiotherapy combined with temozolomide or temozolomide plus bevacizumab in elderly patients with glioblastoma aged ≥ 75 years. Radiation Oncol. (2019) 14:200. doi: 10.1186/s13014-019-1389-7
178. Aalders KC, Tryfonidis K, Senkus E, Cardoso F. Anti-angiogenic treatment in breast cancer: Facts, successes, failures and future perspectives. Cancer Treatment Rev. (2017) 53:98–110. doi: 10.1016/j.ctrv.2016.12.009
179. Nayak L, de Groot J, Wefel JS, Cloughesy TF, Lieberman F, Chang SM, et al. Phase I trial of aflibercept (VEGF trap) with radiation therapy and concomitant and adjuvant temozolomide in patients with high-grade gliomas. J Neurooncol. (2017) 132:181–8. doi: 10.1007/s11060-016-2357-9
180. Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomized phase 3 trial. Lancet Oncol. (2014) 15:1224–35. doi: 10.1016/S1470-2045(14)70420-6
181. Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. (2014) 384:665–73. doi: 10.1016/S0140-6736(14)60845-X
182. Arnold D, Fuchs CS, Tabernero J, Ohtsu A, Zhu AX, Garon EB, et al. Meta-analysis of individual patient safty data from six randiomized, placebo-controlled trials with the antiangiogenic VEGFR2-binding monoclonal antibody ramucirumab. Annals of Oncology. (2017) 28:2932–42. doi: 10.1093/annonc/mdx514
183. Lewin J, Khamly KK, Young RJ, Mitchell C, Hicks RJ, Toner GC, et al. A phase Ib/II translational study of sunitinib with neoadjuvant radiotherapy in soft-tissue sarcoma. Br J Cancer. (2014) 111:2254–61. doi: 10.1038/bjc.2014.537
184. Kleibeuker EA, ten Hooven MA, Verheul HM, Slotman BJ, Thijssen VL. Combining radiotherapy with sunitinib: lessons (to be) learned. Angiogenesis. (2015) 18:385–95. doi: 10.1007/s10456-015-9476-3
Keywords: tumor vasculature, angiogenesis, vasculogenesis, HIV-protease inhibitors, HIV-reverse transcriptase inhibitors, CXCR4 antagonists, AKT
Citation: Barillari G (2020) The Anti-Angiogenic Effects of Anti-Human Immunodeficiency Virus Drugs. Front. Oncol. 10:806. doi: 10.3389/fonc.2020.00806
Received: 09 August 2019; Accepted: 23 April 2020;
Published: 21 May 2020.
Edited by:
Thomas Daubon, UMR5095 Institut de Biochimie et Génétique Cellulaires (IBGC), FranceReviewed by:
Mohane S. Coumar, Pondicherry University, IndiaHrvoje Miletic, University of Bergen, Norway
Copyright © 2020 Barillari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Barillari, YmFyaWxsYXJAdW5pcm9tYTIuaXQ=